
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid synthesis of layered KxMnO2 cathodes from metal–organic frameworks for potassium-ion batteries†
Ang
Li‡
a,
Changfeng
Li‡
a,
Peixun
Xiong‡
 a,
Jinfeng
Zhang
a,
Dongling
Geng
b and
Yunhua
Xu
*a
a,
Jinfeng
Zhang
a,
Dongling
Geng
b and
Yunhua
Xu
*a
aSchool of Materials Science and Engineering, Key Laboratory of Advanced Ceramics and Machining Technology (Ministry of Education), Tianjin Key Laboratory of Composite and Functional Materials, Tianjin University, Tianjin 300072, China. E-mail: yunhua.xu@tju.edu.cn
bSchool of Materials Science and Engineering, Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing 210094, P. R. China
First published on 6th June 2022
Abstract
Layered transition metal oxides (LTMOs) are a kind of promising cathode materials for potassium-ion batteries because of their abundant raw materials and high theoretical capacities. However, their synthesis always involves long time calcination at a high temperature, leading to low synthesis efficiency and high energy consumption. Herein, an ultra-fast synthesis strategy of Mn-based LTMOs in minutes is developed directly from alkali-transition metal based-metal–organic frameworks (MOFs). The phase transformation from the MOF to LTMO is systematically investigated by thermogravimetric analysis, variable temperature optical microscopy and X-ray diffraction, and the results reveal that the uniform distribution of K and Mn ions in MOFs promotes fast phase transformation. As a cathode in potassium-ion batteries, the fast-synthesized Mn-based LTMO demonstrates an excellent electrochemical performance with 119 mA h g−1 and good cycling stability, highlighting the high production efficiency of LTMOs for future large-scale manufacturing and application of potassium-ion batteries.
Introduction
Rechargeable potassium-ion batteries (KIBs) are considered as one of the most promising alternatives to lithium-ion batteries on account of the abundant raw materials and low redox potential of potassium for large-scale energy storage technologies.1–3 However, there exist significant challenges in seeking suitable electrode materials to achieve sufficient electrochemical performance of KIBs because of the large size of K+, which causes huge volume changes and sluggish kinetics, and thus low specific capacity, poor rate capability and fast performance degradation.4–11 In the past few years, numerous efforts have been devoted to the development and optimization of low-cost and high-performance electrode materials, particularly cathode materials.12–17Recently, layered transition metal oxides (LTMOs) have been extensively investigated as cathode materials for KIBs.18–21 They have ionic diffusion paths with a large interlayer distance, benefiting the migration of K+. Among them, layered potassium manganese oxide (KxMnO2) has attracted great attention owing to their high theoretical capacities and abundance of raw materials.22–25 Generally, layered KxMnO2 is synthesized through a long-time high-temperature calcination (≥800 °C), which typically takes more than 10 hours (Table S1†). Apparently, this procedure takes time and consumes energy, leading to low production efficiency and high cost.
It is well known that the atomic distance seriously impacts the diffusion process during pyrolysis treatment, and further influences the formation of a new phase. Aiming at rapid synthesis of LTMO, a short distance between alkali metal ions and transition metal ions as well as a uniform distribution is necessary. Metal–organic frameworks (MOFs) are a kind of porous crystalline material formed by metal ions and organic ligands through coordination bonds, which have a homogenous distribution of different elements at the atomic level.26 With the merits of large surface area, ultra-high porosity, and structural diversity, they have attracted extensive attention in energy fields.27,28 The investigation was mainly focused on MOF-derived anode materials but rarely on cathodes.29–33 A lot of efforts have been devoted to developing transition metal (Fe, Co, Ni, Mn, Cu, and Zn)-based MOF precursors for anode materials, while less on alkali-transition metal based-MOFs. Despite differences in the properties between alkali and transition metal ions, alkali-transition metal based-MOFs can be constructed and are suitable for rapid synthesis of LTMO cathodes due to their atomic level distribution of different elements.
Herein, an ultra-fast synthesis method is developed for preparing a layered cathode material of KxMnO2 directly from the K[Mn(HCOO)3] MOF (KM-MOF). The formate anion (HCOO−) is the simplest and smallest carboxylate ligand and the atomic-level homogenous distributions of elements promises a short migration distance of metal ions, enabling a fast phase formation of layered KxMnO2 in minutes (Fig. 1). The phase transformation process from MOF to layered metal oxide was systematically investigated by thermogravimetric analysis (TGA), variable temperature optical microscopy and X-ray diffraction (XRD). The fast-synthesized layered KxMnO2 achieved excellent electrochemical performance with 119 mA h g−1 and good cycling stability, that surpasses most previously reported LTMO cathodes (Table S2†), highlighting the high production efficiency of LTMOs for future large-scale manufacturing and application of KIBs.
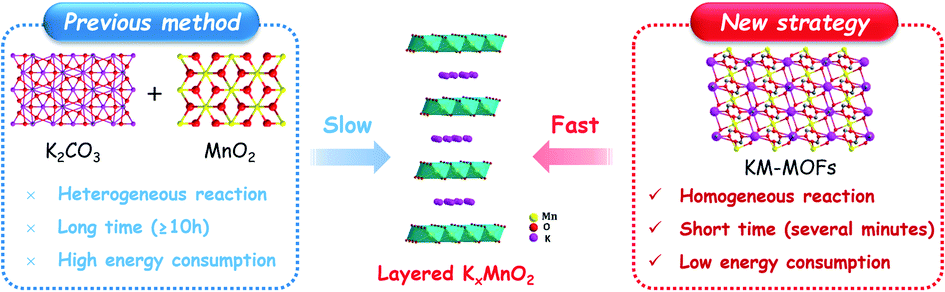 | ||
| Fig. 1 Comparison between the traditional and new synthesis methods of layered KxMnO2. | ||
Experimental
Materials
All the reagents and solvents, including manganese chloride tetrahydrate (MnCl2·4H2O, Aladdin), potassium formate (HCOOK, Acros), formic acid (HCOOH, Tianjin Jiangtian Chemical Technology Co., Ltd) and methanol (CH3OH, Tianjin Jiangtian Chemical Technology Co., Ltd) were commercially obtained and used without further purification.Materials characterization
Single-crystal X-ray diffraction data for the K[Mn(HCOO)3] MOF were collected on a Rigaku XtalAB Pro MM007 DW at 298 K. Room temperature X-ray diffraction (XRD) patterns were recorded on a Rigaku MiniFlex 600 X-ray diffractometer at 40 kV and 15 mA with a Cu-target tube. Thermogravimetric analysis (TGA) was carried out on a Rigaku TG-DTA 8121 analyzer at a rate of 10 °C min−1 from 25 to 900 °C. Scanning electron microscopy (SEM) (JEOL JSM-7500F), transmission electron microscopy (TEM), and high-resolution transmission electron microscopy (HRTEM) (JEM-ARM200F) were used to investigate the material morphologies and structures. Variable temperature optical microscope photograph images were taken on a Linkam THMS600 with a thermal stage. XPS measurements were performed on a Thermo Scientific ESCALAB 250. ICP-OES (PerkinElmer 8300) was used to analyze the composition of elements.Electrochemical tests
The working electrodes were prepared by casting the slurry of layered KxMnO2, Ketjen black (KB) and polyvinylidenefluoride (PVDF) at a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 on aluminum foil. Coin-type cells were fabricated using potassium foil as the counter electrodes, 2.5 M potassium bis(fluorosulfonyl)imide (KFSI) in triethyl phosphate (TEP) as the electrolyte, and a glass fiber membrane in a glove box filled with highly pure argon gas (O2 and H2O levels < 0.1 ppm) as the separator. Galvanostatic discharge/charge tests were performed in a voltage range of 2.0–4.2 V (vs. K/K+) on Neware battery testers. CV profiles were collected on a Solartron 1470 Electrochemical Interface. For operando XRD, an electrolytic cell (Beijing Scistar Technology Co. Ltd) with one side beryllium (Be) window (20 mm in diameter) was employed and cycled at a current density of 50 mA g−1 between 2.0 and 4.2 V (vs. K+/K). The operando XRD patterns were recorded with a scan speed of 3° min−1 and a time step of 0.01 s.
:2:1 on aluminum foil. Coin-type cells were fabricated using potassium foil as the counter electrodes, 2.5 M potassium bis(fluorosulfonyl)imide (KFSI) in triethyl phosphate (TEP) as the electrolyte, and a glass fiber membrane in a glove box filled with highly pure argon gas (O2 and H2O levels < 0.1 ppm) as the separator. Galvanostatic discharge/charge tests were performed in a voltage range of 2.0–4.2 V (vs. K/K+) on Neware battery testers. CV profiles were collected on a Solartron 1470 Electrochemical Interface. For operando XRD, an electrolytic cell (Beijing Scistar Technology Co. Ltd) with one side beryllium (Be) window (20 mm in diameter) was employed and cycled at a current density of 50 mA g−1 between 2.0 and 4.2 V (vs. K+/K). The operando XRD patterns were recorded with a scan speed of 3° min−1 and a time step of 0.01 s.
Results and discussion
The KM-MOF was synthesized by a self-assembly process using precursors of manganese chloride tetrahydrate (MnCl2·4H2O), potassium formate (HCOOK) and formic acid (HCOOH) in methanol at room temperature.34 X-ray crystallographic analysis reveals that the KM-MOF crystallizes in the monoclinic space group C2/c, with Mn(II) ions positioned at an inversion center. Each Mn(II) octahedron is coordinated with six Mn(II) octahedra through four equatorials syn–anti and two axial anti–anti formate bridges. The structure can be regarded as a distorted perovskite anionic framework with zig–zag chains of K ions in the channels (Fig. 2a and S2†). The ultra-small distance (3.599 Å) and uniform distributions of K and Mn ions in the KM-MOF would promote the phase transformation from MOF to LTMO in a short time. The XRD pattern of the as-synthesized KM-MOF is consistent with the simulated data, suggesting a successful synthesis of the KM-MOF (Fig. S1†). A spindle-shaped crystal morphology of KM-MOF was simulated by using the Bravais–Friedel–Donnay–Harker (BFDH) method (Fig. 2b), which is verified by the optical microscope photograph (Fig. S3†).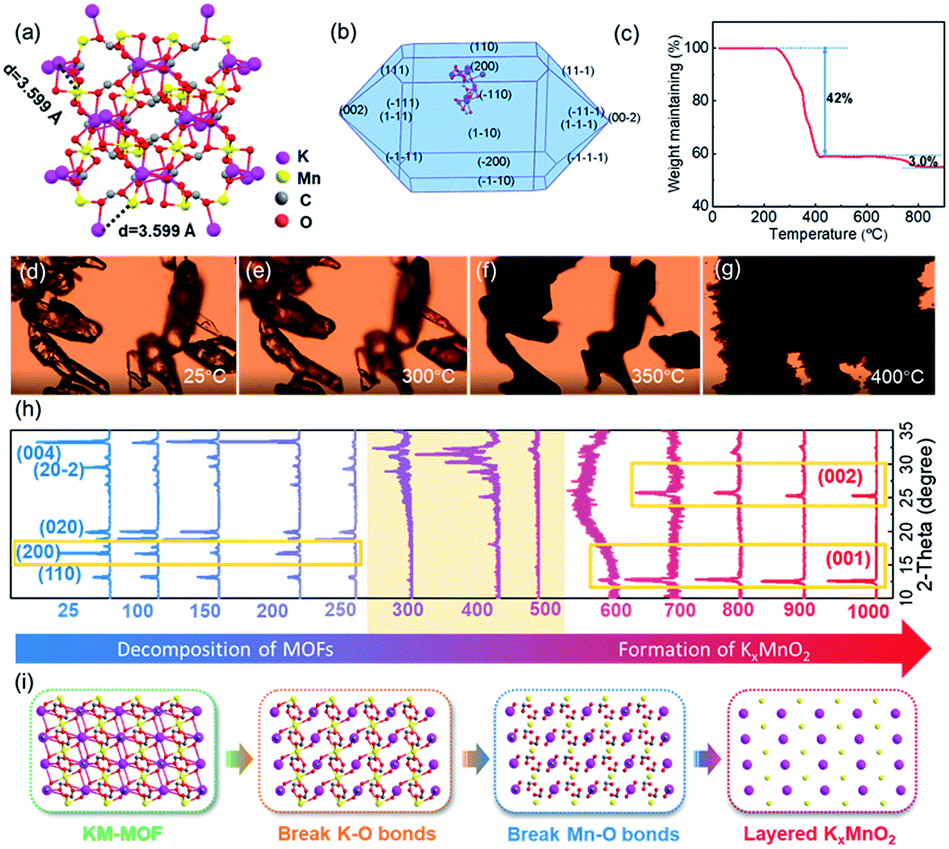 | ||
| Fig. 2 Structural and thermal stability characterization of the KM-MOF. (a) Coordination environments of the metal centers and the ligands of the KM-MOF. (b) Simulated crystal morphology, (c) TGA curve, (d–g) variable temperature optical microscope photograph images, and (h) XRD patterns at different temperatures of the KM-MOF. (i) Schematic structural evolution from the KM-MOF to KxMnO2. | ||
In general, the composition and structure of MOF precursors significantly affect the pyrolysis process and the corresponding derivative. Therefore, an insight into the KM-MOF to layered KxMnO2 transformation process is required to improve the understanding of the rapid synthesis. To probe the transformation process, component evolution was monitored during the calcination process by TGA. TGA measurements were performed from 50 to 900 °C with a heating rate of 10 °C min−1 in air. As shown in Fig. 2c, a remarkable weight loss of 42.0% occurred from 250 to 410 °C, which is assigned to the decomposition of the KM-MOF and the formation of metal oxide. Theoretically, the organic species were burned into CO2, leading to 57.6% weight loss according to the theoretical calculations, while the formation of metal oxide caused 14.0% weight gain upon the uptake of oxygen. The TGA measurement result is consistent with the theoretical values. A small weight loss of 3.0% was displayed from 700 to 800 °C, which should be attributed to the sublimation of potassium because of its low boiling point of 759 °C. The bond lengths of the KM-MOF obtained by using crystalline data are in the order of K–O > Mn–O > C–O (Table S3†), indicating that the bond strengths are in the order of K–O < Mn–O < C–O. Therefore, during the thermal heating process, the K–O and Mn–O bonds would break prior to the C–O bonds. The morphology evolution of the KM-MOF is also monitored by variable temperature optical microscopy, and the optical microscope photographs at different temperatures are shown in Fig. 2d–g. At low temperatures below 300 °C, there is no obvious change to the crystalline morphology (Fig. 2d and e). However, when the temperature increased to 350 °C, the crystals became opaque (Fig. 2f), and molten at 400 °C (Fig. 2g), suggesting damage of the crystalline structure. These results are well consistent with the TGA measurement results.
To gain further insights into the phase conversion process from the KM-MOF to layered KxMnO2, variable temperature XRD was conducted (Fig. 2h). The diffraction peak of the (200) crystal plane that is based on K–O bonds disappeared at 250 °C (Fig. S4†). When the temperature increased to 300∼500 °C, the KM-MOF almost lost its crystallinity. From 600 °C, new peaks appeared at 12.7 and 25.4°, which can be indexed to the planes of (001) and (002) of layered KxMnO2, respectively, indicative of the beginning of layered phase formation. These two peaks slightly shifted to lower diffraction angles of 12.6° for (001) and 25.2° for (002) planes from 800 to 900 °C, indicating that the interlayer spacing became larger. This can be explained by the enhanced coulombic repulsion between the MO6 slabs upon K+ sublimation. The interlayer spacing is calculated to be 0.69 nm based on the diffraction peak of the (001) plane. The variable temperature XRD measurement results show that layered KxMnO2 can be facilely synthesized from the KM-MOF through thermal sintering. Based on the aforementioned analysis, the transformation process from the KM-MOF to layered KxMnO2 is schematically illustrated in Fig. 2i. During the heating process, K–O and Mn–O bonds successively break along with the uptake of oxygen to form K and Mn oxides from 300 to 400 °C, and then the layered phase of KxMnO2 is formed above 600 °C.
An ultra-fast synthesis method was developed to produce layered KxMnO2, by sintering the KM-MOF at 1000 °C for only 8 minutes, and the resulting product is denoted as KMO-F. As a control, layered KxMnO2 was also prepared using a conventional long-time sintering method from the same MOF precursor, denoted as KMO-S. Their layered crystal structures were revealed by the XRD patterns, and they are in good agreement with the previously reported results in the literature.35 The pronounced peaks at 12.7/25.4° of KMO-S and 12.6/25.2° of KMO-F are assigned to the (001)/(002) planes (Fig. 3a and S5†), and the interlayer spacings are calculated to be 0.69 and 0.68 nm, respectively. Rietveld refinement suggests that both KMO-F and KMO-S belong to the hexagonal symmetry with a space group of P63/mmc. The lattice parameters of KMO-F phase are calculated to be a = b = 2.881 Å and c = 14.132 Å, while they are a = b = 2.880 Å and c = 14.062 Å for KMO-S. Therefore, they have the same crystalline structure, and the schematic crystal structure of the layered KxMnO2 is shown in Fig. 3b. Mn ions are octahedrally coordinated with oxygen anions while K ions are located between the layers of MnO6 octahedra. The overall K/Mn atomic ratios were measured by using inductively coupled plasma (ICP) to be 0.53 for KMO-F and 0.51 for KMO-S (Table S4†), corresponding to the molecular formulas of K0.53MnO2 and K0.51MnO2, respectively. The chemical properties of the layered compounds were investigated by X-ray photoelectron spectroscopy (XPS) (Fig. S6†). The high-resolution Mn 3s spectrum of KMO-F shows a peak energy separation (ΔE) of 5.06 eV (Fig. S7†), consistent with the previous reports.35 According to the linear relationship between the chemical valence of Mn and the ΔE value, the average oxidation state of Mn in KMO-F was calculated to be 3.47 (Table S5†). Correspondingly, the Mn 2p1/2 and Mn 2p3/2 peaks in the high-resolution Mn 2p spectrum can be deconvoluted into two peaks of Mn3+ and Mn4+ with equal integral areas (Fig. 3c), suggesting an average valence of 3.5, very close to the value of 3.47. Similar results were obtained for KMO-S.
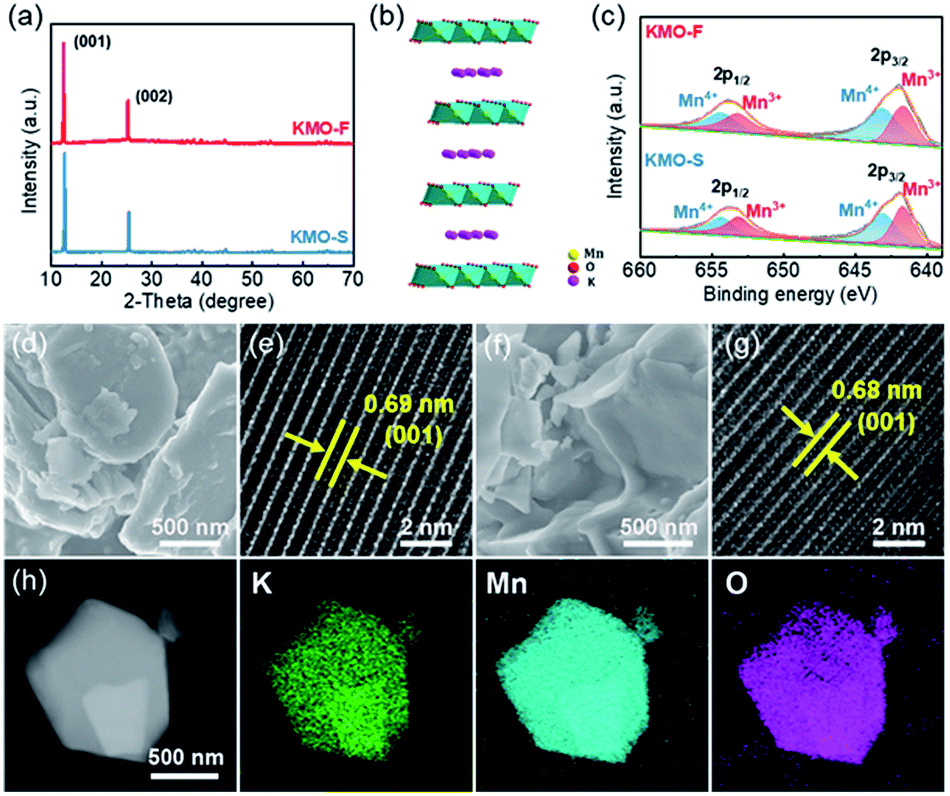 | ||
| Fig. 3 Structure characterization of KMO-F and KMO-S. (a) XRD patterns of KMO-F and KMO-S. (b) Simulated crystal structure of layered KxMnO2. (c) XPS spectra of KMO-S and KMO-F. SEM image of (d) KMO-F and (f) KMO-S. HRTEM images of (e) KMO-F and (g) KMO-S. (h) STEM image of KMO-F with corresponding elemental mappings of K, Mn, and O. | ||
The morphologies and microstructures of the layered KMOs were characterized by scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). The SEM images of KMO-F and KMO-S present similar morphologies with crystal particles in random shapes and sizes (Fig. 3d and f). The HRTEM images reveal highly crystalline structures with interlayer spacings of 0.69 nm for KMO-F and 0.68 nm for KMO-S (Fig. 3e and g), corresponding to the (001) plane. Uniform elemental distributions of K, Mn and O were measured over the whole particles by scanning transmission electron microscopy (STEM) (Fig. 3h and S8†), suggesting a well-formed layered structure. Apparently, well-crystallized layered KxMnO2 can be produced by the fast synthesis method, similar to the conventional long-time synthesis methods from the same MOF precursor, demonstrating the effectiveness and superiority of the new method.
The electrochemical performances of KMO-F and KMO-S were investigated and compared under the same test conditions. Coin-type cells were fabricated using KMO-F or KMO-S cathodes and 2.5 M KFSI/TEP as the electrolyte. The cyclic voltammetry (CV) measurements were carried out at a scan rate of 0.1 mV s−1 in a voltage window of 2.0–4.2 V (vs. K+/K). Similar CV curves were displayed for the two cathodes with five redox couples (Fig. 4a and b), suggesting similar electrochemical behaviors. These current peaks can be assigned to the reversible potassium insertion/extraction into/from layered KxMnO2 with Mn3+/Mn4+ redox reaction. Obviously, the well-overlapped CV curves after the first cycle predict good cycling stability for the KMO cathodes. The galvanostatic charge/discharge profiles at a current density of 50 mA g−1 are shown in Fig. 4c and d. The first charge/discharge capacities of KMO-F and KMO-S are 119/133 and 84/111 mA h g−1, respectively. Although KMO-F delivered a slightly higher initial capacity, similar cycling performance was obtained for them (Fig. 4e). KMO-F and KMO-S retained 80% and 85% of their capacity after 100 cycles at 50 mA g−1, respectively, with high average coulombic efficiencies of about 99%. In addition, the KMO-F cathode delivers an initial discharge capacity of 81.14 mA h g−1 at 300 mA g−1 and retains 86% of its capacity after 120 cycles (Fig. S9†). Good rate performance was obtained for both KMO-F and KMO-S (Fig. 4f). When the current density increased from 50 to 1000 mA g−1, KMO-F maintained a specific capacity of 65 mA h g−1, compared with 73 mA h g−1 for KMO-S. The electrochemical performance once again manifests the feasibility of the fast synthesis method for high performance layered cathode materials.
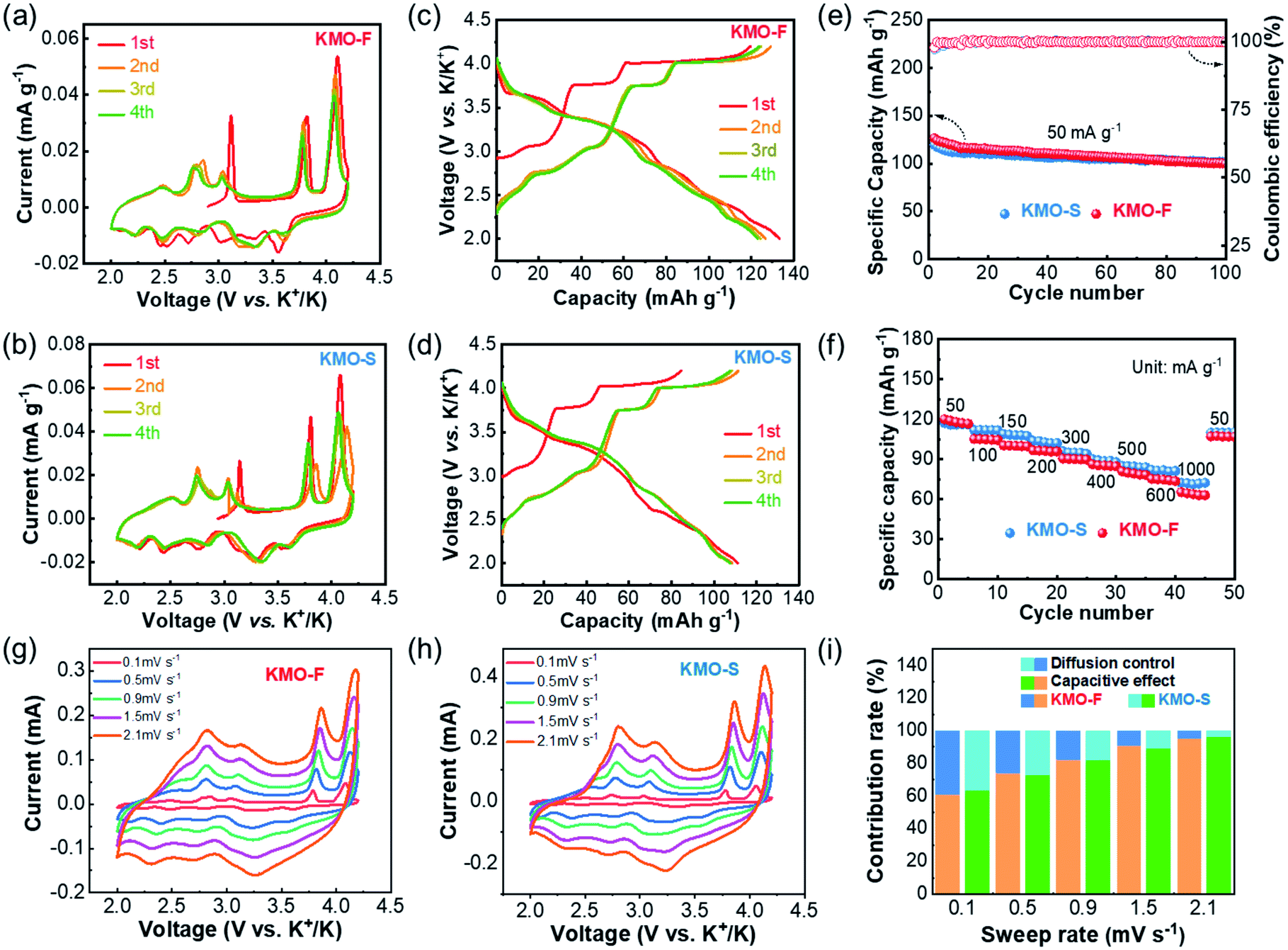 | ||
| Fig. 4 Electrochemical performance of the layered cathodes. CV profiles of (a) KMO-F and (b) KMO-S at a scan rate of 0.1 mV s−1. Galvanostatic charge–discharge profiles of (c) KMO-F and (d) KMO-S at the 1st, 2nd, 3rd, and 4th cycles. (e) Cycling performance and coulombic efficiency of KMO-F and KMO-S at a current density of 50 mA g−1. (f) Rate performance of KMO-F and KMO-S. Storage process characterization, (g) CV curves of the KMO-F cathode at different scan rates, (h) CV curves of the KMO-S cathode at different scan rates, and (i) normalized capacitive-controlled capacities and diffusion-controlled capacities for the cathode electrodes at different scan rates. | ||
To further investigate the K-storage behavior for the KMO cathodes, CV measurements at various sweep rates from 0.1–2.1 mV s−1 were carried out (Fig. 4g and h). The capacity contributions from surface pseudo-capacitance and ion intercalation can be quantified by separating the current response i at a fixed potential V into capacitive-controlled (k1v) and diffusion-controlled (k2v1/2) processes, according to the following equation:36i(V) = k1v1 + k2v1/2. In our work, based on the integration of CV curves, 91.2% of the total charge storage capacity of the KMO-F cathode is capacitive at a sweep rate of 1.5 mV s−1. Such high proportions of pseudocapacitive contribution result from fast kinetics, which should account for the good rate capability of the KMO-F cathode. The surface capacitive-controlled capacities and diffusion-controlled capacities for KMO-F and KMO-S at different scan rates are summarized in Fig. 4i. KMO-S shows slightly higher diffusion-controlled capacities at various sweep rates.
The K storage process of the layered cathodes was investigated by in situ XRD, and the data are shown in Fig. 5. By charging (extraction of K ions), the (001) and (002) peaks shifted to lower 2-theta degrees, indicating the increased interlayer spacing due to enhanced electrostatic repulsion between the adjacent oxygen layers upon K+ extraction. During the discharge process, a reversible peak shift was observed, demonstrating good structural reversibility and cycling stability.
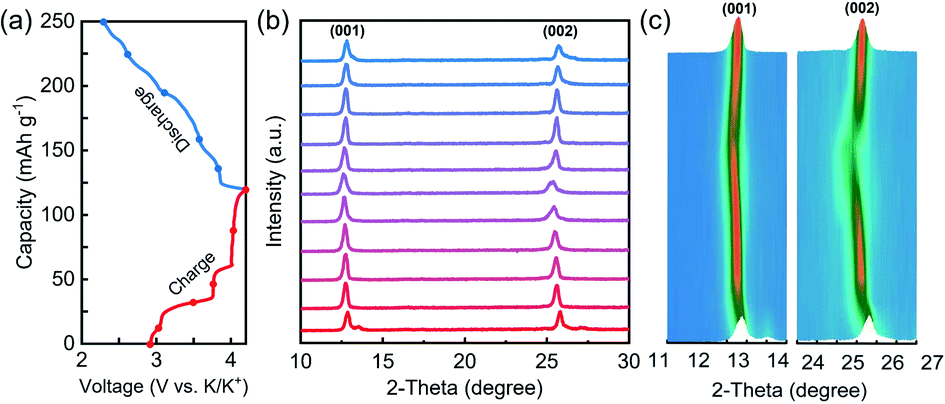 | ||
| Fig. 5 (a) Galvanostatic charge/discharge profiles in the first cycle of layered KxMnO2 at a current density of 50 mA g−1, (b) in situ XRD patterns, and (c) the image plots of the in situ XRD data. | ||
Conclusions
In this work, a fast synthesis method is developed by directly calcining an alkali-transition metal based-MOF of K[Mn(HCOO)3] for preparing layered KxMnO2 cathode materials thanks to the ultra-small distance (3.599 Å) and uniform distributions of K and Mn ions in the KM-MOF. The new method saves time and energy compared with the traditional long-time high-temperature synthesis method. The fast phase transformation process was systematically investigated by multiple analysis techniques and theoretical simulations. The layered KxMnO2 cathode demonstrated excellent potassium storage performance with high reversibility and cycling stability, which is similar to the cathode produced by a long-time-sintering method from the same precursor and is superior to most previously reported LTMO cathodes in KIBs. Our findings provide a new strategy for synthesizing high performance layered cathode materials for KIBs.Data availability
The article and ESI† contain all the experimental and computational data.Author contributions
Y. X. and A. L. designed the research. A. L., C. L., J. Z. and D. G. carried out the experiments and analysis. P. X. and Y. X. revised the manuscript. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support of the National Key Research and Development Program of China (Grant No. 2019YFE0118800).Notes and references
- W. Zhang, Y. Liu and Z. Guo, Sci. Adv., 2019, 5, eaav7412 CrossRef CAS PubMed.
- X. Zou, P. Xiong, J. Zhao, J. Hu, Z. Liu and Y. Xu, Phys. Chem. Chem. Phys., 2017, 19, 26495–26506 RSC.
- Y. Jiang, Y. Yang, F. Ling, G. Lu, F. Huang, X. Tao, S. Wu, X. Cheng, F. Liu, D. Li, H. Yang, Y. Yao, P. Shi, Q. Chen, X. Rui and Y. Yu, Adv. Mater., 2022, 34, 2109439 CrossRef CAS PubMed.
- X. Ren, Q. Zhao, W. D. McCulloch and Y. Wu, Nano Res., 2017, 10, 1313–1321 CrossRef CAS.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- Y. Liu, Y.-X. Lu, Y.-S. Xu, Q.-S. Meng, J.-C. Gao, Y.-G. Sun, Y.-S. Hu, B.-B. Chang, C.-T. Liu and A.-M. Cao, Adv. Mater., 2020, 32, 2000505 CrossRef CAS PubMed.
- Z. Jian, S. Hwang, Z. Li, A. S. Hernandez, X. Wang, Z. Xing, D. Su and X. Ji, Adv. Funct. Mater., 2017, 27, 1700324 CrossRef.
- J. Zhao, X. Zou, Y. Zhu, Y. Xu and C. Wang, Adv. Funct. Mater., 2016, 26, 8103–8110 CrossRef CAS.
- M. Wang, W. Lu, H. Zhang and X. Li, Trans. Tianjin Univ., 2021, 27, 1–23 CrossRef CAS.
- Y. Chen, B. Xi, M. Huang, L. Shi, S. Huang, N. Guo, D. Li, Z. Ju and S. Xiong, Adv. Mater., 2022, 34, 2108621 CrossRef CAS.
- W. Luo, Y. Feng, D. Shen, J. Zhou, C. Gao and B. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 16379–16385 CrossRef CAS PubMed.
- L. Wu, M. Gu, Y. Feng, S. Chen, L. Fan, X. Yu, K. Guo, J. Zhou and B. Lu, Adv. Funct. Mater., 2022, 32, 2109893 CrossRef CAS.
- J. Ge, L. Fan, A. M. Rao, J. Zhou and B. Lu, Nat. Sustain., 2022, 5, 225–234 CrossRef.
- L. Deng, J. Qu, X. Niu, J. Liu, J. Zhang, Y. Hong, M. Feng, J. Wang, M. Hu, L. Zeng, Q. Zhang, L. Guo and Y. Zhu, Nat. Commun., 2021, 12, 2167 CrossRef CAS PubMed.
- S. Xu, Y. Chen and C. Wang, J. Mater. Chem. A, 2020, 8, 15547–15574 RSC.
- B. Ji, W. Yao, Y. Zheng, P. Kidkhunthod, X. Zhou, S. Tunmee, S. Sattayaporn, H.-M. Cheng, H. He and Y. Tang, Nat. Commun., 2020, 11, 1225 CrossRef CAS.
- M. Tang, Y. Wu, Y. Chen, C. Jiang, S. Zhu, S. Zhuo and C. Wang, J. Mater. Chem. A, 2019, 7, 486–492 RSC.
- T. Deng, X. Fan, C. Luo, J. Chen, L. Chen, S. Hou, N. Eidson, X. Zhou and C. Wang, Nano Lett., 2018, 18, 1522–1529 CrossRef CAS PubMed.
- W. Zhong, X. Liu, Q. Cheng, T. Tan, Q. Huang, Q. Deng, J. Hu and C. Yang, Appl. Phys. Rev., 2021, 8, 031412 CAS.
- J.-Y. Hwang, J. Kim, T.-Y. Yu, S.-T. Myung and Y.-K. Sun, Energy Environ. Sci., 2018, 11, 2821–2827 RSC.
- Z. Xiao, F. Xia, L. Xu, X. Wang, J. Meng, H. Wang, X. Zhang, L. Geng, J. Wu and L. Mai, Adv. Funct. Mater., 2022, 32, 2108244 CrossRef CAS.
- B. Lin, X. Zhu, L. Fang, X. Liu, S. Li, T. Zhai, L. Xue, Q. Guo, J. Xu and H. Xia, Adv. Mater., 2019, 31, 1900060 CrossRef PubMed.
- K. Lei, Z. Zhu, Z. Yin, P. Yan, F. Li and J. Chen, Chem, 2019, 5, 3220–3231 CAS.
- A. Gao, M. Li, N. Guo, D. Qiu, Y. Li, S. Wang, X. Lu, F. Wang and R. Yang, Adv. Energy Mater., 2019, 9, 1802739 CrossRef.
- H. Kim, D. H. Seo, J. C. Kim, S. H. Bo, L. Liu, T. Shi and G. Ceder, Adv. Mater., 2017, 29, 1702480 CrossRef PubMed.
- L. J. Kong, M. Zhong, W. Shuang, Y. H. Xu and X. H. Bu, Chem. Soc. Rev., 2020, 49, 2378–2407 RSC.
- X. Zhang, A. Chen, M. Zhong, Z. H. Zhang, X. Zhang, Z. Zhou and X. H. Bu, Electrochem. Energy Rev., 2019, 2, 29–104 CrossRef CAS.
- W. Zhu, A. Li, Z. Wang, J. Yang and Y. Xu, Small, 2021, 17, e2006424 CrossRef PubMed.
- J. Liu, C. Wu, D. Xiao, P. Kopold, L. Gu, P. A. van Aken, J. Maier and Y. Yu, Small, 2016, 12, 2354–2364 CrossRef CAS PubMed.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed.
- A. Li, P. Xiong, Y. Zhang, W. Shuang, Z. Chang, Y. Xu and X.-H. Bu, Chem. Eng. J., 2021, 405, 126638 CrossRef CAS.
- X. X. Li, F. Y. Cheng, S. N. Zhang and J. Chen, J. Power Sources, 2006, 160, 542–547 CrossRef CAS.
- P. Xiong, X. Zhao and Y. Xu, ChemSusChem, 2018, 11, 202–208 CrossRef CAS PubMed.
- E. Eikeland, N. Lock, M. Filsoe, M. Stingaciu, Y. Shen, J. Overgaard and B. B. Iversen, Inorg. Chem., 2014, 53, 10178–10188 CrossRef CAS PubMed.
- Y. Kadoma, Y. Uchimoto and M. Wakihara, J. Phys. Chem. B, 2006, 110, 174–177 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruna, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD, optical microscope photograph, XPS, elemental mapping, CV, and galvanostatic charge–discharge profiles. See https://doi.org/10.1039/d2sc02442j |
| ‡ Ang Li, Changfeng Li and Peixun Xiong contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |