AlBr3·6H2O catalyzed oxidation of benzylic alcohols†
Yun-Mei
Zhong
,
Heng-Chang
Ma
*,
Jin-Xia
Wang
,
Xiao-Jie
Jia
,
Wen-Feng
Li
and
Zi-Qiang
Lei
*
Key Laboratory of Polymer Materials of Gansu Province, Key Laboratory of Eco-Environment-Related Polymer Materials Ministry of Education, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, People's Republic of China. E-mail: leizq@nwnu.edu.cn; Fax: +86 931 7970359; Tel: +86 931 7970359
First published on 22nd June 2011
Abstract
AlBr3·6H2O was found to be an efficient catalyst in the field of oxidation of benzylic alcohols. Primary and secondary benzylic alcohols could be transformed into corresponding aldehydes and ketones with almost complete conversion and high selectivity. The non-transition metal related reaction systems have an incomparable advantage over others, such as easy work-up, excellent selectivity as well as commercial availability.
Selective oxidation of alcohols to carbonyl compounds is one of the most important and challenging transformations in the synthesis of fine chemicals and intermediates.1 Traditionally, chromate and permanganate reagents are employed for this purpose.2 Unfortunately, these oxidations call for the use of at least stoichiometric amounts of oxidants and generate a large quantity of noxious byproducts.3
From an environmental and economic viewpoint, there has been an increasing need for selective oxidation using more green and effective procedures. The transition-metal-catalyzed oxidation of alcohols in the presence of molecular oxygen as the terminal oxidant has received great attention in recent years.4 The metal-catalyzed aerobic oxidation of alcohols has recently been reviewed, and these reviews outline significant progress in the elaboration of homogeneous and/or heterogeneous catalysts in general.3a,5 However, for most cases, the indispensible additives such as N-hydroxyphthalimide,6 diethyl azodicarboxylate,7 or nitrasonium ions8 and some ligands9 were required to accomplish the catalytic cycle, as well as the catalyst separation and recycling of homogeneous metal catalysts often made practical and industrial applications very difficult.10 Mostly, selective methods allowing for oxidation of primary alcohols to aldehydes without overoxidation to carboxylic acids still remain challenging. Particularly, the alternative oxidations in this field are the use of free radical assisted systems, for instance, nitroxyl radical 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)11 and NXS (X = Cl, Br, I),12 as well as allylbromide based oxidation procedures. They are desirable and efficient catalysts for the aerobic oxidation of a broad range of alcohols under mild reaction conditions. However, for these systems, some limitations are obvious, such as substrates containing acid-sensitive functional groups could not be tolerant in some cases,13 the other undesirable auxiliaries, Ru(II),14 Cu(I),15 NaNO2,16 and Br2,16a are absolutely indispensible, and also increased temperature and oxygen pressure are needed. Especially, overoxidation could not be prevented, resulting in carboxylic acid as a major product.17 As a result, these more complicated systems conduct low atom efficiency, waste disposal, and expensive material cost, as well as great energy should be devoted.
Our previous study explored that a variety of alcohols, including inactive aliphatic alcohols, could be converted into aldehydes and ketones in the presence of H2O2 or Oxone® (2KHSO5·KHSO4·K2SO4) with the catalyst of AlCl3.18 Although, these disclosed catalyst systems are efficient, some disadvantages are inevitable, for instance, the high loading amount of catalyst, low oxidant efficiency, and no-tolerance of acid sensitive substituent. In the ongoing research programs, we found that in the presence of air, AlBr3·6H2O exhibits excellent catalysis oxidative behavior for alcohols. Notably, for the primary benzyl alcohol, the overoxidation could be suppressed. Subsequently, the optimization of our initial oxidation system led to an improved and highly efficient protocol for the oxidation of primary and secondary benzylic alcohols under mild conditions. We believe that the newly developed simple oxidation system will be an important component in the field of oxidation of alcohols.
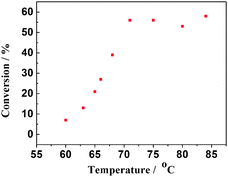
Initially, benzyl alcohol was treated with 30 mol% AlBr3·6H2O in the commonly used solvents under open air conditions. The results confirmed that the more polar solvent 1,4-dioxane significantly enhanced the reaction rate, while, non-polar hexane, CCl4, and toluene were found to be poor media. We can also note that significant improvement was achieved by increasing the reaction temperature up to 70 °C, resulting in clean formation of benzaldehyde with relatively high conversion and high selectivity. Furthermore, conducting the above reaction at 100 °C for a longer reaction time furnished benzoic acid as a major product with almost complete conversion. However, the reaction process could not be performed if the temperature is below 60 °C (Fig. 1). We carried out the catalytic oxidation of benzyl alcohol in a batch reactor by following the reaction with time. The plots of reaction products against time are depicted in Fig. 2. These results show that the yield rates of benzaldehyde are almost constant within 250 min. Although, the dehydrogenation of substrate was effective, the transformation occurred after a variable induction period (within 20 to 30 min). Under an argon atmosphere instead of air or O2, a trace amount of benzaldehyde together with a complicated mixture was observed, and the most amount of substrate could be recovered. This means that the oxygen from air applied as an oxidant was beneficial for both the rate and yield of the product. Herein, the optimum reaction conditions thus far developed employ 1.0 equiv. of substrate, 30 mol% of AlBr3·6H2O in dioxane at 70 °C. Here the doubt of the excessive catalytic amount of AlBr3·6H2O led us to have a more detailed study on the reaction conditions. However the screened results verified that AlBr3·6H2O should be used definitely as we stated 30 mol%, which was possible due to the fact that the solubility of AlBr3·6H2O in the organic solvent was poor. After completion of the reaction, a certain amount of AlBr3·6H2O always precipitated in the reaction vessel, which seems to be wasted. We considered that these precipitates could offer sufficient concentrations of AlBr3·6H2O in the solvent during the reaction, so the amount of AlBr3·6H2O used was more than that of some compounds with better solubility in the organic solvents. What should be declared is that these used precipitates of AlBr3·6H2O could be reused after the pre-cycle reaction. It was also noted that the rigorous exclusion of moisture is not required in any of these transformations, and comparable results are obtained in the presence and absence of O2, as well as in freshly distilled commercial solvents. As such, this represents an exceedingly convenient method for oxidation of alcohols to the corresponding carbonyl compounds.
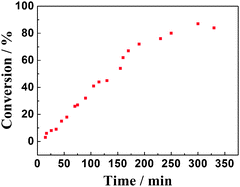 | ||
| Fig. 2 Typical reaction time course for the oxidation of benzyl alcohol by the AlBr3·6H2O system. | ||
Under the reaction conditions mentioned above (0.1 mmol of alcohol, 0.03 mmol of AlBr3·6H2O, 2.0 mL of 1,4-dioxane, 70 °C) several primary benzyl alcohols were tested to explore the scope of this method (Scheme 1). Generally, excellent selectivity for the most of transformations to benzaldehydes was demonstrated, while, the oxidation of benzyl alcohol was somewhat less selective, as benzaldehyde and benzoic acid were obtained with 89% and 11% selectivity. Although different substitution patterns on the aryl ring lead to similar GC yields, different reaction times were required according to the electronic contribution of the substituents on the benzene ring. For example, in the cases of 6a and 7a, methyl and methoxyl groups exhibited an accelerating effect on the oxidation reaction, almost complete conversions and >98% selectivity could be obtained. Electron-withdrawing groups are less reactive, and thus longer reaction times and elevated amount of catalyst were needed to reach good conversions. Nitro moieties tolerated these reaction conditions to furnish the corresponding aldehydes and 3b in 100% conversion. Interestingly, compared to p-methyl benzyl alcohol, the m-methyl and o-methyl benzylic alcohols showed low activity under this condition, but with a higher catalyst loading, the complete oxidation could also be achieved. In the presence of 1,4-dioxane and with a higher ratio of AlBr3·6H2O (0.05 mmol), 4a and 5a were oxidized into corresponding aldehydes in 98% and 100% conversions, respectively.
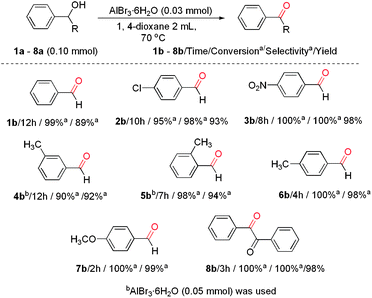 | ||
| Scheme 1 Results of oxidations of various alcohols catalyzed by AlBr3·6H2O. | ||
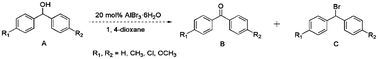
The scope of catalytic oxidation reaction with AlBr3·6H2O was also expended to several representative secondary benzyl alcohols. Different diphenylmethanols could successfully be transformed to the corresponding products with excellent conversions (>94%), but with the mixture of benzophenones and brominated derivatives (Table 1). In most cases, benzophenones were afforded as major products; the highest selectivity was nearly up to 80%. Notably, in the optimized reaction conditions, the bromonation process could not be prevented. To have more insight into the parallel transformations of the substrate to the reaction mechanism, modified reaction conditions were exploited, using quinone or MNP (2-methyl-2-nitrosopropane, C4H9NO) as a free radicals capture agent. Accordingly, the brominated product was formed predominantly, and accompanying with trace amounts of benzophenone. Similar results also could be verified in the case of performing the reaction in the N2 atmosphere, or injecting a small amount of water during the reaction. The above results clearly support the proposal that the oxidation reaction system probably resembles that of an N-bromosuccinimide (NBS) mediated process, which carries a bromine radical as the reaction initiator.
| Entry | Substrate | Additive | Conv.b (%) | Sele.b (%) | Yieldc (%) | ||
|---|---|---|---|---|---|---|---|
| B | C | B | C | ||||
| a Reaction conditions: alcohol (0.10 mmol), 1,4-dioxane 2 mL, AlBr3·6H2O (0.02 mmol), 70 °C, 4 h. b Conversion and selectivity were determined by GC analysis. c All yields are for pure, isolated products. d N2-balloon protection. e Quinone or MNP as a free radicals capture agent. | |||||||
| 1 |
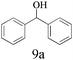
|
None | 100 | 47 | 53 | 44 | 50 |
| 2d | N2 | 93 | — | 99 | |||
| 3e | Quinone | 95 | 6 | 94 | |||
| 4e | MNP | 96 | 4 | 96 | |||
| 5 |

|
None | >99 | 60 | 40 | 56 | 35 |
| 6 |
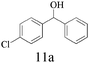
|
None | 94 | 77 | 23 | 73 | |
| 7 |
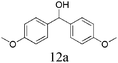
|
>98 | 63 | 37 | |||
In particular, molecular oxygen has received much attention as an ultimate oxidant, since it is photosynthesized by plants, produces little waste, is inexpensive and of larger atom efficiency than that of other oxidants. With this background in mind, in the course of our study, we have found that alcohols were oxidized to the corresponding products under visible light irradiation by a high-pressure xenon lamp and using air as an oxidant. The results clearly demonstrated that excellent yields and selectivity could be obtained in this photocatalyzed oxidation reaction (Scheme 2). More details about the reactions will be reported in our further studies.
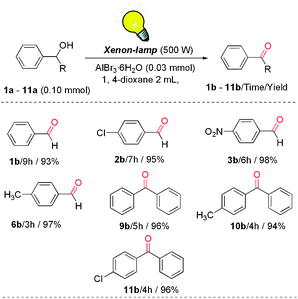
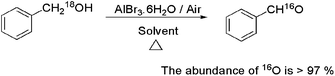
We then studied the aerobic oxidation with the isotope-labeled alcohols, as the substrate has been often used as a mechanistic probe to understand whether the final step of the alcohol oxidation is the dehydrogenation or oxygen transfer from oxidants to yield carbonyl compounds. A GC-MS detector indicated that the abundance of 16O in product was >97%. We also considered that the oxygen exchange between an isotope-labeled alcohol and AlBr3·6H2O occurred during the reaction, then 10 equiv. of H218O was injected, the result demonstrated that the 16O product was found to be >95%. So what should be concerned is that the oxygen exchange was not accessible. That is, in this oxidation process involving a selective cleavage of the C–O bond of the alcohol with concomitant formation of a new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, in the product, the oxygen atoms transfer from the oxidant. This interesting finding verified that in the process of alcohol oxidation, the exchange of oxygen atoms between alcohol and oxidant was taken place (Scheme 3).
O bond, in the product, the oxygen atoms transfer from the oxidant. This interesting finding verified that in the process of alcohol oxidation, the exchange of oxygen atoms between alcohol and oxidant was taken place (Scheme 3).
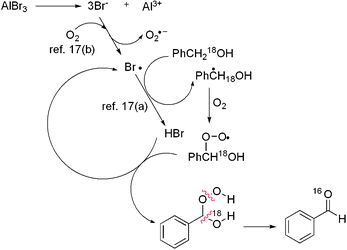
Here, we present in Scheme 4 what we assume is a plausible path of this oxidation. The radical species is thought to be generated by abstraction of a hydrogen radical with a bromine radical, formed by continuous aerobic oxidation of the bromo anion from AlBr3·6H2O. The radical species traps molecular oxygen to afford peroxy radical species, which subsequently transforms to benzaldehyde with the selective C–18O bond cleavage, yielding the no isotope-labeled product.
In summary, we have developed an efficient, inexpensive, clean, free radical initiated oxidation of alcohol protocol, which uses AlBr3·6H2O as the catalyst, the inexhaustible air as an oxidant, and dioxane as the solvent, and this method is insensitive to atmospheric moisture, and neither dried solvent is required, which also could provide the reasonable yields under relatively mild conditions. Most importantly, this protocol demonstrated excellent selectivity of the oxidation of primary benzyl alcohols; yielding benzaldehyde as a major product. This issue is regarded as the greatest obstacle for the bromides involved processes. The new form of oxidation reaction is also interesting in keeping with the notion of green chemistry due to non-use of transition metals and halogenated solvents, waste reduction. The scope, further mechanism, and synthetic application of this reaction are under investigation.
Experimental section
General information
AlBr3·6H2O was obtained from commercial sources. All solvents used were of analytical grade and were used as received. All of the alcohols used in the reaction were obtained from ABCR GmbH & Co. KG and used as received without further treatment. H218O was obtained from Huayi Isotope Co. All NMR spectra are recorded on MERCURY (400 MHz for 1H NMR, 100 MHz for 13C NMR) spectrometers; chemical shifts are expressed in ppm (δ units) relative to a TMS signal as internal reference in CDCl3. Gas chromatography (GC) analysis was performed on a Shimadezu GC-2010 equipped with a 15 m × 0.53 mm × 1.5 μm RTX-1 capillary column and a oxyhydrogen flame detector. GC/MS analysis was carried out on a trace HP GC6890/MS5973 equipped with a 25 m × 0.25 mm SE-54 column and a Shimadzu GC-16A gas chromatograph with a 3 m × 3 mm OV-17 column.General procedure for the oxidation of alcohols with AlBr3·6H2O
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After the first cycle, the reaction mixture was extracted from the reaction vessel by syringes, the yellow precipitates were washed with petroleum ether several times, then fresh solvent 1,4-dioxane (2 mL) and benzyl alcohol (0.10 mmol) were charged. Under the optimized reaction conditions, the conversion and selectivity were detected as 45% and 98% respectively. After the third cycle, the conversion and selectivity were maintained at 18% and 99%.
:1).
:1). After the first cycle, the reaction mixture was extracted from the reaction vessel by syringes, the yellow precipitates were washed with petroleum ether several times, then fresh solvent 1,4-dioxane (2 mL) and benzyl alcohol (0.10 mmol) were charged. Under the optimized reaction conditions, the conversion and selectivity were detected as 45% and 98% respectively. After the third cycle, the conversion and selectivity were maintained at 18% and 99%.
:1).
The NMR data for representative products
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 20674063 and 20774074), Specialized Research Fund for the Doctoral Program of Higher Education (20050736001) and Young Teacher Research Foundation of Northwest Normal University (NWNU-LKQN-08-8, NWNU-kjcxgc-03-73). We also thank Key Laboratory of Eco-Environment-Related Polymer Materials (Northwest Normal University), Ministry of Education, for financial support.Notes and references
- Z. Hou, N. Theyssen, A. Brinkmann and W. Leitner, Angew. Chem., Int. Ed., 2005, 44, 1346 CrossRef CAS.
- (a) G. Cainelli and G. Cardillo, Chromium Oxidants in Organic Chemistry, Springer, Berlin, 1984 Search PubMed; (b) W. J. Mijs and C. R. H. de Jonge, Organic Synthesis by Oxidation with Metal Compounds, Plenum Press, New York, 1986 Search PubMed.
- (a) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS; (b) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; (c) M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414 CrossRef CAS.
- (a) G.-J. ten Brink, I. W. C. E. Arends, M. Hoogenraad, G. Verspui and R. A. Sheldon, Adv. Synth. Catal., 2003, 345, 497 CrossRef CAS; (b) T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Adv. Synth. Catal., 2009, 351, 1890 CrossRef CAS.
- (a) B.-Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917 CrossRef CAS; (b) M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227 CrossRef CAS; (c) M. S. Sigman and D. R. Jensen, Acc. Chem. Res., 2006, 39, 221 CrossRef CAS.
- T. Iwahama, Y. Yosino, T. Keitoku, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2000, 65, 6502 CrossRef CAS.
- V. Kogan, M. M. Quintal and R. Neumann, Org. Lett., 2005, 7, 5039 CrossRef CAS.
- N. Jiang and A. J. Ragauskas, Org. Lett., 2005, 7, 3689 CrossRef CAS.
- (a) M. Colladon, A. Scarso and G. Strukul, Green Chem., 2008, 10, 793 RSC; (b) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636 CrossRef; (c) P. A. Shapley, N. Zhang, J. L. Allen, D. H. Pool and H.-C. Liang, J. Am. Chem. Soc., 2000, 122, 1079 CrossRef CAS.
- M. Guo and H. Li, Green Chem., 2007, 9, 421 RSC.
- (a) A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153 CrossRef CAS; (b) W. Adam, C. R. Saha-Moller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499 CrossRef CAS; (c) R. A. Sheldon, I. W. C. E. Arends, G.-J. ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774 CrossRef CAS.
- (a) Z. Wang, Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2008, 10, 1863 CrossRef CAS; (b) B. Karimi, G. R. Ebrahimian and H. Seradj, Org. Lett., 1999, 1, 1737 CrossRef CAS.
- Y. Xie, W. Mo, D. Xu, Z. Shen, N. Sun, B. Hu and X. Hu, J. Org. Chem., 2007, 72, 4288 CrossRef CAS.
- A. Dijksman, A. Marino-Gonzalez, A. Mairata i Payeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826 CrossRef CAS.
- (a) B. Betzemeier, M. Cavazzini, S. Quicib and P. Knochela, Tetrahedron Lett., 2000, 41, 4343 CrossRef CAS; (b) I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507 CrossRef CAS.
- (a) R. Liu, C. Dong, X. Liang, X. Wang and X. Hu, J. Org. Chem., 2005, 70, 729 CrossRef CAS; (b) R. Liu, X. Liang, C. Dong and X. Hu, J. Am. Chem. Soc., 2004, 126, 4112 CrossRef CAS.
- (a) A. Itoh, S. Hashimoto, K. Kuwabara, T. Kodama and Y. Masaki, Green Chem., 2005, 7, 830 RSC; (b) S. Hirashima and A. Itoh, Green Chem., 2007, 9, 318 RSC.
- (a) Z. Lei and R. Wang, Catal. Commun., 2008, 9, 740 Search PubMed; (b) S. Wu, H. Ma and Z. Lei, Tetrahedron, 2010, 66, 8641 Search PubMed.
- P. Yan, R. Wang, S. Wu and Z. Lei, Catal. Commun., 2008, 9, 406 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1cy00165e |
| This journal is © The Royal Society of Chemistry 2011 |