Green organophotocatalysis. TiO2-induced enantioselective α-oxyamination of aldehydes†
Xuan-Huong
Ho
,
Min-Jung
Kang
,
Seung-Joo
Kim
,
Eun Duck
Park
and
Hye-Young
Jang
*
Division of Energy Systems Research, Ajou University, Suwon, 443-749, Korea. E-mail: hyjang2@ajou.ac.kr; Fax: +82 31 219 1615; Tel: +82 31 219 2555
First published on 11th July 2011
Abstract
Asymmetric C–O bond formation with various aldehydes is examined under conditions of environmentally benign photo-organocatalysis. A heterogeneous TiO2 photocatalyst is used as a photooxidant in place of toxic and expensive chemical oxidants, for the first time in chemo- and stereoselective bond formation.
The environmentally benign TiO2 photocatalyst has been used in a wide range of green processes, including the decomposition of organic pollutants and the production of hydrogen by water photolysis, because of its excellent chemical stability, reusability, low cost, and low toxicity.1 In particular, the highly oxidizing holes produced in photo-activated TiO2 have been used for the simple oxidation of hydrocarbons and alcohols.2 According to an intensive study on TiO2-catalyzed photo-oxidations, the high oxidation potential of the holes in the valence band of TiO2 (2.53 V versusSHE) promotes the efficient and complete oxidation of organic molecules, but control of selectivity in this reaction remains a challenge. Therefore, TiO2-mediated photocatalytic oxidation has not been applied to stereoselective fine chemical synthesis.3
Continuing efforts have been made to find highly stereoselective protocols for the synthesis of fine chemicals under environmentally friendly conditions such as light-induced stereoselective bond formations.4 Recently, photo-induced enantioselective organocatalytic reactions involving single-electron oxidation/reduction have been carried out using homogeneous photoredox inorganic (Ru(II) or Ir(III)) complexes and organic dyes.5,6 For a single-electron-transfer process in asymmetric organocatalytic reactions, stoichiometric amounts of inorganic/organic oxidants (reductants) are generally required;7 however, photoredox materials with remarkable photoreactivity and electrochemical properties can oxidize or reduce organic compounds under light irradiation. Thus, the use of stoichiometric amounts of oxidants or reductants can be avoided in order to carry out green and economical asymmetric reactions with a high level of selectivity.
Our interest in the development of green organocatalytic systems prompted us to explore environmentally friendly oxidation protocols for enantioselective reactions. Therefore, we studied the enantioselective organocatalyzed α-oxyamination and α-alkylation of aldehydes under galvanostatic conditions.8,9 Subsequent to the successful development of electro-organocatalysis reactions, we proposed a novel heterogeneous photo-organocatalytic reaction by combining the environmentally benign TiO2 photo-oxidation and the enantioselective organocatalytic α-oxyamination of aldehydes. Racemic α-oxyamination of aldehydes and asymmetric α-alkylation of aldehydes were conducted under photocatalytic conditions using [Ru(bpy)3]2+,5,10 but enantioselective organocatalysis mediated by the non-hazardous and recyclable photoactivated TiO2 could not be conducted. The present TiO2 photo-organocatalytic reaction shows that an organocatalytic reaction involving single-electron-oxidation can be conducted under cost-effective, nontoxic, and reusable heterogeneous photo-catalytic conditions.
Optimization was first attempted using a chiral sec-amine catalyst (A) and TiO2 (Degussa P25), which promoted the coupling of aldehyde 1a with TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) in dichloromethane under irradiation by a mercury lamp to afford the desired product 3a in 84% yield and 43% enantiomeric excess (ee) (Table 1, entry 1). As commercially available Degussa P25 consists of anatase with a band gap of 3.2 eV (387 nm) and rutile with a band gap of 3.0 eV (415 nm), a mercury lamp generating UV light was used as the light source. The absolute stereochemistry of compound 1c was assigned as S by comparing the HPLC results for this compound with the HPLC data reported in the literature.8a To test the recyclability of the heterogeneous TiO2 photocatalyst, the recovered TiO2 was reused under the reaction conditions mentioned in entry 1. The recovered TiO2 photocatalyst afforded 3a in 71% (1st recycling) and 48% (2nd recycling) yields. The XRPD (X-ray powder diffraction) pattern of the recovered catalyst showed that there was no change in the structure of TiO2 after the catalytic process. The reduced yield obtained with the recovered TiO2 could be attributed to the contamination of the TiO2 surface by organic compounds. With a small amount of TiO2, a decreased yield (58%) with 46% ee of 3a was observed (entry 2). Next, the effect of the solvent was evaluated. Among dichloromethane, dichloroethane, acetonitrile, acetone, DMF, and THF, the highest yield was observed when using dichloromethane, while a high ee with a reasonable yield could be obtained when using acetonitrile (entries 1, 3–7). To improve the enantioselectivity, the reaction was performed at a low temperature (0 °C), resulting in a similar enantioselectivity after a prolonged reaction time (entry 8). Subsequent to solvent screening, a wide range of proline derivatives and imidazolidinone-based catalysts (MacMillan catalysts) were evaluated for the TiO2 photo-organocatalytic coupling reaction in acetonitrile, as shown in entries 9–16. Catalyst E, which had larger aromatic rings than did catalyst A, showed high enantioselectivity (78% ee) and afforded a somewhat low product yield (19% at ambient temperature and 37% at 0 °C). To evaluate the substrate scope, both A and E were tested along with TiO2. Other photoreactive metal oxides (WO3, and ZnO) did not promote the desired α-oxyamination.
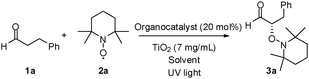
| Entry | Catalyst | Solvent | Temp. | Time/h | Yield (%) | ee (%) |
|---|---|---|---|---|---|---|
a
TiO2 1.7 mg mL−1.
b At 0 °C, 37% yield and 78% ee.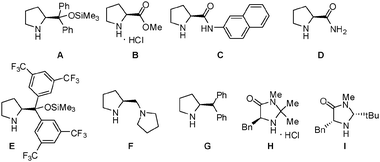
|
||||||
| 1 | A | CH2Cl2 | rt | 6 | 84 | 43 |
| 2 | A | CH2Cl2 | rt | 10 | 58 | 46a |
| 3 | A | Dichloroethane | rt | 10 | 68 | 41 |
| 4 | A | CH3CN | rt | 8 | 60 | 63 |
| 5 | A | Acetone | rt | 9 | 30 | 65 |
| 6 | A | DMF | rt | 9 | 5 | 45 |
| 7 | A | THF | rt | 10 | 53 | 57 |
| 8 | A | CH3CN | 0 °C | 8 | 47 | 61 |
| 9 | B | CH3CN | rt | 9 | 60 | 2 |
| 10 | C | CH3CN | rt | 9 | 65 | 21 |
| 11 | D | CH3CN | rt | 10 | 56 | 3 |
| 12 | E | CH3CN | rt | 10 | 19 | 78b |
| 13 | F | CH3CN | rt | 10 | 15 | 11 |
| 14 | G | CH3CN | rt | 10 | 56 | 37 |
| 15 | H | CH3CN | rt | 6 | 35 | 48 |
| 16 | I | CH3CN | rt | 6 | NR | — |
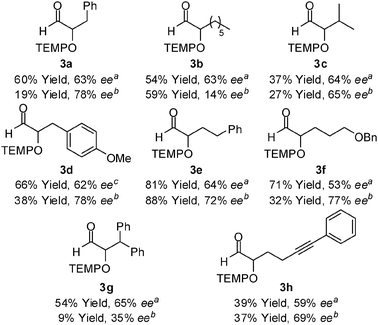
The α-oxyamination of various aldehydes was examined, as shown in Table 2. The aliphatic octylaldehyde 1b and TEMPO were subjected to the conditions specified for catalyst A to obtain the product 3b in 54% yield and with 63% ee. Catalyst E improved the ee of 2a but not that of 3b. In the case of isovaleraldehyde 1c, the reaction was complete in a short time (2 h), but a low yield and enantioselectivity comparable to that of 3a and 3b were observed. With p-methoxy-substituted hydrocinnamaldehyde 1d and TEMPO, the product 3d was obtained in 66% yield and with 62% ee in the presence of catalyst A at 0 °C. At ambient temperature, 3d was decomposed, thereby resulting in low yield (23% yield). Similar to the reaction of 1a, α-oxyamination of 1d with catalyst E afforded a high ee but low yield. Aldehydes 1e and 1f, which had a benzyloxy group, participated in the coupling reaction with TEMPO to form 3e and 3f, respectively, in reasonable yield and ee, in the presence of A as well as E. As shown in the case of 3g, diphenyl substitution at the β-position of the aldehyde did not improve the enantioselectivity of the reaction. The yield of 3h possessing the alkyne which was thought to be susceptible under radical reaction conditions was lower than those of other compounds with similar structure.
In addition to the reaction of aldehydes with TEMPO, a photo-organocatalytic reaction of phenylpropanol with TEMPO was conducted (Scheme 1), under the assumption that alcohols photo-oxidize to form aldehydes and that the aldehydes formed in situ undergo photo-organocatalytic α-oxyamination with TEMPO to afford the desired products.7k It is known that the oxoammonium cation generated from the oxidation of TEMPO or photo-activated TiO2 can oxidize alcohols.2e,2g,11 The ee in the photo-organocatalytic coupling was similar to that in the direct α-oxyamination of aldehydes, but a relatively low yield was obtained in the former case because of the inefficient alcohol oxidation.12 Under our TiO2 photo-oxidation conditions, the conversion of alcohols to aldehydes was low.
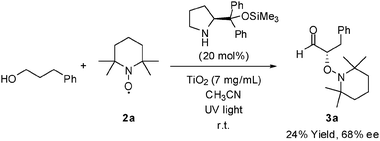 | ||
| Scheme 1 Photo-organocatalytic coupling of phenylpropanol with TEMPO. | ||
To probe the mechanism for the enantioselective organocatalytic α-oxyamination under photo-oxidation conditions, several control experiments were conducted. The desired α-oxyamination of aldehydes did not occur in the absence of TiO2 or organocatalysts, implying that TiO2-catalyzed photo-oxidation and enamine formation from the organocatalyst are essential steps in the reaction. The possibility of occurrence of a photo-reaction induced by UV light in the absence of photocatalysts was excluded.13 Under the conditions of visible-light irradiation or no light irradiation, the desired α-oxyamination did not occur. On the basis of the abovementioned results of the control experiment, we concluded that the enamine intermediate should be formed from the aldehyde and an organocatalyst, and that the photo-activation of TiO2 in the presence of UV light is a prerequisite for this transformation. However, it is not clear whether the reaction proceeds through enamine oxidation or TEMPO oxidation. To verify the oxidized species formed under the photo-oxidation conditions, the previously reported electrochemical data pertaining to the enamine derived from pyrrolidine and hydrocinnamaldehyde, and TEMPO were used.7h,8a,14 During the anodic scan over the potential range 0.0 to 2.5 V (or 3.0 V), the enamine derived from pyrrolidine and hydrocinnamaldehyde, and TEMPO gave oxidation peaks at 0.72 V and 1.03 V vs. a silver wire pseudoreference electrode (scan rate: 0.1 V s−1), implying that TiO2 may effect the oxidation of the enamine rather than that of TEMPO under UV light irradiation.4,7i,10 On the other hand, enamine addition to the oxidized TEMPO could not be ruled out since TEMPO is known to undergo photooxidation in the presence of TiO2.2d Thus, the organocatalytic (catalyst A) reaction of hydrocinnamaldehyde with an oxopiperidinium salt (TEMPO+) in acetonitrile was conducted to afford the desired product in 1% yield (65% ee) in the absence of UV light.7h The time required for the reaction between hydrocinnamaldehyde and TEMPO+ was considerably longer (18 h) than that for the photo-organocatalytic reaction mentioned in Tables 1 and 2 (2–8 h), but the yield obtained in the former case was very low. Overall, the catalytic cycle involving the photo-oxidation of enamine I is considered the major route for the formation of 3a. However, the reaction route involving TEMPO oxidation and subsequent addition to I is not completely excluded (Scheme 2). After the workup, the reduced TEMPO (2,2,6,6-tetramethylpiperidin-1-ol) was detected, implying that some TEMPO molecules function as the electron acceptor in this organophotocatalysis.
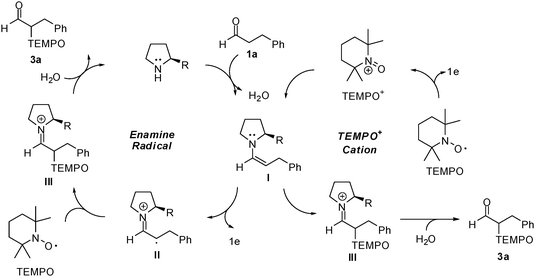 | ||
| Scheme 2 Catalytic cycles. | ||
Conclusions
Organocatalysis involving the single-electron-transfer process can be carried out under heterogeneous photocatalytic conditions to obtain α-oxyaminated aldehydes in good yield and enantioselectivity. This study is the first to show that heterogeneous and recyclable TiO2 photocatalysts can be used for enantioselective organocatalyzed C–O bond formation. Remarkably, enantioselective coupling products are obtained without further oxidation of the α-oxyaminated aldehydes in the presence of highly oxidizing TiO2 holes, whereas most of the TiO2 photo-reactions reported in the past have shown poor selectivity in organic reactions. Furthermore, a tandem alcohol oxidation/asymmetric C–O bond formation process is facilitated under our photo-organocatalytic conditions. On the basis of the results of control experiments and electrochemical analysis, the reaction route involving enamine radicals is concluded to be the working mechanism.Experimental
The mixture of the aldehyde (0.5 mmol), TEMPO (1 mmol), TiO2 (35 mg) and a catalyst (0.1 mmol) in the solvent (0.1 M) was exposed to UV irradiation (200–2500 nm, Hg lamp at distance of 15 cm) at ambient temperature under N2 until the aldehyde was consumed completely. The solvent was removed with a rotary evaporator to produce a residue which was purified by column chromatography on a silica gel eluting with hexane and ethyl acetate for the yield. To determine the enantiomeric excess, the crude materials of α-oxyaminated aldehydes were exposed to the solution containing NaBH4, and the resulting purified alcohols or further modified (benzoylated) alcohols were injected to chiral stationary phase HPLC (Agilent 1100 series). Degussa P25 titanium dioxide nanoparticles were purchased and used as photocatalysts. The mercury-lamp (50–500 W, Newport) was used as a UV light source.Acknowledgements
This study was supported by the Korea Research Foundation (grant No. 2010-0029617, 2010-0016079, and 2010-0002396). The authors wish to thank Korean Basic Science Institute (Deagu) for the mass spectra.Notes and references
- For selected reviews of TiO2 photocatalysis: (a) M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS; (b) M. R. Hoffman, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS; (c) A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS; (d) X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS; (e) K. Hashimoto, H. Irie and A. Fujishima, AAPPS Bull., 2007, 17, 12–28 Search PubMed.
- For recent selected articles of TiO2-photocatalyzed reactions: (a) L. Cermenati, M. Mella and A. Albini, Tetrahedron, 1998, 54, 2575–2582 CrossRef CAS; (b) G. Palmisano, S. Yurdakal, V. Augugliaro, V. Loddo and L. Palmisano, Adv. Synth. Catal., 2007, 349, 964–970 CrossRef CAS; (c) S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568–1569 CrossRef CAS; (d) V. Augugliaro, T. Caronna, V. Loddo, G. Marcì, G. Palmisano, L. Palmisano and S. Yurdakal, Chem.–Eur. J., 2008, 14, 4640–4646 CrossRef CAS; (e) Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976–7979 CrossRef CAS; (f) C.-T. Chen, J.-Q. Kao, C.-Y. Liu and L.-Y. Jiang, Catal. Sci. Technol., 2011, 1, 54–57 RSC. Dye-sensitized TiO2-photocatalyzed reactions: (g) M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS.
- For recent selected articles of non-photocatalytic stereoselective reactions using TiO2 as a hetereogeneous supporter: (a) T. Bürgi and A. Baiker, J. Phys. Chem. B, 2002, 106, 10649–10658 CrossRef; (b) G. Fantin, G. Paolo, A. Guerrini, S. Maietti, A. Medici and P. Pedrini, Biotechnol. Lett., 2006, 28, 805–810 CrossRef CAS; (c) T. Kubota, H. Kubota, T. Kubota, E. Moriyasu, T. Uchida, Y. Nitta, T. Sugimura and Y. Okamoto, Catal. Lett., 2009, 129, 387–393 CrossRef CAS; (d) F. Naso, M. A. M. Capozzi, A. Bottoni, M. Calvaresi, V. Bertolasi, F. Capitelli and C. Cardellicchio, Chem.–Eur. J., 2009, 15, 13417–13426 CrossRef CAS; (e) R. M. D. Nunes, B. F. Machado, M. M. Pereira, M. J. S. M. Moreno and J. L. Faria, J. Mol. Catal. A: Chem., 2010, 333, 1–5 CrossRef CAS.
- For recent reviews of photocatalytic reactions: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS; (b) G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC; (c) P. Renaud and P. Leong, Science, 2008, 322, 55–56 CrossRef CAS; (d) P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 1360–1363 CrossRef CAS; (e) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS; (b) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS.
- M. Newmann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS.
- (a) T. D. Beeson, A. Mastrachhio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS; (b) H.-Y. Jang, J.-B. Hong and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 7004–7005 CrossRef CAS; (c) H. Kim and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 398–399 CrossRef CAS; (d) T. H. Graham, C. M. Jones, N. T. Jul and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 16494–16495 CrossRef CAS; (e) J. E. Wilson, A. D. Casarez and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11332–11334 CrossRef CAS; (f) J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640–11641 CrossRef CAS; (g) M. Amatore, T. D. Beeson, S. P. Brown and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 5121–5124 CrossRef CAS; (h) J. F. Van Humbeck, S. P. Simonovich, R. R. Knowles and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10012–10014 CrossRef; (i) M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 395–405 CrossRef CAS; (j) K. Akagawa, T. Fujiwara, S. Sakamoto and K. Kudo, Org. Lett., 2010, 12, 1804–1807 CrossRef CAS; (k) K. Akagawa, T. Fujiwara, S. Sakamoto and K. Kudo, Chem. Commun., 2010, 46, 8040–8042 RSC; (l) T. Kano, H. Mii and K. Maruoka, Angew. Chem., Int. Ed., 2010, 49, 6638–6641 CAS.
- (a) N.-N. Bui, X.-H. Ho, S.-i. Mho and H.-Y. Jang, Eur. J. Org. Chem., 2009, 5309–5312 CrossRef CAS; (b) X.-H. Ho, S.-i. Mho, H. Kang and H.-Y. Jang, Eur. J. Org. Chem., 2010, 4436–4441 CAS.
- (a) A. M. Costero, G. M. Rodíguez-Muniz, P. S. Gavina, A. Gil and A. Domenech, Tetrahedron Lett., 2007, 48, 6992–6995 CrossRef CAS; (b) K. L. Jensen, P. T. Franke, L. T. Nielsen, K. Daasbjerg and K. A. Jøgensen, Angew. Chem., Int. Ed., 2010, 49, 129–133 CAS.
- T. Koike and M. Akita, Chem. Lett., 2009, 38, 166–167 CrossRef CAS.
- R. A. Sheldon and W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS and references therein.
- The oxidation of phenylpropanol was conducted under TiO2 photo-oxidation conditions with TEMPO and without TEMPO. Under both conditions, hydrocinnamaldehyde was formed with low conversions. Low oxidation yield of aliphatic aldehydes under TiO2/TEMPO photo-oxidation conditions is reported in ref. 2d.
- C. Müller, A. Bauer and T. Bach, Angew. Chem., Int. Ed., 2009, 48, 6640–6642 CrossRef.
- The solution was prepared in dichloromethane using TBAP (tetrabutylammonium perchlorate) as an electrolyte.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental, XRPD data and spectral analysis for new compounds. See DOI: 10.1039/c1cy00183c |
| This journal is © The Royal Society of Chemistry 2011 |