
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrimodal hierarchical porous carbon nanorods enable high-performance Na–Se batteries†
Xiang Long
Huang
 *a,
Xiaofeng
Zhang
a,
Mingjie
Yi
e,
Ye
Wang
a,
Shaohui
Zhang
c,
Shaokun
Chong
d,
Hua Kun
Liu
bg,
Shi Xue
Dou
*bg and
Zhiming
Wang
*af
*a,
Xiaofeng
Zhang
a,
Mingjie
Yi
e,
Ye
Wang
a,
Shaohui
Zhang
c,
Shaokun
Chong
d,
Hua Kun
Liu
bg,
Shi Xue
Dou
*bg and
Zhiming
Wang
*af
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, 610054, China. E-mail: xlhuang@163.com
bInstitute for Superconducting and Electronic Materials, University of Wollongong, North Wollongong, 2500, Australia. E-mail: shi@uow.edu.au
cGuangdong Provincial Key Laboratory of Micro/Nano Optomechatronic Engineering, College of Mechatronics and Control Engineering, Shenzhen University, Shenzhen 518060, China
dFrontiers Science Center for Flexible Electronics, Xi'an Institute of Flexible Electronics, Xi'an Institute of Biomedical Materials & Engineering, Northwestern Polytechnical University, Xi'an 710072, China
eState Key Laboratory of Advanced Welding and Joining, Harbin Institute of Technology, Shenzhen 518055, China
fInstitute for Advanced Study, Chengdu University, Chengdu 610106, P. R. China
gInstitute of Energy Materials Science, University of Shanghai for Science and Technology, 516 Jungong Road, Shanghai, 200093, China
First published on 13th September 2022
Abstract
Technical bottlenecks of polyselenide shuttling and material volume variation significantly hamper the development of emerging sodium–selenium (Na–Se) batteries. The nanopore structure of substrate materials is demonstrated to play a vital role in stabilizing Se cathodes and approaching superior Na-ion storage properties. Herein, an ideal nanorod-like trimodal hierarchical porous carbon (THPC) host is fabricated through a facile one-step carbonization method for advanced Na–Se batteries. The THPC possesses a trimodal nanopore structure encompassing micropores, mesopores, and macropores, and functions as a good accommodator of Se molecules, a reservoir of polyselenide intermediates, a buffer for volume expansion of Se species during sodiation, and a promoter for electron/ion transfer in the electrochemical process. As a result, Na–Se batteries assembled with the Se–THPC composite cathode realize high utilization of Se, fast redox kinetics, and excellent cyclability. Furthermore, the Na-ion storage mechanism of the well-designed Se–THPC composite is profoundly revealed by in situ visual characterization techniques.
1 Introduction
Rapid development and progress in harvesting and utilizing renewable but unstable energies (e.g., solar and wind energy) compels researchers to exploit matching energy storage technologies.1–3 Sodium-based batteries are expected to become scalable, sustainable, and cost-effective stationary energy storage systems, due to wide availability of natural sodium resources and inexpensive material prices.4–6 Although conventional sodium-ion batteries (NIBs), paired with carbon anodes such as hard carbons and intercalated cathodes such as layered oxides and polyanion compounds, have been well developed in recent years and considered as a promising commercial candidate, their overall energy densities are relatively too low to meet the demand for long-distance electric transportation.7,8 In contrast, sodium–selenium (Na–Se) batteries assembled with metallic Na anodes and Se-based cathodes and operated via a multielectron/phase pathway potentially lead to a high-energy-density Na-ion storage system as a result of high theoretical specific capacities of Na (1166 mA h g−1) and Se (675 mA h g−1) and high theoretical volumetric capacity of Se (3253 mA h cm−3).9–12 Therefore, ever-increasing research attention has been paid to emerging Na–Se batteries in recent years, especially on rational design of Se cathode materials.Despite advantages of abundant Na resources and high capacity of Se, the development of Na–Se batteries is plagued by a series of critical challenges, especially on the Se cathode side. To start with, the weak reactivity of Se with metallic Na ions means low atomic utilization and poor capacity contribution;13 then, the multiphase reaction mechanism easily induces dissolution and shuttling of sodium polyselenides (Na2Sex), giving rise to irreversible loss of active materials and degeneration of Na-ion storage capability of Se cathodes;14 otherwise, the dramatic volumetric variation of Se species during the (de)sodiation process brings huge mechanical stress to Se cathodes, resulting in their structural destruction and even failure in electrode function.15 More significantly, electrochemical evolution of Se species is always accompanied by intrinsic sluggish conversion kinetics.16 These issues lead to poor cyclability and rate capabilities of Na–Se batteries, which need to be urgently addressed via efficient strategies.
Dispersing Se into porous carbon substrates is demonstrated as a useful pathway to circumvent the aforementioned problems.17,18 Porous carbons feature distinct effects in Se encapsulation and Na2Sex confinement, related to pore sizes. Micropores have large adsorption volumes and strong confinement ability for polyselenides, mesopores exhibit large contact areas with active materials and short ion diffusion length besides superior confinement capability, and macropores are able to load a large number of active materials, accelerate electrolyte infiltration, and present rich active sites for ion adsorption.19–21 Attributable to restricting dissolution and shuttling of intermediates greatly, unimodal porous carbons (micropores or mesopores) are designed as host materials for Na–Se batteries to address the issue of polyselenide shuttling.22–24 Bimodal hierarchical porous carbons with both micropores and mesopores create an opportunity to overcome the drawback of low loading contents of Se for micropores as well as an approach to couple advantages of two kinds of nanopores.25–27 To our knowledge, unimodal porous carbons and bimodal hierarchical porous carbons are the most mainstream carrier materials for Na–Se batteries at present (Fig. 1), and occupy a quantitative proportion of 41 and 54%, respectively. In contrast, few of the macroporous carbons are directly utilized in Na–Se batteries, since their open pore structures easily lead to rapid dissolution and escape of polyselenides from host frameworks and thus rapid loss of active materials. Actually, incorporation of a handful of macropores into carbons may regulate the electrochemical kinetics via improving Na ion transport.28,29 Thus, trimodal hierarchical porous carbons with micropores, mesopores, and macropores are promising to integrate the multiple functions of improving mass loading of Se, confining dissolution and shuttling of polyselenide intermediates, and regulating electrolyte ion transport.30,31 Unfortunately, the difficult synthesis of trimodal hierarchical porous carbons leads to their research scarcity in Na–Se batteries.
 | ||
| Fig. 1 Statistics of publications on various porous carbons for Na–Se batteries, obtained from Web of Science. | ||
Considering the promise in performance enhancement of multifunctional trimodal hierarchical porous carbons, we elaborately fabricate a nanorod-like trimodal hierarchical porous carbon (THPC) through facile one-step carbonization of a metal–organic framework (MOF) precursor, herein. The micro–mesopores serve as reaction voids for Se, which can physically confine the dissolution and shuttling of polyselenides and adapt the volumetric change of the active materials during (de)sodiation, while the open macropores functioning as ion transport pores are beneficial to electrolyte infiltration and Na ion accessibility to accelerate the redox reaction kinetics. As such, the Na–Se batteries exhibit excellent cyclability (a capacity retention of 386 mA h g−1 over 800 cycles at 1C) and superior rate capability (433 mA h g−1 at 2C). Furthermore, electrochemical analysis and in situ characterization techniques jointly unveil the electrochemical evolution behaviors of Se confined within the THPC.
2 Results and discussion
The synthetic procedure of the Se@THPC composite is schematically illustrated in Fig. S1.† A Zn-based MOF (Zn-MOF) was pre-prepared as the precursor to derive the target porous carbon. The X-ray diffraction (XRD) pattern (Fig. S2†) and field-emission scanning electron microscopy (FESEM) image (Fig. S3†) of the Zn-MOF are in good accordance with the previous reports,32 confirming successful preparation of the nanorod-like precursor. Through a high-temperature carbonization treatment at 900 °C, Zn ions were converted into metallic gas and completely took away by the protection gas and the rest of organic ligands were converted into a porous carbon substrate. Afterwards, pure Se was uniformly infused into nanovoids of the as-obtained porous carbon through a typical molten-diffusion method to produce the target sample Se@THPC.The morphologies and porous structures of the THPC and Se@THPC composite were observed via FESEM and transmission electron microscopy (TEM). It can be witnessed that the THPC obtained after carbonization maintains the overall nanorod shape (Fig. S4†). Its porous nanorod-like structure is further revealed by the TEM images in Fig. 2a and b. And there is no obvious impurity of metallic Zn particles in the internal and on the surface of the THPC, indicating complete removal of metal components. The high-resolution TEM (HRTEM) image in Fig. 2c clearly demonstrates the hollow nanovoids with various pore sizes inside the THPC, which provide storage space for following Se encapsulation. After compositing with Se, there are not any Se particles adhered to the surfaces of the THPC, meaning full impregnation of Se into internal pores of the THPC, and the resultant Se@THPC composite also retains the pristine nanorod morphology of the THPC very well (Fig. S5a†). Elemental linear scan in Fig. S5b† suggests the uniform distribution of Se. It can be clearly seen from TEM images in Fig. 2d and e that after Se encapsulation, the loose porous feature of the THPC vanishes, the color of the material gets evidently darkened at the same magnification, and the Se@THPC composite presents a solid nanorod morphology. These phenomena further verify that Se perfectly fills the nanopores of the THPC. Moreover, EDS mappings of C and Se for the Se@THPC composite (Fig. 2f) further probe the uniform distribution of Se in the entire carbon substrate, which is beneficial for improving the reactivity of Se with Na ions.
 | ||
| Fig. 2 (a–c) TEM images and HRTEM image of the THPC; (d and e) TEM images of the Se@THPC composite; (f) STEM image and corresponding EDS mappings of the Se@THPC composite; XRD patterns (g) and Raman spectra (h) of pure Se, THPC, and Se@THPC; (i) TGA curve of the Se@THPC composite under an argon atmosphere. | ||
Other than morphologies, phase compositions and microstructures of all the samples were jointly characterized via XRD and Raman techniques (Fig. 2g and h). The THPC exhibits two broad characteristic peaks positioned at 25° and 44° without any signal of metallic Zn, which suggests the high purity of the carbon structure. For the Se@THPC composite, a new hump peak located at 52° appears in addition to peaks ascribed to carbon; however, none of strong feature peaks attributed to pure Se can be detected except for some minor impurity peaks. This indicates the dominant amorphous nature of Se confined within the THPC substrate and its uniform dispersion into the THPC.33,34 Moving to Raman spectrum analysis, two clear peaks appear at 1344 and 1602 cm−1 in Raman spectra of the THPC that are assigned to the D band and G band, respectively.35 The former reflects the disorder structure of carbon, while the latter is associated with the graphitized structure of carbon. Most often, their intensity ratio (IG/ID) serves as an indicator of defective degree of carbon materials. The calculated IG/ID value of 0.96 reveals its partially graphitized amorphous nature, which is highly consistent with observation result of the HRTEM image in Fig. 2c. As Se enters into the THPC substrate, its IG/ID value remains unchanged, suggesting its structural stability. At the same time, a strong characteristic peak (260 cm−1) usually regarded as a signal of Se8 rings occurs in Raman spectra of the Se@THPC composite.36 Differently, pure Se has three intrinsic peaks corresponding to ring-like Se12 (144 and 460 cm−1) and chain-structured Se (235 cm−1), respectively.37 The comparison of peaks indicates a possible change in the existing form of Se during the Se encapsulation process. As a whole, Se is well confined within the THPC matrix and present in the form of amorphous Se8 rings. Previous studies unveil that amorphous Se has a higher electrochemical reactivity and better cyclability in Na-ion storage.33,34 The loading content of Se is as high as 53% in the Se@THPC composite, as determined by the thermogravimetric analysis (TGA) in Fig. 2i. Note that the mass ratio of carbon and Se used in the synthetic process (e.g., 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 or 1:3) is generally higher than that of the as-obtained Se/carbon composites, meaning a waste of a part of pristine Se in the preparation process. In this work, a pristine mass ratio of 45 (THPC):55 (Se) was employed, but a final Se content of 53% close to the theoretical value (55%) was successfully obtained. This fully demonstrates the great confinement and accommodation ability of the THPC for Se and its structural superiorities.
:2 or 1:3) is generally higher than that of the as-obtained Se/carbon composites, meaning a waste of a part of pristine Se in the preparation process. In this work, a pristine mass ratio of 45 (THPC):55 (Se) was employed, but a final Se content of 53% close to the theoretical value (55%) was successfully obtained. This fully demonstrates the great confinement and accommodation ability of the THPC for Se and its structural superiorities.
To get further insights into chemical states of elements, X-ray photoelectron spectroscopy (XPS) was conducted on the Se@THPC composite. The existence of elemental C, O, and Se in the composite is testified, as displayed in the XPS survey spectrum of Fig. S6.† In the high-resolution XPS spectra of C 1s (Fig. 3a), two main peaks are located at 284.8 and 286 eV, which correspond to C–C bonds and C–O bonds.38 The O atomic dopant has been demonstrated to enable improved chemical affinity of carbon for Se species.34 As for the XPS spectra of Se 3d (Fig. 3b), there appear two dominant peaks and a minor peak positioned at 55.6, 56.4, and 59.1 eV, ascribed to Se 3d5/2, Se 3d3/2, and Se–C bonds, respectively.34,39 The appearance of Se–C bonds suggests the strong interaction between Se and the carbon substrate, favoring chemical immobilization of Se species during the electrochemical process.
 | ||
| Fig. 3 XPS spectra of C 1s (a) and Se 3d (b); N2 adsorption–desorption isotherms (c) and pore-size distribution (d) of the THPC and Se@THPC composite. | ||
As discussed in the aforementioned parts, the THPC realizes good encapsulation towards the Se guest molecules. It should be actually attributed to the fine porous nanostructure of the THPC. Accordingly, the Brunauer–Emmett–Teller (BET) method was employed to acquire more information about the porosity of the THPC and Se@THPC composite. As shown in Fig. 3c, the THPC exhibits a sharp increase in isotherm in the low-pressure region (P/P0 ≤ 0.015), suggesting the presence of micropores, while its hysteresis loop in the higher-pressure region (P/P0 = 0.4–0.95) is an indicator of mesopores.40 Furthermore, the pore-size distribution acquired via density functional theory (DFT) shows that the size of micropores is centered at 1.93 nm and that the size of mesopores is relatively continuous. The continuous pore structure is proved to enable sufficient Na ion accessibility for better kinetics.41,42 More significantly, a handful of macropores can be witnessed from a broader pore range from 50 to 150 nm (Fig. S7†). As a result, it can be concluded that the as-obtained carbon substrate possesses a trimodal hierarchical porous nature. It possesses a large specific surface area of 928 m2 g−1 and a pore volume of 1.77 cm3 g−1. With incorporation of Se into the THPC, a large amount of micropores, mesopores, and macropores rapidly disappear as these hollow voids are occupied by Se molecules. It finally leads to a significantly reduced specific surface area and pore volume. Despite this, the Se@THPC composite still maintains a relatively high specific surface area of 91 m2 g−1 and pore volume of 0.73 cm3 g−1, conducive to fast contact between the active materials and electrolyte ions.
In consideration of advantages in nanostructures and chemical compositions, the Se@THPC composite was utilized to assemble coin-cells in order to evaluate its Na-ion storage ability (Fig. 4). Cyclic voltammetry (CV) was adopted to examine the Na-ion storage behaviors of the Se electrode with a voltage window ranging from 2.8 to 0.5 V at a sweep rate of 0.1 mV s−1 (Fig. 4a). In the initial cathodic process, two main reduction peaks emerge at 1.78 and 1.25 V, revealing the stepwise sodiation mechanism of Se. Some minor impurity peaks might be caused by electrolyte-associated side reactions and unstable Se activation.13,33 In the following cycles, two stabilized broad cathodic peaks and a minor peak that are located at 1.5, 1.2, and 0.9 V, respectively, can be clearly seen. These peaks reflect the electrochemical evolution from Se8 to long-chain Na2Sex to short-chain Na2Sex and finally to Na2Se. For the minor peak, a slightly decreased current response may be the result of polarization during solid–solid reactions among insoluble products. In all the anodic processes, there is a dominant peak around 1.65 V and a shoulder peak at 0.94 V, indicating the oxidation process from Na2Se to Na2Sex and finally to Se8. The difference in redox behaviors between the first cycle and subsequent cycles may result from the decomposition of the electrolyte and electrochemical activation of the active materials.43
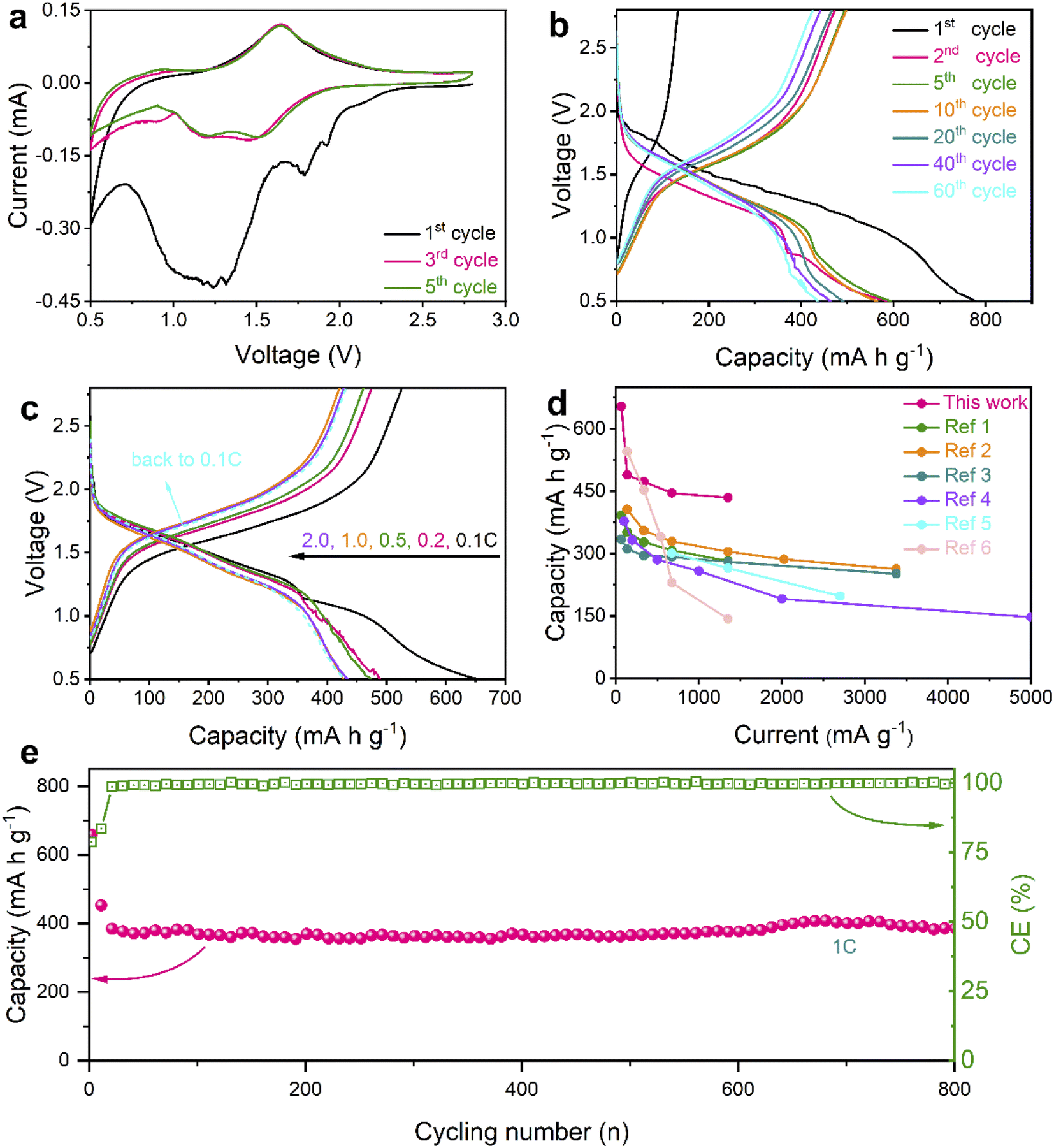 | ||
| Fig. 4 Electrochemical performance of the Se@THPC composite. (a) CV curves at a scan rate of 0.1 mV s−1; (b) discharge–charge profiles at 0.1C; (c) discharge–charge profiles at various rates; (d) a comparison of rate capabilities between this work and other studies (ref. 1–6 are provided in the ESI†); (e) prolonged cycling stability at 1C. | ||
Fig. 4b displays the galvanostatic discharge–charge profiles of the Se@THPC composite at 0.1C. Their voltage platforms are highly consistent with redox peak values of the aforementioned CV curves. The Se@THPC exhibits an initial discharge capacity close to 800 mA h g−1, which is the result of both good electronic conductivity of the THPC and uniform dispersion of Se molecules to render full electrochemical discharging. However, the irreversible capacity and low coulombic efficiency (CE) should be ascribed to the construction of the solid-electrolyte-interface (SEI), as reported in previous literature.44 Afterwards, the well-overlapped discharge–charge profiles suggest the stable and highly reversible electrochemical reactions of the Se cathode. In contrast to the first cycle, the capacity fading in the following cycles could be posed by unavoidable Na2Sex dissolution and shuttling during the redox process.
Various current densities were further utilized to evaluate the rate capabilities of the Se@THPC cathode, as shown in Fig. S8† and 4c. The Se@THPC cathode shows reversible discharge capacities of 654, 497, 473, 445, and 433 mA h g−1 at 0.1C, 0.2C, 0.5C, 1C, and 2C, respectively. Upon returning to 0.1C, it still delivers a high reversible discharge capacity of 429 mA h g−1. Both high capacity delivery and high recovery effectively certify the excellent rate performance of the Se@THPC composite. The ever-increasing CE should be due to the gradual electrochemical activation of Se molecules, while the decrease in capacity at 0.1C may be caused by incomplete utilization of Se in the initial several cycles. Even if at various rates, the discharge–charge profiles are highly overlapped and exhibit identical electrochemical features, which indicates small polarization and fast redox kinetics during the electrochemical reaction process. In contrast, the as-designed Se@THPC cathode presents obviously superior rate performance to other Se cathodes reported (Fig. 4d).
As is known to all, long-term cycling stability is a key parameter of electrode materials for practical applications. Herein, a large current density of 1C was used to test the cyclability of the well-designed Se@THPC cathode. Given the significance of full electrochemical activation for overall properties of Se (as discussed before), the Se@THPC cathode was pre-cycled for 10 cycles at a relatively small rate of 0.1C. The prolonged cycling performance of the Se@THPC is exhibited in Fig. 4e. It retains a high reversible discharge capacity of 385 mA h g−1 over 800 cycles at 1C. Besides, ultrahigh CE exceeding 99% always accompanies the cycling process. Fig. S9† displays CV curves of the THPC at 0.1 mV s−1, where there are no obvious redox peaks and current response, indicating electrochemical inertness of the THPC under the same test condition as the Se@THPC composite. Fig. S10† depicts the cycling performance of the THPC at 675 mA g−1, demonstrating negligible capacity contribution of the THPC. These cases fully make clear that the capacity of the cathode is mainly from the reversible electrochemical reactions of Se instead of the carbon substrate. Outstanding cycling ability should be attributed to structural stability of electrode materials, which enables suppressed shuttle effect of polyselenides and rapid release of mechanical stress posed by the volume change during the (de)sodiation process.
Among most of the reports about Na–Se batteries, understanding and insights of electrochemical evolution of Se and its Na-ion storage mechanism mainly relied on conventional electrochemical analysis techniques such as CV and discharge–charge tests, hitherto. To the best of our knowledge, some advanced visual characterization techniques have been not employed to reveal the electrochemical mechanism of Na–Se batteries. Here, we utilized an in situ XRD technique to identify the electrochemical evolution of the Se@THPC composite for acquiring deeper insights into Na–Se chemistry (Fig. 5). As the fresh Na–Se cell with an open voltage (CV) is discharged, two characteristic peaks positioned at 20.2° and 36.4° appear, which should be attributed to high-order Na2Sen. Further discharging leads to emergence of Na2Se4 (JCPDF no. 86-1846) with three clear characteristic peaks located at 11.3°, 18.1°, and 27°, respectively. Next, a characteristic peak (34.6°) ascribed to Na2Se2 (JCPDF no. 89-2754) is observable, indicating the conversion process from Na2Se4 to Na2Se2. When the Se@THPC cathode is discharged to 0.5 V, a strong characteristic peak emerges around 36.8°, which can be assigned to Na2Se (JCPDF no. 77-2155). In the subsequent charging process, Na2Se completely vanishes as well as Na2Se4 and Na2Se2 can be detected again in turn. Combining in situ XRD patterns with the previous analysis results via CV and discharge–charge tests, a reversible electrochemical evolution mechanism of the Se@THPC cathode during the (de)sodiation process can be proposed: Se8 ↔ Na2Sex (4 < x ≤ 8) ↔ Na2Se4 ↔ Na2Se2 ↔ Na2Se. Although in situ XRD is carried out to identify the phase change mechanism clearly, the nucleation, growth, and deposition mechanism of Na2Se/Se and relevant influence factors are still unclear. In future research, more efforts needs to be made to unlock these unknown aspects (e.g., adopting advanced characterization measures).45
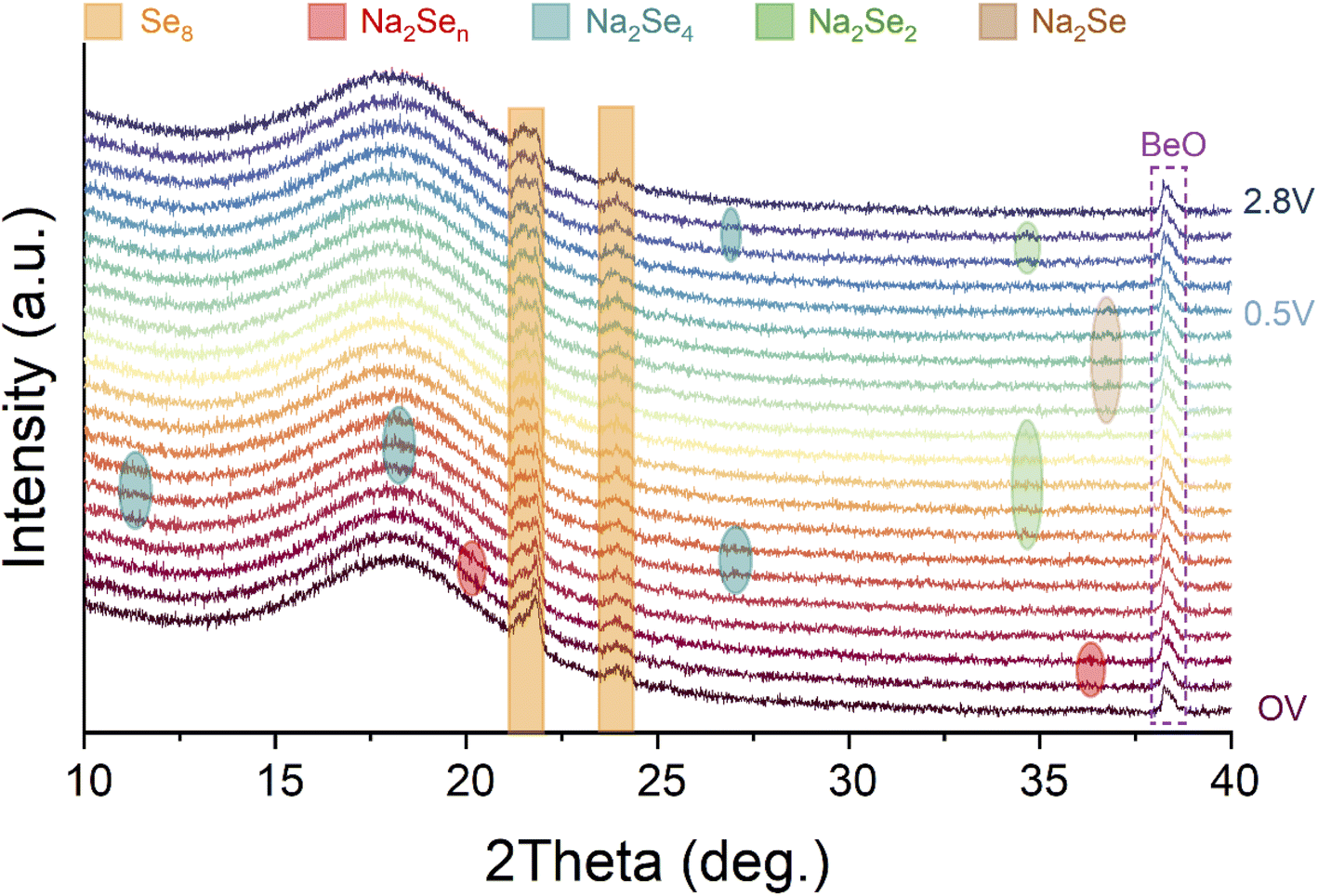 | ||
| Fig. 5 In situ XRD patterns of the Se@THPC cathode during the electrochemical process. | ||
In order to understand why the as-designed THPC can result in superior Na-ion storage capability, a series of post analyses were carried out. The reaction kinetics of the Se@THPC composite was revealed via CV curves obtained at sweep rates ranging from 0.1 to 1.0 mV s−1 (Fig. S11†). With the ever-increasing sweep rate, the CV curves still exhibit clear and strong redox peaks despite shifted positions. Two pair of redox peaks are denoted as P1, P2, P3, and P4, respectively. As known, the power-law equation (i = avb) is a good tool to determine the Na-ion diffusion behaviors, where a and b are the linear-fitted parameters ranging from 0.5 to 1.0, while i and v are redox peak current and sweep rate. The b-value can function as a direct indicator of electrochemical kinetics, since the electrochemical process is diffusion-controlled when b is closer to 0.5 but it is surface capacitance-dominant when b is closer to 1.0.25,46 As shown in Fig. 6a and b, the b values obtained from four peaks of P1–P4 are 0.90, 0.97, 0.96, and 0.88, respectively, adequately suggesting the fast kinetics of the Se@THPC composite during electrochemical reactions. The capacitive Na-ion diffusion behaviors may be the result of the large specific surface area and fine porous nanostructure of the well-designed carbon-based materials. Electrochemical impedance spectroscopy (EIS) was used to examine the charge transfer nature (Fig. S12†). Before cycling, a high charge transfer resistance (Rct) of 1039 Ω can be witnessed, which is due to the considerably limited interface contact. After the cell is cycled, the Rct is significantly reduced to 57 Ω, which is benefitted by the complete activation of the active materials, improved electrolyte infiltration, and shortened Na ion diffusion pathways.47 Furthermore, the TEM image of the Se@THPC composite after cycling was acquired to demonstrate its structural stability. As illustrated in Fig. S13,† the cycled Se@THPC composite still maintains its nanorod morphology very well albeit it looks a little puffed, suggesting its robust ability to accommodate the volume change of Se species in the electrochemical process.
 | ||
| Fig. 6 (a and b) The linear fitted b-values obtained from CV curves at various sweep rates; (c) schematic illustration of the electrochemical reaction process for the Se@THPC composite. | ||
Generally speaking, the excellent electrochemical performance of the Se@THPC composite can be ascribed to several aspects: (1) the fine porous nanostructure can effectively suppress the dissolution and shuttling of Se species as well as accommodate their volume expansion; (3) the one-dimensional rod-like shape is able to offer smooth pathways for electron transfer to lead to fast activation and redox reactions of the active materials; (3) the introduction of macropores, large specific surface area, and abundant active sites can synergistically promote the Na-ion diffusion kinetics. Fig. 6c depicts the advantages of the Se@THPC cathode in redox reactions. Based on the superior structural features of the well-designed THPC, it is expected to play a useful role in other battery systems to broaden its universality. Thus, we utilize it to serve as a host material of the emerging high-capacity and low-cost SeS2 and evaluate the electrochemical performance in Na-ion storage, as displayed in Fig. S14.† The as-synthesized SeS2@THPC composite with a loading content of 45% shows a high capacity retention of 603 mA h g−1 after 280 cycles at 0.2 A g−1 along with a high CE close to 100%.
3 Conclusions
A superior trimodal hierarchical porous carbon was engineered via a facile synthetic method for Na–Se batteries. Micro–mesopores served as the main nanovoids for electrochemical reactions, while macropores played an accelerator role in ion diffusion and accessibility. The Na–Se batteries exhibited fast kinetics, excellent cyclability, and outstanding rate performance, due to the multiple favoring structural advantages of electron/ion transport, physical confinement capability, and stress alleviation ability. The electrochemical mechanism of Se cathodes was profoundly revealed via in situ characterization techniques. This work highlights the significance of macropores in designing porous carbons for Na–Se batteries, provides an easy synthetic method to trimodal hierarchical porous carbon materials, and sheds light on the Na-ion storage principle of high-performance Se cathodes.4 Experimental section
Materials preparation
600 mg of 2,5-dihydroxyterephthalic acid was dissolved in a mixed solution of deionized water and dimethyl formamide. 1.008 g of zinc acetate dihydrate was dissolved in the same mixed solution. And, the two kinds of solutions were quickly mixed and magnetically stirred for 20 min. Then, the brown solution with suspension was transferred into a Teflon-lined autoclave and thermostatically heated for one day at 100 °C. After cooling, the yellow precipitate was centrifuged, washed with ethanol three times at least, and dried overnight at 80 °C to produce yellow Zn-MOF powder. 1 g of Zn-MOF was annealed for 4.5 hours at 900 °C under argon gas to yield the THPC. 90 mg of THPC and 110 mg of Se powder were fully ground and heated at 260 °C for 20 hours under argon to obtain the Se@THPC composite.Materials characterization
The morphologies were analyzed via a field emission scanning electron microscope (ZEISS, GeminiSEM 300) and transmission electron microscope (Tecnai G2 F20 S-TWIN). The elemental distributions and linear scanning were obtained by energy dispersive spectrometry (EDS). The crystal structures of all the samples were investigated via an X-ray diffractometer (XRD) with Cu K-alpha radiation. X-ray photoelectron spectra (XPS, Thermo Scientific ESCALAB 250Xi) were recorded to examine the chemical states of materials. Raman spectra of all the samples were recorded via an InviaReflex (Renishaw, UK) with 532 nm laser light. The specific surface areas and pore distributions were tested by the Brunauer–Emmett–Teller method (BSD PS2, China). The loading content of Se was evaluated via thermogravimetric analysis (TG 209, Germany).Electrochemical measurements
The Se@THPC composite, Ketjen black, and PVDF were completely mixed and ground with a mass ratio of 8:1:1 to form a black slurry, which was further coated on carbon-coated Al foil, and dried overnight at 80 °C. The areal mass loading of each electrode foil is 1.0–1.2 mg cm−2. The diameter of electrode discs was 12 mm. The Se@THPC cathode, Na foil, and glass fiber disc were utilized to assemble 2032-type coin cells in an argon-filled glove box holding an oxygen/water content below 0.01 ppm. The electrolyte used was 1 M NaClO4 salt dissolved in EC–PC (v/v = 1:1) mixed solvent with 5% FEC additive. The amount of electrolyte in every coin-cell was 50 μL. An electrochemical workstation (Chenhua Instrument, CHI 660E) was employed to carry out CV tests with a voltage window of 0.5–2.8 V and EIS tests with a frequency range from 0.01 Hz to 100 kHz. The galvanostatic discharge–charge properties were examined on a LAND instrument testing system. A coin-cell sealed with Be foil was conducted on in situ XRD, which was tested at a specific current of 100 mA g−1 and a voltage window of 2.8–0.5 V. XRD patterns were recorded at 12 minute intervals.
Author contributions
X. L. H. conceived the project, carried out the experiments, and wrote the manuscript. X. Z., M. Y., and Y. W. performed the materials characterization. S. Z., S. C., and H. K. L. read and edited the manuscript. S. X. D., and Z. W. contributed with an advisory role in the project and manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the financial support from the National Key Research and Development Program of China (2019YFB2203400), the “111 Project” (B20030), and ARC DP210102215.References
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- X. L. Huang, Y.-X. Wang, S.-L. Chou, S. X. Dou and Z. M. Wang, Energy Environ. Sci., 2021, 14, 3757–3795 RSC.
- Y. Wu, L. Wu, S. Wu, Y. Yao, Y. Feng and Y. Yu, Small Sci., 2021, 1, 2100059 CrossRef.
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sánchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y.-S. Hu, Science, 2020, 370, 708–711 CrossRef CAS.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- Y. Li, F. Wu, J. Qian, M. Zhang, Y. Yuan, Y. Bai and C. Wu, Small Sci., 2021, 1, 2100012 CrossRef.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 RSC.
- X. L. Huang, C. Zhou, W. He, S. Sun, Y.-L. Chueh, Z. M. Wang, H. K. Liu and S. X. Dou, ACS Nano, 2021, 15, 5876–5903 CrossRef CAS PubMed.
- X. L. Huang, S. X. Dou and Z. M. Wang, Energy Stor. Mater., 2022, 45, 265–280 CrossRef.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef CAS PubMed.
- C. Lu, A. Li, G. Li, Y. Yan, M. Zhang, Q. Yang, W. Zhou and L. Guo, Adv. Mater., 2021, 33, 2008414 CrossRef CAS PubMed.
- Q. Xu, T. Liu, Y. Li, L. Hu, C. Dai, Y. Zhang, Y. Li, D. Liu and M. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 41339–41346 CrossRef CAS PubMed.
- X. Yang, J. Wang, S. Wang, H. Wang, O. Tomanec, C. Zhi, R. Zboril, D. Y. W. Yu and A. Rogach, ACS Nano, 2018, 12, 7397–7405 CrossRef CAS PubMed.
- L. Zeng, W. Zeng, Y. Jiang, X. Wei, W. Li, C. Yang, Y. Zhu and Y. Yu, Adv. Energy Mater., 2015, 5, 1401377 CrossRef.
- J. Ding, H. Zhou, H. Zhang, T. Stephenson, Z. Li, D. Karpuzov and D. Mitlin, Energy Environ. Sci., 2017, 10, 153–165 RSC.
- T. Wu, Z. Ding, M. Jing, G. Zou, H. Hou, Y. Tian, Y. Jiang, W. Hong and X. Ji, Adv. Funct. Mater., 2019, 29, 1809014 CrossRef.
- X. Yang, H. Wang, D. Y. W. Yu and A. L. Rogach, Adv. Energy Mater., 2018, 28, 1706609 CrossRef.
- X. Zhao, L. Yin, Z. Yang, G. Chen, H. Yue, D. Zhang, Z. Sun and F. Li, J. Mater. Chem. A, 2019, 7, 21774–21782 RSC.
- F. A. Perras, S. Hwang, Y. Wang, E. C. Self, P. Liu, R. Biswas, S. Nagarajan, V. H. Pham, Y. Xu, J. A. Boscoboinik, D. Su, J. Nanda, M. Pruski and D. Mitlin, Nano Lett., 2020, 20, 918–928 CrossRef CAS PubMed.
- F. Xiao, X. Yang, T. Yao, H. Wang and A. L. Rogach, Chem. Eng. J., 2022, 430, 132737 CrossRef CAS.
- S. Xin, L. Yu, Y. You, H.-P. Cong, Y.-X. Yin, X.-L. Du, Y.-G. Guo, S.-H. Yu, Y. Cui and J. B. Goodenough, Nano Lett., 2016, 16, 4560–4568 CrossRef CAS.
- W. Dong, H. Chen, F. Xia, W. Yu, J. Song, S. Wu, Z. Deng, Z.-Y. Hu, T. Hasan, Y. Li, H. Wang, L. Chen and B.-L. Su, J. Mater. Chem. A, 2018, 6, 22790–22797 RSC.
- B. Yuan, X. Sun, L. Zeng, Y. Yu and Q. Wang, Small, 2018, 14, 1703252 CrossRef.
- Y. Yao, M. Chen, R. Xu, S. Zeng, H. Yang, S. Ye, F. Liu, X. Wu and Y. Yu, Adv. Mater., 2018, 30, 1805234 CrossRef.
- J. K. Kim and Y. C. Kang, ACS Nano, 2020, 14, 13203–13216 CrossRef CAS PubMed.
- F. Zhang, P. Xiong, X. Guo, J. Zhang, W. Yang, W. Wu, H. Liu and G. Wang, Energy Stor. Mater., 2019, 19, 251–260 CrossRef.
- J. Jin, X. Tian, N. Srikanth, L. B. Kong and K. Zhou, J. Mater. Chem. A, 2017, 5, 10110–10126 RSC.
- M. Adam, P. Strubel, L. Borchardt, H. Althues, S. Dörfler and S. Kaskel, J. Mater. Chem. A, 2015, 3, 24103–24111 RSC.
- X. Hu, J. Li, G. Zhong, Y. Liu, J. Yuan, S. Lei, H. Zhan and Z. Wen, Small, 2020, 16, 2005534 CrossRef CAS PubMed.
- W. D. Dong, C. Y. Wang, C. F. Li, F. J. Xia, W. B. Yu, L. Wu, H. S. H. Mohamed, Z. Y. Hu, J. Liu, L. H. Chen, Y. Li and B. L. Su, Mater. Today Energy, 2021, 21, 100808 CrossRef CAS.
- X. Sui, X. Huang, H. Pu, Y. Wang and J. Chen, Nano Energy, 2021, 83, 105797 CrossRef CAS.
- Q. Xu, H. Liu, W. Du, R. Zhan, L. Hu, S. Bao, C. Dai, F. Liu and M. Xu, Electrochim. Acta, 2018, 276, 21–27 CrossRef CAS.
- Q. Xu, T. Yang, W. Gao, R. Zhan, Y. Zhang, S. Bao, X. Li, Y. Chen and M. Xu, J. Power Sources, 2019, 443, 227245 CrossRef CAS.
- H. Wang, Y. Jiang and A. Manthiram, Adv. Energy Mater., 2018, 8, 1701953 CrossRef.
- X. Zhao, L. Yin, T. Zhang, M. Zhang, Z. Fang, C. Wang, Y. Wei, G. Chen, D. Zhang, Z. Sun and F. Li, Nano Energy, 2018, 49, 137–146 CrossRef CAS.
- W. Sun, K. Guo, J. Fan, Y. Min and Q. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 16558–16566 CrossRef CAS PubMed.
- S.-K. Park, J.-S. Park and Y. C. Kang, J. Mater. Chem. A, 2018, 6, 1028–1036 RSC.
- S. Li, H. Yang, R. Xu, Y. Jiang, Y. Gong, L. Gu and Y. Yu, Mater. Chem. Front., 2018, 2, 1574–1582 RSC.
- L. Shang, H. Yu, X. Huang, T. Bian, R. Shi, Y. Zhao, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 1668–1674 CrossRef CAS PubMed.
- C. Wu, Y. Lei, L. Simonelli, D. Tonti, A. Black, X. Lu, W.-H. Lai, X. Cai, Y.-X. Wang, Q. Gu, S.-L. Chou, H.-K. Liu, G. Wang and S.-X. Dou, Adv. Mater., 2022, 34, 2108363 CrossRef CAS.
- S.-J. Kim, M. Ahn, J. Park, Y. Jeoun, S.-H. Yu, D.-H. Min and Y.-E. Sung, ChemElectroChem, 2020, 7, 3653–3655 CrossRef CAS.
- Y. Qi, Q.-J. Li, Y. Wu, S.-j. Bao, C. Li, Y. Chen, G. Wang and M. Xu, Nat. Commun., 2021, 12, 6347 CrossRef CAS PubMed.
- B.-W. Zhang, T. Sheng, Y.-D. Liu, Y.-X. Wang, L. Zhang, W.-H. Lai, L. Wang, J. Yang, Q.-F. Gu, S.-L. Chou, H.-K. Liu and S.-X. Dou, Nat. Commun., 2018, 9, 4082 CrossRef PubMed.
- J. H. Um, A. Jin, X. Huang, J. Seok, S. S. Park, J. Moon, M. Kim, S. H. Kim, H. S. Kim, S.-P. Cho, H. D. Abruña and S.-H. Yu, Energy Environ. Sci., 2022, 15, 1493–1502 RSC.
- F. Zhang, X. Guo, P. Xiong, J. Zhang, J. Song, K. Yan, X. Gao, H. Liu and G. Wang, Adv. Energy Mater., 2020, 10, 2000446 CrossRef CAS.
- H. Wang, Y. Jiang and A. Manthiram, Energy Stor. Mater., 2019, 16, 374–382 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04648b |
| This journal is © The Royal Society of Chemistry 2022 |