
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScholl reaction as a powerful tool for the synthesis of nanographenes: a systematic review
Rabab S. Jassasa,
Ehsan Ullah Mughal
 *b,
Amina Sadiqc,
Reem I. Alsantalid,
Munirah M. Al-Rooqie,
Nafeesa Naeemb,
Ziad Moussaf and
Saleh A. Ahmed
*egh
*b,
Amina Sadiqc,
Reem I. Alsantalid,
Munirah M. Al-Rooqie,
Nafeesa Naeemb,
Ziad Moussaf and
Saleh A. Ahmed
*egh
aDepartment of Chemistry, Jamoum University College, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
bDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
cDepartment of Chemistry, Govt. College Women University, Sialkot-51300, Pakistan
dDepartment of Pharmaceutical Chemistry, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
eDepartment of Chemistry, Faculty of Applied Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia. E-mail: saahmed@uqu.edu.sa
fDepartment of Chemistry, College of Science, United Arab Emirates University, P.O. Box 15551, Al Ain, United Arab Emirates
gResearch Laboratories Unit, Faculty of Applied Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
hChemistry Department, Faculty of Science, Assiut University, 71516 Assiut, Egypt
First published on 29th September 2021
Abstract
Nanographenes, or extended polycyclic aromatic hydrocarbons, have been attracting increasing attention owing to their widespread applications in organic electronics. However, the atomically precise fabrication of nanographenes has thus far been achieved only through synthetic organic chemistry. Polycyclic aromatic hydrocarbons (PAHs) are popular research subjects due to their high stability, rigid planar structure, and characteristic optical spectra. The recent discovery of graphene, which can be regarded as giant PAH, has further stimulated research interest in this area. Chemists working with nanographene and heterocyclic analogs thereof have chosen it as their preferred tool for the assembly of large and complex architectures. The Scholl reaction has maintained significant relevance in contemporary organic synthesis with many advances in recent years and now ranks among the most useful C–C bond-forming processes for the generation of the π-conjugated frameworks of nanographene or their heterocyclic analogs. A broad range of oxidants and Lewis acids have found use in Scholl-type processes, including Cu(OTf)2/AlCl3, FeCl3, MoCl5, PIFA/BF3–Et2O, and DDQ, in combination with Brønsted or Lewis acids, and the surface-mediated reaction has found especially wide applications in PAH synthesis. Undoubtedly, the utility of the Scholl reaction is supreme in the construction of nanographene and their heterocyclic analogues. The detailed analysis of the progress achieved in this field reveals that many groups have contributed by pushing the boundary of structural possibilities, expanding into surface-assisted cyclodehydrogenation and developing new reagents. In this review, we highlight and discuss the recent modifications in the Scholl reaction for nanographene synthesis using numerous oxidant systems. In addition, the merits or demerits of each oxidative reagent is described herein.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal was born in Wazirabad, Pakistan, in 1978 and obtained his Master's degree from Quaid-e-Azam University, Islamabad, Pakistan, in organic chemistry. He obtained his PhD in 2013 from Bielefeld University, Germany, under the supervision of Prof. Dr Dietmar Kuck. For postdoctoral studies, he joined the group of Prof. Dr Xinliang Feng, Max Planck Institute for Polymer Research, Mainz, Germany. In 2015, he started his independent research group at the Department of Chemistry, University of Gujrat, Gujrat, Pakistan. His current research interests focus on the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metals-based terpyridine complexes, and their fabrication in DSSCs and organic functional materials for applications in organic electronics. |
 Saleh A. Ahmed | Saleh A. Ahmed was born in Assiut, Egypt in 1968 where is undertook undergraduate and postgraduate studies. He received his Bachelor and MSc from Assiut University, Egypt and PhD in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr in Saarland University, Saarbrücken, Germany. He worked as postdoctoral fellow, senior researcher and visiting professor in France (CNRS fellow), Japan (JSPS fellow), Germany (AvH, DFG and DAAD fellows), Italy (TEMPUS fellow) and USA (Arab fund fellow). His current research interests include synthesis and photophysical properties of novel organic compounds, electronic devices and solar energy conversion, fluorophores with unique applications. |
1. Introduction to nanographenes
Over the past decade, polycyclic aromatic hydrocarbons (PAHs) and graphene nanoribbons (GNRs) have gained increasing attention owing to their unique applications in various fields of materials chemistry. As such, they have been used in polymer films,1 sensors,2 electronic devices,3 and photovoltaic studies, and have shown interesting optical and biological properties because of their microporous nature.4–6 The construction of the aryl–aryl bond in the π-extended PAHs and GNRs is undoubtedly the most promising approach to further elaborate the structure of such molecules and comprises a novel and exceptional research endeavor to pursue. This approach has brought about a revolution in materials chemistry as it has rebuilt the typical synthetic routes as well as introduced new synthetic pathways for large organic compounds.Graphene, an allotrope of carbon, can be described as a single layer scaffold of interconnected hexagonal benzene rings having sp2-hybridized carbons and are arranged in the form of a honeycomb pattern. It is a 2D material7 that constitutes the individual layers of graphite. In fact, nanographenes are limited to nanoscale segments of graphene, while PAHs are the distinct patterns of graphene. The basic structure of nanographene is similar to that of PAHs but they differ in properties from PAHs.8 In recent years, nanographenes have attracted great attention in organic chemistry due to their high electron mobility and high elasticity.9 Notably, the Scholl reaction is the most used synthetic methodology for the synthesis of PAHs and GNRs and has been repeatedly used over time ever since it was introduced up to the present day. The most recent form of nanographenes is graphene nanoribbons (GNRs), which is prepared through surface chemistry mediated gold-catalyzed Scholl reaction.
2. Scholl reaction
In 1910, Roland Scholl, a Swiss chemist, found that aromatic moieties can undergo carbon–carbon bond formation in the presence of strong Lewis acids, such as neat aluminum chloride, at elevated temperatures.10,11 Expanded polycyclic arenes became accessible under such harsh conditions.12–14 Firstly, Scholl proposed the chemical syntheses of PAHs using the abovementioned conditions. Later, this oxidative cyclization transformation was named as the “Scholl reaction”.15 In principle, it is an acid-catalyzed oxidative condensation of aryl groups that results in new C–C covalent bonds. The reaction has been extensively utilized for a intramolecular oxidative cyclodehydrogenation of various polybenzenoid hydrocarbons. An intermolecular reaction is a reaction between two or more reactant molecules. In some reactions, two pathways are possible: one via an intramolecular reaction (reacting groups both within the same molecule) and one via an intermolecular reaction (reacting groups in different molecules). Generally, intramolecular reactions are entropically favored. A representative example is depicted in Fig. 1a (intramolecular oxidative aromatic coupling) and 1b (intermolecular oxidative aromatic coupling).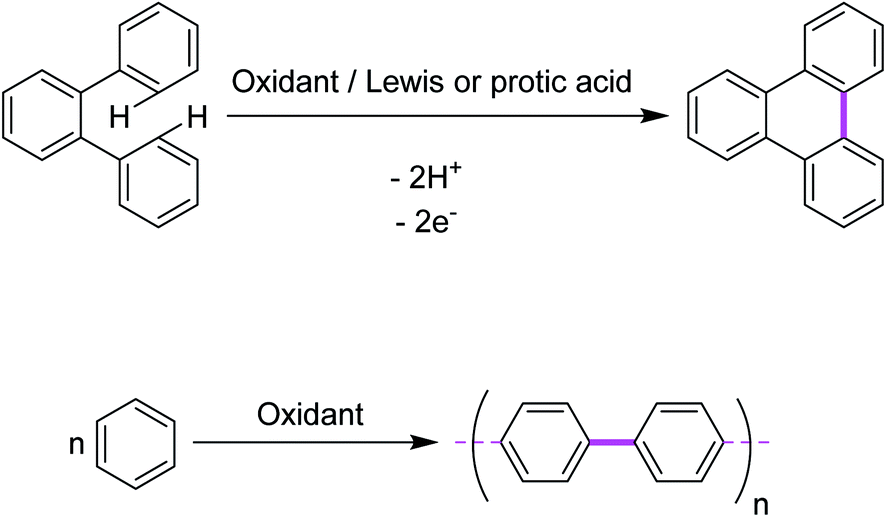 | ||
| Fig. 1 (a) C–C bond formation between the two aryl units (intramolecular) through the Scholl reaction. (b) C–C bond formation between the two different aryl units (intermolecular) through the Scholl reaction. | ||
In this review, we will focus on key achievements in the field of intermolecular and intramolecular oxidative aromatic coupling w.r.t various oxidants that have appeared in the literature.
3. Mechanistic pathways of the Scholl reaction
Oxidative aromatic coupling and the Scholl reaction, both discovered more than 100 years ago,16–19 have not lost their appeal to organic chemists in the last decade despite the appearance of many modern C–H activation protocols and C–C bond-forming reactions. These reactions are often the methods of choice for the synthesis of large π-extended scaffolds as well as smaller benzenoid or heteroaromatic compounds possessing biaryl linkages. Recent findings on the skeletal rearrangement of polycyclic aromatics under oxidative and acidic conditions are envisioned to help the development of these Scholl reactions into a more useful and versatile method for synthesizing polycyclic aromatics based on rational design. Undoubtedly, the utility of Scholl reaction is the greatest in the construction of nanographenes20a–e and their heterocyclic analogues.21 Indeed, in some cases, more than 100 C–C bonds can be formed in one synthetic operation from suitable precursors, furnishing truly amazing PAHs. There is no other reaction known in the literature capable of duplicating this result for a discrete molecule (excluding not a polymer). However, despite the ubiquitous applications of the Scholl reaction, the ability to foresee when and how this reaction will occur is still lacking. Indeed, the complexity of the dehydrogenative coupling reaction is strongly related to the fact that it works with stunning efficiency in some cases but fails in others.Mechanistically, the Scholl reaction mostly proceeds through electrophilic aromatic substitution process (Fig. 2). The reaction is facilitated by the presence of electronically activating substituents on the aromatic rings.22 At the beginning, the mechanism of Scholl reaction was not entirely recognized. Later, Kenner and Baddeley23a proposed two possible mechanisms for this reaction. Kenner, along with Baddeley, firstly reported a radical cation-based mechanism while, later on, Baddeley23b suggested that the reaction proceeds via a mechanism involving the arenium cation as the intermediate. In both the cases, H2 is eliminated as a result of cyclodehydrogenation driven by the oxidant and the catalyst, and a new C–C bond is formed, giving rise to new aryl rings. The exact mechanism of the Scholl reaction is not definite and still controversial. The chosen mechanistic pathway depends upon various factors such as the reagent used, electronic and steric effects, as well as the substitution positions, among others. The dehydrogenation of aromatic nuclei is carried out in the presence of a Lewis acid. As a result, a condensed ring system is formed.24
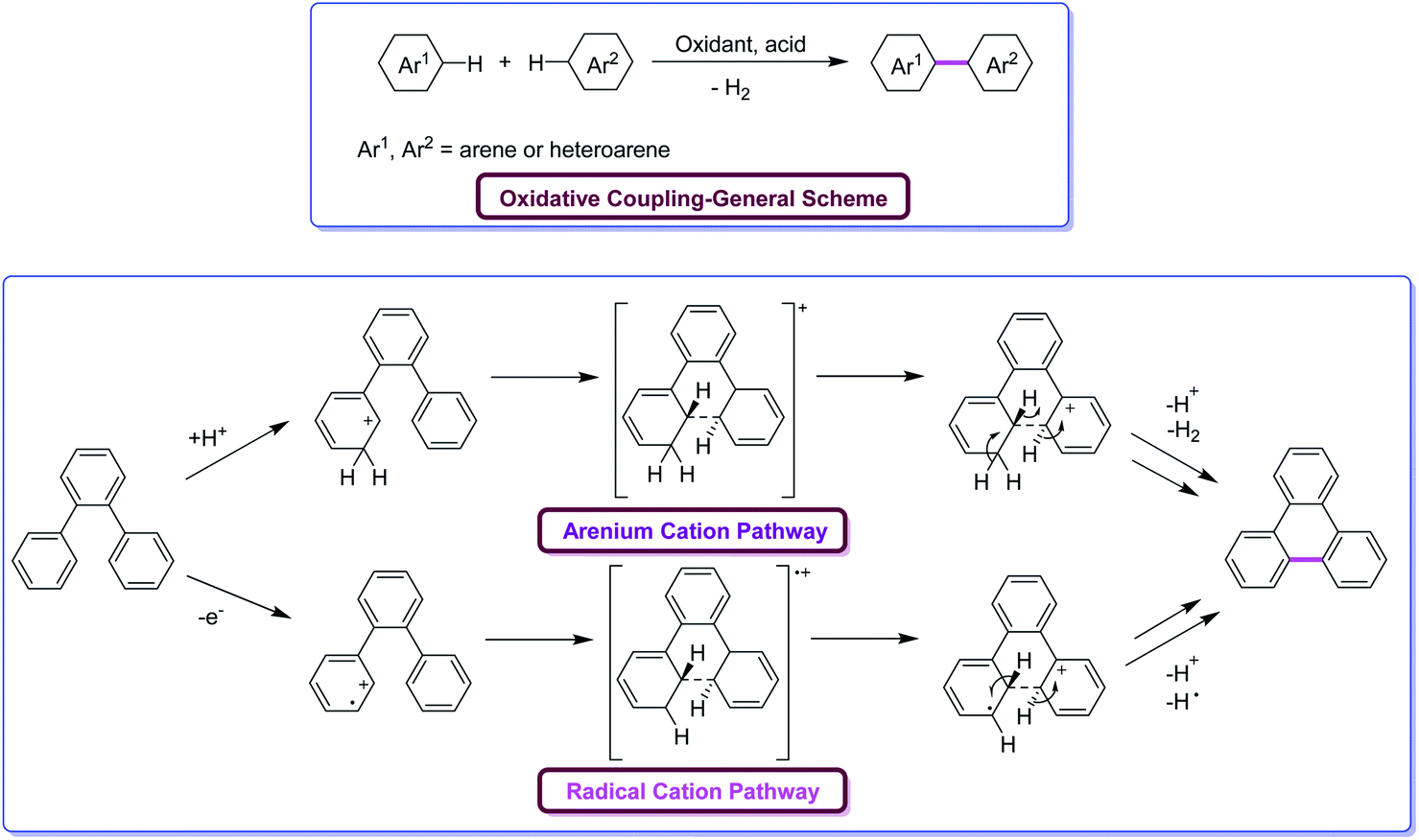 | ||
| Fig. 2 Oxidative coupling of arenes through the two alternative possible pathways under Scholl reaction conditions.24 | ||
Most recently, these controversial mechanisms of the Scholl reaction have been described by King25 and Rathore26 in their research articles. King and co-workers supported the arenium ion mechanism by reporting the synthetic pathway for hexabenzocoronene synthesis from hexaphenylbenzene via the Scholl reaction.27 In particular, a highly efficient synthesis of hexa-peri-hexabenzocoronene (HBC) 2 was established through the oxidative intramolecular cyclodehydrogenation of hexaphenylbenzene 1 (Scheme 1); a wide variety of π-extended PAHs were obtained using tailor-made oligophenylenes as the precursors.28a Such PAHs, consisting of sp2 carbon frameworks extending over 1 nm, can be regarded as the smallest possible nanographenes or graphene molecules.28b,c In the last decade, extended PAHs have thus attracted renewed synthetic interest and more interdisciplinary attention as structurally well-defined graphene molecules with great potential for future applications in nanoelectronics, optoelectronics, and spintronics.28d–i
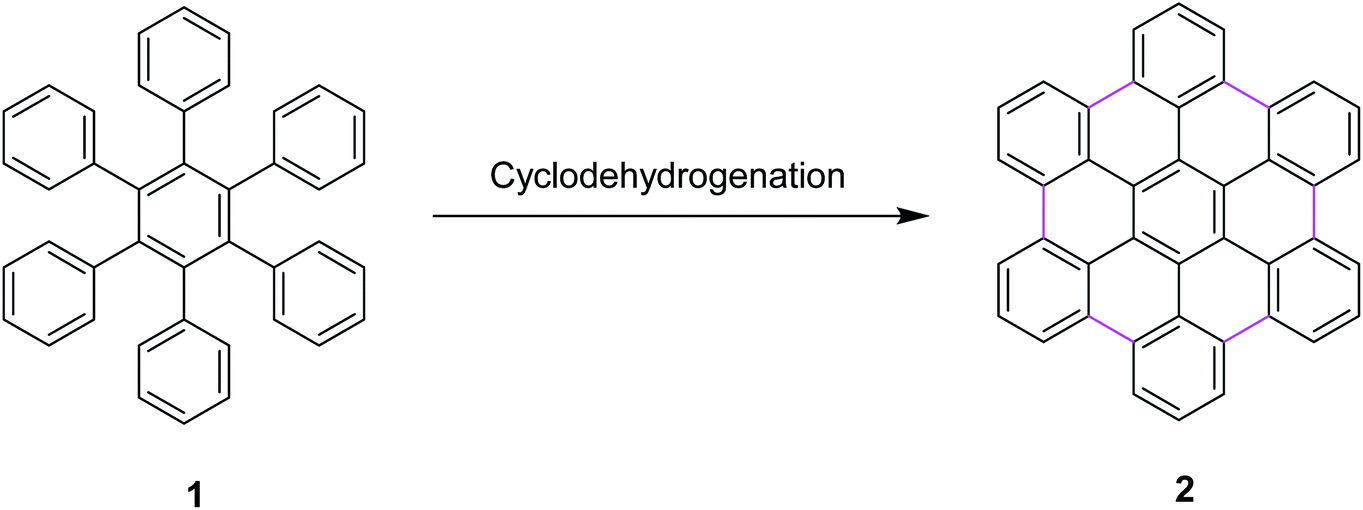 | ||
| Scheme 1 Synthesis of hexa-peri-hexabenzocoronene through intramolecular oxidative cyclodehydrogenation.28a | ||
Rathore and co-workers supported the radical cation mechanism by reporting the synthetic pathways for the synthesis of tetramethoxytriphenylenes 4 from o-terphenyl 3 by cyclodehydrogenation under either strong or mild conditions via the Scholl reaction using various oxidants (Scheme 2).29 It is noteworthy that the Scholl reaction with the DDQ–acid oxidation system is equally effective with substrates undergoing both intramolecular and intermolecular aryl–aryl C–C bond formations. The efficient preparation of a variety of PAHs including tetramethoxytriphenylenes, ease of isolation of the synthesized products, and the ready regeneration of DDQ from easily recovered reduced DDQ–H2 underscores the advantages of DDQ/H+ for the Scholl reaction.29
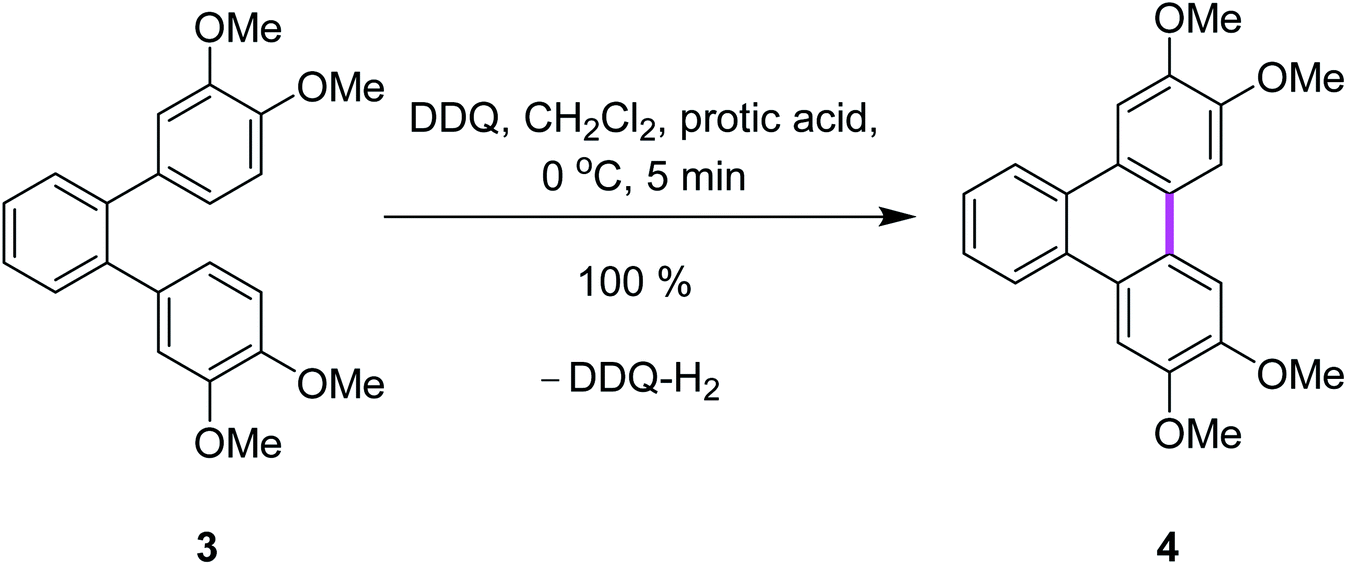 | ||
| Scheme 2 Synthesis of tetramethoxytriphenylene using a combination of DDQ and protic acid.29 | ||
4. Reagent modifications in the Scholl reaction
Over the last century, numerous reagent-mediated coupling reactions have been developed employing oxidizers such as iron(III) chloride, molybdenum(V) chloride, phenyliodobis(trifluoroacetate) (PIFA), and 2,3-dichloro-5,6-dicyano benzoquinone (DDQ) in combination with strong acids (e.g., MeSO3H).30a–c These elite reagents are highly regarded in reagent-mediated dehydrogenative coupling reactions since they replace highly toxic reagents based on thallium, mercury, or lead. The Scholl reaction can be accomplished using a variety of oxidants such as FeCl3, CuCl2 or Cu(OTf)2 and AlCl3, Tl(O2CCF3)3 in CF3CO2H or BF3–OEt2, Pb(OAc)4/BF3–Et2O in MeCN, triethyloxonium hexachloroantimonate (Et3O+SbCl6−), SbCl5, and MoCl5.In this review, we have focused on the development of different reagent systems used in nanographene synthesis via the Scholl reaction. Firstly, we will provide a brief historical perspective to shed light on the Scholl reaction in its earliest phases and subsequent gradual evolution using these reagent systems. Then, we will comprehensively discuss the most advanced developments in nanographene synthesis to date. We will emphasize on the synthetic routes, improvements, merits, and demerits of some prominent reagent systems such as Cu salts, FeCl3, DDQ, PIFA, molybdenum pentachloride (MoCl5), palladium complexes, and gold along with various solvent systems. We will also concisely discuss some miscellaneous reagent systems such as PPA, HTIB, TTFA, VOF3, CoF3, SbCl6, AgSO4, and MnO2 used for the Scholl reaction.
5. Synthetic strategies toward nanographenes by reagent-mediated Scholl reaction
5.1. AlCl3 and Cu(II)-mediated Scholl reaction
Among numerous oxidants typically used for the construction of the new C–C bond in an oxidative manner, the most frequently applied is aluminum(III) chloride. The Scholl reaction was first mentioned as early as 1910, when Scholl and Mansfeld reported a clean conversion of quinone 5 to the π-extended quinone 6 by treatment with an excess of pure anhydrous AlCl3 for 45 min at 140–145 °C, although no yield was given (Scheme 3).31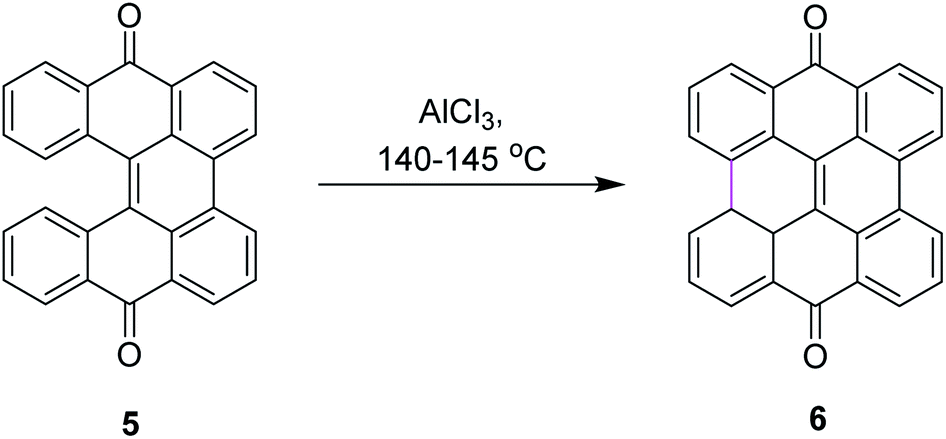 | ||
| Scheme 3 Synthesis of quinones via the Scholl reaction. | ||
In order to further explore the reaction and optimize the yield, Scholl and co-workers performed multiple synthetic reactions. In 1910, Scholl proposed a potential mechanism for perylene 8 synthesis from naphthalene 7 using AlCl3 at a high temperature of 180 °C; however, because of some decomposition, the yield of 8 was also poor. In 1913, the same reaction was repeated under modified conditions using 1,1′-binaphthalene 9 as the substrate. In this case, perylene 8 was synthesized from 9 using the same mechanism of the Scholl reaction at a lower temperature of 140 °C and this time, they obtained a better yield (Scheme 4).32
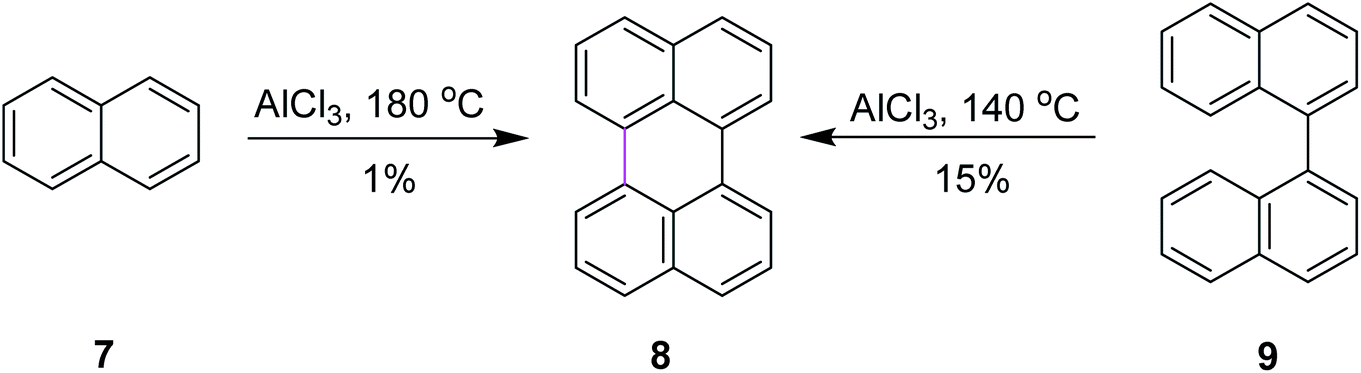 | ||
| Scheme 4 Synthesis of perylene from naphthalene and binaphthalene. | ||
In 1941, Wiener and Mieg used the AlCl3/SO2 complex to promote C–C bond formation between two aryl groups.33 Similarly, many other oxidants were used in combination with AlCl3 but none of them provided the required targets with improved yields. Hence, all these protocols were ruled out. In 1962, Kovacic and Kyriakis synthesized polyphenylene 11 from benzene via oxidative polymerization under AlCl3/CuCl2 conditions (Scheme 5).34 They reported that monomer 10 can be polymerized smoothly in a catalyst–cocatalyst-oxidizing system to a solid product possessing the properties of p-polyphenylene. For example, in the presence of AlCl3 (0.5 moles), water (1 mL), and CuCl2 (0.5 moles), benzene (1 mole) was converted under mild conditions (36–37 °C) over a duration of 30 min to produce a brown solid of the target 11.
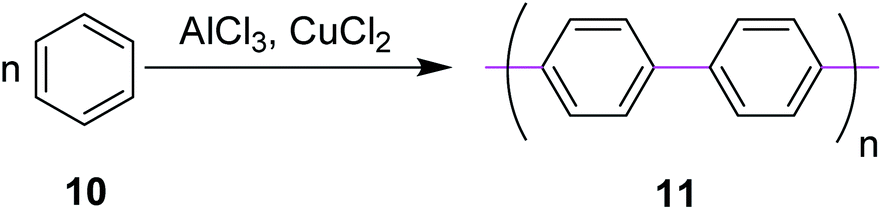 | ||
| Scheme 5 Polyphenyl synthesis via the Scholl reaction. | ||
As a further improvement, Vollmann proposed a better synthetic strategy than the previous methodologies, wherein the C–C bonds were constructed in the presence of an equimolar mixture of AlCl3 and NaCl at a low vapor pressure (Scheme 6).35 Since the early 1920s, this method has been applied in the industrial synthesis of many anthraquinone-derived dyes. This work was used until the 1970s.
 | ||
| Scheme 6 C–C bond formation in anthraquinone-derived dyes. | ||
Scholl and co-workers published numerous other examples following the preceding protocol. Until then, the organic substrate was baked along with AlCl3. However, in 1971, they developed a new synthetic method for the dehydrogenative coupling of aromatic compounds by treatment with anhydrous aluminum chloride and co-catalysts. Mechanistically, the transformation bore close resemblance and was mainly based on the Friedel–Crafts reaction. In this method, tin(IV) chloride (SnCl4) was used along with AlCl3 to synthesize naphthotetraphene 17 from phenyltetraphene 16 in good yields (Scheme 7).36
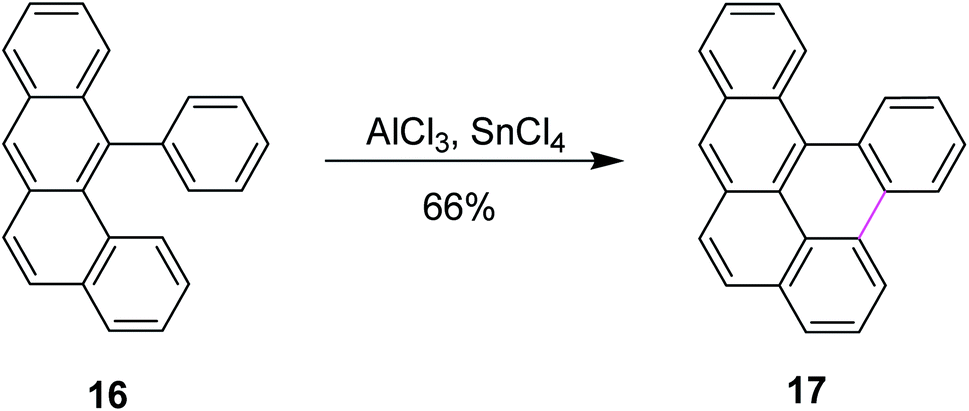 | ||
| Scheme 7 Naphthotetraphene synthesis. | ||
In 1971, Wick utilized the AlCl3–pyridine complex for the synthesis of compound 19 and compared it with the AlCl3/NaCl complex system. He reported that the former complex system is superior to the latter (Scheme 8).37 This new process also triggered the synthetic developments of aromatic hydrocarbon extension.
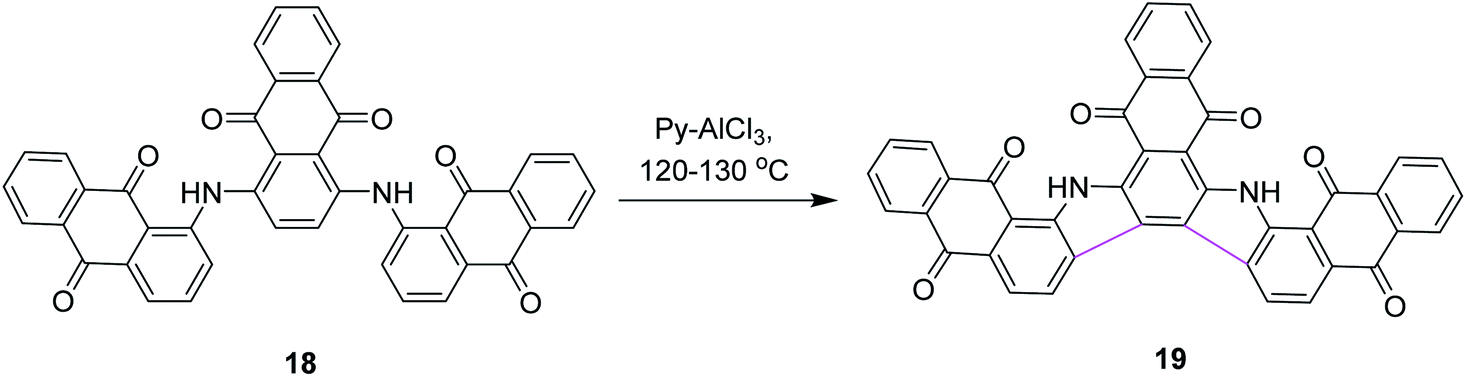 | ||
| Scheme 8 Phthaloyl carbazole synthesis. | ||
In 1990, Zavada and co-workers developed a method for the cross coupling of 2-naphthols 20a and 20b using an oxidant to selectively construct C–C bonds in binaphthyls such as 21. The reaction was carried out in the presence of a Cu(II)–amine complex that provided 97% selective product depending on the substitutions, as depicted in Scheme 9. The reaction evidently proceeds through the radical-cation mechanism. The oxidative coupling of substituted 2-naphthols is now a well-established method for the preparation of l,l′-binaphthalene-2,2′-diols. Various oxidants such as FeC13, K3Fe(CN)6, and Cu(II)–amine complexes have been used so far to promote the coupling reaction. The authors described that Cu(II) mediated-coupling was more efficient as compared to Fe(III) and Mn(III)-mediated coupling. The work of Zavada also explored the cross-coupling reactions as the means to obtain unsymmetrical binaphthols selectively from differently substituted 2-naphthols instead of classical oxidative dimerization, which yielded symmetrical binaphthols.38
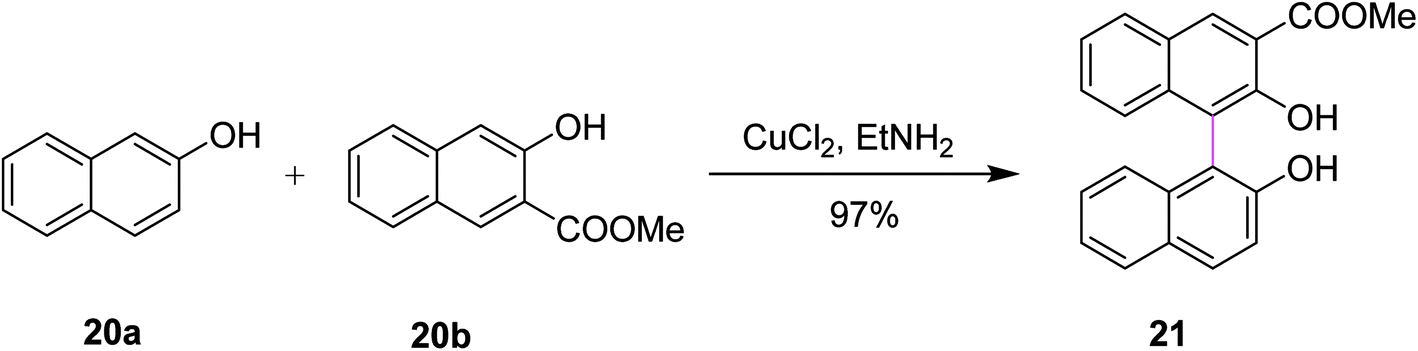 | ||
| Scheme 9 Oxidative cross coupling of 2-naphthols. | ||
In 1992, Smrcina and co-workers reported a simple and effective method for the construction of symmetrically-substituted binaphthyl 23. They developed a process for the mediated oxidative cross-coupling of 2-naphthol units such as 22. They used CuCl2/t-BuNH2 in methanol to generate C–C bonds between two aryl rings, providing enantiomerically pure products, as presented in Scheme 10. 2,2′-Dihydroxy-1,1′-binaphthyl 23 is an established and highly potent chiral ligand, both enantiomers of which have been utilized in a variety of synthetic reactions to induce chirality. In most cases, the chemoselectivity was good and the cross-coupled products were obtained in good to excellent yields.39
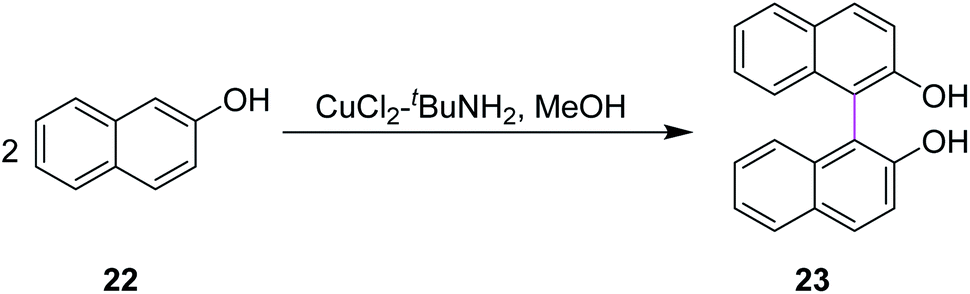 | ||
| Scheme 10 Synthesis of enantiomerically pure binaphthol derivatives. | ||
Similarly, Kovosky and co-workers (1994) worked on the selective cross coupling of 2-naphthol 24a and 2-naphthylamine 24b to yield binaphthyl 25 (Scheme 11). This reaction was carried out in the presence of the highly efficient CuCl2/t-BuNH2 system that provided the product with good yield and exclusive chemoselectivity. They performed these cross-coupling reactions with a chiral complex of Cu(II), which resulted in enantioenriched/enantiomerically enriched binaphthyls.40
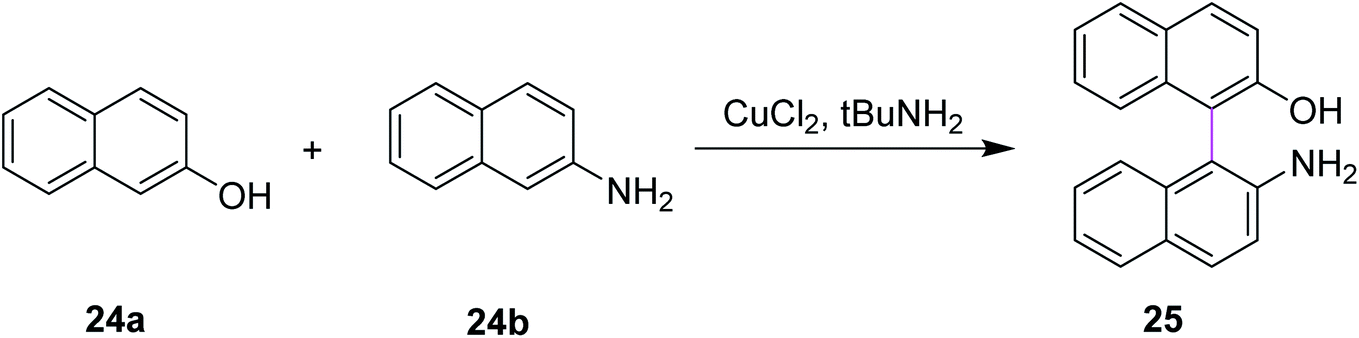 | ||
| Scheme 11 Selective cross coupling induced by CuCl2. | ||
In 1995, Müllen and co-workers introduced new conditions for the oxidative condensation of precursor 26 utilizing AlCl3/CuCl2/CS2 at ambient temperature (Scheme 12).41 This method is quite feasible for the synthesis of a large number of PAHs. The presented cycloaddition–cyclodehydrogenation sequence offers a new and surprisingly simple approach to obtain planar aromatic hydrocarbons. However, the CuCl2/AlCl3 oxidizing system is associated with certain limitations. For example, this reagent leads to the increased formation of chlorinated and partially dehydrogenated side products in addition to the desired products. On the other hand, the use of FeCl3 only yielded partially-cyclized products.
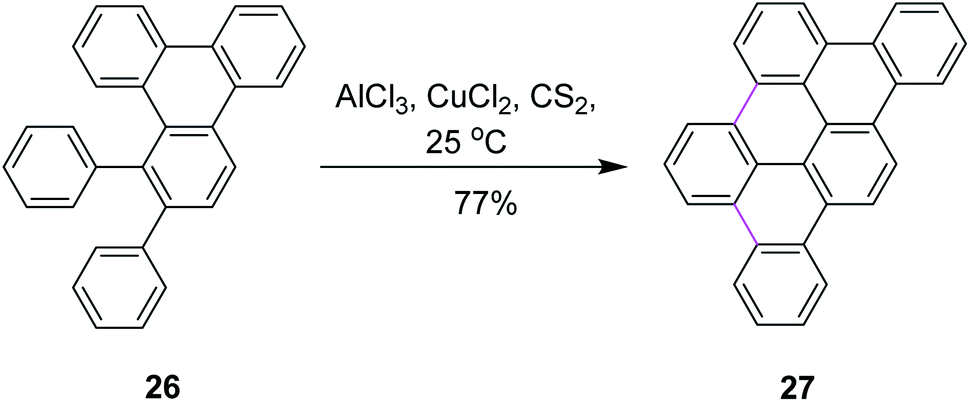 | ||
| Scheme 12 Planar polycyclic aromatic hydrocarbon synthesis.41 | ||
In 2002, the same group reported an improved oxidizing system, in which they replaced CuCl2 by copper(II) trifluoromethanesulfonate salt and thus developed a synthetic method to prepare the largest flat PAHs so far utilizing AlCl3/Cu(SO3CF3)2/CS2 (ref. 42) oxidant system, concluding that the successful synthesis of PAH 29 from oligophenylene 28 represents an important step toward improved graphite model structures. It shows, on the one hand, the efficiency, but on the other hand, also the limits of the synthetic concept employed regarding the planarization of large soluble oligophenylene precursors by the multiple cyclodehydrogenation to PAHs. Due to the extremely low solubility of 29 in all solvents, the successful cyclodehydrogenation could not be proven by conventional spectroscopic methods. Nevertheless, the formation of such planar nanographene was confirmed by mass measurement using MALDI-ToF mass spectrometry. This protocol prompted the scientific community to design and synthesize the large π-extended nanographenes with tailored dimensions and electronic properties (Scheme 13).
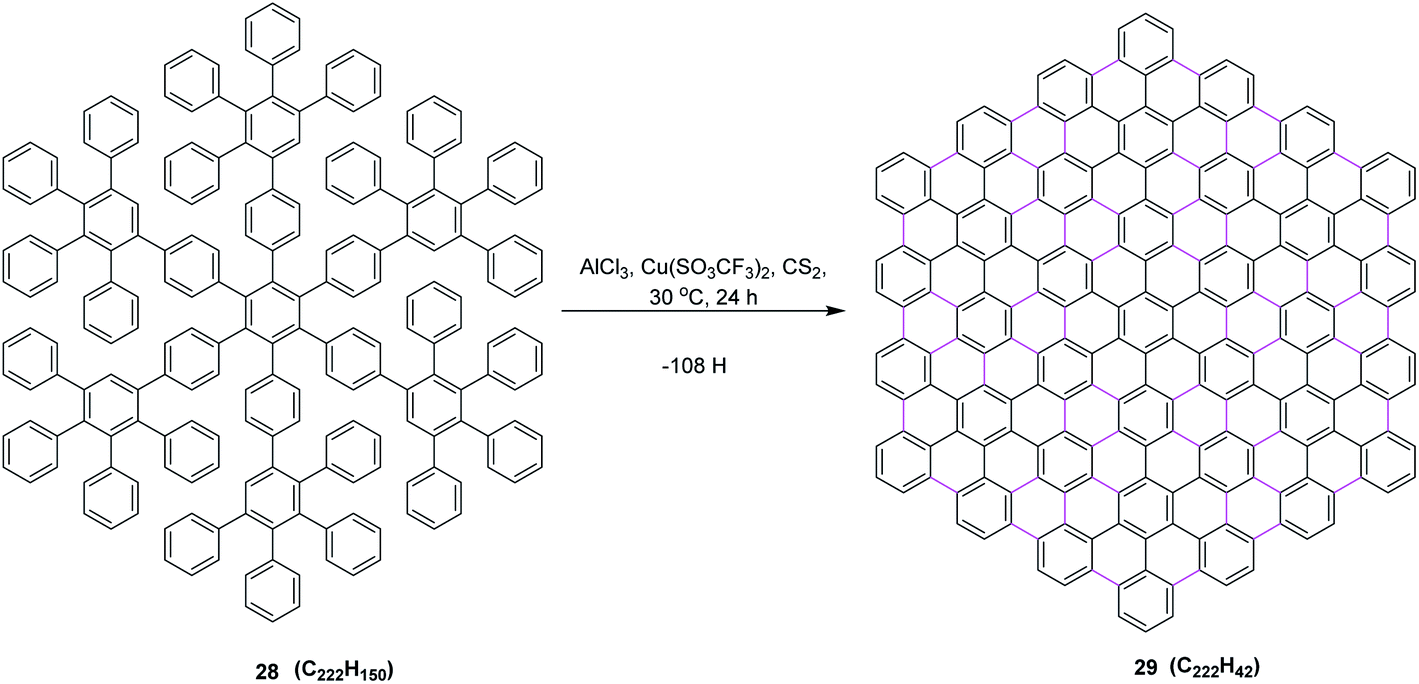 | ||
| Scheme 13 Oxidative cyclodehydrogenation of oligophenylene to the PAH.42 | ||
In 2006, Müllen and co-workers developed a new synthetic route for oxidative cyclodehydrogenation to construct terylene 31, a higher homolog of perylene, over multiple steps. They utilized AlCl3 in chlorobenzene at 80 °C as a mild oxidant to develop the new hexagonal ring in compound 30 (Scheme 14). Here, AlCl3 is in fact a non-oxidizing Lewis acid. This implies oxidation by air at high temperatures. Iron(III) chloride (FeCl3) was also used as the oxidant and provided less chlorinated derivatives as the by-products, although it resulted in a pentagonal ring instead of the hexagonal ring in compound 30.43
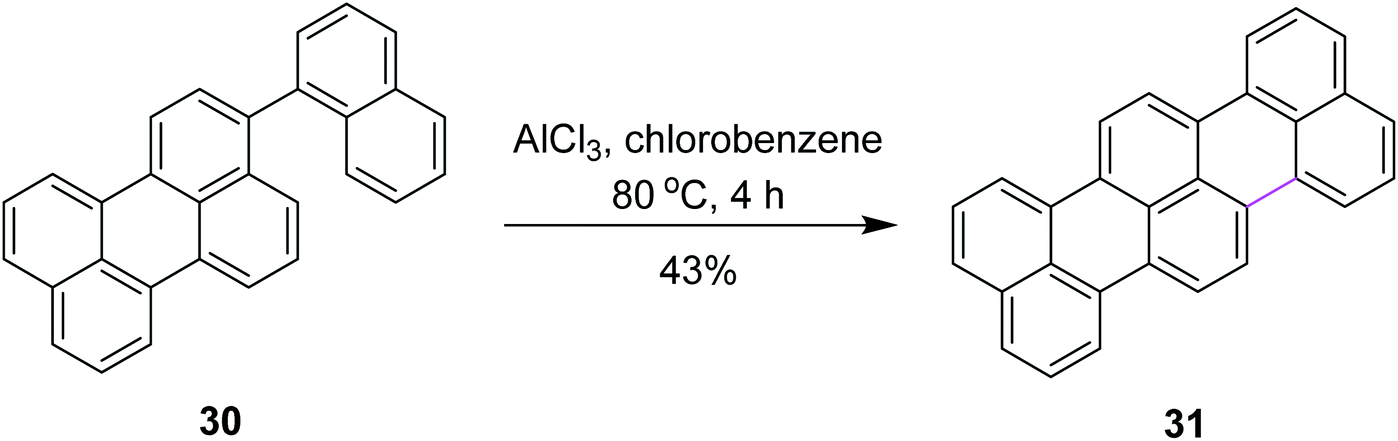 | ||
| Scheme 14 Terylene synthesis via AlCl3. | ||
In 2006, King and co-workers proposed a facile pathway for intramolecular Scholl reaction for reactive phenylbenzenes using CuCl2/AlCl3/CS2 as an efficient catalytic system. They also disclosed a possible mechanism using different oxidants such as PIFA, MoCl5, FeCl3, and CuCl2. The CuCl2/AlCl3/CS2 complex for the synthesis of hexabenzocoronenes (HBC) 33 is shown in Scheme 15. According to thermodynamic assumptions, PIFA and MoCl5 proved to be more efficient than CuCl2 for the Scholl cyclization of phenylbenzenes. Moreover, CuCl2 was not soluble in CS2, which indicated that oxidation occurred on the CuCl2 surface.44
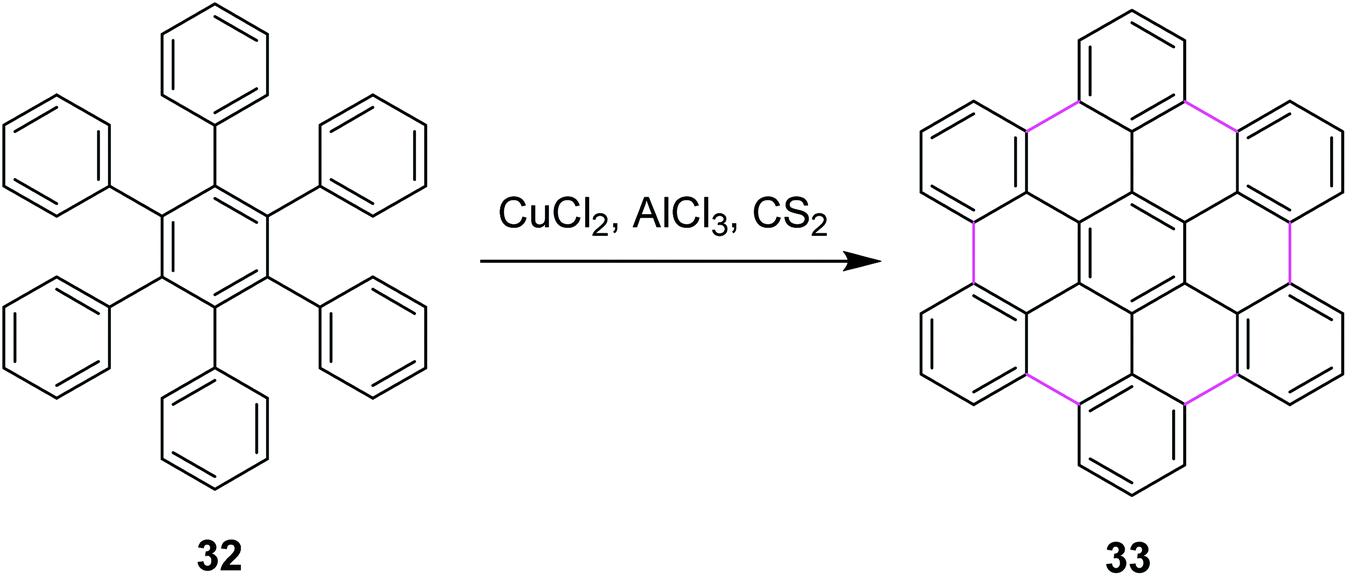 | ||
| Scheme 15 HBC synthesis by the Scholl reaction.44 | ||
In 2010, Wu and Jiao introduced a synthetic methodology for the preparation of polycyclic aromatic near-infrared (NIR) dyes using Scholl reaction as the key step. They implemented oxidative coupling for C–C bond formation in annulated perylene 34 to construct a partially saturated hexagonal ring in perylene 35 by treating the substrate with oxalyl chloride and AlCl3 (Scheme 16). The resulting product was passed through multistep reactions and finally converted into a bisquaterrylene dye.45
 | ||
| Scheme 16 Arene–arene coupling by the Scholl reaction.45 | ||
In 2011, Groth and co-workers introduced a new bottom-up strategy for the synthesis of large PAHs by the Scholl reaction. The AlCl3/NaCl complex was used for the cyclodehydrogenation of perylene 36 to construct giant quaterrylenes 37 (Scheme 17). This reagent system was the most common method used at that time for the Scholl reaction due to its easy availability and low cost. The main factor that controlled the oligomerization trend was temperature. The process of oligomerization was more controllable at a high temperature.46
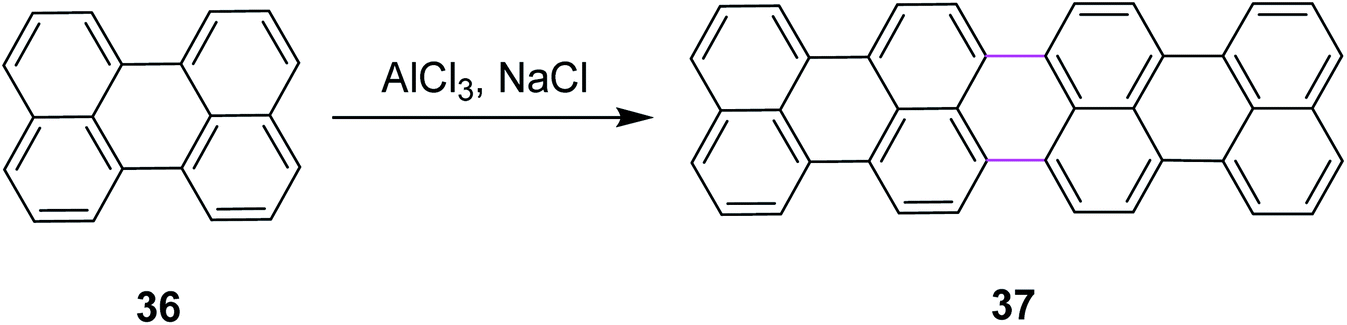 | ||
| Scheme 17 Synthesis of quaterrylenes via the Scholl reaction.46 | ||
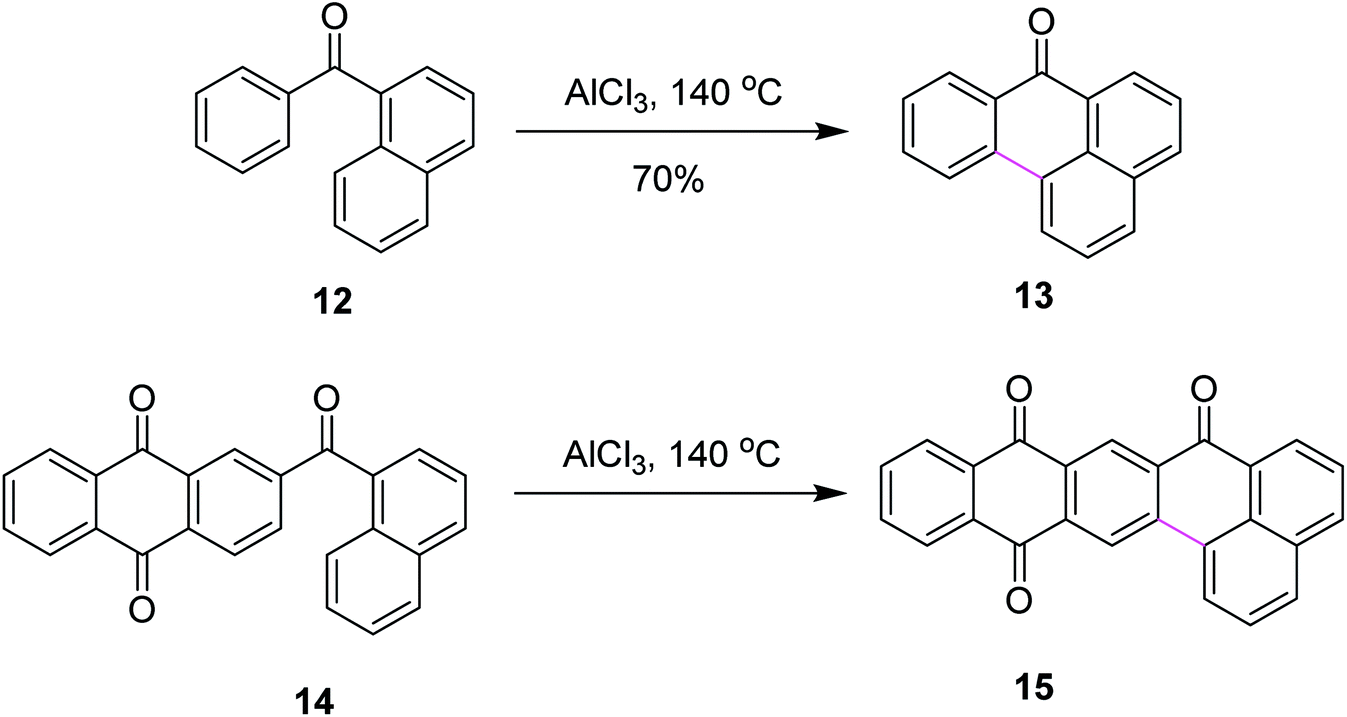
Later on, encouraged by a previous work reported on the Scholl reaction, Kuck and Mughal (2012) introduced a method to merge tribenzotriquinacene (TBTQ) with HBC, generating a cycloheptatriene ring by the Scholl-type reaction (Scheme 18). They constructed the bowl-shaped nanographene 42 utilizing AlCl3/Cu(OTf)2/CS2 as an effective oxidant system for the intramolecular cyclodehydrogenation step. This oxidant system proved more efficient than FeCl3 as it provided the complete condensation product and eliminated all other side reactions. The cycloheptatriene ring was constructed by the incorporation of the 1,2-benzene unit in the rigid diphenylmethane units of the TBTQ-scaffold.47a This reaction was repeated several times under different conditions. However, each attempt gave a mixture of partially reacted products and the starting material. Unfortunately, the complete removal of 48 hydrogen atoms to give the target compound 40 turned out to be unsuccessful and still presents a great challenge. Increasing the reaction time and amount of the reagents did not alter the results; rather, complex mixtures of the products were obtained. The concept to use TBTQ derivatives that bear three oligophenylphenyl 41 residues in the C3-symmetrical orientation at the outer periphery (C-2, C-6, and C-10) of the TBTQ core appeared promising despite the limitations encountered in this work. In conclusion, for the first time, one of the three 3D-bays of the TBTQ scaffold was bridged efficiently through a 1,2-benzeno unit, resulting in the generation of a seven-membered ring bearing PAH 42 within the quasi-planar fused carbon skeletons of TBTQ and hexa-peri-hexabenzocoronene. Encouraged by the abovementioned results, we attempted the threefold extension of compound 43 in order to access the bowl-shaped graphene-like structure 44 (Scheme 18). This reaction was repeated several times under different conditions. However, each attempt gave a mixture of partially reacted products and the starting material, as revealed by MALDI mass spectrometry. Increasing the reaction time and amount of the reagents did not alter the results; rather, complex mixtures of products were obtained. Probably the poor solubility of the intermediates or particular crowdedness of the starting material are major obstacles in the complete intramolecular oxidative coupling, requiring the removal of 48 hydrogen atoms of precursor 43 to form target compound 44 (Scheme 18).47b Therefore, the synthesis of the bowl-shaped graphene-like structure 44 by adapting the above-discussed synthetic plan remains an open challenge to date.
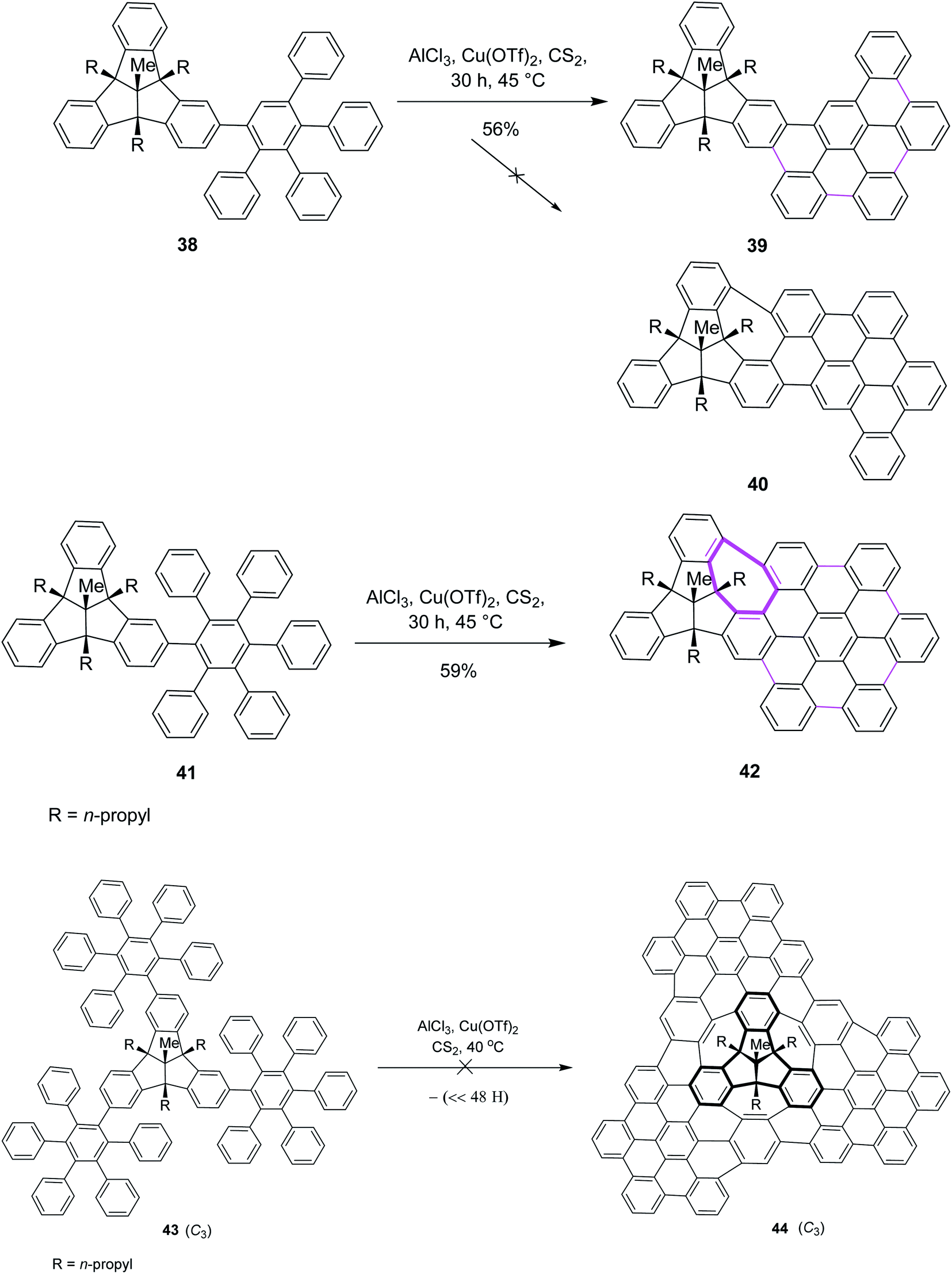 | ||
| Scheme 18 Synthesis of oligophenylphenyl–TBTQ derivatives by the Scholl reaction leading to the condensed and threefold TBTQ hydrocarbons. | ||
In 2012, Tao and co-workers developed a new protocol for the construction of contorted tetrabenzocoronene 46 by the Scholl reaction. They utilized the AlCl3/Cu(OTf)2/CS2 complex for the cyclodehydrogenation of bis-olefin 45 to construct the C–C bonds of tetrabenzocoronene (Scheme 19). They also used FeCl3 as the oxidant for the same reaction but it gave a mixture of the desired product 46 and the chlorinated by-products. Therefore, Cu(OTf)2 proved to be a comparatively better oxidant.48
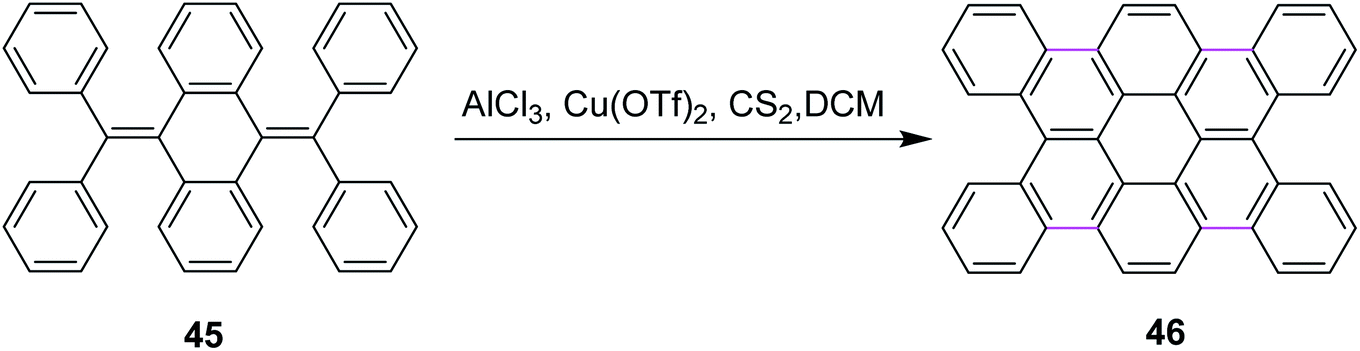 | ||
| Scheme 19 Tetrabenzocronene synthesis by the Scholl reaction.48 | ||
In 2015, Gourdon and co-workers proposed a novel pathway for the formation of 3-fluoroterrylene 48 from perylene 47 (Scheme 20). The resulting product was recognized as a molecular nanoprobe for single-electron optical sensing. The reaction pathway involved the AlCl3-mediated Scholl reaction to form a C–C bond between the two functionalized aryl vertices. The authors used other reagents along with AlCl3 and presented a selectivity comparison using 19F NMR spectroscopic characterization, where AlCl3 proved to be more efficient in constructing the hexagonal ring and DDQ promoted the more efficient construction of the pentagonal ring.49
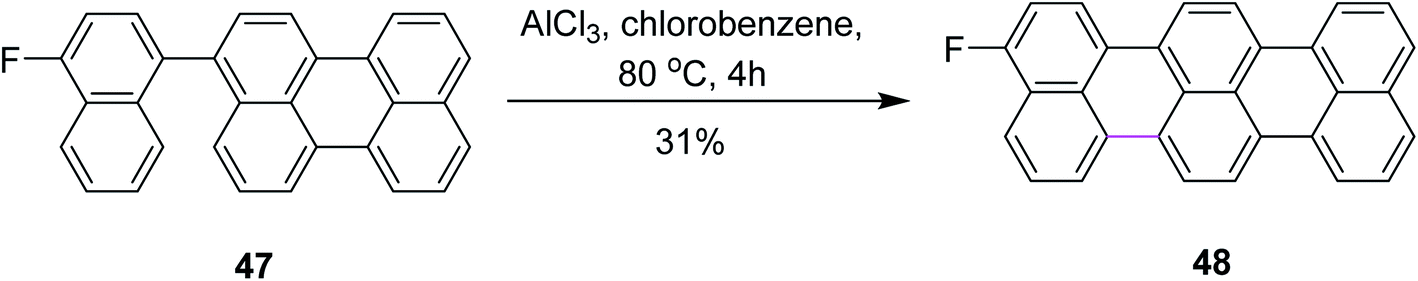 | ||
| Scheme 20 AlCl3-mediated cyclodehydrogenation for C–C bond formation. | ||
In 2016, Agranat and co-workers described the regioselectivity in the Scholl reaction of 1-benzoylpyrene 49. The reaction was carried out under the Scholl reaction conditions, i.e., AlCl3/NaCl at high temperature to obtain fine yield of the product 50 and 51 from Z-49 (Scheme 21). The reaction followed the arenium ion mechanism for C–C bond formation, which proceeded through a lower energetic barrier via the Scholl reaction in acidic conditions, as demonstrated by the quantum mechanical calculations. This reaction proved to he highly regioselective as only one product was formed out of the four expected products.50
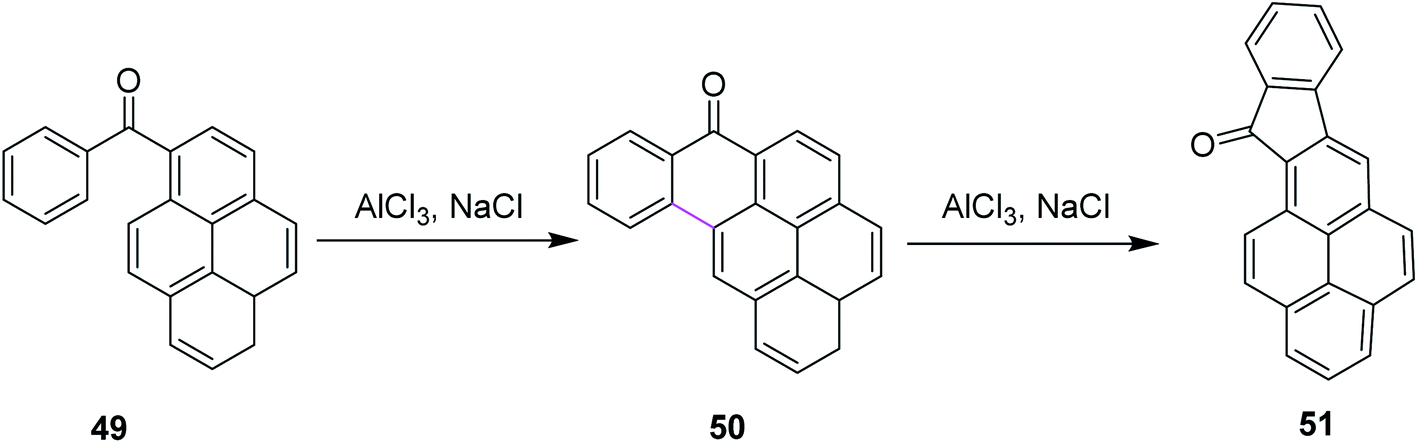 | ||
| Scheme 21 AlCl3-mediated intramolecular Scholl reaction.50 | ||
In 2018, Huang and co-workers introduced a method for the synthesis of microporous organic polymers (MOPs) using the Scholl reaction to promote the key C–C coupling reaction. During this process, two aryl hydrogens were removed from adjacent phenyl rings by treating reactant 52 with anhydrous AlCl3 in chloroform to construct a new aryl–aryl bond in product 53 (Scheme 22). This C–C bond formation between two aryl groups generated diversity in the functional group and the molecular geometry of MOPs, which gave rise to different properties in MOPs such as rigidity and cross-linked network that eventually led to high microporosity.51 The methodology provides a new route for the fabrication of microporous organic nanotubes for various potential applications including energy storage, adsorption, separation, and catalysis.
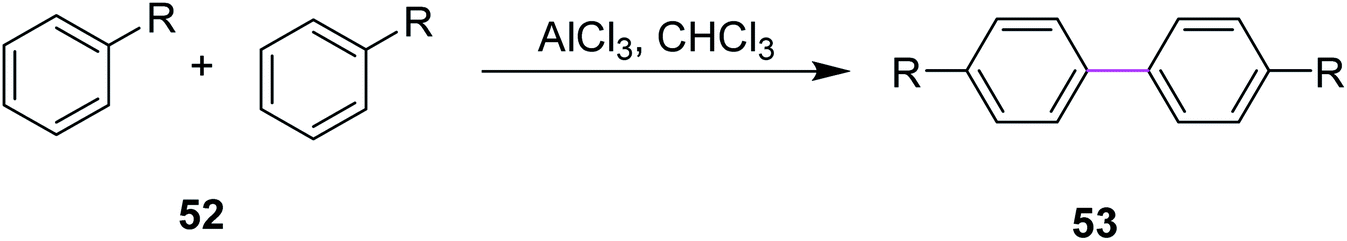 | ||
| Scheme 22 Scholl coupling reaction via AlCl3.51 | ||
In 2019, Agranat and co-workers designed a protocol for C–C bond formation between two aryl systems using excess of AlCl3/NaCl at elevated temperature (140–220 °C). This reaction involved Scholl cyclization of different benzoylnaphthalenes 54 to synthesize 7H-benz[de]anthracen-7-one 55 (Scheme 23). This type of reaction could be extended further into the synthesis of PAHs. This reaction was recognized as similar to the Friedel–Crafts acylation that was carried out in the presence of PPA. AlCl3 promoted the Scholl reaction, whereas PPA promoted the Friedel–Craft acylation.52
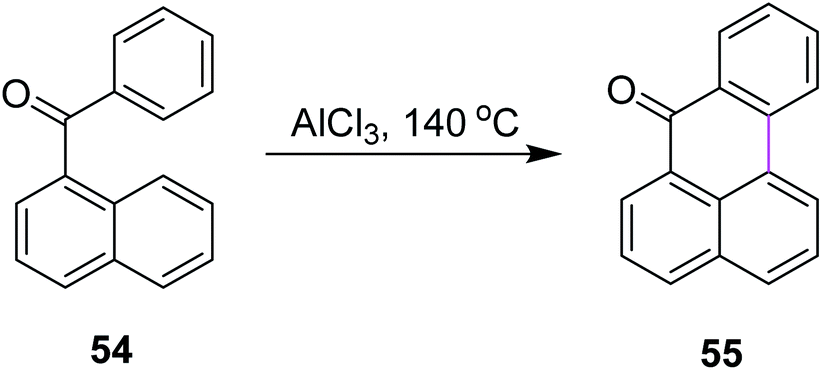 | ||
| Scheme 23 AlCl3-mediated Scholl reaction of benzoylnaphthalene.52 | ||
In 2020, Jimenez and co-workers reported a method for the cyclodehydrogenation reaction of 2-substituted binaphthyls 56a and 56b to afford bay-substituted perylenes. Using AlCl3 as the Lewis acid and high temperatures, the two new products 57a and 57b bearing NH2 and N(CH3)2 groups at position 2 of the perylene ring were synthesized (Scheme 24).53 Under these conditions, the authors were also able to obtain perylene 57c from ternaphthalene in 39% yield after two cyclodehydrogenation reactions in a single step. According to these analyses, within this experimental condition (AlCl3/NaCl, 170 °C), substrates 56a and 56b can generate the σ-complex to initiate the reaction and consequently produce intramolecular cyclization. The attempts to promote the formation of a radical cation using FeCl3 or a strong oxidant such as DDQ did not yield the expected products.
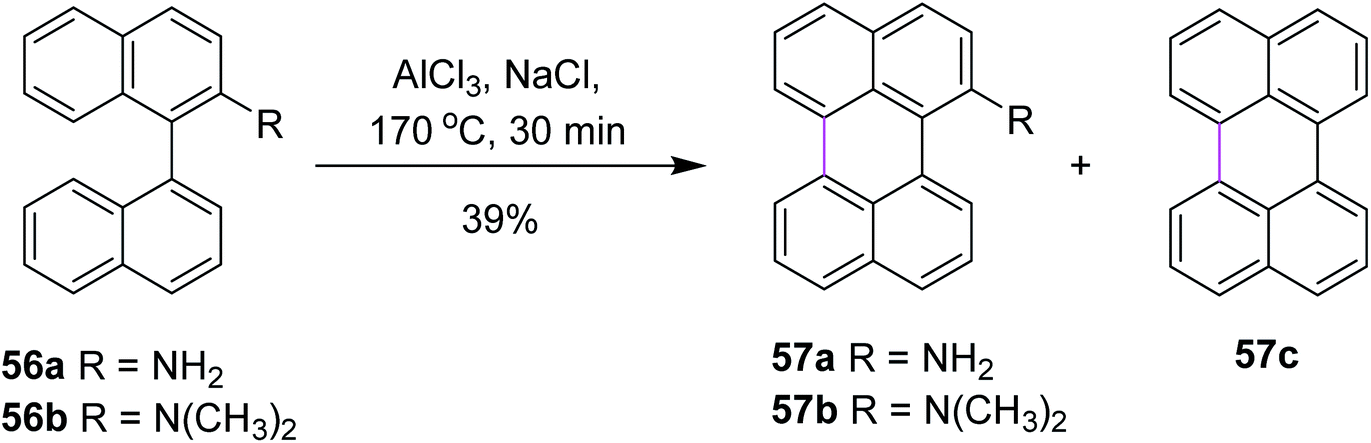 | ||
| Scheme 24 Synthesis of perylene via the Scholl reaction.53 | ||
In 2020, Miao and co-workers presented their work, where they stitched nitrogen-containing aromatic units 58 via easily maneuverable AlCl3-mediated Scholl reaction in a single operation. The mild reaction conditions in this strategy feature moderate temperature and short time. As a result, a highly stable nitrogen-containing porous organic framework (POF) 59 was constructed, as described in Scheme 25. AlCl3 was used as a catalyst due to its lower toxicity, high stability, and high cross-linking ability. POF was then used to capture radioactive volatile iodine by reversible capture.54
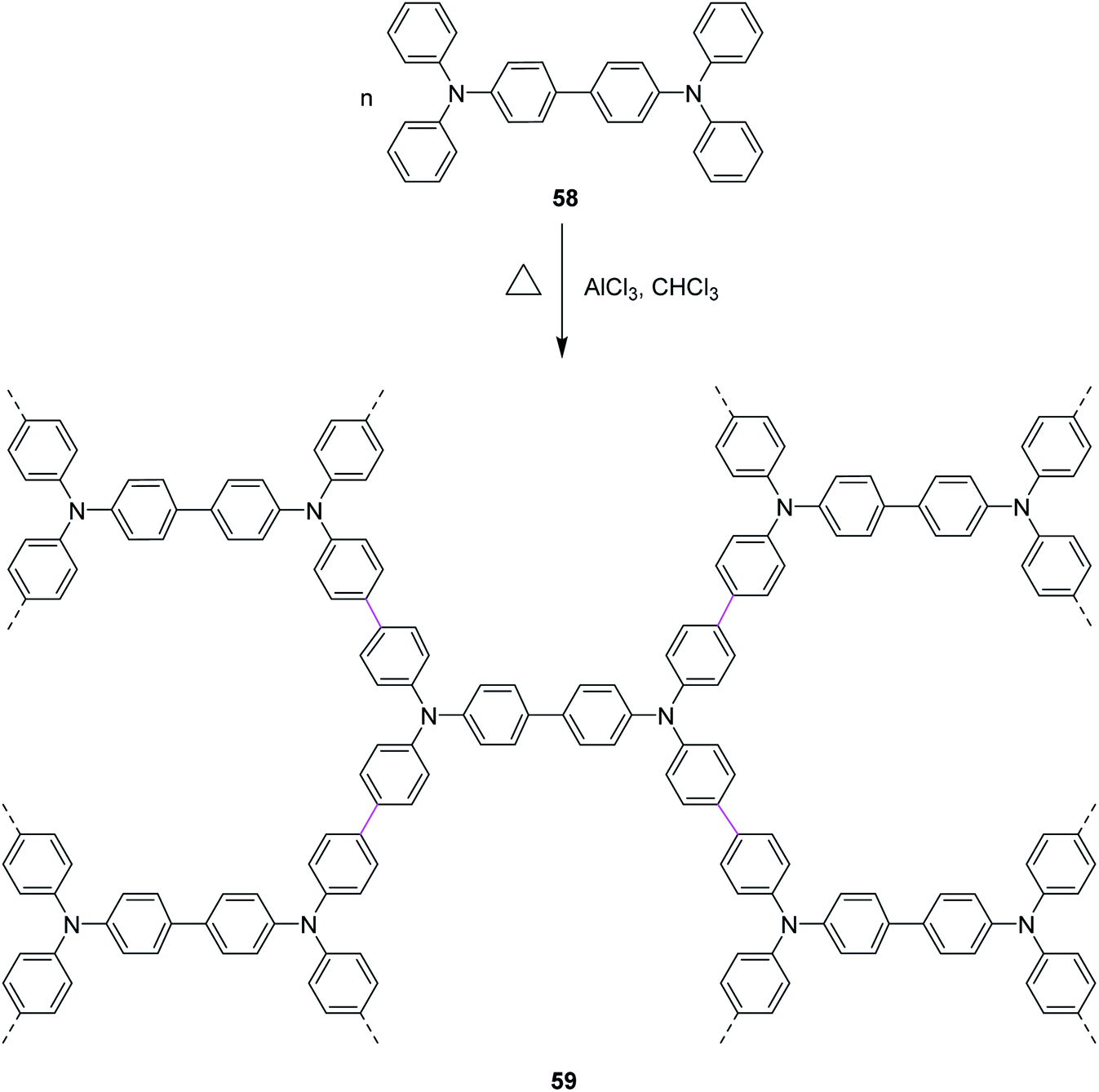 | ||
| Scheme 25 POF synthesis by the AlCl3-mediated Scholl reaction.54 | ||
It is concluded that AlCl3 proved to be a better oxidant due to its better oxidizing ability as compared to the other reagents. Its reduction potential value is −1.667 V.
5.2. FeCl3-mediated Scholl reaction
Iron(III) chloride (FeCl3) simultaneously acts as the oxidant and as the Lewis acid in the Scholl reaction. Previously, FeCl3 was utilized in the Scholl reaction because of its ready availability and eco-friendly nature. Many substituted triphenylenes 61 were synthesized from benzene derivatives 60 through cyclotrimerization using FeCl3 as the oxidant to construct the C–C bonds (Scheme 26).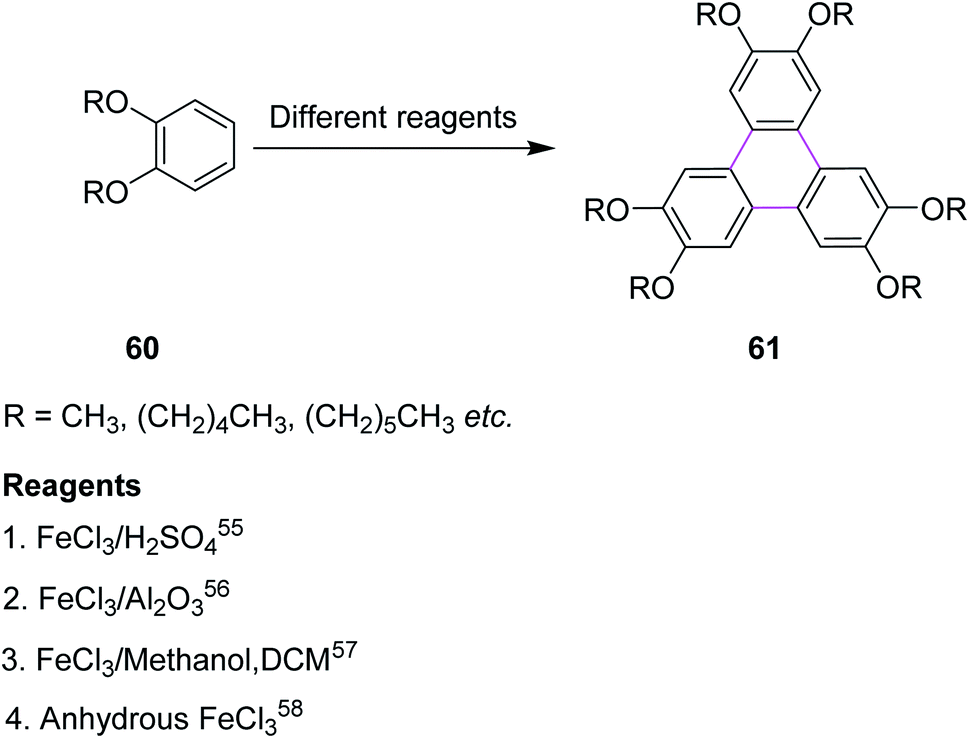 | ||
| Scheme 26 Triphenylene synthesis employing FeCl3. | ||
Mattmer and co-workers (1994) used a mixture of FeCl3 and conc. sulfuric acid (H2SO4) for the synthesis of triphenylene.55 This procedure provided a good yield but its workup was a little strenuous and hazardous to the environment. To overcome this problem, Cooke and co-workers synthesized a triphenylene in 1999 by replacing the abovementioned oxidant system with FeCl3/Al2O3. This process provided quite good yield, which is a safe and environmentally friendly synthetic route for performing the cyclotrimerization of dialkyloxy benzenes 60 to produce triphenylene derivatives such as 61.56
Bushby and co-workers, in 2006, used FeCl3/methanol in dichloromethane (DCM) for triphenylene synthesis. They experimentally proved that this oxidant complex was much better many other oxidant complexes such as FeCl3/Al2O3, MOCl5, and VOCl3.57
Similarly, Bai and Lin (2011) synthesized triphenylene in the presence of anhydrous FeCl3. This method involved a more efficient and selective procedure for the solvent-free oxidative cyclodehydrogenation of arenes. This coupling reaction was cheap and an environmentally friendly “Green Chemistry” method due to the elimination of expensive and hazardous solvent systems. The product obtained by this method was soluble in organic solvents.58
In 2011, Yang and co-workers developed a synthetic protocol for the synthesis of binaphthyls 63 utilizing FeCl3/K2CO3 in dichloroethane (DCE) via Scholl cyclization. Naphthylamines 62 were subjected to oxidative coupling for the construction of C–C bonds in binaphthyl diamines (Scheme 27). The group used FeCl3 as the oxidant owing to its properties as a promoter for the oxidative coupling of electron-rich arenes and more importantly, is non-toxic, less expensive, environmental benign, and easily commercially available. K2CO3 was used as the base for catalyzing the reaction as it neutralizes the additional HCl produced during this process. The reaction is dependent on the nature of the solvent under these reaction conditions. In the presence of DCE as the solvent, maximum yield was obtained.59
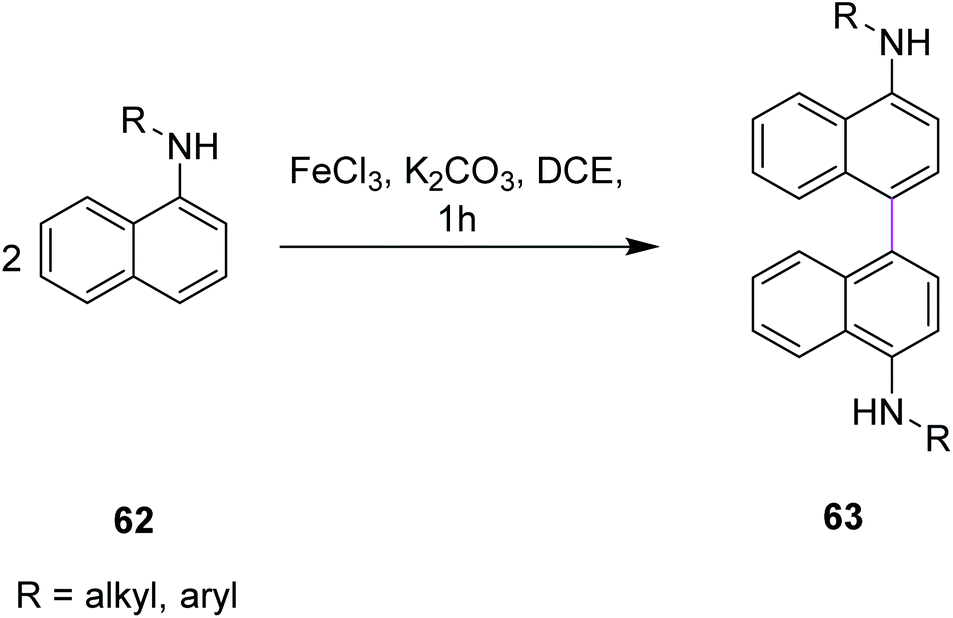 | ||
| Scheme 27 Binaphthyl synthesis by FeCl3 catalysis. | ||
Heteroatom-doped graphenes constitute a new class of materials that possess properties and functions different from those of the parent graphene. In 2013, Müllen and co-workers devised a method for the synthesis of pyrrole-fused azacoronene (65a,b, 67a,b and 69) via the oxidative cyclodehydrogenation of the corresponding hexaarylbenzenes (64a,b, 66a,b, and 68). Bottom-up organic synthesis can provide structurally-defined heteroatom-doped graphene fragments with perfect control of not only the size, periphery, and substituents but also of the doping concentration and position. Final planarization with FeCl3 offered targets (65a,b and 67a,b) in good yields (60–86%). Notably, the 3,5-dialkoxyphenyl group (64a,b and 66a,b) appears to be required for the complete planarization of the azacoronene core/scaffold. However, when the Scholl reaction was performed with 68, which possesses a 4-tertbutylphenyl group instead of a 3,5-dialkoxyphenyl group, partially fused compound 69 was obtained as the main product (91%) even though the completely fused structure was detected by MALDI. In line with their strategy to fuse hybrid aromatics, the further functionalization of PAHs would be beneficial for molecular electronics and spintronics with well-defined structures (Scheme 28).60
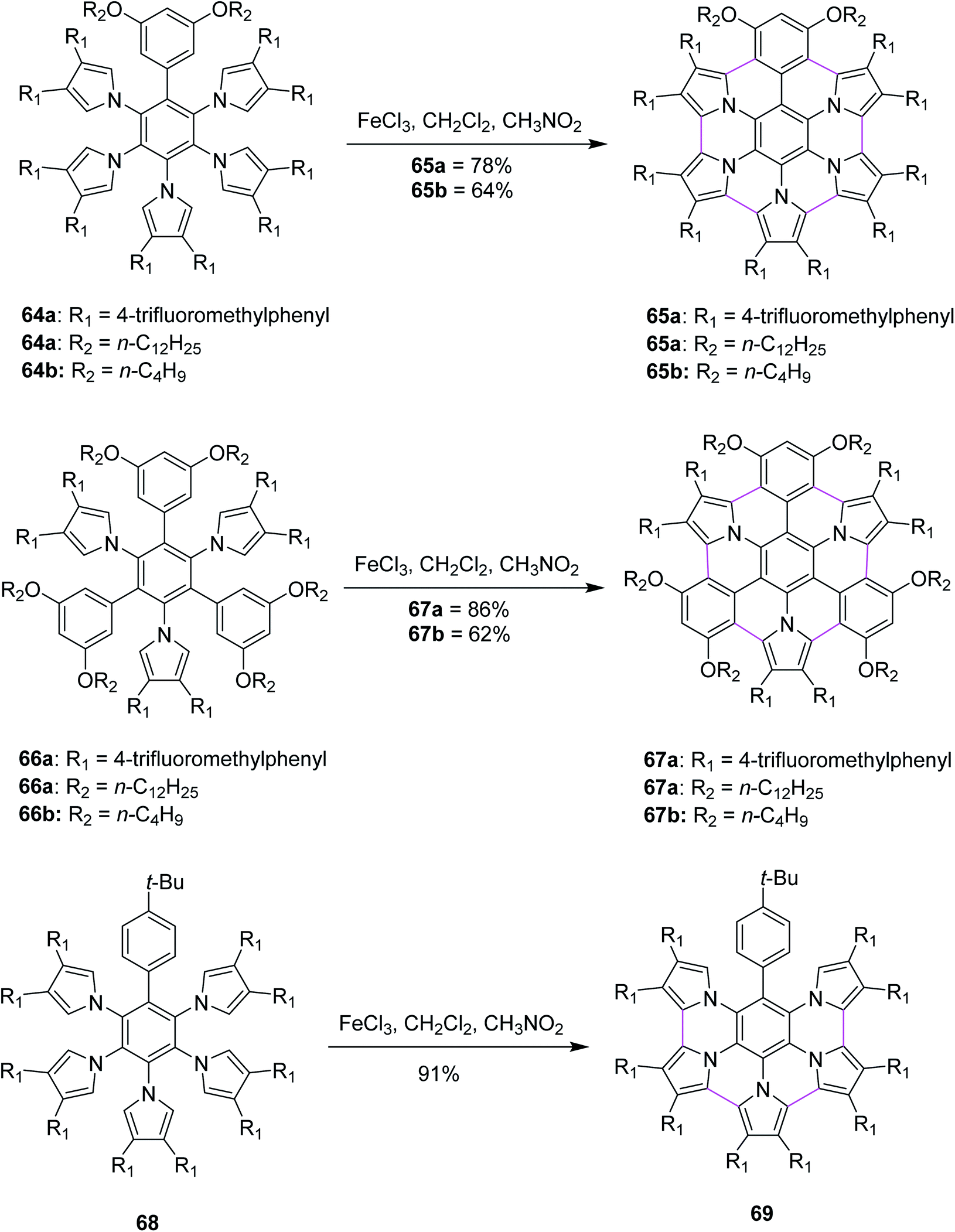 | ||
| Scheme 28 Synthesis of pyrrole-fused azacoronenes employing FeCl3. | ||
In 2014, Feng and co-workers switched their attention to apply the Scholl reaction for the intramolecular condensation of polyphenylene-based precursor 70 to furnish solution-processed GNRs 71 (Scheme 29). For the first time, they synthesized an AB-type Diels–Alder polymer to prepare GNR 71 with a “cove-type” edge structure and desired dimensions. The structurally well-defined GNRs can be prepared using a ‘bottom-up’ organic synthesis method through surface-assisted or solution-mediated cyclodehydrogenation reactions. In addition, non-planar polyphenylene precursors were first ‘built up’ from small molecules, and then ‘graphitized’ and ‘planarized’ to produce GNRs. Furthermore, C–C bond formation at undesired positions during the cyclodehydrogenation of 70 is hindered by the steric repulsion between the benzene rings and the bulky alkyl chains, ensuring the non-kinked and uniform structure of the resulting GNR 71. Moreover, the insertion of various functional groups on the periphery of such GNRs is feasible and may prove to be valuable for the fundamental studies on graphene nanostructures, which can place specific functionalities on them and further enhance their processability to realize wider applications, such as in GNR-based nanocomposites.61
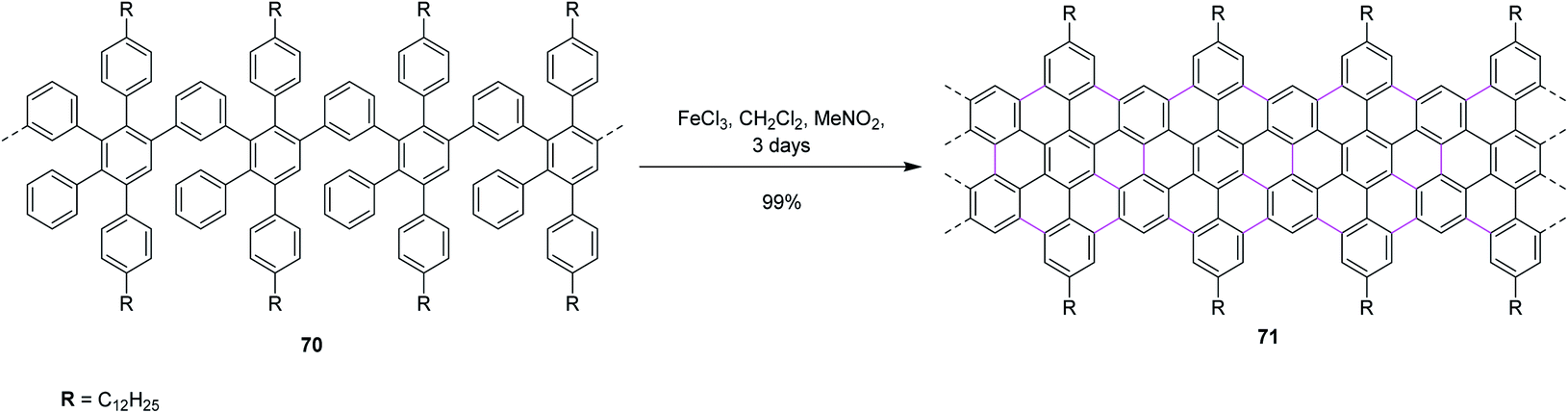 | ||
| Scheme 29 Synthesis of graphene nanoribbons by the Scholl reaction. | ||
In 2014, Laschat and co-workers presented a protocol for the synthesis of the sterically crowded o-terphenyl crown ether 73 using alkoxy substituents from tetrabromodibenzocrown 72 as well as boronic acids or borolanes by Suzuki coupling and consequently converted to triphenylenes applying the Scholl reaction (Scheme 30). Fifteen equivalents of FeCl3 were used for Scholl cyclization in the presence of DCM. Sequentially, the product was dissolved in methanol for quick quenching. The resulting products demonstrated liquid crystalline behavior. The authors proposed that this type of harmonizing of the steric impact could be utilized as a scaffold for many other organic compounds.62
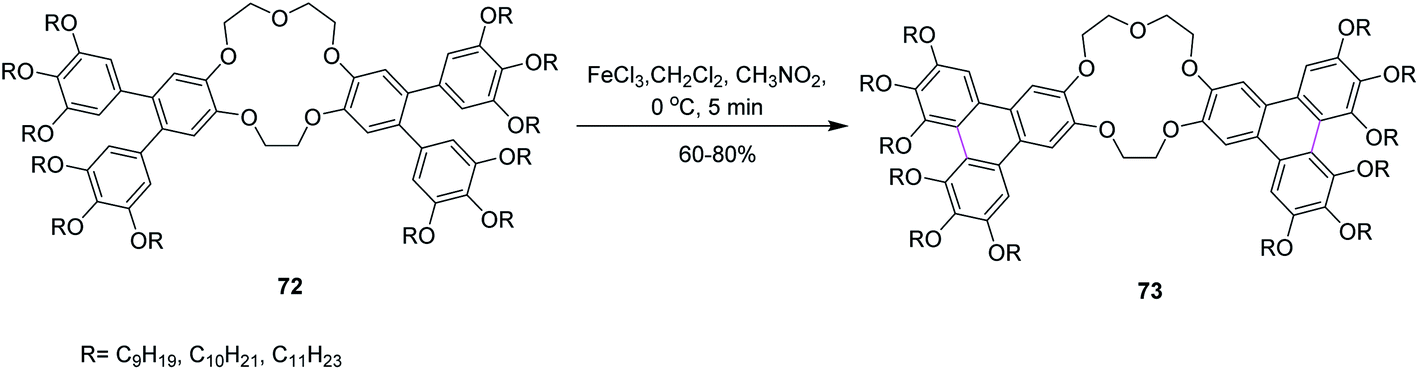 | ||
| Scheme 30 Synthesis of sterically crowded o-terphenyl crown ether. | ||
In 2015, Chi and co-workers reported a method for the synthesis of bisindeno-annulated pentacenes 75 using a facile FeCl3-mediated Scholl reaction of regioselective nature from the pentacene precursor 74 (Scheme 31). FeCl3 as the oxidant here selectively produced the transoid double cyclized product and in turn was used preferentially for such type of reactions. Notably, the high regioselectivity has been attributed to the high strain observed in the cisoid isomers. This combination of indeno-units intensely altered the chemical as well as the electronic properties of pentacene and the resulting products exhibited extraordinary photo-stability in solution. These compounds were used practically in electronic devices such as OFETs. This research offered broad conception to stabilize the π-conjugated systems, which are highly reactive in nature.63
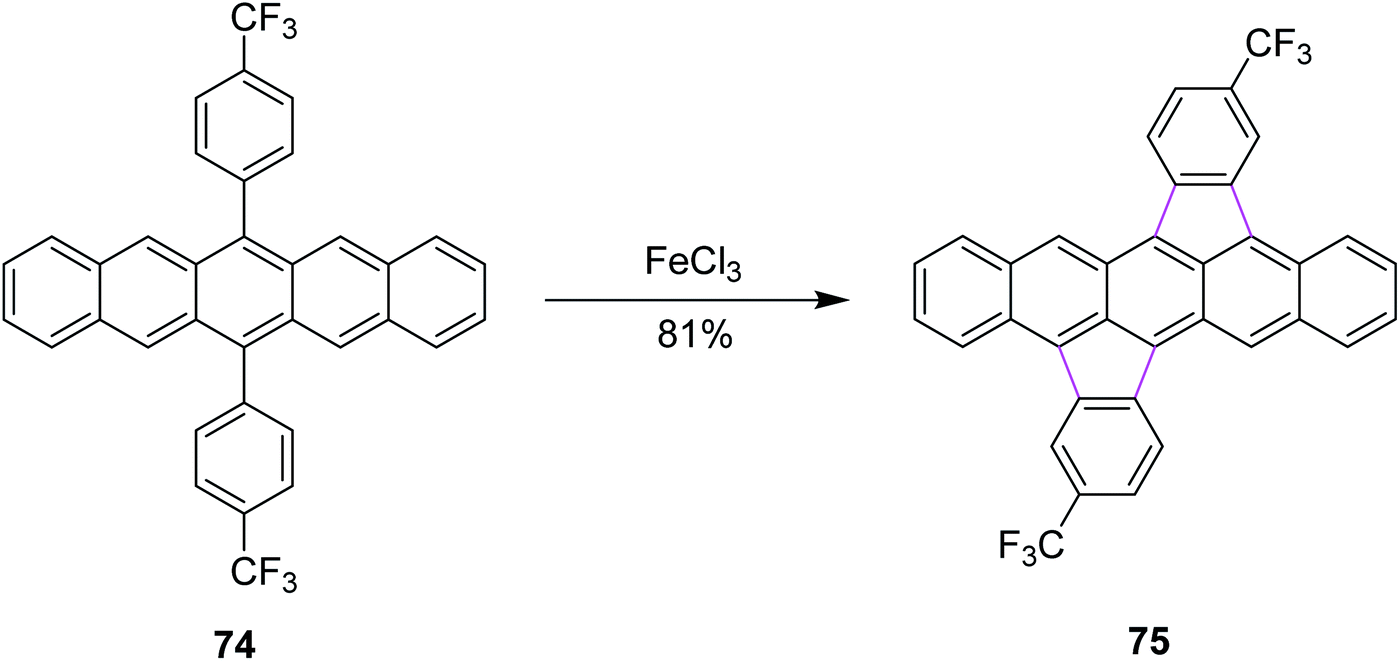 | ||
| Scheme 31 Synthesis of bisindeno-annulated pentacenes.63 | ||
In the same year, Nishiuchi and co-workers established a new post-construction method for the synthesis of GNRs and NGs. In this procedure, the large π-surface was formed using the FeCl3-mediated Scholl reaction after attaining the cyclic structure (Scheme 32). In this method, graphene fragment 77 was constructed from hexaarylbenzene 76 through cyclodehydrogenation under FeCl3-induced Scholl reaction conditions at ambient temperature.64
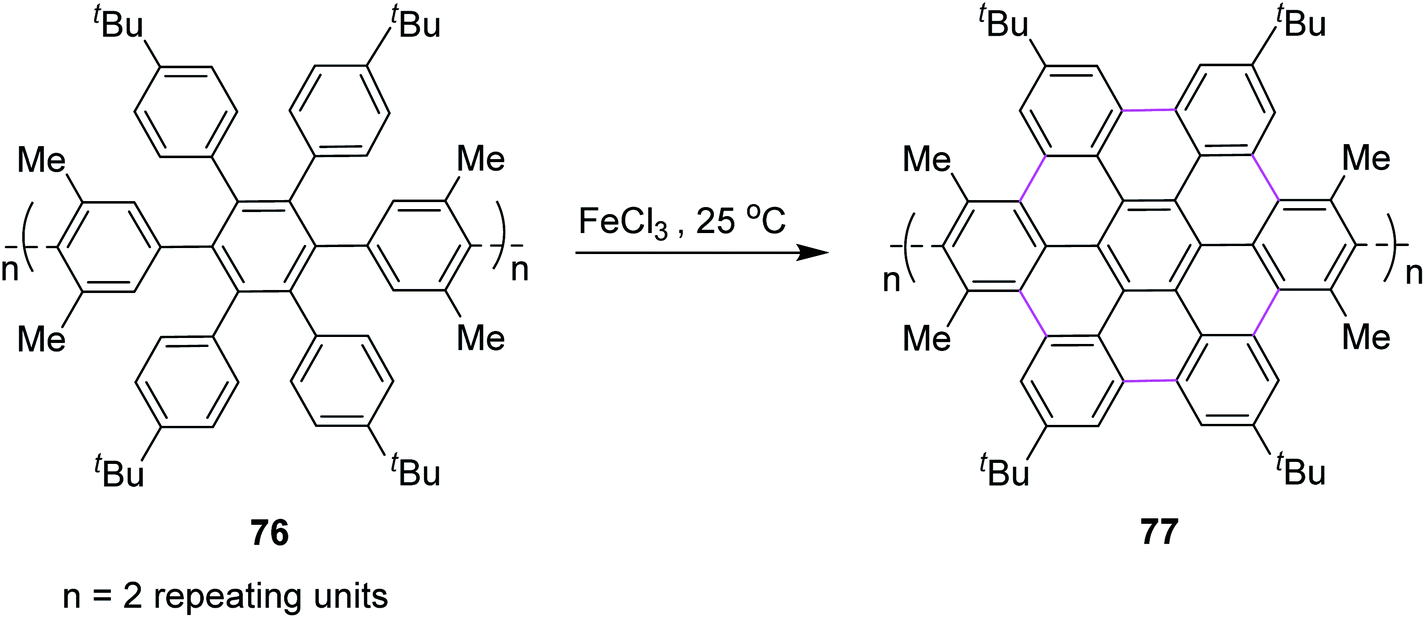 | ||
| Scheme 32 Cyclodehydrogenation of polyphenylenes by FeCl3. | ||
In 2016, Xu and co-workers described the three reaction pathways to synthesize benzoquinones and hydroquinone by following the Scholl reaction protocol. They used DDQ as the oxidant along with toluene in the first reaction pathway to give the intermediate product 79a, which was then reacted with FeCl3 to produce 80a in 82% yield (Scheme 33). In a second reaction pathway, FeCl3 was used directly to obtain product 80a in 34% yield from substrate 78. Lastly, in a third reaction pathway, target compound 80b was obtained in 83% yield from reactant 78 over two steps, as described in Scheme 33. In their work, the authors also reported that the reaction center was located next to the carbonyl groups that influenced the Scholl reactivity. The direct oxidation of 78 with FeCl3 to yield 80a provided less yield (34%) due to the involvement of an active hydroxyl group hydrogen that could potentially initiate free radical reactions. Some by-products were observed in this reaction pathway. Therefore, an alternate pathway was used for the Scholl cyclization of 78 where it was treated with DDQ in the first step to generate an intermediate product 79a, which was eventually converted to 80a, giving much higher yield (82%). Similarly, the related product 80b was obtained via intermediate 79b in good yield (83%).65
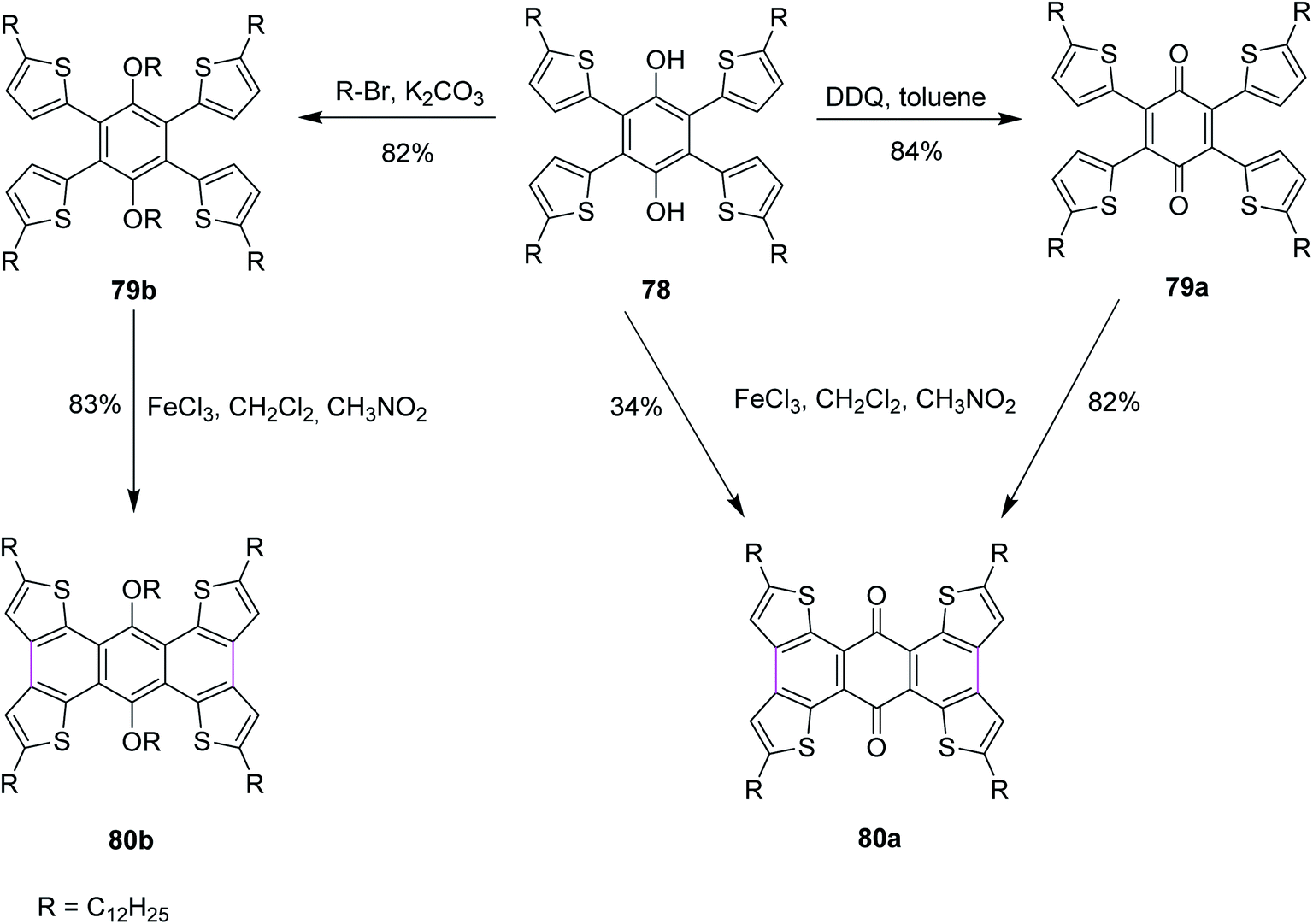 | ||
| Scheme 33 Fused aromatic compound synthesis by the Scholl reaction. | ||
In 2017, Murata and co-workers devised a protocol to induce unsymmetrical two-fold Scholl cyclization for the synthesis of pentagonal and hexagonal rings in the PAH systems such as 5,11-dinaphthyltetracene 81. A FeCl3/CH2Cl2/CH3NO2 reagent system was used for the Scholl cyclization. This reaction was selective as only one type of product 83 was obtained out of the three expected products (Scheme 34). The authors reported that the reaction mechanism of the Scholl cyclization in this type of synthesis was based on the formation of the di-cationic intermediate 82 instead of classical pathways that were based on radical cation or arenium ion formation. To avoid chlorination, they introduced a methyl group at each naphthyl ring. Hence, the choice of the reagent complex was also explained.66
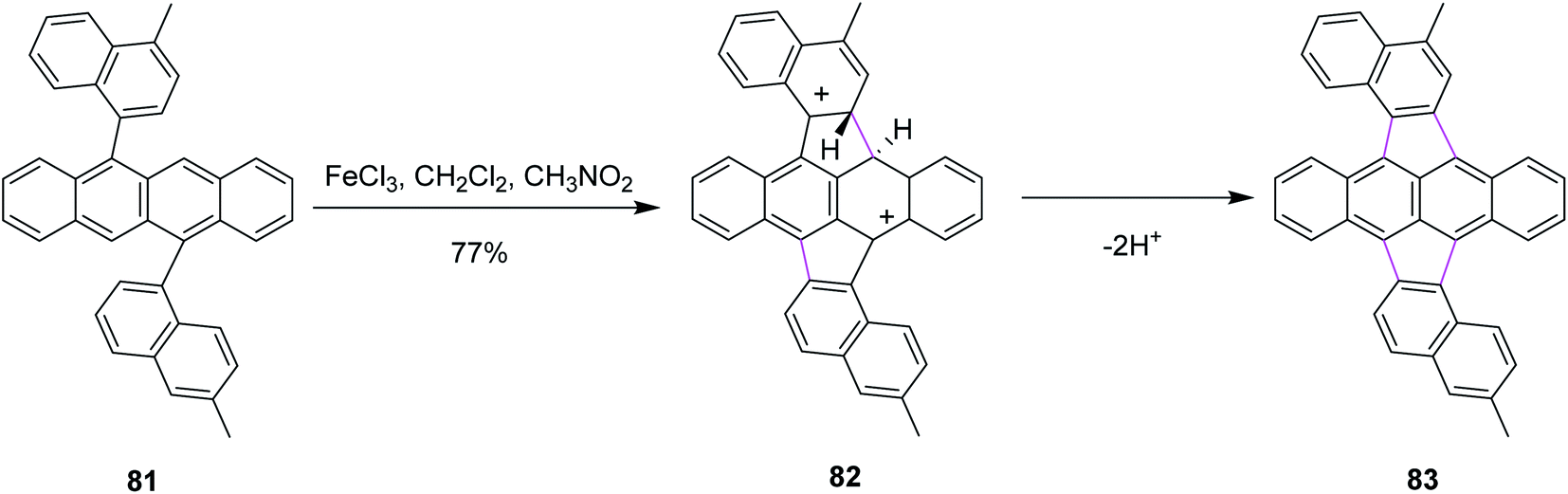 | ||
| Scheme 34 Twofold Scholl cyclization by FeCl3. | ||
In 2018, Borchardt and co-workers developed a solvent-free procedure for mechanochemical Scholl reaction. This procedure was introduced as a versatile graphitization tool for C–C bond formation. Several types of extensive ring systems such as hexabenzocoronene 85 from hexaphenylbenzene 84 were synthesized using this procedure (Scheme 35). Ball mills were used during this procedure to synthesize large-size nanographenes within a very short time due to the absence of extensive solvent systems and elimination of solubility limitation. FeCl3 was used along with sodium chloride to obtain high yields in this process. High input energy from the ball mill and excess of FeCl3 provided excellent yield with full chemical conversion. Furthermore, this pathway was safe to use and also avoided undesired by-product formation.67
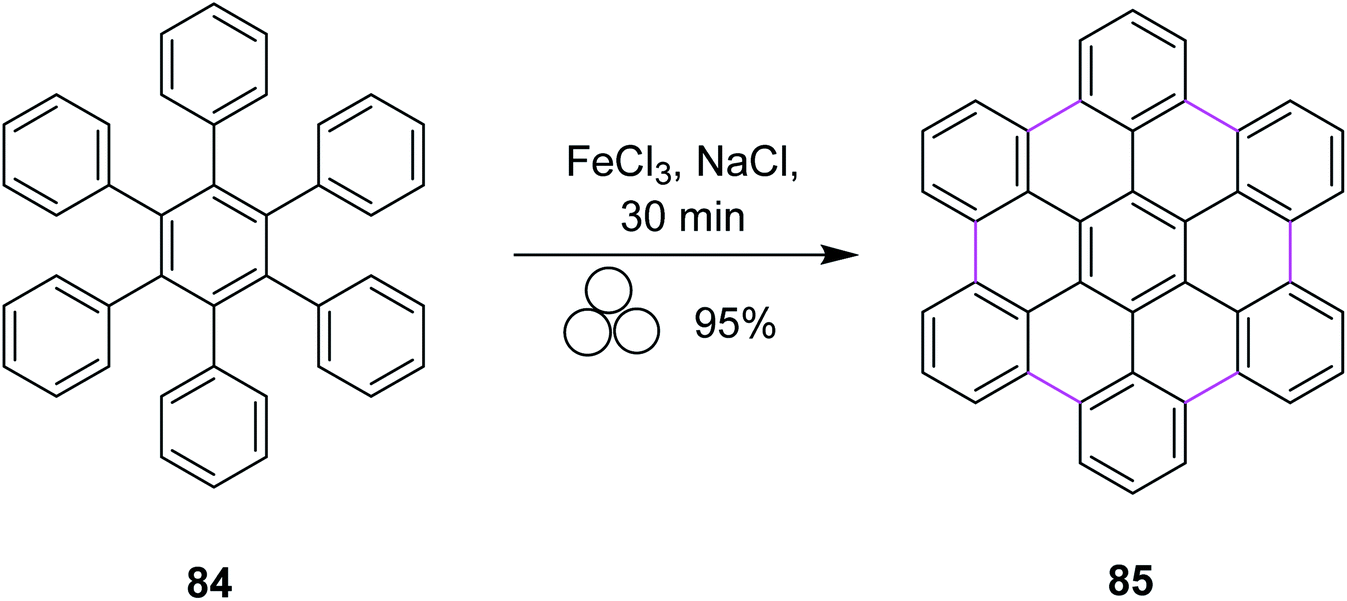 | ||
| Scheme 35 Hexabenzocoronene synthesis via the Scholl reaction. | ||
In 2018, Merner and co-workers introduced a process for the π-extension of the benzenoid macrocycles 86 by the FeCl3-mediated Scholl reaction. They converted a non-planar benzenoid systems to a non-planar PAH system and subsequently subjected the latter to oxidation to promote cyclodehydrogenation. As a result, new hexagonal rings were formed, and the production of undesired products was also controlled. Two products 87a (65%) and 87b (7%) were observed during this reaction (Scheme 36). The reaction yield was quite high because of the suitable oxidant system and reduced the number of by-products.68
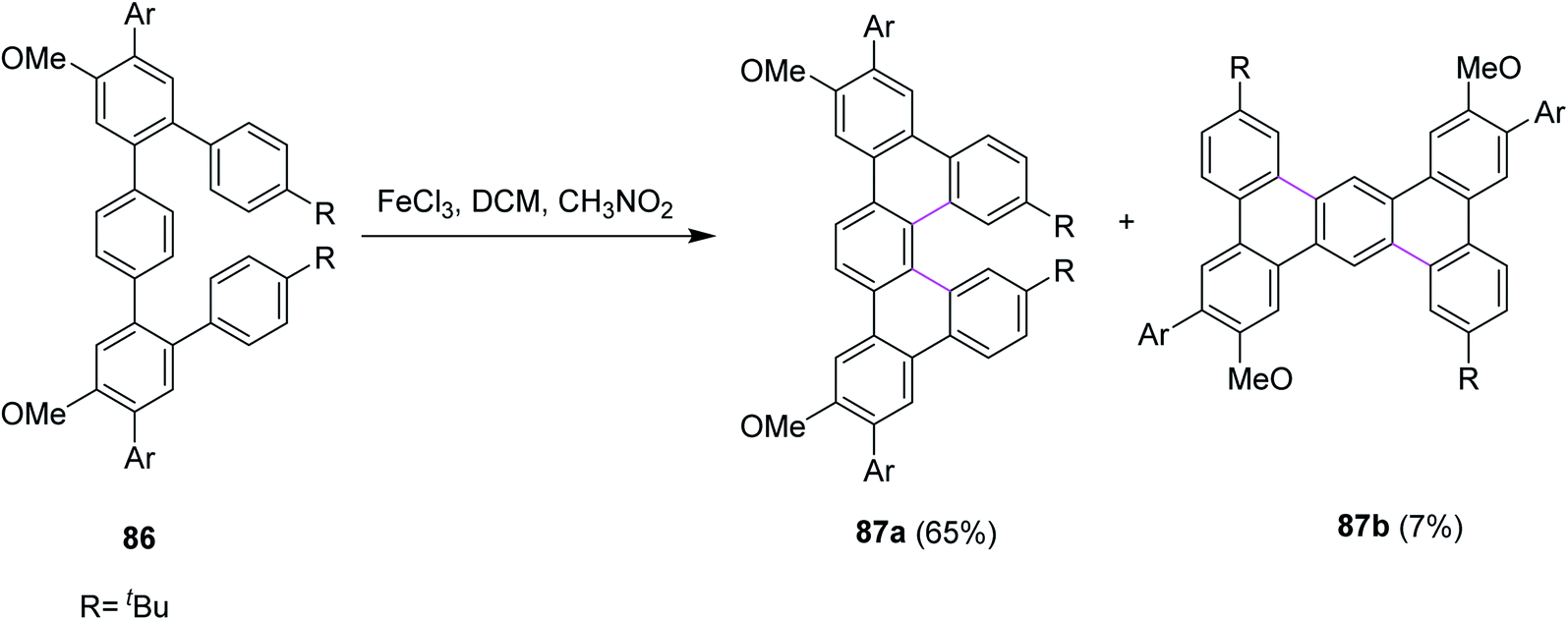 | ||
| Scheme 36 FeCl3-mediated Scholl reaction for π-extension. | ||
In 2020, Martín and co-workers synthesized a novel homo 3D nanographenes, featuring a cyclooctatetraene core. A concise and efficient bottom-up methodology was employed, during which 24 new C–C bonds were formed by means of the Scholl reaction. Nanographenes with 53 fused rings are realized, which exhibited good solubility in common organic solvents. The resulting saddle-like structures are electron-rich and show good chemical and electrochemical stability. Compound 88 subsequently underwent intramolecular cyclodehydrogenation mediated by FeCl3 to generate 89 in moderate yields. Compound 88 was subjected to a further one-fold FeCl3-mediated oxidative coupling in refluxing dichloroethane to produce compound 89. However, under such optimized reaction conditions, 88 was interconverted into the fully coupled 89 (Scheme 37).69
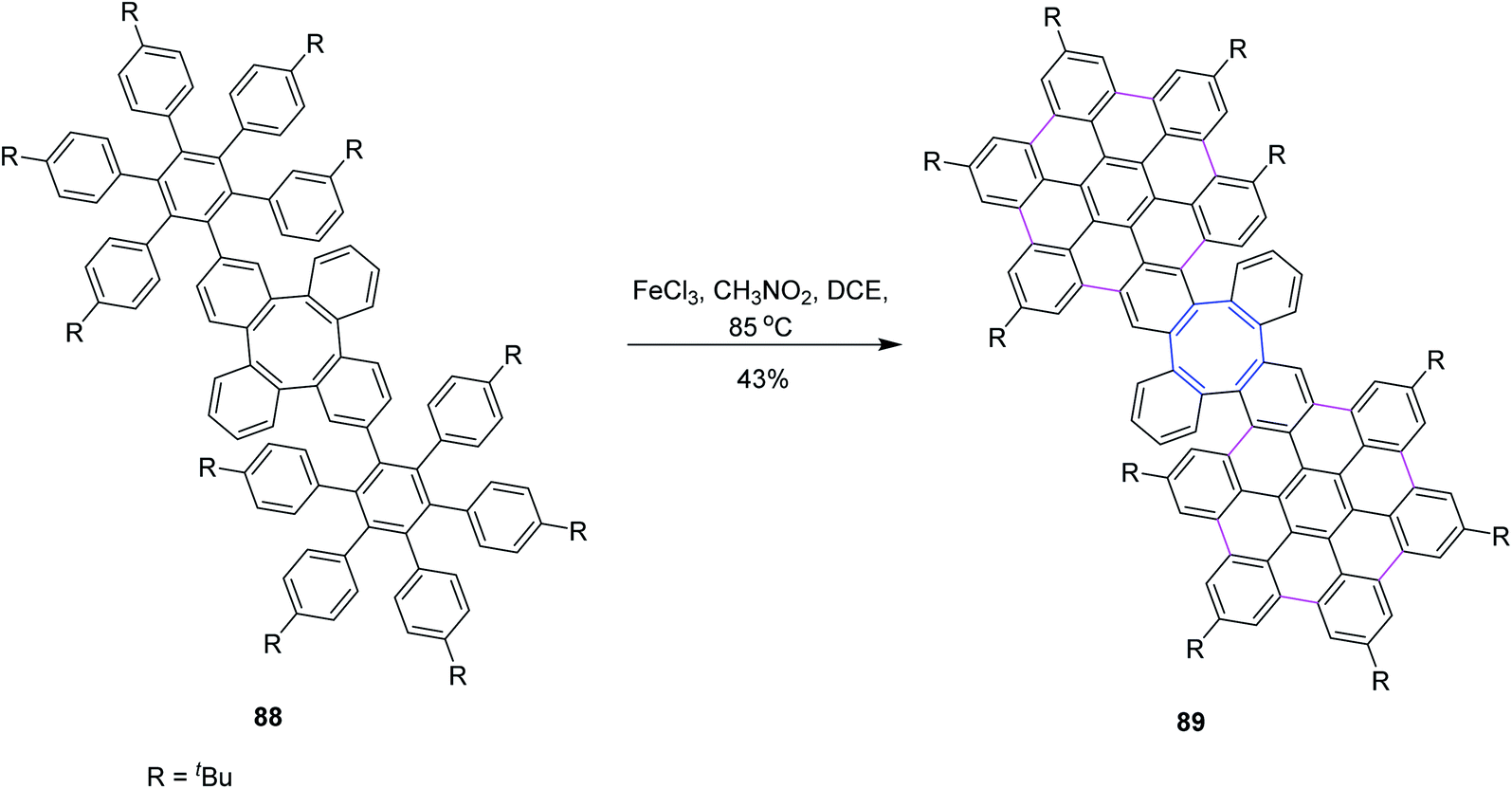 | ||
| Scheme 37 Nanographene synthesis by the Scholl reaction using FeCl3. | ||
Overall, it is concluded that FeCl3 proved to be a better oxidant due to its excellent oxidizing capacity as compared to AlCl3. Its reduction potential value is −0.409 V.
5.3. DDQ-mediated Scholl reaction
In 2009, Rathore and co-workers explored 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as a new oxidant for performing the Scholl reaction with a variety of aromatic donor substrates. The reaction followed a radical cation mechanism to produce triphenylenes and hexabenzocoronene scaffolds under strong and mild conditions.70 Biaryl synthesis has been particularly useful for the oxidative cyclodehydrogenation of a variety of substituted hexaarylbenzenes and o-terphenyls to produce the corresponding planar PAHs, for example, hexa-peri-hexabenzocoronenes (HBC's) and triphenylenes, respectively. The usefulness of the DDQ–acid system for oxidative cyclodehydrogenation in the Scholl reaction was further validated by the formulation of soluble HBC's [91a–c] from hexakis (4-isoalkylphenyl)benzenes [90a–c], where six new C–C bonds were formed in one step to give graphitic products in excellent yields and high purity (Scheme 38).70 The authors have demonstrated that the DDQ/H+ oxidation system, which is known to oxidize a variety of aromatic donors with the oxidation potential as high as ∼1.7 V to the corresponding cation radicals, can be effectively employed for the preparation of a variety of PAHs including graphitic hexa-peri-hexabenzocoronenes.70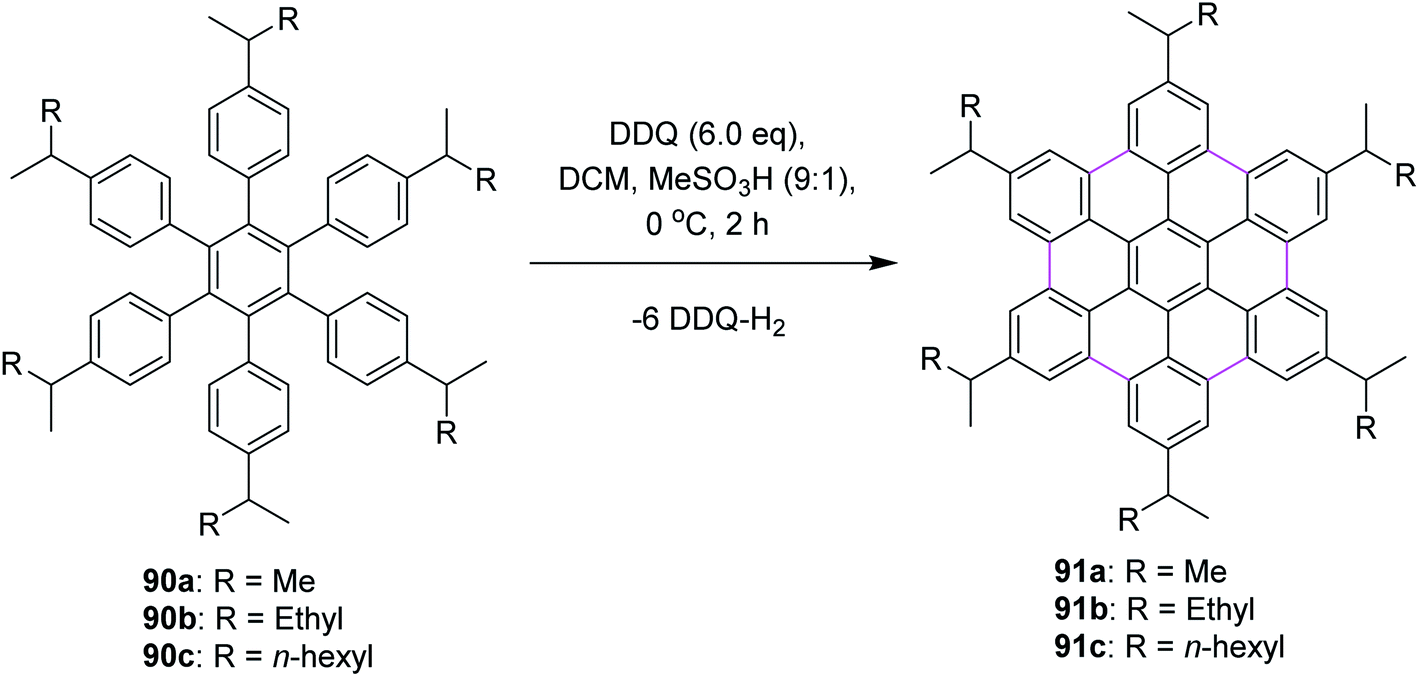 | ||
| Scheme 38 Synthesis of soluble HBCs from hexaarylbenzene precursors. | ||
In a follow-up study, Wong and co-workers (2012) developed a new set of reaction conditions for the oxidative cyclodehydrogenation of electron-poor polyphenylenes with various EWGs groups such as –Br and –F. Their coupling reactions to planar nanographenes was achieved using the DDQ–CF3SO3H oxidative system.71 The –Br and –F substituted HBC derivatives (93a–c) were prepared from hexaphenylbenzene precursors (92a–c) on treatment with a combination of DDQ and CF3SO3H (Scheme 39). It is noteworthy that there are two proposed reaction mechanisms for the oxidative cyclodehydrogenation (Scholl) reaction, proceeding either through an arenium-ion or a radical-cation intermediate. In both the pathways, protic acids play a vital role in the progress of the reaction.
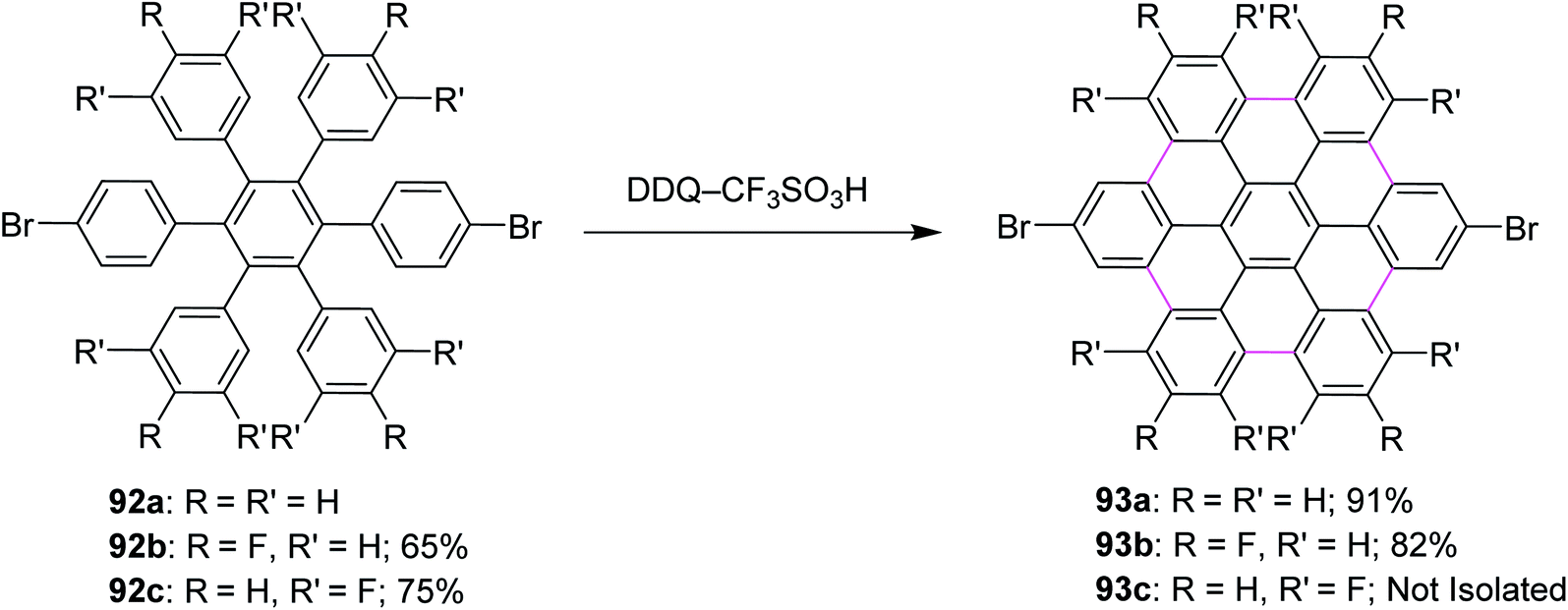 | ||
| Scheme 39 Synthesis of a series of HBC derivatives containing EWGs via DDQ. | ||
Consequently, Itami, Scott, and co-workers (2013) made major progress by achieving a concise two-step synthesis of grossly warped graphene molecule 96 (C80) with five embedded seven-membered rings and one five-membered ring at the center. Starting from corannulene 94, precursor 95 was obtained by Pd-catalyzed direct C–H arylation with arylboroxins, using o-chloranil as an oxidant for the intramolecular cyclization of precursor 95 that efficiently provided grossly warped graphene molecule 96 in good yields, overcoming the high steric demand to form five distorted seven-membered rings (Scheme 40).72 Because of its highly non-planar structure, graphene molecule 96 showed good solubility in common organic solvents, and, surprisingly, graphene molecule 96b with tertiary butyl groups could be dissolved even in hexane. The aforementioned studies proved the success of the current protocol, thus paving the way to synthesize the challenging heptagon-containing non-planar PAHs from polyphenylenes after condensing with the DDQ/H+ oxidant system.
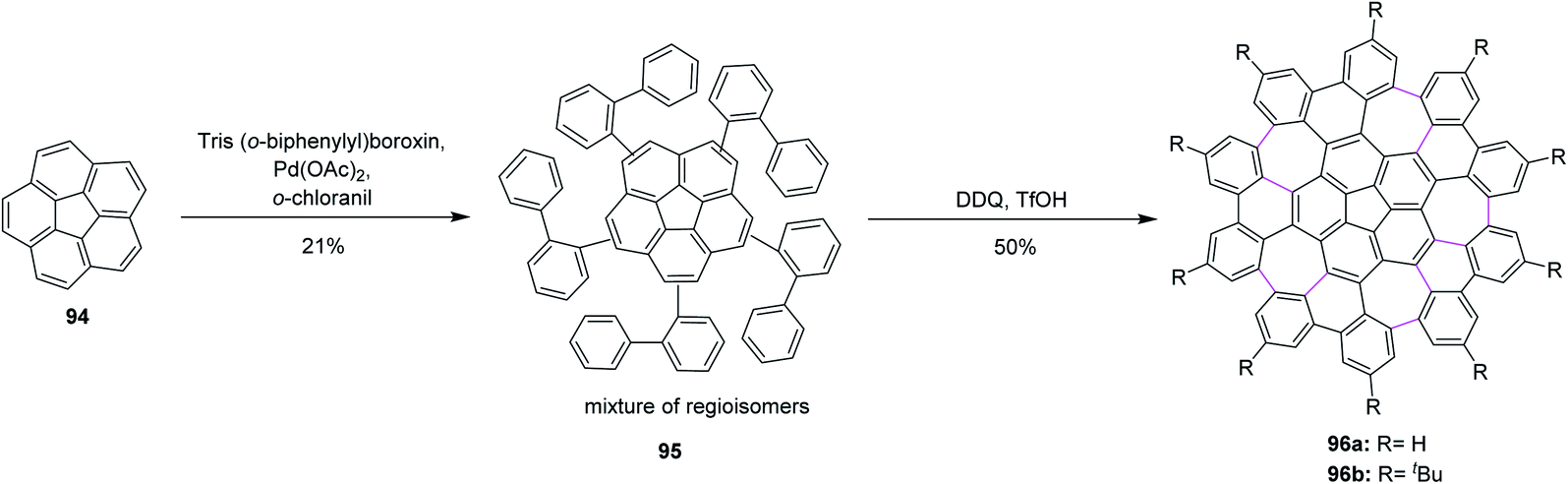 | ||
| Scheme 40 DDQ-induced synthesis of grossly-warped graphene molecules. | ||
Inspired by the previous outcomes, in 2014, Müllen and co-workers explained the synthesis of the biphenylene-based condensed PAH 98 merged with two 8-membered and one 4-membered rings by the cyclodehydrogenation of the precursor octaarylbiphenylene 97 using a mixture of DDQ and MeSO3H (Scheme 41). The cyclodehydrogenation did not proceed any further, possibly due to the increased steric hindrance and selectivity, exclusively producing nanographene 98 in 90% yield.73 Undoubtedly, this was also a breakthrough in showcasing the importance/value of DDQ as an oxidant in the field of nanographene synthesis. Moreover, the authors concluded that the isomeric graphene nanostructures were prepared to demonstrate a combination of four-, six-, and eight-membered rings in a single structure.
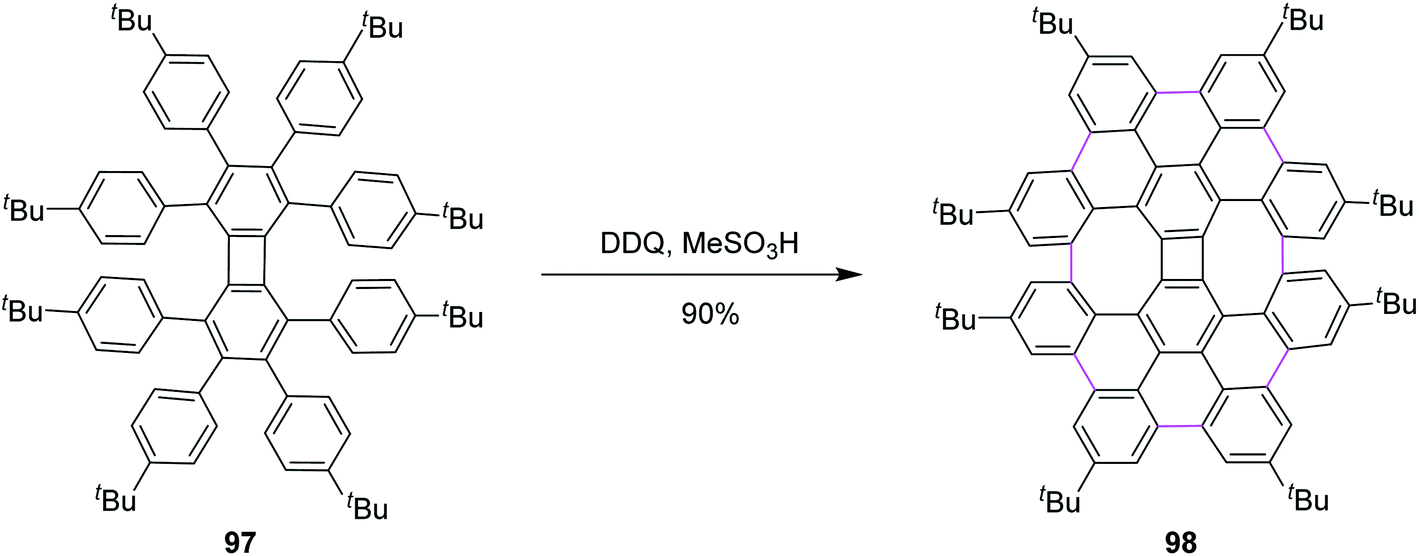 | ||
| Scheme 41 Synthesis of biphenylene-based PAH by DDQ/MeSO3H. | ||
Miao and co-workers (2015) synthesized two new saddle-shaped polycyclic arenes having two heptagons delivered from saddle-shaped diketone 99a,b (Scheme 42). The synthesis of the two novel saddle-shaped PAHs 101a,b and 102a,b with two entrenched heptagons was conducted by the oxidative cyclodehydrogenation of the precursors 100a,b and 103a,b using DDQ and CF3SO3H-initiated oxidation.74 These findings further strengthened the importance of the DDQ/H+ system in the insertion of a seven-membered ring in the nanographene structures, which could offer exciting electronic properties for applications in organic electronics.
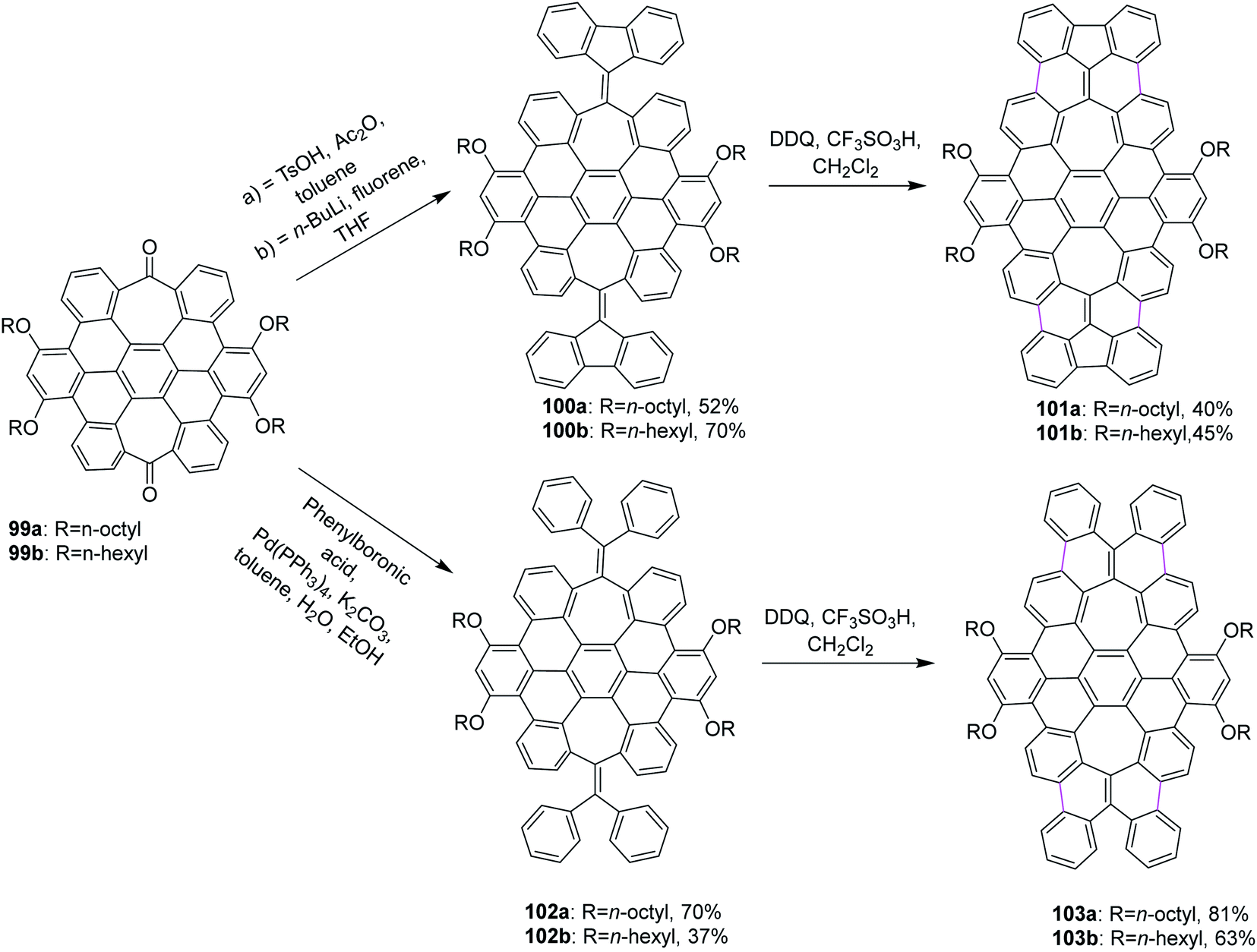 | ||
| Scheme 42 Graphene syntheses with an embedded 7-membered ring by cyclodehydrogenation. | ||
In 2015, Mastalerz and co-workers developed a route for the synthesis of triptycene derivative 105 based on the tris-ortho terphenylene 104 (Scheme 43). They initially implemented Suzuki–Miyaura coupling to construct the basic scaffold of terphenylene from hexabromotriptycene, followed by the Scholl reaction in the presence of DDQ and CH3SO3H in DCM at 0 °C. This route provided high yield as compared to zirconium and stannylene routes. When the classical zirconium route was used for the same type of Scholl cyclization of terphenylene, no intermolecular oxidative coupling was observed.75
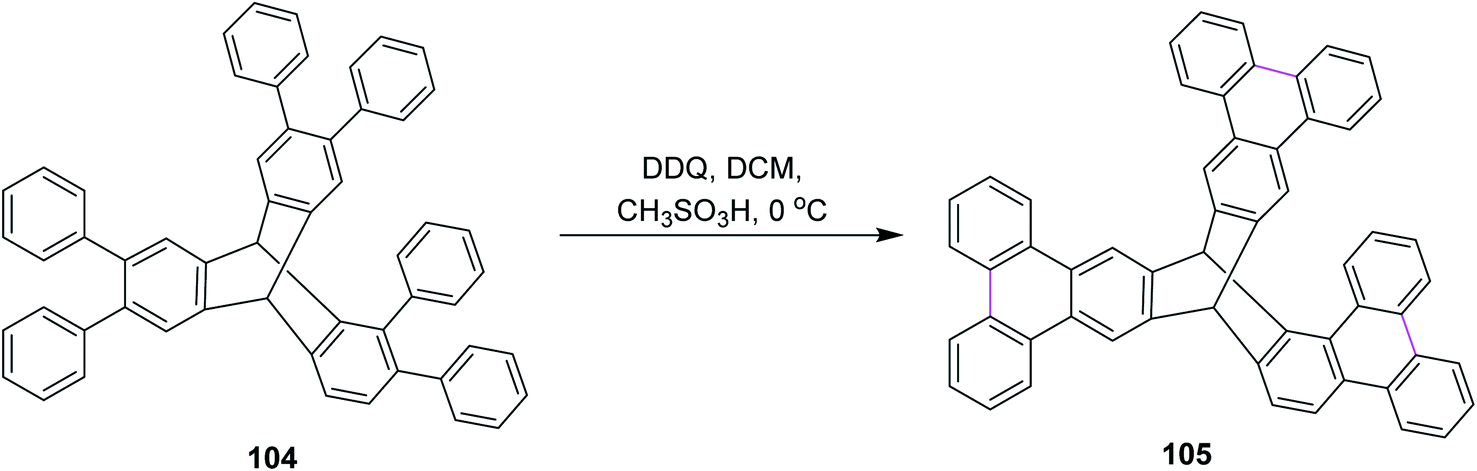 | ||
| Scheme 43 Synthesis of triptycenes by DDQ. | ||
In 2016, Müllen and co-workers developed a novel synthetic strategy for bowl-shaped PAH containing two pentagons such as structure 107, as shown in Scheme 44. They used the bottom-up strategy to construct C70 with two C34 subunits of fullerenes. Numerous reagents were utilized in this synthetic pathway due to its multistep process; however, the DDQ/CF3SO3H set-up was used in the fundamental step where pentagon rings were constructed in the presence of DCM by the Scholl reaction. DDQ provided highly selective 1,2-shifts of aryl groups in 106 during this synthetic strategy.76 However, the main attempts were focused on the synthesis of buckminsterfullerene C60. In contrast, the access to subunits of the C70 homologue remains challenging.
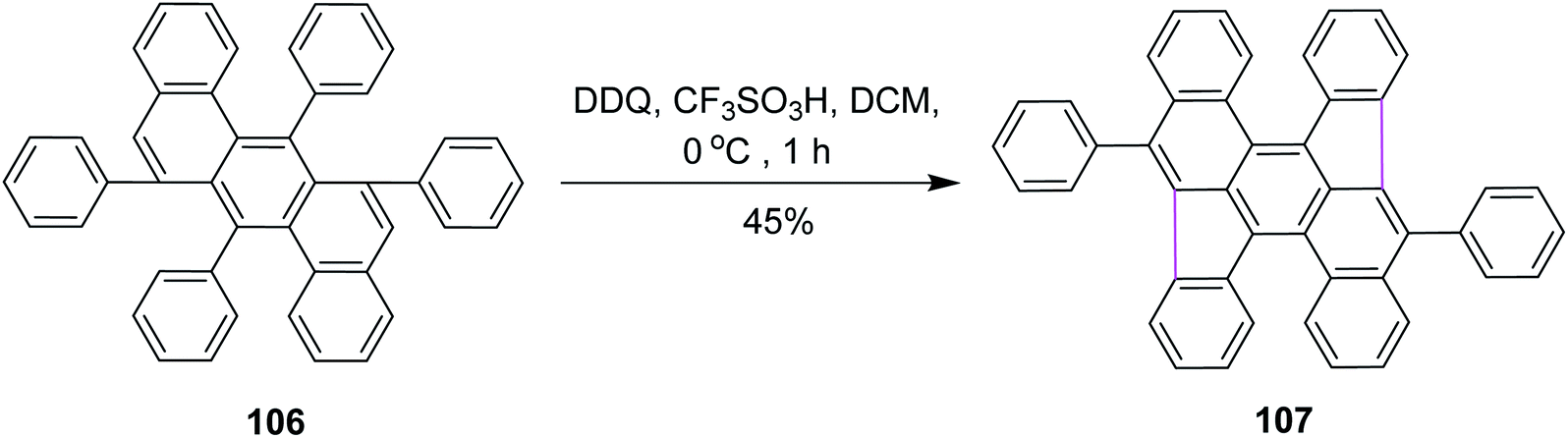 | ||
| Scheme 44 Synthesis of fullerene pentagons with DDQ. | ||
Kuck, Chow, and co-workers (2016) published, for the first time, the challenging approach for the attachment of three o-phenylene units 108a at the bay regions of the C3v-symmetrical hat-shaped polycyclic aromatic hydrocarbon bearing the tribenzotriquinacene (TBTQ) skeleton. The key step in their synthetic sequence includes three Scholl-type cycloheptatriene ring formation around the TBTQ core. Over the past years, they employed several oxidants to promote these oxidative cyclization reactions and were ultimately successful in fulfilling their goals, utilizing the DDQ/TfOH oxidant system. In this respect, they treated hydrocarbon 108a with stoichiometric amounts of DDQ and TfOH in DCM, as displayed in Scheme 45. These oxidative conditions produced intermediate product 108b with incomplete cyclodehydrogenation in good yields. Obtaining multiple functionalization around the peripheral arene positions of TBTQ is itself quite challenging. Besides, upon increasing the amount of DDQ to 3.0 equivalents, they observed the attachment of two 1,2-arylene units around the TBTQ core in the form of an unwanted product 109a (Scheme 45) produced in moderate yield. Nevertheless, the desired target compound 109b could be obtained in 37% yield using 5.0 equivalents of DDQ along with TfOH. Despite the relatively lower yield, TBTQ merged with three cycloheptatriene units represents a promising key intermediate for the construction of non-planar nanographene molecules bearing TBTQ as the central core.77 These intriguing results opened the door for future design and to prepare organic functional materials based on the TBTQ motif.
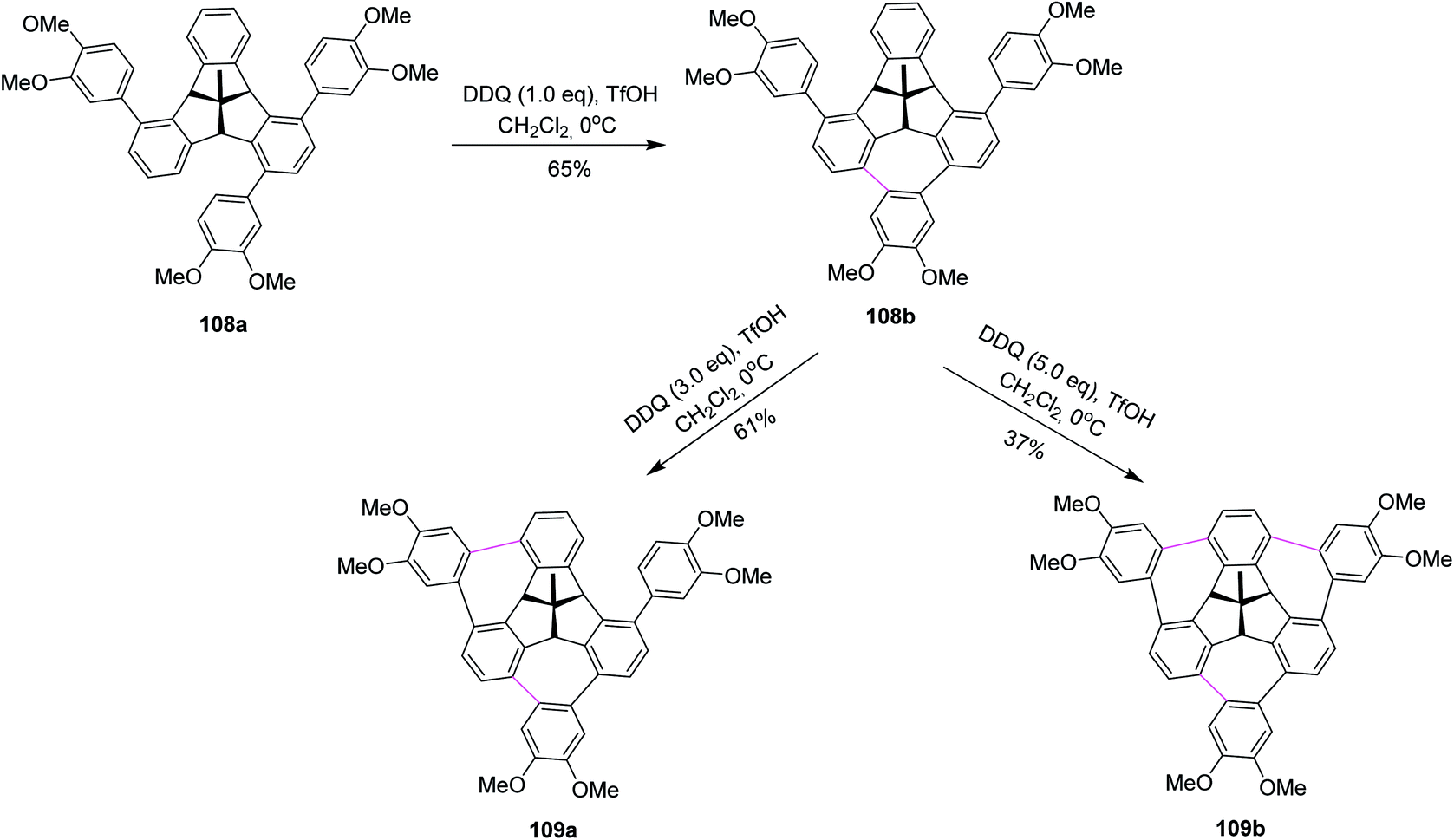 | ||
| Scheme 45 DDQ-promoted oxidative cycloheptatriene ring formation around the TBTQ core. | ||
In the following years, Kuck, Chow, and co-workers (2017) extended their research endeavors to apply the previously reported oxidative conditions (DDQ and TfOH) in the synthesis of cycloheptatriene-merged fenestrindane derivatives, as shown in Scheme 46. Promoted by DDQ/TfOH, the key step featured four Scholl-type cycloheptatriene formation steps, starting from the corresponding tetraarylfenestrindane derivatives 110 and 111 bearing electron-rich substituents. Consequently, the precursor hydrocarbons 110 and 111 were reacted with DDQ/TfOH in slight excess to produce a mixture of two-fold and four-fold cyclization reactions to afford the products 112a (52%) and 113 (20%), respectively, in moderate yields (Scheme 46). In other attempts, the products of neither single nor triple cyclization were observed. With the peripheral –OCH3 groups (114, 115, and 116) being available for further functionalization and C–C cross-coupling reactions, these molecules should provide access to various novel π-extended saddle-shaped nanographenes.78
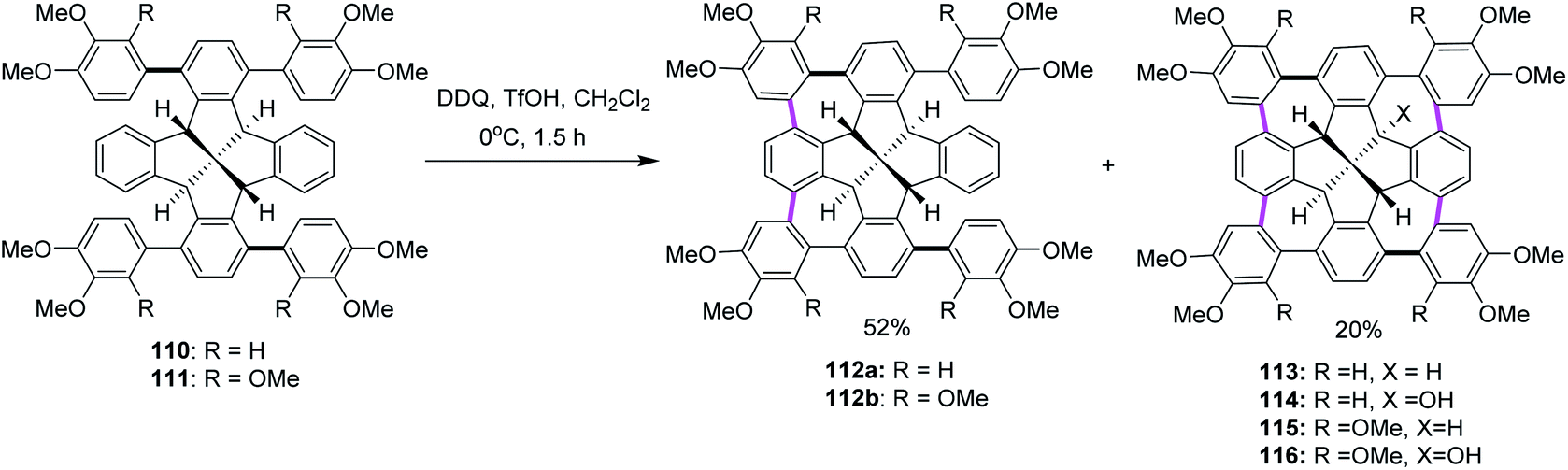 | ||
| Scheme 46 DDQ-mediated Scholl-type oxidative cycloheptatriene formation; a fourfold bridging of the fenestrindanes core with the electron-rich aryl units. | ||
In 2017, Tobe and co-workers proposed a procedure where a spiral-polycyclic hydrocarbon 117 was treated with DDQ/Sc(OTf)3 in DCM at different temperatures. An unexpected skeletal rearrangement was observed instead of cyclodehydrogenation. The eight-membered ring rearranged into a seven-membered ring that was further converted into a six-membered ring 119 (Scheme 47). They also used FeCl3 for the synthesis of 118. FeCl3 provided high yield as compared to DDQ.79 The reaction of 117 with DDQ and Sc(OTf)3 also gave 118, whereas at higher temperature, tetrabenzocoronene 119 was obtained via 2-fold isomerization. Compound 118 adopts a twisted geometry to avoid intramolecular steric repulsions. It exhibits weak aggregation behavior in solution via the stacking of the π-conjugated framework. Theoretical calculations suggested that the activation barrier for skeletal rearrangements in cationic intermediates is lower than that of intramolecular bond formation for oxidative dehydrogenation reactions.
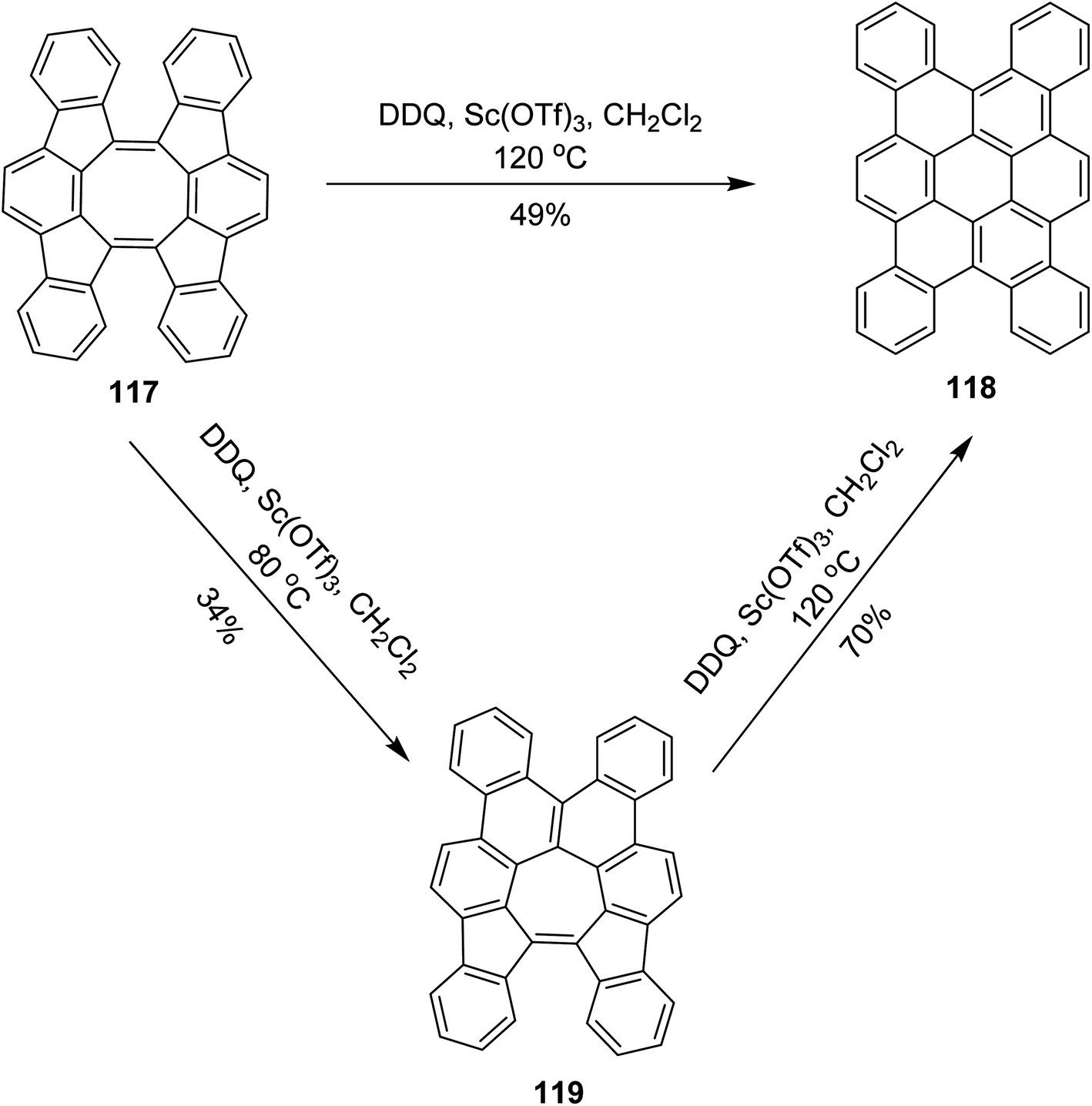 | ||
| Scheme 47 Skeletal rearrangement of PAH by DDQ. | ||
Narita and co-workers, in the same year, disclosed a new protocol for the synthesis of benzo-fused carbohelicene 121 by the regioselective cyclodehydrogenation of PAHs 120 (Scheme 48). DDQ, along with TfOH/CH2Cl2, was utilized in the Scholl cyclization step to construct π-extended C–C bond in carbohelicene. DDQ provided highly regioselective products with multi-helical structures and promoted the selective formation of sterically-hindered double helicene in good yield.80
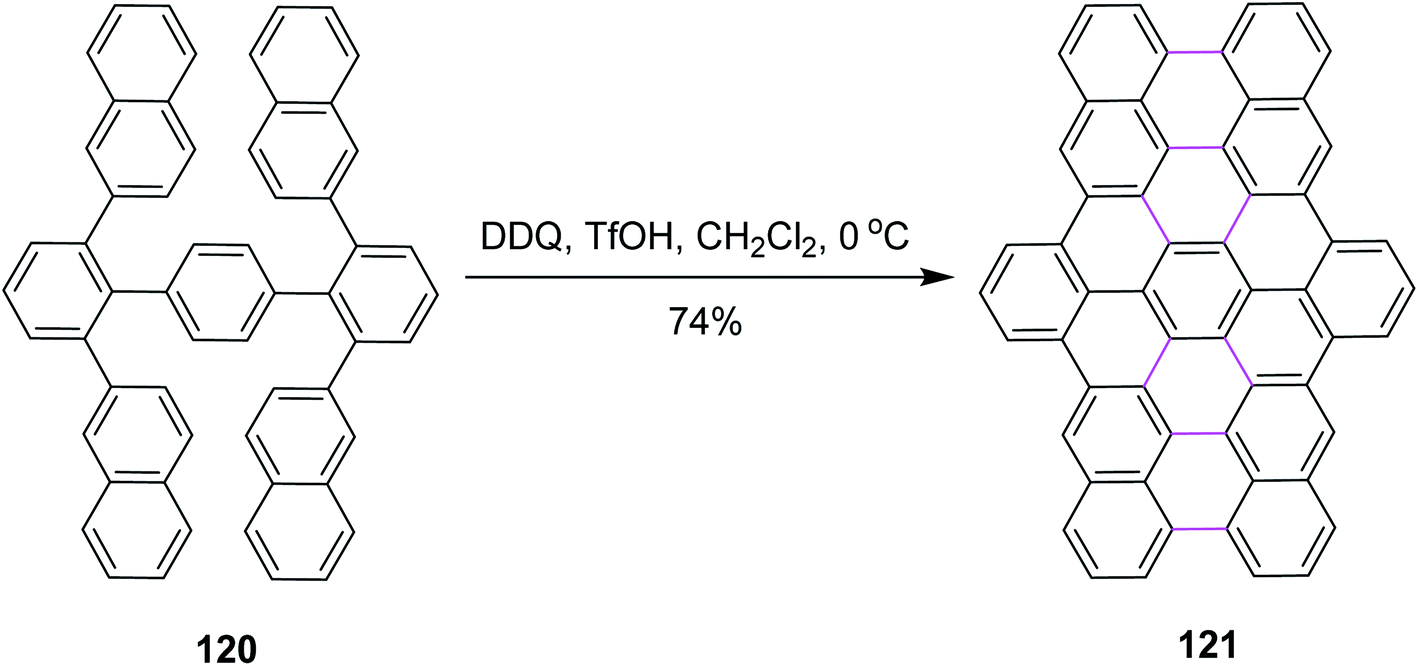 | ||
| Scheme 48 Synthesis of benzo-fused carbohelicene by DDQ. | ||
In 2018, Feng and co-workers developed a method for the development of zigzag-edged nanographenes. They performed oxidative cyclodehydrogenation of open-shell oligophenylenes and polyphenylenes such as 122 via the DDQ/CF3SO3H set-up in DCM to synthesize tetracenes. The reaction pathway was multistep and Scholl cyclization was the dynamic step to construct the π-extended arene rings in 123 (Scheme 49). DDQ here provided a good yield (70%) despite the high steric hindrance created by the iodine atoms. This approach offered access to the higher PAHs under mild conditions, yielding a novel electron-deficient tetracyano-circumanthracene with a full zigzag-edged structure.81
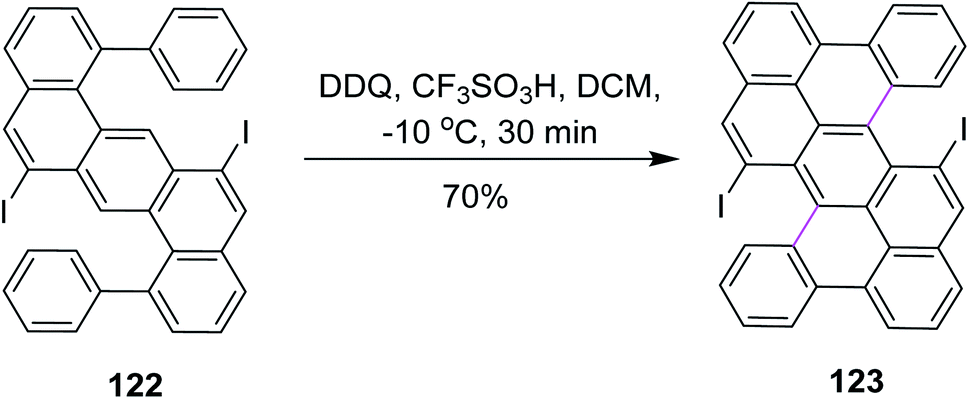 | ||
| Scheme 49 Synthesis of tetracene via the Scholl reaction. | ||
Recently, Kuck, Chow, and co-workers (2018) described the formation of porous and complex bowl-shaped TBTQ-based nanographenes (Scheme 49). Precursor 124 was synthesized over several steps from easily accessible starting materials82 and was treated with different equivalents of DDQ–TfOH to promote the Scholl-type macrocyclization to produce the porous nanographene molecules 125–127 in different yields (Scheme 50). However, the authors concluded that to achieve threefold oxidative cyclization, 4.5 equivalents of DDQ and TfOH mixture were needed. The further extension of this cyclic porous nanographene 127 by Scholl's macrocyclization using DDQ/TfOH is still underway by researchers to prepare several novel PAHs.82
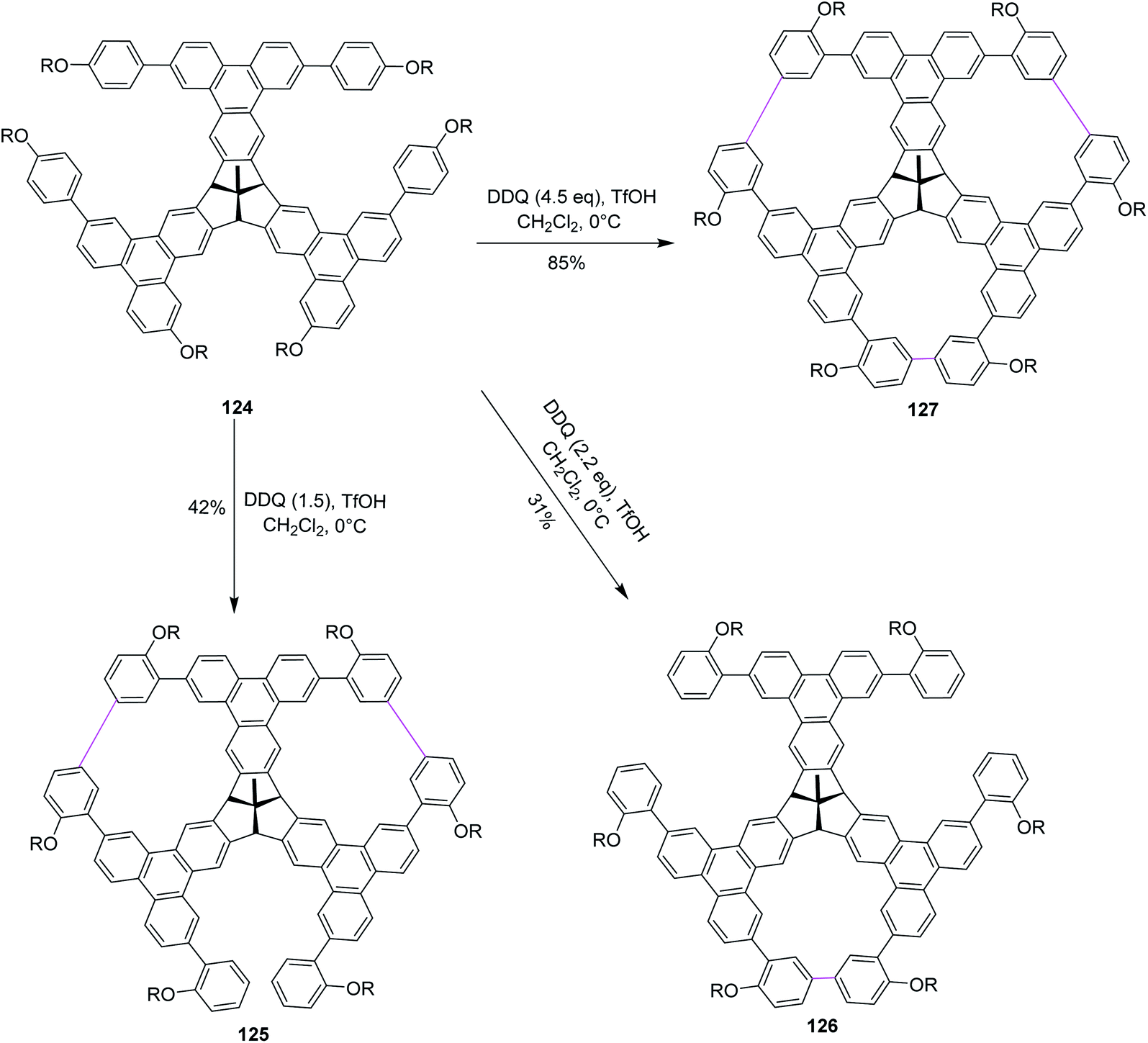 | ||
| Scheme 50 DDQ–TfOH-promoted Scholl macrocyclization for porous nanographene syntheses. | ||
In 2020, Chalifoux and co-workers introduced a novel methodology for the construction of contorted nanographenes. It was a novel method for the development of contorted nanographenes other than typical planar nanographenes by ruthenium-catalyzed benzo-annulation of alkynes, followed by Scholl cyclization (Scheme 51) using bottom-up synthesis. The DDQ/CH3SO3H set-up in DCM was used as the oxidant for the introduction of a new arene ring. Here, DDQ provided the double annulated product 129 by the oxidative coupling of isolated compound 128. They also utilized FeCl3 but DDQ provided the desired product selectively.83
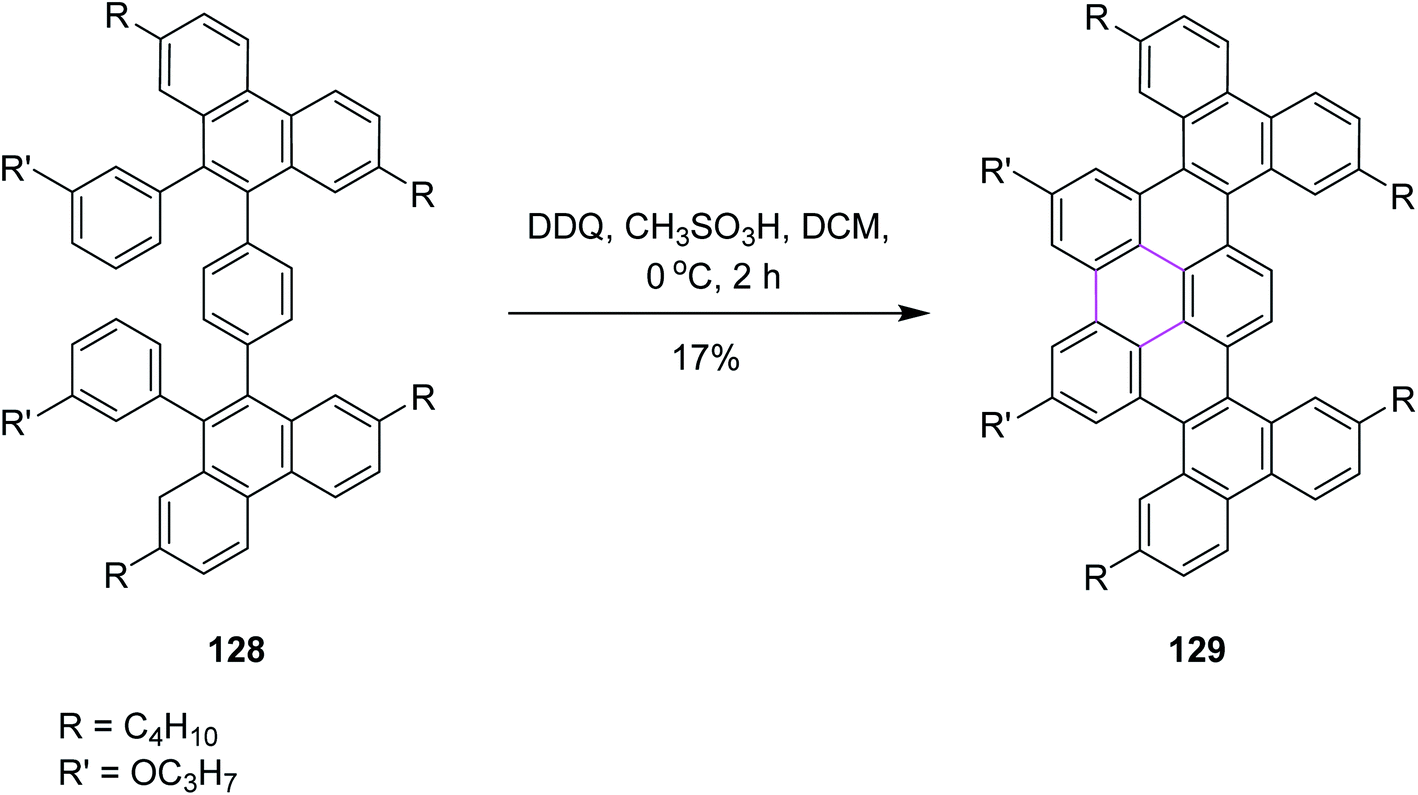 | ||
| Scheme 51 Synthesis of nanographene unit via the Scholl reaction. | ||
In the same year, Chi and co-workers developed a synthetic pathway for azulene-embedded nanographenes 131 by implementing the Scholl reaction. The naphthalene rings of 130 were rearranged into azulene rings via DDQ and triflic acid in DCM, as depicted in Scheme 52. Hence, the incorporation of a non-hexagonal ring into a nanographene framework can lead to new electronic properties. Initially, they tested the approach with FeCl3 in DCM but it only gave a complex mixture of products. However, DDQ was used preferentially to obtain the desired product regioselectively.84 This research represents the first experimental example of the thermodynamically unfavorable naphthalene–azulene rearrangement and may lead to new azulene-based molecular materials.
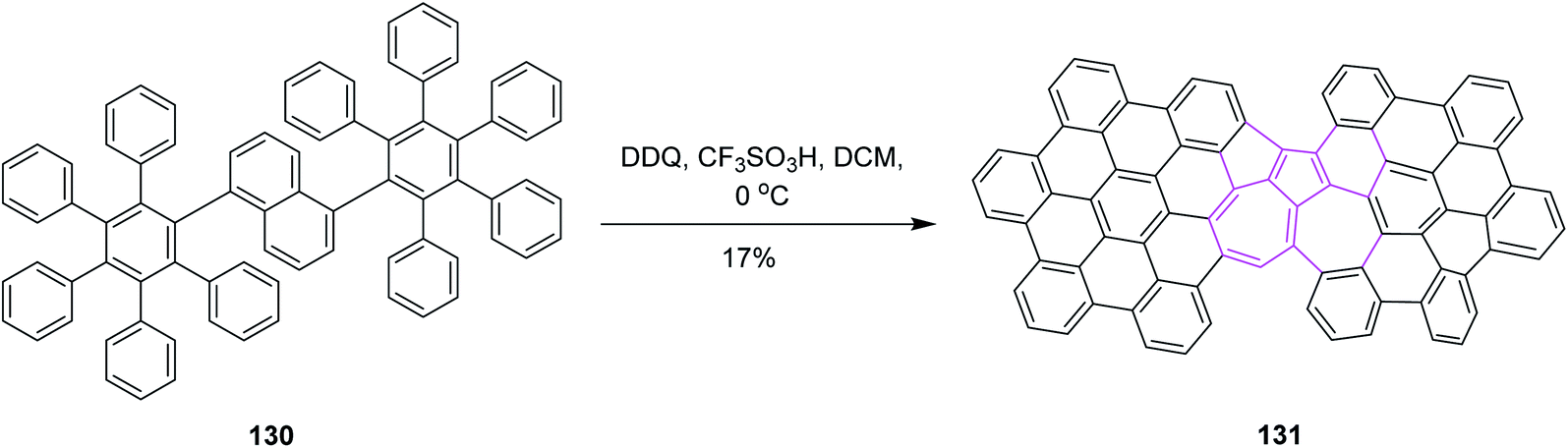 | ||
| Scheme 52 Synthesis of azulene nanographene via the Scholl reaction. | ||
Later on, Feng and co-workers (2020) prepared helical azulene-embedded nanographenes 133 from precursor 132. They added three helical units in each nanographene. This was also a non-hexagonal addition of the rings. The synthetic pathway consisted of multiple steps and Scholl cyclization occurred in the presence of DDQ/TfOH in DCM. Here, DDQ provided the highest yield of the azulene-embedded compound via the oxidative coupling of diiodo-7,7′-bibenzochrysene 132 (Scheme 53). However, there was also a drawback associated with this type of cyclization as a lot of unidentified by-products were also formed.85
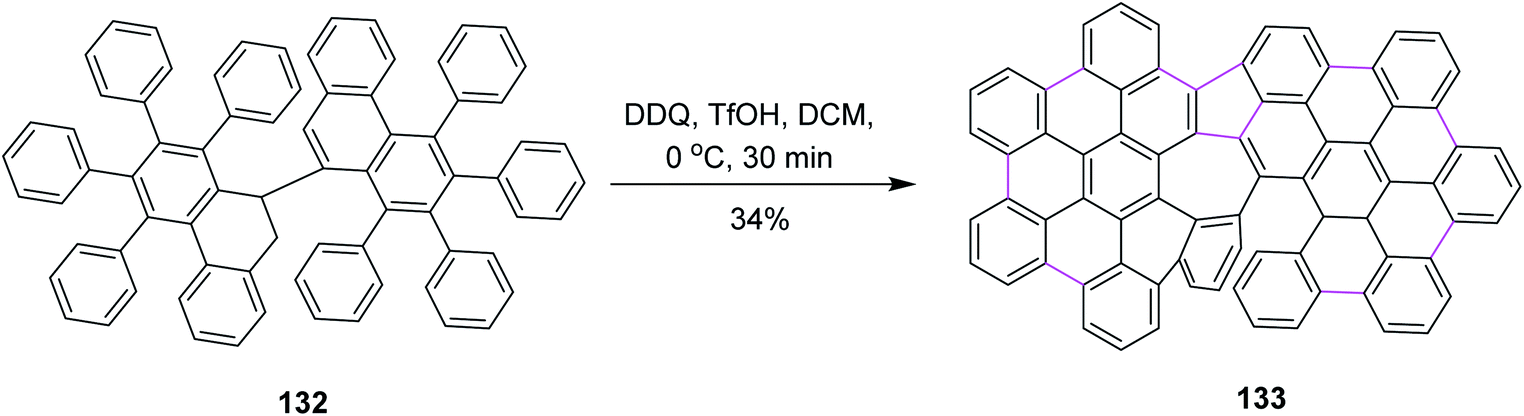 | ||
| Scheme 53 Synthesis of helical azulene-based nanographene via the Scholl reaction. | ||
In 2021, Wu and co-workers (2021) described various conditions under the Scholl reaction to prepare nanographenes containing hexagonal and octagonal rings. Their procedure was mainly focused on the extension of graphene such as PAHs. They used FeCl3 and DDQ in their reported procedure. The use of iron(III) chloride produced hexagonal rings while DDQ generated octagonal rings in compounds such as 135 via the oxidative cyclodehydrogenation of dibenzo-peri-hexacene 134 (Scheme 54). The DFT calculations suggested that the DDQ-mediated Scholl reaction occurred by the arenium ion mechanism, while the FeCl3-mediated reaction took place via the radical cation mechanism to build the hexagonal ring. Thus, DDQ was preferably used to obtain eight-membered ring-embedded nanographene 135.86
 | ||
| Scheme 54 Synthesis of eight-membered ring-embedded nanographene by the Scholl reaction. | ||
Recently, Miao and co-workers (2021) developed the zigzag nanobelts containing the wave-like arrangement of hexagonal aromatic rings. Hybrid cyclic arene oligomers 137 containing phenylene units were added to build nanographenes in the presence of the DDQ/TfOH set-up in DCM (Scheme 55). These hybrids and highly-strained nanobelts were part of the giant loop system of the polycyclic framework and comprise the advanced form of nanographenes. This area is still under development and offers ample room for future work. In the above study, the authors used both FeCl3 and DDQ simultaneously. The use of DDQ provided impure product, while the use of FeCl3 provided pure ones.87a The successful synthesis of these nanobelts demonstrates the power and efficiency of the Scholl reactions in synthesizing strained polycyclic aromatics. Previously, in 2019, Miao and co-workers reported the synthesis of carbon nanobelts from 2,5-di(benzyloxy)-1,4-benzoquinone in six steps by the π-expansion of the corresponding polyarylated carbon nanorings through the Scholl reaction.87b
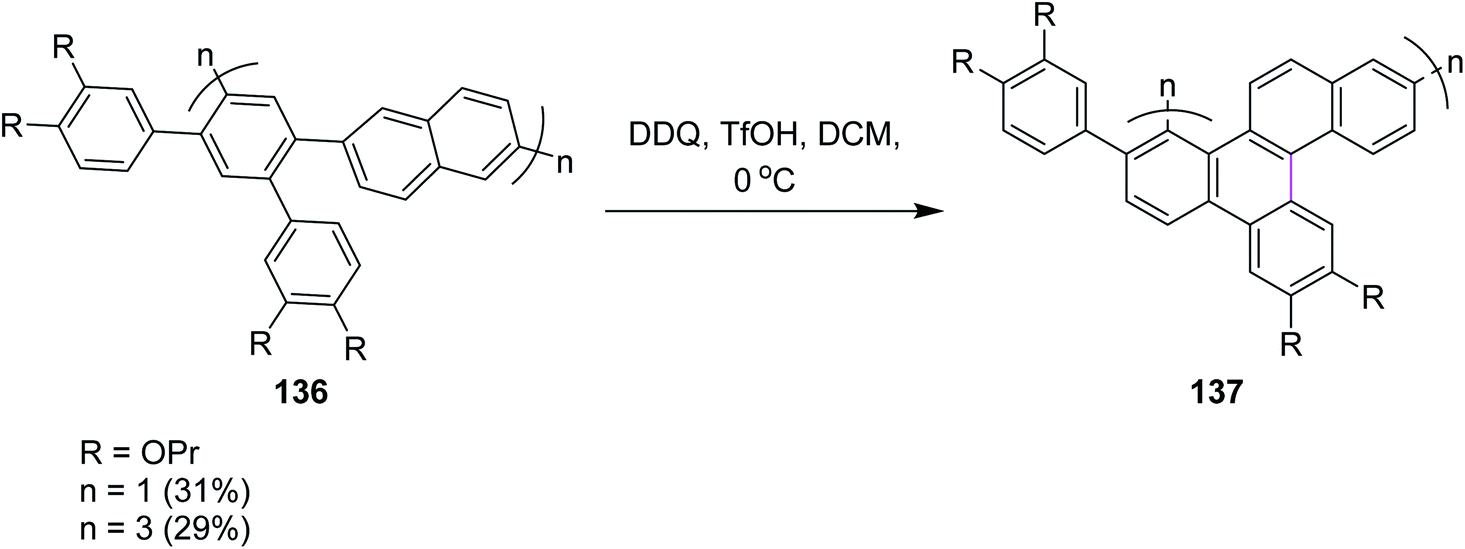 | ||
| Scheme 55 Synthesis of graphene nanobelts via the Scholl reaction. | ||
Recently, Kuck, Chow, and co-workers (2021) disclosed the synthesis of TBTQ-based wizard hat-shaped π-extended nanographene 138 from the precursor polycyclic aromatic compound 138 through the Scholl type dehydrocyclization (Scheme 56). The key step involves the Scholl-type cyclodehydrogenation of precursor 138 upon treatment with the DDQ–TfOH/CH2Cl2 oxidative system to afford the unique non-planar PAH 139 in 38% isolated yield. This DDQ-induced methodology has paved the way to design and synthesize electron-rich π-conjugated aromatics with the combination of five-, six-, and seven-membered carbocycles with interesting optical and electronic properties.88
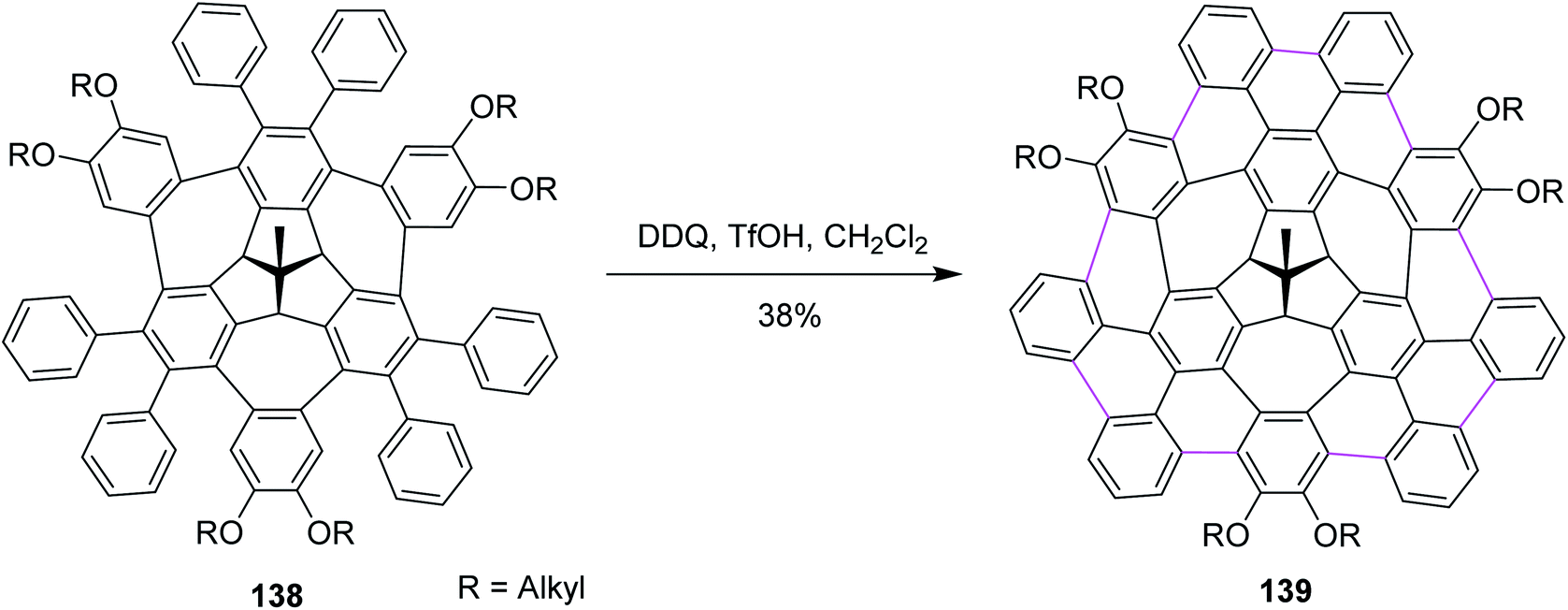 | ||
| Scheme 56 Synthesis of the π-extended nanographene from the three-fold bay-bridged TBTQ derivative by DDQ/TfOH. | ||
From the abovementioned discussion, it is concluded that DDQ has excellent oxidant oxidizing capacity as compared to FeCl3 and AlCl3. Its reduction potential value is +0.50 V.
5.4. Molybdenum pentachloride (MoCl5)-mediated Scholl reaction
Over the past two decades, molybdenum pentachloride (MoCl5) was explored as an eco-friendly and efficient catalyst to perform the Scholl reaction. In this context, Kumar and Manickam (1997) developed a synthetic methodology for the synthesis of substituted triphenylene 141 via the oxidative trimerization of substituted benzene 140. They constructed the C–C bonds using molybdenum pentachloride (MoCl5) in DCM, affording the Scholl reaction, as depicted in Scheme 57. These sophisticated FeCl3-based methods are useful for the preparation of low degree-substituted triphenylenes but are difficult to apply for large-scale production, while MoCl5 was used for large-scale production. The reaction occurs under very mild conditions with or without an acid catalyst and in a very short time at room temperature. Moreover, the workup for this reaction was quite simple and provided high yield.89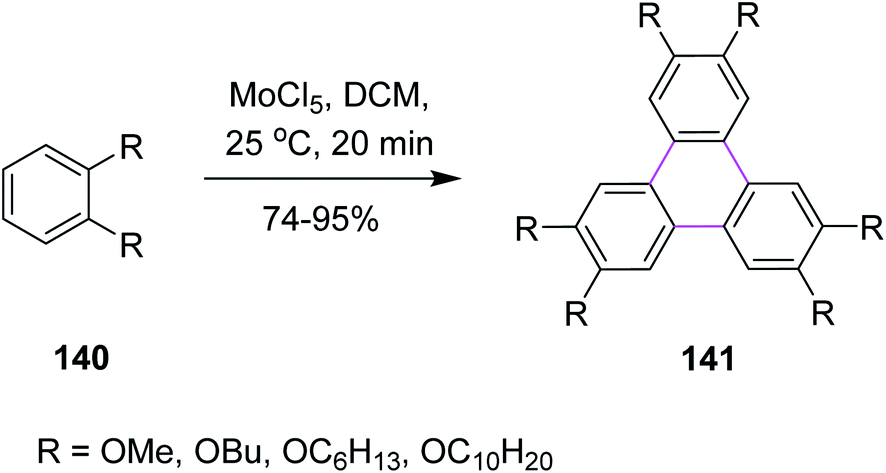 | ||
| Scheme 57 Synthesis of substituted triphenylenes using MoCl5. | ||
In 2004, Kramer and Waldvogel proposed a novel protocol to prepare heptagonal ring-containing hydrocarbon 143 via the Scholl reaction. They utilized a combination of MoCl5 and TiCl4 in DCM to construct the seven-membered ring between two benzene rings from the precursor 142, as shown in Scheme 58. The synthetic potential of MoCl5 in oxidative cyclization is clear as it enabled the construction of a seven-membered ring. This type of reagent system gave typical moderate yields. The potential of MoCl5 was enhanced significantly by Lewis acid additives such as TiCl4. This reagent system provided comparatively high yields and proved to be efficient for this type of synthesis.90
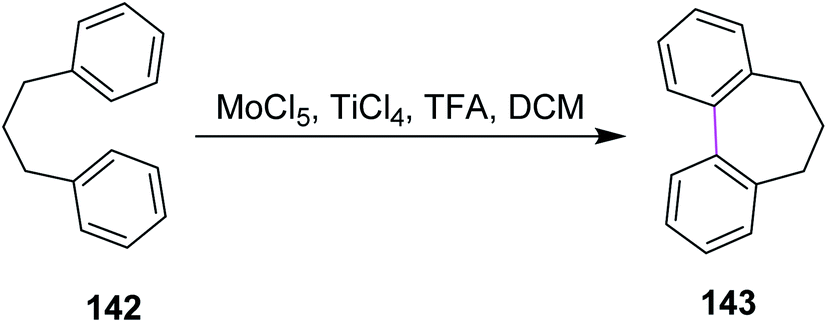 | ||
| Scheme 58 Construction of a heptagonal ring using MoCl5. | ||
In 2009, Waldvogel and co-workers devised a procedure for the preparation of thianthrene 145a and 145b by the oxidative coupling of the arylsulfides 144 with molybdenum pentachloride-induced Scholl reaction (Scheme 59). In such types of reactions, a strong oxidizing agent was required to promote the insertion of the sulfur–arene bond. Therefore, MoCl5 was used for this synthetic pathway, which features one-electron oxidation and successfully accomplished Scholl cyclization in the presence of TiCl4 (Lewis acidic additive).91 Hence, the oxidative treatment of 1,2-diaryldisulfides using MoCl5 or mixtures thereof with TiCl4 gives quick access to thianthrenes in moderate to excellent yields. Due to the cationic exchange of the aryl moiety, the cross-coupling of mixed disulfides does not proceed cleanly.
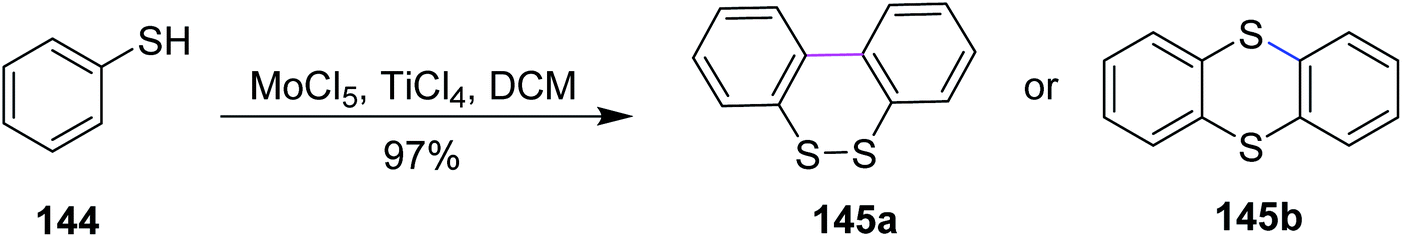 | ||
| Scheme 59 Synthesis of thianthrene with MoCl5. | ||
In 2011, Waldvogel and co-workers followed the same synthetic pathway for the synthesis of product 147. They employed MoCl5-mediated oxidative coupling to construct the C–C bond in 146 by the Scholl reaction, as described in Scheme 60. Due to the presence of multiple functional groups, MoCl5 was used as a strong oxidizing agent and was shown to tolerate these functional groups to achieve the particular substitution pattern on the aryl groups. The authors successfully produced selective heptagonal rings bearing the carboxymethyl moiety without oxidizing any other functional group in good yield (72%).92 Notably, the insertion of the carboxymethyl moiety on the seven-membered ring can only be achieved by the MoCl5/TiCl4 mixture.
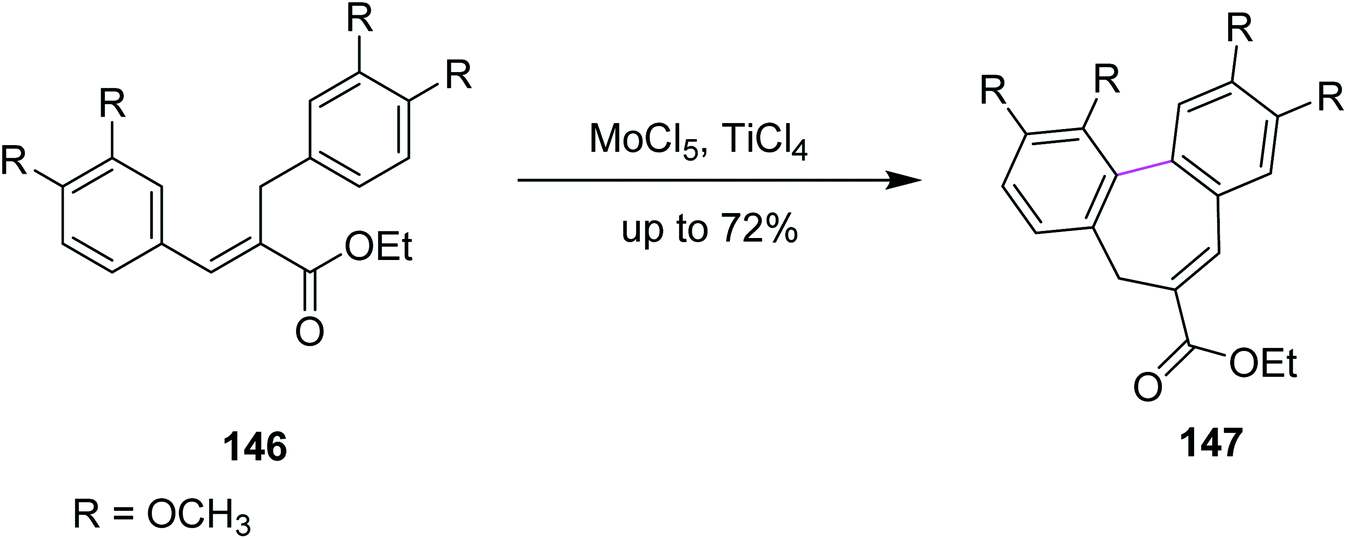 | ||
| Scheme 60 Synthesis of metasequirin via MoCl5. | ||
In 2013, Ishikawa and co-workers presented a direct bio-inspired dimerization reaction, in which the amine-free tryptophan derivatives 148 were used in the presence of an aqueous acidic medium to give dimeric compounds having different symmetries. Different oxidants were utilized where MoCl5 showed quick decomposition when mixed with simple Nb-methyltryptamine in aq. HCl solution and gave two symmetrical compounds, i.e., chimonanthine 149a, a naturally occurring compound, naseseazine 149b, and one unsymmetrical dimeric compound 149c (Scheme 61).93
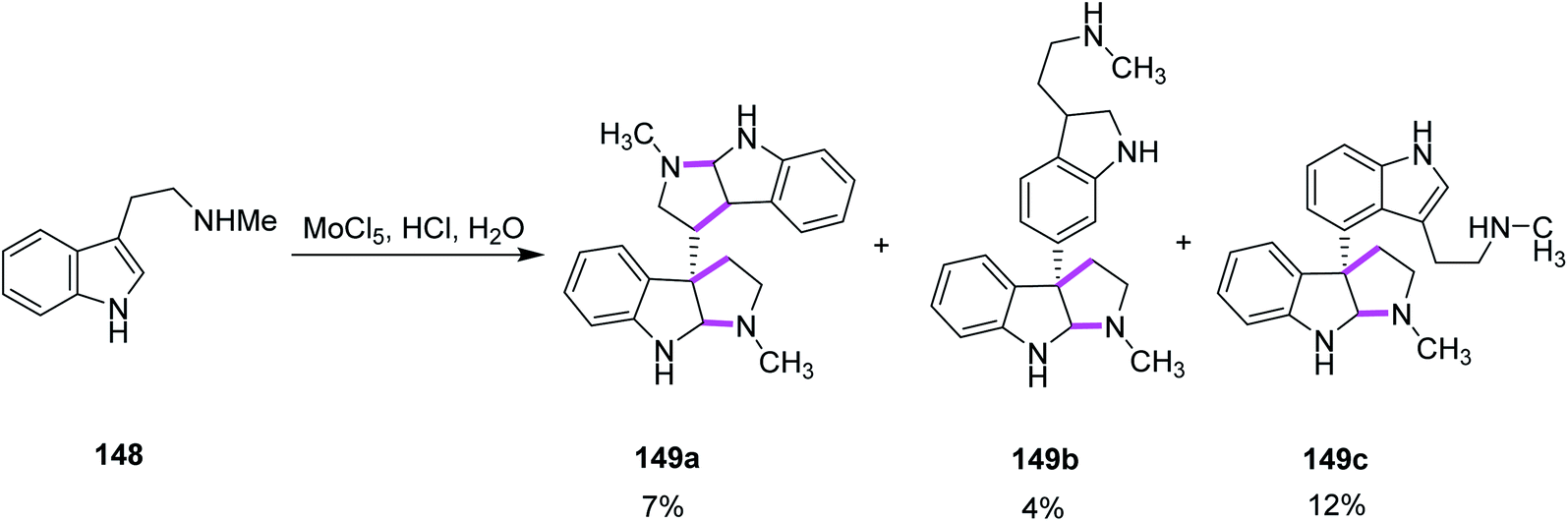 | ||
| Scheme 61 Screening of the MoCl5 oxidant for the oxidative dimerization reaction. | ||
In 2015, Sperry and Boyd proposed a mechanism for the biomimetic synthesis of dendridine 151 involving the intermolecular Scholl-type oxidation reaction of 7-isopropoxytryptamine with MoCl5, following the desired para–para coupling. Various oxidants were used initially for this synthetic route but none except (BuO)2 and MoCl5 provided the dimeric product. The 7-isopropoxy substituent 150, when treated with MoCl5 (generating a radical cation to form the C–C bond) and TiCl4 (a Lewis acid), produced 4,4′-bistryptamine 151 in an impressive ∼32% yield while some non-symmetrical compounds appeared as the minor product (Scheme 62). Therefore, MoCl5 was preferred over other reagents.94
 | ||
| Scheme 62 Biomimetic synthesis of dendridine. | ||
In 2017, Itami and co-workers worked on different parameters to form novel π-extended double [6]helicene and a dithia[6]helicene having unique 2D structure; here again, the Scholl reaction was carried out with MoCl5. For this purpose, naphthalene derivative 152 was treated with 20 equiv. of MoCl5 in DCM, resulting in the formation of tetrachlorinated π-extended PAH 153 (26% yield), where reduced amounts of the oxidant did not give any fruitful results (Scheme 63). MoCl5 resulted in the production of twisted isomers in this synthetic pathway.95
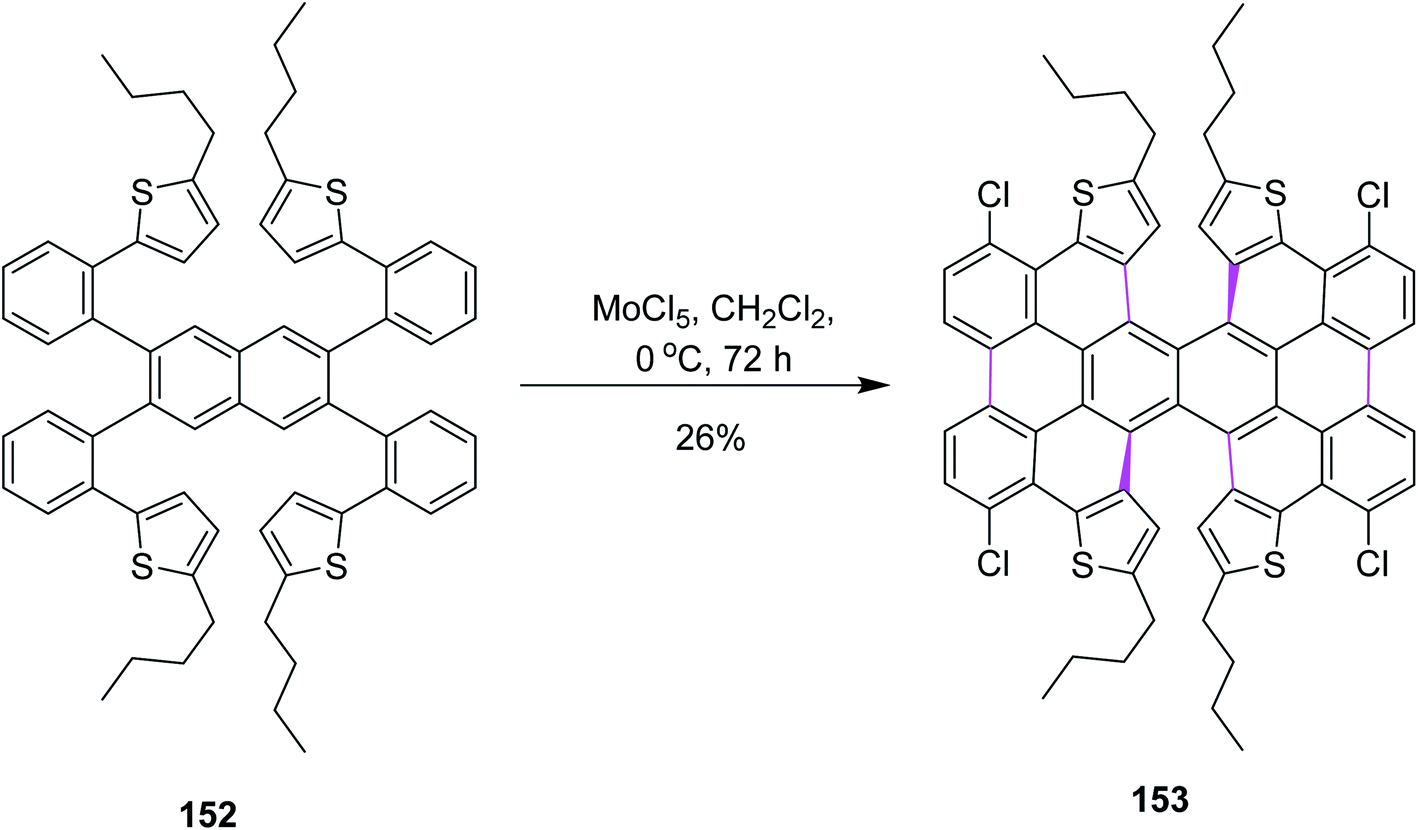 | ||
| Scheme 63 Formation of π-extended dithia[6]helicene. | ||
In 2019, Waldvogel and co-workers studied some intramolecular cyclization reactions by comparing them with conventional reagent-mediated coupling reactions. They used olefin 154 as a substrate as it produces five- and six-membered ring-containing compounds, as illustrated in Scheme 64. Surprisingly, different results were obtained under different conditions as under the anodic conditions, the only observed product was a six-membered ring 155a formed in 64% yield, while the MoCl5-mediated reaction formed product 155b containing both rings in 87% yield. This method proved to be important because electrochemical intramolecular coupling with high yield was observed with MoCl5 reagent.96
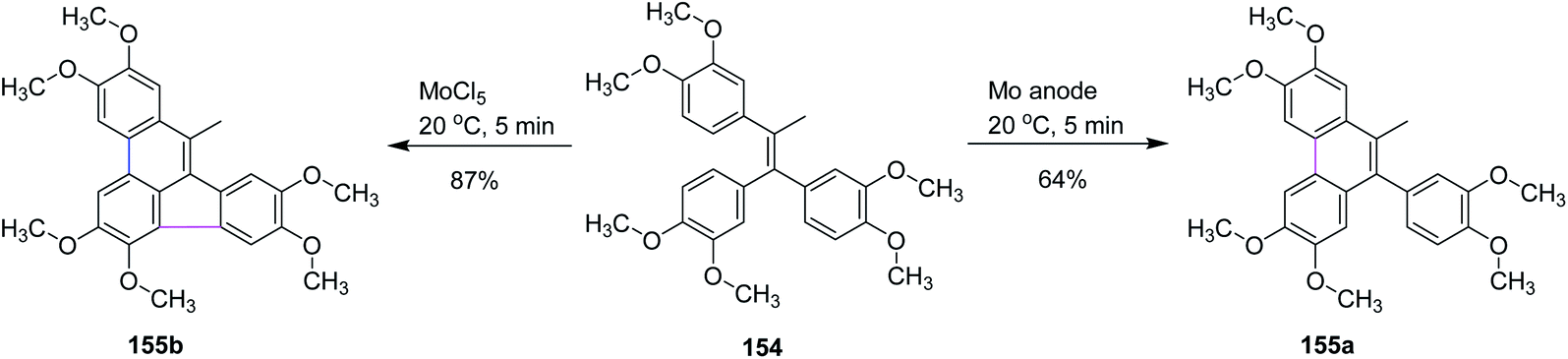 | ||
| Scheme 64 Oxidative coupling of olefin. | ||
5.5. Phenyliodinebis(trifluoroacetate) (PIFA)-mediated Scholl reaction
In 1998, Kita and co-workers employed the oxidative coupling of arene derivatives 156 by the Scholl reaction using a unique oxidation agent such as PIFA. This hypervalent iodine reagent PIFA induced the intramolecular oxidative coupling reaction of the biaryls to generate seven-membered rings in biphenyls 157 (Scheme 65). Notably, the previously used reagents comprised of heavy and toxic metals, which did not give satisfactory products. Therefore, PIFA was proved to be superior to metals for performing the Scholl reaction.97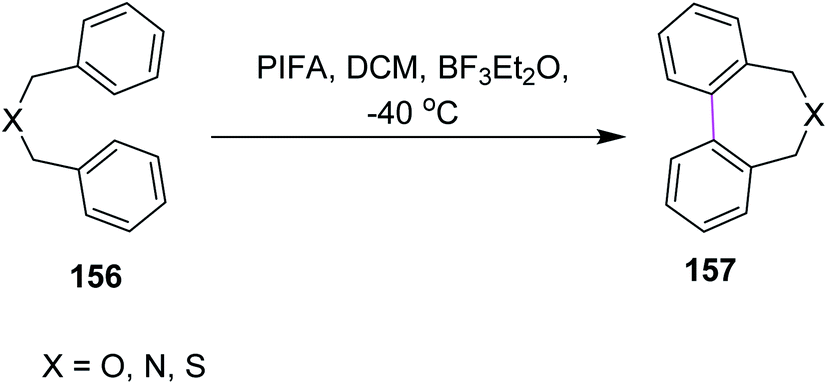 | ||
| Scheme 65 Oxidative coupling of benzyl ethers with PIFA. | ||
Kita and co-workers (2003) followed the same protocol of PIFA oxidative coupling for the synthesis of the bithiophenes/dithienyl derivative 159. The substituted thiophenes 158 were subjected to the PIFA/BF3·Et2O complex to generate the C–C bond between the two aryl units (Scheme 66). The reaction featured the selective and direct oxidative coupling reaction of alkyl thiophene derivatives without any activation of the thiophene monomers using the PIFA–BF3·Et2O reagent system.98
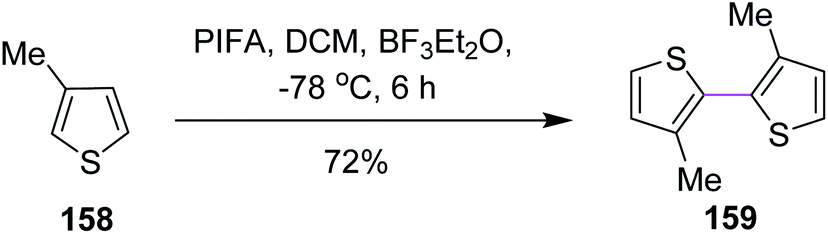 | ||
| Scheme 66 Oxidative coupling of bithiophenes with PIFA. | ||
In 2006, Kita and co-workers synthesized bipyrroles by the oxidative coupling of substituted pyrroles 160. They utilized PIFA/TMSBr in DCM to construct the C–C bond of bipyrroles 161 under the Scholl reaction (Scheme 67). Palladium salts were used previously for the oxidative coupling of pyrroles. However, Pd salts produced many unwanted by-products during this reaction. Hence, the reagent system was shifted toward the direct coupling strategy using PIFA.99
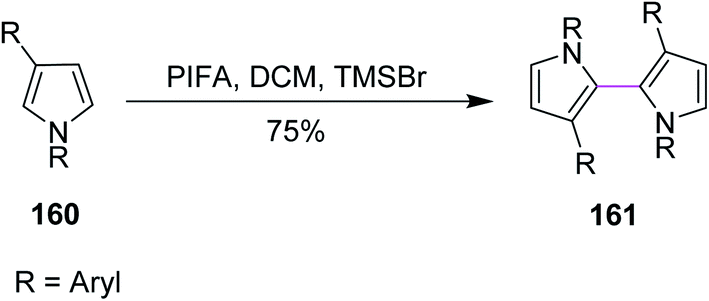 | ||
| Scheme 67 Oxidative coupling of bipyrroles. | ||
In 2009, Kita and co-workers used the same earlier approach for bithiophenes and bipyrroles synthesis using the PIFA/BF3·Et2O complex for the Scholl reaction as an extension of their previous work. The group synthesized compound 163 from pyrrole 162 (Scheme 68). The authors favored PIFA over other reagents due to its high reactivity and the possibility to recycle iodine. In addition, PIFA is a valuable reagent for the oxidative coupling of heterobiaryls.100
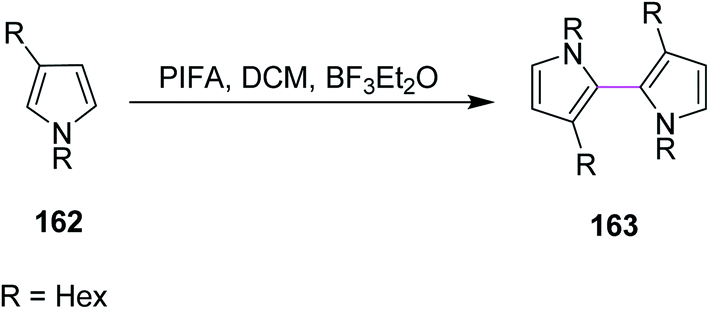 | ||
| Scheme 68 Oxidative coupling of bipyrroles. | ||
In 2013, Shafir and co-workers performed direct dehydrogenative coupling reaction under Kita's conditions using hypervalent PIFA–BF3. The oxidative arylation of simple ternaphthalene 164 Nap3 with pentamethylbenzene (path I) and triethylbenzene (path II) was achieved using the PIFA/BF3·Et2O reagent system and produced excellent yield of the double arylation products 165a and 165b, respectively; however, such results were not observed for bulkier arenes as they lowered the reaction efficiency (Scheme 69).101
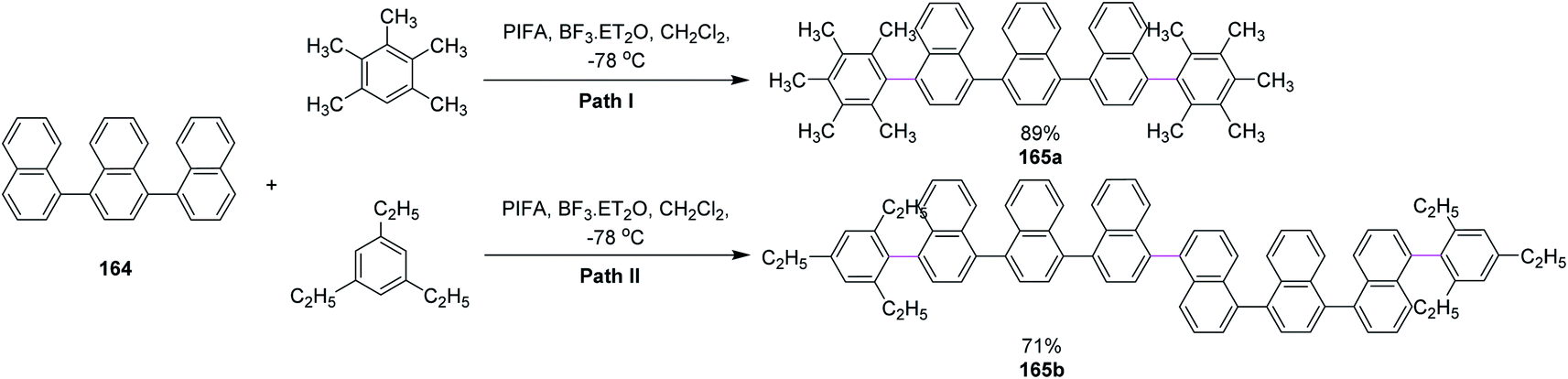 | ||
| Scheme 69 Oxidative arylation of ternaphthyls. | ||
In 2014, Pelkey and co-workers synthesized dibenzo[e,g]isoindol-1-ones following the Scholl-type cyclodehydrogenation pathway with the PIFA reagent. They investigated the effect of different methoxy groups, which were substituted on 3,4-diaryl-3-pyrrolin-2-one substrates. Thus, substrates bearing one 3,4-dimethoxyphenyl group afforded 60–90% product yield while those bearing two 3,4-dimethoxyphenyl groups as in 166 gave 167 in 96% yield and substrates without such a moiety did not give desired product in yields higher than 50% (Scheme 70). The PIFA-mediated oxidative cyclization offered both symmetrical and unsymmetrical products in good yield and the reagent system was favored due to its facile and mild oxidative transformations.102
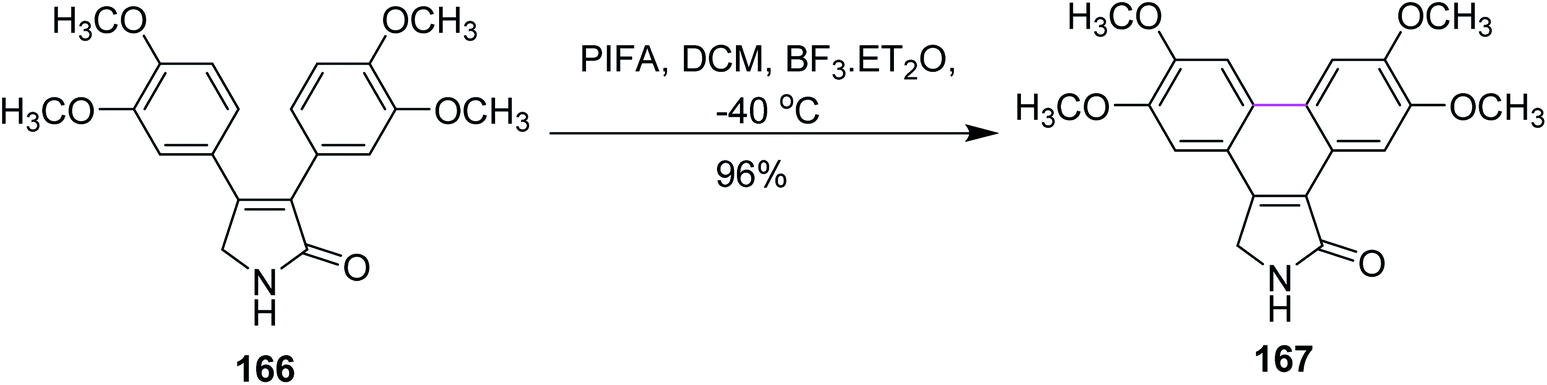 | ||
| Scheme 70 Oxidative cyclization using PIFA–BF3·ET2O. | ||
In 2016, Breder and co-workers established a mechanism for the synthesis of substituted phenanthrene analogs by selective oxidative intramolecular arene–alkene coupling (a Scholl-type reaction). They used different iodine(III)-mediated reagents such as PIFA, (diacetoxyiodo)benzene (PIDA), and N-chlorosuccinimide (NCS) to achieve this goal. While the addition of PIDA into 2,3,4,5-tetrahydroterphenyl derivative 168 did not setup the required reaction, PIFA (1 equiv.) at 10 °C produced product 169 in 80% yield in the presence of CH3NO2 (Scheme 71). PIFA, in this case, offered more consistent chemical conversion and isolated products in high yields.103
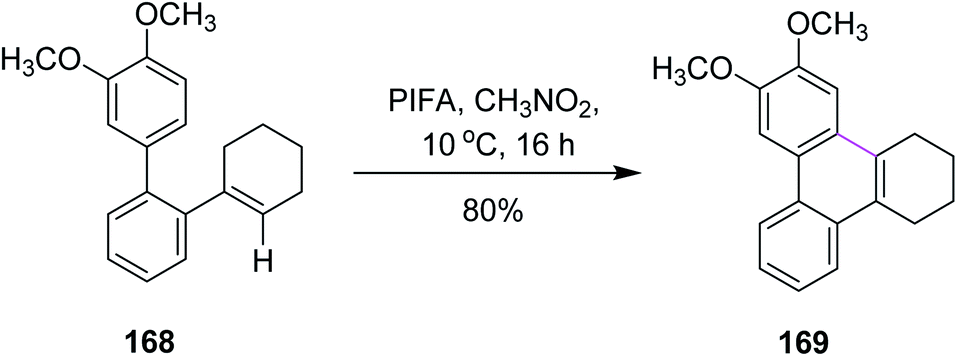 | ||
| Scheme 71 A controlled oxidative intramolecular arene–alkene coupling reaction. | ||
In the same year, Pelkey and co-workers prepared benzocarbazoles and an indolocarbazole using 3-aryltetramic acid as the substrate and the BF3–PIFA reagent system. The respective fused carbazole 171 (83%) was obtained by the Scholl-type reaction by reacting 4-indolylpyrrolone 170 in the presence of PIFA and BF3·Et2O (Scheme 72). A similar method was applied to the synthesis of indolocarbazole 173, which was obtained in 69% yield through the oxidative cyclization of 172 (Scheme 73).104
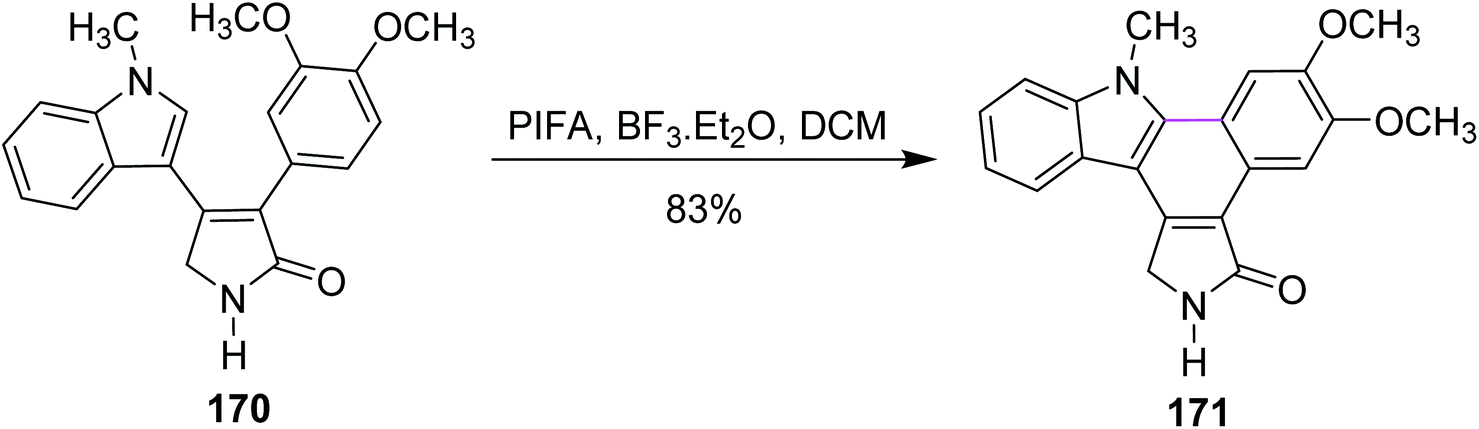 | ||
| Scheme 72 An example of benzocarbazole synthesis. | ||
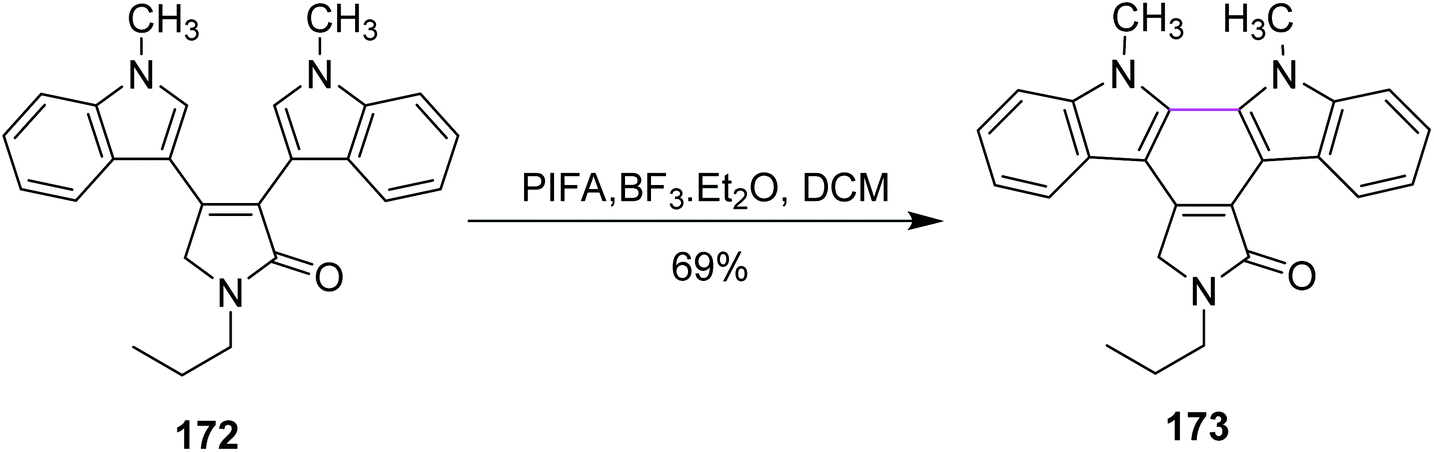 | ||
| Scheme 73 Indolcarbazole synthesis. | ||
In 2017, Li and co-workers devised a novel strategy to produce highly conjugated acenes, an important material to be used in electronics and optical devices, through two pathways, namely, Kita-coupling and Scholl type oxidative cyclodehydrogenation. Firstly, the precursors for acenes 175a and 175b were formed using ter-174a and quaternaphthyls 174b and mesitylene under mild oxidation conditions (PIFA/BF3·ET2O in CH2Cl2). Then, the prepared precursors 175a and 175b were subjected to reaction with FeCl3 (oxidant and Lewis acid) to form promising products 176a and 176b in good yields (Scheme 74).105
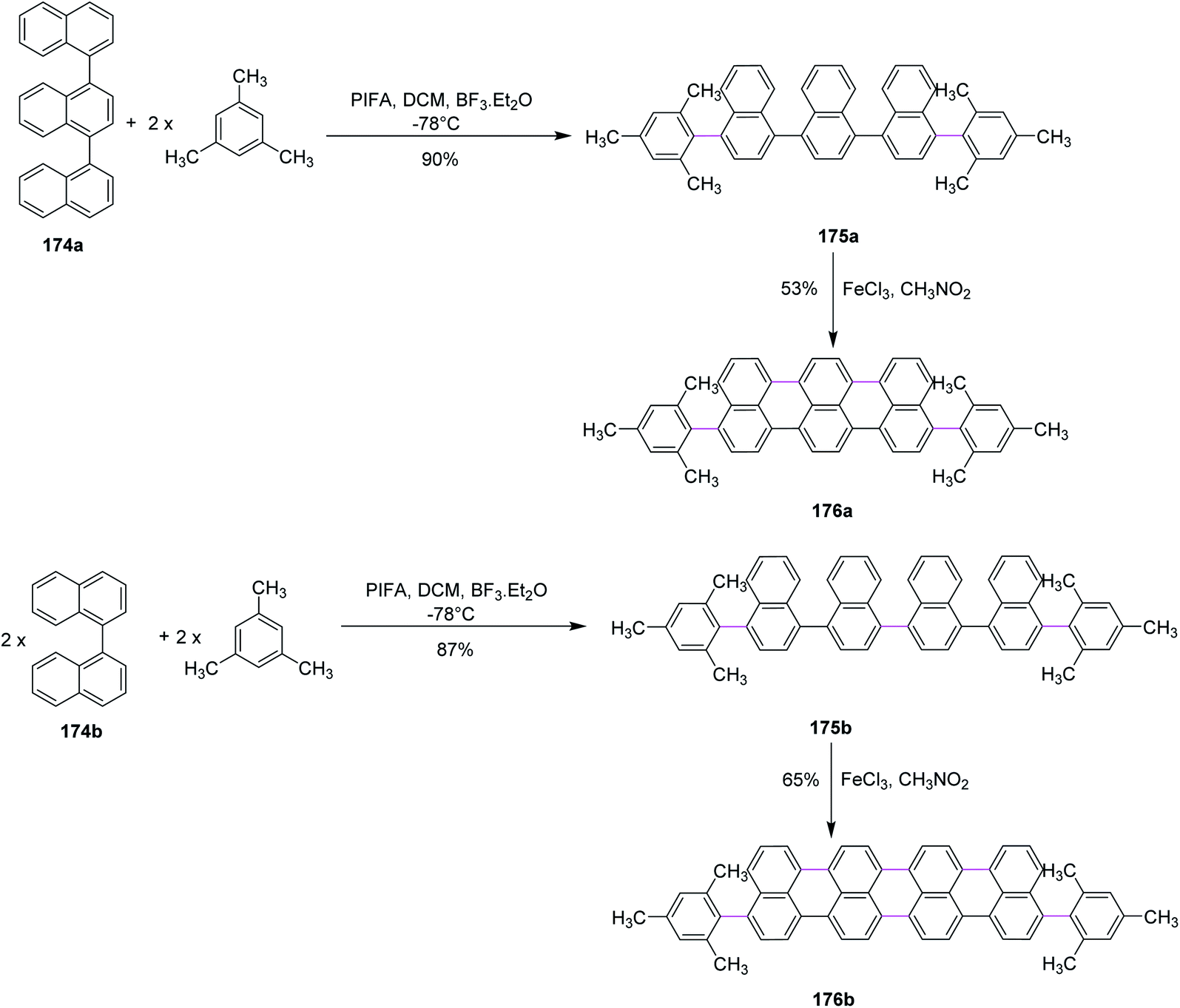 | ||
| Scheme 74 Synthesis of highly conjugated acenes. | ||
5.6. Palladium- and chloranil-mediated Scholl reaction
In 2015, Guan and co-workers introduced new conditions comprising of a mixture of palladium (Pd) and chloranil to carry out the intermolecular Scholl reaction. They reported a variety of para-substituted biaryl units 178 synthesized from the coupling of tertiary benzamides 177 with arenes through palladium-catalyzed oxidative coupling reaction using dipotassium peroxodisulfate as the oxidant (Scheme 75). This was a potentially favored approach toward the selective arylation of mono-substituted arenes with high yield due to easily accessible reaction conditions. Due to the mild conditions and the easy availability of substrates and oxidant, this method could potentially provide a practical approach for the synthesis of p-substituted biaryl compounds.106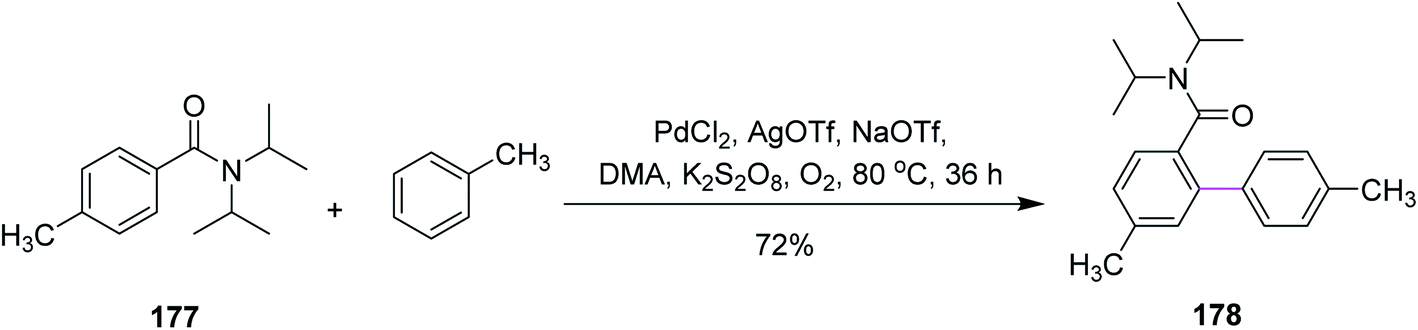 | ||
| Scheme 75 Oxidative coupling of a tertiary benzamide with toluene. | ||
In 2017, Itami and co-workers proposed a synthetic pathway for the annulative π-extension (APEX) of alkyne 179a via Pd/o-chloranil catalysis. The regioselective APEX of PAHs could provide structurally uniform nanographenes such as 180b by the Scholl reaction (Scheme 76). CuCl2 was initially used for this purpose but it proved to be ineffectual. On the other hand, Pd promoted this reaction more efficiently. This reagent system expanded the APEX reaction considerably. In addition, the double APEX reaction of alkynes was also established as an efficient route to obtain sterically-hindered diphenanthrylenes.107
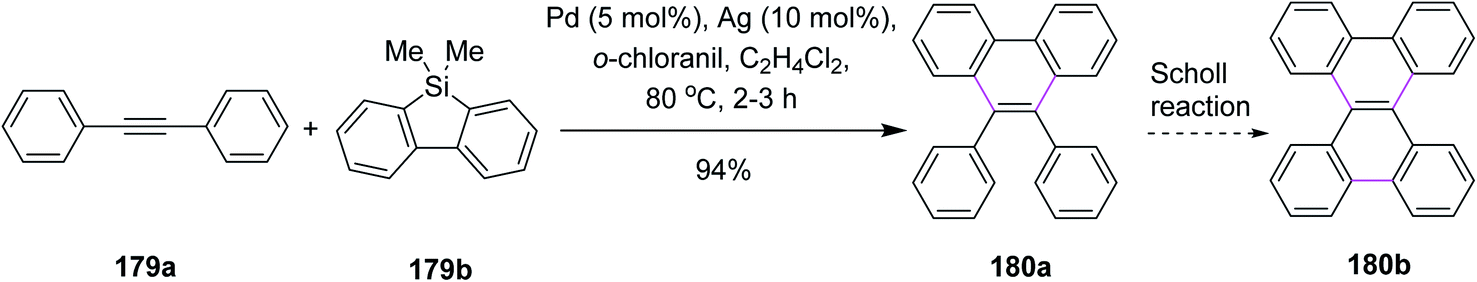 | ||
| Scheme 76 Pd catalyzed APEX of PAHs. | ||
In the same year, Itami and co-workers developed a protocol for nanographene synthesis by the Scholl reaction. The APEX reaction was carried out in the presence of o-chloranil to produce compound 181. Subsequently, this precursor was subjected to SR in the presence of FeCl3 to furnish hexabenzotetracene 182 in good yield (Scheme 77). Metallic Pd catalyzed the regioselective construction of clover-shaped nanographenes in this method.108
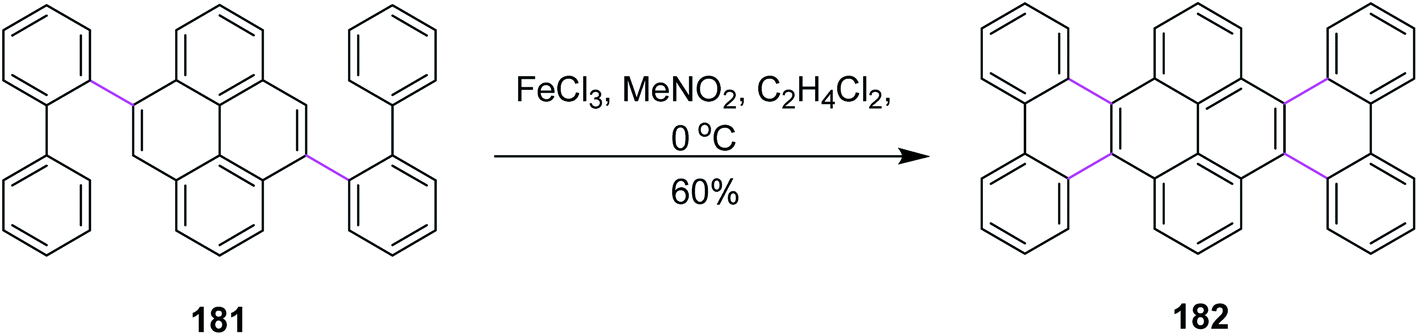 | ||
| Scheme 77 π-extension of perylene by the Scholl reaction. | ||
In 2017, Venkatakrishnan and co-workers revealed a specific type of intermolecular oxidative C–C coupling of arylamines 183a and 183b for the formation of benzidines or naphthidines 184a and 184b, respectively, using the quinone-based organic reagents, i.e., chloranil, instead of using any metal-containing oxidant. The aim was to learn about the chemistry of both the reaction mechanism and the preferred oxidative dimerization of substituted arylamines (either secondary or tertiary). The synthetic perspective (Scheme 78) showed that the methodology worked not only for substrates with various functional groups but also for ones with sterically hindered reactive sites.109
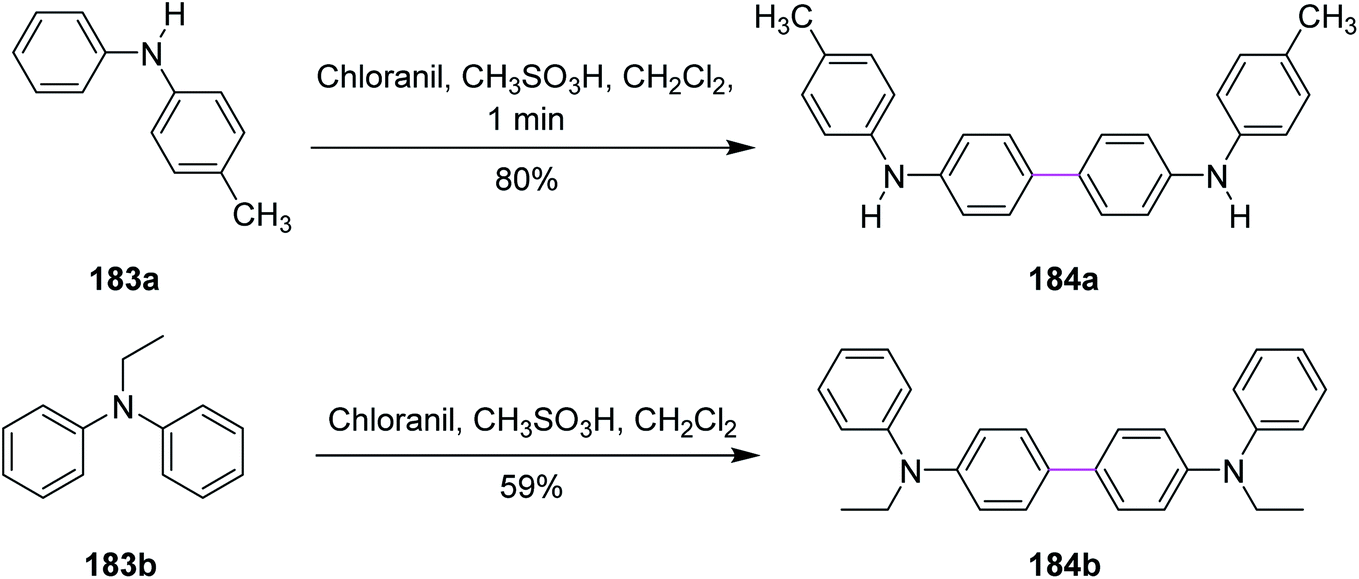 | ||
| Scheme 78 Oxidative coupling of secondary and tertiary aromatic amines under the Scholl reaction. | ||
5.7. Other miscellaneous reagents for the Scholl reaction
In 1980, Taylor and co-workers developed a new synthetic approach for the regiospecific oxidative coupling of substituted benzenes 185. The reaction was carried out in the presence of thallium trifloroacetate (TTFA) under Scholl reaction conditions to synthesize substituted biaryls 186, as shown in Scheme 79. The authors reported that TTFA provided better yield as compared to FeCl3 and Hg(OCOCF3)2 for biaryl synthesis while Pb(OCOCH3)4 provided either equal of better yields than TTFA. Thus, this reagent was selected and used preferentially over all other reagent systems to prepare, in particular, biaryls in good yields.110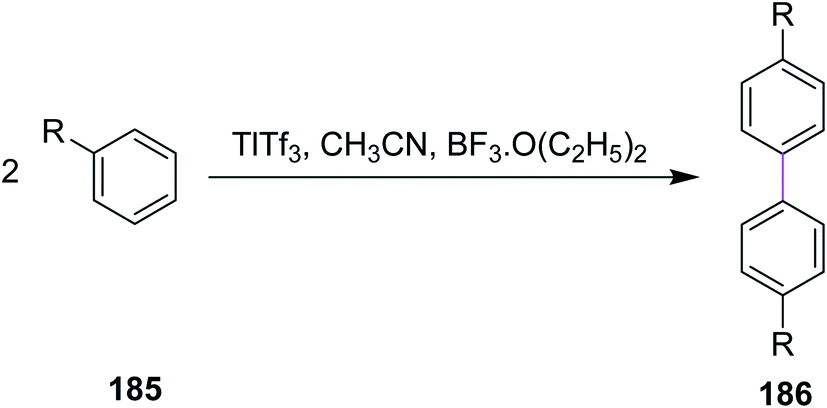 | ||
| Scheme 79 Biaryl construction via Scholl reaction with TlTf3. | ||
In 1994, Wegner and co-workers devised a protocol for oxidative biaryl coupling using vanadium oxyfluoride to construct the C–C bonds by the Scholl reaction. The new hexagonal ring in compound 188 was formed between two substituted aryl rings of the benzil derivatives 187 mediated by the VOF3/BF3·Et2O complex in DCM with excellent yield (Scheme 80). Such oxidizing agents such as vanadium oxyfloride and other transition metal oxides were innovative and effective means for intermolecular coupling, which are frequently utilized in the synthesis of various substituted benzils. The group used two oxidants simultaneously for this reaction, namely, thallium oxide and vanadium oxyfloride. The latter reagent gave the highest yield.111 Substituted benzils and phenanthrene-9,10-diones constitute the key precursors for ligands in the field of discotic metallomesogens or polymeric mesogens.
 | ||
| Scheme 80 Biaryl coupling with vanadium oxyfloride. | ||
In 1996, Bard and co-workers disclosed a new procedure for the synthesis of dibenzo-tetraphenylperiflanthene 190 by the CoF3-induced Scholl reaction. The C–C bonds of PAH 190 were constructed in floranthene 189 by fissure coupling in TFA (Scheme 81). The notable advantages associated with the procedure involving the CoF3/TFA reagent system is the flexible and short pathway for the synthesis of a wide range of non-benzenoid aromatics prone to dehydrogenation to highly fused systems by fissure, fjord, and core couplings.112
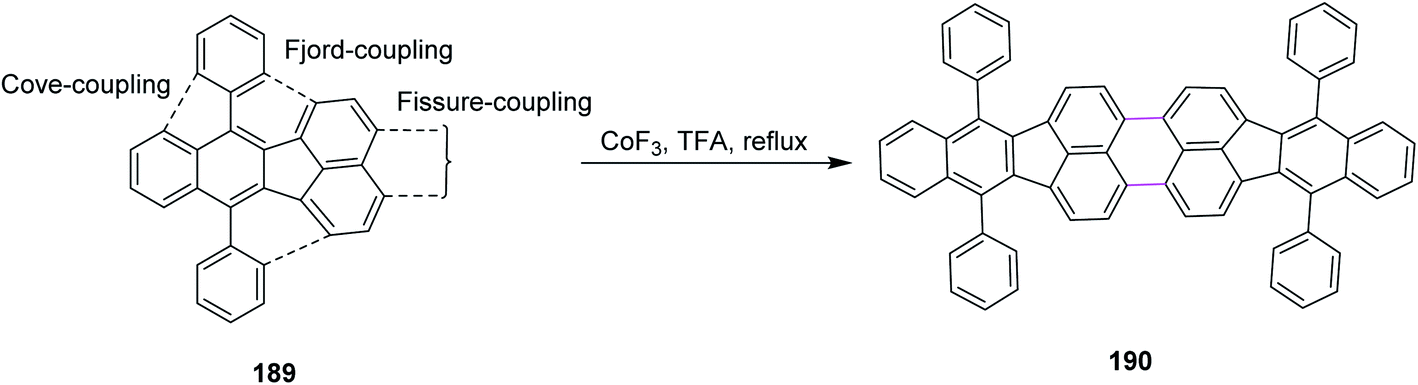 | ||
| Scheme 81 PAHs extension with CoF3. | ||
In 1998, Kochi and co-workers expanded the role of hexachloroantimonate in the oxidative coupling of naphthalene derivative 191 to generate the substituted binaphthyl 192 (Scheme 82). This reagent system provided the selective oxidation of aromatic donors (ArH) and permitted the easy synthesis and isolation of the pure biaryl product, successfully avoiding the chlorinated by-products.113
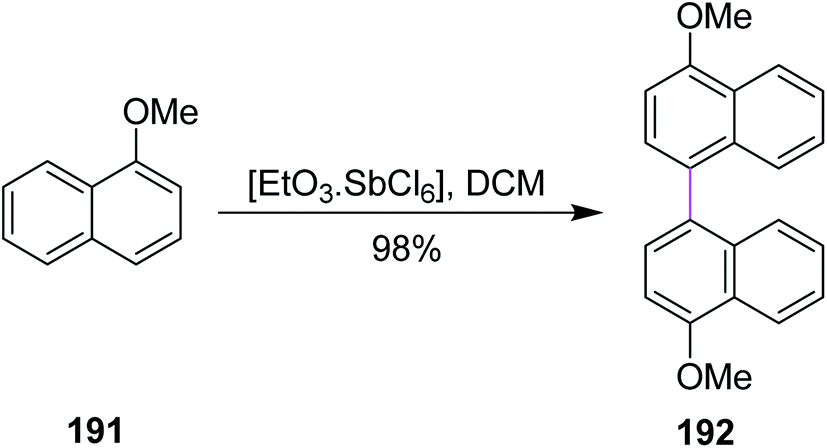 | ||
| Scheme 82 Biaryl coupling via the Scholl reaction. | ||
In 1999 and 2001, Kumar and Varshney introduced a synthetic pathway for the oxidative trimerization of substituted benzenes 193. They synthesized unsymmetric triphenylene 194 using vanadium oxytrichloride in DCM and following the Scholl reaction, as shown in Scheme 83. VOCl3 was the only liquid reagent used in the oxidative cyclization reaction and is soluble in common organic solvents. Therefore, handling the reaction under anhydrous conditions for large-scale synthesis was quite facile. It is noted though that some side products were also observed during this reaction.114,115
 | ||
| Scheme 83 Synthesis of triphenylenes with vanadium oxytrichloride. | ||
In 2008, Kita and co-workers developed a C–C bond formation reaction for phenols 195. The reaction employed the H2O2/anhydrous acid complex for an iodoarene-catalyzed reaction to construct the seven-membered ring 196 by Scholl cyclization, as shown in Scheme 84. This metal-free reagent system provided an environmentally-benign synthetic pathway. An outstanding issue was that strong oxidizing agents could oxidize the iodoarenes to the iodine (I2) at a low temperature. To circumvent this drawback, TFAA-modified with H2O2 was used during the spirocyclization of phenols to obtain high yields of novel aryl systems.116
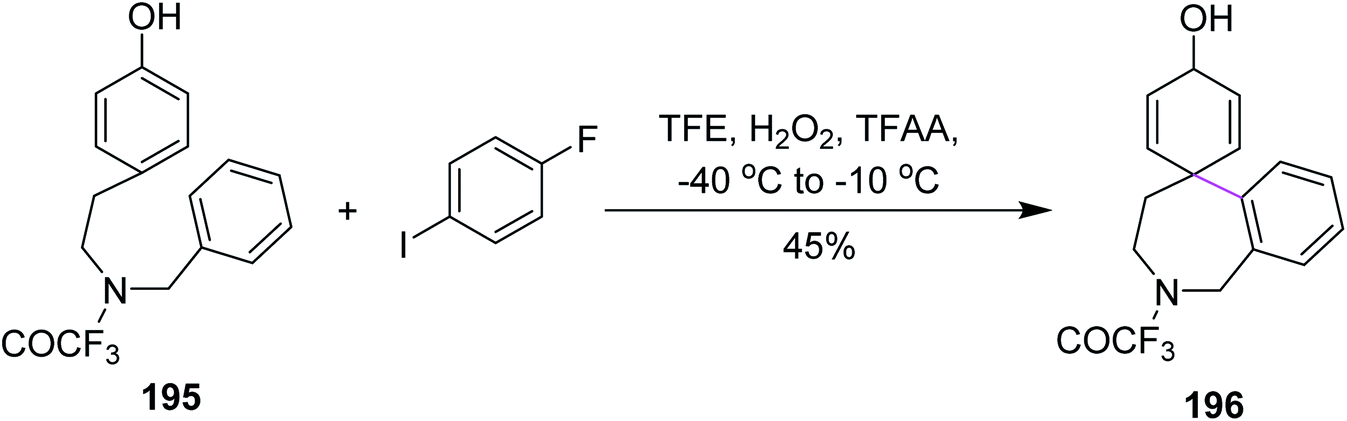 | ||
| Scheme 84 Synthesis of the heptagonal ring by iodoarene using the Scholl reaction. | ||
In 2009, Yang and co-workers described the methodology behind the oxidative biaryl coupling of esters 197. They reported the atropselective synthesis of decinine 198 using vanadium oxyfloride following non-phenolic oxidative Scholl cyclization, as depicted in Scheme 85. Decinine consisted of a macrocyclic ring, a quinolizidine ring, and a biphenyl moiety. The strained 12-membered macrocyclic ring that connected the two aryl rings was constructed by oxidative cyclodehydrogenation. VOF3 played a key role in realizing the total synthesis of racemic decinine.117
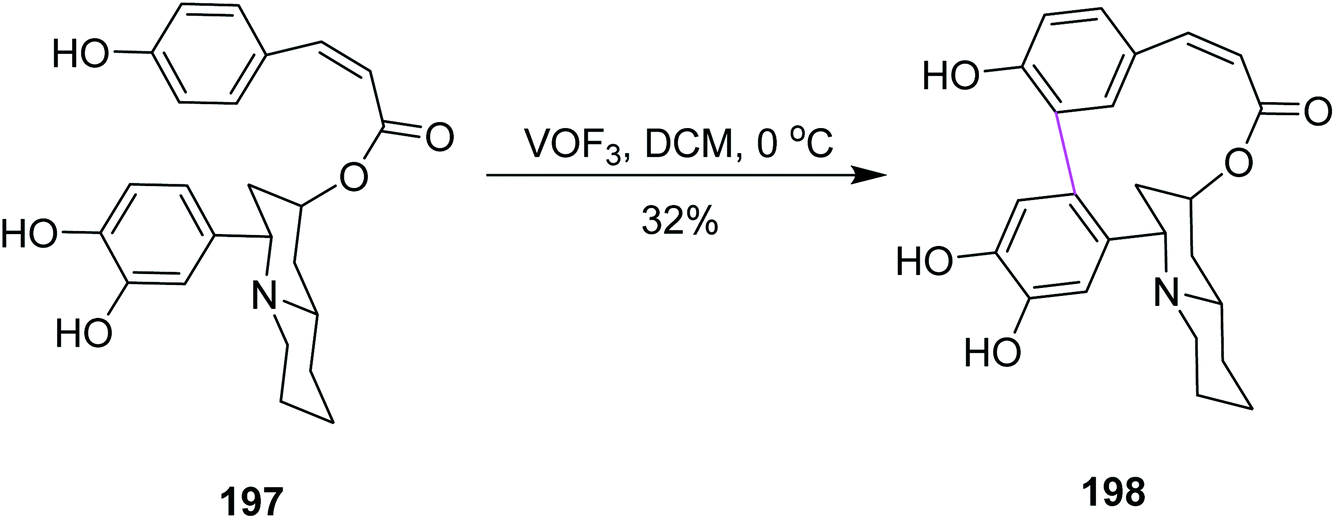 | ||
| Scheme 85 Synthesis of decinine with vanadium oxyfloride. | ||
In 2010, Wang and co-workers revealed a novel methodology for the oxidative coupling of polycyclic aromatic hydrocarbons via manganese dioxide-induced Scholl reaction. The phenanthrene 200 was synthesized using the MnO2/acid system for the coupling of stilbenes 199 (Scheme 86). They used readily available manganese oxide as a mild oxidizing agent to create intermolecular and intramolecular C–C bonds in unfunctionalized arenes in the presence of protic acid. Excellent yields were obtained with less tedious experimental work.118
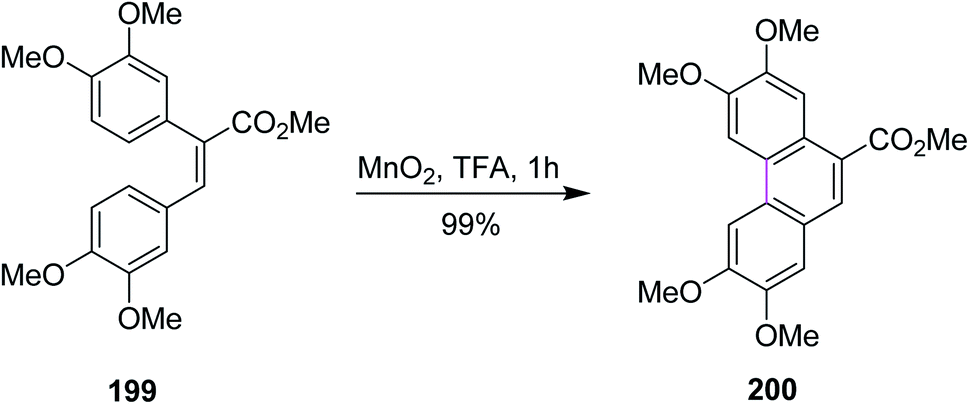 | ||
| Scheme 86 Synthesis of phenanthrene with MnO2/TFA. | ||
In 2010, Kita and co-workers developed a protocol for the regioselective oxidative coupling of thiophenes 201 by the Scholl reaction. Bithienyls 202a (64%) and 202b (>5%) were synthesized in the presence of HTIB- and HFIP-activators, as shown in Scheme 87. The typical metal oxidants were replaced with hypervalent iodine reagent (HTIB) to provide the regioselective products. This particular approach was a novel, selective, facile, and direct route for the coupling of thiophenes to obtain bithiophenes.119 On the basis of this strategy, a novel direct method for the synthesis of head-to-tail bithiophenes using hypervalent iodine(III) reagents has been developed.
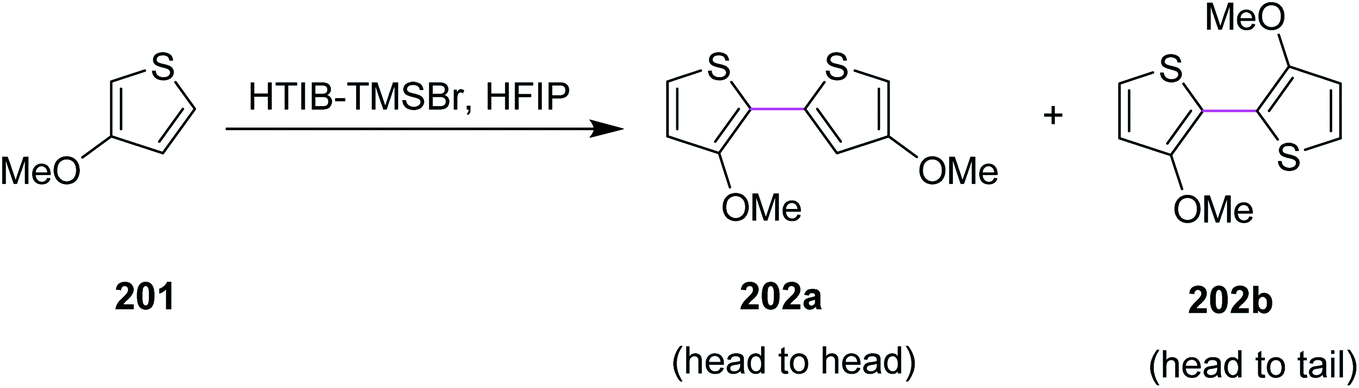 | ||
| Scheme 87 Bithiophene synthesis by HTIB-activators. | ||
In 2017, Leszczynski and co-workers reported a synthetic approach for the oxidative coupling of aromatic hydrocarbons 203 using silver sulphate as a novel reagent for intermolecular Scholl cyclization to construct C–C bonds between two aryl groups in 204a (Scheme 88). The chemical transformation of precursor 204a into the coupled product 204b is still challenging. This synthetic methodology was economical and cost-effective. AgSO4 could simply be regenerated electrochemically from AgHSO4 (side-product). This particular coupling technique does not provide high yield and still requires further optimization.120
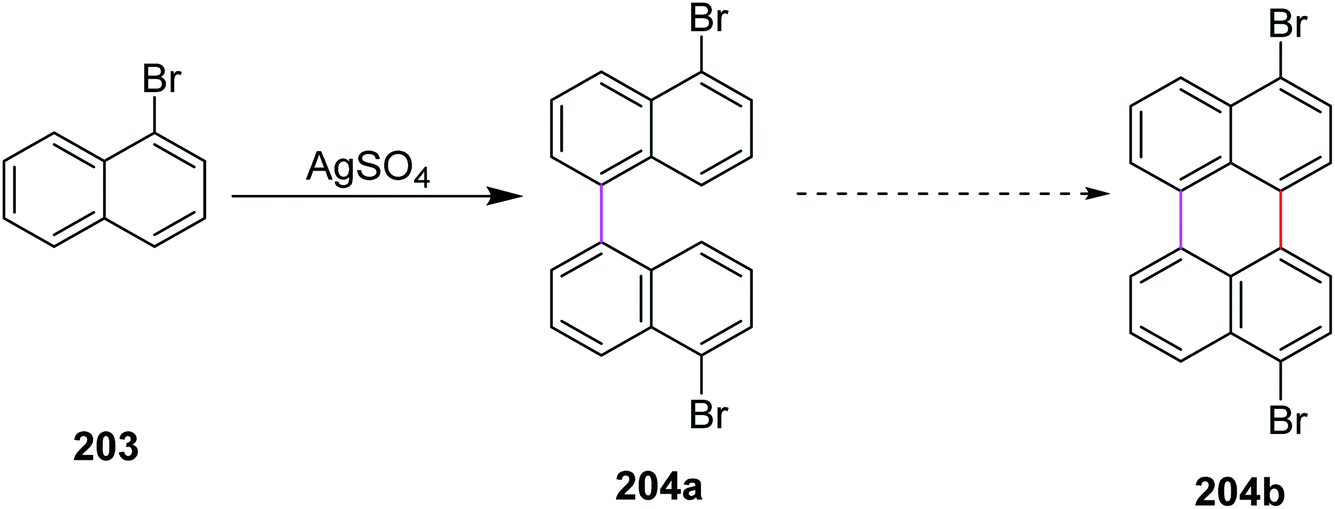 | ||
| Scheme 88 C–C coupling of naphthalene units with AgSO4. | ||
In 2020, Borchardt and co-workers introduced a novel and alternative reaction strategy to synthesize nanographenes, for example, hexabenzocoronene 206. They used a combination of elemental copper and sodium chloride for the mechanochemical cyclodehydrogenation of hexaphenylbenzene 205 to generate six new C–C bonds in HBC 206 (Scheme 89). The procedure was carried out under solvent-free conditions in a ball-mill apparatus. This methodology proved to be more efficient, cheap, and easy compared to the previous ones. Using this method, other PAHs such as triphenylene were also obtained without any side products. Unlike FeCl3, elemental copper did not give any chlorinated or dimerized products.121
 | ||
| Scheme 89 Mechanochemical cyclodehydrogenation of hexaphenylbenzene by the Scholl reaction. | ||
Moreover, the researchers used electrochemistry to perform the Scholl reaction using several different substrates. Previously, the involvement of cation radical intermediates (formed by single electron oxidation) in oxidative C–C bond formations or biaryl syntheses has been carefully probed by Evrard,122a Rathore,122b and co-workers.
In 2021, Müllen and co-workers explored a one-step electrochemical method to synthesize unsubstituted PAHs, starting from twisted oligophenyl precursors and depositing their films with highly ordered and tightly packed structures. In this methodology, anodic oxidation was used in order to replace already existing chemical oxidizing agents used in the Scholl reaction for the formation of a new C–C bond between the two arene units. The electrochemical oxidative characteristics of the triangle-shaped precursor 207 and the ribbon-shaped precursor 209 (Scheme 90)123 were first studied by cyclic voltammetry (CV) at a scan rate of 0.05 V s−1 using glassy carbon as a working electrode. The formation of π-conjugated discs of PAHs dramatically decreased the solubility, causing precipitation under the formation of homogeneous thin films in the neutral state. This electrochemical deposition method substantially simplifies the film-forming process and produces high quality thin PAH films with a controlled thickness and doping level. Furthermore, the group/authors enlarged the size to precursor 215 to produce PAH 212 (C-96). The envisaged product that formed at a potential of 1.65 V appeared to be incompletely cyclodehydrogenated. This behavior was contradictory to the formation of nanographenes 208 and 210. Nonetheless, the complete dehydrogenation of precursor 211 into the flat disc 212 was achieved at a potential of 1.50 V. In fact, it is supposed that the large molecular size of precursor 211 increases the degree of charge delocalization over the large oligoaryl moieties and, in turn, the less reactive phenonium (or radical) cations hinder the intramolecular oxidative cyclodehydrogenation under mild conditions, which are some limitations associated with electrochemistry used in lieu of the oxidant in the Scholl reaction.
 | ||
| Scheme 90 Oxidative cyclodehydrogenation by electrochemical synthesis. | ||
5.8. Surface-assisted synthesis of nanographenes under the Scholl reaction
The oxidative cyclodehydrogenation method is efficient for the synthesis of nanographenes and GNRs in solution. However, the bottom-up chemical synthesis of these graphene materials in solution suffers from the re-aggregation of graphitic structures during the cyclodehydrogenation of the polyphenylene precursors, which renders the further processing to regenerate single-layer graphenes immensely problematic.Recently, researchers involved in the chemical synthesis of nanographenes through the Scholl reaction have shifted their attention toward the use of new protocols aimed at the direct synthesis of graphene materials on the surface via the cyclodehydrogenation of pre-deposited or newly established precursors. This technique has proved to be robust and represents a promising route for the generation of structurally well-defined nanographenes for applications in electronic devices.
In 2009, Wegner introduced a novel synthetic methodology for the gold-catalyzed oxidative coupling of unfunctionalized aromatic compounds utilizing stoichiometric amounts of gold(111) for the construction of C–C bonds in porphyrin derivatives 213 by the Scholl reaction. Different equivalents of reagents gave different products 214a and 214b, as described in Scheme 91. The use of Au(111) during the pathway was convenient because precautions to remove moisture or air were not required. Moreover, oxidative coupling catalyzed by gold enabled the fast and effective construction of complex molecules.124
 | ||
| Scheme 91 C–C coupling of porphyrin units using Au(111). | ||
In the follow-up study, Cai and co-workers (2010) introduced a new strategy to construct graphene nanoribbons 216a and 216b from precursor 215 at a very high temperature. With a gradual increase in the temperature, the tendency to form the C–C bonds between two aryl groups also increased. The whole process shown in Scheme 92 was carried out on an Au(111) surface. The oxidative cyclodehydrogenation of the precursor monomer 215 was achieved by annealing it on the gold surface by the bottom-up approach. The presence of the gold surface fostered a significant increase in the length of the nanoribbons with ease.125 In this case, gold offered an alternative to the oxidant used in the Scholl reaction. The polymerization of monomer 215 on the gold surface proceeded through two steps. This involved dehalogenation, followed by cyclodehydrogenation under the Scholl reaction.
 | ||
| Scheme 92 Graphene nanoribbon synthesis on gold surface. | ||
In 2016, Ruffieux and Fasel developed a synthetic route toward cove-edged GNR 218b employing 11,11′-dibromo-5,5′-bischrysene 217 as the monomer (Scheme 93).126 Although a variety of GNRs with armchair-type edges have been prepared, GNR 218b is the first GNR with the cove-edge structure. Short dimers and tetramers of GNR 218b were separately synthesized in solution as the model compounds, and the single-XRD analysis of the tetramers revealed non-planar structures of such cove-edged GNRs with alternate “up-down” conformations of the peripheral benzene rings. The DFT calculations predicted a bandgap of 1.70 eV for cove-edged GNR 218b. The successful bottom-up synthesis of atomically precise GNRs creates ample opportunities and challenges for the analysis of their physical properties (for example, the magnetism and charge/spin transport) and for the fabrication of GNR-based devices such as the proposed spin valves.
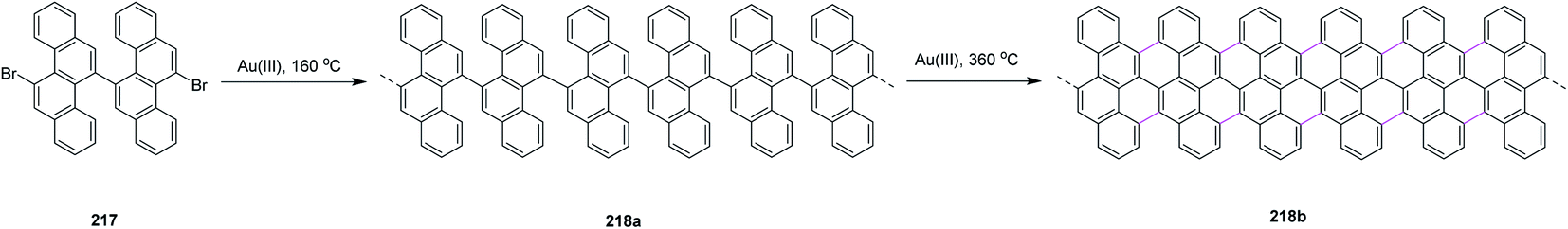 | ||
| Scheme 93 Synthetic strategy for GNRs with zigzag edges by gold catalysis on the surface. | ||
In 2017, Meunier and co-workers revealed a new approach for the on-surface formation of C–C bonds by the formal cycloaddition of the precursor monomer, which are halogen-functionalized at the ortho (2,3) positions of arenes 219a. Choosing a 2,3-dibromotetracene precursor, the authors achieved the formation of tetracene linear dimers 220, cyclo-trimers 221, and X-shaped tetramers 222 (Scheme 94) under the Scholl reaction conditions.127 This product variety is possible due to the availability of two different coupling scaffolds, namely, the formation of four-membered rings (cyclobutadiene units) via the [2+2] cycloaddition 220 and the formation of six-membered rings 221 and 222 via the [2+2+2] cycloaddition, respectively. Ortho-halogen functionalization and the resulting products provide new access to the emerging field of on-surface synthesis, and a prospective pathway to the synthesis of intriguing 1-D structures, such as biphenylene-bridged acene ribbons.
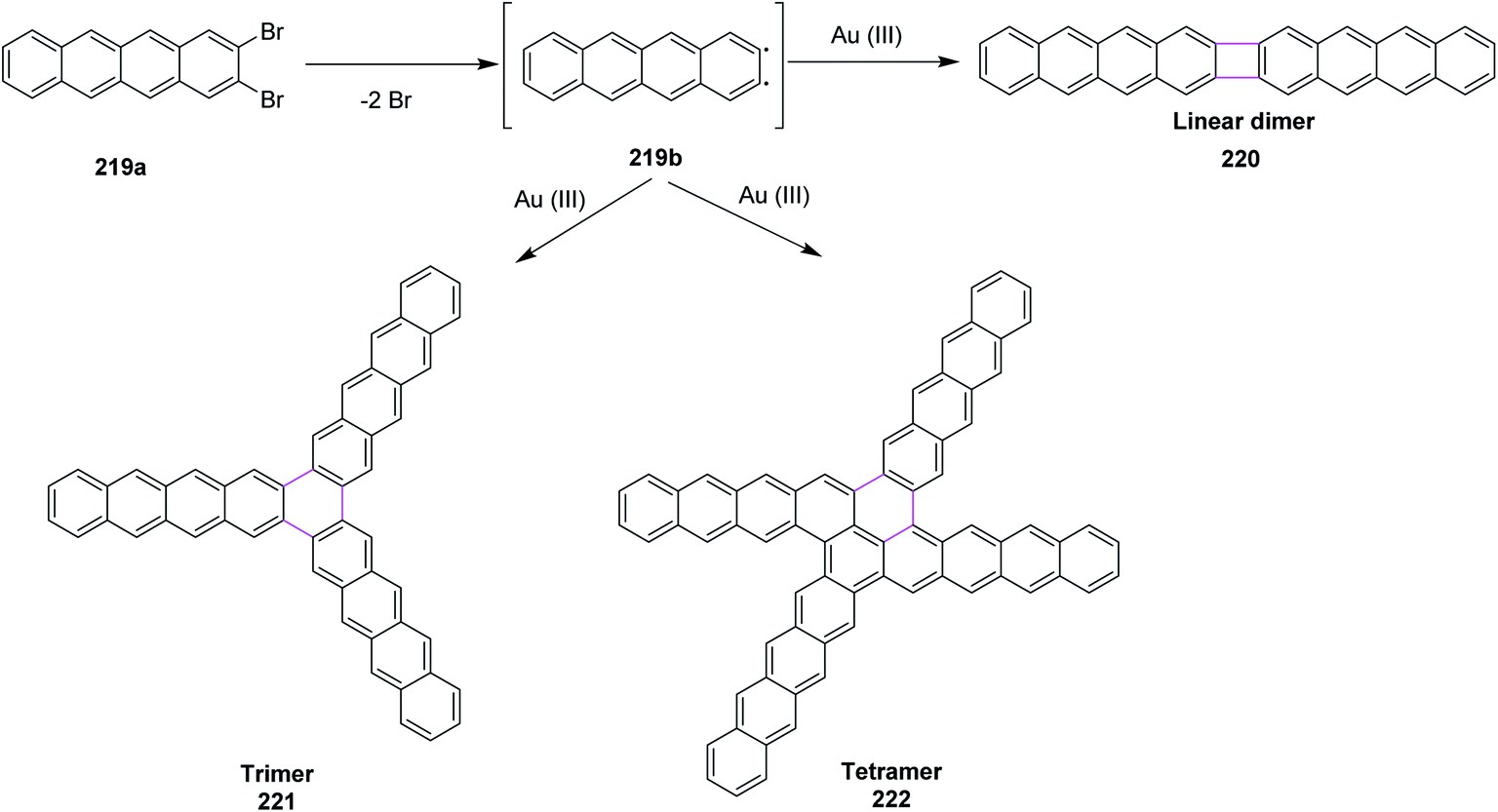 | ||
| Scheme 94 On-surface cyclization of o-dihalotetracenes to four and six-membered rings. | ||
In 2017, Feng and co-workers reported that peri-pentacene 224 could be obtained under ultra-high vacuum (UHV) conditions on an Au(111) surface by the thermally induced cyclodehydrogenation of the precursor 6,6′-bipentacene 223 (Scheme 95).128 The fully cyclized peri-pentacene 224 was produced by annealing 223 at 200 °C for 30 min. The interaction of peri-pentacene with the free valences of the Au surface stabilizes this highly reactive molecule and prevents undesired radical-induced side reactions.
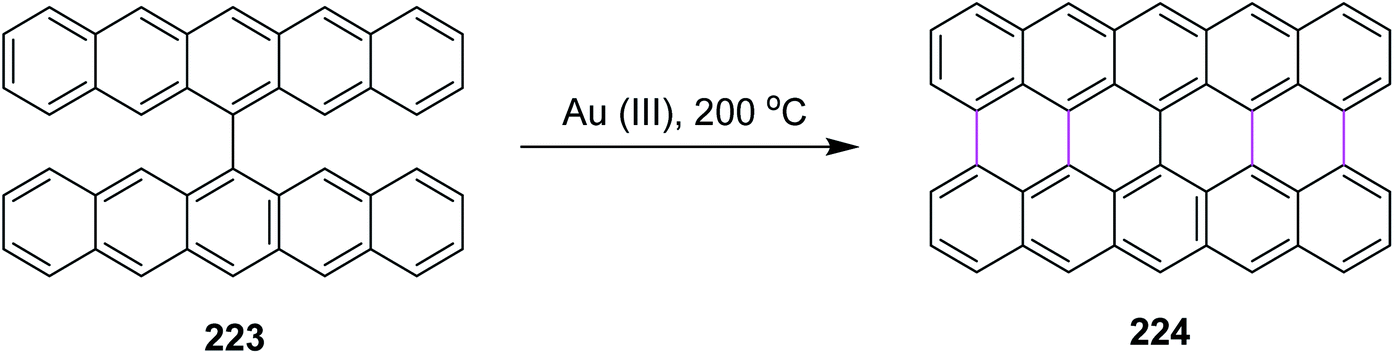 | ||
| Scheme 95 Synthetic route toward peri-pentacene on the Au(111) surface. | ||
In 2017, Narita and co-workers revealed the on-surface synthesis of planarized heteroatom-doped (oxygen–boron–oxygen or OBO) peri-hexacene 226 by thermally annealing the substrate at 380 °C (Scheme 96).129 Surface-catalyzed cyclodehydrogenation successfully transformed the double helicene 225 into the planar peri-hexacene 226. Furthermore, the non-planar structure 225 underwent a conformational change upon the adsorption on surfaces, and 1D self-assembled superstructures were observed for both 225 and 226 due to the presence of OBO units along with zigzag edges. This work demonstrates a potential edge-heteroatom-doping strategy of the edges for fabricating tailor-made graphene nanoarchitectures and highlights the significance of on-surface chemistry to achieve an increased variety of nanographene structures.
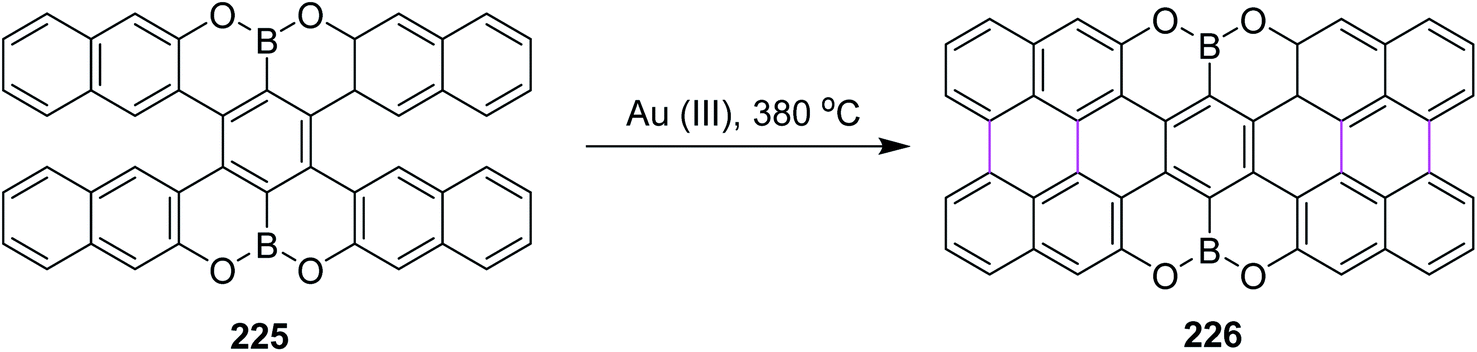 | ||
| Scheme 96 Synthetic route to the peri-hexacene derivative on gold surface. | ||
In the follow-up study, Fasel and co-workers (2018) demonstrated that on-surface synthesis is a powerful route toward the fabrication of specific graphene-like nanostructures confined in 2D. This strategy was successfully applied to the growth of graphene nanoribbons 229 of diverse width and edge morphology. Particular attention was given to the role of halogen functionalization (Br or I) of the molecular precursors 227 (Scheme 97).130 The researchers showed that the use of di-iodinated monomers fosters the growth of longer graphene nanoribbons (average length of 45 nm) due to a larger separation of the polymerization and cyclodehydrogenation temperatures for the conversion of 228 into 229. The reaction was thermally activated on Au(111) and proceeds via two steps (with T1 < T2), involving aryl–aryl coupling (after dehalogenation) and cyclodehydrogenation.
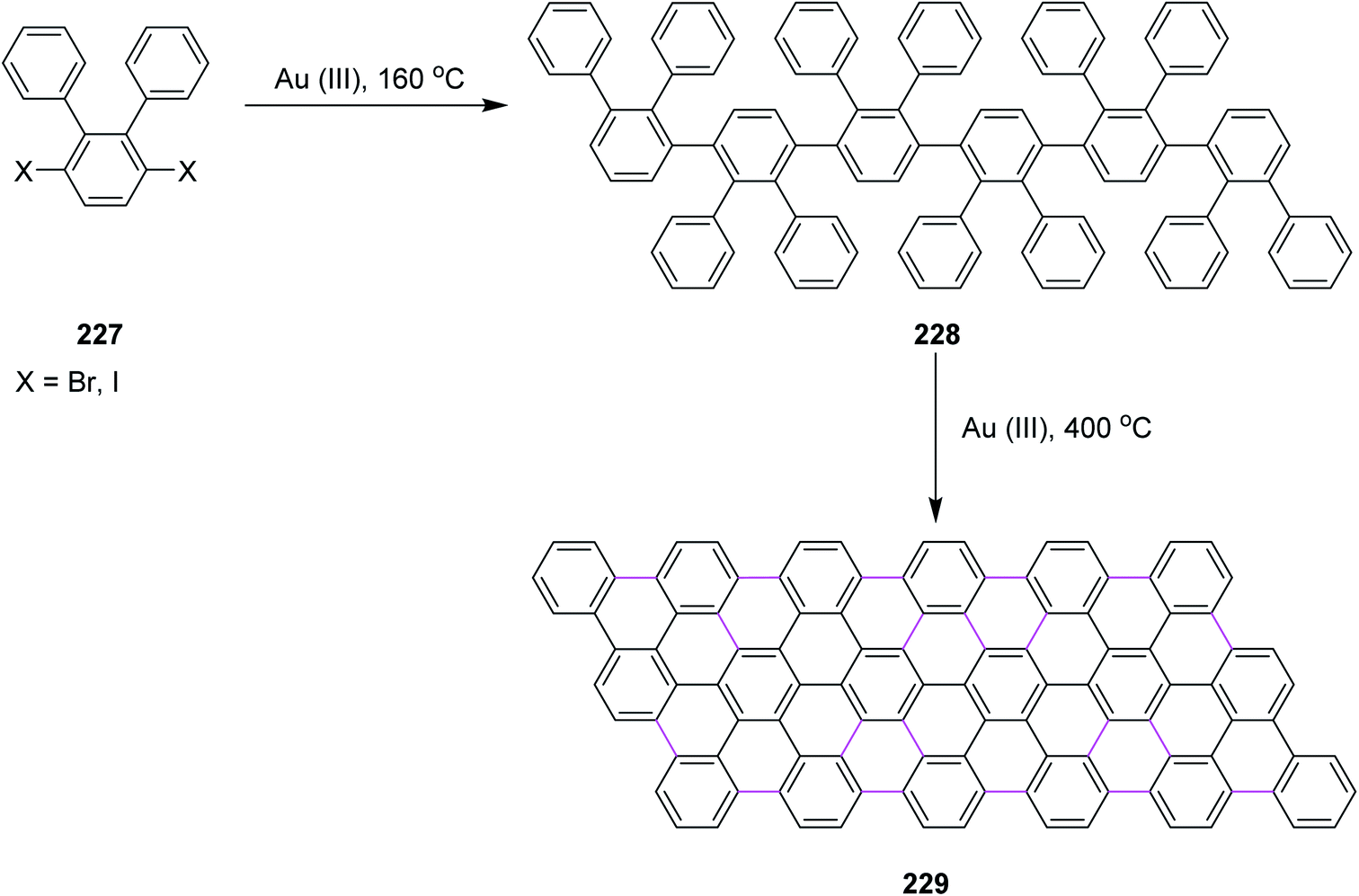 | ||
| Scheme 97 On-surface growth dynamics of graphene nanoribbons. | ||
In 2019, Pena and co-workers synthesized the trigonal porous nanographenes 231 on gold surface, combining the solution chemistry and surface chemistry under the Scholl reaction. They constructed inter-blade and intra-blade C–C bonds for annular π-extension of PAHs. They implemented on-surface cyclodehydrogenation on the gold surface at elevated temperatures (Scheme 98). They initially used other typical oxidants such as DDQ and Pd(PPh3)4 but did not get satisfactory results due to incomplete cyclization. The complete cyclization of PAH 230 was realized in the presence of a gold catalyst at enhanced annealing temperature and 3 new C–C bonds 231 were formed.131
 | ||
| Scheme 98 Trigonal porous nanographene synthesis. | ||
In 2019, Feng and co-workers introduced a process for the synthesis of non-planar porous nanographenes. They implemented on-surface synthetic methodology for the construction of C–C bonds between the aryl rings upon heating on a gold surface. Notably, this type of cyclization of porous compounds 232 is not possible to carry out in solution; therefore, the reaction was carried out via surface chemistry. The well-developed non-planar nanographenes 233 were constructed by annealing the PAHs 232 on the gold surface (Scheme 99).132
 | ||
| Scheme 99 Non-planar porous nanographene synthesis. | ||
In 2020, Torres and co-workers introduced a new protocol for the construction of π-extended boron-dipyrromethene (BODIPY) derivatives 235 such as by gold-catalyzed cycloisomerization. The reaction was carried out under mild Scholl conditions, which provided excellent yields of 235 in a short time. The resulting product was formed regioselectively and formed to be kinetically and thermodynamically stable (Scheme 100). Gold showed high affinity for the π-bonds of alkynes 234 and constructed various η2-alkyne complexes. These complexes further reacted with different heteroatom- or carbon-based nucleophiles under mild reaction conditions.133
 | ||
| Scheme 100 Synthesis of π-extended BODIPYs. | ||
In 2021, Itami and co-workers published their most recent work on gold-catalyzed heptagon-containing molecular nanocarbons. The synthetic methodology behind their work was Scholl cyclization involving the on-surface synthesis of azulene-embedded nanocarbons 237. The cyclodehydrogenation of PAHs 236 resulted in the formation of giant nanographenes (Scheme 101). They established a novel pathway for the construction of abnormal 5–7 defects in N-doped nanographene by the on-surface cyclodehydrogenation reaction. The reaction pathway consisted of many intermediate states but the fundamental step, where the final product combining the azulene motifs was formed, occurred on the Au(111) surface.134
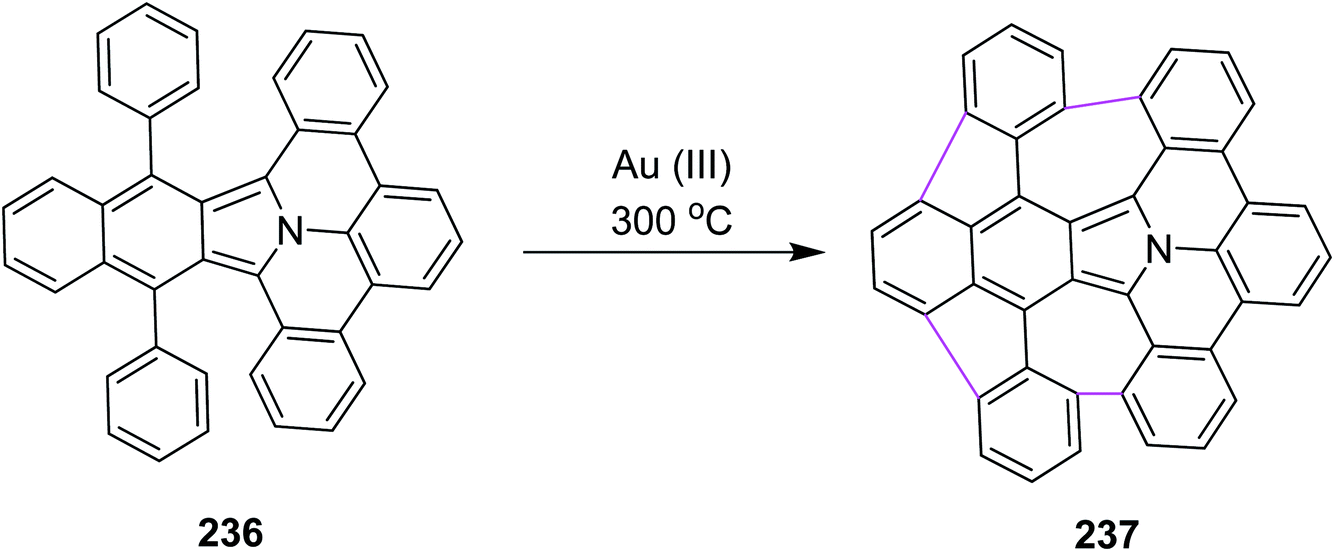 | ||
| Scheme 101 Synthesis of molecular nanographenes. | ||
Overall, it is concluded that AuCl3 proves to be a better oxidant due to its excellent oxidizing capacity as compared to all the abovementioned oxidants. Its reduction potential value is +1.50 V.
6. Comparative summary of various reagents used in the Scholl reaction
Thus far, we have discussed in detail the implementations, merits, and demerits of various reagents used in the Scholl reaction. Herein, we will briefly compare these reagents with one another. With time and more demand for improved conditions, reagents have been modified gradually from the simplest reagent system comprising AlCl3 (proposed by Scholl) to the most advanced recent oxidizing reagent systems utilizing gold and electrochemistry. Each reagent system has some benefits as well as drawbacks associated with it and the choice of the reagent system is dictated by the synthetic target. For example, copper salts are prominent due to their better yields, low cost, easy availability, and less or non-toxic properties. Iron chloride is eco-friendly, plays the dual role of oxidant and Lewis acid, and provides excellent yields. Moreover, it is non-hazardous and provides a lesser number of side products. Similarly, DDQ is readily available, cost effective, and provides high yield. It offers pure and highly selective products. Molybdenum pentachloride offers a facile and efficient route, which provides high yields of symmetrical, regioselective, and twisted isomers of nanographenes. PIFA provides excellent yield and eliminates any pre-activation of the monomers. It is useful for heteroatomic binary coupling under mild oxidation conditions, providing a less toxic and direct synthetic pathway. Palladium complexes give selective arylations of sterically congested substrates, providing high yields as well. Finally, though still the most valuable, electrochemical oxidation and gold are the utmost advanced reagent systems; they involve surface chemistry, eliminating the complex and extensive solution systems, is less hazardous, quick, and provides pure products. The higher reduction potential of various oxidants used in the Scholl reactions indicate that these are the best oxidizing agents and thus have the potential to dehydrogenase the aromatics. For example, the reduction potential of various metals/reagents is in the following order, i.e., Pd2+ > Mn2+> Au3+ > Ag2+ > I− > DDQ > Cu2+ > Co2+ > Tl3+ > Fe3+ > Al3+ > Mo2+ (+0.951 V > +1.54 V > +1.50 V > +0.80 V > +0.54 V > +0.50 V > +0.34 V > −0.28 V > −0.34 V > −0.409 V > −1.667 V > −1.75 V). This information reveals that the metals such as Ag, Cu, Fe, and Mn, and DDQ reagent have high affinity for hydrogen removal. They ensure complete cyclization to provide extended nanographenes. Moreover, many of the structures that are accessible by intramolecular oxidative coupling, in particular, large planar and curved PAHs, would be nearly impossible to synthesize using other methods for forming biaryl bonds because of the required pre-functionalization. Although a variety of valuable reactants are being developed for oxidative coupling, intramolecular oxidative coupling is dominated by two oxidants, namely, FeCl3 and DDQ/acid (Rathore conditions). These two reagents have been used by the Müllen, Itami, Miao, and Wu groups for the preparation of the most spectacular PAH molecules to date, including nanographenes and nanobelts. Overall, it is concluded that the oxidants AlCl3, FeCl3, DDQ, and Au(111) have stronger oxidizing capacity as compared to the other various reagents. The electrochemical deposition method substantially simplifies the film-forming process and produces high-quality thin PAH films with a controlled thickness and doping level. Notably, these PAHs self-assemble to form discrete nanofibers with a defined molecular alignment. This discovery offers a new method to align insoluble polycyclic aromatic materials with strong π–π interactions into thin films. The latter hold promises for further incorporation into photovoltaic cells and field-effect transistors. Thus far, we have compared the merits of each reagent system, although there are certainly some shortcomings associated with them as well. These have been already discussed in our sequential development of the reagent system.7. Conclusion and future perspectives
Over the years, tremendous knowledge has been collected regarding the dehydrogenative inter and intramolecular C–C coupling of aromatic compounds. These economically advantageous processes attract warranted attention as the ultimate method for the formation of complex biaryl units. Electron-rich and electron-neutral compounds/moieties can both react to form one or more carbon–carbon single bonds. The presentation of various aspects of oxidation leading to the formation of aryl–aryl linkages has allowed us to state the hypothesis that depending on the structure of the substrates, two mechanisms must be considered. The fact that this process can be mediated by both a strong Lewis acid at high temperature and by mild oxidants under mild conditions has caused certain confusion in recent years. Therefore, we propose to call this process dehydrogenative aromatic coupling when the mechanism is unclear. We also propose to reserve the name “Scholl reaction” only for those cases when non-oxidizing Brønsted or Lewis acids are used to perform the reaction (with or without the use of an additional oxidant). Moreover, we have explained above that the Scholl reaction can occur in many cases with substrates having low oxidation potentials but are still able to provide the final products in good yield. To conclude, we have described some advanced synthetic methods, employing the Scholl reaction in the development of planar and non-planar nanographenes. Among all the methods, cyclodehydrogenation via on-surface gold catalysis appears to be advanced and efficient to push multiple dehydrogenations, and thus needs to be explored further in future. Furthermore, a great deal of additional effort, particularly focusing on electrochemically-mediated oxidative aromatic coupling, experiments with Brønsted acids, and computational studies, is needed to clarify the mechanisms of these processes. We hope that this review, in addition to providing and organizing the overview of the topic, will serve as a catalyst to spark further studies.Abbreviations
| APEX | Annulative p-extension |
| BIC | Bicyclic |
| BODIPY | Boron dipyrromethenes |
| CPME | Cyclopentyl methyl ether |
| CV | Cyclic voltammetry |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DFT | Density functional theory |
| GNRs | Graphene nanoribbons |
| HBC | Hexabenzocoronene |
| HFIP | Hexafluoroisopropanol |
| HITAB | Hydroxy(tosyloxy)iodobenzene |
| HPB | Hexaphenylbenzene |
| MALDI | Matrix-assisted laser desorption ionization |
| MOPs | Microporous organic polymers |
| NCS | N-Chlorosuccinimide |
| NIR | Near infrared |
| OFETs | Organic field-effect transistors |
| PAHs | Polycyclic aromatic hydrocarbons |
| PIDA | Phenyliodine(III) diacetate |
| PIFA | (Bis(trifluoroacetoxy)iodo)benzene |
| PPA | Polyphosphoric acid |
| POF | Porous organic framework |
| TTFA | Thenoyltrifluoroacetone |
| TBTQ | Tribenzotriquinacene |
| TMSBr | Trimethylsilyl bromide |
| TFA | Trifluoroacetic acid |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their sincere appreciation to Taif University Researchers Supporting Project number (TURSP-2020/312), Taif University, Taif, Saudi Arabia. The authors are also highly grateful to the Higher Education Commission of Pakistan (HEC) for providing financial assistance under Project No. (NRPU-6484). The authors are highly thankful to Prof. Dr Dietmar Kuck, Department of Chemistry, Bielefeld University, Germany for his kind help in proof-reading and editing this review.References
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- J. Zhao, C. He, R. Yang, Z. Shi, M. Cheng, W. Yang, G. Xie, D. Wang, D. Shi and G. Zhang, Appl. Phys. Lett., 2012, 101, 063112 CrossRef.
- L. Zhi and K. Müllen, J. Mater. Chem., 2008, 18, 1472–1484 RSC.
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed.
- M. C. Drummer, V. Singh, N. Gupta, J. L. Gesiorski, R. B. Weerasooriya and K. D. Glusac, Photosynth. Res., 2021, 1–22 CAS.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson and E. W. Hill, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- M. Quernheim, F. E. Golling, W. Zhang, M. Wagner, H. J. Räder, T. Nishiuchi and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 10341–10346 CrossRef CAS PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschoen and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- R. Scholl and C. Seer, Justus Liebigs Ann. Chem., 1912, 394, 111–177 CrossRef.
- E. Faggi, R. M. Sebastián, R. Pleixats, A. Vallribera, A. Shafir, A. Rodríguez-Gimeno and C. Ramirez de Arellano, J. Am. Chem. Soc., 2010, 132, 17980–17982 CrossRef CAS PubMed.
- M. L. Rahman, G. Hegde, M. M. Yusoff, M. N. F. A. Malek, H. T. Srinivasa and S. Kumar, New J. Chem., 2013, 37(8), 2460–2467 RSC.
- T. M. Swager and B. Koo, Synfacts, 2014, 10, 0581 CrossRef.
- Y. Yang, L. Yuan, B. Shan, Z. Liu and Q. Miao, Chem.–Eur. J., 2016, 22, 18620–18627 CrossRef CAS PubMed.
- A. Balaban and C. Nenitzescu, Friedel-Crafts Relat. React., 1964, 2, 979–1047 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, in Nanoscience and technology: a collection of reviews from nature journals, World Scientific, 2010, pp. 11–19 Search PubMed.
- (a) X. Dou, X. Yang, G. J. Bodwell, M. Wagner, V. Enkelmann and K. Müllen, Org. Lett., 2007, 9, 2485–2488 CrossRef CAS PubMed; (b) C. Shen, G. Zhang, Y. Ding, N. Yang, F. Gan, J. Crassous and H. Qiu, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed; (c) Z. Qiu, S. Asako, Y. Hu, C.-W. Ju, T. Liu, L. Rondin, D. Schollmeyer, J.-S. b. Lauret, K. Müllen and A. Narita, J. Am. Chem. Soc., 2020, 142, 14814–14819 CrossRef CAS PubMed; (d) M. Krzeszewski, K. Sahara, Y. M. Poronik, T. Kubo and D. T. Gryko, Org. Lett., 2018, 20, 1517–1520 CrossRef CAS PubMed; (e) X. Zhang, Z. Xu, W. Si, K. Oniwa, M. Bao, Y. Yamamoto and T. Jin, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed; (f) A. Pradhan, P. Dechambenoit, H. Bock and F. Durola, J. Org. Chem., 2013, 78, 2266–2274 CrossRef CAS PubMed.
- D. J. Jones, B. Purushothaman, S. Ji, A. B. Holmes and W. W. Wong, Chem. Commun., 2012, 48, 8066–8068 RSC.
- X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203–2224 CAS.
- (a) G. Baddeley and J. Kenner, J. Chem. Soc., 1935, 303–309 RSC; (b) G. Baddeley, J. Chem. Soc., 1950, 994–997 RSC.
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- B. T. King, J. Kroulík, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, J. Org. Chem., 2007, 72, 2279–2288 CrossRef CAS PubMed.
- L. Zhai, R. Shukla, S. H. Wadumethrige and R. Rathore, J. Org. Chem., 2010, 75, 4748–4760 CrossRef CAS PubMed.
- P. Rempala, J. Kroulík and B. T. King, J. Am. Chem. Soc., 2004, 126, 15002–15003 CrossRef CAS PubMed.
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (b) K. Müllen, ACS Nano, 2014, 8, 6531–6541 CrossRef PubMed; (c) L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed; (d) X. Yan and L.-s. Li, J. Mater. Chem., 2011, 21, 3295–3300 RSC; (e) K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2013, 52, 5436–5438 CrossRef CAS PubMed; (f) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed; (g) W. W. Wong, J. Subbiah, S. R. Puniredd, B. Purushothaman, W. Pisula, N. Kirby, K. Müllen, D. J. Jones and A. B. Holmes, J. Mater. Chem., 2012, 22, 21131–21137 RSC; (h) Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197 CrossRef CAS PubMed; (i) Z. Sun, Z. Zeng and J. Wu, Chem.–Asian J., 2013, 8, 2894–2904 CrossRef CAS PubMed.
- L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed.
- (a) A. Ronlan, O. Hammerich and V. D. Parker, J. Am. Chem. Soc., 1973, 95, 7132–7138 CrossRef CAS; (b) A. Ronlan and V. D. Parker, J. Org. Chem., 1974, 39, 1014–1016 CrossRef CAS; (c) R. Rathore and J. K. Kochi, J. Org. Chem., 1995, 60, 7479–7490 CrossRef CAS.
- R. Scholl and J. Mansfeld, Ber. Dtsch. Chem. Ges. A, 1910, 43, 1734–1746 CrossRef CAS.
- R. Scholl, C. Seer and R. Weitzenböck, Ber. Dtsch. Chem. Ges. A, 1910, 43, 2202–2209 CrossRef CAS.
- F. Wiener and W. Mieg, CA, 1941, 35, 1240 Search PubMed.
- P. Kovacic and A. Kyriakis, Tetrahedron Lett., 1962, 3, 467–469 CrossRef.
- H. Vollmann, Justus Liebigs Ann. Chem., 1963, 669, 22–44 CrossRef CAS.
- R. Scholl and C. Seer, J. Org. Chem., 1971, 36, 2053 CrossRef.
- A. Wick, Helv. Chim. Acta, 1971, 54, 769–782 CrossRef CAS.
- M. Hovorka, J. Günterova and J. Zavada, Tetrahedron Lett., 1990, 31, 413–416 CrossRef CAS.
- M. Smrčina, M. Lorenc, V. Hanus, P. Sedmera and P. Kocovsky, J. Org. Chem., 1992, 57, 1917–1920 CrossRef.
- M. Smrčina, S. Vyskocil, B. Maca, M. Polasek, T. A. Claxton, A. P. Abbott and P. Kocovsky, J. Org. Chem., 1994, 59, 2156–2163 CrossRef.
- M. Müller, H. Mauermann-Düll, M. Wagner, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed., 1995, 107, 1751–1754 CrossRef.
- C. D. Simpson, J. D. Brand, A. J. Berresheim, L. Przybilla, H. J. Räder and K. Müllen, Chem.–Eur. J., 2002, 8, 1424–1429 CrossRef CAS.
- Y. Avlasevich, C. Kohl and K. Müllen, J. Mater. Chem., 2006, 16, 1053–1057 RSC.
- P. Rempala, J. Kroulík and B. T. King, J. Org. Chem., 2006, 71, 5067–5081 CrossRef CAS PubMed.
- C. Jiao and J. Wu, Curr. Org. Chem., 2010, 14, 2145–2168 CrossRef CAS.
- F. Cataldo, O. Ursini, G. Angelini and S. Iglesias-Groth, Fullerenes, Nanotubes, Carbon Nanostruct., 2011, 19, 713–725 CrossRef.
- (a) E. U. Mughal and D. Kuck, Chem. Commun., 2012, 48, 8880–8882 RSC; (b) E. U. Mughal, B. Neumann, H. G. Stammler and D. Kuck, Eur. J. Org. Chem., 2014, 7469–7480 CrossRef CAS.
- S. Pola, C.-H. Kuo, W.-T. Peng, M. M. Islam, I. Chao and Y.-T. Tao, Chem. Mater., 2012, 24, 2566–2571 CrossRef CAS.
- M. S. Markoulides, C. Venturini, D. Neumeyer and A. Gourdon, New J. Chem., 2015, 39, 6498–6503 RSC.
- Y. N. Oded, S. Pogodin and I. Agranat, J. Org. Chem., 2016, 81, 11389–11393 CrossRef CAS PubMed.
- Z. He, T. Wang, Y. Xu, M. Zhou, W. Yu, B. Shi and K. Huang, J. Phys. Chem. C, 2018, 122, 8933–8940 CrossRef CAS.
- I. Agranat, Y. N. Oded, T. Mala'bi, S. Pogodin and S. Cohen, Struct. Chem., 2019, 30, 1579–1610 CrossRef CAS.
- P. C. Solórzano, M. T. Baumgartner, M. Puiatti and L. B. Jimenez, RSC Adv., 2020, 10, 21974–21985 RSC.
- J. Wang, C. Wang, H. Wang, B. Jin, P. Zhang, L. Li and S. Miao, Microporous Mesoporous Mater., 2021, 310, 110596 CrossRef CAS.
- H. Naarmann, M. Hanack and R. Mattmer, Synthesis, 1994, 477–478 CrossRef CAS.
- G. Cooke, V. Sage and T. Richomme, Synth. Commun., 1999, 29, 1767–1771 CrossRef CAS.
- R. C. Borner, N. Boden, R. J. Bushby, R. Borner, A. N. Cammidge, R. Bushby, A. Cammidge and M. Jesudason, Liq. Cryst., 2006, 33, 1439–1448 CrossRef CAS.
- W. Bai and J. Lin, Synth. Commun., 2011, 41, 903–906 CrossRef CAS.
- X.-L. Li, J.-H. Huang and L.-M. Yang, Org. Lett., 2011, 13, 4950–4953 CrossRef CAS PubMed.
- M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nishinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031–8040 CrossRef CAS PubMed.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen and A. H. Koch, Nat. Chem., 2014, 6, 126–132 CrossRef CAS PubMed.
- T. Wöhrle, J. Kirres, M. Kaller, M. Mansueto, S. Tussetschläger and S. Laschat, J. Org. Chem., 2014, 79, 10143–10152 CrossRef PubMed.
- A. N. Lakshminarayana, J. Chang, J. Luo, B. Zheng, K.-W. Huang and C. Chi, Chem. Commun., 2015, 51, 3604–3607 RSC.
- M. Quernheim, F. E. Golling, W. Zhang, M. Wagner, H. J. Räder, T. Nishiuchi and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 10341–10346 CrossRef CAS PubMed.
- Q. Ye, Z. Zhang, Z. M. Png, W. T. Neo, T. Lin, H. Zeng, H. Xu and J. Xu, J. Org. Chem., 2016, 81, 9219–9226 CrossRef CAS PubMed.
- M. Murata, A. Wakamiya and Y. Murata, Angew. Chem., Int. Ed., 2017, 129, 5164–5168 CrossRef.
- S. Grätz, D. Beyer, V. Tkachova, S. Hellmann, R. Berger, X. Feng and L. Borchardt, Chem. Commun., 2018, 54, 5307–5310 RSC.
- N. K. Saha, N. K. Mitra, K. F. Johnson and B. L. Merner, Org. Lett., 2018, 20, 6855–6858 CrossRef CAS PubMed.
- J. Urieta-Mora, M. Krug, W. Alex, J. Perles, I. Fernández, A. Molina-Ontoria, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2019, 142, 4162–4172 CrossRef PubMed.
- L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed.
- D. J. Jones, B. Purushothaman, S. Ji, A. B. Holmes and W. W. Wong, Chem. Commun., 2012, 48, 8066–8068 RSC.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- F. Schlütter, T. Nishiuchi, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed., 2014, 126, 1564–1568 CrossRef.
- K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910–3914 CrossRef CAS PubMed.
- D. Reinhard, F. Rominger and M. Mastalerz, J. Org. Chem., 2015, 80, 9342–9348 CrossRef CAS PubMed.
- J. Liu, S. Osella, J. Ma, R. Berger, D. Beljonne, D. Schollmeyer, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 8364–8367 CrossRef CAS PubMed.
- H.-W. Ip, C.-F. Ng, H.-F. Chow and D. Kuck, J. Am. Chem. Soc., 2016, 138, 13778–13781 CrossRef CAS PubMed.
- W. S. Wong, C. F. Ng, D. Kuck and H. F. Chow, Angew. Chem., Int. Ed., 2017, 56, 12356–12360 CrossRef CAS PubMed.
- S. Nobusue, K. Fujita and Y. Tobe, Org. Lett., 2017, 19, 3227–3230 CrossRef CAS PubMed.
- Y. Hu, X. Y. Wang, P. X. Peng, X. C. Wang, X. Y. Cao, X. Feng, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef CAS PubMed.
- M. Ajayakumar, Y. Fu, J. Ma, F. Hennersdorf, H. Komber, J. J. Weigand, A. Alfonsov, A. A. Popov, R. Berger and J. Liu, J. Am. Chem. Soc., 2018, 140, 6240–6244 CrossRef CAS PubMed.
- L. He, C. F. Ng, Y. Li, Z. Liu, D. Kuck and H. F. Chow, Angew. Chem., Int. Ed., 2018, 57, 13635–13639 CrossRef CAS PubMed.
- K. M. Magiera, V. Aryal and W. A. Chalifoux, Org. Biomol. Chem., 2020, 18, 2372–2386 RSC.
- Y. Han, Z. Xue, G. Li, Y. Gu, Y. Ni, S. Dong and C. Chi, Angew. Chem., Int. Ed., 2020, 132, 9111–9116 CrossRef.
- J. Ma, Y. Fu, E. Dmitrieva, F. Liu, H. Komber, F. Hennersdorf, A. A. Popov, J. J. Weigand, J. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 132, 5686–5691 CrossRef.
- Y. Zou, Y. Han, S. Wu, X. Hou, C. H. E. Chow and J. Wu, Angew. Chem., Int. Ed., 2021, 60(32), 17654–17663 CrossRef CAS PubMed.
- (a) Z. Xia, S. H. Pun, H. Chen and Q. Miao, Angew. Chem., Int. Ed., 2021, 133, 10399–10406 CrossRef; (b) K. Y. Cheung, S. Gui, C. Deng, H. Liang, Z. Xia, Z. Liu, L. Chi and Q. Miao, Chem, 2019, 5, 838–847 CrossRef CAS.
- H.-W. Ip, Y. Li, D. Kuck and H.-F. Chow, J. Org. Chem., 2021, 86, 5546–5551 CrossRef CAS PubMed.
- S. Kumar and M. Manickam, Chem. Commun., 1997, 1615–1666 RSC.
- B. Kramer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2004, 116, 2501–2503 CrossRef.
- A. Spurg, G. Schnakenburg and S. R. Waldvogel, Chem.–Eur. J., 2009, 15, 13313–13317 CrossRef CAS PubMed.
- K. Hackeloer, G. Schnakenburg and S. R. Waldvogel, Org. Lett., 2011, 13, 916–919 CrossRef CAS PubMed.
- S. Tadano, Y. Mukaeda and H. Ishikawa, Angew. Chem., Int. Ed., 2013, 125, 8148–8152 CrossRef.
- E. M. Boyd and J. Sperry, Org. Lett., 2015, 17, 1344–1346 CrossRef CAS PubMed.
- T. Fujikawa, N. Mitoma, A. Wakamiya, A. Saeki, Y. Segawa and K. Itami, Org. Biomol. Chem., 2017, 15, 4697–4703 RSC.
- S. B. Beil, P. Franzmann, T. Müller, M. M. Hielscher, T. Prenzel, D. Pollok, N. Beiser, D. Schollmeyer and S. R. Waldvogel, Electrochim. Acta, 2019, 302, 310–315 CrossRef CAS.
- T. Takada, M. Arisawa, M. Gyoten, R. Hamada, H. Tohma and Y. Kita, J. Org. Chem., 1998, 63, 7698–7706 CrossRef CAS.
- H. Tohma, M. Iwata, T. Maegawa, Y. Kiyono, A. Maruyama and Y. Kita, Org. Biomol. Chem., 2003, 1, 1647–1649 RSC.
- T. Dohi, K. Morimoto, A. Maruyama and Y. Kita, Org. Lett., 2006, 8, 2007–2010 CrossRef CAS PubMed.
- T. Dohi, K. Morimoto, C. Ogawa, H. Fujioka and Y. Kita, Chem. Pharm. Bull., 2009, 57, 710–713 CrossRef CAS PubMed.
- W. Guo, E. Faggi, R. M. Sebastián, A. Vallribera, R. Pleixats and A. Shafir, J. Org. Chem., 2013, 78, 8169–8175 CrossRef CAS PubMed.
- A. A. Van Loon, M. K. Holton, C. R. Downey, T. M. White, C. E. Rolph, S. R. Bruening, G. Li, K. M. Delaney, S. J. Pelkey and E. T. Pelkey, J. Org. Chem., 2014, 79, 8049–8058 CrossRef CAS PubMed.
- C. Depken, F. Krätzschmar and A. Breder, Org. Chem. Front., 2016, 3, 314–318 RSC.
- N. J. Truax, F. Banales Mejia, D. O. Kwansare, M. M. Lafferty, M. H. Kean and E. T. Pelkey, J. Org. Chem., 2016, 81, 6808–6815 CrossRef CAS PubMed.
- Z. Jia, T. Jiu, Y. Li and Y. Li, Mater. Chem. Front., 2017, 1, 2261–2264 RSC.
- Z. Yang, F.-C. Qiu, J. Gao, Z.-W. Li and B.-T. Guan, Org. Lett., 2015, 17, 4316–4319 CrossRef CAS PubMed.
- K. Ozaki, K. Murai, W. Matsuoka, K. Kawasumi, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2017, 129, 1381–1384 CrossRef.
- H. Ito, K. Ozaki and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11144–11164 CrossRef CAS PubMed.
- S. Maddala, S. Mallick and P. Venkatakrishnan, J. Org. Chem., 2017, 82, 8958–8972 CrossRef CAS PubMed.
- A. McKillop, A. G. Turrell, D. W. Young and E. C. Taylor, J. Am. Chem. Soc., 1980, 102, 6504–6512 CrossRef CAS.
- B. Mohr, V. Enkelmann and G. Wegner, J. Org. Chem., 1994, 59, 635–638 CrossRef CAS.
- J. D. Debad, J. C. Morris, V. Lynch, P. Magnus and A. J. Bard, J. Am. Chem. Soc., 1996, 118, 2374–2379 CrossRef CAS.
- R. Rathore, A. Kumar, S. V. Lindeman and J. K. Kochi, J. Org. Chem., 1998, 63, 5847–5856 CrossRef CAS PubMed.
- S. Kumar and S. K. Varshney, Liq. Cryst., 1999, 26, 1841–1843 CrossRef CAS.
- S. Kumar and S. K. Varshney, Synthesis, 2001, 1, 0305–0311 CrossRef.
- T. Dohi, Y. Minamitsuji, A. Maruyama, S. Hirose and Y. Kita, Org. Lett., 2008, 10, 3559–3562 CrossRef CAS PubMed.
- Z.-H. Shan, J. Liu, L.-M. Xu, Y.-F. Tang, J.-H. Chen and Z. Yang, Org. Lett., 2012, 14, 3712–3715 CrossRef CAS PubMed.
- K. Wang, Y. Hu, Z. Li, M. Wu, Z. Liu, B. Su, A. Yu, Y. Liu and Q. Wang, Synthesis, 2010, 10, 1083–1090 CrossRef.
- K. Morimoto, N. Yamaoka, C. Ogawa, T. Nakae, H. Fujioka, T. Dohi and Y. Kita, Org. Lett., 2010, 12, 3804–3807 CrossRef CAS PubMed.
- A. K. Budniak, M. Masny, K. Prezelj, M. Grzeszkiewicz, J. Gawraczyński, Ł. Dobrzycki, M. K. Cyrański, W. Koźmiński, Z. Mazej and K. J. Fijałkowski, New J. Chem., 2017, 41, 10742–10749 RSC.
- S. Grätz, M. Oltermann, C. G. Vogt and L. Borchardt, ACS Sustainable Chem. Eng., 2020, 8, 7569–7573 CrossRef.
- (a) D. A. Evrard, Studies toward a biomimetic oxidative macrocyclization approach to the synthesis of the vancomycin family of antibiotics, Harvard University, 1994 Search PubMed; (b) R. Rathore and J. K. Kochi, J. Org. Chem., 1995, 60, 7479–7490 CrossRef CAS.
- C. Zeng, B. Wang, H. Zhang, M. Sun, L. Huang, Y. Gu, Z. Qiu, K. Müllen, C. Gu and Y. Ma, J. Am. Chem. Soc., 2021, 143, 2682–2687 CrossRef CAS PubMed.
- H. A. Wegner, Chimia, 2009, 63, 44–48 CrossRef CAS.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh and X. Feng, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli and D. Passerone, Nature, 2016, 531, 489–492 CrossRef CAS PubMed.
- C. Sanchez-Sanchez, A. Nicolaï, F. Rossel, J. Cai, J. Liu, X. Feng, K. Müllen, P. Ruffieux, R. Fasel and V. Meunier, J. Am. Chem. Soc., 2017, 139, 17617–17623 CrossRef CAS PubMed.
- J. Liu, R. Berger, K. Müllen and X. Feng, From Polyphenylenes to Nanographenes and Graphene Nanoribbons, 2017, pp. 1–32 Search PubMed.
- X.-Y. Wang, T. Dienel, M. Di Giovannantonio, G. B. Barin, N. Kharche, O. Deniz, J. I. Urgel, R. Widmer, S. Stolz, L. H. De Lima, M. Muntwiler, M. Tommasini, V. Meunier, P. Ruffiuex, X. Feng, R. Fasel, K. Mullen and A. Narita, J. Am. Chem. Soc., 2017, 139, 4671–4674 CrossRef CAS PubMed.
- M. Di Giovannantonio, O. Deniz, J. I. Urgel, R. Widmer, T. Dienel, S. Stolz, C. S. Sánchez, M. Muntwiler, T. Dumslaff, R. Berger, R. Berger, A. Narita, X. Feng, K. Mullen, P. Ruffiuex and R. Fasel, ACS Nano, 2018, 12, 74–81 CrossRef CAS PubMed.
- R. Zuzak, I. Pozo, M. Engelund, A. Garcia-Lekue, M. Vilas-Varela, J. M. Alonso, M. Szymonski, E. Guitián, D. Pérez, S. Godlewski and D. Peña, Chem. Sci., 2019, 10, 10143–10148 RSC.
- K. Xu, J. I. Urgel, K. Eimre, M. Di Giovannantonio, A. Keerthi, H. Komber, S. Wang, A. Narita, R. Berger, P. Ruffieux, C. A. Pignedoli, J. Liu, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 7726–7730 CrossRef CAS PubMed.
- J. Labella, G. Durán-Sampedro, M. V. Martínez-Díaz and T. Torres, Chem. Sci., 2020, 11, 10778–10785 RSC.
- C. Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2021, 133, 2–27 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |