Drug-loaded nanoparticles induce gene expression in human pluripotent stem cell derivatives†
Virendra
Gajbhiye‡
a,
Leah
Escalante‡
a,
Guojun
Chen
b,
Alex
Laperle
a,
Qifeng
Zheng
b,
Benjamin
Steyer
a,
Shaoqin
Gong
*ab and
Krishanu
Saha
*a
aDepartment of Biomedical Engineering and Wisconsin Institute for Discovery, University of Wisconsin-Madison, Madison, WI 53715, USA. E-mail: sgong@engr.wisc.edu; ksaha@wisc.edu
bMaterial Science Program and Wisconsin Institute for Discovery, University of Wisconsin-Madison, Madison, WI 53715, USA
First published on 6th November 2013
Abstract
Tissue engineering and advanced manufacturing of human stem cells requires a suite of tools to control gene expression spatiotemporally in culture. Inducible gene expression systems offer cell-extrinsic control, typically through addition of small molecules, but small molecule inducers typically contain few functional groups for further chemical modification. Doxycycline (DXC), a potent small molecule inducer of tetracycline (Tet) transgene systems, was conjugated to a hyperbranched dendritic polymer (Boltorn H40) and subsequently reacted with polyethylene glycol (PEG). The resulting PEG–H40–DXC nanoparticle exhibited pH-sensitive drug release behavior and successfully controlled gene expression in stem-cell-derived fibroblasts with a Tet-On system. While free DXC inhibited fibroblast proliferation and matrix metalloproteinase (MMP) activity, PEG–H40–DXC nanoparticles maintained higher fibroblast proliferation levels and MMP activity. The results demonstrate that the PEG–H40–DXC nanoparticle system provides an effective tool to controlling gene expression in human stem cell derivatives.
Introduction
Human pluripotent stem cells (hPSCs) can mature into any part of the body and constitute an attractive cell source for tissue engineering, cell therapies, drug discovery and disease modeling.1 In addition to deriving hPSCs from embryos, recent advances in inducing hPSCs from routine blood samples and biopsies allow researchers to create patient-matched stem cells from virtually any patient.1 To capitalize on these expanded sources of pluripotent stem cells, new tools are required to rapidly induce hPSCs and precisely guide stem cell behavior through development to more mature cell types.2Tools to guide human stem cell behavior typically involve controlling gene expression in a spatiotemporal fashion. In development, complex spatiotemporal gene expression in stem cells is orchestrated by complex signaling interactions between cells in unique tissue-specific cell populations and microenvironments.2,3 While engineering extracellular cues in synthetic or natural biomaterials can lead to cell signaling that controls spatiotemporal gene expression,2 emerging techniques in genetic engineering provide a complementary route to spatiotemporal control triggered by small molecules or light.4–7 In some of these techniques, small molecules actuate proteins that bind DNA or RNA to alter transcription or translation thereby controlling gene expression. These triggers and genetic elements have been combined by synthetic biologists in microorganisms and mammalian cells to achieve complex spatiotemporal control of gene expression and cell transitions.8
Doxycycline (DXC) is a small molecule routinely used to induce transgene expression in cell culture, tissues, and whole organisms.9–11 DXC is the most potent analogue for the tetracycline (Tet) inducible system and permeates through the cell and nuclear membranes.12,13 Inside the cell, DXC binds to modified versions of the Tet repressor fused to a transcriptional activation domain (rtTA) to induce transgenes under the control of the Tet DNA promoter.12,13 In addition to binding to Tet proteins, DXC can bind to matrix metalloproteinases (MMPs) and is one of the more potent matrix metalloproteinase inhibitors in the Tet family.14 MMPs control diverse biological processes, ranging from extracellular matrix remodeling to autocrine signaling by growth factors,15 and these processes may enhance or interfere with DXC-induced transgene function.
Nanoparticle-conjugation presents an attractive alternative to current routes of controlling the activity of DXC in time and space. Spatiotemporal control has been achieved with light by “caging” DXC with a photosensitive protecting group.16,17 DXC has also been loaded into microparticles and hydrogels18 to achieve controlled release in various settings but not to gain specific control of transgene expression. In contrast to microparticles, hydrogels and implants, nanoparticles can be judiciously engineered to provide multiple functionalities including controlled drug release, specific cell targeting, and enhanced cellular uptake. These functionalities have already been combined in drug nanocarriers to treat major health threats such as cancers19 and vascular diseases,20 and to promote tissue regeneration.21 In addition, various reactive functional groups can also be conveniently conjugated onto the surface of the nanoparticles allowing further immobilization of these nanoparticles on various templates. Spatiotemporal chemistry utilizing such functionality could be utilized in advanced manufacturing or solid tissue engineering applications where light or other stimuli offer poor penetration depth or control.22
To gain additional functional handles on drug release and thus gene expression, here we report the conjugation of DXC to a hyperbranched biodegradable and biocompatible polymer nanoparticle, i.e., Boltorn H40. The fourth-generation hyperbranched polymer nanoparticle offers a large number of peripheral functional groups which can be used to conjugate a large number of DXC molecules as well as PEG arms, thereby making the drug–polymer conjugate water soluble. The PEG–H40–DXC drug–polymer conjugate nanoparticle demonstrated pH-sensitive drug release behavior and was shown to control transgene expression in stem cell derived fibroblasts. The PEG–H40–DXC nanocarrier provides a novel tool to controlling gene expression in human stem cells and their derivatives. Further work with these nanoparticle systems could produce patterned hPSC-derived cell cultures, offering researchers the ability to investigate these cells in environments that optimally recapitulate complexities found within developing and diseased human tissues.
Experimental
Materials
Hyperbranched polymer, Boltorn H40 (64 terminal hydroxyl groups per molecule theoretically), was obtained from Perstorp Polyols Inc., USA. N-Hydroxy succinimide (NHS), 1,3-dicyclohexylcarbodiimide (DCC), succinic anhydride, and 4-dimethylamino pyridine (DMAP) were purchased from Acros and used without further purification. Doxycycline hyclate was purchased from Sigma-Aldrich, USA. MPEG–COOH (MW, 3500) was obtained by JenKem Technology, USA. All other chemicals used were of analytical reagent grade.Purification of H40–(OH)64
1 g of H40 was dissolved in 10 mL acetone overnight. Then, 10 mL of ethyl ether was poured into it and allowed to precipitate for 12 h and re-crystallized three times using a mixture of ethyl ether and THF.23 Purified H40 was vacuum-dried.Synthesis of 32(HOOC)–H40–(OH)32 or carboxylation of H40–OH
Half of the hydroxyl groups per H40–OH (i.e., 50% in molar, or 32 in theory) were carboxylated through a reaction with succinic anhydride using DMAP as the catalyst (Scheme 1). H40–OH (28.3 mg, 1 μmol) and succinic anhydride (38.4 mg, 38.4 μmol) were dissolved in anhydrous THF (5 mL) and reacted in presence of DMAP.24 The molar ratio between succinic anhydride and DMAP was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1. The reaction was carried out in the mixture of TEA and anhydrous THF (the volume ratio between TEA:THF was 0.01) for 48 h at room temperature under inert atmosphere. The product was precipitated using diethyl ether and vacuum-dried.
:0.1. The reaction was carried out in the mixture of TEA and anhydrous THF (the volume ratio between TEA:THF was 0.01) for 48 h at room temperature under inert atmosphere. The product was precipitated using diethyl ether and vacuum-dried.
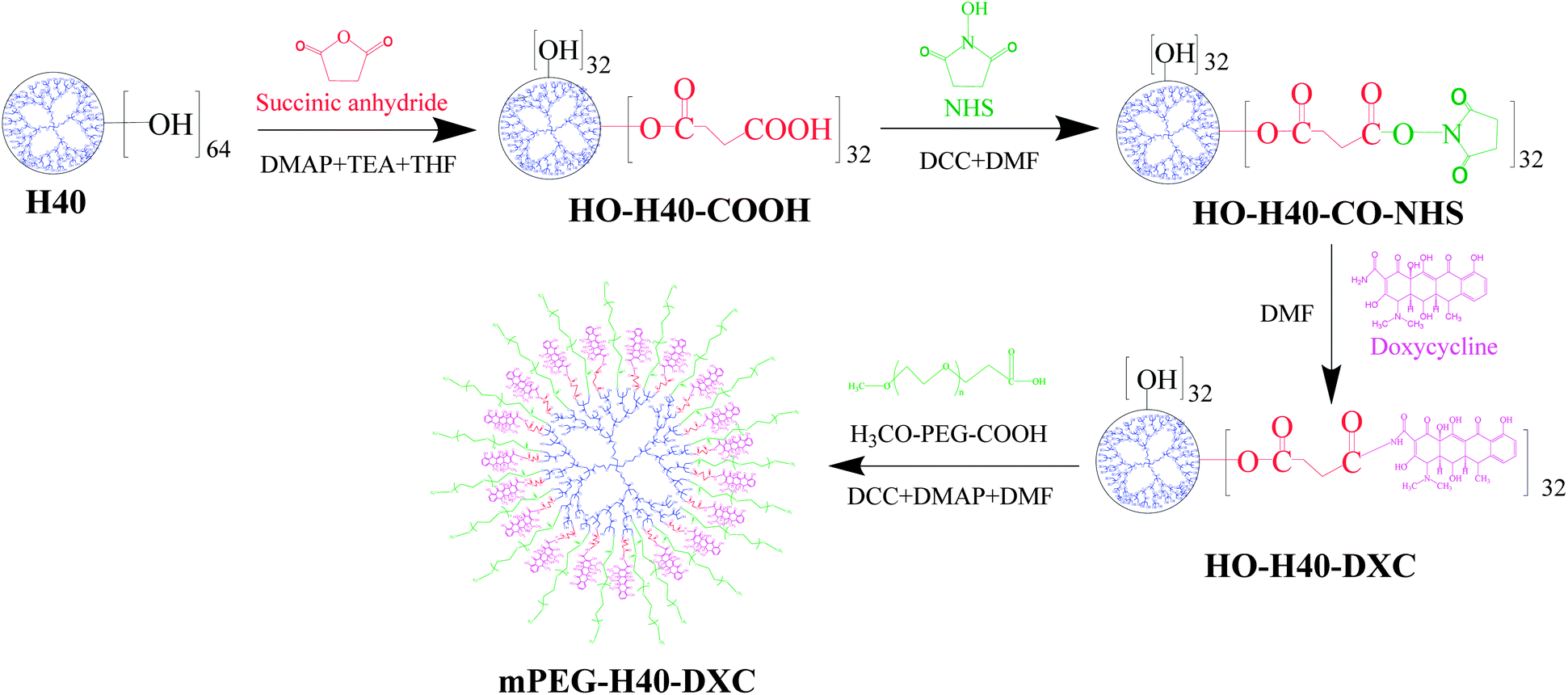 | ||
| Scheme 1 Synthesis scheme of PEG–H40–DXC. | ||
Synthesis of 32(OH)–H40–NHS ester
N-Hydroxysuccinimide ester of 32(HOOC)–H40–(OH)32 was prepared according to a reported method.2532(HOOC)–H40–(OH)32 (28.3 mg, 1 μmol), DCC (7.8 mg, 38 μmol) and NHS (4.3 mg, 38 μmol) was dissolved in anhydrous DMF and stirred for 48 h. After removing dicyclohexylurea by filtration, the product was vacuum-dried to remove solvent.Conjugation of doxycycline (DXC) or synthesis of 32(OH)–H40–DXC
Doxycycline hyclate (51.2 mg, 0.1 mM) was stirred with TEA (1:1.2) in 5 mL DMF for 2 h. Then, H40–NHS ester (7 mg, 2.5 μM) was added and stirred for 48 h at room temperature and under an inert atmosphere. Thereafter, the product was dialyzed against 2 L DMF for 48 h using a dialysis membrane (MWCO 5000 Da) to remove un-conjugated drug and TEA salt. The product was then dried using a rotary evaporator.
Synthesis of PEG–H40–DXC
mPEG–COOH (140 mg, 40 μmol), DCC (8.21 mg, 40 μmol) and DMAP (4.7 mg, 40 μmol) were dissolved in anhydrous DMF (10 mL). Then 32(OH)–H40–DXC (13 mg, 1 μmol) in anhydrous DMF (5 mL) was added drop-wise into the reaction mixture. The reaction was carried out at room temperature for 24 h under stirring. After the by-product, dicyclohexylcarbodiurea, was removed by filtration, the product was precipitated with cold diethyl ether. The precipitate was then dialyzed against 2 L of de-ionized water for 48 h using a dialysis membrane (MWCO 15 kDa) and then freeze dried.Nanoparticle characterization
1H NMR spectra of the various samples were recorded on a Bruker DPX 300 spectrometer at 25 °C using deuterated acetone (acetone-d6) and deuterated DMSO (DMSO-d6) as the solvents with tetramethylsilane as an internal standard. The DXC concentration in various samples was measured and quantified at 273 nm using Varian Cary 100 Bio UV-visible spectrophotometer based on a DXC calibration curve. Nanoparticle size was determined by dynamic light scattering (DLS, Beckman Coulter PCS submicron particle size analyzer, USA) with angle detection of 90° and at a concentration of 0.05 mg mL−1. For transmission electron microscope (TEM) analysis, a drop of the nanoparticle solution (0.05 mg mL−1) containing 1.0 wt% phosphotungstic acid was deposited on a 200 mesh copper grid coated with carbon and dried at room temperature. The shape and size of the nanoparticles were observed at 75 kV with a TEM (Hitachi H-600, Japan).In vitro drug release study
In vitro drug release studies were carried out using a glass jar at 37 °C in either acetate buffer (pH 5.0) or phosphate buffer (pH 7.4) solutions. 5 mg of PEG–H40–DXC was dispersed in 5 mL of medium and placed in a dialysis bag. The dialysis bag was immersed in 50 mL of the release medium and kept in a horizontal laboratory shaker (100 rpm) under constant temperature (37 ± 1 °C). Samples of 3 mL volume were periodically removed and the same volume of fresh medium was replaced. The amount of released DXC was assayed using a UV-visible spectrophotometer at 273 nm. The drug release experiments were carried out in triplicate.Cell culture
Human stem-cell-derived secondary C1 fibroblasts, containing DXC-inducible lentiviral transduction of transcription factors Oct4, Sox2 and Klf4,26 and IMR90 fibroblasts (ATCC) were cultured in fibroblast medium [DMEM supplemented with 10% fetal bovine serum (FBS, Invitrogen), 1 mM L-glutamine (Invitrogen), 1% nonessential amino acids (Invitrogen) and Penicillin/Streptomycin (Invitrogen)]. Cells were maintained between 15 and 25 passages with media changes every 2 days, and passaging every 6–7 days with TrypLE (Invitrogen). DXC- or PEG–H40–DXC-treated cells were cultured by adding 5 μM DXC or PEG–H40–DXC solution, respectively, to medium prior to media changes. PEG–H40–DXC concentrations listed in all cell studies denote the concentration of DXC only without incorporating the concentration of the nanoparticle. Hence, to add 5 μM of drug, we added 2 μg mL−1 of DXC or 18.2 μg mL−1 of PEG–H40–DXC, which contains the same amount of DXC at 11% drug loading (see Results for drug loading data). The DXC and PEG–H40–DXC PBS stock solutions with a DXC concentration of 2 mg mL−1 (or 5 mM) were prepared via 5 min sonication (Fischer Scientific FS30D) at room temperature shortly before use. To ensure uniform distribution, the PEG–H40–DXC solution was vortexed for 2 min prior to addition.Flow cytometry
Cells were passaged with TrypLE then fixed in 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.1% Triton X-100 (Invitrogen) and 10% FBS in PBS for 30 min, and then stained overnight at 4 °C with fluorophore-conjugated primary antibodies: Alexa 488 anti-Sox2 [pre-diluted by BD Biosciences for use of 5 μL for 106 cells in a 100 μL experimental sample (BD Biosciences)], and PE anti-Oct3/4 [Pre-diluted by BD Biosciences for use of 20 μL for 106 cells in a 100 μL experimental sample (BD Biosciences)]. Data was acquired with an Accuri CSampler C6 (BD Biosciences). Isotype control antibodies PE-IgG1 and Alexa 488-IgG2a (BD Biosciences) were used as negative controls. Filter compensations were set from single-stained non-treated cells and validation beads (BD Biosciences). Plots were generated in FlowJo (Tree Star, Inc.).Imaging
Samples were washed with PBS, fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.1% Triton X-100 and 10% FBS in PBS for 30 min, and then stained overnight at 4 °C with fluorophore-conjugated primary antibodies: Alexa 488 anti-Sox2 and PE anti-Oct3/4 (dilutions as previously stated). The nuclei were stained with Hoechst 33342 (Life Technologies). Images were taken with Nikon Eclipse Ti.Cell proliferation analysis
Secondary C1 and IMR90 fibroblasts were plated on a 24-well plate (75000 cells per well), cultured in fibroblast medium, and, upon reaching 60% confluence, treated with 5 μM DXC, PEG–H40, or PEG–H40–DXC, respectively, for 72 h. Identical cell densities were observed at the center to the periphery of the wells without any aggregation. A Click-iT EdU cell proliferation assay (Life Technologies) was performed, according to the manufacturer's protocol, followed by staining overnight with Alexa 488 anti-Sox2 and PE anti-Oct3/4 (dilutions as previously stated), and Hoechst 33342. EdU positive and negative cells were determined by fluorescent imaging.
MMP assays
Fluorescent MMP2/9 substrate I (EMD Millipore # 444215) was re-suspended in DMSO to generate a stock solution of 1 mM. 3 μL of this stock solution was added to 300 μL of 0.5 mg mL−1 collagenase type IV (Invitrogen 17104-019) solution in PBS with 5 μM DXC, PEG–H40, or PEG–H40–DXC (with an equivalent amount of 5 μM DXC) for a final fluorescent substrate concentration of 10 μM. PBS was used here in place of fibroblast media because the media contained components that interfered with fluorescent measurements. The resulting mixtures were then incubated for 18 hours at 37 °C in a 96-well plate (Corning; black with clear bottom). Sample fluorescence was measured using a Tecan Infinite M1000 microplate reader at 290/360 nm.Results
Synthesis and characterization of PEG–H40–DXC
Methoxy–PEG–H40–DXC was synthesized via a two-step procedure as shown in Scheme 1. In the first step, H40–OH was partially converted to carboxylated H40 followed by conversion into NHS ester and then conjugated with DXC molecules. Every reaction step was followed by a purification step to remove un-reacted chemicals and by-products. To confirm conjugation, 1H NMR spectra was collected for H40–OH and 32(HOOC)–H40–(OH)32 (Fig. S-1a and b in the ESI†). Compared with H40–OH, a new proton peak around 2.7 ppm appeared in the 1H NMR spectrum of carboxylated H40–OH, which can be assigned to the methylene protons from the succinic acid groups (Fig. S-1 in the ESI,† peak d). In addition, the proton peak around 3.5 ppm assigned to terminal –CH2–OH groups (peak b) of the H40 molecules noticeably decreased. These results indicate that the esterification reaction shown in Scheme 1 between succinic anhydride and the hydroxy groups of H40 occurred successfully.Synthesis of 32(OH)–H40–NHS and 32(OH)–H40–DXC was also confirmed via1H NMR spectroscopy. The 1H NMR spectrum of 32(OH)–H40–NHS ester shows the proton peak at 2.6 ppm (Fig. S-1c in the ESI,† peak e), which clearly suggested the formation of NHS ester of the partially carboxylated H40–OH. Fig. S-2 in the ESI† shows the 1H NMR spectrum of 32(OH)–H40–DXC. The peak around 1.0 ppm was attributed to the H40 core. The peaks of DXC were assigned as follows: DXC-1.54 ppm (C-6, CH3), 2.56 ppm (H-5a), 2.71 (H-6A), 2.87 ppm (C-4 N(CH3)2), 3.39–3.43 ppm (H-5), 4.62 ppm (H-4), 5.7 ppm (C-5, OH), 6.86 ppm (H-9), 6.98 ppm (H-7), 7.60 ppm (H-8), 9.15 ppm (C-2, CONH), 9.36 ppm (C-2, CONH).16 All the peaks shown in Fig. S-2 in the ESI† demonstrate the formation of H40–DXC.
mPEG–COOH was subsequently conjugated with 32(OH)–H40–DXC to make the resulting PEG–H40–DXC water-soluble. In the 1H NMR spectrum of PEG–H40–DXC, the peaks at 3.52 ppm (Fig. 1 peak e) and 3.4 ppm were observed due to the methylene protons of oxyethylene units and methyl protons of MPEG, respectively, suggesting the successful conjugation of PEG onto 32(OH)–H40–DXC. The DXC loading content of the PEG–H40–DXC nanoparticles, determined by the UV absorption of DXC at 273 nm,27 was 11 wt%. The morphology and size distribution of the PEG–H40–DXC nanoparticles were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). Fig. 2a shows the distribution of the hydrodynamic diameter of the PEG–H40–DXC nanoparticles measured by DLS, with an average diameter of 77 nm. Fig. 2b displays a representative TEM image of the PEG–H40–DXC nanoparticles. The nanoparticles exhibited a spherical shape with a diameter ranging from 30 to 40 nm. The size measured via TEM was smaller than that measured by DLS, because TEM measures the diameter of the dried nanoparticles, whereas DLS determines the hydrodynamic diameters of the nanoparticles in solution.
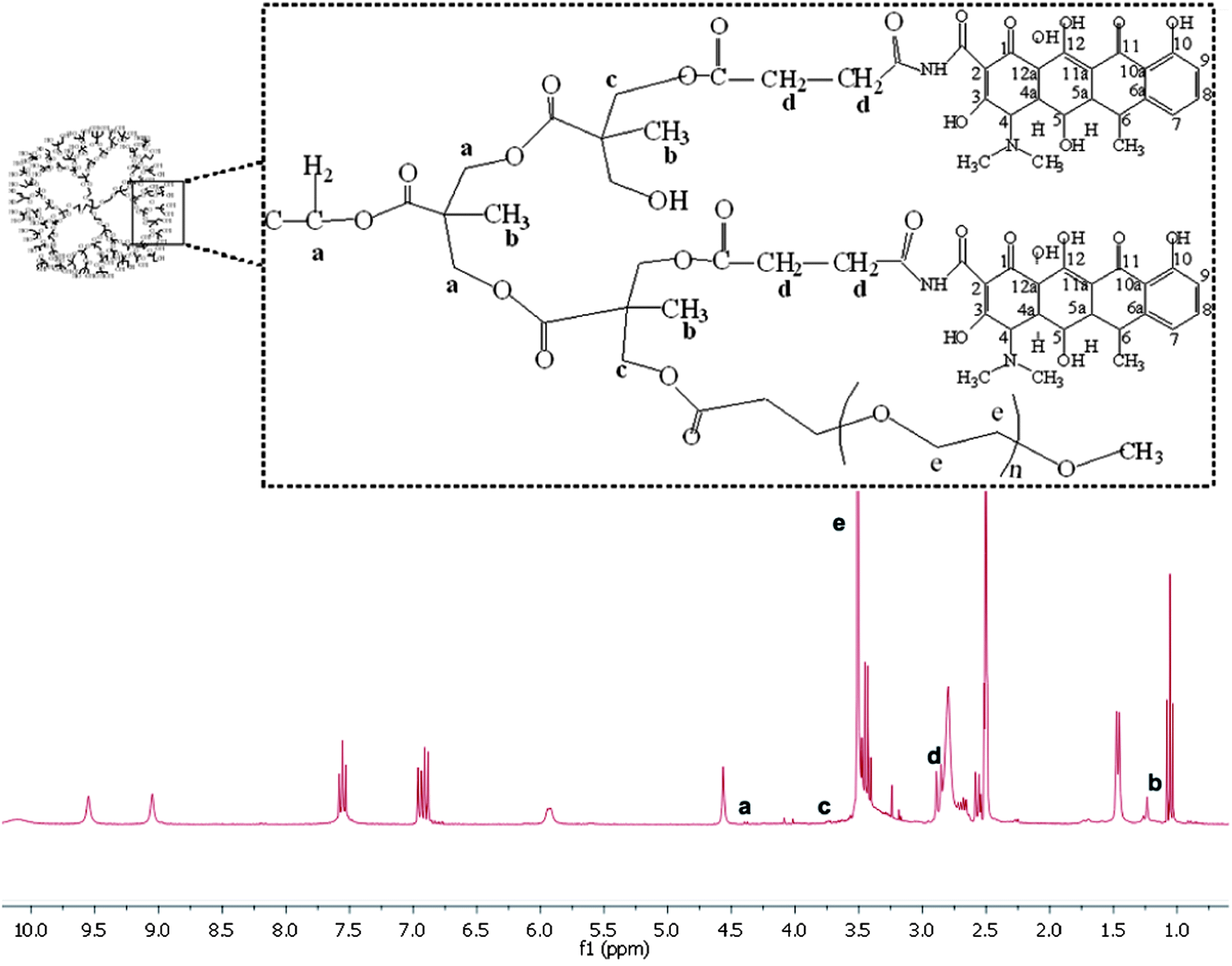 | ||
| Fig. 1 1H NMR spectra of PEG–H40–DXC in DMSO-d6. | ||
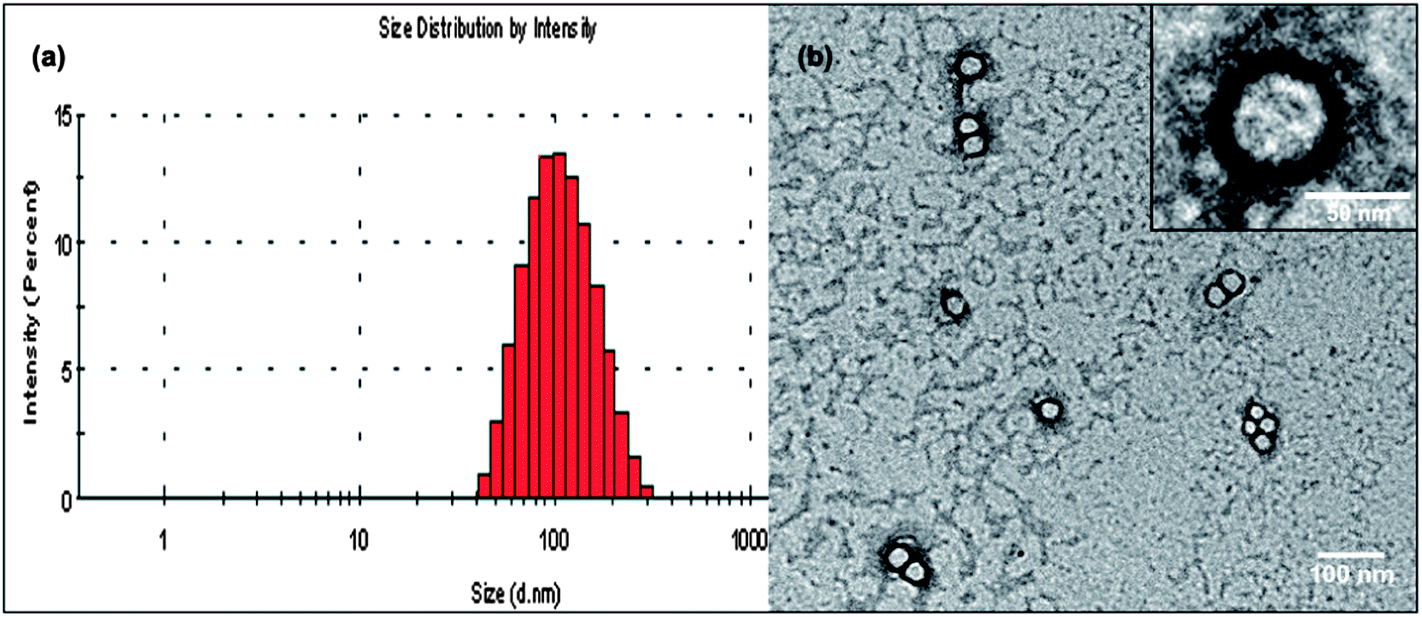 | ||
| Fig. 2 (a) Size distribution of the PEG–H40–DXC nanoparticles as measured by DLS. (b) TEM image of the PEG–H40–DXC nanoparticles. | ||
In vitro drug release behavior of the PEG–H40–DXC nanoparticles
In order to analyze the pH-sensitive drug release behaviors of the PEG–H40–DXC nanoparticles, in vitro drug release studies were carried out under simulated physiological conditions at pH 7.4 in PBS buffer and in intracellular acidic conditions at pH 5.0 in acetate buffer at 37 °C. Fig. 3 shows the pH-dependent drug release profile of the PEG–H40–DXC nanoparticles. The DXC release rate was significantly lower at pH 7.4 than pH 5.0. At pH 5.0, 28.8 ± 0.6% of DXC release was observed after 12 h, however, at pH 7.4, only 5.6 ± 0.2% of DXC release was observed. At 168 h, DXC release was found to be 72.4 ± 1.01% at pH 5.0, whereas only 24.3 ± 0.85% DXC release was found at pH 7.4. These results indicate low pH-triggered release of DXC in acidic conditions.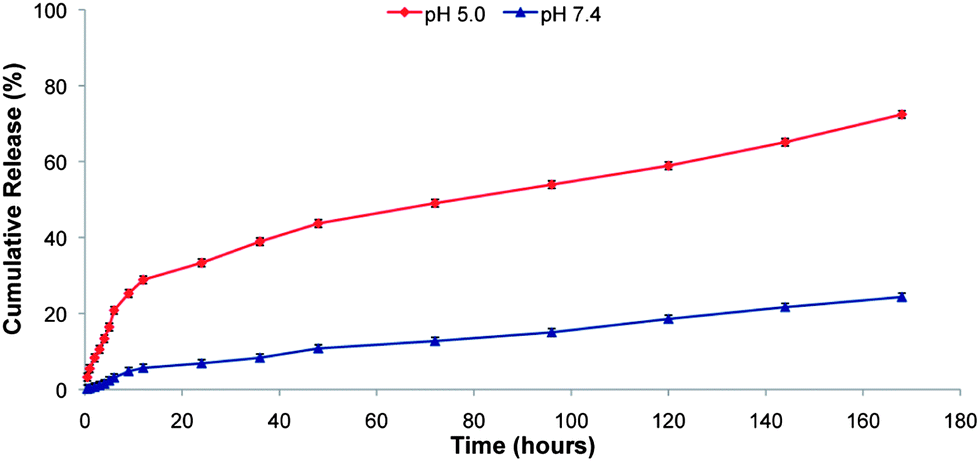 | ||
| Fig. 3 In vitro DXC release profiles of the PEG–H40–DXC nanoparticles measured at pH 5.0 and 7.4. | ||
Extracellular MMP inhibition assay
In the extracellular space, DXC binds MMPs causing inhibition of proteolytic activity (Fig. 4a, right). To test whether DXC in media outside of cells could inhibit MMPs, an MMP2/9 activity assay was conducted in the presence of DXC or PEG–H40–DXC. Incubation of MMPs with free DXC at 5 μM significantly inhibited MMP activity to a greater extent than incubation with 5 μM PEG–H40–DXC or PEG–H40 (Fig. 4b). Similar results were seen at 2 and 18 hours of incubation (data not shown). These results indicate that DXC conjugated onto the nanoparticles allows for greater MMP activity and protects extracellular MMPs from inhibition by free DXC.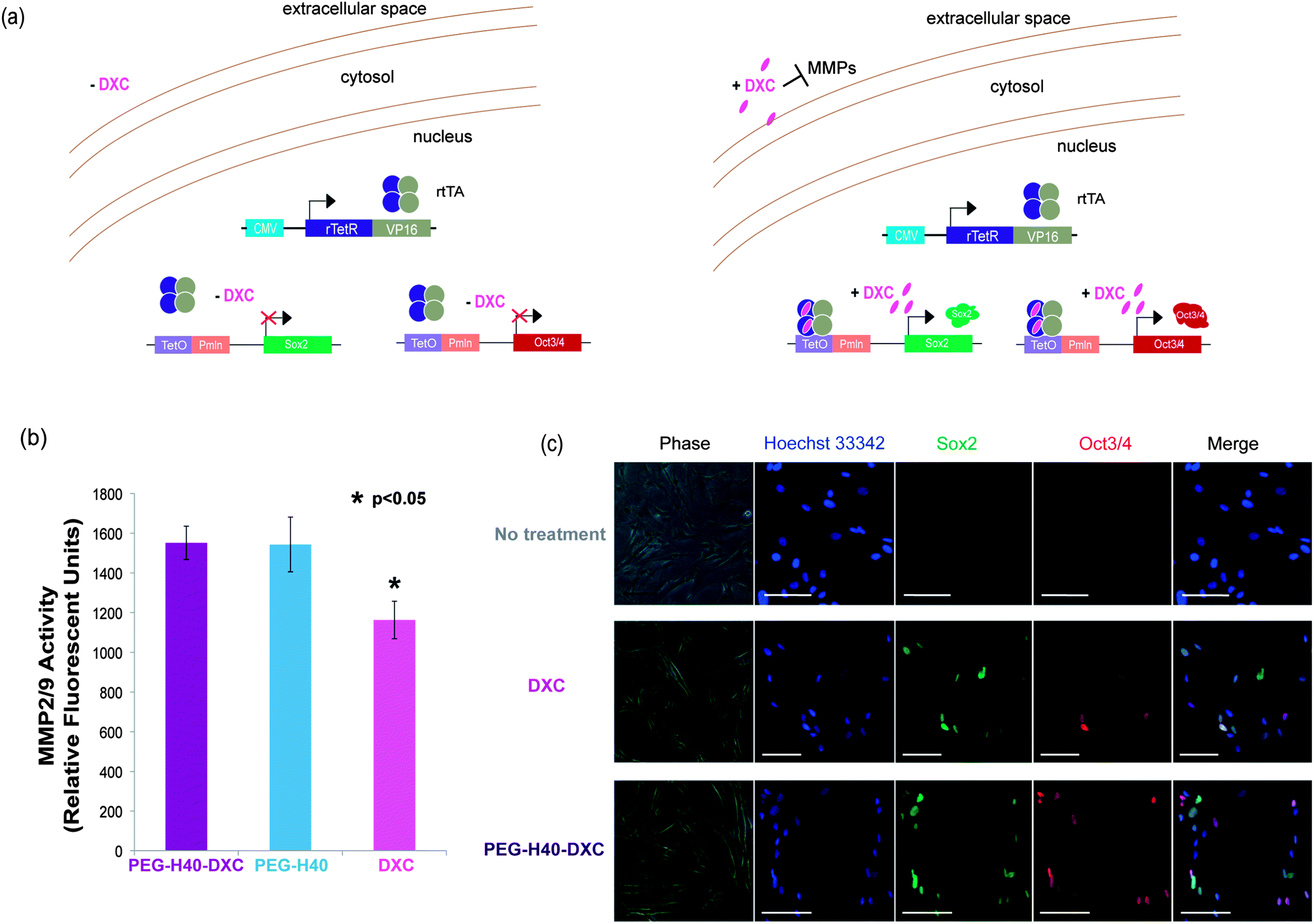 | ||
| Fig. 4 DXC conjugated onto the PEG–H40–DXC nanoparticles specifically activates intracellular targets. (a) Schematic of Tet-on DXC-inducible gene circuit for expression of Oct3/4 and Sox2 proteins in secondary C1 fibroblasts, noting MMP inhibition due to additions of DXC. (b) MMP2/9 activity as measured by fluorescence intensity of a cleavable, fluorogenic substrate. The substrate is fluorescent only after being cleaved by an MMP. Asterisk represents a statistically significant difference between DXC-treated and PEG–H40–DXC treated secondary C1 fibroblasts with a p < 0.05, two-tailed Student t-test. (c) Induced protein expression in stem-cell derived fibroblasts is localized to nucleus. Fibroblasts harbor inducible gene circuits that express Oct3/4 and Sox2 proteins upon addition of DXC or PEG–H40–DXC. Fibroblasts were treated with normal media, DXC, or PEG–H40–DXC with an equivalent DXC concentration of 5 μM. Immunocytochemistry against Oct3/4 and Sox2 were performed prior to imaging to distinguish induced gene protein products. Scale bar represents 50 μm. | ||
Gene expression studies
To test the intracellular activity of the PEG–H40–DXC nanoparticles, human stem cell-derived secondary C1 fibroblasts26 were cultured with DXC or PEG–H40–DXC. These fibroblasts contain a Tet-on DXC-inducible gene circuit for expression of Oct3/4 and Sox2 proteins (Fig. 4a). Treatment of fibroblasts with DXC or PEG–H40–DXC at an equivalent DXC concentration of 5 μM for 24 h resulted in high levels of properly-localized, nuclear Oct3/4 and Sox2 proteins in a subset of cells, as determined by immunocytochemistry (Fig. 4c). The variable level of expression within each fibroblast cell population reflects previously described noise in gene expression upon addition of exogenous inducer.26,28 No significant difference in signal intensity or prevalence was observed between DXC- and PEG–H40–DXC-treated cells. Oct3/4 and Sox2 proteins were not detected in cells cultured with fibroblast media alone (Fig. 4c).Flow cytometry was used to quantify levels of protein produced from induced gene expression within single cells. The distribution of Oct3/4 and Sox2 proteins induced in non-treated, PEG–H40 treated, free DXC-treated, and PEG–H40–DXC treated cells (Fig. 5a and b) was monitored by plotting the number of single cells that stained positive for Oct3/4 only or Sox2 only (“single positive”, upper left and lower right quadrants in Fig. 5a and b) or both Oct3/4 and Sox2 (“double positive”, upper right quadrant). Thresholds for positive gating were set from cells cultured in normal fibroblast medium (Fig. 5a). Similar percentages of cells contained induced gene protein products after 24 h of incubation with DXC or PEG–H40–DXC (equivalent DXC concentration 5 μM), regardless of whether single-positive, double-positive or double-negative cell populations were compared (Fig. 5b). For instance, 86.96 ± 3.56% cells were found positive for one or both induced gene protein product after DXC treatment, while 86.20 ± 1.65% cells were found positive after PEG–H40–DXC treatment (Fig. 5b). No significant difference in signal intensity was observed between DXC and PEG–H40–DXC treated cells.
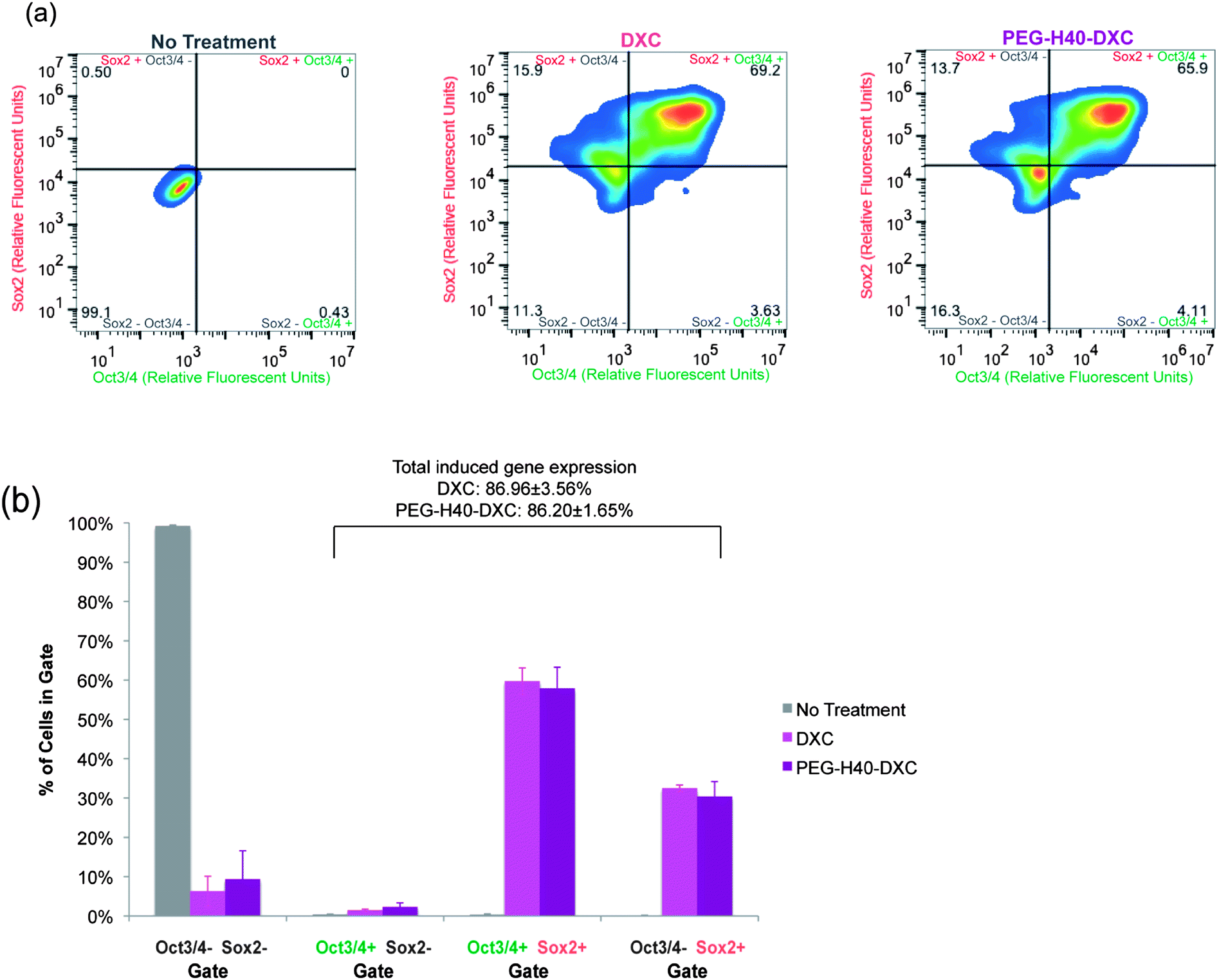 | ||
| Fig. 5 Quantification of DXC and PEG–H40–DXC nanoparticle-induced gene expression. (a) Representative heat maps of induced protein expression in stem-cell derived human fibroblasts as measured by flow cytometry, which allows single-cell quantification of protein levels. Immunocytochemistry as described in Fig. 4c was performed prior to analysis. Lines indicate thresholds for positive and negative signals above background staining, and colors indicate number of cells illustrated by pseudocolor dot plots smoothened by locally weighted polynomial regression, which allows simultaneous information of rare events (dots) and high-frequency areas with dots of different color. Secondary C1 fibroblasts were treated with normal media, DXC, or PEG–H40–DXC with an equivalent DXC concentration of 5 μM for 24 h prior to immunocytochemistry. (b) Percent of cells in each quadrant as described in part (a). | ||
Free DXC or PEG–H40–DXC treatment for 12 to 144 h generated similar dynamics in gene expression and dose response (Fig. 6). The percent of cells positive for one or both induced gene protein product over time was similar, as determined by flow cytometry (Fig. 6a). Peak percent positive was observed at 24 hours, with a dip observed at 48 hours followed by a rise again at 72 hours (Fig. 6a). These dynamics were similar between DXC and PEG–H40–DXC, likely reflecting intrinsic properties of the Tet-on system in the C1 fibroblasts. PEG–H40 nanoparticles without any DXC conjugation showed no expression of either induced gene protein product. Treated cells exhibited no statistically significant difference in dose response to DXC and PEG–H40–DXC, with an IC50 of 0.1 μM for both DXC and PEG–H40–DXC (Fig. 6b). Significant cell toxicity was observed at 400 μM of PEG–H40–DXC (data not shown).
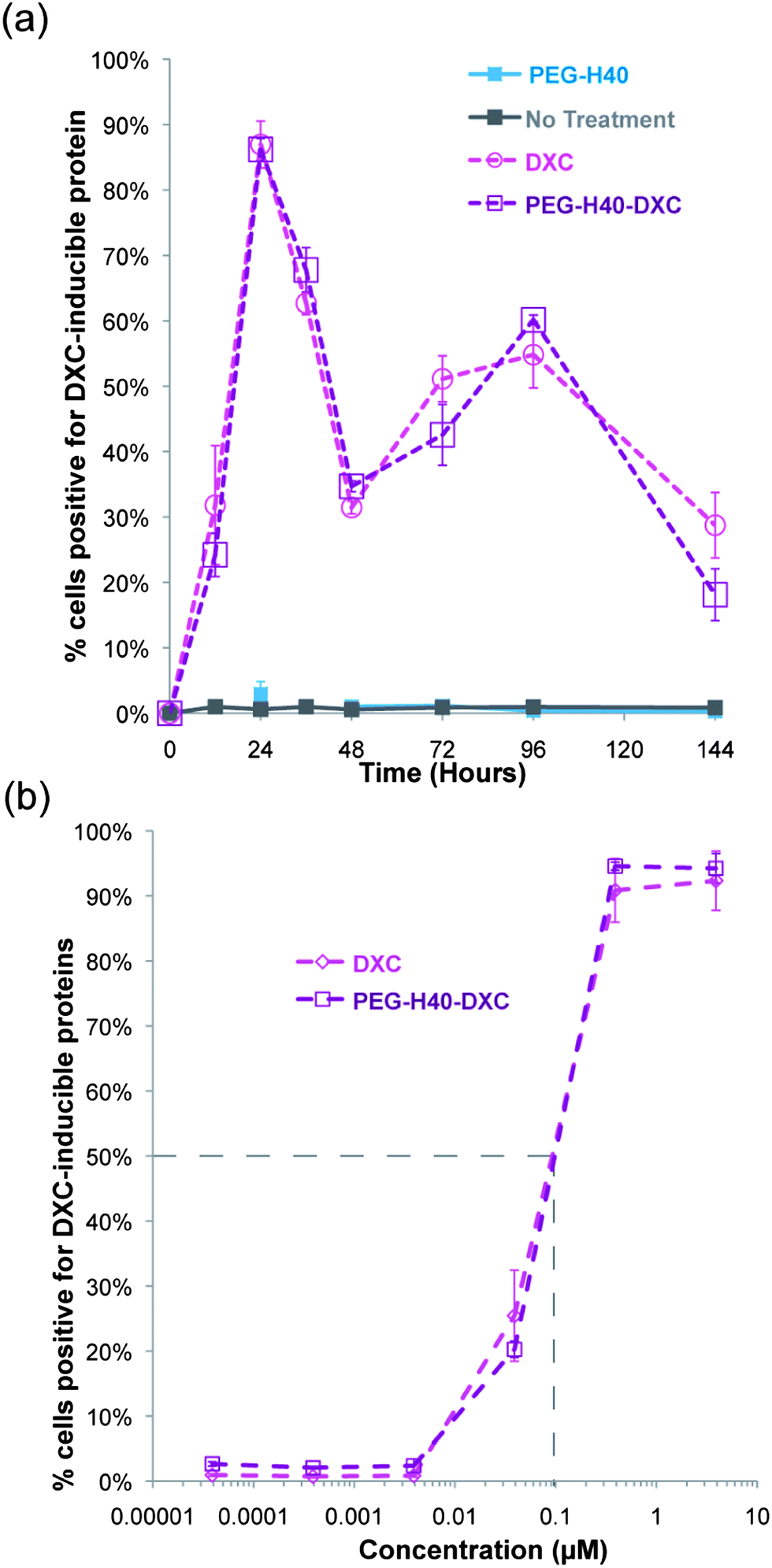 | ||
| Fig. 6 Temporal and dose dependence of DXC and PEG–H40–DXC. (a) Percent of fibroblasts with induced gene expression over time. Summary of flow cytometry data on fibroblasts treated with normal media, PEG–H40, DXC, or PEG–H40–DXC with an equivalent DXC concentration of 5 μM. Positive indicates shift out of lower left quadrant in Fig. 5a and possession of at least one positive signal for either Sox2 or Oct4. Thresholds are indicated in Fig. 5a. (b) Dose response curve of percent positive secondary C1 fibroblasts after 24 h incubation with DXC or PEG–H40–DXC. IC50 values indicate a similar response for DXC and PEG–H40–DXC. | ||
Cell proliferation assays
Poor fibroblast cell morphology was observed at prolonged exposures to DXC compared to cells treated with conventional media without drug (Fig. 7a). In the DXC treated condition, fibroblasts lost their fusiform morphology and produced long, thin processes (indicated by arrowheads in Fig. 7a), which are characteristic of cell stress and senescence. These effects on cell morphology were independent of transgene expression, as primary fibroblasts without the Tet-on system also exhibited poor morphology upon DXC treatment, similar to the stem-cell-derived fibroblasts that induced Oct3/4 and Sox2. Fibroblasts treated with PEG–H40–DXC or PEG–H40, however, exhibited cell morphology similar to control cells (Fig. 7a). For all the samples, no elevated cell death was observed.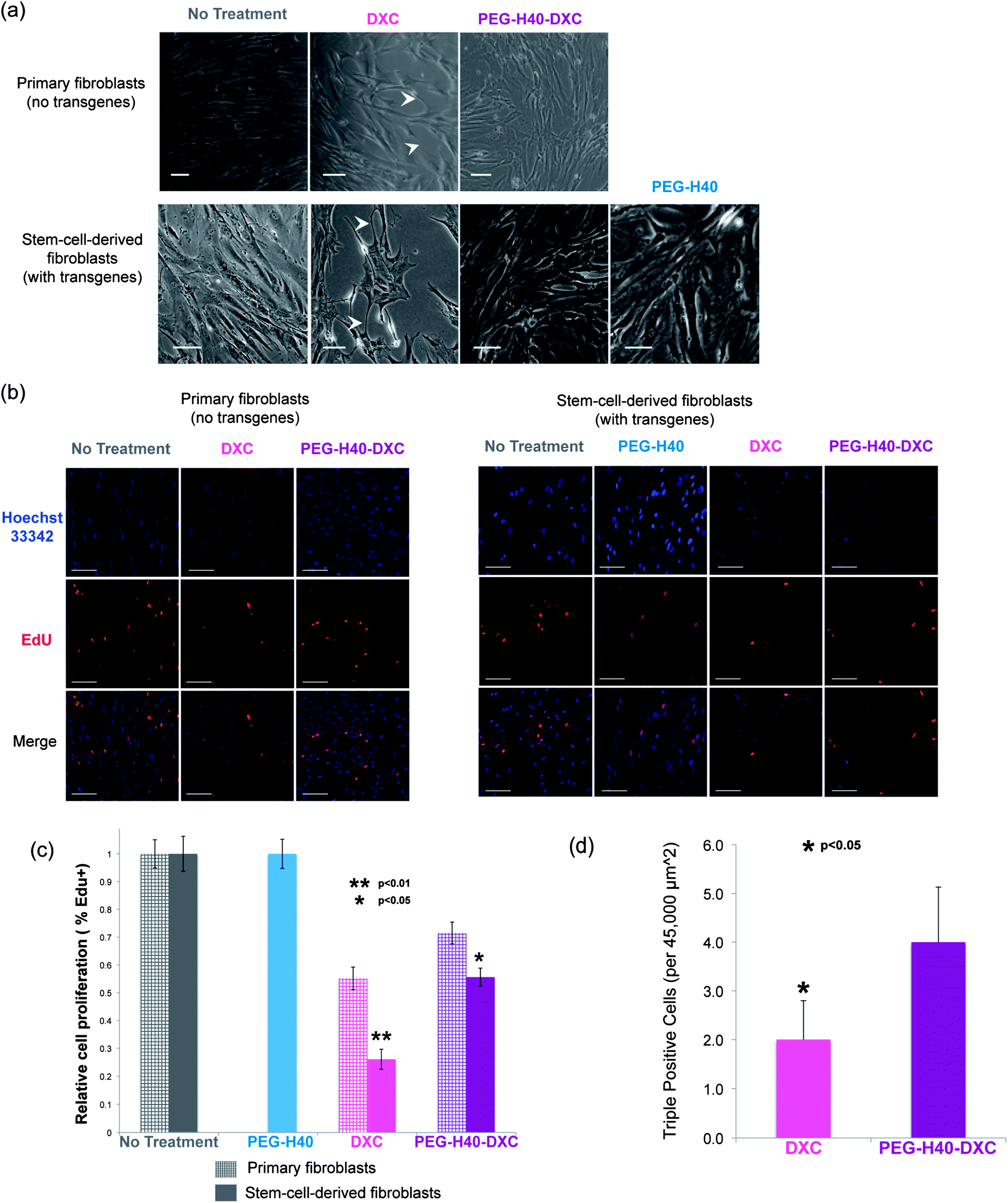 | ||
| Fig. 7 Fibroblasts treated with PEG–H40–DXC grow faster in comparison with free DXC. (a) Phase contrast images of two different fibroblast lines showing different morphology with DXC treatment, as compared to non-treated, PEG–H40 treated, and PEG–H40–DXC treated cells. White arrowheads indicate long, thin processes that connect fibroblasts in the DXC condition. Cell proliferation increases and more closely resembles non-treated cells with PEG–H40–DXC. Fibroblasts were treated for 72 h. Scale bar represents 50 μm. Treatment of primary fibroblasts (IMR90) with PEG–H40 was not performed. (b) Fluorescent images of proliferative cells of the fibroblast lines in part (a). A Click-iT EdU assay was performed prior to imaging. EdU is incorporated during DNA synthesis, and Click chemistry is utilized for fluorescent detection. Scale bar represents 50 μm. (c) Proliferation quantification via Click-iT EdU assay for the fibroblasts lines in part (a). Double asterisk represents a statistically significant difference between DXC-treated secondary C1 fibroblasts and all other conditions with a p < 0.01, two-tailed Student t-test. Single asterisk represents a statistically significant difference between IMR90 and secondary C1 fibroblasts treated with PEG–H40–DXC with a p < 0.05, two-tailed Student t-test. (d) Bar graph of the number of “triple-positive” cells per representative fluorescent image (45000 μm2) that are EdU+ (fluorescently detected EdU via Click-iT EdU assay) and OS+ (positive staining for Oct3/4 and Sox2). Asterisk represents a statistically significant difference between DXC and PEG–H40–DXC treatment with a p < 0.05, two-tailed Student t-test. | ||
Proliferation of DXC-treated cells also decreased significantly compared to PEG–H40–DXC-treated cells (Fig. 7b and c). Proliferative cells were determined by their incorporation of a modified nucleoside, 5-ethynyl-2′-deoxyuridine (EdU), during DNA synthesis. Using immunocytochemistry, Click chemistry reactions were employed to fluorescently tag incorporated EdU for visualization. In primary fibroblasts, cell proliferation decreased by 44.8 ± 4.1% with DXC treatment versus 28.5 ± 3.9% with PEG–H40–DXC treatment. A similar trend was observed in stem-cell-derived fibroblasts: cell proliferation decreased by 73.9 ± 3.6% with DXC treatment versus 44.4 ± 3.3% with PEG–H40–DXC treatment (Fig. 7c). In both fibroblast cell types, PEG–H40–DXC produced higher percentage of proliferative cells compared to DXC alone. Primary fibroblasts proliferated more than stem-cell-derived fibroblasts upon treatment by DXC or PEG–H40–DXC (Fig. 7c), indicating that the extent of the proliferation decrease is cell type specific and likely depends on transgene expression.
In stem cell derived fibroblasts, treatment with PEG–H40–DXC produced more cells with phenotypes important for epigenetic, reprogramming transitions – proliferative cells that express all the reprogramming factors (Fig. 7d). Through immunocytochemistry, proliferation and Oct4/Sox2 transgene expression were simultaneously monitored in the same cell (Fig. 7d and S-3 in the ESI†). Almost all of the proliferative cells (97.1 ± 5.6%) in culture stained positively for Oct3/4 and/or Sox2. This proliferative phenotype of cells with induced proteins is consistent with recent reports that note fast-dividing cells are created early in reprogramming.29,30 Only a fraction of cells were positive for all three stains: Oct3/4-Sox2-Edu (“triple-positive”). Triple-positive cells mark rare, proliferative cells that express both reprogramming factors, and these cells are destined to successfully complete the epigenetic reprogramming at high efficiencies.29–33 Nearly 2-fold more triple-positive cells were found in PEG–H40–DXC treated cultures than in DXC cultures (4.0 ± 1.1 vs. 2.0 ± 0.8, p < 0.05 in Fig. 7d).
Together, the cell assays indicate that PEG–H40–DXC protects MMPs in the extracellular space to preserve robust cell proliferation while retaining the ability to properly activate intracellular targets to induce transgene expression.
Discussion
Boltorn H40 is a biodegradable and biocompatible hyperbranched polyester. As shown in Scheme 1, DXC was conjugated onto H40 via biodegradable amide and ester bonds. The resulting PEG–H40–DXC nanoparticles exhibited a strong pH-dependent drug release profile (Fig. 3), because both amide bonds and ester bonds break down faster under acidic conditions. Such pH-dependent drug release behavior is highly desirable for many applications including targeted cancer therapy as well as tissue engineering. More specifically, in this case, DXC conjugated onto the PEG–H40–DXC nanoparticles was quite stable in the cell culture medium or extracellular environment (pH ∼ 7.0–7.4); however, once the PEG–H40–DXC nanoparticles are taken up by fibroblasts into acidic, endocytic compartments (i.e., endosomes/lysosomes), DXC will be released relatively quickly albeit controllably into the cytosol. The specific internalization pathways of nanoparticles after being taken up by cells via endocytosis depend on many factors including the nature of the nanoparticles and the types of cells. Thus, the exact internalization pathways of PEG–H40–DXC in C1 fibroblasts merit further studies in the future.PEG–H40–DXC activates the Tet-on system present in secondary C1 fibroblasts as efficiently as DXC alone, while reducing off-target effects like inhibition of MMPs and decreased cell growth. DXC has been shown, both in this study (Fig. 4b) and previously,34–37 to inhibit MMP activity, affecting varied and far-reaching cellular processes.15 This inhibition has been partly attributed to the blocking of MMP-9 cleavage of TGF-β1 which activates Smad, JNK, ERK, and p38 MAPK pathways.37 PEG–H40–DXC likely decreases this inhibition, and its subsequent effects on kinase pathways, by protecting MMPs in the extracellular space (pH ∼7.0–7.4) from DXC due to the slow release of DXC from the PEG–H40–DXC at pH 7.4.
The intracellular degradation and release of DXC inside the cell is efficient and rapid enough to induce the same level of expression of transgenes as the current system of treatment with DXC alone (Fig. 4c and 5a and b). The Tet-on system exhibited the same complex dynamics and dose response to PEG–H40–DXC as it did to DXC alone (Fig. 6a and b), extending the similarities in the targeted in vitro response between the two treatments. However, the reduction of extracellular off-target effects by PEG–H40–DXC treatment in place of DXC treatment substantially boosts the ability of PEG–H40–DXC to be used in gene expression applications productively. For fibroblasts that rely on MMP activity in culture for robust growth, treatment with PEG–H40–DXC exhibited cell growth comparable to cells cultured in normal fibroblast media with higher absolute numbers of cells producing the transgene products, as opposed to cells treated with DXC (Fig. 4c and 7a–c). Differences in cell morphology could not be detected visually between PEG–H40–DXC-treated cells and non-treated cells, in contrast to the stressed morphology seen soon after DXC treatment (Fig. 7a).
The ability of PEG–H40–DXC to facilitate stem cell transitions was studied in the process of inducing pluripotent stem cells from fibroblasts with Oct3/4 and Sox2 reprogramming factors. The process of cellular reprogramming requires a gain of proliferation, activation of embryonic stem cell genes, followed by epigenetic remodeling that together culminates in the activation of stem cell circuitry.31 Efficiency of this process is improved by high levels of reprogramming factors31 and/or with an increased rate of cell division.29,30 Specifically, Sox2 has been shown to initiate a chain of events that enable the cell to enter a pluripotent state; while Oct3/4 assists in the reactivation of the pluripotency circuitry;28 and, increased cellular proliferation plays an important role in raising the probability for the stochastic events characteristic of successful reprogramming attempts.29 Coordinating these three mechanisms within cells in reprogramming cultures has been a challenge in the stem cell field. Treatment with H40–DXC–PEG increased the number of triple-positive cells (Fig. 7d), indicating a significant advance over the state-of-the-art means of generating these rare and important cells.
Conclusions
The PEG–H40–DXC nanoparticle serves as a versatile nanoplatform with the ability to control gene expression in human stem cell derivatives through the use of Tet-on transgene systems. Gene expression with the nanoparticle occurs as effectively as DXC, the currently utilized Tet inducer, while minimizing off-target effects that decrease cell health, experimentation productivity, and important reprogramming processes. The synthesis and characterization of the PEG–H40–DXC nanoparticle indicated successful conjugation of the drug at 11% loading levels, a 50–100 nm hydrodynamic diameter, and a strong pH-dependent drug release profile. The intensity, temporal dynamics, and dose-dependence of gene expression in PEG–H40–DXC-treated human stem-cell-derived fibroblasts were similar to treatment with free DXC. More importantly, the use of PEG–H40–DXC for induction of the Tet-on system led to a reduction of negative off-target effects including a return of MMP activity and healthy morphology, and an increase in cellular proliferation in comparison with free DXC. These effects combine to facilitate stem cell-related transitions within fibroblasts creating more triple-positive-cells in culture than free DXC. Subsequent work with these nanoparticle systems could be used to intricately control Tet-on gene circuits developed by synthetic biologists8 in hPSC-derived cells to model and treat diseases in innovative ways.1Acknowledgements
We thank Sean Palecek, Kristyn Masters, Travis Cordie, Max Salick, Gavin Knight, Lydia Ashton, and Carlos Marti-Figueroa for support and technical help. We would also like to acknowledge the partial financial support for this work from the Society in Science Foundation, the National Institutes for Health (1K25CA166178, T32GM008692), and the National Science Foundation (DMR 1032187).References
- K. Saha and R. Jaenisch, Cell Stem Cell, 2009, 5, 584–595 CrossRef CAS PubMed.
- K. Saha, J. F. Pollock, D. V. Schaffer and K. E. Healy, Curr. Opin. Chem. Biol., 2007, 11, 381–387 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- D. J. Sauers, M. K. Temburni, J. B. Biggins, L. M. Ceo, D. S. Galileo and J. T. Koh, ACS Chem. Biol., 2010, 5, 313–320 CrossRef CAS PubMed.
- L. J. Bugaj, A. T. Choksi, C. K. Mesuda, R. S. Kane and D. V. Schaffer, Nat. Methods, 2013, 10, 249–252 CrossRef CAS PubMed.
- L. R. Polstein and C. A. Gersbach, J. Am. Chem. Soc., 2012, 134, 16480–16483 CrossRef CAS PubMed.
- S. Konermann, M. D. Brigham, A. Trevino, P. D. Hsu, M. Heidenreich, L. Cong, R. J. Platt, D. A. Scott, G. M. Church and F. Zhang, Nature, 2013, 500, 472–476 CAS.
- W. C. Ruder, T. Lu and J. J. Collins, Science, 2011, 333, 1248–1252 CrossRef CAS PubMed.
- M. Gossen and H. Bujard, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5547–5551 CrossRef CAS.
- X. Zhou, M. Vink, B. Klaver, B. Berkhout and A. T. Das, Gene Ther., 2006, 13, 1382–1390 CrossRef CAS PubMed.
- U. Baron and H. Bujard, in Methods in Enzymology, ed. J. Thorner, S. D. Emr and J. N. Abelson, Academic Press, Massachusetts, 2000, vol. 327, ch. 33, pp. 401–421 Search PubMed.
- A. T. Das, X. Zhou, M. Vink, B. Klaver, K. Verhoef, G. Marzio and B. Berkhout, J. Biol. Chem., 2004, 279, 18776–18782 CrossRef CAS PubMed.
- P. Orth, D. Schnappinger, W. Hillen, W. Saenger and W. Hinrichs, Nat. Struct. Biol., 2000, 7, 215–219 CrossRef CAS PubMed.
- L. H. Nip, V. J. Uitto and L. M. Golub, J. Periodontal Res., 1993, 28, 379–385 CrossRef CAS.
- A. Page-McCaw, A. J. Ewald and Z. Werb, Nat. Rev. Mol. Cell Biol., 2007, 8, 221–233 CrossRef CAS PubMed.
- S. B. Cambridge, D. Geissler, S. Keller and B. Cürten, Angew. Chem., Int. Ed. Engl., 2006, 45, 2229–2231 CrossRef CAS PubMed.
- S. B. Cambridge, D. Geissler, F. Calegari, K. Anastassiadis, M. T. Hasan, A. F. Stewart, W. B. Huttner, V. Hagen and T. Bonhoeffer, Nat. Methods, 2009, 6, 527–531 CrossRef CAS PubMed.
- E. K. Moioli, P. A. Clark, X. Xin, S. Lal and J. J. Mao, Adv. Drug Delivery Rev., 2007, 59, 308–324 CrossRef CAS PubMed.
- C.-M. J. Hu and L. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS PubMed.
- A. S. Gupta, Nanomedicine, 2011, 7, 763–779 CrossRef PubMed.
- L. Zhang and T. J. Webster, Nano Today, 66–80 Search PubMed.
- P. J. Keller, Science, 2013, 340, 1234168 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071–3082 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 3009–3019 CAS.
- D. Hockemeyer, F. Soldner, E. G. Cook, Q. Gao, M. Mitalipova and R. Jaenisch, Cell Stem Cell, 2008, 3, 346–353 CrossRef CAS PubMed.
- S. Shanmuganathan, N. Shanumugasundaram, N. Adhirajan, T. S. Ramyaa Lakshmi and M. Babu, Carbohydr. Polym., 2008, 73, 201–211 CrossRef CAS PubMed.
- Y. Buganim, D. A. Faddah, A. W. Cheng, E. Itskovich, S. Markoulaki, K. Ganz, S. L. Klemm, A. van Oudenaarden and R. Jaenisch, Cell, 2012, 150, 1209–1222 CrossRef CAS PubMed.
- J. Hanna, K. Saha, B. Pando, J. van Zon, C. J. Lengner, M. P. Creyghton, A. van Oudenaarden and R. Jaenisch, Nature, 2009, 462, 595–601 CrossRef CAS PubMed.
- Z. D. Smith, I. Nachman, A. Regev and A. Meissner, Nat. Biotechnol., 2010, 28, 521–526 CrossRef CAS PubMed.
- J. M. Polo, E. Anderssen, R. M. Walsh, B. A. Schwarz, C. M. Nefzger, S. M. Lim, M. Borkent, E. Apostolou, S. Alaei, J. Cloutier, O. Bar-Nur, S. Cheloufi, M. Stadtfeld, M. E. Figueroa, D. Robinton, S. Natesan, A. Melnick, J. Zhu, S. Ramaswamy and K. Hochedlinger, Cell, 2012, 151, 1617–1632 CrossRef CAS PubMed.
- T. Kawamura, J. Suzuki, Y. V. Wang, S. Menendez, L. B. Morera, A. Raya, G. M. Wahl and J. C. I. Belmonte, Nature, 2009, 460, 1140–1144 CrossRef CAS PubMed.
- A. Banito, S. T. Rashid, J. C. Acosta, S. Li, C. F. Pereira, I. Geti, S. Pinho, J. C. Silva, V. Azuara, M. Walsh, L. Vallier and J. Gil, Genes Dev., 2009, 23, 2134–2139 CrossRef CAS PubMed.
- V. J. Uitto, J. D. Firth, L. Nip and L. M. Golub, Ann. N. Y. Acad. Sci., 1994, 732, 140–151 CAS.
- R. Hanemaaijer, H. Visser, P. Koolwijk, T. Sorsa, T. Salo, L. M. Golub and V. W. van Hinsbergh, Adv. Dent. Res., 1998, 12, 114–118 CrossRef CAS PubMed.
- G. N. Smith Jr, E. A. Mickler, K. A. Hasty and K. D. Brandt, Arthritis Rheuma, 1999, 42, 1140–1146 CrossRef CAS.
- H.-S. Kim, L. Luo, S. C. Pflugfelder and D.-Q. Li, Invest. Ophthalmol. Visual Sci., 2005, 46, 840–848 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI containing 1H NMR spectra and additional fibroblast characterization data. See DOI: 10.1039/c3nr04794f |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2014 |