
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected evolution of a cyclodipeptide synthase with new activities via label-free mass spectrometric screening†
Songya
Zhang‡
a,
Jing
Zhu‡
a,
Shuai
Fan
b,
Wenhao
Xie
a,
Zhaoyong
Yang
*b and
Tong
Si
 *a
*a
aCAS Key Lib Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
bThe Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 1000050, China
First published on 2nd June 2022
Abstract
Directed evolution is a powerful approach to engineer enzymes via iterative creation and screening of variant libraries. However, assay development for high-throughput mutant screening remains challenging, particularly for new catalytic activities. Mass spectrometry (MS) analysis is label-free and well suited for untargeted discovery of new enzyme products but is traditionally limited by slow speed. Here we report an automated workflow for directed evolution of new enzymatic activities via high-throughput library creation and label-free MS screening. For a proof of concept, we chose to engineer a cyclodipeptide synthase (CDPS) that synthesizes diketopiperazine (DKP) compounds with therapeutic potential. In recombinant Escherichia coli, site-saturation mutagenesis (SSM) and error-prone PCR (epPCR) libraries expressing CDPS mutants were automatically created and cultivated on an integrated work cell. Culture supernatants were then robotically processed for matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) MS analysis at a rate of 5 s per sample. The resulting mass spectral data were processed via custom computational algorithms, which performed a multivariant analysis of 108 theoretical mass-to-charge (m/z) values of 190 possible DKP molecules within a mass window of 115–373 Da. An F186L CDPS mutant was isolated to produce cyclo(L-Phe–L-Val), which is undetectable in the product profile of the wild-type enzyme. This robotic, label-free MS screening approach may be generally applicable to engineering other enzymes with new activities in high throughput.
Mimicking random mutagenesis and natural selection in the laboratory,1–3 directed enzyme evolution is valuable in improving a variety of properties of biocatalysts (i.e., catalytic activity, stability, substrate specificity, and enantioselectivity).4–6 While it is straightforward to generate thousands of enzyme variants using established methods such as error-prone PCR (epPCR), site-saturation mutagenesis (SSM), and DNA shuffling, phenotypic screening remains a major bottleneck due to the substrate and product versatility of biocatalysts.7,8 Optical assays are widely applied in high-throughput screening (HTS) during directed evolution, often requiring a surrogate substrate or a coupling assay with a colorimetric or fluorogenic readout.9,10 However, it is very challenging to apply such approaches to discover new catalytic activities. On the other hand, mass spectrometry (MS) is label-free and suitable for untargeted molecular profiling.11 The high sensitivity and chemical resolution of MS further highlight its potential in discovering new products, because new catalytic activities are often weak when first emerging from enzyme promiscuity.12
We and others have recently developed a range of high-throughput MS screening methods for directed protein evolution.13–16 However, the label-free advantage of MS has not been fully demonstrated in engineering new enzymatic activities, possibly due to the difficulties in untargeted MS screening. Particularly, it requires careful standardization and optimization of sample preparation, MS acquisition, and data processing, which are necessary to minimize experimental noise and spot the weak signals of a new product. On the other hand, biofoundries provide an emerging infrastructure to assist the design–build–test–learn (DBTL) cycles in biological engineering via robotic standardization and parallelization.17–19 Using an integrated biofoundry, here we report a workflow for unlabeled MS screening of recombinant libraries to rapidly isolate enzyme mutants that catalyze the formation of new products. This new workflow extends our previous matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) MS-based screening approach from agar colonies16 to liquid cultures in industry-standard microplates for better uniformity. Unlike colony biomass, liquid culture media often contain high concentrations of nonvolatile components, which interfere with MALDI matrix crystallization and cause severe ion suppression during MS analysis. Therefore, sample preparation steps, such as liquid–liquid extraction and solid-phase extraction, need to be incorporated and automated.
Cyclodipeptide synthases (CDPSs) are a family of enzymes that catalyze the formation of a diketopiperazine (DKP) from two aminoacyl-tRNA substrates (aa-tRNAs), which is an important pharmacophore for modern drug development20–22 (Fig. 1A and S1†). To produce new DKP derivatives for therapeutic and industrial applications, the catalytic mechanism of CDPSs has been studied23,24 to guide engineering. AlbC (239 aa) is the first identified CDPS protein, taking phenylalanyl-tRNAPhe (Phe-tRNAPhe) and leucyl-tRNALeu (Leu-tRNALeu) as substrates to synthesize cyclo(L-Phe–L-Leu) (cFL) in its native host Streptomyces noursei.25,26 Recombinant E. coli with AlbC overexpression is able to produce more cyclodipeptide derivatives in addition to cFL.27 To date, eight CDPSs have been structurally characterized,26,28–32 and mutagenesis studies reveal that the residues within the P1 and P2 catalytic pockets are the key determinants of substrate specificity. However, the detailed mechanism of substrate recognition and catalysis is still elusive, and CDPSs are recognized as recalcitrant targets for rational engineering.33,34 To our knowledge, large-scale screening of CDPS variants has not been reported, possibly due to the lack of applicable HTS assays.
 | ||
| Fig. 1 (A) Scheme of diketopiperazine (DKP) biosynthesis catalyzed by cyclodipeptide synthase (CDPS). (B) Workflow of the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF MS)-based high-throughput screening method for directed evolution of AlbC protein in E. coli. | ||
Here we develop and apply untargeted MS screening on a biofoundry for directed evolution of AlbC mutants that form new CDP products. The workflow consists of strain library creation, colony picking, microtiter cultivation, organic solvent extraction, transfer of the sample to a MALDI target and acquisition of mass spectra, followed by data processing and visualization (Fig. 1B). For library creation, AlbC variants were generated by either SSM or epPCR approaches and expressed from a pET28a plasmid under control of a T7 promoter. Upon genetic transformation of E. coli Rosetta(DE3), individual clones were transferred by a colony picker into microtiter plates. Subsequent cultivation, inducible production by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG), ethyl acetate extraction, and MS sample preparation were performed using an integrated robotic workcell in the Shenzhen Synthetic Biology Infrastructure. This workcell integrates common instruments for synthetic biology, including a liquid-handling station, shaking incubators, centrifuge, plate reader, and so on. Organic extracts of the liquid cultures were spotted onto a MALDI target and overlaid with 4-CHCA matrix solution. MS targets were then manually transferred to a stand-alone MALDI mass spectrometer in this study, although a robotic configuration has been previously reported to automate this step.35
We first applied the workflow to analyze the wild-type (WT) AlbC-expressing strain, Rosetta(DE3)/pET28-AlbC(WT). After cell cultivation, inducible production, and sample preparation, MS analysis was performed using an Autoflex MALDI-ToF mass spectrometer in the reflector positive ion mode (Fig. 1). In the resulting MALDI mass spectra, we observed [M + H]+ ion peaks with m/z values corresponding to cFL (m/z 261.17), cFY (m/z 311.15), cFM (m/z 279.12), cYL (m/z 277.16), cFF(cYM) (m/z 295.15), cLL (m/z 227.16) and cMM (m/z 263.12), all of which were absent from the control strain Rosetta(DE3)/pET28 (Fig. S2†). Production of these CDP molecules was further confirmed by examining tandem MS results using LC-MS in the multiple reaction monitoring (MRM) mode (Table S3†), where ion fragmentation patterns were consistent with the literature.26,27 Moreover, the observed production profile was also consistent with previous studies.26,27 These results validated our workflow to detect CDP products from microplate cultures of AlbC-expressing E. coli using MALDI-ToF MS.
To engineer AlbC mutants that synthesize new products, we first focused on the key residues in the substrate-binding pockets. AlbC employs a “ping-pong” catalytic mechanism: the initial step transfers the aminoacyl moiety of the first aa-tRNA onto a conserved serine, leading to the formation of an aminoacyl enzyme intermediate; then, the aminoacyl enzyme reacts with the aminoacyl moiety of the second aa-tRNA to form a dipeptidyl enzyme.32 Previous biochemical experiments demonstrated that two binding pockets P1 and P2 of AlbC accommodate the aminoacyl moieties of the two aa-tRNA during the biosynthesis24 (Fig. 2). Only a limited number of mutations were examined on P1 and P2 residues,25,26 possibly due to the lack of HTS assays for large-scale analysis. Instead, we applied our workflow to create and screen the SSM libraries of select residues, including 10 residues in the binding pockets (L33, V65, L119, L185, L200, M152, M159, I204, T206 and P207) and 4 residues (R99, R101, R102 and D205) outside the pockets which have interactions of the tRNA moiety reported in the literature (Fig. 2A).
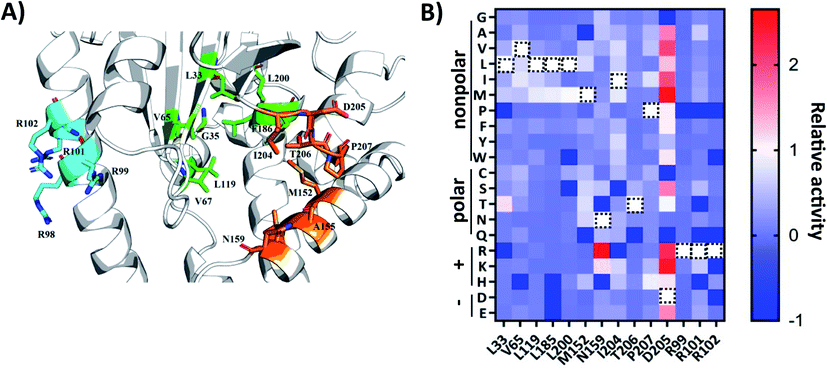 | ||
| Fig. 2 (A) Structural analysis of the AlbC (PDB ID, 3OQV). Enlarged view of the catalytically active pocket. The possible catalytic residues are shown in green (pocket-1) and orange (pocket-2) and basic residues on helix α4 are labelled in cyan. (B) Heatmap of the relative activity change in the cFL production levels of mutation of 14 amino acid residues in AlbC. WT residues are labeled with a black dashed rectangle. Dark blue boxes indicate that a specific mutant was not covered in randomly picked clones (activity assigned as −1). | ||
For SSM libraries, we adopted the “22c-trick” strategy to design degenerative primers, and 94 library clones were randomly picked for each residue library to reach a >98.6% probability of full library coverage of the 20 canonical amino acids.36 In addition to library variants, the WT and control strains were also included in the same 96-well plate. Overall, the abovementioned workflow (Fig. 1B) takes approximately 5 s for sample preparation and MS analysis for each mutant culture. Tentative ion peaks of cyclodipeptides were assigned based on theoretical m/z values (ESI Section 1†) using the MetaboAnalyst webserver.37 For each ion peak, one-way analysis of variance (ANOVA) was used to evaluate statistical differences between the mean peak areas of a library variant and those of the control group. AlbC gene mutations were revealed by Sanger sequencing for all 14 SSM library members.
To visualize the MALDI-ToF MS screening results, a heatmap was generated based on the cFL production of library members relative to that of WT (Fig. 2B), and we observed a general consistency between our results and literature data (Table 1). For example, the L200N mutation almost abolished cFL production (Table 1) but increased cYL (m/z 277.16, [M + H]+), cYY (m/z 327.16, [M + H]+), and cYM (m/z 295.18 [M + H]+) production by about 1.2-fold, 5-fold, and 3-fold (Fig. 3A), respectively, which was similar to a previous study.26 Also, mutagenesis of basic residues (R99, R101 and R102) outside the pockets led to the decline of cFL production in the mutants, although R101 was more tolerant to mutations than R99 and R102. Furthermore, the substitution of D205 in the β6-α8 loop to alanine enhanced AlbC catalytic activity as observed previously.25 On the other hand, some previously unnoticed phenomena were observed. For example, in the D205 SSM library, relative ion intensities of cFL (m/z 261.17) in many mutants are higher than that of WT (Fig. S3†). The top 3 mutants, D205M, D205K and D205R, were subsequently analyzed by LC-MS/MS. The results not only confirmed augmented synthesis of cFL, but also revealed increased production of other leucine-containing products including cYL, cLL and cML (Fig. 3B). Also interestingly, the discovery that most mutations of the T206 residue in the P2 pocket abolished the biosynthesis of the main product cFL was unprecedented (Fig. S3 and ESI Section 2†). When examining MALDI mass spectra in detail, we noticed that although the T206F mutation greatly reduced cFL production, cFF and cFY products were largely not affected. Replacing the small threonine side chain with a bulky aromatic phenylalanine side chain could affect the activity of WT, because the phenyl ring structure increased the steric hindrance of T206F, thus impairing binding with its substrate and causing loss of its enzyme activity for cFL (Fig. 5A). These results suggested that T206 is also a key residue for the selectivity of the second Leu-tRNA substrate binding in the pocket. Unfortunately, we did not observe any new CDP molecules produced from the 14 SSM library mutants.
| Target residue | Mutants and production levels relative to WT (literature) | Mutants and production levels relative to WT (this study) |
|---|---|---|
| a Calculated from MALDI-TOF MS data. b Calculated from LC-MS/MS data. | ||
| WT: cFL (set as 100%) | WT: cFL (set as 100%) | |
| Within binding pockets | ||
| L33/L185D | L33Y/L185D: cFL (0)26 | L33Y: cFL (n.d.)a |
| L200 | L200N: cFL (<10%)26 | L200N: cFL (n.d.,a 5.6%)b |
| N159 | N159A: cFL (45%)25 | N159A: cFL (60%)a |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Outside the pockets | ||
| R98 | R98A: cFL (20%)26 | |
| R99 | R99A: cFL (<10%)26 | R99A: cFL (n.d.)a |
| R98A/R99A: cFL (0) | ||
| R101/R102 | R101A/R102A: cFL (20%)26 | R101A: cFL (>50%)a |
| R102A: cFL (23%)a | ||
| D205 | D205A: cFL (>100%)25 | D205A: cFL (>100%)a |
 | ||
| Fig. 3 Cyclodipeptide product analysis of AlbC mutants. (A) LC-MS characterization of the L200N mutant; (B) LC-MS characterization of select mutants in the D205 saturation mutagenesis library. Normalized production levels relative to WT (set as 1) are reported. Error bars represent the standard deviations of three biological replicates. | ||
The failure of isolating AlbC mutants with new substrate specificities from the SSM libraries highlights the limitation of semi-rational approaches that target manually selected residues. Therefore, we further turned to profile epPCR libraries that contain random mutations throughout the whole protein sequence. Under optimized conditions using the Agilent GeneMorph II Random Mutagenesis Kit, on average 2 nucleotides were introduced per gene variant so that most epPCR library members contain no more than one amino acid change. In total around 4500 independent clones were screened using the above workflow. The peak intensities at the theoretical m/z values of predicted cyclodipeptide ions in each MALDI mass spectrum were analyzed by the one-way ANOVA with Tukey's multiple comparison test. A new peak (m/z 247) absent from WT that corresponds to the [M + H]+ ion of cFV was observed with three clones (Fig. 4A), all of which harbored the F186L mutation as revealed by Sanger sequencing results. Product identification was performed using LC-MS/MS and high-resolution (HR)-MS, and the retention time (5.7 min, Fig. 4B), exact mass (C14H19N2O2 [M + H]+m/z: 247.1441, Table S3†), and MS/MS daughter ions23 (Fig. S4†) of the new product were consistent with that of the chemically synthesized cFV standard. When comparing LC-MS/MS traces, we also noticed substantial reduction of the native main product cFL in the F186L variant relative to WT, and a slight enhancement in production of other products including cFM, cFF, and cFY, indicating a shift towards more bulky substrates with the F186L mutant (Fig. 4B). Another round of epPCR library screening was performed to further extend the AlbC substrate scope using the F186L variant as a parent, but unfortunately, no mutants were isolated to produce new cyclodipeptides.
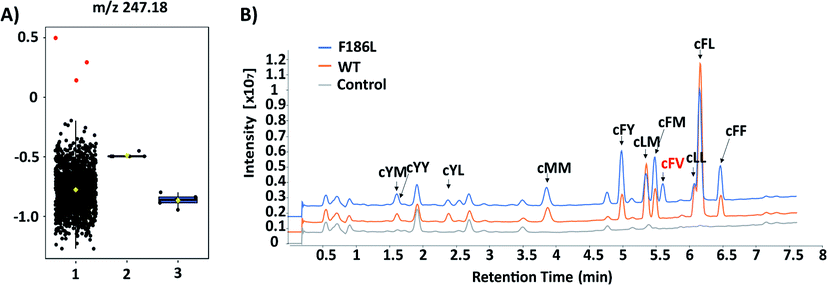 | ||
| Fig. 4 (A) Box–whisker plots of 1152 samples at m/z 247.18 based on ANOVA analysis. Group 1, epRCR variants; 2, Rosetta(DE3)/pET28a-AlbC (WT); 3, Rosetta(DE3)/pET28a (control). Three clones containing F186L were labeled in red. ANOVA, analysis of variance. (B) LC-MS traces of WT and F186L mutant. | ||
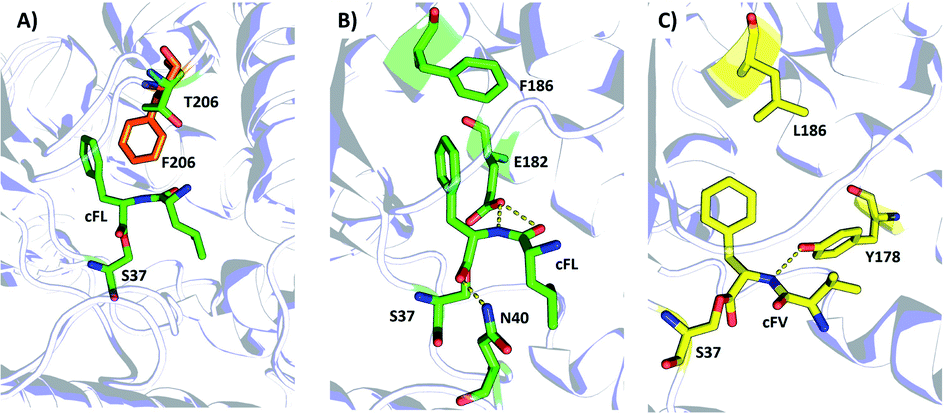 | ||
| Fig. 5 Structural illustration of the substrate binding pocket of the wildtype AlbC and the mutants F186L and T206. Note: Panel (A) Superimposition of the WT with T206F. Panels (B and C) Schematic representation of the hydrogen-bond and hydrophobic interaction between cFL·WT (B) and cFV·F186L (C). Dashed lines indicate hydrogen bond (yellow) interactions. The key amino acid residues of WT, F186L, and T206F are drawn as sticks in green, yellow and orange, respectively. | ||
Then, we investigated possible mechanisms underlying new substrate specificity of the F186L variant from structural aspects using computational modeling. In the substrate binding pocket, the cFL substrate formed three hydrogen bonds with two amino acid residues (N40 and E182) in the WT AlbC. The phenyl side chain of cFL forms a π–π stacking interaction with F186 (Fig. 5B and S5†). However, when the residue at position 186 was mutated to leucine, this π–π stacking interaction was abolished. This leads to a change in the conformation of the Phe1 ring and thereby the hydrogen bond between Phe1 and N40 is abolished (Fig. 5C and S5†), which ultimately results in outward movement of loop G33-S44 (Fig. S6†). Therefore, the volume of the substrate binding cavity, which was measured to be 197 Å3, increased up to 288 Å3 when the residue at position 186 of WT was mutated to leucine (Table S4†). Overall, it is possible that an increase in the production of cFV occurs precisely due to the larger volume of the substrate-binding cavity in F186L. Moreover, cFL and the new derivative cFV were separately docked into the binding pockets of the WT and F186L variant for 100 ns MD simulations (Fig. S5†). The results showed that the binding energy of F186L was significantly increased when docking with the substrate cFV (Table 2). The root-mean-square deviations (RMSD) were calculated in 100 ns to investigate the stability of the WT/F186L-L-lysine complexes (Fig. S5†). Both WT and F186L reached the equilibrium state from an early stage.
| Binding energya (kcal mol−1) of WT | Binding energyb (kcal mol−1) of F186L | Binding energyc (kcal mol−1) of F186L | |
|---|---|---|---|
| a WT-cFL. b F186L-cFV. c F186L-cFL. d van der Waals energy. e Electrostatic energy. f Polar-solvation energy. g Nonpolar solvation energy. h ΔGbinding = ΔGVDW + ΔGEt + ΔGpolar + ΔGapolar. | |||
| ΔGVDWd | −35.01 | −23.67 | −34.27 |
| ΔGEte | −31.95 | −8.70 | −29.37 |
| ΔGpolarf | 48.71 | 31.08 | 47.60 |
| ΔGapolarg | −5.60 | −4.08 | −5.50 |
| ΔGbindingh | −23.85 | −5.37 | −21.54 |
In conclusion, we developed a robotic assay for directed evolution of AlbC, a model CDPS, using MALDI-ToF MS for label-free screening. Compared with conventional LC-MS (typically more than 5 min per sample), our HTS workflow represents a two-magnitude reduction of analytical time (5 s per sample). Contrary to previous reports that only study limited mutations of select residues, 14 SSM libraries were created and profiled for sequence-activity profiling, which not only confirmed the impact of known mutations (Table 1), but also revealed new specificity-modulating mutations (i.e., T206F reduced preference torwards Leu-tRNA as the second substrate). Notably, by creating and screening ∼4500 epPCR library members within a week using unlabeled MS on an integrated, robotic workcell, an AlbC mutant producing a new cyclopeptide product was identified, revealing a previously unknown residue (F186) that exerts a substantial impact on substrate specificity. Other MS modalities, such as droplet microfluidics coupled with ESI-MS,38,39 may also serve as label-free HTS assays, but further development is needed to address ion suppression issues caused by direct infusion of complex culture media into a mass spectrometer. On the other hand, the scarcity of positive hits in the single-residue SSM and epPCR libraries in this study confirmed AlbC as a difficult target for evolving new activities. Therefore, it is desirable to create and screen new AlbC mutant libraries that are comprehensive (i.e., deep mutational scanning40), combinatorial (i.e., combinatorial active site saturation test/iterative saturation mutagenesis, CAST/ISM41), or data-driven (i.e., machine learning-assisted directed evolution, MLDE42,43) in the future. Together, we envision that the label-free MS screening method should be generally applicable to engineering other enzymes with new activities.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
T. S., J. Z. and S. Y. Z. conceived the project and experimental design; J. Z. and S. Y. Z. conducted all the wet experimental work; S. Y. Z., S. F. and Z. Y. Y. performed the molecular docking and the dynamic simulation calculation; W. H. X. assisted with sample preparation for MALDI-ToF MS analysis. S. Y. Z., J. Z., S. F. and T. S. prepared the manuscript, and all authors refined the draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA0910800), National Natural Science Foundation of China (32071428 to T. S., 82104044 to S. Y. Z. and 81872782 to Z. Y. Y.), and the Chinese Postdoctoral Grant (2019M663193 to S. Y. Z.). We thank Zhiyu Zhang and Yunquan Lan at the Shenzhen Synthetic Biology Infrastructure for help with robotic workflow development.Notes and references
- F. H. Arnold, Angew. Chem., Int. Ed. Engl., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- Y. Wang, P. Xue, M. Cao, T. Yu, S. T. Lane and H. Zhao, Chem. Rev., 2021, 121, 12384–12444 CrossRef CAS.
- G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, Angew. Chem., Int. Ed. Engl., 2020, 59, 13204–13231 CrossRef CAS.
- H. Y. Choi, H. S. Lim, K. H. Park, J. Kim and W. G. Kim, Appl. Microbiol. Biotechnol., 2021, 105, 4599–4607 CrossRef CAS PubMed.
- B. Morawski, S. Quan and F. H. Arnold, Biotechnol. Bioeng., 2001, 76, 99–107 CrossRef CAS PubMed.
- R. Carr, M. Alexeeva, A. Enright, T. S. Eve, M. J. Dawson and N. J. Turner, Angew. Chem., Int. Ed. Engl., 2003, 42, 4807–4810 CrossRef CAS.
- H. Yu, J. Li, D. Zhang, Y. Yang, W. Jiang and S. Yang, Appl. Microbiol. Biotechnol., 2009, 82, 279–285 CrossRef CAS.
- M. L. Geddie and I. Matsumura, J. Biol. Chem., 2004, 279, 26462–26468 CrossRef CAS.
- N. N. Rosic, Methods Mol Biol, 2013, 987, 205–224 CrossRef CAS PubMed.
- V. Tarallo, K. Sudarshan, V. Nosek and J. Misek, Chem. Commun., 2020, 56, 5386–5388 RSC.
- T. X. Si, P. K. Choe, H. Zhao and J. V. Sweedler, in Protein Engineering, 2021, pp. 57–79 Search PubMed.
- T. de Rond, J. Gao, A. Zargar, M. de Raad, J. Cunha, T. R. Northen and J. D. Keasling, Angew. Chem., Int. Ed. Engl., 2019, 58, 10114–10119 CrossRef CAS PubMed.
- P. Xue, T. Si, S. Mishra, L. Zhang, K. Choe, J. V. Sweedler and H. Zhao, Biotechnol. Bioeng., 2020, 117, 2131–2138 CrossRef CAS PubMed.
- C. Yan, F. Parmeggiani, E. A. Jones, E. Claude, S. A. Hussain, N. J. Turner, S. L. Flitsch and P. E. Barran, J. Am. Chem. Soc., 2017, 139, 1408–1411 CrossRef CAS PubMed.
- A. J. Pluchinsky, D. J. Wackelin, X. Huang, F. H. Arnold and M. Mrksich, J. Am. Chem. Soc., 2020, 142, 19804–19808 CrossRef CAS PubMed.
- T. Si, B. Li, T. J. Comi, Y. Wu, P. Hu, Y. Wu, Y. Min, D. A. Mitchell, H. Zhao and J. V. Sweedler, J. Am. Chem. Soc., 2017, 139, 12466–12473 CrossRef CAS.
- N. Hillson, M. Caddick, Y. Cai, J. A. Carrasco, M. W. Chang, N. C. Curach, D. J. Bell, R. Le Feuvre, D. C. Friedman, X. Fu, N. D. Gold, M. J. Herrgard, M. B. Holowko, J. R. Johnson, R. A. Johnson, J. D. Keasling, R. I. Kitney, A. Kondo, C. Liu, V. J. J. Martin, F. Menolascina, C. Ogino, N. J. Patron, M. Pavan, C. L. Poh, I. S. Pretorius, S. J. Rosser, N. S. Scrutton, M. Storch, H. Tekotte, E. Travnik, C. E. Vickers, W. S. Yew, Y. Yuan, H. Zhao and P. S. Freemont, Nat. Commun., 2019, 10, 2040 CrossRef PubMed.
- J. Zhang, Y. Chen, L. Fu, E. Guo, B. Wang, L. Dai and T. Si, Curr. Opin. Biotechnol., 2021, 67, 88–98 CrossRef CAS PubMed.
- R. Chao, S. Mishra, T. Si and H. M. Zhao, Metab. Eng., 2017, 42, 98–108 CrossRef CAS PubMed.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS PubMed.
- N. Canu, M. Moutiez, P. Belin and M. Gondry, Nat. Prod. Rep., 2020, 37, 312–321 RSC.
- A. Ortiz and E. Sansinenea, Curr. Med. Chem., 2017, 24, 2773–2780 CrossRef CAS PubMed.
- I. B. Jacques, M. Moutiez, J. Witwinowski, E. Darbon, C. Martel, J. Seguin, E. Favry, R. Thai, A. Lecoq, S. Dubois, J. L. Pernodet, M. Gondry and P. Belin, Nat. Chem. Biol., 2015, 11, 721–727 CrossRef CAS PubMed.
- M. Gondry, I. B. Jacques, R. Thai, M. Babin, N. Canu, J. Seguin, P. Belin, J. L. Pernodet and M. Moutiez, Front. Microbiol., 2018, 9, 46 CrossRef PubMed.
- M. Moutiez, J. Seguin, M. Fonvielle, P. Belin, I. B. Jacques, E. Favry, M. Arthur and M. Gondry, Nucleic Acids Res., 2014, 42, 7247–7258 CrossRef CAS PubMed.
- L. Sauguet, M. Moutiez, Y. Li, P. Belin, J. Seguin, M. H. Le Du, R. Thai, C. Masson, M. Fonvielle, J. L. Pernodet, J. B. Charbonnier and M. Gondry, Nucleic Acids Res., 2011, 39, 4475–4489 CrossRef CAS PubMed.
- M. Gondry, L. Sauguet, P. Belin, R. Thai, R. Amouroux, C. Tellier, K. Tuphile, M. Jacquet, S. Braud, M. Courcon, C. Masson, S. Dubois, S. Lautru, A. Lecoq, S. Hashimoto, R. Genet and J. L. Pernodet, Nat. Chem. Biol., 2009, 5, 414–420 CrossRef CAS.
- M. W. Vetting, S. S. Hegde and J. S. Blanchard, Nat. Chem. Biol., 2010, 6, 797–799 CrossRef CAS.
- L. Bonnefond, T. Arai, Y. Sakaguchi, T. Suzuki, R. Ishitani and O. Nureki, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3912–3917 CrossRef CAS PubMed.
- G. Bourgeois, J. Seguin, M. Babin, P. Belin, M. Moutiez, Y. Mechulam, M. Gondry and E. Schmitt, J. Struct. Biol., 2018, 203, 17–26 CrossRef CAS PubMed.
- C. J. Harding, E. Sutherland, J. G. Hanna, D. R. Houston and C. M. Czekster, RSC Chem. Biol., 2021, 2, 230–240 RSC.
- M. Moutiez, E. Schmitt, J. Seguin, R. Thai, E. Favry, P. Belin, Y. Mechulam and M. Gondry, Nat. Commun., 2014, 5, 5141 CrossRef CAS PubMed.
- K. Brockmeyer and S. M. Li, J. Nat. Prod., 2017, 80, 2917–2922 CrossRef CAS.
- T. Yao, J. Liu, Z. Liu, T. Li, H. Li, Q. Che, T. Zhu, D. Li, Q. Gu and W. Li, Nat. Commun., 2018, 9, 4091 CrossRef PubMed.
- S. Lin, S. Dikler, W. D. Blincoe, R. D. Ferguson, R. P. Sheridan, Z. Peng, D. V. Conway, K. Zawatzky, H. Wang, T. Cernak, I. W. Davies, D. A. DiRocco, H. Sheng, C. J. Welch and S. D. Dreher, Science, 2018, 361 Search PubMed.
- S. Kille, C. G. Acevedo-Rocha, L. P. Parra, Z. G. Zhang, D. J. Opperman, M. T. Reetz and J. P. Acevedo, ACS Synth. Biol., 2013, 2, 83–92 CrossRef CAS PubMed.
- Z. Pang, J. Chong, G. Zhou, D. A. de Lima Morais, L. Chang, M. Barrette, C. Gauthier, P. E. Jacques, S. Li and J. Xia, Nucleic Acids Res., 2021, 49, W388–W396 CrossRef CAS.
- X. W. Diefenbach, I. Farasat, E. D. Guetschow, C. J. Welch, R. T. Kennedy, S. Sun and J. C. Moore, ACS Omega, 2018, 3, 1498–1508 CrossRef CAS PubMed.
- D. J. Steyer and R. T. Kennedy, Anal. Chem., 2019, 91, 6645–6651 CrossRef CAS PubMed.
- D. M. Fowler and S. Fields, Nat. Methods, 2014, 11, 801–807 CrossRef CAS PubMed.
- G. Li, M. Furst, H. R. Mansouri, A. K. Ressmann, A. Ilie, F. Rudroff, M. D. Mihovilovic, M. W. Fraaije and M. T. Reetz, Org. Biomol. Chem., 2017, 15, 9824–9829 RSC.
- B. J. Wittmann, Y. Yue and F. H. Arnold, Cell Syst., 2021, 12, 1026–1045 CrossRef CAS PubMed.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01637k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |