
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective addition of cyclosilanes to alkynes enabling new conjugated materials†
Qifeng
Jiang
 ,
Alexandra F.
Gittens
,
Sydnee
Wong
,
Maxime A.
Siegler
and
Rebekka S.
Klausen
*
,
Alexandra F.
Gittens
,
Sydnee
Wong
,
Maxime A.
Siegler
and
Rebekka S.
Klausen
*
Department of Chemistry, Johns Hopkins University, 3400 N. Charles St, Baltimore, MD 21218, USA. E-mail: Klausen@jhu.edu
First published on 6th June 2022
Abstract
Main group organometallic compounds can exhibit unusual optical properties arising from hybrid σ,π-conjugation. While linear silanes are extensively studied, the shortage of methods for the controlled synthesis of well-defined cyclic materials has precluded the study of cyclic conjugation. Herein we report that Ru-catalyzed addition of cyclosilanes to aryl acetylenes (hydrosilylation) proceeds with high chemoselectivity, regioselectivity, and diastereoselectivity, affording complex organosilanes that absorb visible light. We further show that the hydrosilylation products are useful building blocks towards novel conjugated polymers.
Introduction
We report the synthesis of visible-light absorbing organocyclosilanes via Ru-catalyzed chemoselective, regioselective, and diastereoselective hydrosilylation. While extraordinary progress in predicting and controlling selectivity in organic reactions has been made, the development of similar insights for main group molecules is in its infancy. This lack of synthetic control limits the discovery of structure–property relationships in main group materials. For example, while linear oligosilanes (e.g., Ar(SiMe)nAr) capped with aromatic end groups have been extensively studied for molecular electronics,1,2 metal–organic framework (MOF) secondary building units,3 charge transport,4 photoinduced charge transfer,5–7 and nonlinear optical properties,8–10 there has been very limited investigation of cyclic σ,π-conjugation,11 as cyclosilanes are typically synthesized by methods12–14 that restrict functional group diversity on an organic fragment. Nonetheless, the well-established conformation-dependence of σ-conjugation15 strongly suggests that the restricted degrees of freedom in cyclosilanes should give rise to properties distinct from linear oligosilanes.Our group is interested in the synthesis and properties of (macro)molecular mimics of crystalline silicon and we have described new approaches to the selective synthesis of materials based on cyclohexasilanes. We reported the five-step synthesis and chemoselective polymerization of the cyclohexasilane building blocks 1,4Si6 and 1,3Si6,16–21 as well as the seven-step stereocontrolled synthesis of cis- and trans-Si10H4 (Fig. 1).22 These works have led to new insights, such as the discovery that the diastereomeric siladecalins have vastly different absorption spectra arising from configuration- and conformation-dependent σ-conjugation.
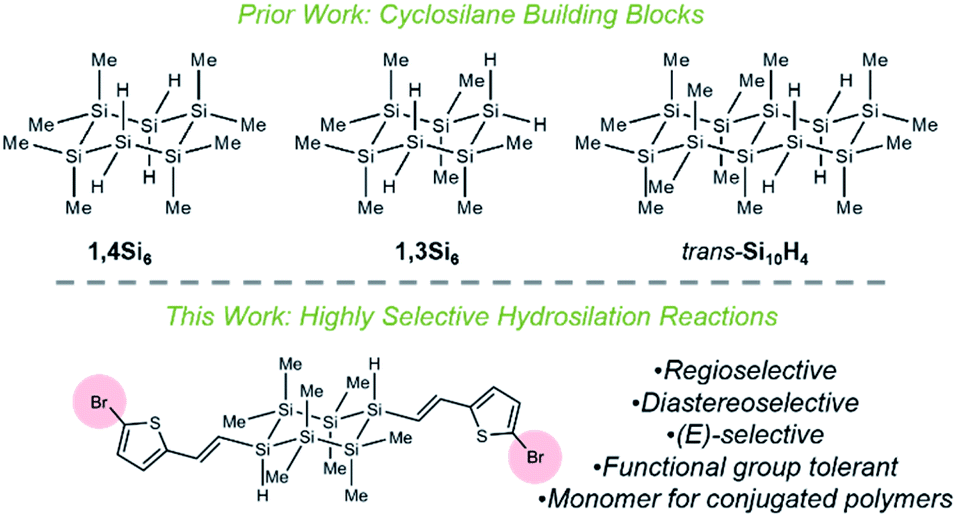 | ||
| Fig. 1 Highly selective reactions between cyclosilane building blocks and functionalized acetylenes. | ||
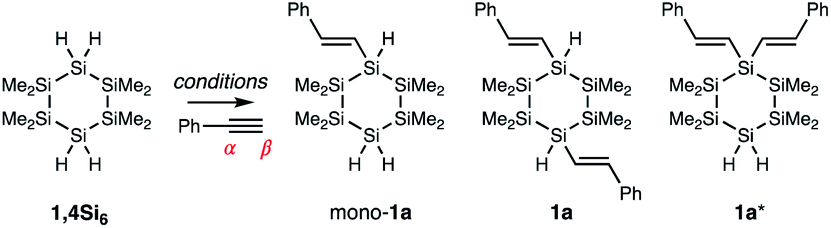
We envisioned an alternative approach to functionalized cyclosilanes via derivatization of the Si–H bond of our building blocks,23 but confronted several challenges. First, the Me3Si–SiMe3 bond is weaker than a H3C–CH3 bond (BDE: 79 vs. 90 kcal mol−1)24 and cross-reactive with reagents for Si–H functionalization; Pd- and Pt-complexes like Karstedt's catalyst fragment Si–Si bonds.25–27 Second, while the targeted cyclosilanes exhibit cis/trans diastereoisomerism, Si–C bonds are longer than C–C bonds,28 potentially diminishing the steric factors often implicated in asymmetric stereoinduction. Third, the tetrafunctional cyclosilanes pose a significant selectivity challenge: the reaction of 1,4Si6 and phenylacetylene could provide more than 40 distinct products arising from mono-, di-, tri-, or tetra-functionalization (E)/(Z)-geometric isomers, α/β-addition, or cis/trans diastereoisomerism.
We report herein the extraordinarily selective outcome of the RuHCl(CO)(PPh3)3-catalyzed29 reaction between cyclosilanes and aryl acetylenes. We observed perfect regioselectivity for 1,4-difunctionalization and β-addition and perfect (E)-selectivity. Products were isolated as a single diastereomer or as a mixture (ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) favoring the thermodynamic isomer. The hydrosilylation is highly functional group tolerant: no Si–Si bond cleavage occurred, and functional group tolerance was high. These syntheses enabled a broad probe of the influence of substituent effects, structural isomerism, and configuration on conjugation in organocyclosilanes, which revealed bathochromic shifts relative to starting materials. We also expect the work herein to expand additional frontiers for the study of conjugated materials. As initial examples, we report the synthesis of novel thienyl-silane conjugated polymers derived from a trans-1,4-bis(thienylvinyl)cyclosilane copolymerized with other dibromo building blocks. The polymers not only absorb visible light, but are also an entry to stereoregular polysilanes, a class of materials where control of tacticity has been a long-standing challenge.
:1) favoring the thermodynamic isomer. The hydrosilylation is highly functional group tolerant: no Si–Si bond cleavage occurred, and functional group tolerance was high. These syntheses enabled a broad probe of the influence of substituent effects, structural isomerism, and configuration on conjugation in organocyclosilanes, which revealed bathochromic shifts relative to starting materials. We also expect the work herein to expand additional frontiers for the study of conjugated materials. As initial examples, we report the synthesis of novel thienyl-silane conjugated polymers derived from a trans-1,4-bis(thienylvinyl)cyclosilane copolymerized with other dibromo building blocks. The polymers not only absorb visible light, but are also an entry to stereoregular polysilanes, a class of materials where control of tacticity has been a long-standing challenge.
Results and discussion
Investigation of regio- and diastereoselectivity
Hydrosilylation is widely used in the industrial preparation of silicones (e.g., Si–O polymers).30–33 The application of hydrosilylation to oligo- and polysilanes (e.g., Si–Si polymers) has been of interest since at least the 1960's,34 but as documented by Rosenberg,35 free-radical36–38 and many transition-metal catalyzed34,39 hydrosilylations are accompanied by partial to extensive Si–Si cleavage. Borane Lewis acids are useful for Si–H/X–H dehydrocoupling and Si–H/X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrosilylation without skeletal fragmentation.23,40,41 Recently, Kyushin reported Ru-catalyzed (E)- or (Z)-selective hydrosilylation of alkynes and hydrooligosilanes without Si–Si bond cleavage.29,42
C hydrosilylation without skeletal fragmentation.23,40,41 Recently, Kyushin reported Ru-catalyzed (E)- or (Z)-selective hydrosilylation of alkynes and hydrooligosilanes without Si–Si bond cleavage.29,42
We envisioned that (E)-selective addition of cyclosilanes to alkynes could yield σ,π-conjugated cyclosilanes. As Kyushin's study focused on linear monohydrosilanes like H–SiMe2SiMe2Ph, our initial focus was regioselectivity with tetrafunctional 1,4Si6 (Table 1 and Fig. 2a). It was unclear if replacement of one Si–H bond with a vinyl group would enforce selectivity for the 1,1′- or 1,4-functionalized product in the second Si–H replacement. The success of monohydrosilanes suggested the feasibility of fully alkylated Si centers, such as 1a*. A first attempt with 2.50 equivalents of phenylacetylene and 5 mol% RuHCl(CO)(PPh3)3 resulted in mono-1a (22%) and 1,4-difunctionalized 1a (9%), as determined by 1H NMR spectroscopy. No 1,1′-difunctionalized 1a* was observed. Further optimization of solvent and catalyst loading led to increased conversion to 1a without residual mono-1a (entry 6). 1,1′-Difunctionalization was not observed even with excess phenylacetylene (entry 7). The mechanistic origin of the selectivity for monofunctionalization could have steric or electronic origins, but the mechanism is not yet clear. In all cases, the 1H NMR spectra were consistent with Si-addition to the phenylacetylene β-position (Table 1), as α-addition would result in a 1,1-disubsituted alkene.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Catalyst]b | Solvent | Time | Conversionc | ||
| Mono-1a | 1a | 1a* | ||||
| a The reactions were carried out with 0.171 mmol of 1,4Si6 and 0.428 mmol of phenylacetylene in 1 mL of solvent at room temperature under nitrogen protection unless noted. b RuHCl(CO)(PPh3)3 loading was relative to 1,4Si6. c The conversion was determined by 1H NMR spectra of reaction aliquots. d The reaction was carried out at 60 °C. e The reactions were carried out with 0.171 mmol of 1,4Si6 and 0.171 mmol of phenylacetylene. f The reactions were carried out with 0.171 mmol of 1,4Si6 and 1.026 mmol of phenylacetylene. | ||||||
| 1 | 5 mol% | Toluene | 7 d | 22% | 9% | 0% |
| 2d | 10 mol% | Toluene | 24 h | 42% | 50% | 0% |
| 3 | 5 mol% | DCM | 24 h | 61% | 39% | 0% |
| 4 | 10 mol% | DCM | 24 h | 17% | 83% | 0% |
| 5e | 10 mol% | DCM | 24 h | 45% | 25% | 0% |
| 6 | 20 mol% | DCM | 2 h | 0% | 100% | 0% |
| 7f | 20 mol% | DCM | 2 h | 0% | 100% | 0% |
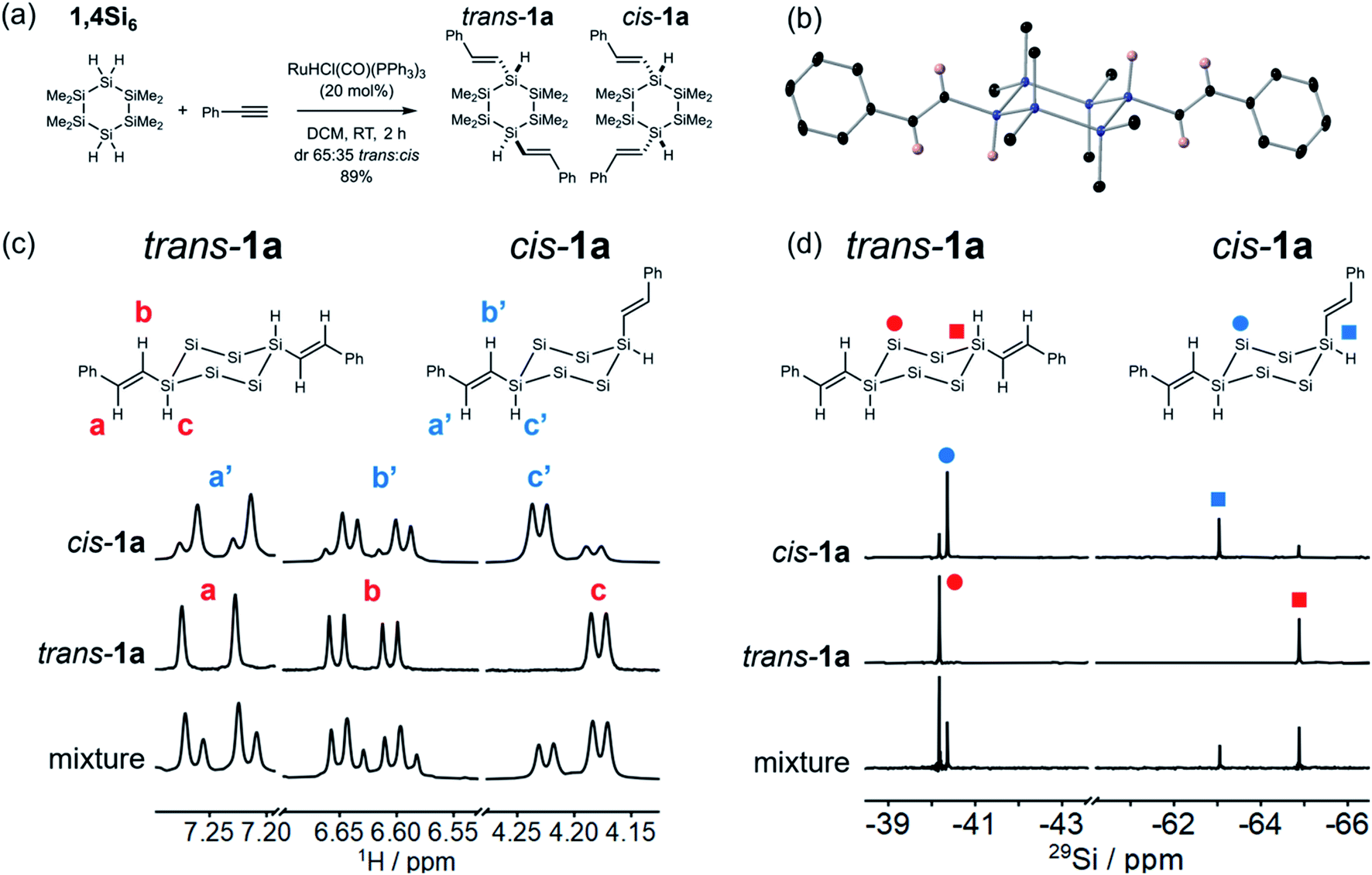 | ||
| Fig. 2 (a) Synthesis of trans and cis-1avia hydrosilylation of phenylacetylene with 1,4Si6 catalyzed by RuHCl(CO)(PPh3)3. (b) Displacement ellipsoid plot (30% probability level) of trans-1a at 110(2) K. Blue = Si, black = C, pink = H. Hydrogens on methyl and phenyl groups were omitted for clarity. Ellipsoids 30% probability. (c) Cropped 1H NMR spectra (400 MHz, C6D6) comparing (top to bottom) cis-1a (dr 25:75 trans:cis), trans-1a, and the as-isolated mixture of isomers. (d) Cropped 29Si {1H} DEPT spectra (79 MHz, C6D6) comparing (top to bottom) cis-1a (dr 25:75 trans:cis), trans-1a, and the as-isolated mixture of isomers. DEPT = distortionless enhancement by polarization transfer. The methyl groups are omitted for clarity. | ||
Having established regioselectivity, we investigated (E)-selectivity and diastereoselectivity under the optimized conditions. 1a was isolated in 89% yield as a 65:35 mixture of two isomeric products, assigned to the trans and cis diastereomers (Fig. 2a). The isomers were not separable by silica gel chromatography, but recrystallization afforded pure trans-1a (X-ray crystal structure, Fig. 2b), which was confirmed as the major diastereomer. The X-ray crystal structure also confirmed that hydrosilylation proceeded without skeletal rearrangement and with (E)- and β-selectivity. In the solid state, trans-1a adopted a chair conformation with both styryl groups at equatorial positions. This was the expected thermodynamic isomer, in which 1,3-diaxial interactions were minimized.
The recrystallization filtrate was enriched in the minor product, which was assigned to cis-1a based on symmetry consistent with 1,4-difunctionalization. The ca. 1:3 trans:cis ratio was stable over time but could not be further increased by silica gel column chromatography. The coupling constants of the vinylic peak a/a′ (d, J = 18.7 Hz, Fig. 2c) and b/b′ (dd, J = 5.2, 18.7 Hz) were consistent with an (E)-alkene. 1H–29Si HSQC and 29Si INEPT + spectroscopy (Fig. S and S2†) allowed assignment of the SiMe2 and SiH resonances (Fig. 2d).
Substrate scope
Having established selectivity with 1,4Si6, we investigated substrate scope (Fig. 3). With a variety of 4-substituted phenylacetylenes, 1,4-bis(styryl)cyclohexasilanes were obtained in high isolated yield and without erosion in regio- or diastereoselectivity, as shown by X-ray crystallography (Fig. S3†). This provided σ,π-hybrids functionalized with halogens (1b–c), electron donating groups (1d–f) and moderately electron withdrawing groups (1g–h).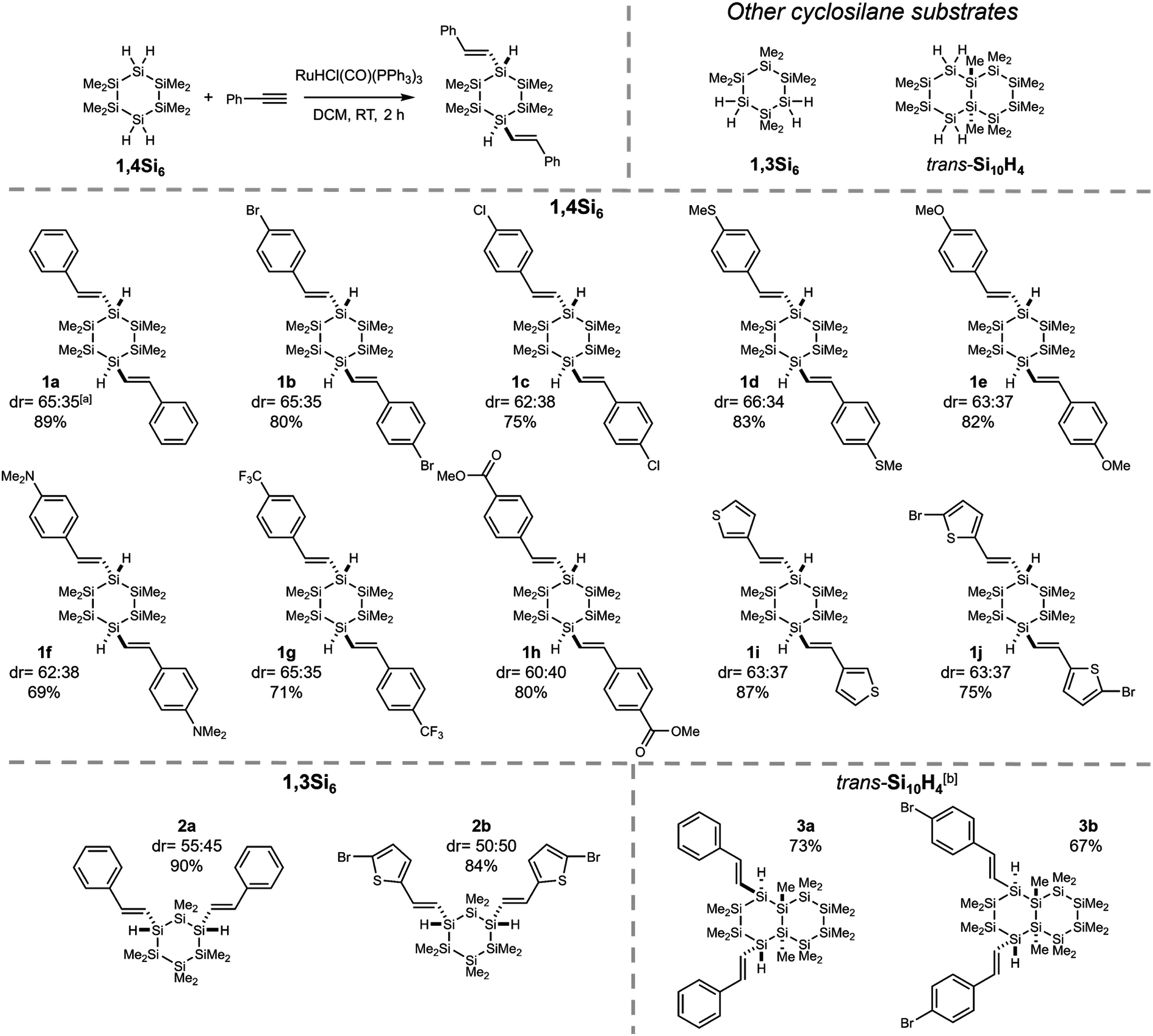 | ||
| Fig. 3 Substrate scope of hydrosilylation of alkynes with cyclosilanes. For 1,4Si6, major diastereomers are shown. adr determined by 1H NMR spectroscopy. Isolated yields are reported. bOnly the trans diastereomer was detected. | ||
The functional groups selected for investigation in Fig. 3 were chosen on the basis of potential applications. Sulfur was tolerated in thiomethyl 1d and thienyl 1i–j, of relevance to molecular electronics.43 Functional groups that would not be tolerated by the reductive coupling methods traditionally used to prepare cyclosilanes from dihalosilanes were also successfully incorporated, such as methyl ester 1h. Benzoate esters are useful intermediates to a variety of other oxygenated functionality, such as carboxylic acids that have been employed as MOF ligands.3 In addition, halogenated products (e.g., 1b and 1j) are suitable for catalytic cross-coupling (vide infra). Low conversion was observed with strongly electron-withdrawing substituents (e.g., 4-nitro and 4-cyanophenylacetylene) and an electron-poor heterocycle (4-ethynylpyridine).
We also explored the cyclosilane scope. Regioselectivity was maintained (Fig. 3), yielding 1,3- or 1,4-difunctionalized products instead of 1,1′-difunctionalized cyclosilanes. No skeletal decomposition was observed and perfect (E)- and β-selectivities were retained. Interestingly, cis-Si10H4 underwent slow reaction with phenylacetylene (<5% conversion in 24 hours).
The cyclosilane exerted a striking effect on diastereoselectivity. While diastereomeric ratios (dr) ca. 2:1 were observed for all 1,4Si6-derived products, the dr decreased to ca. 1:1 for 1,3Si6 and increased dramatically for trans-Si10H4, where only one diastereomer could be detected by 1H NMR spectroscopy (Fig. S4†). An X-ray crystal structure of 3a confirmed assignment to the trans diastereomer (Fig. 4) with diequatorial styryl groups. Like trans-decalin, trans-Si10H4 is conformationally locked,22 suggesting that hydrosilylation is selective for the equatorial Si–H bond. The reduced diastereoselectivity with 1,3Si6 and 1,4Si6 was attributed to rapid ring inversion44–46 in a mono-functionalized intermediate (Fig. 5). Therefore, we expect that the equilibrium between equatorial and axial conformers in mono-functionalized intermediates should determine the dr in difunctionalized products.
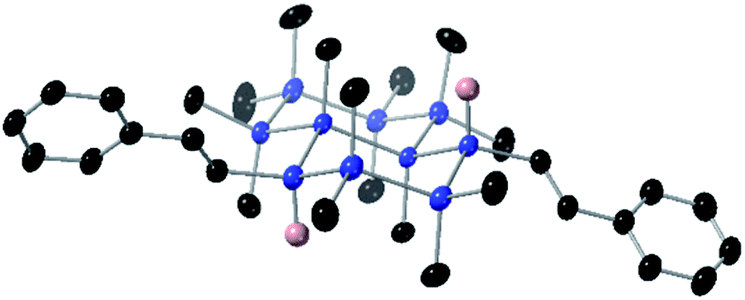 | ||
| Fig. 4 Displacement ellipsoid plot (30% probability level) of 3a at 173(2) K. Blue = Si, black = C, pink = H. Hydrogens on methyl and phenyl groups were omitted for clarity. | ||
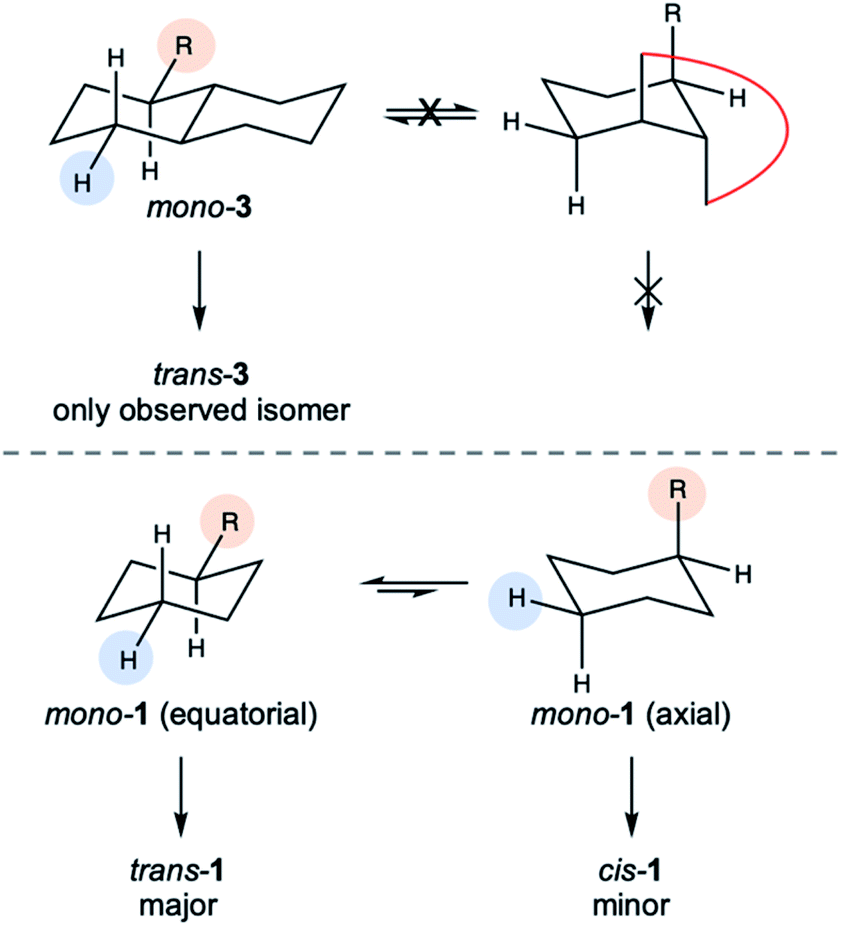 | ||
| Fig. 5 Model for stereoinduction. Hydrosilylation is highly selective for the equatorial proton (blue circle). Interconversion of axial and equatorial conformers (substituent labelled with orange circle) of a mono-functionalized intermediate determines the trans:cis ratio of difunctionalized products. | ||
UV-vis absorbance spectra
We obtained UV-vis spectra of the bis(styryl)cyclosilanes. Comparison of trans-1a to starting materials showed a 50- to 100 nm red-shift relative to purely π- or σ-conjugated materials (Fig. 6a). DFT calculations showed that the trans-1a HOMO was delocalized across the entire molecule (Fig. S5†). We also compared the UV-vis spectra of trans- and cis-1a, finding that relative configuration did not strongly influence absorbance in solution (Fig. 6b). This observation facilitated comparison of all three cyclosilanes (Fig. 6c). While 1,4- and 1,3-difunctionalized 1a and 2a were similar in onset of absorption, siladecalin-derived 3a was red-shifted by ca. 30 nm. We also noted the bathochromic influence of thienyl relative to phenyl substitution (Fig. 6d). Additional UV-spectra for selected compounds can be found in the ESI (Fig. S6†).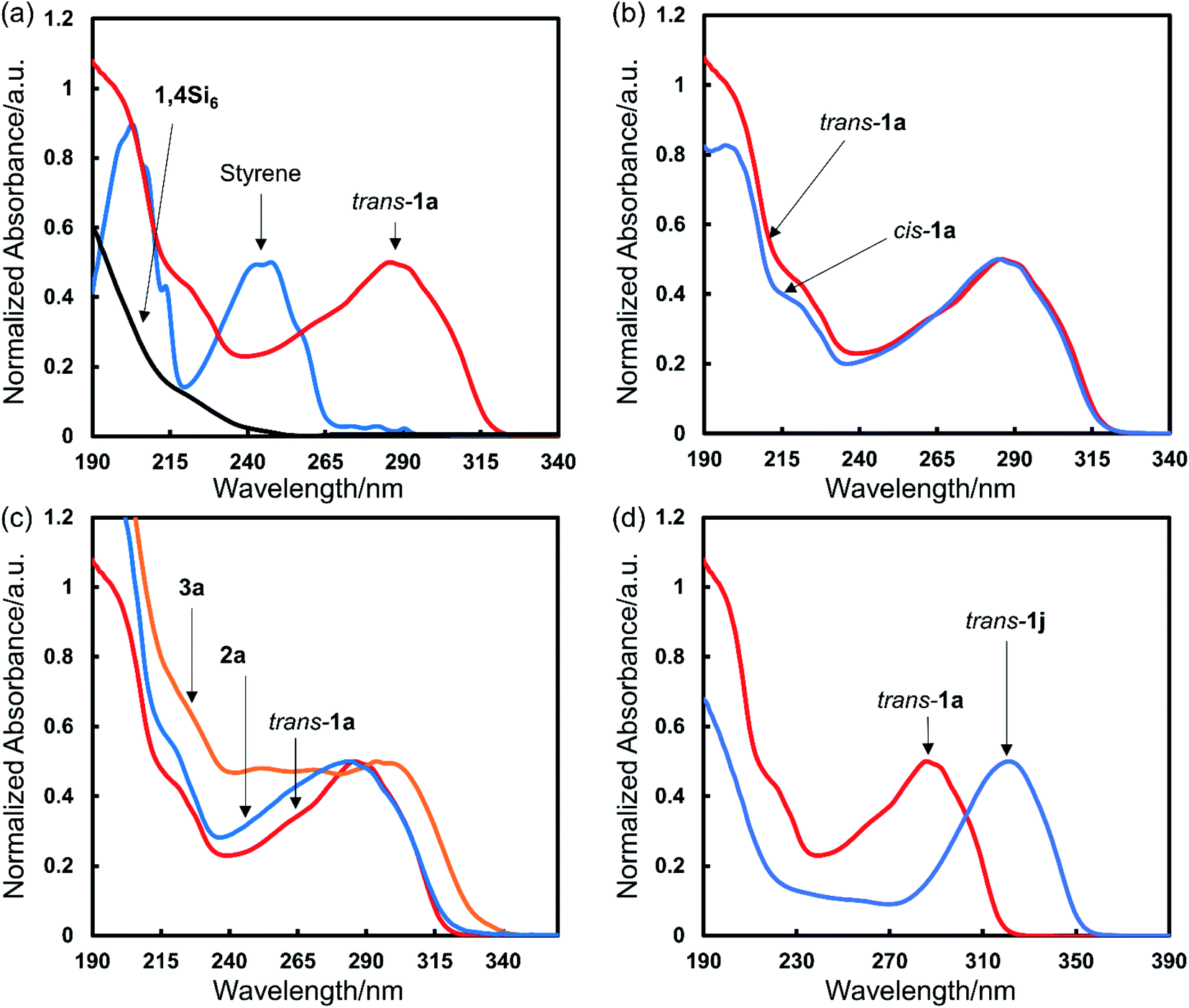 | ||
| Fig. 6 UV-vis spectra with normalized absorbance ([compound] = 10−5 M, in n-pentane). (a) Comparison to starting material. Red: trans-1a; black: 1,4Si6; blue: styrene. (b) Influence of configuration. Red: trans-1a; blue: cis-1a (dr 25:75 trans:cis). (c) Cyclosilane comparison. Red: trans-1a; blue: 2a (dr 55:45 trans:cis); orange: 3a. (d) Heterocycle effect. Red: trans-1a; blue: 1j (dr 90:10 trans:cis). | ||
Stereoregular conjugated polymer synthesis
The extended conjugation observed in hybrid molecules suggested a new conjugated polymer design: stereoregular copolymers alternating in σ- and π-conjugated motifs could be prepared via copolymerization of a dibrominated building block (e.g., trans-1b or trans-1j) with an appropriate comonomer. Polymers containing Si–Si bonds can be largely divided into two types: polysilanes (e.g., poly(SiR2))47,48 and σ,π-copolymers (e.g., poly(Ar(SiMe2)nAr)).49,50 Polysilanes in which the alkyl substituents are not the same (e.g., poly(SiR′R′′), R′ ≠ R′′) are stereogenic at each Si atom, but assignment and control of polysilane tacticity is a long-standing problem with no general solution.51–56 Use of a stereochemically pre-defined monomer appeals to avoid issues of control of relative configuration during polymerization.We found that treatment of dibromo trans-1j (dr 90:10 trans:cis) with stoichiometric i-PrMgCl resulted in quantitative Mg–halogen exchange.57 The intermediate α,ω-dimagnesiocyclosilane underwent Kumada cross-coupling with 2-bromothiophene, yielding trans-1k in 100% conversion (86% isolated yield) and without cyclosilane skeletal rearrangement or erosion of relative configuration (Fig. 7a). The novel polymer P1 (Mn = 6706 g mol−1, Mw/Mn = 2.63, Fig. S7†) was prepared by Kumada polycondensation of 2,5-dibromothiophene with the α,ω-dimagnesiocyclosilane derived from trans-1j (Fig. 7b). The dibromo building blocks could in principle serve as either a precursor to a nucleophile for Kumada polymerization, as showed above in the synthesis of P1, but also as the electrophile. We therefore also synthesized copolymer P2 in which Mg–halogen exchange of trans-1j was followed by Kumada polycondensation with dibromo trans-1b (Mn = 5075 g mol−1, Mw/Mn = 2.43, Fig. 7c). We note that residual catalysts were observed in both P1 and P2 by NMR, and further purification will be investigated in the future.
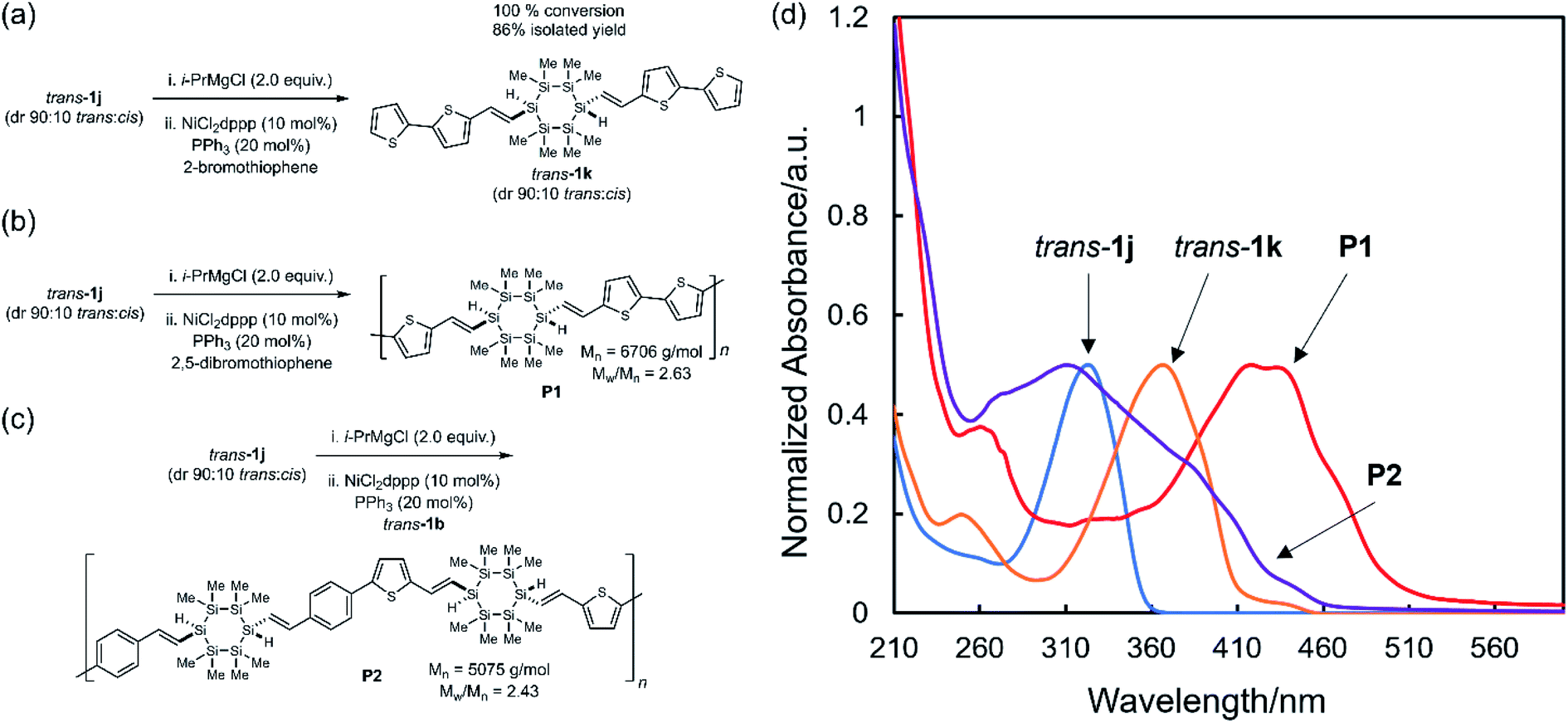 | ||
| Fig. 7 (a) Model reaction: Kumada cross-coupling of trans-1j and 2-bromothiophene. (b) Kumada polycondensation synthesis of P1. (c) Kumada polycondensation synthesis of P2. (d) Comparison of UV-vis spectra of trans-1j (blue), trans-1k (orange), P1 (red) and P2 (purple) in THF. [trans-1j] = 10−5 M, [trans-1k] = 10−5 M, [P1] = 0.03 g L−1, [P2] = 0.03 g L−1. | ||
The P1 onset of absorption (510 nm) was 140 nm red-shifted from trans-1j itself and 50 nm from trans-1k (Fig. 7d), validating the hypothesis that σ,π-mixing could be extended across a conjugated polymer chain. Comparison of the UV-vis spectra showed that P1 was red-shifted relative to P2. This was attributed to the difference between dithienyl and phenyl-thienyl units, as we showed (Fig. 6d) that molecular thienyl cyclosilanes were red-shifted relative to phenylsilanes. The all organic conjugated polymer PTVT (poly(thienylenevinylenethienyl)) exhibited a similar absorbance spectrum relative to P1 despite a much longer degree of polymerization (PTVT: λmax = 430 nm, λonset = 550 nm, Mn = 21000 g mol−1).58
These initial studies on conjugated polymer synthesis highlight the utility of stereodefined σ,π-hybrid materials, as well as the stability of relative configuration to subsequent chemical functionalization. Future work will examine the impact of stereoregularity on physical properties, as tactic polymers can demonstrate enhanced mechanical, thermal, and other properties relative to atactic.59
Conclusions
We demonstrated that Ru-catalyzed addition of tetrafunctional cyclosilanes to functionalized alkynes proceeded with high selectivity across several parameters: chemoselectivity for Si–H over Si–Si bonds, regioselectivity with respect to both cyclosilane and alkyne, and high (E)/(Z) and cis/trans diastereoselectivity. Broad functional group tolerance was exhibited, providing a complex organocyclosilanes bearing reactive functional groups challenging to incorporate by other means. The diverse functional groups incorporated open up several new possible directions, from MOF synthesis to molecular electronics. We also demonstrated that these synthetic achievements enabled the first broad investigation of structure–property relationships in σ,π-hybrid cyclosilanes, which revealed visible light absorption dependent on both the cyclosilane and arene identity. As exemplified by the Kumada polycondensation herein, we expect the results of this study to inspire new approaches to stereoregular conjugated materials combining σ- and π-conjugated motifs. We believe this contribution will continue to expand possibilities for the selective synthesis of complex materials from the main group.Author contributions
R. S. K. conceived and supervised the work. Q. J., A. F. G., S. W. and M. A. S. carried out the investigation. All authors contributed to manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was primarily supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, under Award No. DE-SC0020681 (building block synthesis, structural characterization). DFT calculations were conducted using scientific computing services at the Maryland Advanced Research Computing Center (MARCC). CPMAS spectroscopy was made possible through the National Science Foundation Major Research Instrumentation grant CHE-2018176.Notes and references
- R. S. Klausen, J. R. Widawsky, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, J. Am. Chem. Soc., 2012, 134, 4541–4544 CrossRef CAS PubMed.
- T. A. Su, H. Li, R. S. Klausen, J. R. Widawsky, A. Batra, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, J. Am. Chem. Soc., 2016, 138, 7791–7795 CrossRef CAS PubMed.
- D. A. Burns, E. M. Press, M. A. Siegler, R. S. Klausen and V. S. Thoi, Angew. Chem., Int. Ed., 2020, 59, 763–768 CrossRef CAS PubMed.
- S. Surampudi, M. L. Yeh, M. A. Siegler, J. F. M. Hardigree, T. A. Kasl, H. E. Katz and R. S. Klausen, Chem. Sci., 2015, 6, 1905–1909 RSC.
- J. Zhou, S. K. Surampudi, A. E. Bragg and R. S. Klausen, Chem. –Eur. J., 2016, 22, 6204–6207 CrossRef CAS PubMed.
- J. Zhou, C. P. Folster, S. K. Surampudi, D. Jimenez, R. S. Klausen and A. E. Bragg, Dalton Trans., 2017, 46, 8716–8726 RSC.
- B. J. Barrett, D. Jimenez, R. S. Klausen and A. E. Bragg, J. Phys. Chem. B, 2021, 125, 8460–8471 CrossRef CAS PubMed.
- H. K. Sharma, K. H. Pannell, I. Ledoux, J. Zyss, A. Ceccanti and P. Zanello, Organometallics, 2000, 19, 770–774 CrossRef CAS.
- M. Shimada, Y. Yamanoi, T. Matsushita, T. Kondo, E. Nishibori, A. Hatakeyama, K. Sugimoto and H. Nishihara, J. Am. Chem. Soc., 2015, 137, 1024–1027 CrossRef CAS PubMed.
- M. Shimada, M. Tsuchiya, R. Sakamoto, Y. Yamanoi, E. Nishibori, K. Sugimoto and H. Nishihara, Angew. Chem., Int. Ed., 2016, 55, 3022–3026 CrossRef CAS PubMed.
- R. Emanuelsson, H. Löfås, A. Wallner, D. Nauroozi, J. Baumgartner, C. Marschner, R. Ahuja, S. Ott, A. Grigoriev and H. Ottosson, Chem. –Eur. J., 2014, 20, 9304–9311 CrossRef CAS PubMed.
- E. A. Carberry and R. West, J. Am. Chem. Soc., 1969, 91, 5440–5446 CrossRef CAS.
- R. Fischer, T. Konopa, S. Ully, J. Baumgartner and C. Marschner, J. Organomet. Chem., 2003, 685, 79–92 CrossRef CAS.
- D. Y. Larkin, A. A. Korlyukov, E. V. Matukhina, M. I. Buzin, N. A. Chernyavskaya, M. Y. Antipin and A. I. Chernyavskii, Russ. Chem. Bull., 2005, 54, 1612–1622 CrossRef CAS.
- A. Bande and J. Michl, Chem. –Eur. J., 2009, 15, 8504–8517 CrossRef CAS PubMed.
- E. M. Press, E. A. Marro, S. K. Surampudi, M. A. Siegler, J. A. Tang and R. S. Klausen, Angew. Chem., Int. Ed., 2017, 56, 568–572 CrossRef CAS PubMed.
- E. A. Marro and R. S. Klausen, Chem. Mater., 2019, 31, 2202–2211 CrossRef CAS.
- E. A. Marro, E. M. Press, M. A. Siegler and R. S. Klausen, J. Am. Chem. Soc., 2018, 140, 5976–5986 CrossRef CAS PubMed.
- C. P. Folster and R. S. Klausen, Polym. Chem., 2018, 9, 1938–1941 RSC.
- Q. Jiang, S. Wong and R. S. Klausen, Polym. Chem., 2021, 12, 4785–4794 RSC.
- R. W. Dorn, E. A. Marro, M. P. Hanrahan, R. S. Klausen and A. J. Rossini, Chem. Mater., 2019, 31, 9168–9178 CrossRef CAS.
- E. A. Marro, C. P. Folster, E. M. Press, H. Im, J. T. Ferguson, M. A. Siegler and R. S. Klausen, J. Am. Chem. Soc., 2019, 141, 17926–17936 CrossRef CAS PubMed.
- D. J. Harrison, D. R. Edwards, R. McDonald and L. Rosenberg, Dalton Trans., 2008, 3401–3411 RSC.
- R. Walsh, Acc. Chem. Res., 1981, 14, 246–252 CrossRef CAS.
- M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed.
- H. K. Sharma and K. H. Pannell, Chem. Rev., 1995, 95, 1351–1374 CrossRef CAS.
- C. He, S. Chen, J. Zhu, J. Ke and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202117820 Search PubMed.
- G. K. Min, D. Hernández and T. Skrydstrup, Acc. Chem. Res., 2013, 46, 457–470 CrossRef CAS PubMed.
- K. ichiro Kanno, Y. Aikawa and S. Kyushin, Tetrahedron Lett., 2020, 61, 152274 CrossRef.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567–570 CrossRef CAS PubMed.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- H.-H. Moretto, M. Schulze and G. Wagner, Ullmann's Encycl. Ind. Chem., 2012, 675–712 Search PubMed.
- K. Yamamoto, M. Kumada, I. Nakajima, K. Maeda and N. Imaki, J. Organomet. Chem., 1968, 13, 329–341 CrossRef CAS.
- K. C. Song, K. M. Lee, N. Van Nghia, W. Y. Sung, Y. Do and M. H. Lee, Organometallics, 2013, 32, 817–823 CrossRef CAS.
- J. P. Banovetz, Y. L. Hsiao and R. M. Waymouth, J. Am. Chem. Soc., 1993, 115, 2540–2541 CrossRef CAS.
- Y. L. Hsiao and R. M. Waymouth, J. Am. Chem. Soc., 1994, 116, 9779–9780 CrossRef CAS.
- R. Shankar and V. Shahi, J. Organomet. Chem., 2008, 693, 307–315 CrossRef CAS.
- L. Sacarescu, A. Siokou, G. Sacarescu, M. Simionescu and I. Mangalagiu, Macromolecules, 2008, 41, 1019–1024 CrossRef CAS.
- P. T. K. Lee, M. K. Skjel and L. Rosenberg, Organometallics, 2013, 32, 1575–1578 CrossRef CAS.
- P. T. K. Lee and L. Rosenberg, Dalton Trans., 2017, 46, 8818–8826 RSC.
- K. ichiro Kanno, S. Noguchi, Y. Ono, S. Egawa, N. Otsuka, M. Mita and S. Kyushin, J. Organomet. Chem., 2022, 961, 122234 CrossRef.
- T. A. Su, H. Li, R. S. Klausen, N. T. Kim, M. Neupane, J. L. Leighton, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Acc. Chem. Res., 2017, 50, 1088–1095 CrossRef CAS PubMed.
- G. Tekautz, A. Binter, K. Hassler and M. Flock, ChemPhysChem, 2006, 7, 421–429 CrossRef CAS PubMed.
- D. Casarini, L. Lunazzi and A. Mazzanti, Tetrahedron, 1998, 54, 13181–13184 CrossRef CAS.
- D. Casarini, L. Lunazzi and A. Mazzanti, J. Org. Chem., 1998, 63, 9125–9127 CrossRef CAS.
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS.
- R. S. Klausen and E. Ballestero-Martínez, in Comprehensive Organometallic Chemistry IV, Elsevier, 2021 Search PubMed.
- M. C. Fang, A. Watanabe and M. Matsuda, Macromolecules, 1996, 29, 6807–6813 CrossRef CAS.
- Y. Morisaki, F. Fujimura and Y. Chujo, Organometallics, 2003, 22, 3553–3557 CrossRef CAS.
- J. P. Banovetz, K. M. Stein and R. M. Waymouth, Organometallics, 1991, 10, 3430–3432 CrossRef CAS.
- N. Choi, S. Y. Onozawa, T. Sakakura and M. Tanaka, Organometallics, 1997, 16, 2765–2767 CrossRef CAS.
- B. J. Grimmond and J. Y. Corey, Organometallics, 2000, 19, 3776–3783 CrossRef CAS.
- V. K. Dioumaev, K. Rahimian, F. Gauvin and J. F. Harrod, Organometallics, 1999, 18, 2249–2255 CrossRef CAS.
- E. Fossum and K. Matyjaszewski, Macromolecules, 1995, 28, 1618–1625 CrossRef CAS.
- J. Maxka, F. K. Mitter, D. R. Powell and R. West, Organometallics, 1991, 10, 660–664 CrossRef.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- R. Toyoshima, K. Akagi and H. Shirakawa, Synth. Met., 1997, 84, 431–432 CrossRef CAS.
- J. C. Worch, H. Prydderch, S. Jimaja, P. Bexis, M. L. Becker and A. P. Dove, Nat. Rev. Chem., 2019, 3, 514–535 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2158546–2158549. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01690g |
| This journal is © The Royal Society of Chemistry 2022 |