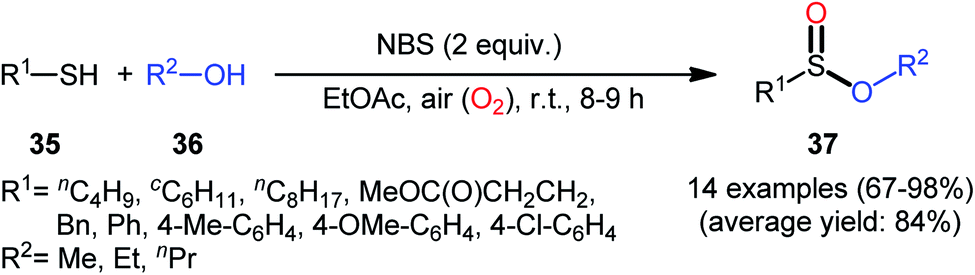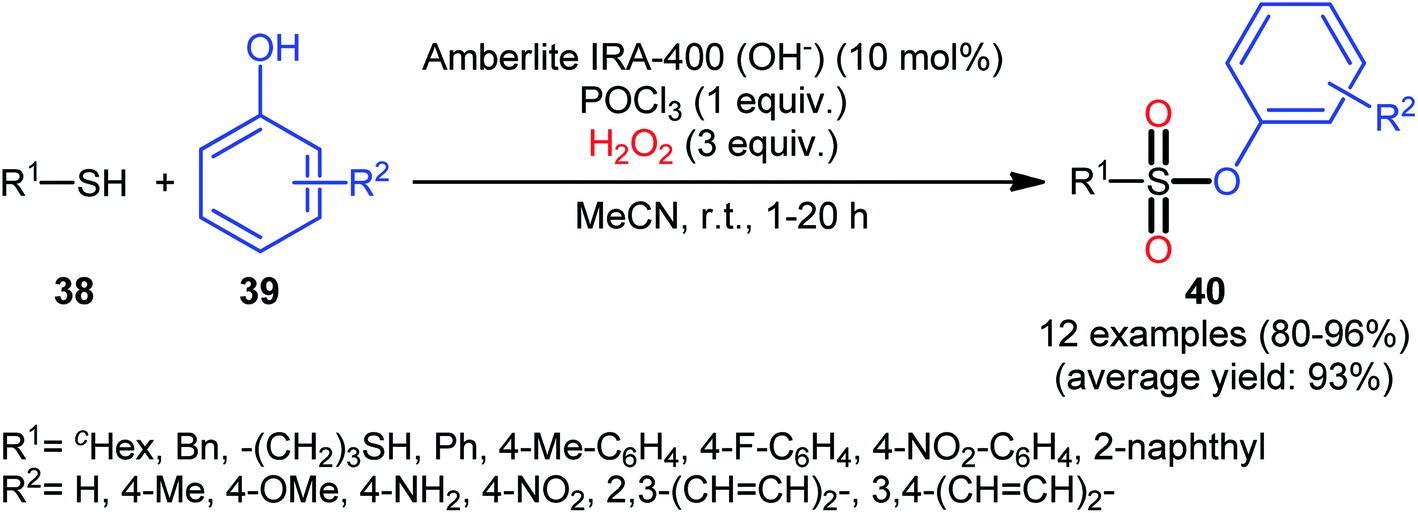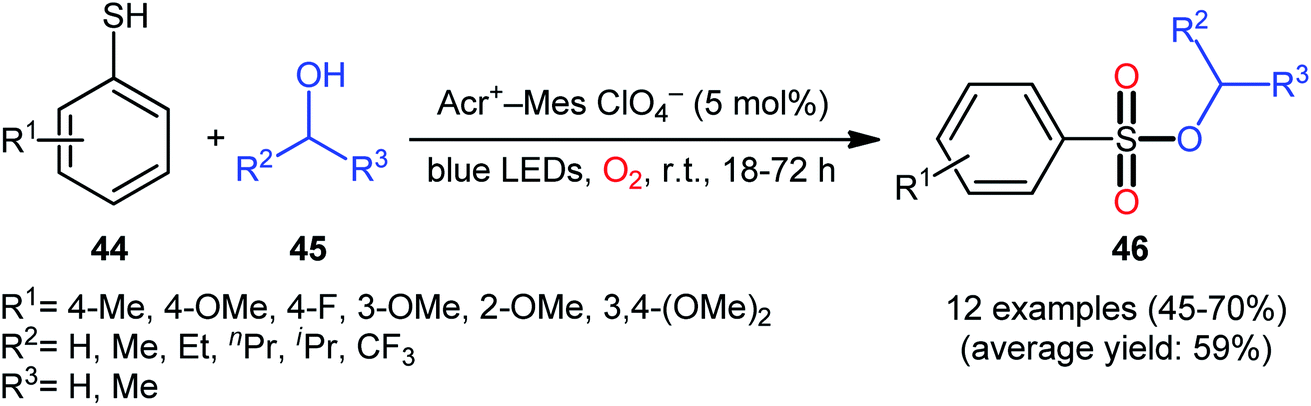DOI:
10.1039/D1RA08058J
(Review Article)
RSC Adv., 2022,
12, 14521-14534
Strategies for the direct oxidative esterification of thiols with alcohols
Received
2nd November 2021
, Accepted 12th April 2022
First published on 13th May 2022
Abstract
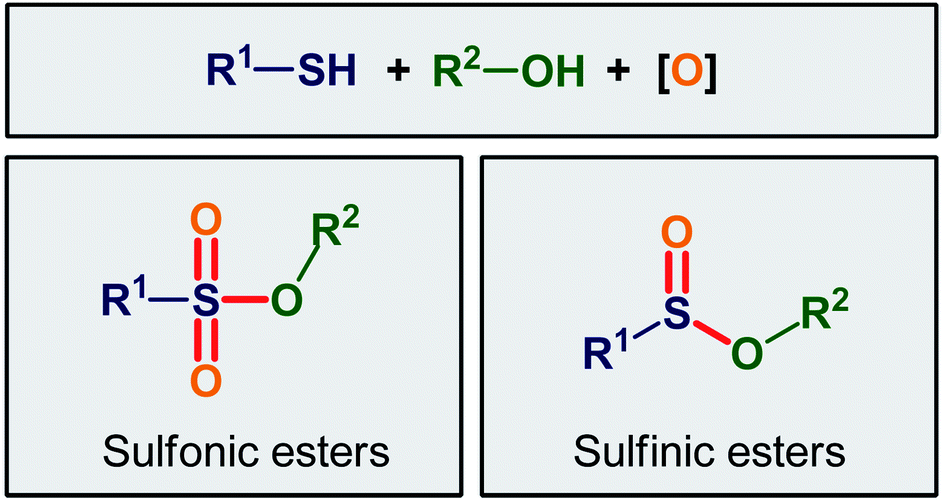
This review paper provides an overview of the main strategies for the oxidative esterification of thiols with alcohols. The review is divided into two major parts according to final products. The first includes the methods for the synthesis of sulfinic esters, while the second contains the procedures for the fabrication of sulfonic ester derivatives.
1. Introduction
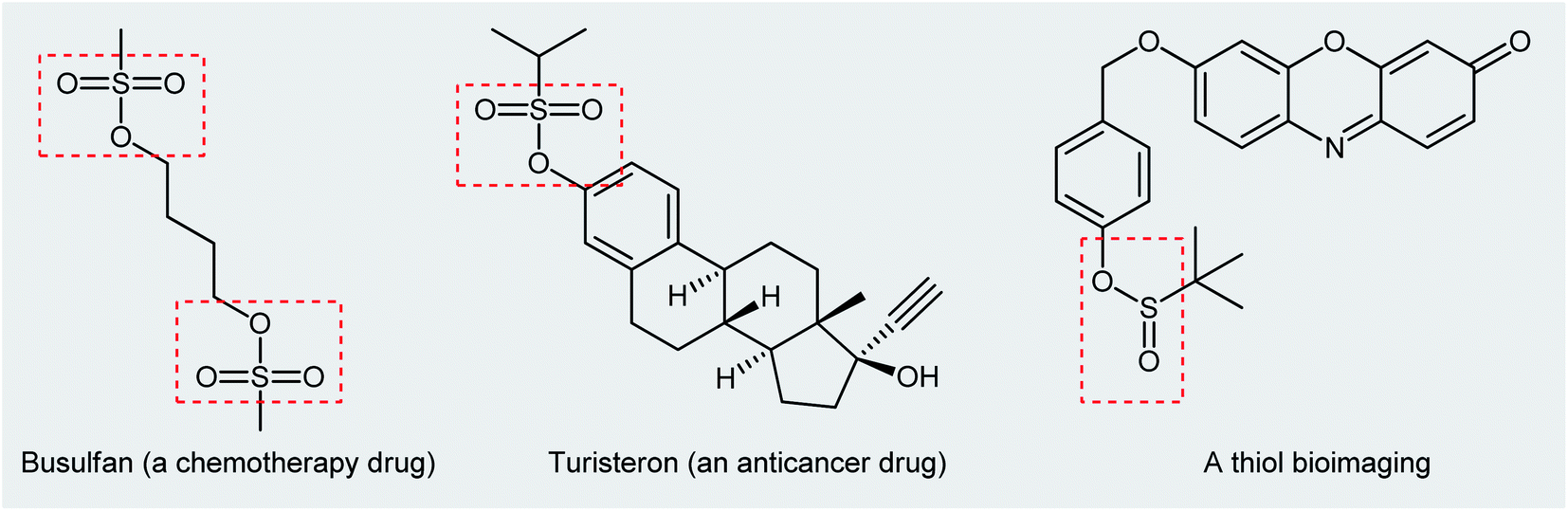
Organosulfur compounds are some of the most versatile building blocks in organic chemistry and occupy a very special place in medicinal, pharmaceutical, and agricultural science.1,2 In this family of compounds, sulfinic and sulfonic esters are of interest due to their biological and therapeutic activities3,4 as well as their application as chemical probes in living cell imaging (Scheme 1).5,6 Furthermore, the title compounds are highly useful synthetic precursors and intermediates in organic synthesis. For example, sulfinic esters were recently applied as efficient sulfenylating agents7 and sulfonic esters as sulfonylating agents.8 The classical methods for the synthesize of these classes of organosulfur compounds involve the reaction of sulf(o,i)nyl chlorides,9,10 sulf(o,i)nic acids,11,12 or sodium sulfonates13 with alcohols. However, corrosion, instability, and/or difficult preparability of some of the sulf(i,o)nylating agents limited the utility of these methods.
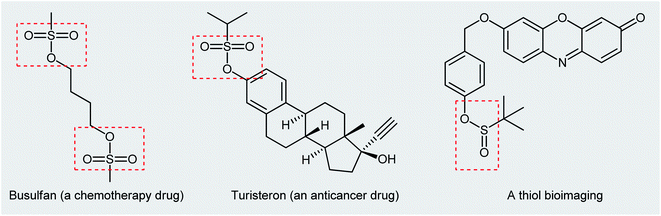 |
| | Scheme 1 Selected examples of commercial sulfonic ester drugs and a thiol bioimaging sulfinic ester compound. | |
In the light of the above limitations and due to the versatile synthetic and biological activity of the titled compounds, their synthesis from easily accessible basic starting materials under mild conditions is a field of growing research interest. In this context, recently, the direct oxy-esterification of thiols with alcohols has emerged as an ideal atom- and step-economic strategy for the construction of these compounds which makes the synthetic route more quick and clean than conventional pathways that relay on the use of pre-functionalized starting materials. As a continuation of our previous works on modern synthetic methods in organic chemistry14 and novel strategies in the preparation of organosulfur compounds,15 in this review, we will highlight the most important developments on the synthesis of sulfinic and sulfonic esters through the direct oxidative esterification of thiols with alcohols (Fig. 1) with emphasis on the mechanistic features of the reactions, by hoping that it further stimulating research and development of this interesting field. It is noteworthy that, despite wide biological and synthetic importance of sulfenic ester derivative,16,17 to date no reporting guideline exists for their direct construction from the corresponding thiols and alcohols. Therefore, in this review no comment was made regarding the cross-dehydrogenative coupling between thiols and alcohols.
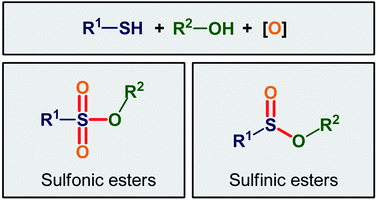 |
| | Fig. 1 Direct oxidative esterification of thiols with alcohols. | |
2. Synthesis of sulfinic esters
In this section we would like to focus on the oxidative esterification of thiols with alcohols into sulfinic esters. To aid the reading, the section is divided into three sub-sections according to catalytic systems. The first includes the electrochemical reactions, the second contains the metal-catalyzed reactions, and the third covers metal-free reactions.
2.1. Electrochemical reactions
Electrochemistry provides some of the greenest, cleanest, and more cost-efficient synthetic strategies, as the use of often hazardous or waste-generating chemical reductants or oxidants can be substituted by electrical power.18 In this context, electrochemical oxidative cross-coupling reactions have recently attracted tremendous attention as cleaner and more sustainable synthetic alternative to traditional coupling procedures which rely on the use of external oxidants, catalysts or ligands.19
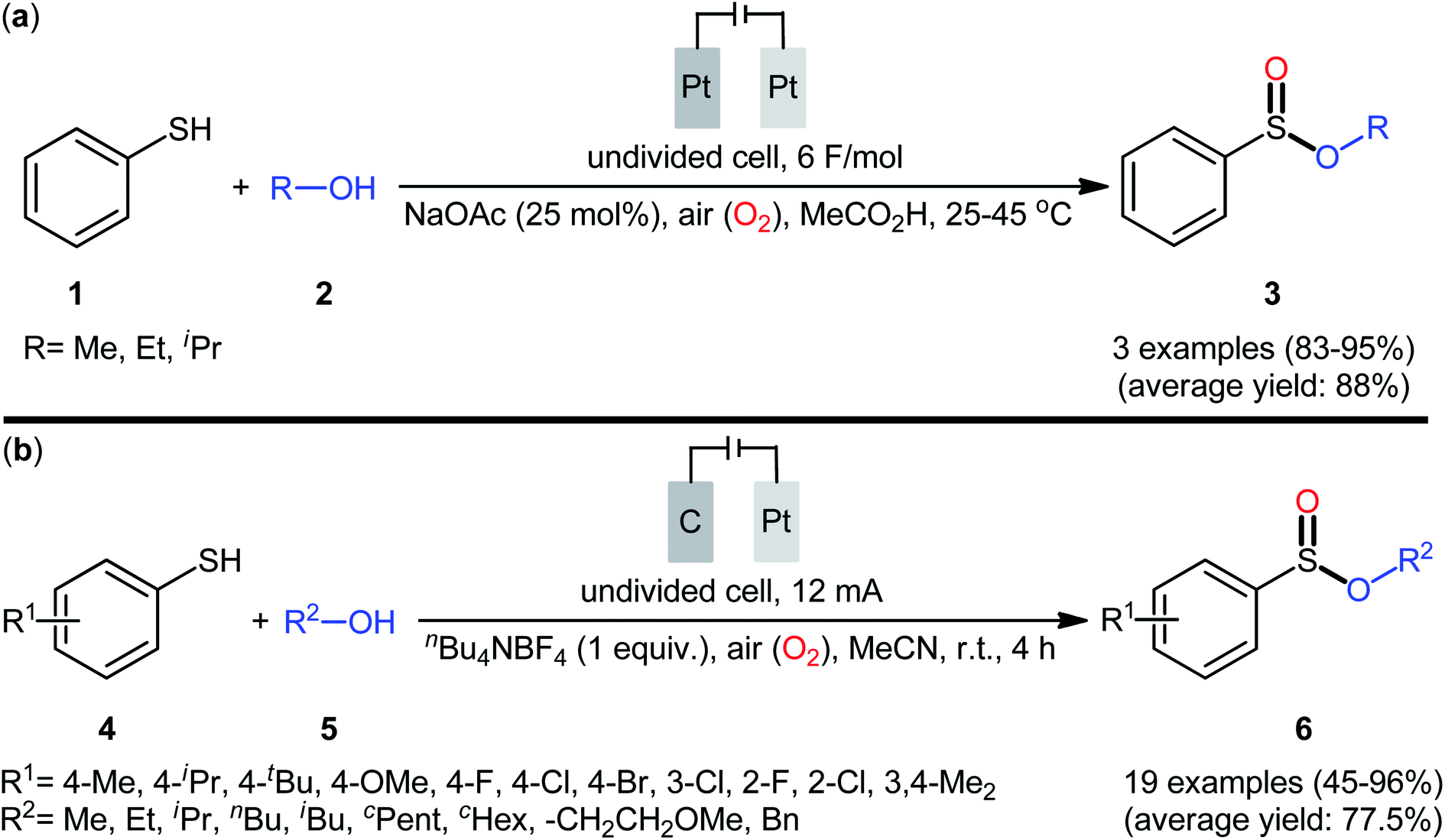
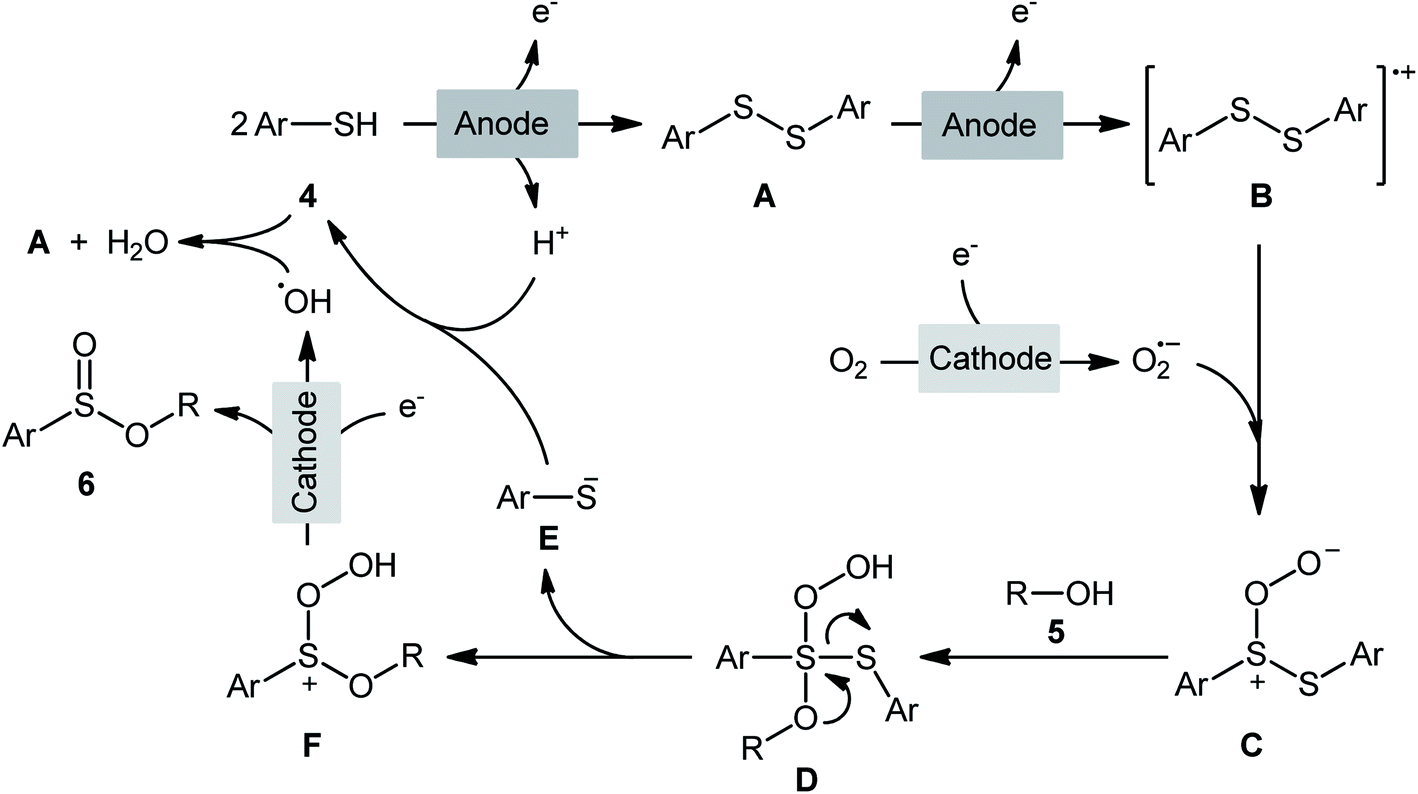
The first report on the electrochemical oxidative esterification of thiols with alcohols was published by Nokami and co-workers in 1979,20 who showed that the treatment of thiophenol 1 with aliphatic alcohols 2 in an undivided cell with platinum electrodes, employing NaOAc as the supporting electrolyte in acetic acid, resulted in the formation of alkyl phenylsulfinates 3 in good to excellent yields (Scheme 2a). The diphenyldisulfide was also examined under the identical conditions and the desired products were obtained in satisfactory yields. However, the excess use of alcohols would be a significant drawback for this protocol and may limit its range of applications. Moreover, in this preliminary work, only one thiol was employed, without any substrate scope exploration. Four decades later, Gong and Lu along with their co-workers developed a robust electrochemical oxidative cross-coupling between thiophenols 4 and aliphatic alcohols 5 for the synthesis of alkyl benzenesulfinates 6.21 In an undivided cell assembled with a graphite rod anode and platinum plate cathode, the best reaction conditions were achieved with tetrabutylammonium tetrafluoroborate (nBu4NBF4) as the electrolyte and acetonitrile as the solvent, with a constant current of 12 mA under air atmosphere at room temperature (Scheme 2b). A broad reaction scope was evaluated by testing different groups on the thiophenol ring (e.g., OMe, F, Cl, Br), as well as a variety of aliphatic (primary and secondary) alkyl/benzyl alcohols. Notably, the authors demonstrated the scalability of the reaction since methyl 4-methylbenzenesulfinate could be obtained in 1.62 g scale in excellent yield of 95%. The importance of air atmosphere was evaluated and in the presence of N2 atmosphere no product was formed. Therefore, they suggested that the source of sulfoxides oxygen comes from air oxygen. The radical trapping experiments with (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) confirmed that this reaction most likely proceeds via a radical pathway (Scheme 3). Initially, a disulfide intermediate A was formed via homocoupling of in situ generated thiyl radical by the anodic oxidation of starting thiol 4. In the next step, this intermediate A underwent a further single-electron oxidation to form a disulfide radical cation B. After that, the newly formed radical B was reacted with superoxide radical anion (O2˙−), generated from the reduction of O2 at the cathode, and provided the intermediate C. Thereafter, the addition of alcohol 5 to this species C gave intermediate D which subsequently delivered sulfide anion E and intermediate F. Finally, reduction of F at the cathode gave the desired product 6.
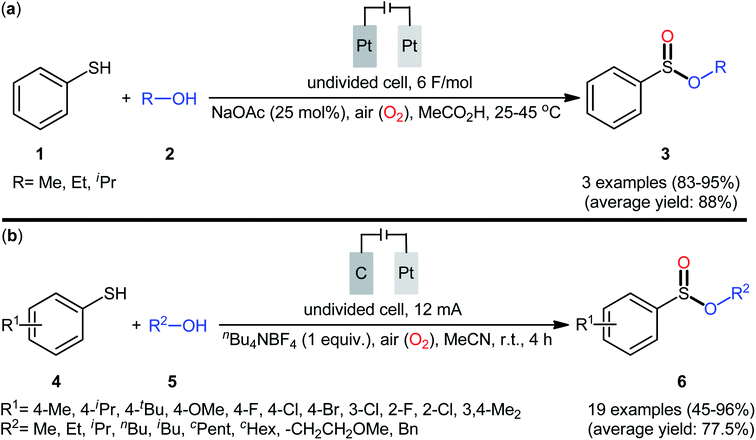 |
| | Scheme 2 (a) Electrooxyesterification of thiophenol 1 with aliphatic alcohols 2 to alkyl phenylsulfinates 3; (b) electrochemical synthesis of alkyl benzenesulfinates 6 from thiophenols 4 and alcohols 5. | |
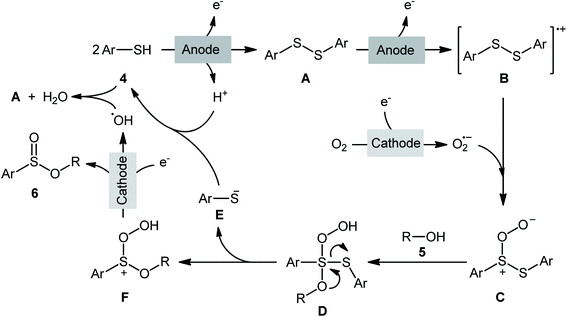 |
| | Scheme 3 Proposed mechanism for electrosynthesis of sulfinic esters 6. | |
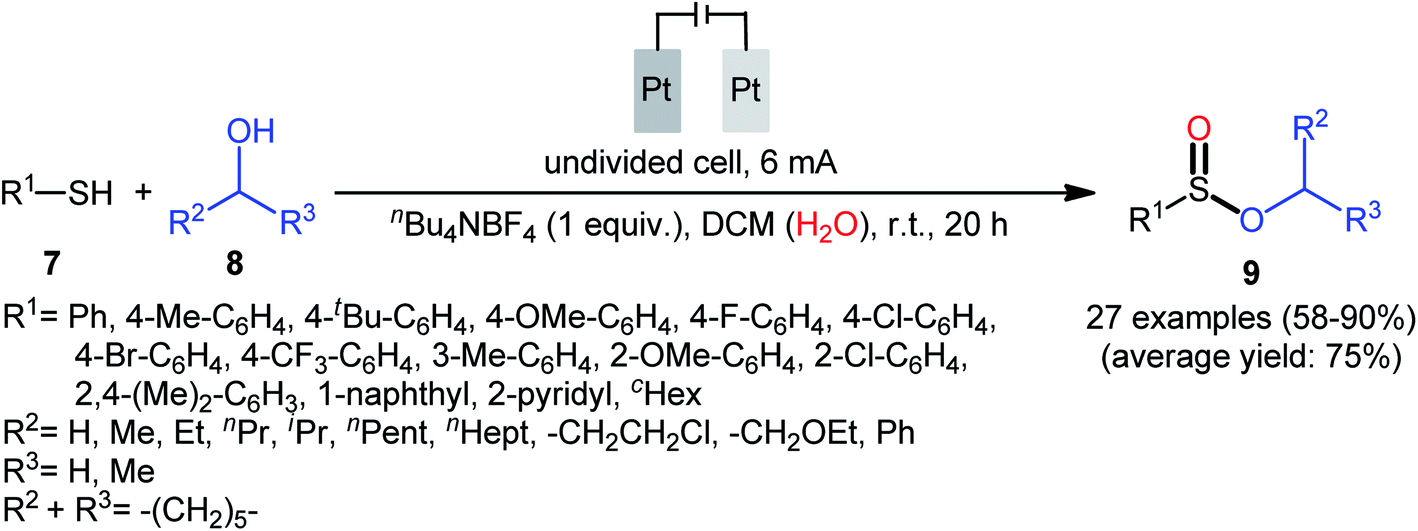
Shortly afterwards, Ling–Zhong's research group disclosed a similar electrocatalytic oxidative esterification of thiols 7 with alcohols 8 under mild conditions which exhibited considerably better substrate scope when compared to the previous works.22 The transformation was performed in an undivided cell under a constant current of 6 mA with platinum plates as electrodes employing nBu4NBF4 as the electrolyte and DCM as the solvent under an inert atmosphere, which afforded the sulfinic ester products 9 in 58–90% yields. Noteworthy, the constant current affected the reaction dramatically; either increasing or decreasing the constant current led to decreased efficiency. As shown in Scheme 4 this electrochemical reaction is applicable for not only aromatic and heteroaromatic thiols but also aliphatic thiols. However, like previous works, the scope of alcohols was restricted to the use of aliphatic and benzylic alcohols. Based on the experimental results, the authors suggested that alcohols may act not only as reactants but also as oxidants to oxidize the S(II) to S(IV) species. The isotopic labelling experimental using H218O indicated that oxygen in sulfur could also come from water.
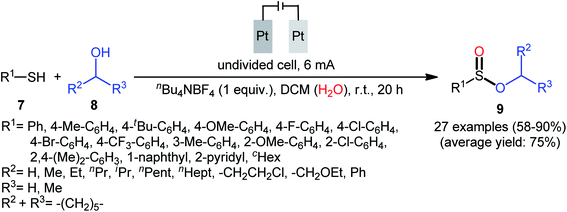 |
| | Scheme 4 Ling–Zhong's synthesis of sulfinic esters 9. | |
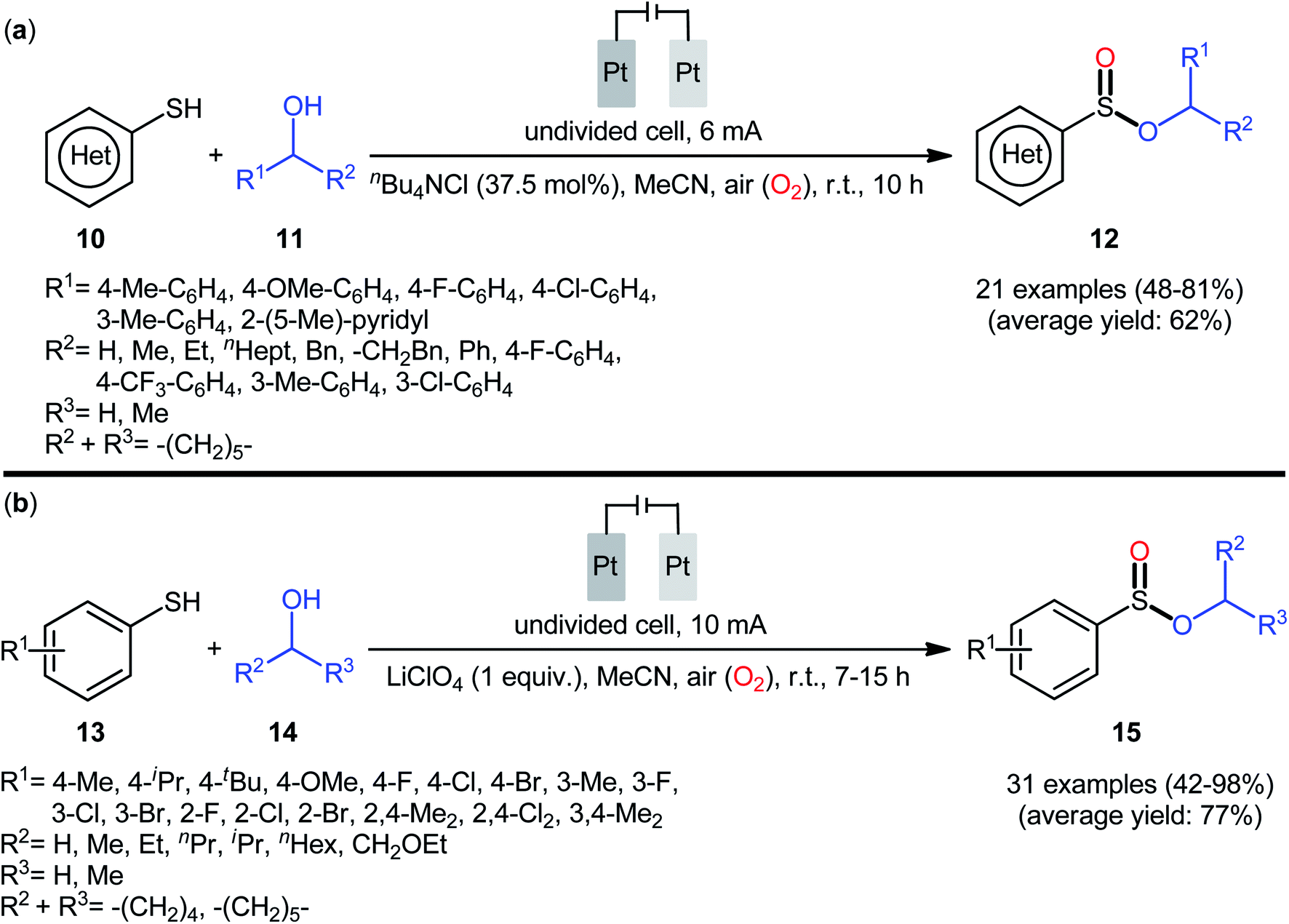
In 2020, Wei and co-workers reported a closely related electrochemical oxidative cross-coupling reaction of thiols 10 with alcohols 11 employing an undivided cell with platinum electrodes and tetrabutylammonium chloride (nBu4NCl) as the supporting electrolyte.23 The reaction was conducted in MeCN under ambient conditions and yields of up to 81% were obtained (Scheme 5a). The reaction, however, appears to be limited to aromatic thiols and aliphatic alcohols. The synthetic applicability of the reaction was nicely demonstrated by converting some of the prepared sulfinic esters to thioether, diaryl sulfoxide, trifluoromethyl sulfoxide, and thiosulfonate derivatives. Notably, when the reaction was performed under O2 atmosphere, sulfonates were obtained instead of the desired sulfinates, however, this transformation proceeded more slowly. In the same year, Yang and Wang along with their co-workers demonstrated the similar esterification under electrochemical and catalyst-free conditions.24 The transformation was carried out in an undivided cell with platinum plate electrodes at room temperature by using LiClO4 as the supporting electrolyte, under 10 mA in MeCN. Under these conditions several thiophenol derivatives 13 carrying various substituents (e.g., OMe, F, Cl, Br) on different positions of phenyl rings were converted to the corresponding alkyl phenylsulfinates 15 in moderate to excellent yields by treatment with aliphatic alcohols 14 (Scheme 5b). However, benzyl thiols were inert under the standard conditions and applicability of aliphatic thiols as starting materials was not investigated in this study. Regarding the reactivity of alcohols, primary alcohols were found to be more reactive than the secondary alternatives. The results also proved that the lengths of carbon chain in alcohols had no impact on the outcome of the reaction. Of note, the authors demonstrated the scalability of the reaction since methyl benzenesulfinate could be obtained in 1.43 g scale in high yield of 84%. Based on some control experiments, the O2 in air proved to be the sole oxygen source.
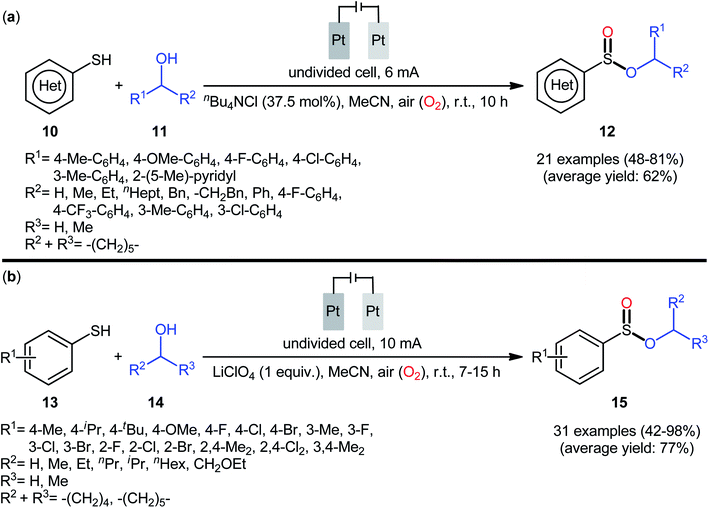 |
| | Scheme 5 (a) Electrochemical oxidative cross-coupling reaction of thiols 10 with alcohols 11 to sulfinic esters 12; (b) Yang–Wang's synthesis of sulfinic esters 15. | |
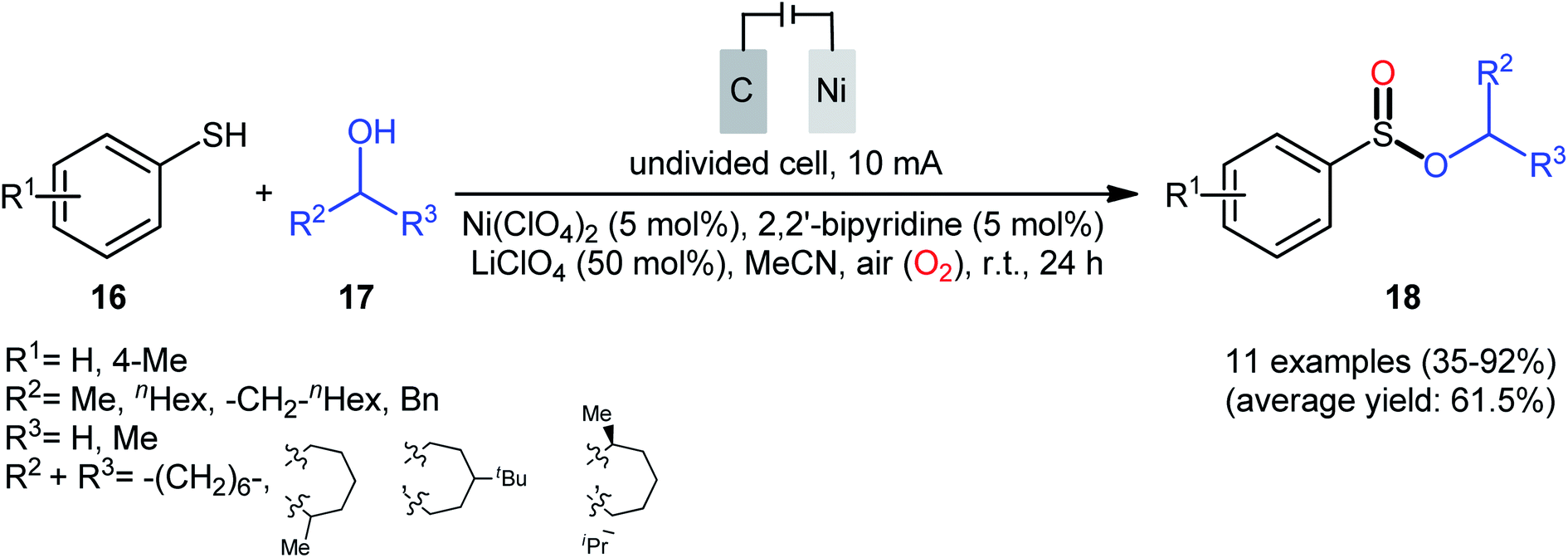
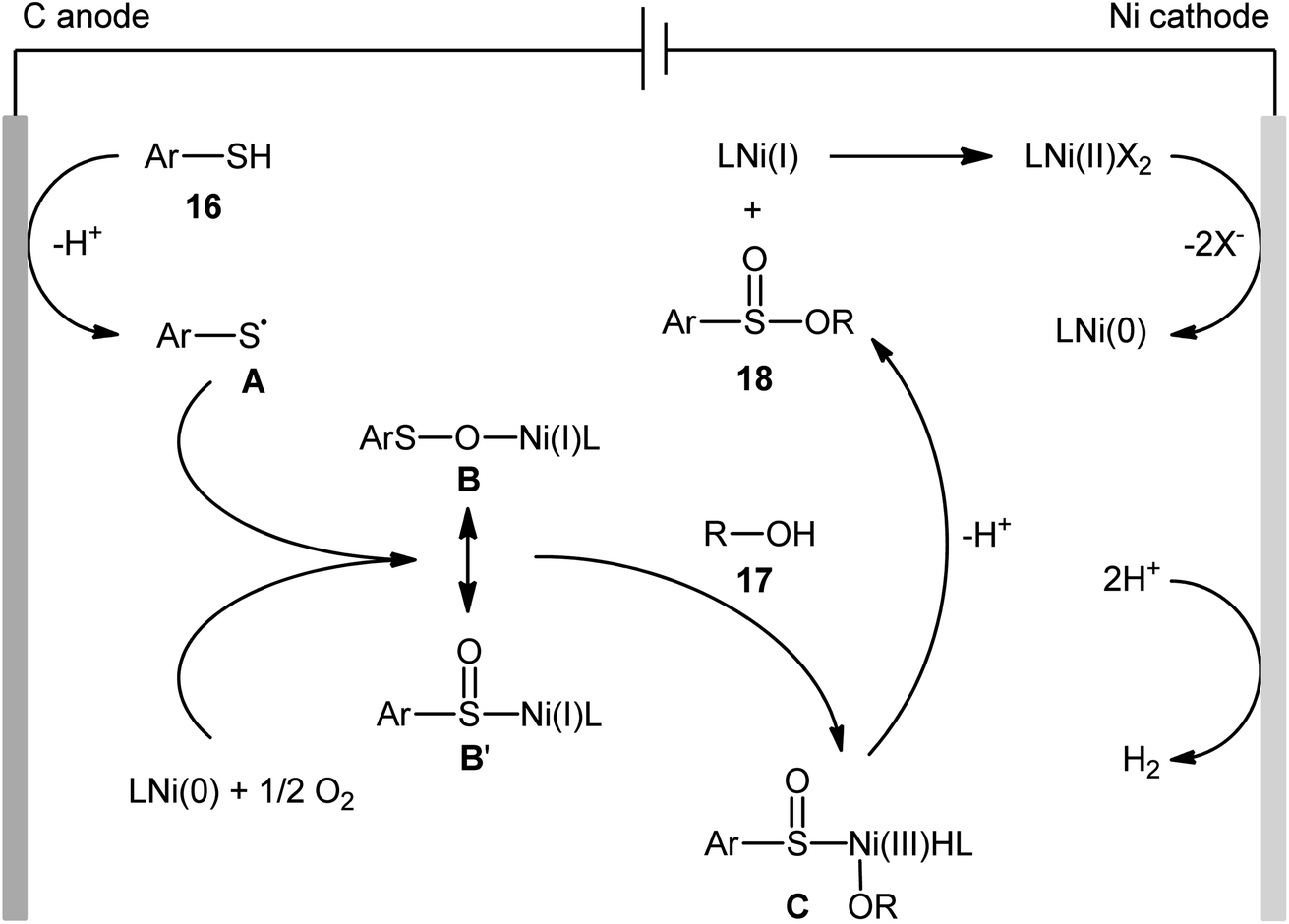
Concurrently, Kaboudin and co-workers developed an interesting electrochemically enabled nickel-catalyzed methodology for oxidative dehydrogenative coupling between thiols 16 and alcohols 17 under ambient conditions.25 The efficient combination of Ni(ClO4)2, 2,2′-bipyridine and LiClO4 with a modified graphite anode and nickel foam cathode in an undivided cell, under constant current of 10 mA, provided the sulfinic esters 18 in modest to high yields, ranging from 35% to 92% (Scheme 6). It is noteworthy that the presence of both catalyst and electricity were crucial for the success of this reaction. No transformation was observed without any of them. Concerning the substrate scope, the protocol limited to the use of only (4-methyl)benzenethiols and simple aliphatic alcohols. Based on a series of control experiments and literature, a plausible mechanism was suggested for the formation of sulfinate esters 18, as illustrated in Scheme 7. The reaction starts with the formation of a thiyl radical A via anodic oxidation of the starting thiol 16 and Ni(0) by cathodic reduction of the nickel complex. Next, the interaction of the thiyl radical A with Ni(0) in the presence of oxygen gives rise a thiyloxy-Ni(I) complex B. Subsequently, the newly formed complex (or its isomer) undergoes an oxidative addition with alcohol 17 to provide a sulfinate-Ni(III) complex C. Finally, the reductive elimination of this complex C leads to the desired sulfinate ester 18 and regenerates the catalyst.
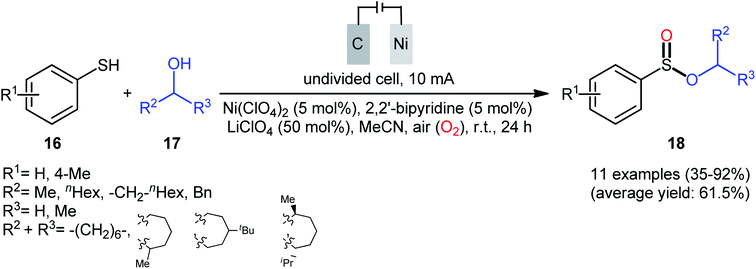 |
| | Scheme 6 Electrochemical nickel-catalyzed oxidative cross-coupling between thiols 16 and alcohols 17. | |
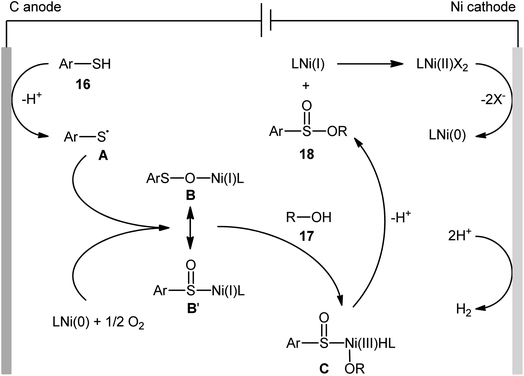 |
| | Scheme 7 Proposed mechanism for reaction in Scheme 6. | |
In the same year, with the aim of designing a metal-free protocol to sulfinic esters through the electrochemical oxidative esterification of thiols and alcohols, Dai and co-workers were able to reveal that a divers set of functionalized sulfinic esters (27 examples) could be prepared in moderate to excellent yields (up to 92%) from the reaction of corresponding (hetero)aromatic thiols with aliphatic alcohols under mild conditions employing an undivided cell with graphite plates as electrodes.26
2.2. Metal-catalyzed reactions
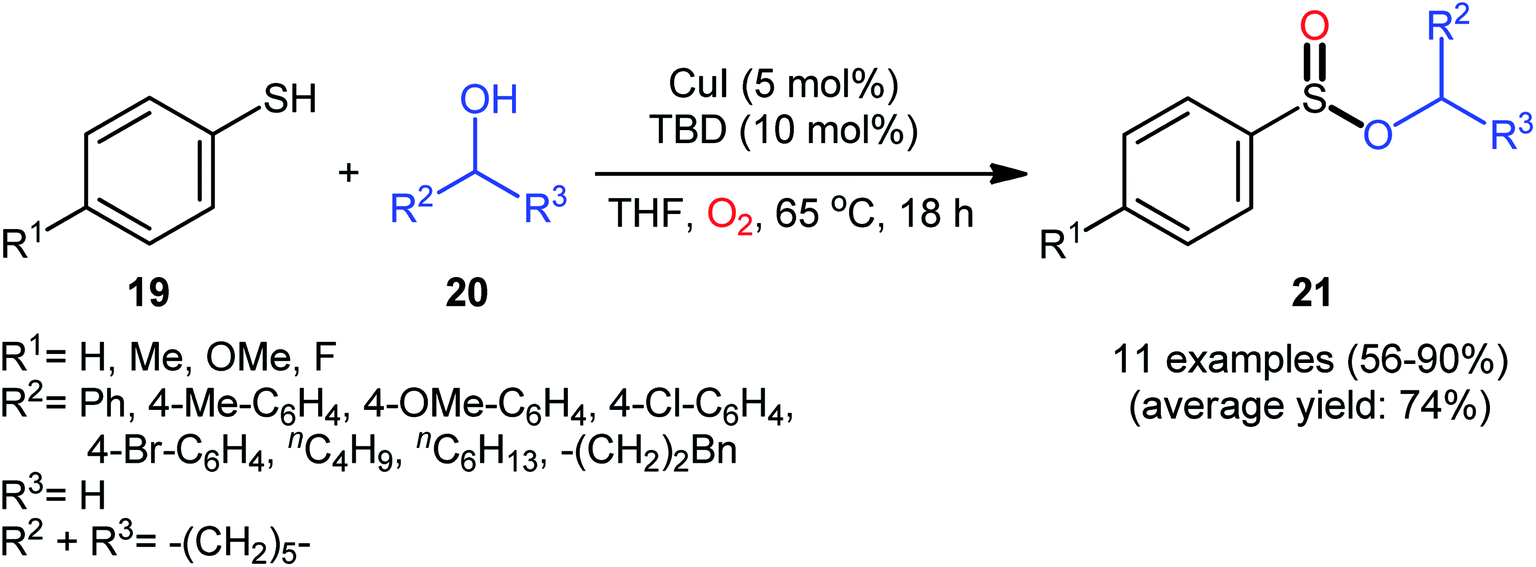
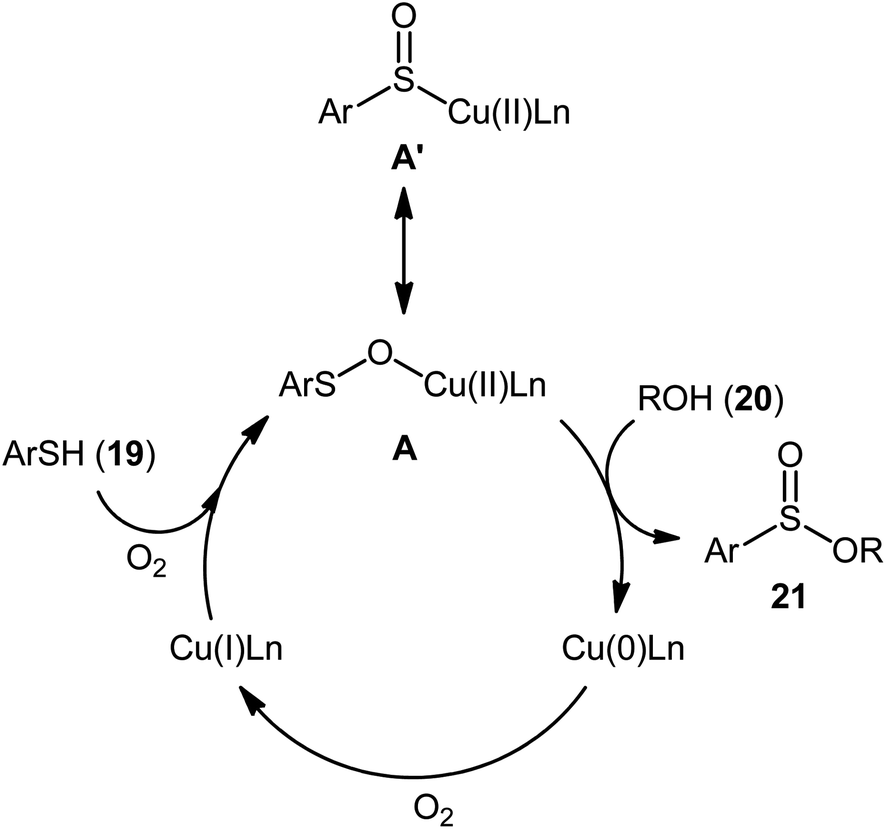
Drawing inspiration from the pioneering work by Field and co-workers on Pb(OAc)4-catalyzed one-pot preparation of sulfinic esters from disulfides and alcohols.27 In 2016, Jang's research group studied the possibility of synthesizing sulfinic esters from the corresponding thiols and alcohols under transition-metal-catalyzed conditions.28 By considering the coupling of thiophenol with benzyl alcohol as the model reaction, the reaction variables such as catalysts, ligands, additives, and solvents were carefully screened. The results indicated that the merge of 5 mol% of CuI with 10 mol% of triazabicyclodecene (TBD) was the most appropriate catalytic system for this transformation and among the various common solvents (i.e., toluene, dioxane, THF, DMF, DMSO, MeCN); THF was found to be the most suitable solvent. Under optimum conditions, a library of alkyl/benzyl phenylsulfinates 21 were selectively obtained in modest to excellent yields by reaction of various thiophenol derivatives 19 with alkyl/benzyl alcohols 20 under 1 atmosphere of oxygen at 65 °C (Scheme 8). The results indicated that the electron-deficient thiophenols afforded better yields compared to the electron-rich ones and benzylic alcohols gave higher yields than aliphatic alcohols. It should be mentioned that in the case of aliphatic alcohols, the amount of TBD was increased to 1 equivalent. Under the identical conditions, the reactions of aliphatic thiols with benzyl alcohols were also investigated. However, in these cases, benzyl benzoates were obtained instead of the desired sulfinic esters through the oxidation of starting thiols to form the respective thioaldehydes, followed by reaction with benzyl alcohols to afford benzyl benzoates via oxidative esterification followed by S–O exchange. Moreover, in this study the authors found some other limitation in their procedure, when they used heteroaromatic thiols (e.g., 2-mercaptobenzimidazole, 2-mercaptopyrimidine, and 4-mercaptopyridine) as substrates. Unfortunately, in all cases no desired product was observed. Based on the experimental results, a plausible mechanism was proposed by the authors for this esterification protocol which involves the following key steps (Scheme 9): (i) oxidation of thiophenol 19 in the presence of the copper catalyst to generate thiyl radical; (ii) formation of thiyloxy-Cu(II) complex A (or its isomer) via interaction of in situ generated thiyl radical with the Cu(I) catalyst; (iii) addition of alcohol 20 to complex A (or A′) to give the observed product 21 and Cu(0) species; and (iv) oxidation of Cu(0) with O2 to recover the Cu(I) catalyst and completes the catalytic cycle.
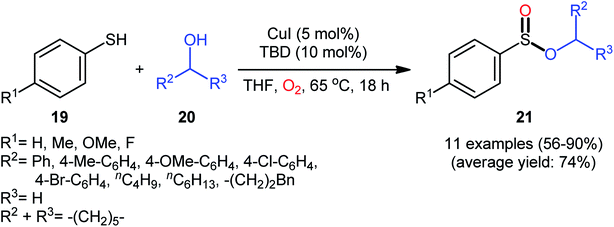 |
| | Scheme 8 Synthesis of phenylsulfinates 21 via Cu-catalyzed oxidative coupling of thiophenols 19 and alcohols 20. | |
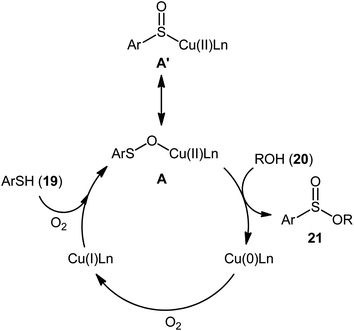 |
| | Scheme 9 Proposed pathway for the formation of sulfinic esters 21. | |
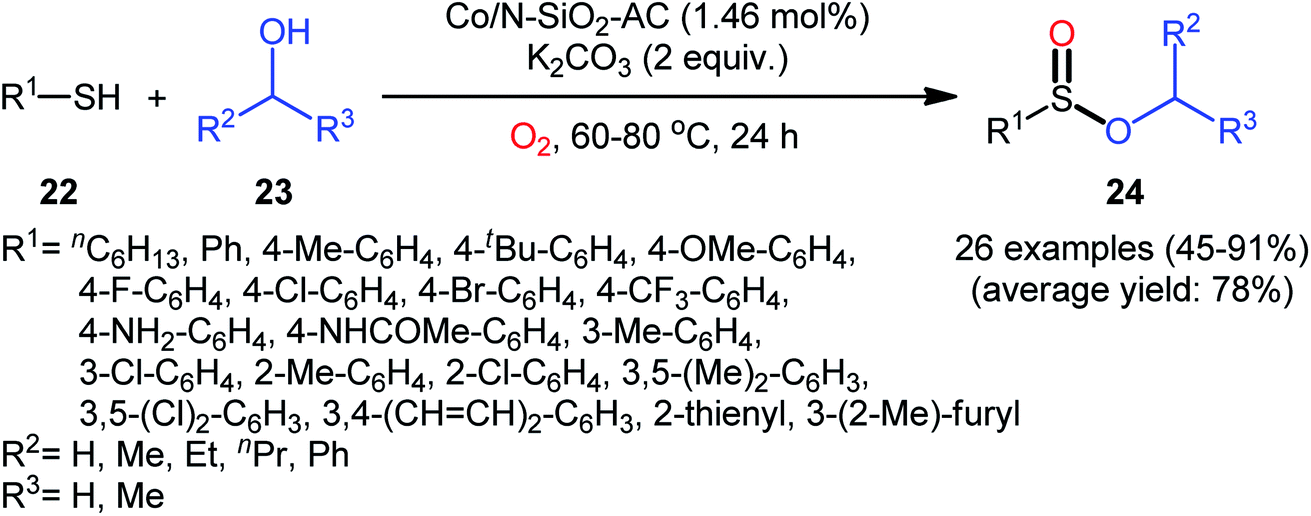
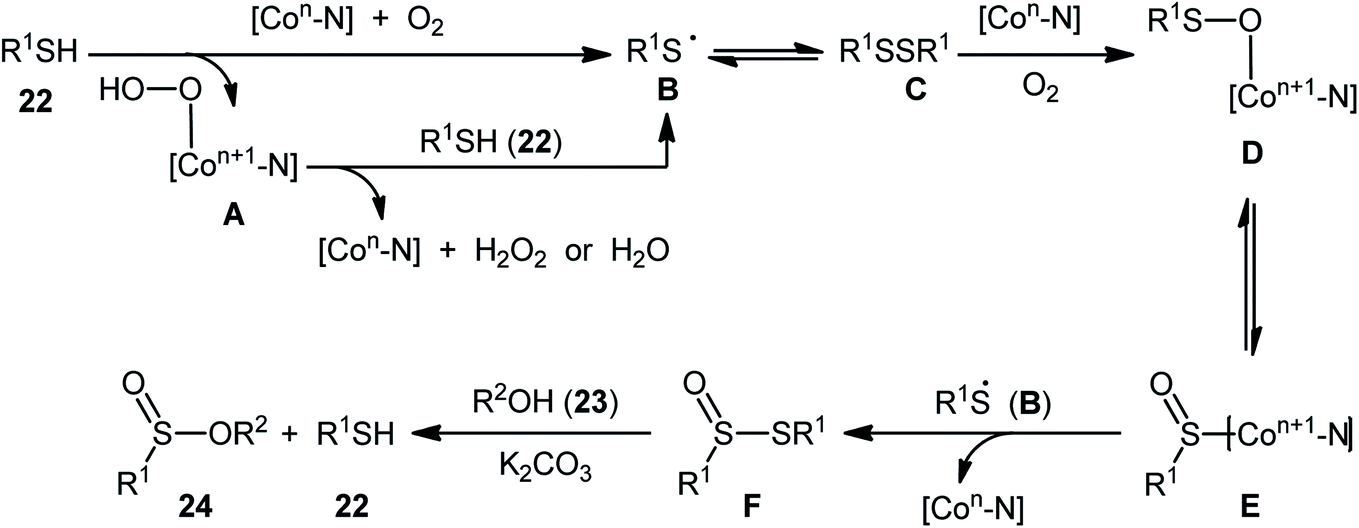
Two years later, Zhang and co-workers introduced ultrafine cobalt nanoparticles supported on N–SiO2-doped activated carbon (Co/N–SiO2-AC) as an efficient catalyst for oxidative esterification of thiols with alcohols.29 Hence, a diverse range of functionalized sulfinic esters 24 were obtained in moderate to excellent yields through the reaction of corresponding thiols 22 with alkyl/benzyl alcohols 23 in the presence of 1.46 mol% Co/N–SiO2-AC as catalyst and 2 equiv. of K2CO3 as a base under O2 atmosphere at 60–80 °C. As shown in Scheme 10, apart from aromatic and heteroaromatic thiols, aliphatic thiols were also compatible with this scenario. The reaction also showed good tolerance to a number of important functional groups such as the methoxy, amino, fluoro, chloro, bromo, trifluoromethyl, and amid functionalities, and promised its potential applications in the post-functionalization of the end product. It is worthwhile to note that in this methodology alcohols not only were served as substrates but also as solvents. The recycling test established that the catalyst could be recovered and reused for six consecutive reaction runs without loss in catalytic performance. After six recycles, the inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis showed negligible decrease of Co content from 1.08 wt% to 1.05 wt%. The mechanism proposed by the authors to explain the formation of sulfinic esters 24 is depicted in Scheme 11.
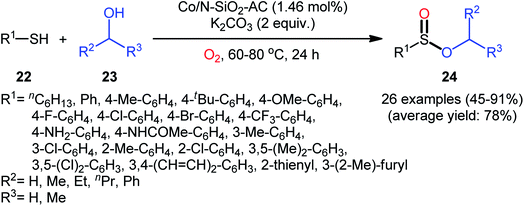 |
| | Scheme 10 Cobalt NPs-catalyzed oxidative esterification of thiols 22 with alcohols 23. | |
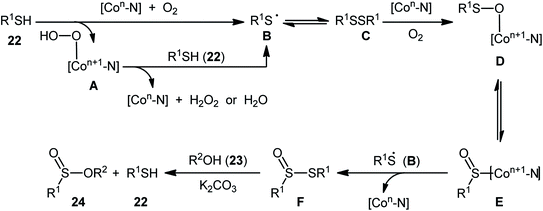 |
| | Scheme 11 The proposed pathway for Co NPs-catalyzed oxidative cross-coupling of thiols 22 with alcohols 23. | |
2.3. Metal-free reactions
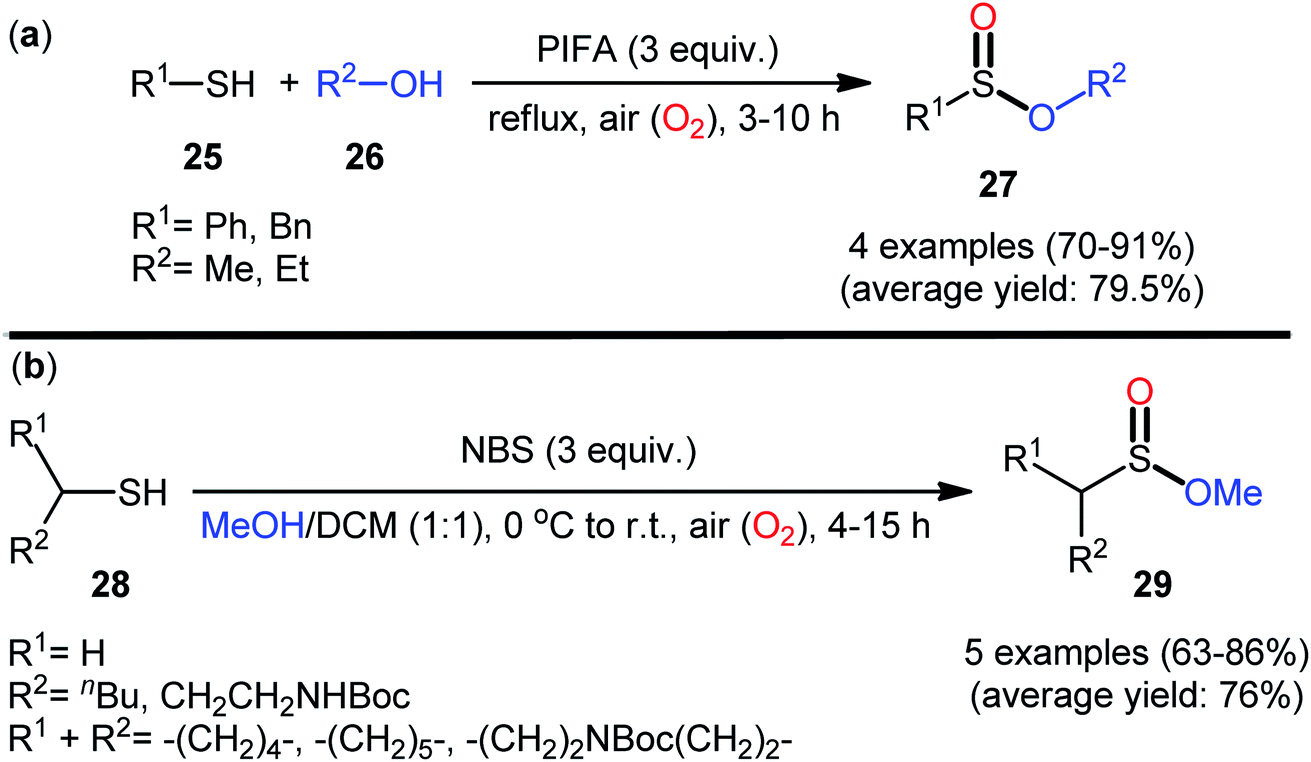
In 1997, Xia and Chen published one of the earliest reports of the metal-free oxidative esterification of thiols with alcohols employing inexpensive phenyliodine(III) bis(trifluoroacetate) (PIFA) as an oxidant under neat conditions.30 The reactions were carried out under the open air in the absence of any catalyst or ligand, tolerated both aromatic and benzylic thiols 25 and a small library of aliphatic alcohols 26, and provided the corresponding sulfinic ester products 27 with yield ranging from 70% to 91% yield (Scheme 12a). However, in this seminal work only four examples with limited scope of the substrates were disclosed and no comment was made regarding the mechanism of the transformation. It is worthwhile to note that sulfinic esters can also be achieved in satisfactory yields when disulfides were employed instead of thiol substrates. Twenty-one years later, a similar principle was used by Qin's research team to the synthesis of a series of methyl alkylsulfinates 29 from the respective alkyl thiols 28 using N-bromosuccinimide (NBS) as a mediator in the binary solvent MeOH/DCM with ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at room temperature (Scheme 12b).31,32 The prepared sulfinic esters were nicely applied as starting materials to the preparation of primary sulfinamides through reaction with LiNH2 or LHMDS.
:1 at room temperature (Scheme 12b).31,32 The prepared sulfinic esters were nicely applied as starting materials to the preparation of primary sulfinamides through reaction with LiNH2 or LHMDS.
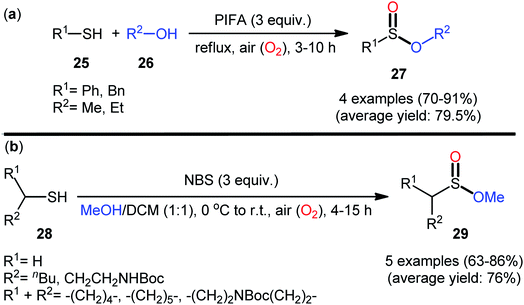 |
| | Scheme 12 (a) Xia's synthesis of sulfinic esters 27; (b) Qin's synthesis of sulfinic esters 29. | |
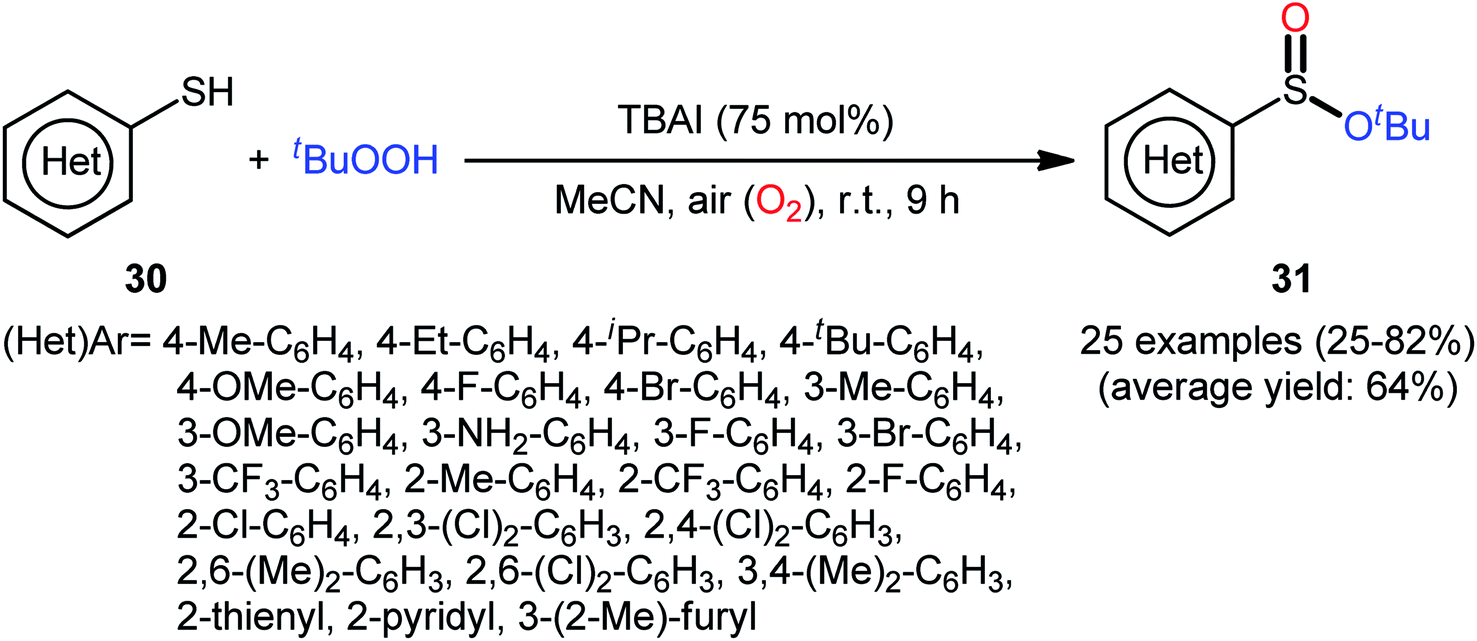
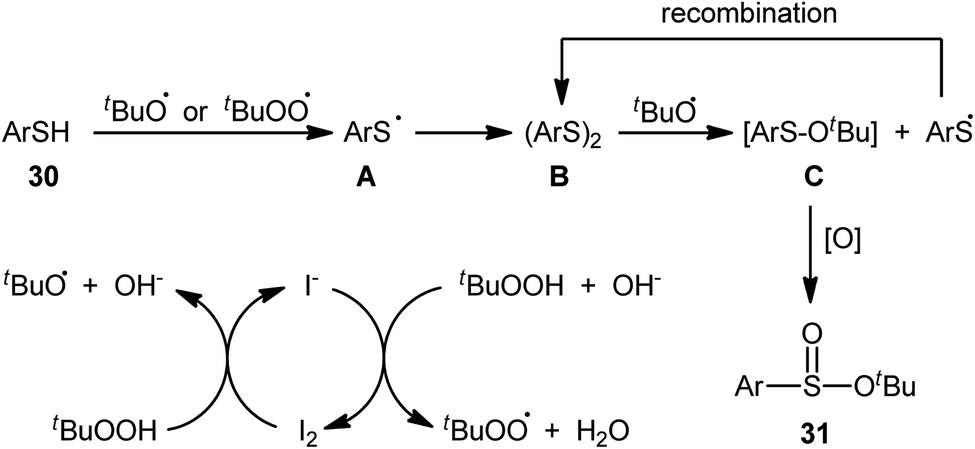
In 2018, Zhang and Chen along with their co-workers unfolded the tetra-n-butylammonium iodide (TBAI)-mediated oxidative esterification of (hetero)aromatic thiols 30 with tert-butyl hydroperoxide (TBHP) to synthesize tert-butyl (hetero)arylsulfinate derivatives 31 under metal-free condition (Scheme 13).33 The reaction exhibited broad substrate scope and functional group tolerance, irrespective of whether electron-donating (e.g., Me, Et, iPr, tBu, OMe and NH2) or – withdrawing groups (e.g., F, Cl, Br and CF3) were at different positions of aromatic rings. Moreover, a tolerance for naphthalene-2-thiol (a bicyclic thiol) was also demonstrated. However, aliphatic thiols (e.g., benzylthiol, cyclohexylthiol) were not suitable substrates for this transformation. Unfortunately, the scope and limitation of peroxides were not investigated in this study so that the reaction appears to be limited to only TBHP. It is noteworthy that other iodine sources such as NaI and KI were also effective in this esterification reaction but gave lower yield of products. To gain mechanistic insights, several preliminary experiments were performed and it was confirmed that this reaction most likely proceeds via a radical pathway (Scheme 14). Initially, TBHP undergoes decomposition in the presence of iodides to generate tert-butoxyl and tert-butylperoxy radicals. Next, homolytic bond breakage of thiols 30 under the oxidative conditions leads to thiyl radicals A, which after homocoupling affords disulfides B. Thereafter, tert-butoxyl radical reacts with disulfides B to yield tert-butoxyl aryl sulfides C via a radical propagation and regenerate thiyl radicals A, which could recombine to disulfides B. Finally, the oxidation of the sulfide intermediates C delivers the desired (hetero)arylsulfinates 31.
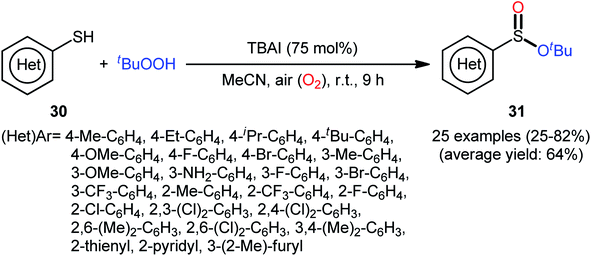 |
| | Scheme 13 TBAI-mediated oxidative esterification of (hetero)aromatic thiols 30 with tBuOOH developed by Zhang–Chen. | |
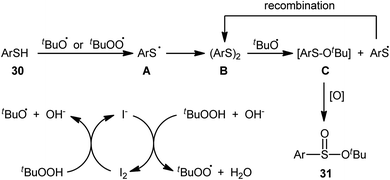 |
| | Scheme 14 The proposed pathway for the formation of (hetero)arylsulfinates 31. | |
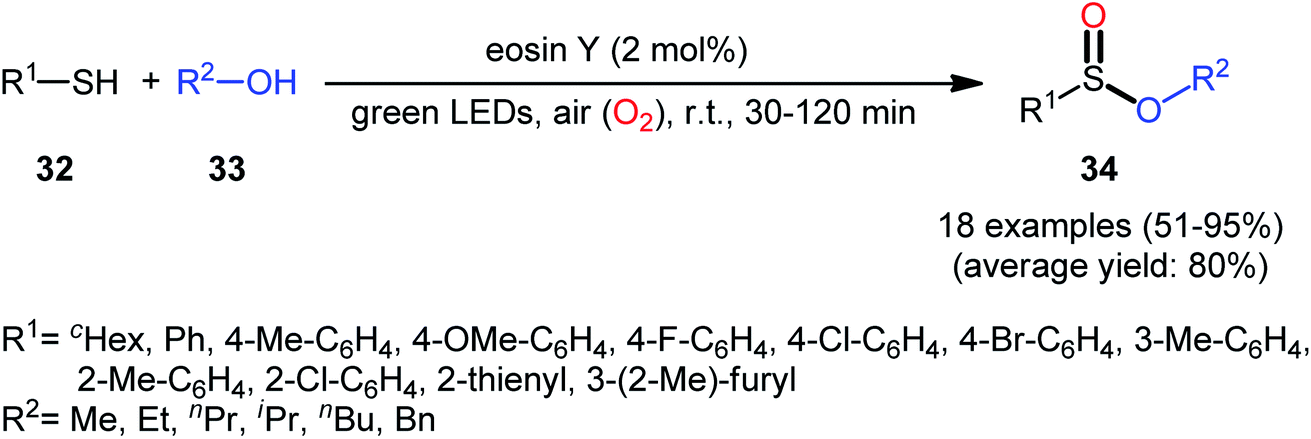
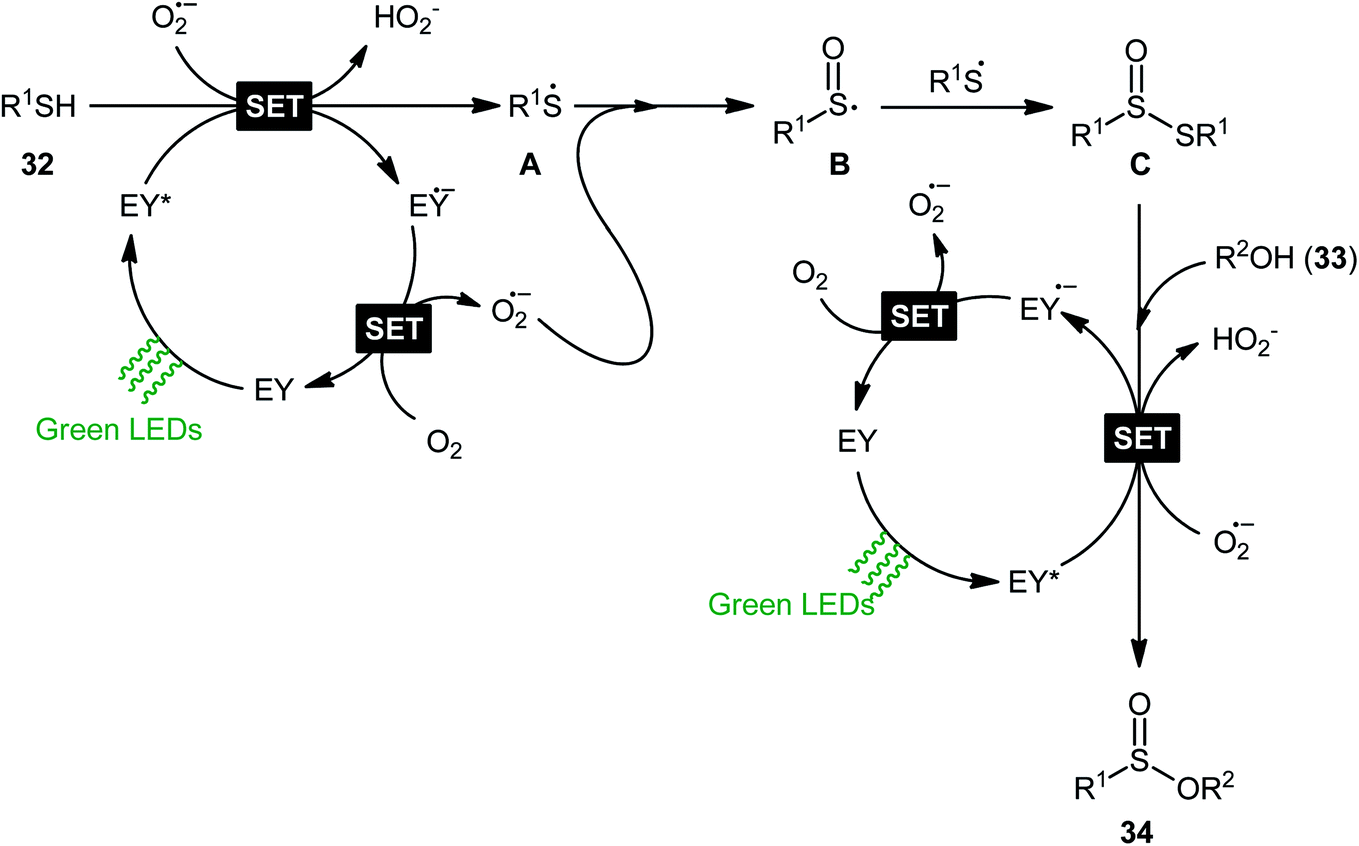
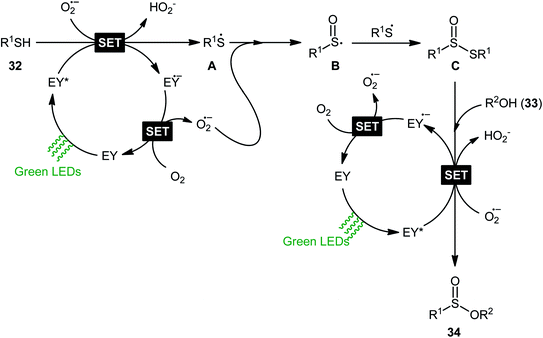
Concurrently, Srivastava and co-workers disclosed a metal-free, visible-light-induced aerobic oxyesterification of various aliphatic, aromatic and heteroaromatic thiols 32 with alkyl/benzyl alcohols 33 as both substrates and solvents.34 No additive was used and only 2 mol% of easily available eosin Y organophotoredox catalyst was enough for preparation of a variety of sulfinic esters 34 in 51–95% yields (Scheme 15). The results indicated that aromatic and heteroaromatic thiols were high yielding compared to aliphatic ones and aliphatic alcohols gave higher yields than benzylic ones. Based on some control experiments, the authors suggested a plausible mechanism as shown in Scheme 16. The transformation may start with the formation of excited state of eosin (EY*) via the excitation of eosin (EY) under visible light irradiation, which reacts via a single electron transfer (SET) process with starting thiol 32 to produce thiyl radical A. Subsequently, this highly active radical interactions with superoxide anion radical (generated by one-electron reduction of O2) to form sulfinyl radical B, which after coupling with another molecule of thiyl radical A affords sulfinothioate intermediate C. Finally, EY – promoted nucleophilic substitution of sulfinothioate C by the alcohol 33 affords the expected products 34.
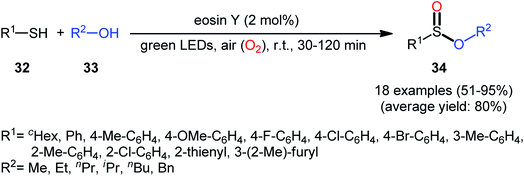 |
| | Scheme 15 Visible light-induced aerobic oxyesterification of thiols 32 with alkyl/benzyl alcohols 33. | |
 |
| | Scheme 16 Mechanistic proposal for the formation of sulfinic esters 34. | |
Based on these developments, very recently, Nguyen and co-workers developed a metal-free oxidative esterification reaction of thiols 35 with alcohols 36 using NBS as the mediator and ethyl acetate as the solvent at room temperature (Scheme 17).35 Although both aromatic and aliphatic thiols were well tolerated under the reaction conditions, the alcohol partners were limited to the use of short-chain alcohols (i.e., MeOH, EtOH, PrOH) as evidenced that butanol totally failed to produce any product under the optimized conditions. Furthermore, under the identical conditions benzyl alcohols were oxidized into benzaldehydes. Note that in some cases performing the process under irradiation of ultrasound slightly improved the efficiency of this reaction in the term of product yields within the shorter times.
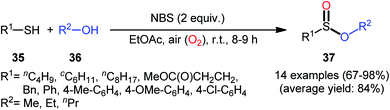 |
| | Scheme 17 NBS-mediated oxidative esterification of thiols 35 with alcohols 36. | |
3. Synthesis of sulfonic esters
Compared with the relatively well-developed oxy-esterification of thiols with alcohols for the synthesis of sulfinate esters, the direct fabrication of sulfonic esters from those easily available commodity chemicals is still uncommon. In fact, only a handful of reports of such a reaction was published in the literature till date.
One of the earliest reports on this chemistry was published by Bahrami and co-workers in 2012,36 who described that the treatment of thiols 38 with phenol derivatives 39 in the presence of a combination of Amberlite IRA-400/POCl3/H2O2 as the catalytic system resulted in the formation of aryl sulfonic esters 40 in high to almost quantitative yields within 1–20 h (Scheme 18). The reaction is noteworthy in that various aliphatic, benzylic, and aromatic thiols with either electron-donating or electron-withdrawing substituents were well tolerated. Moreover, the procedure allowed to synthesis of disulfonate esters through double dioxy-esterification reaction of corresponding dithiols. Of note, besides alcohols, amines also could be applied as nucleophiles under the identical conditions to form biologically valuable sulfonamide derivatives. Interestingly, a competition experiment using phenol and aniline revealed that phenol reacted preferentially and aniline remained untouched. Similarly, sulfonylation of 4-aminophenol produced the respective sulfonate ester with free NH2 moiety. Regarding the plausible mechanistic pathway of this transformation, after NMR spectroscopy investigations, it was confirmed that the reaction most likely proceeds via a sulfonyl chloride intermediate.
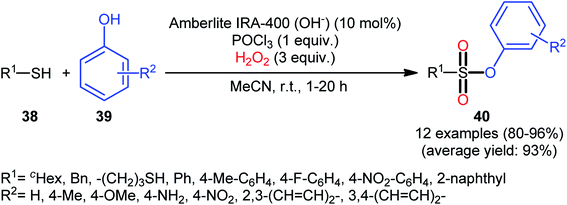 |
| | Scheme 18 Amberlite IRA-400/POCl3-mediated oxidative esterification of thiols 38 with phenols 39 in the presence of H2O2. | |
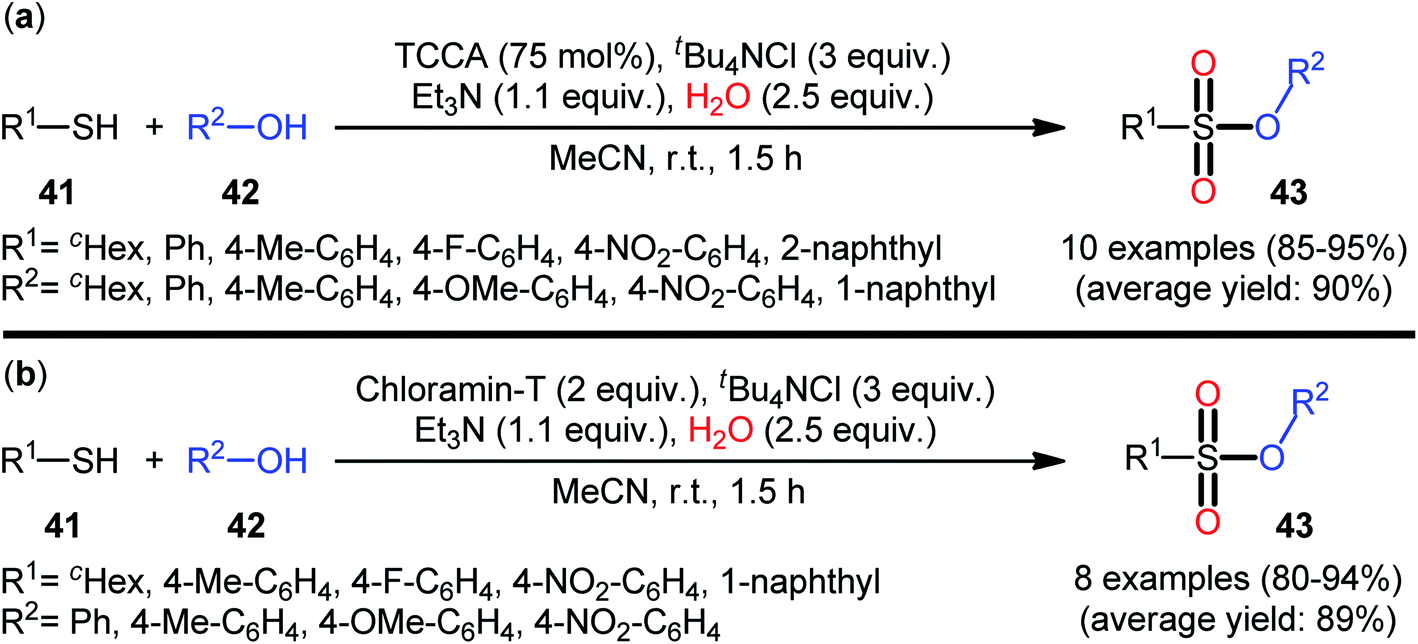
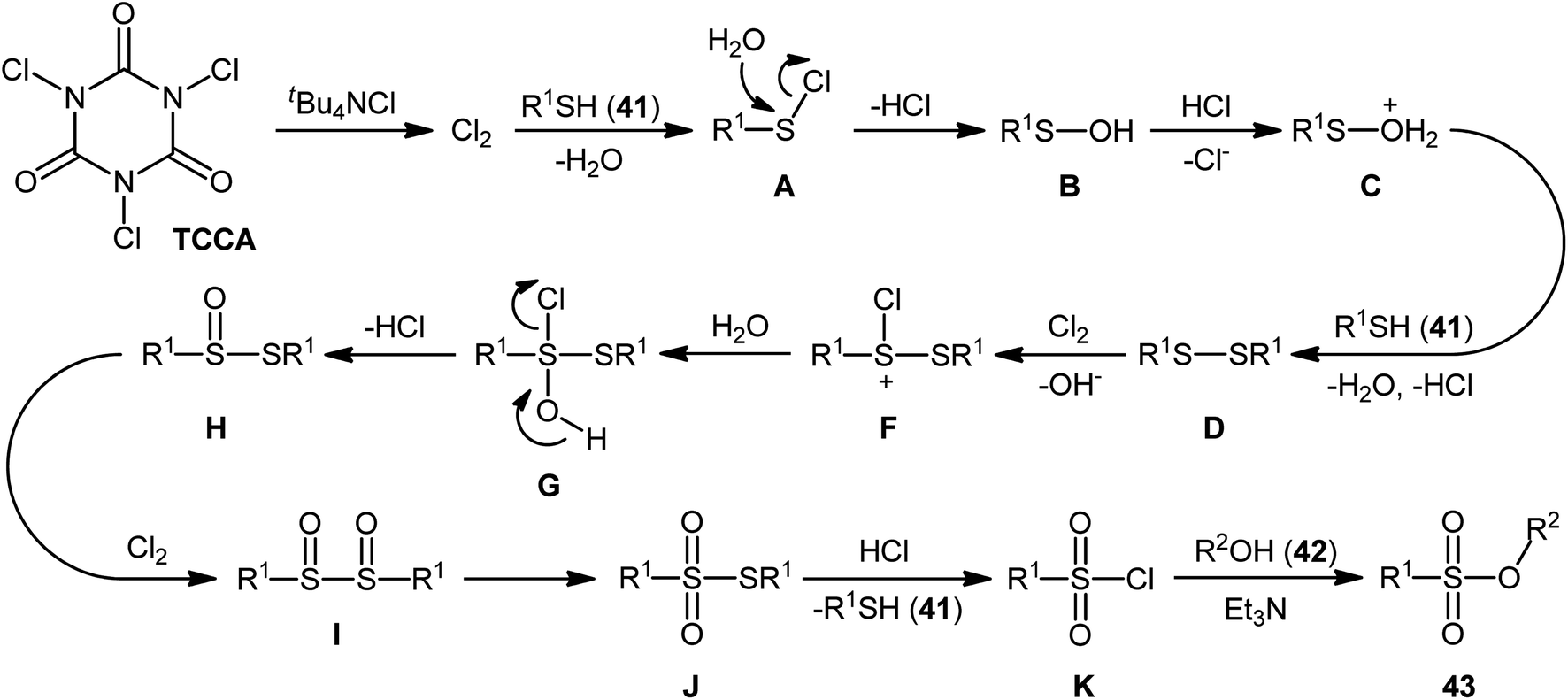
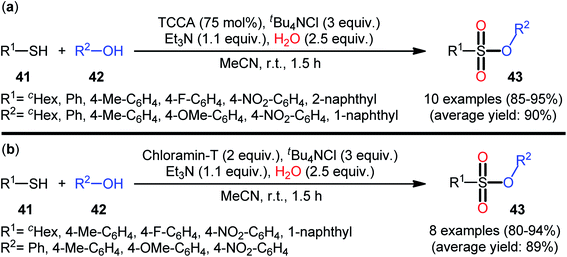
In 2013, Hemmati and co-workers developed a two-step one-pot method for the preparation of sulfonic ester derivatives 43 from the respective thiols 41 and alcohols 42 through oxidative chlorination of thiols to sulfonyl chlorides in the presence of trichloroisocyanuric acid (TCCA), tetrabutylammonium chloride (tBu4NCl), and water followed by reaction with alcohols under basic conditions (Scheme 19a).37 Although either aromatic and aliphatic derivatives of both starting materials exhibited good applications under standard conditions and provided the desired products in high to excellent yields, the toxicity of TCCA might limit the application profile of this synthetic strategy. Noteworthy, when disulfides were subjected to the reaction in place of thiols, the same set of sulfonic ester products were obtained in close yields. According to the authors proposed mechanism (Scheme 20), the oxygen atom of products originated from water. Another independent one-pot two-step method was published by Veisi and co-workers using Chloramin-T as a chlorinating agent under the same conditions (Scheme 19b).38 The employed substrates in this work are the same as the ones reported by Hemmati–Mojtahedi group and the product yields are almost similar.
 |
| | Scheme 19 (a) Hemmati's synthesis of sulfonic esters 43; (b) Mojtahedi's synthesis of sulfonic esters 43. | |
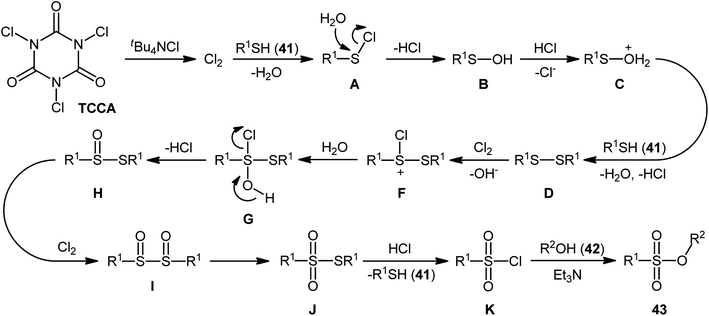 |
| | Scheme 20 The plausible mechanism for the reactions in Scheme 19a. | |
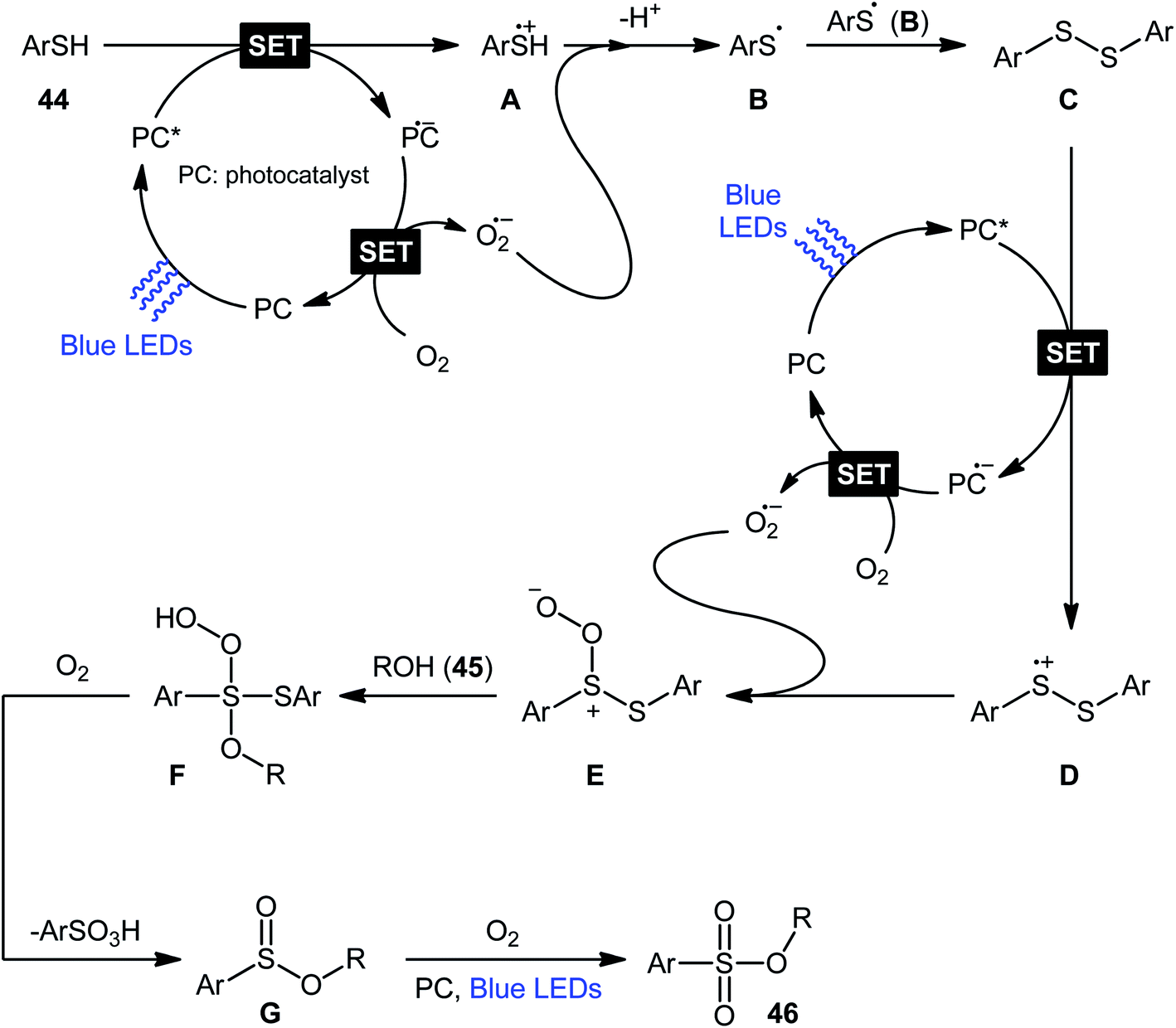
Following these works, Lei and co-workers disclosed an interesting photoinduced oxidative cross-coupling of thiophenols 44 with aliphatic alcohols 45 using 9-mesityl-10-methylacridinium ion (Acr+–Mes) as a photocatalyst under metal-free conditions at room temperature.39 The reactions proceed under O2 atmosphere and blue light irradiation providing the alkyl benzenesulfonate compounds 46 in moderate to good yields, ranging from 45% to 70% yield (Scheme 21). Either primary or secondary alcohol substrates were successfully reacted in this system. However, tertiary alcohols failed to produce any product under the conditions employed. Moreover, the scope of thiophenols appears to be restricted to electron-rich ones. To gain mechanistic insights, several preliminary experiments were performed. No products were obtained in the lack of light, photocatalyst, or oxygen. The possibility of a radical pathway was confirmed since the reaction with TEMPO completely prevent the product formation. Furthermore, an isotope labelling experiment with 18O2 indicated that the oxygen atoms of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds originated from the dioxygen. Based on the above results, the authors speculated that the reaction starts with the formation of Acr˙–Mes˙+ by intramolecular photoinduced electron transfer from the mesitylene moiety to the singlet excited state of the Acr+ moiety of the catalyst. Subsequently, the Mes˙+ moiety oxidizes the thiophenol 44 to form the radical cation A, whereas the Acr˙ moiety reduces O2 to O2˙−. Next, deprotonation of the radical cation A provides the corresponding thiyl radical B, which undergoes homocoupling to yield a disulfide intermediate C. Thereafter, the newly formed intermediate undergoes a further one-electron oxidation to form a disulfide radical cation D that, after oxidization affords the thiopersulfinate E. Later, nucleophilic attack of the alcohol 45 on the sulfur atom of E leads to the intermediate F, which then undergoes photooxidative fragmentation to yield alkyl benzenesulfinate G and benzenesulfonic acid H. Finally, further oxidation of benzenesulfinate G results in the formation of the expected benzenesulfonate product 46 (Scheme 22).
O bonds originated from the dioxygen. Based on the above results, the authors speculated that the reaction starts with the formation of Acr˙–Mes˙+ by intramolecular photoinduced electron transfer from the mesitylene moiety to the singlet excited state of the Acr+ moiety of the catalyst. Subsequently, the Mes˙+ moiety oxidizes the thiophenol 44 to form the radical cation A, whereas the Acr˙ moiety reduces O2 to O2˙−. Next, deprotonation of the radical cation A provides the corresponding thiyl radical B, which undergoes homocoupling to yield a disulfide intermediate C. Thereafter, the newly formed intermediate undergoes a further one-electron oxidation to form a disulfide radical cation D that, after oxidization affords the thiopersulfinate E. Later, nucleophilic attack of the alcohol 45 on the sulfur atom of E leads to the intermediate F, which then undergoes photooxidative fragmentation to yield alkyl benzenesulfinate G and benzenesulfonic acid H. Finally, further oxidation of benzenesulfinate G results in the formation of the expected benzenesulfonate product 46 (Scheme 22).
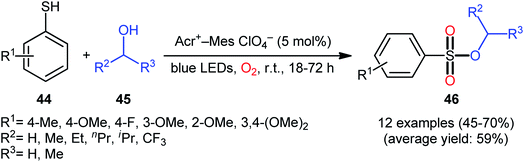 |
| | Scheme 21 Photoinduced oxidative cross-coupling between thiophenols 44 and aliphatic alcohols 45. | |
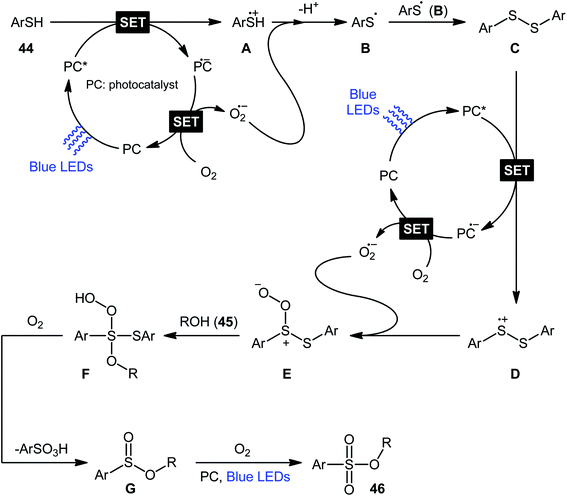 |
| | Scheme 22 Mechanistic proposal for the formation of alkyl benzenesulfonates 46. | |
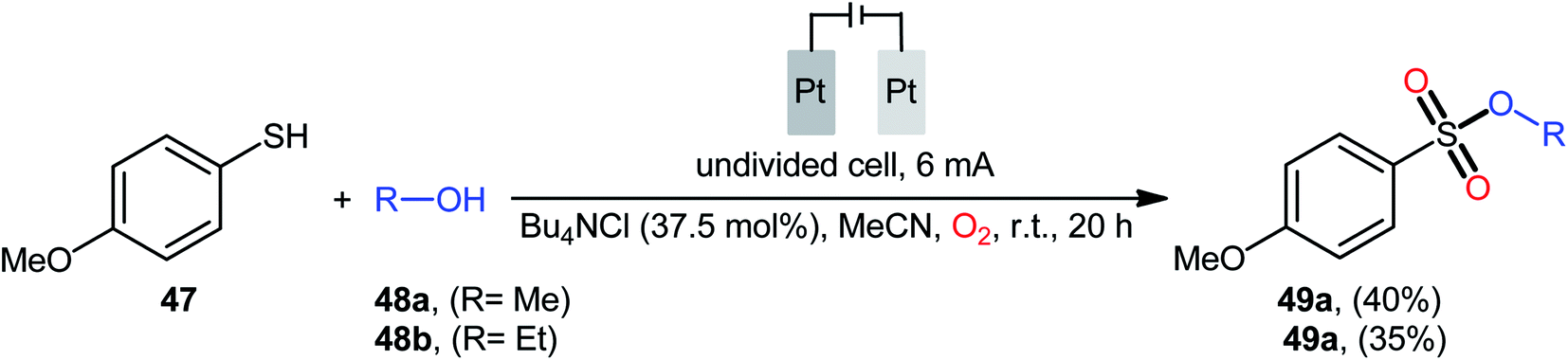
Recently, in the same paper describing the electrochemical oxidative esterification of thiophenols and alcohols to sulfinic esters employing an undivided cell with platinum electrodes, the group of Xu–Wei reported the successful electrocatalyzed preparation of sulfonic esters 49 through the reaction between 4-methoxybenzenethiol 47 and aliphatic alcohols 48 under O2 atmosphere (Scheme 23).23 Although only two low yields examples were disclosed, this paper represents the first example of electrocatalyzed direct dioxy-esterification of thiols. As indicated in previous section, when this reaction was performed in open air the corresponding sulfinic esters were obtained as the sole products. Therefore, of course, O2 has the key role in the further oxidation of sulfur atom.40–47
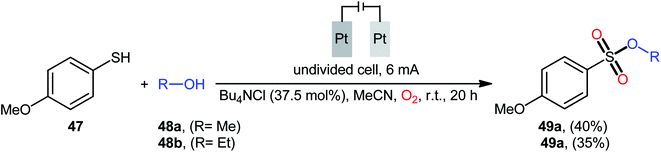 |
| | Scheme 23 Electrosynthesis of sulfonic esters 49 from 4-methoxybenzenethiol 47 and aliphatic alcohols 48 under O2 atmosphere. | |
4. Conclusion
Sulfinic and sulfonic esters, two major kinds of organosulfur compounds containing SO bonds, have drawn a lot of interest in diverse fields due to their crucial applications in organic synthesis, medicinal chemistry, and cell imaging. Therefore, it is always desirable to develop facile and efficient approaches to the synthesis of these special classes of organosulfur compounds from inexpensive starting materials. As summarized in this review, recently direct oxidative esterification of thiols with alcohols has emerged as an atom-efficient new strategy for the one-pot synthesis of sulfinic and sulfonic esters which beside avoids time and cost consuming prefunctionalization steps merits from low-cost easily accessible starting materials. Nevertheless, despite these advances, there are still many unsolved problems and challenges that need to be addressed. Some of these are listed below: (i) the scope of alcohols was mainly limited to aliphatic and benzylic alcohols. Therefore, of course, further research is needed to development of efficient procedures that are compatible with phenol derivatives; (ii) the synthesis of sulfenic esters via cross-dehydrogenative coupling of thiols and alcohols has not been explored thus far. Thus, the development of efficient methods for the direct construction of sulfenic ester derivatives from thiols and alcohols would be highly desirable; and (iii) the number of reported examples on the fabrication of sulfonic esters are narrow and there is an urgent need to study the scope and limitations of this page of sulfonic ester synthesis.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed;
(b) F. Chen, J. Ma, Y. Zhu, X. Li, H. Yu and Y. Sun, J. Hazard. Mater., 2022, 426, 128064 CrossRef CAS PubMed;
(c) M. Cao, Z. Chang, J. Tan, X. Wang, P. Zhang, S. Lin and A. Li, ACS Appl. Mater. Interfaces, 2022, 14, 13025–13037 CrossRef CAS PubMed;
(d) H. Wang, J. Cui, Y. Zhao, Z. Li and J. Wang, Green Chem., 2021, 23, 405–411 RSC;
(e) M. A. I. Molla, M. Furukawa, I. Tateishi, H. Katsumata, T. Suzuki and S. Kaneco, Water Conservation and Management, 2018, 2, 1–5, DOI:10.26480/wcm.02.2018.01.05.
- P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed.
- J. Blackinton, M. Lakshminarasimhan, K. J. Thomas, R. Ahmad, E. Greggio, A. S. Raza, M. R. Cookson and M. A. Wilson, J. Biol. Chem., 2009, 284, 6476–6485 CrossRef CAS PubMed.
-
(a) I. Buggia, F. Locatelli, M. B. Regazzi and M. Zecca, Ann. Pharmacother., 1994, 28, 1055–1062 CrossRef CAS PubMed;
(b) G. Dörner, D. Schnorr, F. Stahl and W. Rohde, Exp. Clin. Endocrinol. Diabetes, 1985, 86, 190–196 CrossRef PubMed.
-
(a) S. R. Malwal, A. Labade, A. S. Andhalkar, K. Sengupta and H. Chakrapani, Chem. Commun., 2014, 50, 11533–11535 RSC;
(b) M. Hemmi, Y. Ikeda, Y. Shindo, T. Nakajima, S. Nishiyama, K. Oka, M. Sato, Y. Hiruta, D. Citterio and K. Suzuki, Chem.–Asian J., 2018, 13, 648–655 CrossRef CAS PubMed.
- S. Wang, Y. Huang and X. Guan, Molecules, 2021, 26, 3575 CrossRef CAS PubMed.
-
(a) X. Yang, Y. Bao, Z. Dai, Q. Zhou and F. Yang, Green Chem., 2018, 20, 3727–3731 RSC;
(b) L. Chen, J. Zhang, Y. Wei, Z. Yang, P. Liu, J. Zhang and B. Dai, Tetrahedron, 2019, 75, 130664 CrossRef;
(c) Y. Wei, J. He, Y. Liu, L. Xu, L. Vaccaro, P. Liu and Y. Gu, ACS Omega, 2020, 5, 18515–18526 CrossRef CAS PubMed;
(d) A. Kobayashi, T. Matsuzawa, T. Hosoya and S. Yoshida, Chem. Lett., 2020, 49, 813–816 CrossRef CAS.
-
(a) M. Ratushnyy, M. Kamenova and V. Gevorgyan, Chem. Sci., 2018, 9, 7193–7197 RSC;
(b) Q. Q. Ge, J. S. Qian and J. Xuan, J. Org. Chem., 2019, 84, 8691–8701 CrossRef CAS PubMed.
-
(a) Y. Watanabe, N. Mase, M.-A. Tateyama and T. Toru, Tetrahedron: Asymmetry, 1999, 10, 737–745 CrossRef CAS;
(b) J. W. Evans, M. B. Fierman, S. J. Miller and J. A. Ellman, J. Am. Chem. Soc., 2004, 126, 8134–8135 CrossRef CAS PubMed;
(c) S. Nakamura, M. Tateyama, H. Sugimoto, M. Nakagawa, Y. Watanabe, N. Shibata and T. Toru, Chirality, 2005, 17, 85–88 CrossRef CAS PubMed.
-
(a) G. A. Meshram and V. D. Patil, Tetrahedron Lett., 2009, 50, 1117–1121 CrossRef CAS;
(b) F. Tamaddon, M. R. Sabeti, A. A. Jafari, F. Tirgir and E. Keshavarz, J. Mol. Catal. A: Chem., 2011, 351, 41–45 CrossRef CAS;
(c) J. G. Kim and D. O. Jang, Synlett, 2007, 2501–2504 CAS.
-
(a) A. R. Hajipour, A. R. Falahati and A. E. Ruoho, Tetrahedron Lett., 2006, 47, 2717–2719 CrossRef CAS;
(b) S. H. Gafur, S. L. Waggoner, E. Jacobsen, C. G. Hamaker and S. R. Hitchcock, Synthesis, 2018, 50, 4855–4866 CrossRef CAS.
-
(a) S. Caddick, J. D. Wilden and D. B. Judd, J. Am. Chem. Soc., 2004, 126, 1024–1025 CrossRef CAS PubMed;
(b) N. Vignola, S. Dahmen, D. Enders and S. Bräse, Tetrahedron Lett., 2001, 42, 7833–7836 CrossRef CAS.
-
(a) M. Huang, L. Hu, H. Shen, Q. Liu, M. I. Hussain, J. Pan and Y. Xiong, Green Chem., 2016, 18, 1874–1879 RSC;
(b) E. Jacobsen, M. K. Chavda, K. M. Zikpi, S. L. Waggoner, D. J. Passini, J. A. Wolfe, R. Larson, C. Beckley, C. G. Hamaker and S. R. Hitchcock, Tetrahedron Lett., 2017, 58, 3073–3077 CrossRef CAS;
(c) Y. Z. Ji, H. J. Li, J. Y. Zhang and Y. C. Wu, Eur. J. Org. Chem., 2019, 1846 CrossRef CAS.
- Selected reviews:
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 1–19 CrossRef PubMed;
(b) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS PubMed;
(c) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 1–32 CrossRef PubMed;
(d) Z. He, D. Wu and E. Vessally, Top. Curr. Chem., 2020, 378, 1–30 CrossRef PubMed;
(e) L. Feng, X. Li, B. Liu and E. Vessally, J. CO2 Util., 2020, 40, 101220 CrossRef CAS;
(f) W. Xu, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2020, 43, 101358 CrossRef;
(g) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2020, 11, 470–483 RSC;
(h) R. T. Kareem, B. Azizi, M. Asnaashariisfahani, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 14941–14955 RSC;
(i) Y. Cao, N. Y. Xu, A. Issakhov, A. G. Ebadi, M. R. P. Heravi and E. Vessally, J. Fluorine Chem., 2021, 109901 CrossRef CAS;
(j) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett., 2020, 3, 2–8, DOI:10.22034/crl.2020.219565.1036;
(k) M. R. J. Sarvestani, N. Mert and E. Vessally, J. Chem. Lett., 2020, 1, 93–102, DOI:10.22034/jchemlett.2020.120304;
(l) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31, DOI:10.22034/jchemlett.2020.107760.
- Selected reviews:
(a) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(b) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef PubMed;
(c) Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC;
(d) Y. Cao, S. Soleimani-Amiri, R. Ahmadi, A. Issakhov, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 32513–32525 RSC;
(e) Y. Cao, S. Abdolmohammadi, R. Ahmadi, A. Issakhov, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 32394–32407 RSC;
(f) Y. Zhang and E. Vessally, RSC Adv., 2021, 11, 33447–33460 RSC;
(g) B. Hashemzadeh, L. Edjlali, P. D. Kheirollahi Nezhad and E. Vessally, Chem. Rev. Lett., 2021 DOI:10.22034/crl.2020.187273.1087;
(h) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18, DOI:10.22034/jchemlett.2020.106645;
(i) S. Majedi and S. Majedi, J. Chem. Lett., 2020, 1, 2–8, DOI:10.22034/jchemlett.2020.106084;
(j) N. Shajari, H. Yahyaei and A. Ramazani, Chem. Rev. Lett., 2021, 4, 21–29, DOI:10.22034/crl.2020.250849.1081;
(k) M. Kamel and K. Mohammadifard, Chem. Rev. Lett., 2021, 4, 54–65, DOI:10.22034/crl.2020.259697.1093.
- V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 847–875 CrossRef CAS PubMed.
- Chemistry of Sulphenic Acids and Esters, ed. D. R. Hogg, John Wiley and Sons Ltd., New York, 1990, pp. 361–402 Search PubMed.
-
(a) H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 CrossRef;
(b) G. M. Martins, A. G. Meirinho, N. Ahmed, A. L. Braga and S. R. Mendes, ChemElectroChem, 2019, 6, 5928–5940 CrossRef CAS.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- J. Nokami, Y. Fujita and R. Okawara, Tetrahedron Lett., 1979, 20, 3659–3660 CrossRef.
- F. Gong, F. Lu, L. Zuo, Q. Wang, R. Li, J. Hu, Z. Li, A. Takfaoui and A. Lei, J. Chin. Chem. Soc., 2020, 67, 192–196 CrossRef CAS.
- C. Ai, H. Shen, D. Song, Y. Li, X. Yi, Z. Wang, F. Ling and W. Zhong, Green Chem., 2019, 21, 5528–5531 RSC.
- Y. He, J. Zhang, L. Xu and Y. Wei, Tetrahedron Lett., 2020, 61, 151631 CrossRef CAS.
- H. Zhou, J. Duan, D. Xie, J. Yang, B. Ma, G. Wang, C. Wu and X. C. Wang, Synthesis, 2020, 52, 2705–2712 CrossRef CAS.
- B. Kaboudin, L. Behrouzi, F. Kazemi, M. M. Najafpour and H. Aoyama, ACS Omega, 2020, 5, 17947–17954 CrossRef CAS PubMed.
- C. H. Yang, C. Wu, J. M. Zhang, X. Z. Tao, J. Xu, J. J. Dai and H. J. Xu, Curr. Org. Synth., 2020, 17, 540–547 CrossRef CAS PubMed.
- L. Field, C. B. Hoelzel, J. M. Locke and J. E. Lawson, J. Am. Chem. Soc., 1961, 83, 1256–1257 CrossRef CAS.
- P. K. Shyam, Y. K. Kim, C. Lee and H. Y. Jang, Adv. Synth. Catal., 2016, 358, 56–61 CrossRef CAS.
- C. Zhou, Z. Tan, H. Jiang and M. Zhang, Green Chem., 2018, 20, 1992–1997 RSC.
- M. Xia and Z. C. Chen, Synth. Commun., 1997, 27, 1321–1326 CrossRef CAS.
- F. Xue, F. Wang, J. Liu, J. Di, Q. Liao, H. Lu, M. Zhu, L. He, H. He, D. Zhang and H. Song, Angew. Chem., Int. Ed., 2018, 130, 6777–6781 CrossRef.
- J. Di, H. He, F. Wang, F. Xue, X. Y. Liu and Y. Qin, Chem. Commun., 2018, 54, 4692–4695 RSC.
- C. Wen, J. Wu, Y. Ou, Y. Huang, K. Zhang and Q. Chen, Tetrahedron Lett., 2018, 59, 3609–3611 CrossRef CAS.
- P. K. Singh, P. P. Singh and V. Srivastava, Croat. Chem. Acta, 2018, 91, 383–388 CrossRef.
- L. A. T. Nguyen, T. N. Le, C. T. Duong, C. T. Vo, F. Duus and T. X. T. Luu, J. Sulfur Chem., 2021, 42, 519–528 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and J. Abbasi, Tetrahedron, 2012, 68, 5095–5101 CrossRef CAS.
- S. Hemmati, M. M. Mojtahedi, M. S. Abaee, Z. Vafajoo, S. G. Saremi, M. Noroozi, A. Sedrpoushan and M. Ataee, J. Sulfur Chem., 2013, 34, 347–357 CrossRef CAS.
- H. Veisi, M. Ataee, L. Fatolahi and S. Lotfi, Lett. Org. Chem., 2013, 10, 111–117 CrossRef CAS.
- A. K. Singh, H. Yi, G. Zhang, C. Bian, P. Pei and A. Lei, Synlett, 2017, 28, 1558–1563 CrossRef CAS.
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS.
- M. Yang, C. Li, Y. Zhang, D. Jia, R. Li, Y. Hou, H. Cao and J. Wang, Ceram. Int., 2019, 45, 14908–14920 CrossRef CAS.
- A. Gupta, V. Dangi, M. Baral and B. Kanungo, Iran. J. Chem. Chem. Eng., 2019, 38, 141–156 CAS.
- Y. Wang, C. Li, Y. Zhang, M. Yang, B. Li, L. Dong and J. Wang, Int. J. Precis. Eng. Man., 2018, 5, 327–339 CAS.
- H. Ahmadizadegan, Iran. J. Chem. Chem. Eng., 2020, 39, 33–47 Search PubMed.
- J. Zhang, C. Li, Y. Zhang, M. Yang, D. Jia, G. Liu, Y. Hou, R. Li, N. Zhang, Q. Wu and H. Cao, J. Cleaner Prod., 2018, 193, 236–248 CrossRef CAS.
- M. Jamil, N. Sultana, M. Sarfraz, M. N. Tahir and M. I. Tariq, Iran. J. Chem. Chem. Eng., 2020, 39, 45–57 CAS.
- N. Salih, M. Jumaah and J. Salimon, Iran. J. Chem. Chem. Eng., 2022 DOI:10.30492/ijcce.2021.521586.4481.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Elham Mohammadid,
Sattar Arshadic and
Esmail Vessally
c,
Elham Mohammadid,
Sattar Arshadic and
Esmail Vessally