
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereomutation and chiroptical bias in the kinetically controlled supramolecular polymerization of cyano-luminogens†
Lucía
López-Gandul
a,
Cristina
Naranjo
a,
Cecilia
Sánchez
a,
Rafael
Rodríguez
b,
Rafael
Gómez
a,
Jeanne
Crassous
 *b and
Luis
Sánchez
*a
*b and
Luis
Sánchez
*a
aDepartamento de Química Orgánica, Facultad; de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: lusamar@ucm.es
bUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000, Rennes, France. E-mail: jeanne.crassous@univ-rennes1.fr
First published on 7th September 2022
Abstract
The synthesis of two pairs of enantiomeric cyano-luminogens 1 and 2, in which the central chromophore is a p-phenylene or a 2,5-dithienylbenzene moiety, respectively, is described and their supramolecular polymerization under kinetic and thermodynamic control investigated. Compounds 1 and 2 form supramolecular polymers by quadruple H-bonding arrays between the amide groups and the π-stacking of the central aromatic moieties. In addition, the peripheral benzamide units are able to form intramolecularly H-bonded pseudocycles that behave as metastable monomer M* thus affording kinetically and thermodynamically controlled aggregated species AggI and AggII. The chiroptical and emissive features of compounds 1 and 2 strongly depend on the aggregation state and the nature of the central aromatic unit. Compounds 1 exhibit a bisignated dichroic response of different intensity but with similar sign for both AggI1 and AggII1 species, which suggests the formation of helical aggregates. In fact, these helical supramolecular polymers can be visualized by AFM imaging. Furthermore, both AggI and AggII species formed by the self-assembly of compounds 1 show CPL (circularly polarized light) activity of opposite sign depending on the aggregation state. Thienyl-derivatives 2 display dissimilar chiroptical, morphological and emissive characteristics for the corresponding kinetically and thermodynamically controlled aggregated species AggI and AggII in comparison to those registered for compounds 1. Thus, a stereomutation phenomenon is observed in the AggI2 → AggII2 conversion. In addition, AggI2 is arranged into nanoparticles that evolve to helical aggregates to afford AggII2. The dissimilar chiroptical and morphological features of AggI2 and AggII2 are also appreciated in the emissive properties. Thus, whilst AggI2 experiences a clear AIE (aggregation induced emission) process and CPL activity, the thermodynamically controlled AggII2 undergoes an ACQ (aggregation caused quenching) process in which the CPL activity is cancelled.
Introduction
Self-assembly of suitable building blocks in an organized fashion is an attractive approach to achieve functional materials holding applicability in a number of research areas like biomedicine, photocatalysis, adhesives or energy conversion.1 All these applications rely on the organized arrangement of the constitutive units to finally yield the functional aggregate. In this regard, supramolecular polymerization has emerged as a useful strategy to achieve the challenging goal of attaining complex, functional materials.2 Especially relevant in the field of supramolecular polymers is the incorporation of asymmetry elements in the self-assembling units. The supramolecular polymerization of these chiral monomeric species results in the efficient transfer of asymmetry from the monomers to the aggregated species that finally yield helical, and therefore chiral aggregates.3 These helical aggregates have been exploited to perform detailed studies on transfer and amplification of asymmetry,4 as well as in the preparation of optical and electronic devices.5 It is worth mentioning that a vast majority of these applications of functional supramolecular polymers are studied under thermodynamic control. However, kinetically controlled chemical processes are common in synthetic protocols, material science and in biological phenomena and have been also reported for supramolecular polymers.6 Thus, kinetically controlled supramolecular polymerization has derived in differentiation events,7 living and seeded processes,7 and also in stereomutation phenomena.7a,9 Importantly, a key issue on the characterization and study of the thermodynamic or kinetic supramolecular polymers decorated with elements of asymmetry is the study of their chiroptical properties, the most common being electronic circular dichroism (CD). However, in the case of chiral, emissive supramolecular polymers, the generation of efficient circularly polarized light (CPL) could open new avenues for their use in technological applications such as data storage, biological sensing and next-generation displays.10 In fact, a number of recent examples report on the behaviour of emissive supramolecular polymers as efficient CPL-emitters with remarkable values of luminescence dissymmetry factors (glum)11 comparable to those reported for molecular systems.12To expand the applicability of CPL-emitters, biasing the CPL response by external stimuli like solvent, temperature, concentration, or chemical complexation, is a challenging issue not largely investigated.13 It is in this context in which this work is dovetailed with. Thus, herein, we report on the supramolecular polymerization of chiral cyano-substituted divinylene arene-based luminogens (compounds 1 and 2 in Fig. 1) able to form two well-defined aggregated species AggI and AggII. The chiroptical features, both CD and CPL, of these highly emissive self-assembling units are kinetically biased due to the formation of intramolecular H-bonded species (Fig. 2). The CD studies performed with these luminogens demonstrate the evolution of the dichroic response depending on time and solvent composition. Whilst p-phenylene cyano-derivatives 1 experience a clear variation of the dichroic response without stereomutation, the thienyl derivatives 2 show a clear increase of the dichroic response accompanied by stereomutation. The CPL characteristics of these luminogens are also kinetically biased. The initially formed aggregated species AggI1 and AggI2 show a CPL response that decreases in intensity and experiences a sign switch upon forming the thermodynamically aggregated species for AggII1 and it is cancelled for AggII2. Atomic force microscopy (AFM) imaging demonstrates the changes associated to the kinetic evolution of AggI and AggII. Both thermodynamic AggII show the formation of thick filaments that involves the generation of helical aggregates. However, AggI1 and AggI2 present dissimilar morphology, the former forming thin helical filaments but the latter giving rise to nanoparticles (Fig. 1). Hence, the results presented herein complement the formation of highly luminescent organogels previously reported for the achiral congeners of cyano-luminogens 1 and 2 and contribute to expand the knowledge and potential applicability of kinetically-controlled supramolecular polymers.14
 | ||
| Fig. 1 Chemical structures of the described chiral cyano-luminogens (central part) and schematic representation of the CPL-emitting kinetic and thermodynamically controlled aggregated structures formed by 1 and 2. | ||
 | ||
| Fig. 2 (a) Partial FTIR spectra of (R)-1 in CHCl3 and MCH; (b) schematic illustration of the equilibrium between the two monomeric species, the totally unbounded and the intramolecularly H-bonded, of cyano-luminogens 1; (c) partial 1H-NMR spectra of (S)-1 at different temperatures (300 MHz, CDCl3, cT = 1 mM). | ||
Results and discussion
Synthesis and self-assembly in solution
The synthesis of the cyano-luminogens 1 and 2 was accomplished by following a similar protocol to that carried out for the achiral congeners.14 Thus, the alkylation reaction of methyl 3,4,5-trihydroxybenzoate with the chiral (S or R)-3,7-dimethyloctyl 4-methylbenzenesulfonate, subsequent basic hydrolysis of the ester group and amidation reaction with ethane-1,2-diamine, yields the monoamides 3. These monoamides are reacted with 4-(cyanomethyl)benzoic acid 4 to yield the corresponding cyanomethyl derivatives 5. Finally, a double Knoevenagel reaction of these cyano compounds 5 with terephthaldehyde or thiophene-2,5-dicarbaldehyde readily yields the chiral, cyano-luminogens 1 and 2 (Scheme S1†). All the new compounds have been characterized by utilizing the usual spectroscopic techniques (see, ESI†).The self-assembly of compounds 1 and 2 has been investigated by utilizing a combination of spectroscopic techniques and taking into account that the supramolecular polymerization of the achiral congeners of the reported cyano-derivatives has been described as a cooperative process yielding highly stable, fibrillar aggregates.14 Thus, we have firstly registered the UV-vis spectra of (S)-1 and (R)-2 in a good solvent (CHCl3) and in a bad solvent (methylcyclohexane, MCH). As expected, the absorption pattern of these chiral compounds shows maxima at λ = 371 and 411 nm for (S)-1 and (R)-2, respectively, in CHCl3. A clear hypsochromic effect is observed in MCH with maxima at λ = 338 and 386 nm for (S)-1 and (R)-2, respectively, that suggests the formation of H-type aggregates and in good agreement with that reported for the achiral congeners (Fig. S1†).14,15 The formation of these H-type aggregates could be justified by considering the non-covalent interaction of the self-assembling units by the π-stacking of the aromatic units and the formation of H-bonding arrays between the four amide groups. In fact, concentration dependent 1H-NMR spectra for (S)-1 and (R)-2 in CDCl3 as solvent show the shielding of the corresponding resonances upon increasing the concentration and the downfield shift of the amide protons, that reinforces this hypothesis (Fig. S2†). To further demonstrate the formation of these H-bonding arrays, we have also registered FTIR spectra in these good and bad solvents (CHCl3 and MCH, respectively). In the latter, the bands corresponding to the N–H stretching and amide I appear at ∼3290 and ∼1634 cm−1, corroborating the formation of the intermolecular H-bonds between the amide functional groups (Fig. 2a and S3†).16 In the former, however, two N–H stretching bands are observed at ∼3453 and ∼3343 cm−1 due to the presence of free monomeric species and also metastable monomeric species M* formed by the intramolecular H-bonding of the inner or outer carbonyls of the amides and their outer and inner N–H (Fig. 2 and S3†).16
The formation of the intramolecularly H-bonded 7-membered pseudocyles has been corroborated by variable temperature (VT) 1H-NMR spectroscopy in CDCl3. In these experiments, all the resonances observed in the spectra experience no shift upon heating the sample. This effect, however, is the contrary for those resonances corresponding to the inner and outer amides (filled and hollow red circles in Fig. 2c and S3c†). These resonances shift upfield upon heating the sample due to the rupture of the two possible intramolecular H-bonds between the amide functional groups of each peripheral side chain. The formation of the possible M* can be justified by considering that both amides shield upon heating (Fig. 2c and S3c†).
The formation of these metastable monomeric species M* has been reported to have a strong influence on the kinetics of the supramolecular polymerization of those self-assembling units endowed with the peripheral N-(2-benzamidoethyl)-3,4,5-trialkoxybenzamide moieties.7c,8b,17 In fact, Fernández and coworkers have very recently demonstrated the ability of self-assembling units decorated with these peripheral moieties to generate supramolecular polymers with intermolecular H-bonding interactions in a consecutive manner, the first step being the formation of dimers in which intra- and intermolecular H-bonding interactions between the amide groups are operative.18 Consequently, we have not used the commonly utilized variable temperature spectroscopic (UV-vis or CD) techniques to elucidate the supramolecular polymerization mechanism, but we have utilized denaturation experiments to derive, if possible, the thermodynamic parameters associated to the supramolecular polymerization of the reported cyano-luminogens (vide infra).
Chiroptical properties and morphology of the aggregated species of the cyano-luminogens 1 and 2
To elucidate the chiroptical properties of the cyano-luminogens 1 and 2, we have firstly carried out CD measurements in MCH as solvent. The high stability of the aggregated species of these compounds in MCH has been demonstrated by registering CD spectra at a total concentration, cT, as low as 0.5 μM. Only at these highly diluted conditions, the CD response is cancelled at high temperatures (Fig. S4†). Both compounds 1 and 2 present clear bisignated CD spectra, with maxima at λ = 348 and 325 nm and a zero-crossing point at λ = 335 nm for 1 and λ = 396 and 371 nm and a zero-crossing point at λ = 379 nm for 2, that suggest the formation of helical aggregates of opposite handedness for each enantiomer (Fig. S5†).The high stability of the supramolecular polymers formed by cyano-derivatives 1 and 2 prompted us to utilize the denaturation (SD) model to achieve a detailed investigation of the supramolecular polymerization mechanism. In this SD model, diluted solutions at equal cT of the investigated self-assembling units in a good (CHCl3) and in a bad (MCH) solvent, are mixed together to achieve a complete binding isoterm.19 During the application of the denaturation model, we have noticed an uncommon pattern in the corresponding CD spectra of the mixtures that follow a dissimilar trend for compounds 1 and 2. In the case of p-phenylenevinylenes 1, the dichroic response of freshly prepared solutions in pristine MCH is less intense than that registered for MCH/CHCl3 mixtures with a low ratio of the good solvent. The dichroic response gradually increases until the MCH/CHCl3 mixtures reaches around a 25% of the good solvent, the dichroic sign being the same in all the experimental conditions (Fig. 3a and S6†). Increasing the percentage of CHCl3 gradually decreases the intensity of the dichroic signal, that is completely cancelled when the ratio of the good solvent is higher than 50% (Fig. 3a and S6†). The corresponding UV-vis spectra at these conditions show the formation of three different species: the monomeric species (red line in Fig. 3b) and two different aggregated species (green and blue lines in Fig. 3b). Interestingly, aging these solutions for 24 h affords a gradual decreasing of the dichroic response upon adding increasing amounts of CHCl3 (Fig. 3c and S6†). In addition, the corresponding UV-vis spectra show only the absorption pattern ascribable to the monomeric (red line in Fig. 3d) and aggregated (green and blue lines in Fig. 3d) species. This behaviour is diagnostic of the operation of a kinetically controlled supramolecular polymerization that, in certain conditions of solvent composition and time, yields an aggregated species AggI that evolves with time to a different aggregated species AggII (Fig. 1).
 | ||
| Fig. 3 CD (a and c) and UV-vis (b and d) spectra of luminogen (S)-1 in MCH/CHCl3 mixtures freshly prepared (a and b) and upon aging for 24 h (c and d) (20 °C; cT = 10 μM). Arrows indicate the changes upon adding the solutions of 1 in CHCl3 (e and f) AFM images of the kinetically controlled AggI (e) and thermodynamically controlled AggII (f) of compound (S)-1 (MCH/CHCl3 9/1 as solvent; HOPG as surface; cT = 10 μM). | ||
Upon reaching the thermodynamic equilibrium, it is possible to apply the SD model to derive the thermodynamic parameters. Thus, plotting the degree of aggregation α, calculated form the variation of the intensity of the dichroic response, observed from the corresponding CD spectra versus the molar fraction of CHCl3 affords a non-sigmoidal curve that implies a cooperative supramolecular polymerization with a relatively large degree of cooperativity σ and a large value of m, that expresses the dependence of ΔG on the solvent (Fig. 3a and S7a† and Table 1). Similar findings have been extracted from the SD model applied to the UV-vis spectra (Fig. 3b and S7b†).
To visualize the morphologic differences of AggI and AggII, we have registered the corresponding AFM images. Thus, isolated fibrillar aggregates of several micrometers of length and typical height of ∼3 nm are visualized by spin-coating a freshly prepared solution of (S)-1 in a 9/1 MCH/CHCl3 mixture at cT = 10 μM (Fig. 3e and S8†). In the case of AggII, the morphology visualized by AFM imaging demonstrates the bundling effect experienced by the initially formed fibers, corresponding to AggI. Thus, AggII appears as thicker fibers of several micrometers of length and typical height of ∼15 nm that would imply a helix to super-helix transition to afford chiral structures (Fig. 3f and S9†).20 In both cases, the rope-like morphology of the fibrillar aggregates corroborates the helical character of both AggI and AggII formed by p-phenylenevinylenes 1.
Upon investigating the dynamic character of the supramolecular polymerization of cyano-luminogens 1, we have also investigated the kinetically-controlled self-assembly of the thienyl-based luminogens 2. In good analogy to that observed for compounds 1, the denaturation experiments performed with 2 evidence drastic spectroscopic changes, especially in the dichroic response. Noteworthy, a first dichroic characteristic of compounds 2 in comparison to 1 is that the CD response is much less intense in pristine MCH (Fig. S5†). Initially, the addition of increasing amounts of 2 in the good solvent CHCl3 to a solution in the bad solvent MCH provokes a slight increase in intensity of the CD response. However, upon adding around 30% of solutions of 2 in CHCl3, a complete inversion of the dichroic response is clearly observed (Fig. 4 and S10†). This stereomutation, observed for both enantiomers (S)-2 and (R)-2, could be ascribable to the kinetically controlled self-assembly of these cyano-luminogens (Fig. S11†).9 In fact, the dichroic response of compounds 2 experiences a signal inversion concomitant with a strong increase in intensity only after 30 min upon mixing both solutions (Fig. S11a and S11b†) or after heating the solutions up to 60 °C, to favour the complete disassembly, and cooling them down to 10 °C to induce the reassembly (Fig. S10b and S11c†). In all the samples with relatively low percentage of CHCl3 (10–30%), the intensity of the dichroic response drastically increases in comparison to that registered for pristine MCH. The evolution of the dichroic response versus time is fairly visualized by registering the CD spectra of (S)-2 and (R)-2 in a 7/3 MCH/CHCl3 mixture that reaches the maximum intensity upon aging the sample for ∼30 min (Fig. S13†). As expected, the addition of higher amounts of chloroform solutions of 2 provokes the gradual disassembly of the aggregated species (Fig. S12†). Unfortunately, the wavy variation of the dichroic response versus the molar fraction of CHCl3 impedes to utilize these data for deriving the corresponding thermodynamic parameters by applying the SD model. However, it is possible to apply the SD model to the denaturation experiments performed upon 24 h by using the corresponding UV-vis spectra. In good accord to compounds 1, the UV-vis spectra of the freshly prepared solutions show the presence of three different species: the monomers (red line in Fig. 4b and d), AggI, that is not detected upon 24 h (green line in Fig. 4b) and AggII (blue line in Fig. 4b and d). The hipsochromic effect observed in the absorption spectra upon increasing the fraction of the bad solvent MCH suggests the formation, as in the case of compounds 1, of H-type aggregates. Plotting the variation of the degree of aggregation α versus the molar fraction of CHCl3 displays a non-sigmoidal curve that can be fitted to the SD model (Fig. S14†). The corresponding thermodynamic parameters are collected in Table 1. In this case, the ΔG′ value is higher to that derived for compound (S)-1, but the degree of cooperativity, σ, is higher.
 | ||
| Fig. 4 CD (a and c) and UV-vis (b and d) spectra of luminogen (R)-2 in MCH/CHCl3 mixtures freshly prepared (a and b) and upon aging for 24 h (c and d) (20 °C; cT = 10 μM). Arrows indicate the changes upon adding the solutions of 2 in CHCl3. (e and f) AFM images of the kinetically controlled AggI (e) and thermodynamically controlled AggII (f) of compound (R)-2 (MCH/CHCl3 9/1 as solvent; HOPG as surface; cT = 10 μM). | ||
We have also visualized the morphology of the aggregated species generated from compounds 2 by AFM imaging onto HOPG as surface. Unlike the fibrillar nature of AggI formed by compounds 1, the AFM images of AggI of compound (R)-2 exhibits the formation of a large number of nanoparticles (Fig. 4e and S15†) that evolve with time to form long fibrillar aggregates ascribable to AggII species (Fig. 4f and S16†). These fibrillar aggregates present several micrometers length and heights of ∼2.5 and ∼5 nm, the latter implying the intertwining effect also observed in the AggII species from phenylenevinylenes 1.
The spectroscopic and morphologic differences found for AggI and AggII species of luminogens 1 and 2 could be justified by considering the above-mentioned ability of the metastable monomeric species M*, in which the intramolecularly H-bonded pseudocycle is able to form intermolecular H-bonding interactions, to generate the corresponding AggI species.18 These AggI species would be characterized by the intermediate spectra observed in the freshly prepared mixtures (green lines in Fig. 3b and 4b). Furthermore, the presence of two pseudocycles per monomeric unit, and the subsequent intermolecular interaction between them, would yield the kinetically controlled AggI species (Fig. S17†). The further evolution of the AggI species relies on the rupture of the intramolecular H-bonding interactions to form highly stable tetrafold intermolecular H-bonding arrays that afford to AggII species (Fig. S17†).
Emissive properties of the cyano-luminogens 1 and 2
The achiral congeners of cyano-luminogens 1 and 2 have been reported to behave as highly emissive soft-materials with color-tunable features and aggregation-induced enhanced emission (AIE).14,21 Noteworthy, the emission studies developed for such achiral cyano-luminogens were carried out in mixtures 7/3 MCH/1,2-dichloroethane (DCE) without detecting the formation of the two aggregated species AggI and AggII.14 Taking into account these emissive characteristics, we have also registered the emission spectra of kinetically and thermodynamically aggregated species formed by compounds 1 and 2. Similarly to that observed for the dichroic features described in the previous part, the emissive features of the p-phenylenevinylenes 1 differ to those observed for the thienyl-derivatives 2. Fig. 5a displays the photoluminescence (PL) spectra of freshly prepared and aged diluted solutions (cT = 10 μM) of compound (S)-1 in a 9/1 MCH/CHCl3 mixture, together with the PL spectrum registered in CHCl3, that corresponds to the monomeric species. The latter spectrum shows two peaks at λ = 486 and 436 nm, in good agreement with that previously described for the achiral congener (Fig. 5a).14 The supramolecular polymerization of (S)-1 provokes a strong increase of the PL intensity accompanied by a bathochromic shift, diagnostic of the operation of an AIE phenomenon.21 Importantly, the PL spectrum of both AggI1 and AggII1 differs in shape but not in intensity. Thus, the PL spectrum of AggI1 species shows a broad emission band centered at λ = 543 nm. Aging this sample to generate AggII1 changes the PL spectrum which, for this aggregated species, displays a fine structure with an intense band centered at λ = 490 nm and two shoulders at λ = 524 and 466 nm (Fig. 5a). The different emissive properties are also well observed in the corresponding Commission International de l' Eclairage (CIE) that present CIE values for the monomeric, AggI1 and AggII1 species of (0.15, 0.15), (0.39, 0.52) and (0.21, 0.40), respectively (Fig. S18†).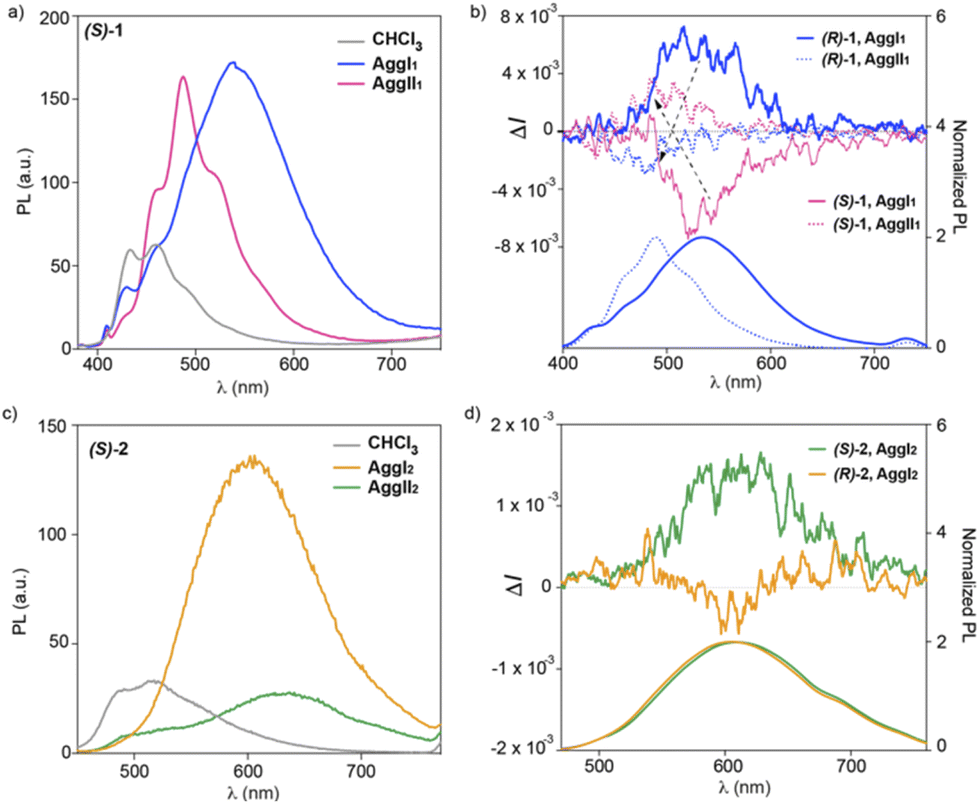 | ||
| Fig. 5 PL (a and c) and CPL/PL (b and d) of (S)-1 (a and b) and (S)-2 (c and d) spectra in monomeric and aggregated states (CHCl3 and 9/1 MCH/CHCl3 mixture, respectively) (experimental conditions: cT = 10 μM; λexc = 365 nm for (S)-1 and 390 nm for (S)-2). Dashed arrows in panel (b) indicate the inversion of the CPL sign. | ||
The emissive characteristics of the chiral cyano-derivatives (S)-1 and (R)-1 prompted us to register the CPL activity displayed by both the monomeric and aggregated species. A first remarkable finding is that, whilst the monomeric species shows no CPL activity in solution, the supramolecular polymerization of (S)-1 and (R)-1 results in intense CPL spectra with opposite sign for AggI and AggII species (Fig. 5b). Thus, the CPL spectra of the kinetically controlled AggI species shows a broad CPL band centered at λ ∼ 560 nm with a positive sign for (R)-1 and a negative sign for (S)-1, in good agreement to that observed in the CD spectra (Fig. S5†). These kinetically controlled AggI species exhibit a remarkable dissymmetry factor glum ∼ 0.007 (λexc = 365 nm) (Fig. 5b). Interestingly, aging the 9/1 MCH/CHCl3 solution of AggI for 24 h, to ensure the complete conversion to AggII, results in CPL active supramolecular aggregates in which a sign inversion is detected. Thus, the AggII species of compound (R)-1 shows negative CPL signal, the opposite being detected for (S)-1. Furthermore, the glum slightly decreases to values ∼ 0.004 (Fig. 5b). The CPL spectra demonstrate that the CPL activity of the kinetically controlled AggI species follows the helicity dictated by the stereogenic centres located at the peripheral side chains. The intertwining effect responsible for the formation of the AggII species changes the CPL sign. This CPL sign inversion, well documented for molecular species,22 is rarely observed for supramolecular polymers.23
Finally, we have also monitored the emissive features of thienyl-derivatives 2 that, similarly to that observed in the CD spectra, present noticeable changes regarding compounds 1. The PL spectra of compounds 2 in the molecularly dissolved state present two maxima of low intensity at λ = 520 and 488 nm that becomes a very broad and intense band centered at λ = 607 nm for the kinetically controlled AggI2 species (Fig. 5c). This strong increase in the PL intensity is diagnostic of an AIE process as it has been reported for the achiral congener.14 To our surprise, the evolution of AggI2 species to the thermodynamically controlled AggII2 species results in a significant quenching of the intensity that implies an aggregation quenching effect (ACQ) related to the kinetically aggregated species (Fig. 5c). In this case, the colour changes are less pronounced with CIE coordinates closer for the monomeric, AggI2 and AggII2 species [(0.51, 0.32), (0.42, 0.52) and (0.41, 0.67), respectively] (Fig. S18†).
The emissive properties of compounds 2 are also reflected in the CPL activity of the aggregated species of these cyano-luminogens. Thus, neither the monomeric species nor the thermodynamically controlled supramolecular polymers AggII2 present CPL activity. In fact, the CPL spectra of AggII2 for both (R)-2 and (S)-2 are very small and not mirror images and only a positive activity is observed for these aggregates ascribable to potential artefacts, linear anisotropic effects or photoselection, derived from the aggregation process (Fig. S19†).24 Nevertheless, the large increase of the PL observed for the kinetically controlled supramolecular polymers formed by compounds 2 is also observed in the CPL activity shown by these aggregates whose CPL spectra are almost mirror images for (R)-2 and (S)-2 with glum ∼ 0.002 (λexc = 390 nm) (Fig. 5d).
Conclusions
In conclusion, we report herein the synthesis of two pairs of enantiomeric cyano-luminogens 1 and 2 in which the central chromophore is a p-phenylene or a 2,5-dithienylbenzene moiety, respectively. The decoration of these chromophoric units with four benzamide units per monomeric unit favours their supramolecular polymerization in paraffinic solvents like MCH and also in mixtures of MCH/CHCl3 by the formation of quadruple H-bonding arrays between the amide groups and the π-stacking of the central aromatic moieties. These two benzamide units, separated by two methylenes, are able to form intramolecularly H-bonded pseudocycles that behave as metastable monomeric species M*. These M* species generate kinetically and thermodynamically controlled aggregated species AggI and AggII. The presence of point chirality in the peripheral side chains gives rise to helical aggregates exhibiting rich chiroptical features depending on the aggregation state and the central aromatic unit. The p-phenylene-based derivatives 1 exhibit a clear bisignated dichroic response of different intensity but similar sign for both AggI and AggII species and remarkable emissive features. The formation of helical (AggI1) and superhelical (AggII1) aggregates is confirmed by AFM imaging. Furthermore, both AggI1 and AggII1 species show CPL activity of opposite sign depending on the aggregation state. Despite the structural similarity between compounds 1 and 2, the latter displays a dissimilar chiroptical, morphological and emissive characteristics for the corresponding kinetically and thermodynamically controlled aggregated species AggI and AggII in comparison to those registered for compounds 1. Thus, a stereomutation phenomenon is observed in the AggI2 → AggII2 with opposite dichroic response of each enantiomer depending on the aggregation state. In addition, AggI2 is arranged into nanoparticles that evolve to helical aggregates to afford AggII2. The dissimilar chiroptical and morphological features of AggI2 and AggII2 species are also appreciated in the emissive properties. Thus, whilst AggI2 experiences a clear AIE process and CPL activity, the thermodynamically controlled AggII2 undergoes an aggregation caused quenching (ACQ) process regarding AggI2 in which the CPL activity vanishes. Therefore, the results shed light on the modulation of relevant optical and chiroptical properties of luminescent self-assembling units able to form highly organized supramolecular structures depending on the aggregation state and contribute to expand the knowledge and potential applicability of kinetically and thermodynamically-controlled supramolecular polymers.Data availability
All data associated with this article have been included in the main text and ESI.†Author contributions
Lucía López-Gandul, Cristina Naranjo and Cecilia Sánchez: synthesis, spectroscopic characterization, supramolecular polymerization mechanisms, AFM imaging and writing. Rafael Rodríguez: chiroptical and emissive properties and writing; Rafael Gómez, Jeanne Crassous and Luis Sánchez: conceptualization, writing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the MCIU of Spain (PID2020-113512GB-I00 and RED2018-102331-T) and the Comunidad de Madrid (S2018/NMT-4389) is acknowledged. R. R. thanks Xunta de Galicia for postdoctoral fellowship. J. C. acknowledges the Ministère de l'Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS). J. C. warmly thanks Dr Kais Dhbaibi for his technical assistance.Notes and references
- (a) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed; (b) Z. Álvarez, A. N. Kolberg-Edelbrock, I. R. Sasselli, J. A. Ortega, R. Qiu, Z. Syrgiannis, P. A. Mirau, F. Chen, S. M. Chin, S. Weigand, E. Kiskinis and S. I. Stupp, Science, 2021, 374, 848 CrossRef PubMed; (c) O. Dumele, L. Đorđević, H. Sai, T. J. Cotey, M. H. Sangji, K. Sato, A. J. Dannenhoffer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 3127 CrossRef CAS; (d) W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579 CrossRef CAS; (e) A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H. W. Schmidt and R. Hildner, Nature, 2015, 523, 196 CrossRef CAS PubMed.
- (a) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed; (b) M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38 CrossRef CAS.
- (a) A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS PubMed; (b) Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem.–Eur. J, 2019, 25, 5848 CrossRef CAS PubMed.
- (a) E. E. Greciano, J. Calbo, J. Buendía, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2019, 141, 7463 CrossRef CAS PubMed; (b) F. García and L. Sánchez, J. Am. Chem. Soc., 2012, 134, 734 CrossRef PubMed; (c) M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. van der Schoot, A. R. A. Palmans, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 611 CrossRef CAS PubMed.
- (a) R. Sethy, J. Kumar, R. Métivier, M. Louis, K. Nakatani, N. M. T. Mecheri, A. Subhakumari, K. G. Thomas, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2017, 56, 15053 CrossRef CAS; (b) A. K. Mondal, M. D. Preuss, M. L. Ślęczkowski, T. K. Das, G. Vantomme, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2021, 143, 7189 CrossRef CAS PubMed.
- (a) J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730 CrossRef CAS PubMed; (b) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476 RSC.
- (a) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. de Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed; (b) T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493 CrossRef CAS PubMed; (c) E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517 CrossRef CAS PubMed.
- (a) J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646 CrossRef CAS PubMed; (b) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed; (c) J. S. Valera, R. Gómez and L. Sánchez, Small, 2018, 14, 1702437 CrossRef PubMed; (d) S. Sarkar, A. Sarkar, A. Som, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2021, 143, 11777 CrossRef CAS PubMed.
- J. S. Valera, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2019, 58, 510 CrossRef CAS PubMed.
- (a) J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef; (b) M. Schulz, F. Balzer, D. Scheunemann, O. Arteaga, A. Lützen, S. C. J. Meskers and M. Schiek, Adv. Funct. Mater., 2019, 29, 1900684 CrossRef; (c) L. Wan, J. Wade, F. Salerno, O. Arteaga, B. Laidlaw, X. Wang, T. Penfold, M. J. Fuchter and A. J. Campbell, ACS Nano, 2019, 13, 8099 CrossRef CAS PubMed; (d) Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- (a) E. E. Greciano, R. Rodríguez, K. Maeda and L. Sánchez, Chem. Commun., 2020, 56, 2244 RSC; (b) J. S. Kang, S. Kang, J.-M. Suh, S. M. Park, D. K. Yoon, M. H. Lim, W. Y. Kim and M. Seo, J. Am. Chem. Soc., 2022, 144, 2657 CrossRef CAS PubMed.
- (a) E. S. Gauthier, L. Abella, N. Hellou, B. Darquié, E. Caytan, T. Roisnel, N. Vanthuyne, L. Favereau, M. Srebro-Hooper, J. A. G. Williams, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 8394 CrossRef CAS PubMed; (b) E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346 CrossRef PubMed; (c) M. A. Medel, R. Tapia, V. Blanco, D. Miguel, S. P. Morcillo and A. Gónzalez Campaña, Angew. Chem., Int. Ed., 2021, 60, 6094 CrossRef CAS PubMed.
- (a) H. Maeda, K. Bando, K. Shimomura, I. Yamada, M. Naito, H. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS PubMed; (b) H. Fan, H. Jiang, X. Zhu, Z. Guo, L. Zhang and M. Liu, Nanoscale, 2019, 11, 10504 RSC; (c) M. Louis, R. Sethy, J. Kumar, S. Katao, R. Guillot, T. Nakashima, C. Allain, T. Kawai and R. Metivier, Chem. Sci., 2019, 10, 843 RSC; (d) J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783 CrossRef CAS PubMed; (e) D. Niu, Y. Jiang, L. Ji, G. Ouyang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 5946 CrossRef CAS.
- F. Aparicio, S. Cherumukkil, A. Ajayaghosh and L. Sánchez, Langmuir, 2016, 32, 284 CrossRef CAS.
- (a) M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2021, 143, 13281 CrossRef; (b) N. Bäumer, K. K. Kartha, S. Buss, I. Maisuls, J. P. Palakkal, C. A. Strassert and G. Fernández, Chem. Sci., 2021, 12, 5236 RSC; (c) H. Wang, Y. Zhang, Y. Chen, H. Pan, X. Ren and Z. Chen, Angew. Chem., Int. Ed., 2020, 59, 5185 CrossRef CAS PubMed.
- (a) M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092 CrossRef CAS PubMed; (b) E. E. Greciano, S. Alsina, G. Ghosh, G. Fernández and L. Sánchez, Small Methods, 2020, 4, 1900715 CrossRef CAS; (c) E. E. Greciano, M. A. Martínez, S. Alsina, A. Laguna and L. Sánchez, Org. Chem. Front., 2021, 8, 5328 RSC.
- E. E. Greciano and L. Sánchez, Chem.–Eur. J., 2016, 22, 13724 CrossRef CAS PubMed.
- J. Matern, Z. Fernández, N. Bäumer and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202203783 CAS.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed.
- M. Hifsudheen, R. K. Mishra, B. Vedhanarayanan, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2017, 56, 12634 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- (a) K. Takaishi, K. Iwachido and T. Ema, J. Am. Chem. Soc., 2020, 142, 1774 CrossRef CAS PubMed; (b) M.-Y. Zhang, X. Liang, D.-N. Ni, D.-H. Liu, Q. Peng and C.-H. Zhao, Org. Lett., 2021, 23, 2 CrossRef CAS PubMed; (c) K. Takaishi, M. Yasui and T. Ema, J. Am. Chem. Soc., 2018, 140, 5334 CrossRef CAS PubMed; (d) K. Takaishi, K. Iwachido, R. Takehana, M. Uchiyama and T. Ema, J. Am. Chem. Soc., 2019, 141, 6185 CrossRef CAS PubMed; (e) E. Yen-Pon, F. Buttard, L. Frédéric, P. Thuéry, F. Taran, G. Pieters, P. A. Champagne and D. Audisio, JACS Au, 2021, 1, 807 CrossRef CAS PubMed.
- (a) R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709 CrossRef PubMed; (b) S. Fa, T. Tomita, K. Wada, K. Yasuhara, S. Ohtani, K. Kato, M. Gon, K. Tanaka, T. Kakuta, T. Yamagishi and T. Ogoshi, Chem. Sci., 2022, 13, 5846 RSC; (c) F. Gan; C. Shen and H. Qiu, Circularly Polarized Luminescence of Aggregation-induced Emission Materials, Handb. Aggregation-Induced Emiss., 2022, vol. 3, pp. 27–56 Search PubMed.
- (a) I. Tinoco Jr, B. Ehrenberg and I. Z. Steinberg, J. Chem. Phys., 1977, 66, 916 CrossRef; (b) H. P. J. M. Dekkers, P. F. Moraal, J. M. Timper and J. P. Riehl, Appl. Spectrosc., 1985, 39, 818 CrossRef CAS; (c) E. Castiglioni, S. Abbate, F. Lebon and G. Longhi, Chirality, 2012, 24, 725 CrossRef CAS PubMed; (d) G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: These might include comments relevant not central to the matter under discussion, limited experimental and spectral data, and crystallographic data. See https://doi.org/10.1039/d2sc03449b |
| This journal is © The Royal Society of Chemistry 2022 |