
Electrochemically mediated deionization: a review†
Adarsh
Suresh‡
,
Grant T.
Hill‡
,
Eli
Hoenig‡
and
Chong
Liu
 *
*
Pritzker School of Molecular Engineering, University of Chicago, 5640 S Ellis Ave, Chicago, IL 60637, USA. E-mail: chongliu@uchicago.edu
First published on 23rd December 2020
Abstract
Electrochemical deionization technologies allow generation of potable water from contaminated sources and extraction of valuable resources from seawater, brines and industrial wastewater—all in an environmentally friendly and energy efficient manner. In this review, we detail existing electrode materials, cell architectures, and charge transfer mechanisms related to electrochemically driven desalination and selective element extraction in aqueous environments. More specifically, we address capacitive and faradaic charge-transfer processes—ion electrosorption and ion intercalation, respectively. We also address selective electrosorption/electrodeposition at functionalized electrode surfaces. The selectivity is driven by ion specific interactions at the electrode, such as chelation and redox activity. Electrochemically mediated deionization strategies can help address the global need for potable water. Their widespread adoption hinges on a thorough understanding of the range of available deionization methods—which we attempt to provide in this review.
Design, System, ApplicationGlobal water shortages and strategic resource scarcities have spurred efforts to efficiently purify water and extract valuable elements from aqueous solutions. The growing market penetration of renewably generated electricity provides new opportunities to develop electrochemical methods for both applications. Here, we review the science and engineering principles of electrochemically mediated water desalination and selective ion extraction. We divide our review into four areas: capacitive deionization, faradaic desalination, depletion zone desalination, and selective electrosorption. In each area, we detail electrode materials and device designs of novel and/or high-performing methods. Fundamental charge transfer mechanisms and ion interactions at the solid–water interface are also discussed in order to provide insights for further technology development. |
1. Introduction
As a result of rapid population growth and global development, the lack of clean and accessible water has become a critical international concern.1 A small fraction of the earth's water is freshwater and much of this supply is inaccessible: <1% of all the water on the planet is readily available for human use.2 Some arid, coastal and arctic regions are geographically limited in their access to freshwater, while other regions are plagued by environmental pollution from heavy metal contaminants such as lead, chromium, arsenic. With ongoing climate change, uncertainties concerning potable water access will only grow more severe.Amid the limited supply of freshwater, the mounting pressures of a rising population, fast paced economic growth and technological development are pushing demand for crucial resources such as lithium (battery applications), uranium (nuclear energy production) and rare-earth elements (advanced consumer electronics and renewable energy technology). Extracting such sought after resources from seawater, brines and wastewater can mitigate the negative environmental impacts of traditional mining methods. Therefore, we are not only faced with the significant task of efficiently sourcing, treating and distributing water to high-stress regions of the world but also of securing critical elements from dilute sources with minimal impact to environment.
All of the current desalination technologies are energy and capital intensive:3 thermal distillation, one of the most common desalination methods, uses ∼50 times more energy than the fundamental thermodynamic limit.4 Non-thermal alternatives for desalination (e.g. reverse osmosis) are more efficient but remain costly to operate and maintain. While seawater reverse osmosis (SWRO) currently holds a majority share of the desalination market, likely a result of the >5 fold increase in energy efficiency since the 1970s,5 it still requires, on average, an order of magnitude more energy than freshwater treatment.6 Environmentally, there are serious concerns surrounding the impacts of brine discharge on coastal ecosystems near SWRO plants, and the CO2 emissions from SWRO plants are significant and unavoidable.7
Electrochemical deionization processes that produce clean water from seawater, brackish water and brines are environmental-friendly and cost-effective: they can operate directly off renewably generated electricity and can operate efficiently at small scales.8 At salt concentrations below ∼10 g L−1, the theoretical efficiency of electrochemically driven desalination surpasses that of reverse osmosis (RO).9 Moreover, certain applications such as toxic waste remediation and resource mining demand ion-selectivity. Ionic selectivity can be induced in electrochemical deionization processes with much higher capacity and kinetics relative to that of physicochemical adsorption, and with more flexibility than in pressure-driven separations due to the vast array of ion-selective electrodes and electrode coatings (described in section 5 of this review).
In this review, the most prominent electrochemical deionization technologies are categorized into non-faradaic, faradaic and ion-depletion systems (Fig. 1), in addition to selective electrosorption/electrodeposition. Capacitive deionization (CDI) and electrodialysis (ED) are among the earliest and most important examples of non-faradaic processes. In CDI, ions and counterions are electrostatically attracted to their respective electrodes and adsorb on the surface to form regions of excess charge known as electrical double layers (EDL); this depletes the remaining solution of charged species and results in a deionized water stream (Fig. 1a). On the other hand, ED relies on the transport of ions through ion-exchange membranes (IEMs) that form deionized and brine streams partitioned by the membrane. In this review, we focus on CDI and refer the reader to several comprehensive reviews on the more mature ED systems.10–12
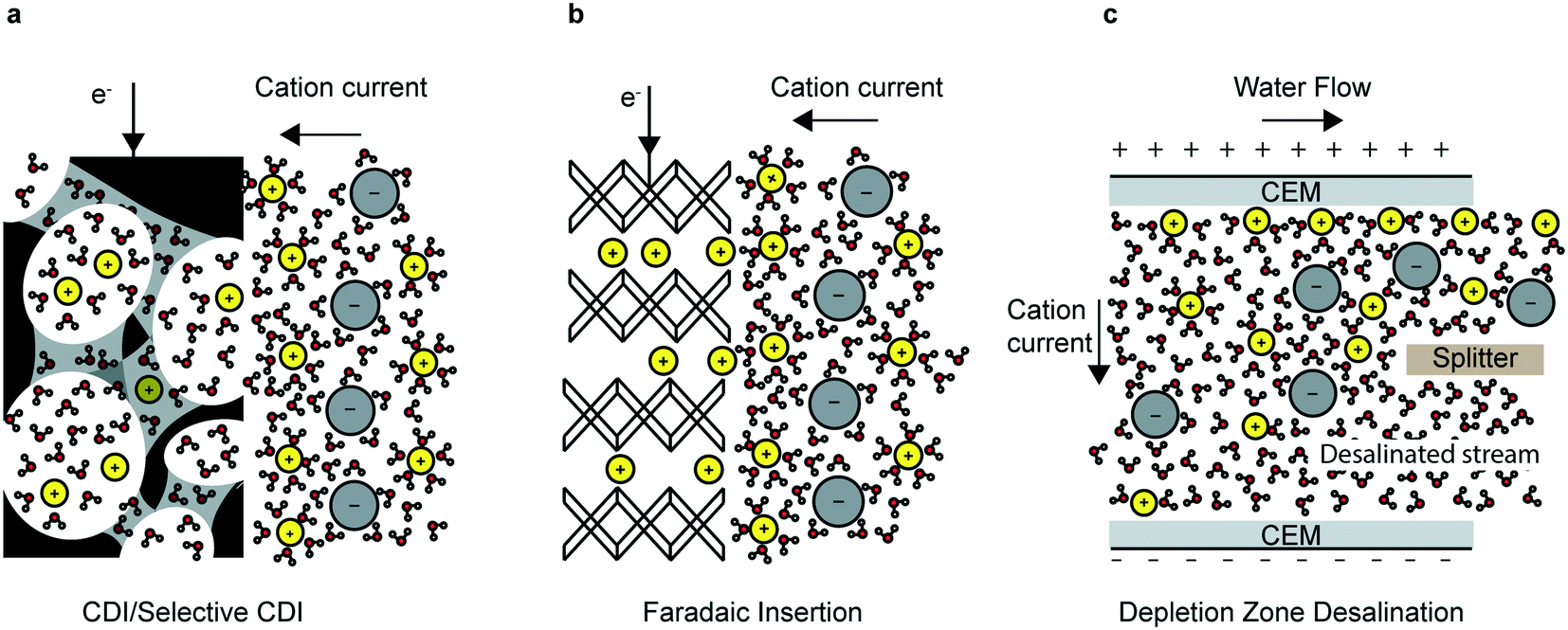 | ||
| Fig. 1 Electrochemically mediated deionization mechanisms (a) capacitive immobilization of ions in the electrical double layer. (b) Faradaic mediated intercalation of ions. (c) Deionization “shock” or depletion zone mediated ion immobilization in porous media. | ||
All electrode materials have intrinsic physicochemical properties that determine their performance in CDI applications: conductivity, surface area, pore-distribution and surface charge. Significant theoretical and experimental work has correlated microscopic physicochemical properties to CDI performance, leading to great improvements in CDI technology.13–15 Researchers have also altered and optimized CDI cell-architectures—such as flow through,8 flow-between16–18 and flow electrode19 type cells—each of which comes with their own tradeoffs, covered in detail in section 2.3 of this review. The addition of ion-selective membranes to the CDI electrodes—the combination is called membrane CDI (MCDI)—has further driven the capacity and kinetics of CDI systems, as have direct chemical modifications of the electrode material.20,21 While CDI is typically associated with desalination, selective CDI systems have also been developed to separate monovalent and divalent ions based on principles of electrostatic interaction and size exclusion.22 Modified CDI processes such as MCDI and hybrid CDI (HCDI, discussed below) have also been used for selective electrosorption of nitrates.23–26
By using the bulk of the electrode material, instead of just the surface (as is the case for CDI), the total capacity can be increased and efficiency in highly concentrated feed solutions can be improved.27 The desalination battery was the first faradaic intercalation desalination system (Fig. 1b). Since the invention of the desalination battery in 2012,28 researchers have identified novel insertion electrode materials based on three main criteria: electrochemical stability of the electrode and the surrounding electrolyte; chemical and mechanical robustness; and the size of the interstitial sites and the insertion pathways. Several novel high-performing faradaic desalination processes such as HCDI,27 cation insertion desalination (CID)29 and redox flow desalination30 have since been developed.
Beyond capacitive and faradaic ion immobilization mechanisms, depletion zone desalination, e.g. shock electrodialysis (SED),31 is an alternative technology examined in this review for its potential to treat ion-contaminated water. Depletion zone desalination relies on a deionized region that forms at the boundary of an ion-selective element when an overlimiting current is driven through that element (Fig. 1c).31 The mechanisms of this process are covered in detail in section 4 of this review.
Electrochemical deionization processes have found many applications in selective electrosorption/electrodeposition of ions, such as remediation of toxic ions from contaminated freshwater and resource mining from seawater. Tailored electrode coatings have been especially instrumental in advancing the field of selective electrosorption; researchers have used electroactive polymers, chelating polymers and redox-active polymers (conjugated and pendant-bearing) to coat electrodes for high capacity, highly selective separations.32,33 Some prominent examples of the latter include uranium extraction from seawater34 and chromium/arsenic oxyanion removal35 from wastewater. Such electrode modifications coupled with modulated electric field techniques have resulted in further enhanced selectivity. The mechanisms of this process, while capacitive/pseudocapacitive in nature (Fig. 1a), are covered in more detail in section 5 of this review.
It is important, for further development of electrochemical deionization technology, that there be standardization of experimental conditions and consistent reporting of performance metrics. Considering the vast amounts of capital resources and time currently being dedicated to the development of new electrode materials, cell architectures and novel selective chemistries, this field only stands to gain popularity in the future. This review aims to highlight the key methods, developments, and future opportunities in prominent electrochemical deionization methods.
2. Capacitive deionization
CDI is an electrochemical technique employing high-surface area, conductive materials that adsorb ions from saline water. In a CDI system, two electrodes are submerged in an electrolyte; on the application of a voltage bias between the electrodes, cations migrate towards the negatively charged cathode and anions towards the anode, where the ions are stored in the electrical double layer formed at the liquid–solid interface, effectively desalinating the feed solution. Once saturated with ions, the electrodes are short circuited or a reverse bias is applied, discharging the stored ions into a brine stream and regenerating the electrodes.16,36 The relative advantages of CDI are detailed in Table 2, but in short, CDI is particularly energy efficient at low salt concentrations; it is characterized by fast adsorption rates, but relatively low salt adsorption capacities compared to faradaic insertion (see Fig. 8).2.1 CDI performance metrics
A single metric quantifying CDI effectiveness can vary with many system parameters, and optimization of one metric often occurs at the detriment of another. In order to accurately describe a given system's CDI performance, multiple metrics must be reported in tandem. As we outline below, reporting a rate vs. capacity diagram37 and specific energy efficiency, allows new research to be easily comparable to existing literature (which often just report rate and/or capacity), while providing a thorough assessment of performance. Additionally, recent research on CDI metrics demonstrates that reporting the salt removal fraction (calculated from the concentration ratio of the deionized permeate to the saline feed solutions) along with energy efficiency at standardized testing conditions provides direct system-level information on how the CDI device will perform in practice.38 Given that performance is highly dependent on test conditions (e.g. feed concentration, as shown in Fig. 8c), experiment parameters should be standardized and always reported in full.Researchers often quote the charge capacitance of electrodes, easily calculable from cyclic voltammetry curves,39 which is an appropriate metric only if the charge efficiency—i.e. number of electrons exchanged per ion adsorbed—is also provided.40 If the charge efficiency is omitted, the quoted performance could be artificially high due to co-ion expulsion from the electrode. More appropriate measures of electrode performance are the energy efficiency with which a system operates, the rate at which salt is removed from the feed solution and the amount (capacity) of salt that can be removed in a single charging step. High performance for one parameter often comes at the cost of another, so all three should be reported in tandem.
The energy efficiency of CDI is one of the primary ways to evaluate a system's usefulness, but it is an insufficient metric if reported in isolation. The total specific energy consumption (SEC) is commonly quoted in kW h m−3 (energy cost per volume of desalinated product) or J mg−1 (energy cost per mass of salt removed). The former is used frequently in reverse osmosis systems but the latter is more relevant for CDI research, where the permeate concentration varies from experiment to experiment.36 Given that the energy required for a given separation process depends on the feed concentration, brine concentration, permeate concentration and water recovery, SEC must be paired with these parameters, or normalized by the theoretical minimum energy cost, for this metric to be meaningful.41 Furthermore, a direct tradeoff exists between SEC—often quoted as its inverse, specific energy efficiency (SEC−1)—and deionization rate and capacity41 (Fig. 2a).. Thus rate vs. capacity diagrams, paired with energy efficiency, captures the real performance of a CDI system. The rate of salt removal is typically recorded as the average salt adsorption rate (ASAR) of an electrode: the total mass of salt adsorbed, normalized by the sampling interval and the combined mass of the cathode and anode (commonly quoted in units of mg g−1 s−1). ASAR reported together with the salt adsorption capacity (SAC), commonly quoted in units of mg g−1, accurately quantifies electrode performance given no efficiency constraints.37 “ASAR vs. SAC” plots, shown in Fig. 2b and c, have been employed by many publications in the CDI field.36,41,42 Other practical performance metrics are CDI cycling stability, anti-fouling capability, water recovery percentage and round-trip efficiency/energy recovery.43
 | ||
| Fig. 2 (a) ASAR vs. SEC−1 plot showing the trade-off between energy efficiency and rate. Both experimental and simulated data are depicted.42 Reproduced with permission from ref. 42. Copyright 2018 Elsevier. (b) ASAR vs. SAC (rate vs. capacity) plot for generic electrodes, showing the trade-off between parameters. Dashed lines represent the time required for complete charging.37 Reproduced with permission from ref. 37. Copyright 2014 Royal Society of Chemistry. (c) Rate vs. capacity plot for recently developed mesoporous block copolymer electrodes (described in detail in section 2.2). In showing the entirety of this rate vs. capacity plot, instead of a single point, the higher performance of this block copolymer electrode is definitively established. References for data displayed in this plot can be found in the original study.44 Reproduced with permission from ref. 44. Copyright 2020 AAAS. | ||
2.2 CDI electrode materials
The majority of CDI systems incorporate carbon electrodes given their intrinsic conductivity, high porosity and low cost.45 Other classes of electrode materials for CDI include metal-oxides, metal-oxide carbon composites, and polymer carbon or polymer metal-oxide composites. In the following, we outline only carbon-based and two-dimensional materials, referring the reader to existing reviews on metal oxide and composite polymer materials.45,46 To compare electrode materials described in this section, we rely on ASAR vs. SAC diagrams due to their broad adoption and direct relevance to CDI performance; see Fig. 8a for a performance summary of existing CDI electrodes.Examples of modifications to activated carbon include the addition of carbon black with a polymeric binder (more than doubling total salt removal without changing system conditions),49 KOH etching and CO2 gasification of coconut shell-based activated carbon to tune the pore size distribution; an increased density of micropores led to enhanced SAC relative to most other activated carbon electrodes.47 By tuning the pore size distribution of activated carbon synthesized by the template method, a subsequent study measured a slightly larger SAC.50 An increased SAC (∼a factor of two over bare activated carbon) and CDI performance was also observed in reduced graphene-oxide activated carbon composites.51 In these electrodes, the graphene oxide acts as a bridge between activated carbon particles, simultaneously preventing restacking—increasing specific surface area—and enhancing electrode conductivity. Employing doped activated carbon electrodes in an asymmetric CDI cell has proven an effective method to enhance the SAC, while still employing relatively low-cost materials, as in a p-toluenesulfonate doped carbonized polyaniline electrode,52 and more recently a N-doped activated carbon electrode (see Fig. 8a).53
To further improve the performance of graphene electrodes, several groups prepared three-dimensional graphene structures by using sponge61 or polysterene39 templates, increasing the accessible surface area. In the former, the specific surface area reached 305 m2 g−1 leading to greater ion adsorption capacity of 4.95 mg g−1 for a 0.5 M NaCl solution. The total electrosorption capacity of graphene-based electrodes was pushed beyond that of activated carbon and carbon aerogels by increasing the frequency of defects in the graphene sheets, which effectively increases the density of micropores and dramatically increases the ion adsorption capacity (see Fig. 8a).62,63
The performance of graphene CDI electrodes was also enhanced via the addition of carbon nanotubes to graphene oxide sheets. The composite demonstrated higher conductivity and porosity relative to pure graphene oxide electrodes; the study reported high adsorption capacity (Fig. 8a), a 98% salt removal efficiency of 1500 ppm NaCl solution, all with low energy requirements (0.8 kW h m−3).64 Recently, two-dimensional materials beyond graphene have captured significant interest for a variety of applications, from semiconductors to catalysts and membranes; examples include MXenes65,66 and MoS2/graphene67 composite materials. MoS2, chemically exfoliated in its metallic 1 T phase, was shown to act as high-performing CDI electrode.68 By increasing the frequency of defect sites by thermally treating flower-like MoS2, the specific capacity was increased nearly threefold, approaching the capacity of the highest-performing graphene-based CDI electrodes, and surpassing that of carbon aerogels and activated carbon (see Fig. 8a).
2.3 CDI systems
Several CDI architectures have been developed, each of which have their unique tradeoffs, as described in Table 1. As a significant portion of CDI research focuses on electrode material (as opposed to system-level innovation) and CDI technology remains in the research phase, the simplest systems – flow-between CDI and membrane CDI – are the default. Flow electrode CDI, invented only recently, shows great promise as a new technique that transcends the capacity limitations of more standard configurations.| CDI configurations | Advantages | Disadvantages |
|---|---|---|
| Flow-between | • Simple and economical | • Co-ion expulsion leads to reduced capacity and efficiency |
| • High water flow rate possible (water does not pass through any active element) | • Diffusion-limited desalination (incomplete desalination for large volumes or high flow rates) | |
| • Isolates electrode characteristics – useful for fundamental studies | ||
| Membrane CDI | • Co-ion expulsion eliminated | • High cost of ion-selective membrane |
| • High water flow rate possible | • Diffusion limited desalination | |
| • Tailored ion-selective separation possible with existing technology | ||
| Flow-through CDI | • Close contact of feed with electrode at all points (fast adsorption rates) | • Electrode fouling is exacerbated |
| • Water flow rate is limited by the pore size distribution of the electrode | ||
| • Ion selective membrane is difficult to adopt due to water flux constraints | ||
| Flow-electrode CDI | • Continuous desalination possible (no pause for desorption step needed) | • Complicated implementation with limited architectures (flow-through not feasible) |
| • Effective capacity (mass of salt removed, normalized by the contact area between the feed solution and electrode compartment) can be increased simply by increasing electrode flow rate | • Electrode must be “flowable”, limiting material choice |
 | ||
| Fig. 3 (a) Example of MCDI applied to selective removal of divalent cations.77 This setup is in the “flow-between” geometry, as is standard for MCDI systems. Reproduced with permission from ref. 77. Copyright 2019 American Chemical Society. (b) An FCDI setup, wherein not only the feed solution, but the electrodes themselves, are continuously replenished. In this work, the electrodes are cycled from one half-cell to the other to allow continuous reuse of the active material.78 Reproduced with permission from ref. 78. Copyright 2020 American Chemical Society. | ||
In a typical FCDI electrode, where a mixture of activated carbon and a carbon black is used, the conductivity is only 0.1–1 mS cm−1, several orders of magnitude lower than conventional CDI electrodes.84 By sacrificing SAC for electrical conductivity (using only carbon black as the electrode material) a recent study demonstrated enhanced ASAR, scaled by the membrane area, by a factor of five.78 Consequently, the total salt removal increased without any decrease in energy efficiency. The absolute value of energy efficiency reported in this same study, ∼8 μmol J−1, is on the same order of magnitude as that of a standard MCDI system,41 but we remind the reader that such comparisons are complicated by variation of experimental conditions (see section on CDI metrics).
2.4 CDI theory
Over the past two decades, accurate and tractable models describing CDI have been developed, the bulk of which rely on a sophisticated theoretical understanding of the electrical double layer in porous structures on multiple length scales.85 The usual model system is comprised of an aqueous electrolyte solution embedded in a conductive porous electrode. In non-faradaic CDI, charge is stored exclusively in the electrical double layer (EDL) at the interface of the electrode and electrolyte in the form of adsorbed ions. The classical description of the EDL, which is applicable for mesoporous structures or highly concentrated electrolytes, is the Guoy–Chapman–Stern (GCS) model. Here, the EDL is composed of the Stern layer—a fixed capacity region at the electrode surface stemming from the finite size of adsorbed ions—and a diffuse region with a mixture of co-ions and counterions.86,87The GCS model system applies for both CDI electrodes and supercapacitors, with a few crucial differences: in the former, the bulk electrolyte concentration changes appreciably with time (whereas for supercapacitors a negligible fraction of the ions from the bulk solution are adsorbed). Further, the ratio of counterion adsorption and co-ion expulsion is, for CDI, both important (whereas for supercapacitors only the overall charge matters) and, in useful systems, always >1. As a result, the standard transmission line model (an RC circuit where the ionic resistance of the electrolyte is added in series to the capacitance of the EDL) used to describe supercapacitors and the original CDI systems,88 fail to describe CDI behavior.86 The GCS model has been used to describe systems in which the EDL is small compared to the size of the pores; it parameterizes the Nernst–Planck equations describing ion adsorption dynamics for a range of applied voltages and feed concentrations.87
When the Debye length is larger than the size of the electrode micropores (<2 nm in diameter), the GCS model breaks down.89 The EDL, instead of being a smooth layer on a flat or gently curving surface, has discontinuities at the location of micropores, where the EDL on the sides of the pores overlap. To account for these discontinuities, the modified Donnan (mD) model was developed. The mD model is an extension of the Donnan model,90,91 which assumes complete overlap of the Debye layer within pores, thereby creating a step-function potential over the porous electrode surface. The mD model adds two important factors: first, it includes the Stern layer at the pore surface; second, it adds an attractive chemical potential in order to describe data showing salt adsorption in microporous structures without an applied voltage.14 The ion concentration in micropores can then be described according to the Boltzmann equilibrium relation.
The mD model was further improved by allowing the chemical potential term to vary with the micropore salt concentration, thereby eliminating the prediction of unrealistically large adsorption capacities at high salt concentrations.14 It has also been extended from the one-dimensional case to cell-level, two-dimensional systems,92 and has been corroborated by molecular dynamic simulations.93 The mD model was applied to describe a series of CDI developments such as “inverted CDI” by fixing charge in the micropores to emulate chemical treatment,15 inclusion of surface transport94 and an explanation of the benefits of pulsed-flow CDI over continuous flow systems.95
Directly consequential for materials design, the mD model incorporates – although it fails to fully explain—the effect of micropores on electrode capacity through the non-electrostatic potential term. The effect of micropores on CDI performance has been subsequently quantified through the fitting of pore size distributions,96 with the strict condition that capacity increases as micropore fraction increases, but decreases as mesopore fraction increases (it is important to note that although capacity may suffer with increasing mesoporosity, adsorption rate will benefit from the increased density of ion pathways in the electrode96). As shown in Fig. 4, this guided fitting procedure leads to an accurate predictor of electrode capacity. The importance of micropore fraction in electrode capacitance has been corroborated repeatedly and is a standard guideline for electrode materials design.97
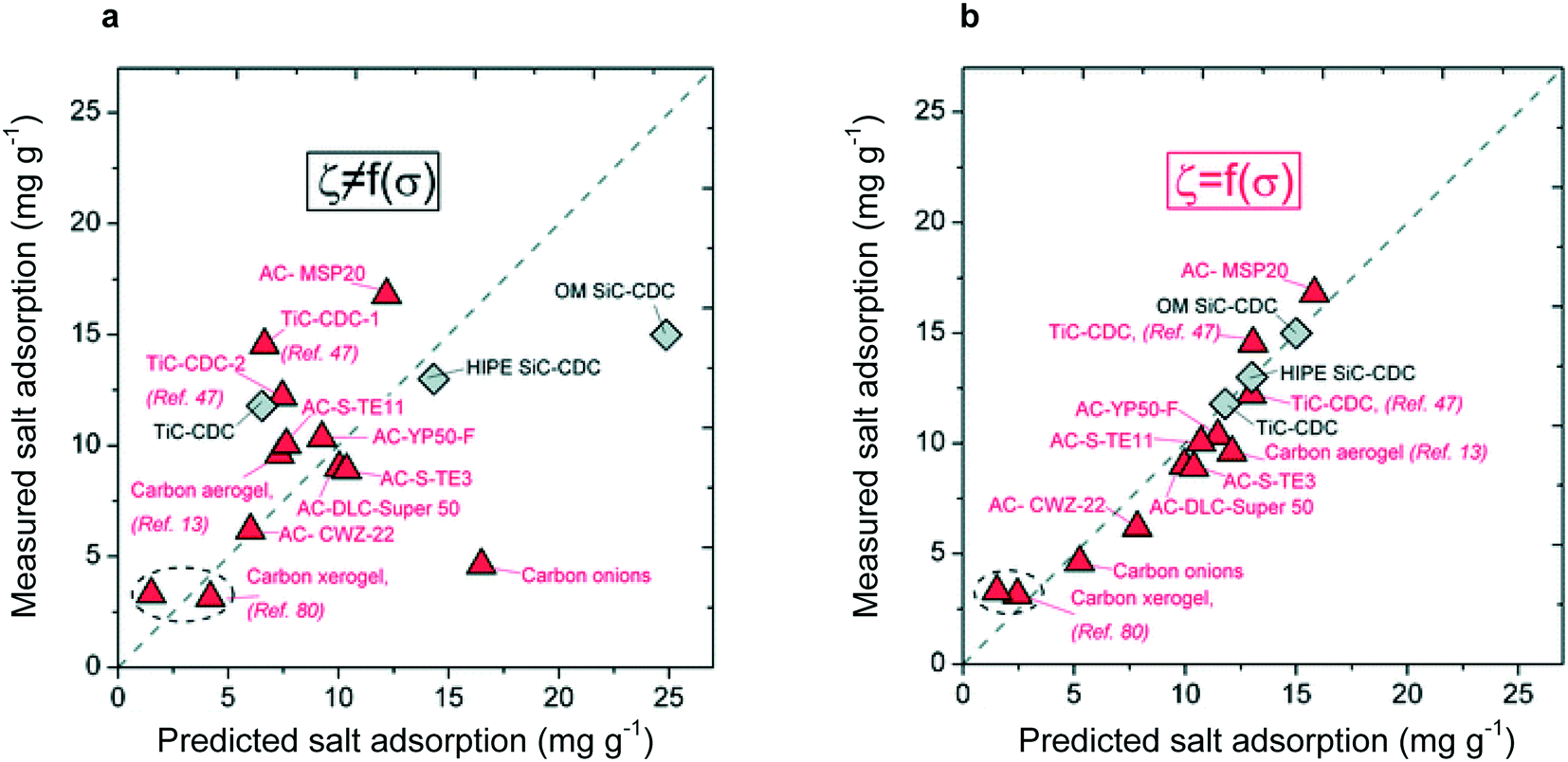 | ||
| Fig. 4 (a) Adsorption capacities predicted through an empirically based model without consideration of pore size distribution. No relationship between predicted and measured adsorption capacity is observed. (b) Adsorption capacity predicted through empirical model considering only the pore size distribution, leading to a one-to-one correspondence between predicted and measured values.96 This work displayed the importance of the pore size distribution, over just frequency of pores, in determining adsorption capacity. Reproduced with permission from ref. 96. Copyright 2013 American Chemical Society. | ||
Recently, a simplified approach based on the Langmuir isotherm (as opposed to a physics-based description of the EDL) has been developed and named the dynamic Langmuir isotherm.98 The use of the Langmuir isotherm has been criticized in existing reviews,9,36 which claim that the underlying physics are based on the adsorption of uncharged gasses, and as such cannot incorporate the effects of co-ion expulsion (charge efficiency), asymmetric electrodes, or voltage dependence.
The dynamic Langmuir isotherm accounts for these factors, however, as well as ion composition; the model accurately fits experimental CDI data, taken under a range of experimental conditions. A major benefit of this model is that it can describe and predict, among other things, selective adsorption from mixed ion solutions for all possible ion combinations.99 However, because the model omits the details of electrode structure or material, it must be calibrated for each new material used, a drawback that will surely limit the applicability of this theoretical development.
2.5 Selective CDI
The selective capabilities of pristine carbon surfaces in CDI cells have been demonstrated in several studies involving lithium,100,101 nitrate24–26,102 and fluoride removal.103 In general, two factors intrinsic to all CDI processes affect selectivity of one ion over another: steric hinderance and electrostatic attraction. These two effects often work in opposition to each other: for small pore sizes, the diffusion of large and multi-valent ions is hindered, giving rise to selectivity towards monovalent ions;22,104 multivalent ions, however, experience a larger attraction to the charged electrode surface, leading to displacement of monovalent ions with multivalent species.105Depending on the application, selectivity towards monovalent or multivalent ions is preferred. Relying only on the steric effects of micropores, and without employing an IEM or functionalizing electrode surfaces, monovalent ions are typically selectively adsorbed. In one study,22 researchers pursued selectivity towards monovalent ions by continuously tuning the pore size of activated carbon via chemical vapor deposition. At pore diameters falling between the hydrated radii of alkaline earth cations such as Ca2+ and Mg2+ and that of monovalent cations such as Na+, electrodes only adsorbed the monovalent ions, leaving the divalent cations in solution. Size selectivity was also employed to demonstrate selective removal of nitrates over chloride and sulfates in feed solutions, as the nitrate ion is only weekly hydrated.23
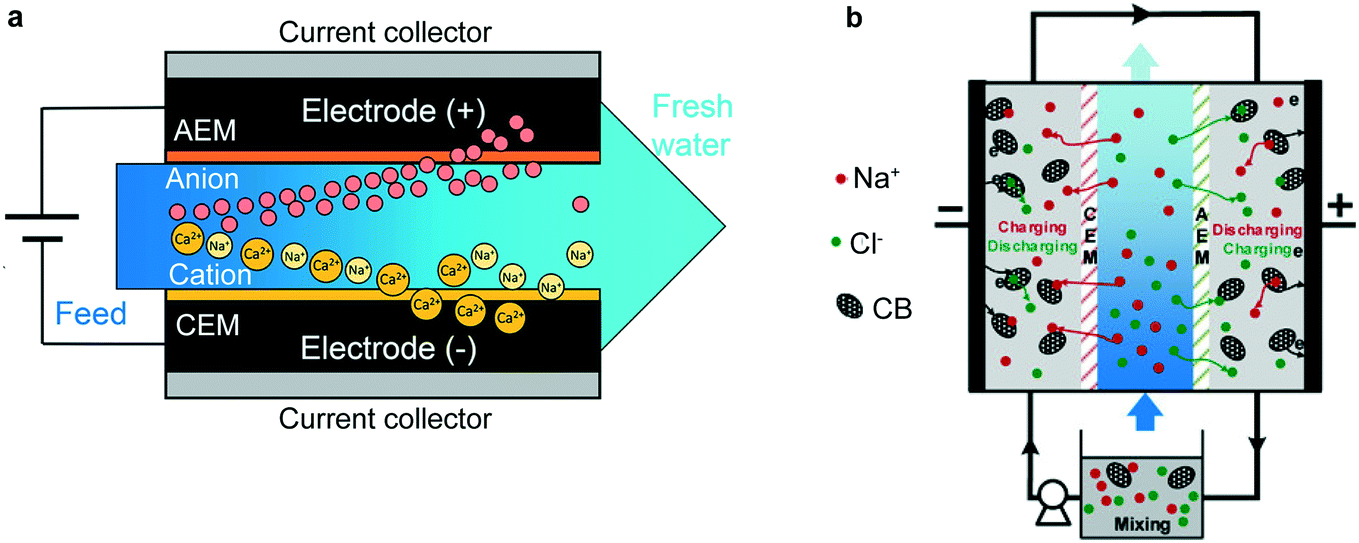
Covered in great detail in section 5, coatings and membranes can be applied to electrode surfaces in order to induce selectivity, as opposed to relying on selectivity intrinsic to the electrode material.77 For example, a recent study employed a cation exchange membrane in an FCDI system to selectively remove Ca2+ over Na+ with applications in water softening. This study demonstrated 6-fold selectivity of Ca2+ over Na+ at low (10 mA m−2) current densities in a 2000 mg L−1 NaCl and 150 mg L−1 CaCl2 solution. Energy consumption was only 0.44 kW h to achieve an effluent salinity of 500 mg L−1 and Ca2+ concentration of 15 mg L−1.
3. Faradaic desalination
Electrochemically driven desalination based on faradaic reactions has been a growing field of study since the creation of the desalination battery by Pasta et al. in 2012.28 The desalination battery system is like CDI, only charge is stored in the chemical bonds in the bulk of the electrode material instead of the electric double layer on the surface of the material. Ion storage occurs through insertion of the ion into a lattice site of a host crystal structure coupled with a redox reaction of an element contained in the host. The low fixed voltages for driving the insertion mechanism results in a low energy input system. Also, insertion materials have high specific capacities for ion removal, but have a slower ion removal rate compared to standard CDI for desalination.106,107 Faradaic desalination performance has been summarized in Fig. 8b and S1, and Table S2.† These electrode materials greatly benefit from the vast amount of research in energy storage, especially the development of aqueous Na-ion batteries, which are governed by the same parameters.108–1103.1 Materials
Extensive research into energy storage materials has elucidated a set of constraints for batteries used in aqueous electrolytes; these constraints transfer directly to faradaic desalination. First, the operating potential of the material must fall within the redox potential window of the electrolyte. In the case of aqueous electrolyte, the window is set by the evolution potentials of H2 and O2via electrochemical water splitting.111,112 Electrode materials that meet this constraint, along with the pH dependence of the aqueous redox potential, are shown in Fig. 5.108 Second, aqueous battery materials must be chemically stable in the given pH of the aqueous electrolyte and must avoid electrode dissolution and side reactions with dissolved O2.112 Third, materials must have interstitial sites and migration pathways that are large enough to allow ion storage while minimizing volume change during cycling.113–115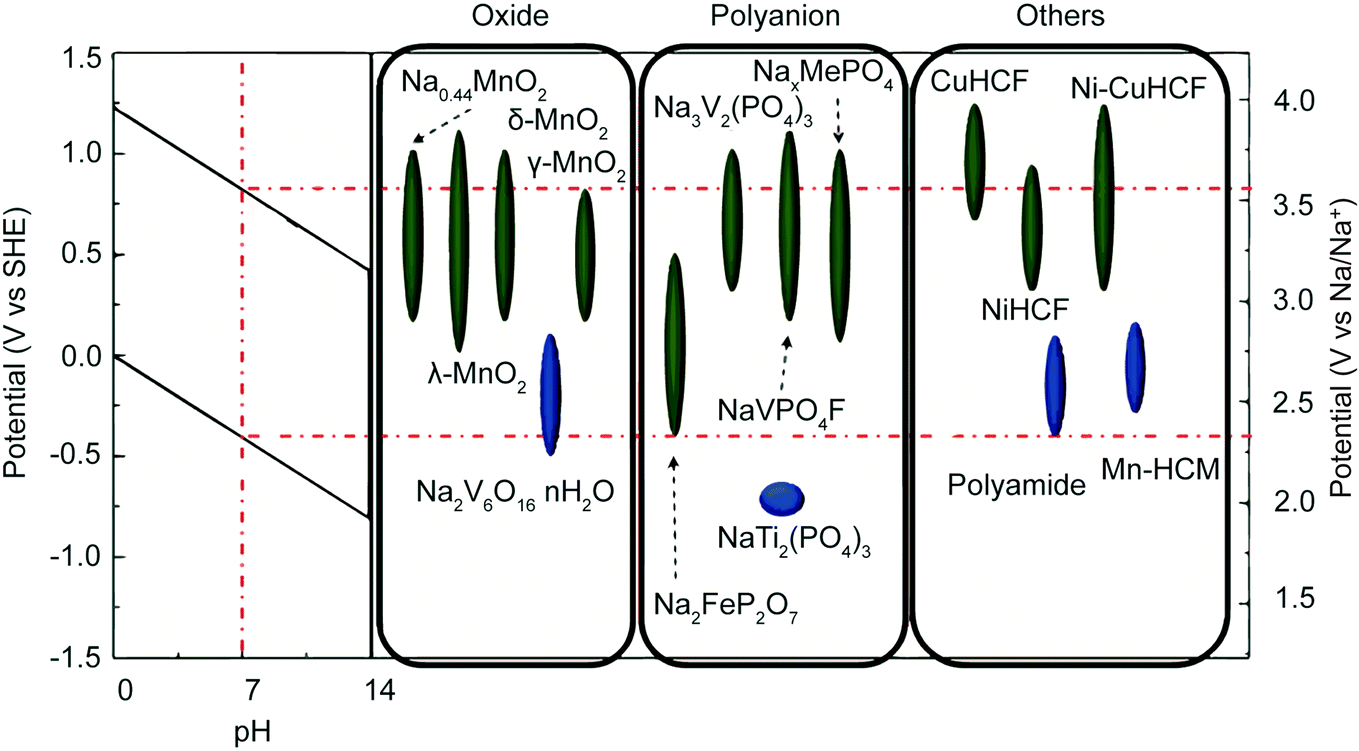 | ||
| Fig. 5 Electrode material operating potentials vs. SHE and Na/Na+. The red dotted line indicates the H2 and O2 evolution limits at neutral pH. The leftmost inset shows the evolution voltage limits as a function of pH. Reproduced with permission from ref. 108. Copyright 2014 American Chemical Society. | ||
Na-ion insertion electrodes used for desalination of aqueous NaCl solutions include different phases of NaxMnO2 (NMO),28,116–118 Na2FeP2O7,119,120 NASICON-type Na3V2(PO4)3,121,122 NASICON-type NaTi2(PO4)3 (NTP),123,124 and Prussian blue and its analogues.125–128 The mechanisms for Cl− capture include conversion reactions with silver/silver chloride and bismuth/bismuth oxychloride;28,123,129 oxidation of Cl− to form chloride species (Cl2, HOCl, radical Cl);130 and intercalation into MXenes, which can host a wide range of cations and anions.131–133
3.2 Desalination batteries
The first desalination battery in 2012 used Na2Mn5O10 (NMO) nanorods for Na+ removal and Ag for Cl− removal, demonstrating membrane-free desalination with battery materials.28 As shown in Fig. 6a, desalination follows a four-step process. The electrode selectivity for intercalation of Na+ in NMO and reaction of Cl− with Ag prevent the co-ion expulsion effect; an ion selective membrane is therefore not necessary. This process required 0.29 kW h m−3 for 25% salt removal, approaching the reverse osmosis energy requirement of 0.20 kW h m−3 for similar conditions. The key limitations were the low NMO electrode capacity of 35 mA h g−1 relative to the average capacity of 160 mA h g−1 of Li-intercalation electrodes, the high cost of Ag, and the kinetic limitations of AgCl due to its poor electronic conductivity.28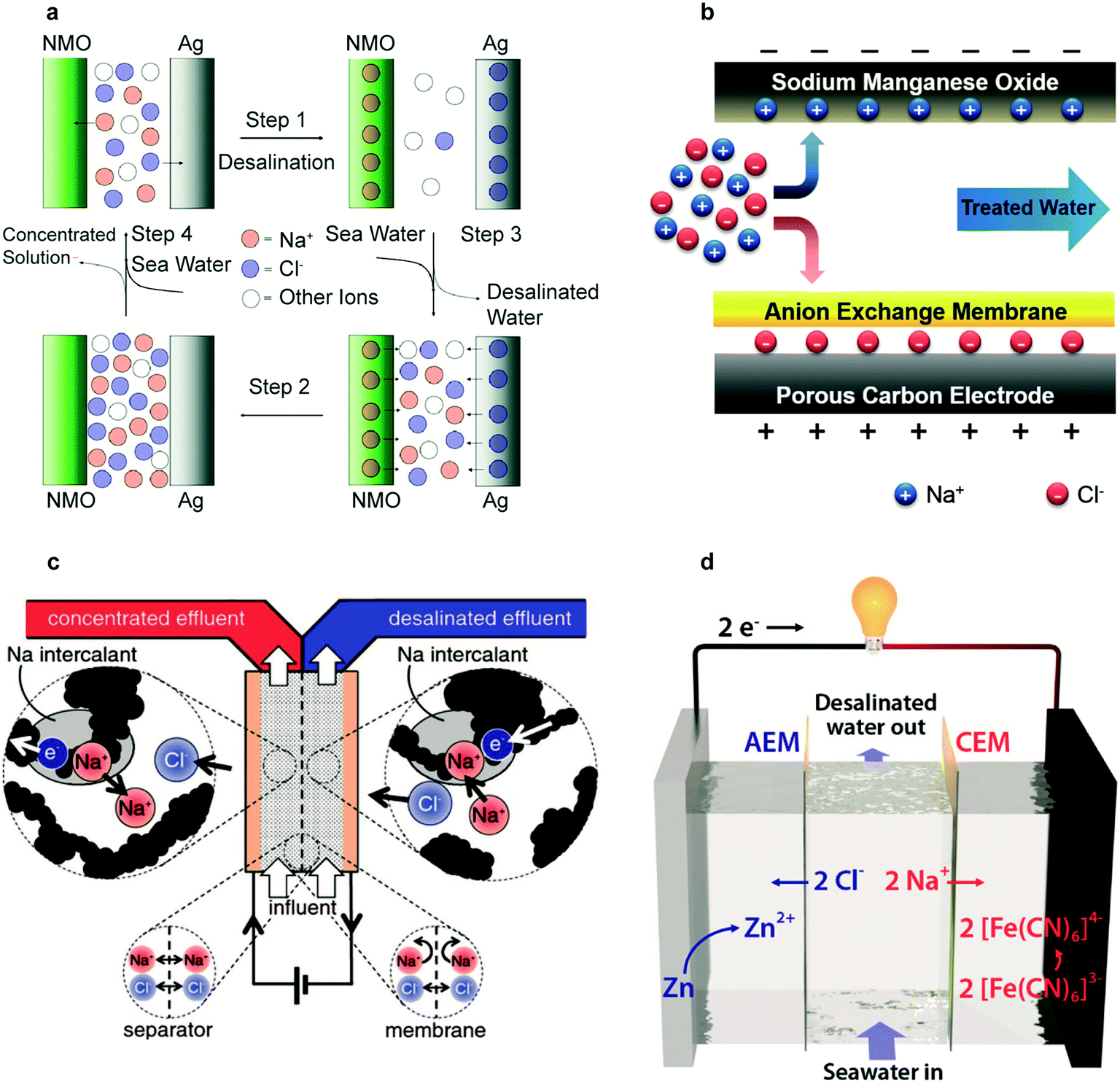 | ||
| Fig. 6 (a) Schematic representation of the complete cycle of the desalination battery. Reproduced with permission from ref. 28. Copyright 2012 American Chemical Society. (b) Schematic of HCDI during the desalination process. Reproduced with permission from ref. 118. Copyright 2014 Royal Society of Chemistry. (c) Schematic of the symmetric CID cell during desalination of the right stream and concentration of the left stream. Reproduced with permission from ref. 29. Copyright 2016 The Electrochemical Society. (d) Schematic of the flow battery desalination cell during the desalination process. Reproduced with permission from ref. 30. Copyright 2018 American Chemical Society. | ||
The promising results of this seminal study have motivated further research of intercalation materials for desalination batteries, focused mainly on development of high capacity Na-insertion electrodes and alternatives for the Ag electrode. A potential alternative to Ag electrodes for Cl− storage, bismuth electrodes exploit the conversion of Bi to BiOCl.123 The primary motivation for this alternative is its lower cost.134,135 Unfortunately, Bi electrodes suffer from both H+ production during oxidation of Bi to BiOCl, which lowers the solution pH, and slow kinetics of the reduction reaction in non-acidic conditions. The Na+ removal was performed with a NASICON-type NaTi2(PO4)3 electrode due to its high theoretical capacity. This electrode paring with Bi removes 3 Na+ for every Cl−, which decreases the pH. Due to the imbalance of the ion removal and the decreasing pH, the NaTi2(PO4)3 was replaced with a Pt electrode in a second deionization step to reduce the H+ ions to H2 while the remaining Cl− were removed, thus neutralizing the desalinated water. A salination step must be performed in a waste stream to regenerate the original electrode structures. NaTi2(PO4)3 will spontaneously oxidize in acidic conditions, however. To prevent this, a Nafion membrane was added to protect the Na-electrode from the acidic electrolyte required to lower the kinetics for the BiOCl reduction.
With a 0.6 M NaCl feed solution, the process required 5.9 kW h m−3 for removal of 21.23 g of Cl−, a higher value than the 1.3 kW h m−3 required for the NMO/Ag battery under similar conditions.28 Further research replaced NAISCON-type NaTi2(PO4)3 with the Prussian blue analogue, Cu3[Fe(CN)6]2·nH2O, which is chemically stable in acidic conditions but has a lower capacity.136 This electrode change allows for a membrane-free desalination and salination process but with an energy cost increase. The increase originates in the voltage difference between the two electrodes where the NAISCON-type electrode has nonspontaneous desalination and spontaneous salination while the Prussian blue analogue has spontaneous desalination and nonspontaneous salination when coupled with the Bi electrode.
Modified cell architecture can improve the desalination performance of brackish feed solutions. One design created a three-stream system by including both a cation selective membrane (CEM) and an anion selective membrane (AEM). The center stream contains the desalinated water and two side streams at the insertion electrode interfaces are held at higher concentration than the feed. This increases the mass transfer limit at the electrode–electrolyte interface, which prevents kinetic limitations and improves the ion removal rate when dealing with lower concentration feed solutions.137
3.3 Hybrid capacitive deionization
The desalination battery and CDI systems both have inherent limitations, which can be surpassed when the systems are combined. The incorporation of a Na+ insertion electrode allows an increase of the cation storage capacity and induces cation selectivity. CDI outperforms the desalination battery design in salt removal rate, which in a desalination battery is limited by the slow kinetics of the Cl− capture electrodes. Hybrid systems were developed by replacing one of the carbon electrodes from CDI with an insertion electrode from the desalination battery, thus combining the high rates of CDI with the large capacity and selectivity of the desalination battery. The seminal work on hybrid CDI (HCDI) incorporated a Na4Mn9O18 electrode into a CDI system. In this system, Na4Mn9O18 captures Na+via intercalation while Cl− crosses an AEM for storage in the EDL of an activated carbon electrode (Fig. 6b).118 The total capacity of the HCDI was much higher than their own results for CDI and MCDI tested under the same conditions. The peak ion removal rate for HCDI was higher than their results for CDI but slightly lower than MCDI tested under the same conditions. The desalination performance in this work demonstrated the benefits of HCDI and motivated further development of the system with new cathode materials.HCDI has been successfully demonstrated using a suite of cathode materials: Na2FeP2O7,120 Prussian blue NaxFe2(CN)6,127 and different phases of MnO2.138–140 Key results from these works include the establishment of ion removal capacity and ion removal rate relationships, identification of materials with open frameworks resulting in faster kinetics, and elucidation of phase changes due to multi-ion insertion. For establishing ion removal capacity and ion removal rate relationships, results from desalination with Na2FeP2O7 indicate that with slower ion removal rates ion removal capacity is higher relative to MCDI (20% increased capacity at 10 mM NaCl feed solution and 50% at 100 mM NaCl feed solution).120 At faster ion removal rates, however, the ion removal capacity is equivalent to MCDI at 10 mM NaCl feed solution and larger by only 20% at 100 mM NaCl.
For materials with open frameworks, Prussian blue NaxFe2(CN)6 was studied for its theoretically high capacity and fast insertion kinetics.127 Prussian blue electrodes were synthesized as nanocubes incorporated into a nanoporous, reduced graphene oxide framework (PB@NPG). PB@NPG achieves a high ion removal rate of 0.5430 mg g−1 s−1 with a capacity of 20 mg g−1 at a 40 C applied current. The increased capacity of PB@NPG is possibly due to the NPG protecting the PB from flowing electrolyte, which can remove particles adhered to the surface of graphene oxide. The porous electrode structure also improves the rate capabilities. PB@NPG sustained an ion removal rate of 0.2925 mg g−1 s−1 at a capacity of 40.8 mg g−1 for 600 cycles at a 20 C applied current. The incorporation of insertion electrodes into porous structures enables improvements of both capacity and rate.141
For investigating the phase changes resulting from multi-cation insertion, layered manganese oxides containing hydrated cations were employed in an HCDI system. These structures included Na-birnessite and Mg-buserite in solutions of NaCl and MgCl2.140 The structure and phase transformations of insertion electrodes due to such mixed electrolytes is relevant for HCDI systems operating in real brackish water and seawater, which can contain Na+, K+, Mg2+, and Ca2+.138,140 Electron dispersion spectroscopy and X-ray diffraction (XRD) measurements indicate that insertion of Mg2+ into Na-birnessite and Na+ into Mg-buserite is accompanied by material phase transformations. XRD measurements revealed that Mg-buserite has a lower crystallinity than Na-birnessite, which may cause poor stability in the Mg-buserite. The degree of crystallinity was determined by the full width half max of the (001) peaks and the presence of XRD peak shoulders upon cycling. From this work it is reasonable to infer that materials with higher crystallinity provide longer cycling stability. Further understanding of insertion material changes due to multi-ion insertion and its effects on desalination performance will be necessary for using faradaic insertion desalination in real conditions.
3.4 Cation insertion desalination
Insertion electrodes have demonstrated high capacities for Na+ removal in desalination batteries and HCDI. Taking advantage of this high capacity, Smith and Dmello proposed a symmetric cell design, currently called cation insertion desalination (CID), and simulated the local electrochemical processes in which both electrodes are built from cation insertion material, only one is filled to capacity with Na+ and the other is empty.29 Inspired by the rocking-chair battery concept in Li-ion batteries, the symmetric design takes advantage of the salt depletion effect, the simultaneous accumulation and depletion of salt ions in solution at opposing electrodes.142 As shown in Fig. 6c, Na+ intercalates into the anode from a NaCl influent stream producing a desalinated effluent and Na+ deintercalated from the cathode into a parallel influent stream producing a concentrated effluent that is separated by an anion selective membrane. The charge imbalance between the two streams causes Cl− ions in the anode-side stream to diffuse across the anion selective membrane into the cathode-side stream, creating a freshwater stream and brine stream. Once the capacity of the electrodes is filled or exhausted, the effluent tanks are switched, allowing the process to continue. An NMO CID cell operating at a C/2 rate was predicted to desalinate a 700 mM NaCl solution by 63% with energy consumption of 0.74 kW h m−3 and 50% water recovery. The work also predicted 59–64% drop in influent salinity for 70 mM and 700 mM with water recovery levels of 95% and 80%, respectively. These optimistic simulation results have motivated additional development of porous electrode theory coupled with fluid flow and ion transport across anion selective membranes for desalination with intercalation materials.143–145In experimental settings, the rocking chair design was implemented with two Prussian blue electrodes, NaNiHCF and NaFeHCF.146 The Prussian blue electrodes were chosen for their higher capacity relative to that of NMO and their ability to reversibly intercalate Na+, K+ Mg2+, and Ca2+.126,147 In seawater conditions, the Prussian blue system removed 40% of Na+ with an energy consumption of 0.34 kW h m−3 and 50% water recovery. For 25% Na+ removal, the energy consumption is 0.14 kW h m−3 which is much lower than the 0.29 kW h m−3 for the original desalination battery.28 These results demonstrated high desalination capacity at a low energy cost, but further optimization is needed to approach water recovery levels of 95%, as proposed by Smith and Dmello.29 Analogous to the CID system, anion desalination has been experimentally demonstrated with symmetric Ag/AgCl electrodes. The Ag electrode benefits from a high theoretical capacity, but has kinetic limitations, especially in brackish conditions.148
Recent related designs have taken advantage of the salt depletion effect to lower energy consumption, improve mass-limiting current, and desalinate highly concentrated influents. Adding cation and anion selective membranes in a CID cell creates new brine streams and freshwater streams without adding additional electrodes. Scaling up the system to include at least three stacks reduces the total energy consumption (Fig. 7a). For a triple stacked cell, a brackish stream of 25 mM NaCl desalinated to 17 mM NaCl requires only 0.02 kW h m−3.149
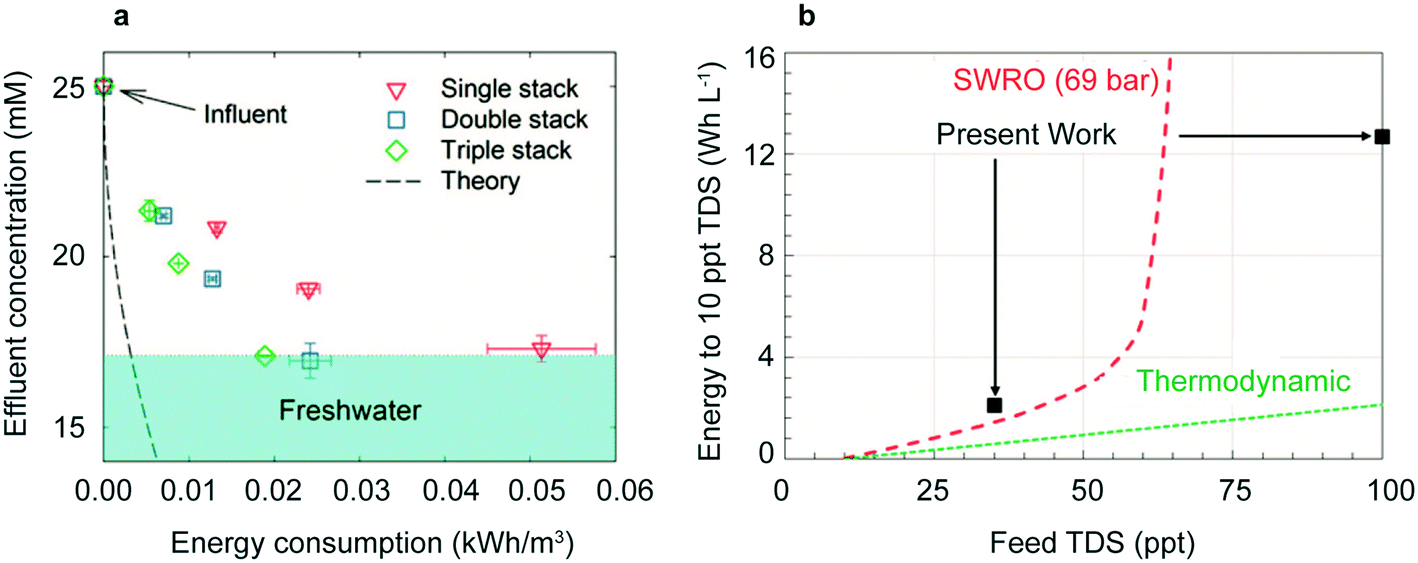 | ||
| Fig. 7 (a) Energy consumption to desalinate 25 mM NaCl for single, double, and triple stacks of membranes. Reproduced with permission from ref. 149. Copyright 2017 American Chemical Society. (b) Energy consumption of redox flow battery design compared to seawater reverse osmosis (SWRO). Reproduced with permission from ref. 30. Copyright 2018 American Chemical Society. | ||
3.5 Redox flow desalination
Redox flow desalination batteries operate without insertion electrodes but instead uses storage into a solution phase via aqueous reactants, which decouples the capacity from the rate. This also improves round trip efficiency, the ratio of energy released from discharge over the energy input from charging and increases capacity for desalination of higher salinity influents.30 During operation, a Zn anode is oxidized to Zn2+, driving Cl− into the analyte tank; at the cathode, ferricyanide is reduced to ferrocyanide, driving Na+ into the catholyte tank (Fig. 6d). This design requires an energy consumption of 2.11 kW h m−3 for 85% desalination of 35 part per thousand (ppt) NaCl (seawater). When combining secondary brackish water reverse osmosis with this design, hypersaline brines (100 ppt salinity) can be made potable with an energy consumption of 14.06 kW h m−3 as shown in Fig. 7b. Subsequent research provided a list of potential solution phase reactants for desalination150 and an improved cell architecture with the addition of a CEM and second AEM.151 With this cell architecture, the desalination of 203.8 ppt NaCl to 0.192 ppt was achieved over a seven stage system at an energy consumption of 84.37 kW h m−3, demonstrating desalination of hypersaline brines currently not practical by non-thermal methods.1513.6 CDI and faradaic insertion desalination comparison
Despite significant effort and real progress in the last five decades, electrochemical deionization strategies must be further improved to supplant existing technology. While the choice of electrochemical deionization technology for a given application certainly depends on performance, it is noteworthy that “performance” is quantified by a variety of metrics, typically decided by the authors, such as ion removal rate, ion removal capacity, energy efficiency and selectivity, to name a few. Therefore, in an effort to compare deionization performance across faradaic and non-faradaic deionization mechanisms as well as feed concentration, herein we report data from several prominent references in the form of capacity (mg g−1) vs. rate (mg g−1 min−1) (Fig. 8a and b). It is important to note that non-faradaic deionization mechanisms display a roughly linear correlation between capacity and feed concentration (Fig. 8c) while faradaic mechanisms do not present such a relationship (Fig. S1†).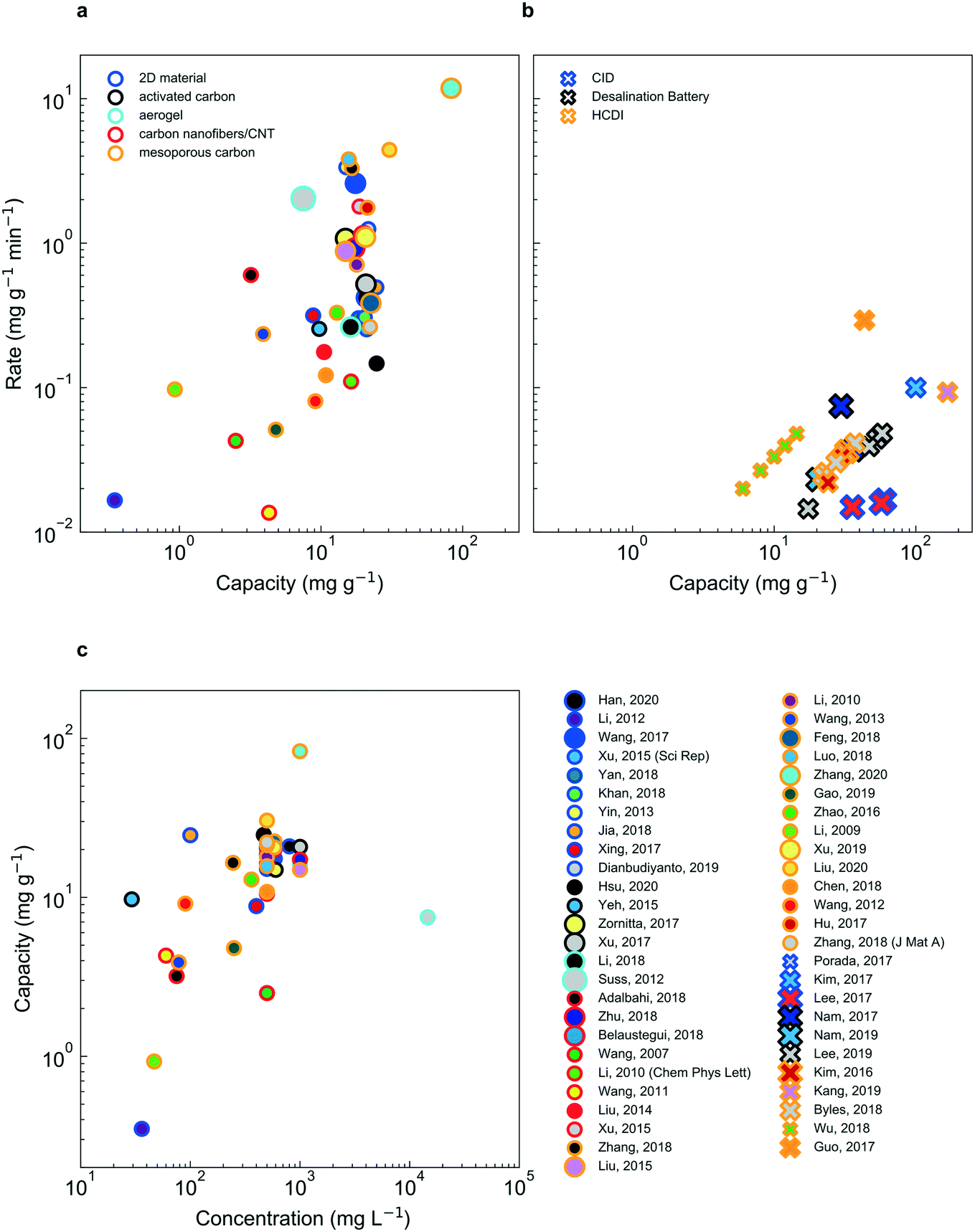 | ||
| Fig. 8 Rate vs. capacity plots for CDI (a) and faradaic insertion (b) technologies. Here, the material class is indicated by the dot outline, the reference by the unique combination of the outline and the fill color. The concentration is divided up into three categories: tap water (smallest marker size), brackish (intermediate marker size) and sea water (largest marker size). Experimental parameters, performance and references are listed in Tables S1 and S2.† Average rate is calculated by dividing 95% of the electrode capacity by the time it takes to charge the electrode to the same point. CDI systems show consistently higher salt adsorption rates, relative to faradaic insertion systems; the latter demonstrates higher capacities, however. In (c), the strong dependence of capacity on feed concentration is shown (a dependence which is not as apparent for faradaic insertion, shown in Fig. S1†). | ||
4. Depletion zone desalination
A new procedure for desalination of water eschews adsorbing electrodes; instead it relies on depletion zones formed by the complex and dynamic interplay of applied electric fields, ionic diffusion and bulk water flow. Two designs of this new method, coined shock electrodialysis, have been studied to-date: one design employs an ion selective membrane or channel and the other relies purely on reaction chemistry at the electrodes.4.1 Shock electrodialysis
Shock electrodialysis (SED) is not limited by the flow-through membranes of electrodialysis or the finite adsorption capacity of CDI. A novel method to separate ions from saline water, SED is a fundamentally nonlinear and electrokinetic phenomenon characterized by a sharp depletion region at the surface of an IEM or charge-selective nanochannel.31,152–157 In macro-scale systems, a saline feed solution flows between two electrodes, at least one of which is coated with an IEM (usually selective for cations). By applying a voltage between these electrodes, cations flow through the IEM towards the cathode and anions are repulsed from the surface of the IEM. To retain charge neutrality at the IEM surface, a depletion region forms and at “over-limiting” currents—corresponding to faster-than-diffusion-limited ionic current—rapidly propagates outward. By placing a splitter at the boundary of the depletion region, the de-ionized water in the depletion region is permanently separated from the concentrated saline solution, effectively creating brine and deionized streams.The phenomenon of ion-depletion near an ionically selective element was first discovered in nanofluidic structures at the interface of a microchannel and a glass nanonchannel,158,159 and was employed as a proof-of-concept desalination system in a single lithographically patterned device, shown in Fig. 9a.160 This proof-of-concept displayed 99% rejection of salt in seawater with 50% of total volume conserved—i.e. the brine concentration was a factor two more concentrated than the incoming feed solution. Although this study serves as an excellent proof-of-concept, the device desalinated only tens of μL of solution and the energy efficiency of the system was not optimized.
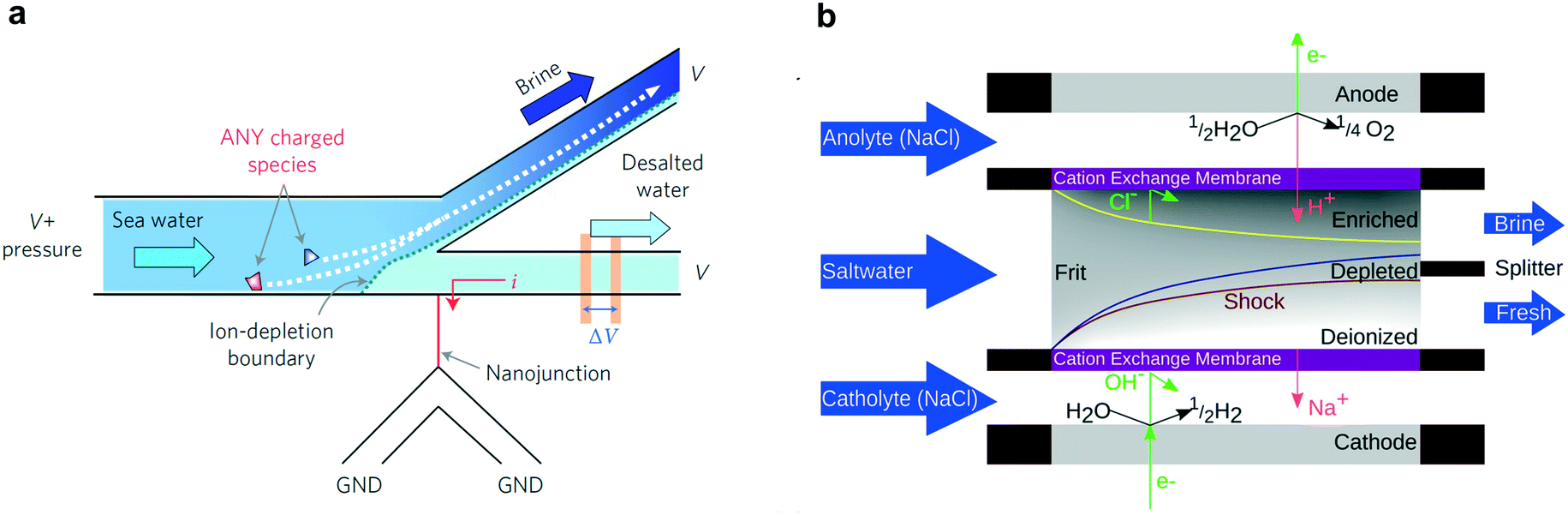 | ||
| Fig. 9 (a) Nanoscale SED design where the depletion region is formed at the junction of a nanochannel and microchannel.160 Reproduced with permission from ref. 160. Copyright 2010 Springer Nature. (b) SED device design for continuous desalination of macroscopic volumes of water between two IEMs.161 Reproduced with permission from ref. 161. Copyright 2015 American Chemical Society. | ||
Subsequent studies turned towards larger systems with macroscopic electrodes and ion exchange membranes separated by glass frits, as shown in Fig. 9b. The negative charge on the glass frits enables the transport of ions faster than diffusion-limited speeds, without pushing the electrode voltage bias beyond 1.5 V.162 The first of such systems effectively separated positive and negative ionic species but was not used as a desalination device. Following this initial demonstration, the same group developed a SED device that could continuously deionize a brackish feed stream,161 schematized in Fig. 9b, with a salt rejection approaching 100% and a maximum water recovery rate of 79%. The energy efficiency of this device, however, was quite poor at 10−1 to 103 kW h m−3, or 0.3 to 0.003% of the thermodynamic limit. By scaling arguments, increasing the size of the device could lower energy requirement and improve efficiency.163 While this type of device may not be applicable to large-scale desalination, it has the potential to provide small-scale desalination for various applications, which is an underdeveloped area.
A similar prototype device was shown to selectively remove (divalent) magnesium cations above (monovalent) sodium.164 This selectivity was loosely attributed to the enhanced interaction between the divalent magnesium ions to the glass frit, slowing diffusion in the brine solution but enhancing diffusion in the depletion zone due to the surface charge mediated transport in that region. In a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NaCl:MgCl2 mixture, a continuous removal of 99.06% Mg2+ was demonstrated, corresponding to 41.3 fold removal of Mg2+ over Na+. The energy requirements remained quite high relative to reverse osmosis systems; however, these results have motivated work on the separation of multivalent ions along monovalent ions from waste streams. For the removal of radionuclides from nuclear waste streams, a three stage SED process could remove 96.3% Ce+ and 99.6% Co2+ with Co2+ being primarily removed in the third stage.165 Parameter tuning allows for the optimization of Co removal, total cation removal, or water recovery ratio from a waste stream containing lithium, cobalt, and cesium. Additional water purification steps are necessary to remove uncharged species such as boric acid.
:1 NaCl:MgCl2 mixture, a continuous removal of 99.06% Mg2+ was demonstrated, corresponding to 41.3 fold removal of Mg2+ over Na+. The energy requirements remained quite high relative to reverse osmosis systems; however, these results have motivated work on the separation of multivalent ions along monovalent ions from waste streams. For the removal of radionuclides from nuclear waste streams, a three stage SED process could remove 96.3% Ce+ and 99.6% Co2+ with Co2+ being primarily removed in the third stage.165 Parameter tuning allows for the optimization of Co removal, total cation removal, or water recovery ratio from a waste stream containing lithium, cobalt, and cesium. Additional water purification steps are necessary to remove uncharged species such as boric acid.
4.2 Faradaic depletion zone desalination
Rather than employing an ion exchange membrane or channel, one can utilize the faradaic reactions at the surface of an electrode to form an ion depletion zone. In the first experiment of its kind, the desalination device comprised a bipolar electrode (pyrolyzed photoresist carbon) placed at the junction of a branched microchannel.166,167 One end of the bipolar electrode acts as the cathode for water reduction and the other end as the anode for chloride oxidation (at an applied potential of 3.0 V). The chloride oxidation neutralizes the Cl−, creating a depletion zone and a local electric field gradient at the branched junction. While pressure driven flow dominates the ionic transport, ions that approach the local electric field will experience an increase in their electrophoretic velocity that drives them to the brine stream at the junction. The process was confirmed with conductivity measurements and monitoring of fluorescent dyes. This process allows for membrane-free rejection of 25 ± 5% salt rejection in seawater for 50% water recovery at an energy consumption of 25 W h m−3 thus approaching the theoretical minimum of 17 W h m−3.168,169 Numerical simulations have also corroborated the mechanism by coupling the mass–charge transport with the surface and volume reactions in a 3D system.170However, a recent study has revealed that the instability of Cl2 leads to rapid hydrolysis; this quenches the depletion zone, and prevents its use for seawater desalination.171 The process of forming a depletion zone via a faradaic reaction can still be utilized in other separations where a more stable product can be formed, such as in a recent study on microplastic separations. Here, the neutralization of the TrisH+ buffer to the stable TrisH species is used to form a depletion zone.172 This setup demonstrated that microplastics with different electrophoretic mobilities can be sorted by using a multi-channel design at the depletion zone. This platform offers many opportunities to develop membrane-free separations of charged species beyond desalination.
5. Selective electrosorption
The systems discussed in previous sections (excluding that of selective CDI) have applications related to complete deionization of water, e.g. desalination with the goal of making potable water from seawater and brackish sources. There is, however, a rising demand for selective electrosorption processes in which a subset of ions is adsorbed preferentially over other competitive ionic species, often present at much higher concentrations, in the same solution. These selective separations include targeted resource mining, such as uranium extraction from seawater where the relevant species, UO22+, is present at only 3 ppb,173 and toxic water remediation. In selective electrosorption/electrodeposition, electron transfer at the electrode–electrolyte interface is coupled with a number of slower processes such as mass transport of ions into the electrical double layer (EDL), adsorption of ions and formation of an expanding ion depletion zone around the electrode before ions can be replenished near the surface. Hence, in order to achieve high capacity, fast kinetics and perhaps most importantly, high selectivity in ion extraction processes, it is important to enhance mass transport into and out of the EDL, promote specific electrode–ion interactions and mitigate depletion zone formation around the adsorbent.Towards this common goal of efficient and selective ion extraction, researchers have developed electric field modulation techniques such as the half-wave rectified alternating current electrochemical method (HW-ACE)34 to mitigate diffusion-limited mass transport and ion depletion zone formation. Alternating current methods are able to achieve high degrees of selectivity by exploiting a plethora of physical properties such as ionic mass, charge, radius and mobility; however, it is important to note in addition to alternating current mediated selectivity, chemical functionalization of electrodes impart selectivity to electrosorption/electrodeposition processes in different and equally important ways. For example, polymer-based electrode coatings have received a great deal of attention in recent years owing to their ability to facilitate highly specific ion interactions inside the EDL. Hence, researchers have combined the merits of alternating current methods with synthetic chemical modifications of electrodes such as the addition of chelating polymers, redox-active polymers and electroactive polymers alike.
Chemical electrode coatings are typically easily synthesized, solution processible, electronically tunable and have molecular selectivity enabled mostly by non-covalent interactions such as electrostatic forces, Van Der Waals forces, hydrophobic effects, hydrogen bonding and π interactions. Different classes of polymers such as electroactive polymers, chelating polymers and redox-active polymers inhabit crucial roles in emerging selective electrochemical ion adsorption technologies such as the HW-ACE. This section focuses on selective electrosorption mediated by alternating current methods, which take care of mass transport and depletion zone limitations, as well as the vast array of chemical functionalities, which facilitate highly specific ion adsorption inside the electrical double layer. While the aforementioned modes of selectivity will be addressed separately in this section, it is important to note that the merits of alternating current techniques (section 5.1) and chemical functionalization (section 5.2) are often coupled to enable the high capacity, kinetically fast and highly selective electrosorption/electrodeposition behavior highlighted below.
5.1 Alternating current mediated selectivity
Physicochemical adsorption methods share three intrinsic limitations: slow diffusion of ions present at low concentrations, coulombic repulsion of incoming ions by adsorbed ions of like charge and saturation of sorbent surface active sites by competing ions present at much higher concentrations than those of interest. Alternating current methods solve the conventional drawbacks of physicochemical adsorption and achieve high performance selective ion extraction. The voltage bias increases the ion collision rate in the double layer and leads to electrodeposition (neutralization) of the charged ions, which mitigates coulombic repulsion of incoming ionic species. The alternating current decreases the adsorption of unwanted species and circumvents water splitting. While the selectivity of such systems is indeed driven by the alternating nature of the applied current, chemical functionalization of the electrode surface, to be discussed in further detail in a later section (section 5.2), is also a significant contributor towards ion selectivity.HW-ACE was the first example of applying an alternating current to leverage highly selective ion extraction.34 Through clever combination of alternating current and chemically induced selectivity, Liu et al. used HW-ACE in tandem with a poly(amidoxime) modified carbon electrode (C-Ami) as a means of extracting uranyl ions from seawater in the form of charge neutral uranium crystals (Fig. 10). HW-ACE/C-Ami achieved unprecedented uranyl selectivity versus sodium ions, nine-fold higher adsorption capacity (1932 mg g−1) without saturation and four-fold faster kinetics compared to conventional diffusion-limited physicochemical adsorption.
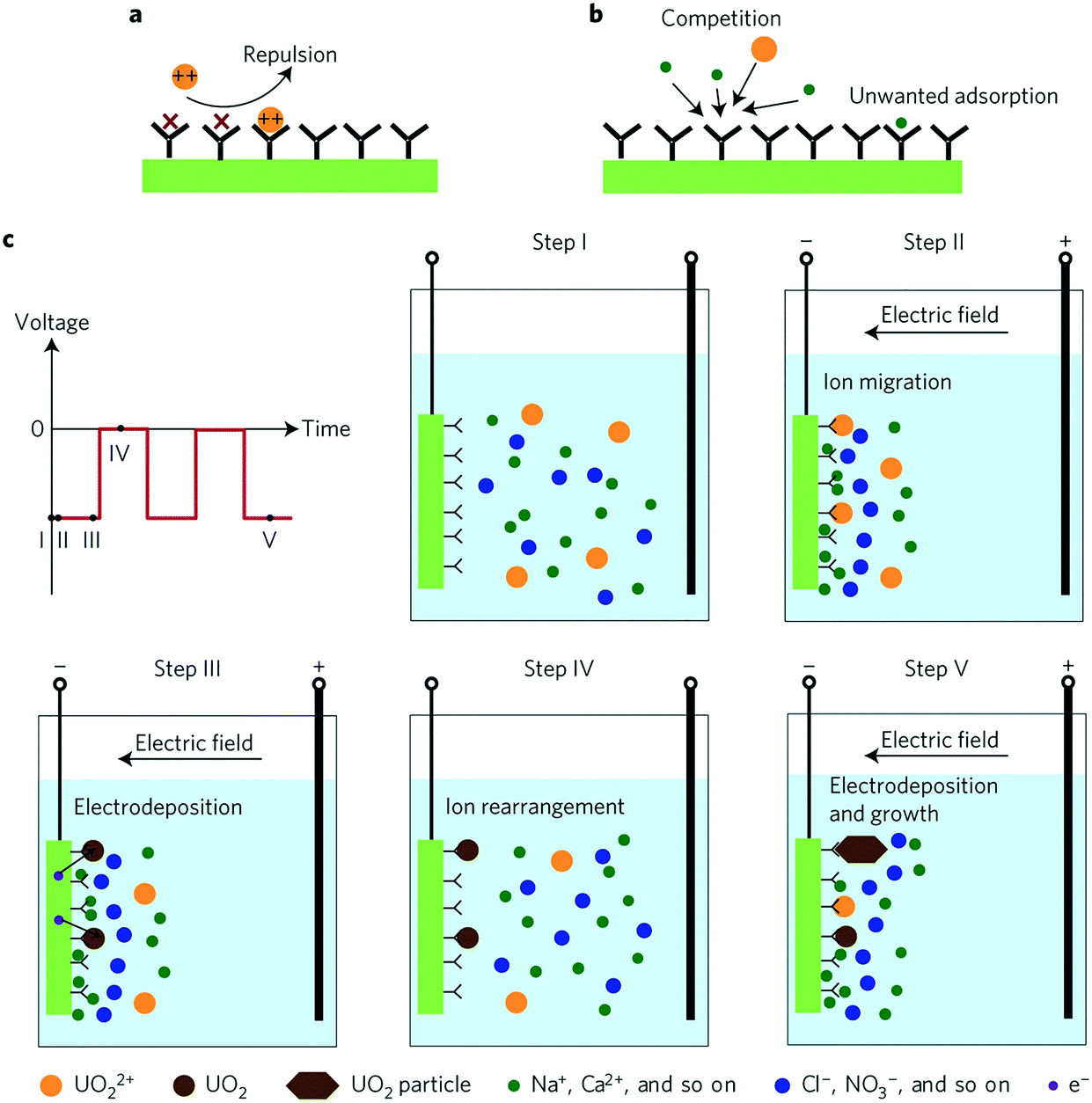 | ||
| Fig. 10 Schematics of physisorption and HW-ACE (a) schematic of coulombic repulsion of incoming ions mediated by adsorbed ions. (b) Schematic of competition between uranyl and other ions that results in blocking of active sites. (c) Outline of the extraction steps of HW-ACE. In step 1, ions are dispersed in the bulk solution; in step 2, ions migrate, form a double layer and uranyl is adsorbed; in step 3, adsorbed ions are reduced to charge-neutral UO2; in step 4 ions are expelled when the voltage bias is removed; in step 5, crystals grow through repetition of steps 3–5.34 Reproduced with permission from ref. 34. Copyright 2017 Springer Nature. | ||
In an effort to extend the success of HW-ACE/C-Ami, Liu et al. developed a reduced graphene-oxide-modified carbon felt electrode (C-rGO) based electrodeposition method that can treat both low-concentration and high-concentration heavy-metal pollution using a direct/alternating current (DC/AC).174 DC electrodeposition with a CF-rGO electrode (DC/C-rGO) can reduce low concentrations (∼300 ppb) of toxic ion mixtures (Cu, Cd, Pb) to ion concentrations within the safe drinking water range, as defined by the U.S. Environmental Protection Agency. Moreover, AC electrodeposition with a CF-rGO electrode (AC/C-rGO) can capture all three ions with capacities exceeding 29 g g−1 without saturation. More interestingly, AC/C-rGO can also be used to selectively extract Cu, Cd and Pb in series by leveraging the inherent differences in electrochemical reduction potentials of different ions. Through the tuning of the applied electrodeposition/desorption voltages and the AC frequency, Pb, Cu and Cd (∼1000 ppm each) can be sequentially extracted, in that order, to concentrations of ∼1 ppm. This AC electrochemical method has also been demonstrated for successful removal of heavy metal ions from elution solutions used in soil decontamination.175
During the performance evaluation of this device, three synthetically contaminated soil samples were prepared with 10000 ppm Cu, 1000 ppm Pb, and 100 ppm Cd, respectively (based on typical concentrations found in contamination sites) and the asymmetrical alternating current electrochemistry (AACE) method successfully reduced the metal concentrations to below their respective California Human Health Screening Levels (CHHSL). It is important to note that the current state-of-the-art electrochemical soil remediation method applies a DC electric field to the soil to transport heavy metal species by electroosmosis and electrodeposits them to metallic states on the negative electrode. However, field application of this technology is limited by the high DC voltage required to maintain a strong electric field (∼100 V m−1) for electroosmosis, the low ion migration speed in soil and the large energy losses associated with water splitting at electrodes. More importantly, if a DC voltage were applied, the negative charge of the ligand complexed ionic species, metal ion-ethylenediaminetetraacetic acid (MEDTA2−), would repel the negatively charged working electrode. This example especially highlights the benefits of leveraging AC, as opposed to DC methods, in tandem with chemical electrode functionalization (to be discussed further in section 5.2) towards highly selective electrosorption and electrodeposition.
5.2 Chemical functionalization
In a prominent example wherein only chelating polymer electrode coatings were responsible for selective ion electrodeposition, polyamidoxime modified carbon felt (PACCF) electrodes, were used for selective electrosorption of heavy metal pollutants (Pb2+, Cd2+, Cu2+ mixture, 1000 ppm each) from point-of-use water and industrial wastewater.180 In addition to providing for strong chelation sites, the amidoxime functionalization also helped modify the inherently hydrophobic carbon surface to hydrophilic, thus allowing for complete utilization of the carbon felt's extensive surface area. In this study, a direct current was applied to a PACCF working electrode that purified heavy metal ion contaminated water to concentrations below 5 ppb with high rates of 3000 L h−1 m−2 and displayed high capacities of 2300 mg g−1.
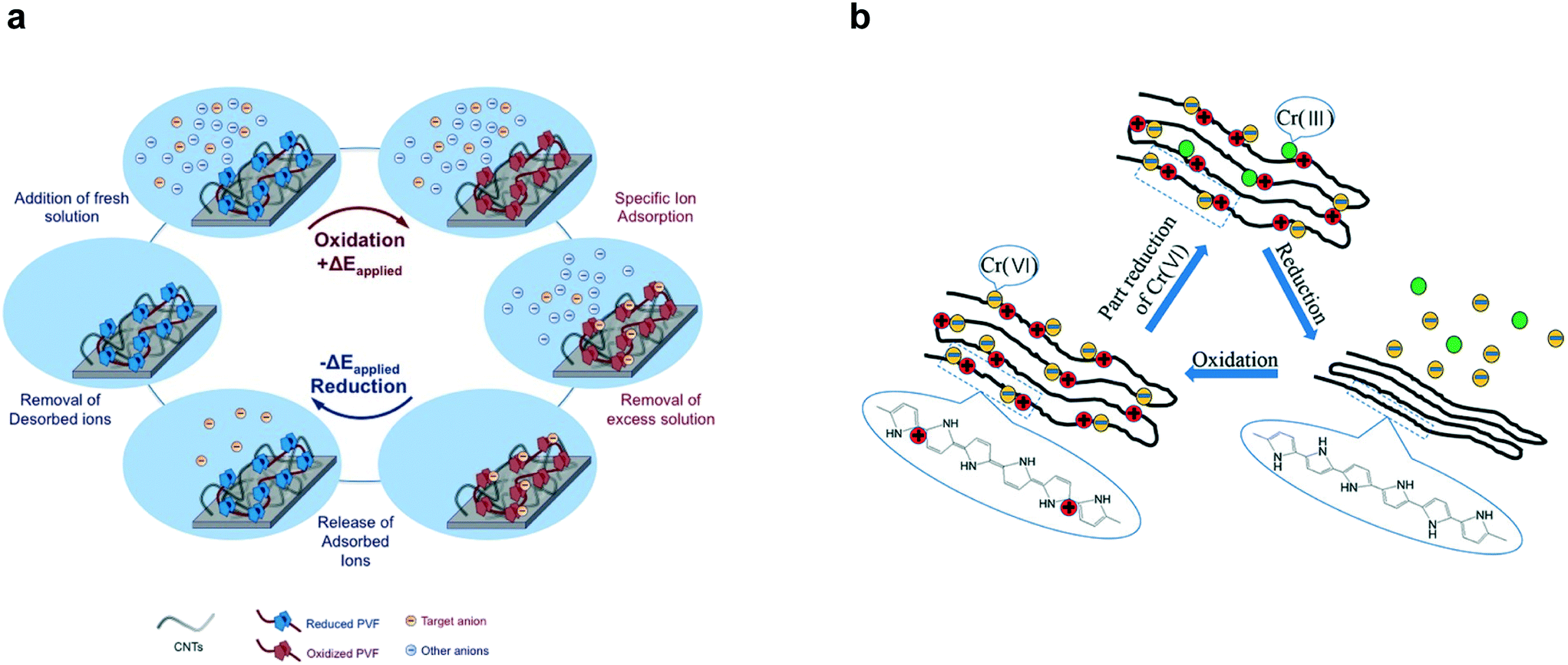 | ||
| Fig. 11 (a) Redox species (PVF) mediated electrosorption and stripping of specific anions by reversible oxidation and reduction respectively.185 Reproduced with permission from ref. 185. Copyright 2016 John Wiley and Sons. (b) Reversible electrosorption of redox-active conducting polymer polypyrrole (PPy).191 Reproduced with permission from ref. 191. Copyright 2018 American Chemical Society. | ||
While PPy is historically the most studied conducting polymer, redox-active electrosorption has been extended to PANI203–206 and PEDOT207 in the context of uranium separations. Pt electrodes coated with PEDOT have been for selective electrodeposition of uranium from a mixed solution of UO22+ and Ln3+ (Ln = La, Ce, Sm) resulting in ∼94 ± 10% stripping efficiency208 and PANI/graphene oxide composites with large adsorption capacities of ∼1960 mg g−1 have been used to preconcentrate uranium from highly saline aqueous solutions.209
The PVF-CNT electrode achieved >100 mg g−1 capacity at saturation while retaining fast adsorption rates with 99% reversible working capacity. During adsorption, hexavalent chromium was captured at the anode surface through PVF oxidation mediated binding and during desorption, the bound chromium is reduced to the less harmful trivalent species (Fig. 12a). Remarkably higher selectivity factors and uptake capacities of chromium from water were achieved by redox-active metallopolymer electrodes relative to traditional carbon electrodes (Fig. 12b).220 Chromium and arsenic at 100 ppb concentrations were selectively remediated in the presence of over 200-fold excess (∼2000 ppb) competing salts (NaCl). The fundamental mechanism for the selectivity of PVF towards oxyanions was elucidated via density functional theory (DFT) calculations. These calculations indicate that charge-transfer is the underlying electronic effect driving selectivity; electronic structure analysis (Fig. 12c) showed that the oxygen atoms in the chromium and arsenic anions (CrO42−, HAsO42−) are more susceptible to losing electrons to Fc+ than are those in non-heavy metal oxyanions (ClO4−). Zero-temperature DFT calculations were corroborated by finite-temperature calculations that showed similar trends in selectivity with respect to heavy-metal oxyanions.
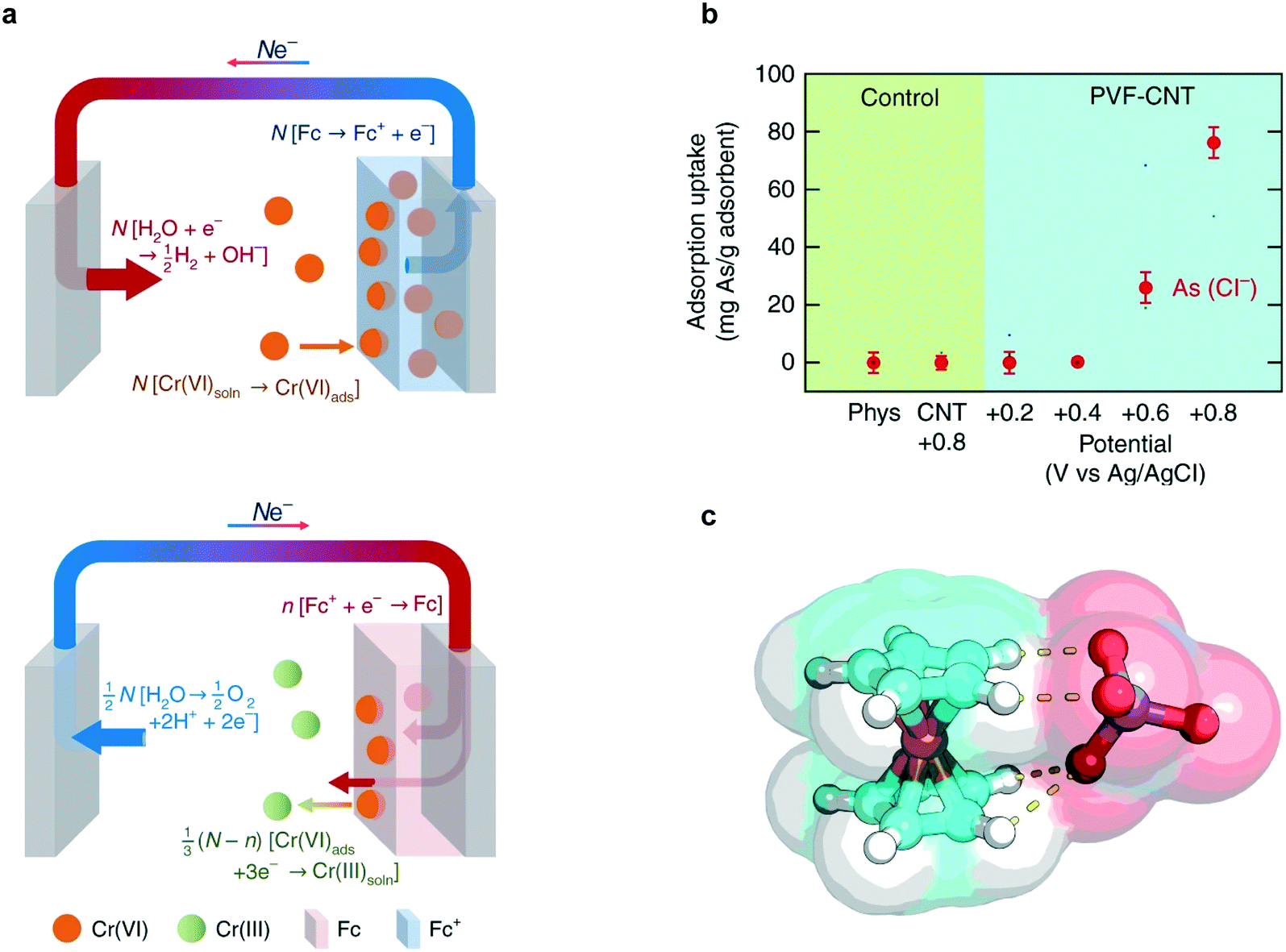 | ||
| Fig. 12 (a) Faradaic half-reactions occurring at electrode surface during charging/adsorption (top figure) and discharge/desorption (bottom figure).219 (b) Adsorption capacity of arsenate ions at 1 mM KH2AsO4 in the presence of 20 mM NaCl using PVF-CNT under different applied potentials. (c) Electronic structure optimization of chromate (CrO42−) with ferrocenium (Fc+).219 Reproduced with permission from ref. 219. Copyright 2018 Springer Nature. | ||
6. Summary and outlook
Electrochemical ion separation is a growing field well-positioned to exploit the development of cheaper, renewable energy to fulfill the global need for potable water. In this review, we have brought together key electrochemical deionization techniques, including CDI desalination, faradaic desalination, depletion zone desalination, and selective electrosorption. As discussed in this review, developments of these technologies have improved salt removal capacity, salt removal rate, energy consumption, and water recovery for commercial desalination. Table 2 shows the advantages and disadvantages of each desalination technology. Electrochemical ion separation methods must still overcome significant challenges to surpass existing separation technologies in industry. The long-term stability of non-faradaic CDI must be improved by incorporating new protective materials and cell architectures to suppress side reactions and corrosion. For faradaic CDI, protective materials must be incorporated to prevent undesired ion insertion, which can initiate irreversible phase changes leading to a loss in capacity and even material failure. While we have not addressed corrosion and fouling in this review, it is a persisting issue in systems that use membranes in seawater and must be addressed when implementing a desalination technology.| Technology | Advantages | Disadvantages |
|---|---|---|
| Common characteristics of CDI, faradaic desalination and depletion zone desalination | • High energy efficiency at low salt concentrations | • Energy efficiency decreases at a faster rate than RO with increasing salt concentrations |
| • All-electric operation suitable for integration with renewable energy | ||
| • Modular and scalable | ||
| CDI | • Fast salt adsorption rate | • Capacity limited by accessible surface area |
| • High adsorption capacity (for advanced materials) | ||
| • High reversibility and stability | ||
| Faradaic desalination | • High volumetric capacity | • Slow (compared to CDI) salt adsorption rate |
| • Confinement-driven ion selectivity is feasible | • Most energy efficient designs use multiple layers of ion exchange membranes | |
| • Low energy consumption | ||
| • Redox flow design can efficiently desalinate hypersaline brines | ||
| Depletion zone desalination | • Ultrahigh (>99%) salt rejection | • Energy efficiency remains low |
| • Robust and portable for use in small-scale desalination (i.e. crisis situations) | • Difficult to deploy for large-scale desalination | |
| • Not limited by finite capacity (faradaic/non-faradaic) | ||
| • Continuous desalination possible | ||
| Reverse osmosis | • Most optimized technology for brackish and seawater desalination | • Membranes susceptible to fouling and degradation |
| • Low energy consumption | • Brine disposal harmful to the environment | |
| Electrodialysis | • High water recovery | • Membranes susceptible to fouling and degradation |
| • Longer membrane lifetime than RO | • High energy consumption | |
| • Selective separation of monovalent and multivalent ions | ||
| Thermal distillation | • Practical method for desalination of hypersaline brines | • High energy consumption |
In forming this review, we found that a standard reporting of key metrics could improve comparison of desalination performance. The reporting of all pairs of cation removal capacity (mg of salt per g of electrode material) and average rate of ion removal (mg of salt per g of electrode per second) will facilitate easier comparison between technologies. Additionally, converting data reported as figure to both a table and figure would make easier accessibility for the reader. Regarding energy consumption, both the gravimetric specific energy consumption (kW h per kg of salt removed) and volumetric specific energy consumption (kW h per L of desalinate output) can help compare operation cost for technologies with the same feed concentration, water recovery, and salt removal fraction. Additional metrics to include are active material mass, total electrode mass, geometric area, flow rate, feed concentration, coulombic efficiency, salt removal fraction, and water recovery. The use of standard conditions for brackish desalination, as proposed by Hawks et al., would also be beneficial for the field.38
Beyond desalination, toxic water remediation and resource mining have been enabled by the development of selective electrosorption techniques. Ion selectivity and removal performance is enhanced for these applications via modification of standard electrodes with polymer coatings. A growing library of selective electrosorptive materials include electroactive polymers for selective CDI, chelating polymers for uranium recovery, redox-active polymers for heavy metal ion extraction, and charged surfactants for nitrate removal. Improved understanding of ion transport mechanisms and interfacial interactions will guide material selection and system design for better electrochemical deionization. This is necessary for the development of new chemistries and methods that allow for selective extraction of more dilute and uncharged species in complex solutions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Pritzker School of Molecular Engineering at the University of Chicago and the Advanced Materials for Energy-Water-Systems (AMEWS) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences.References
- UN DESA Population Division, World population prospects 2019: highlights, 2019 Search PubMed.
- P. H. Gleick and C. W. Howe, Water in crisis: a guide to the world's fresh water resources, Climatic Change, 1993 Search PubMed.
-
B. F. Dodge, Review of distillation processes for the recovery of fresh water from saline waters, in Saline Water Conversion—II, American Chemical Society, 1963, vol. 38, pp. 1–26 Search PubMed
.
- D. S. Sholl and R. P. Lively, Seven chemical separations to change the world, Nature, 2016, 532, 435–437 CrossRef
- M. W. Shahzad, M. Burhan and K. C. Ng, A standard primary energy approach for comparing desalination processes, npj Clean Water, 2019, 2, 1 CrossRef CAS
- WWAP (United Nations World Water Assessment Programme). The United Nations world water development report 2014: water and energy. (UNESCO, 2014).
- M. Elimelech and W. A. Phillip, The future of seawater desalination: energy, technology, and the environment, Science, 2011, 333, 712–717 CrossRef CAS
- Y. Oren, Capacitive deionization (CDI) for desalination and water treatment — past, present and future (a review), Desalination, 2008, 228, 10–29 CrossRef CAS
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Review on the science and technology of water desalination by capacitive deionization, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS
- S. Al-Amshawee,
et al. Electrodialysis desalination for water and wastewater, Chem. Eng. J., 2020, 380, 122231 CrossRef CAS
- A. Campione,
et al. Electrodialysis for water desalination: A critical assessment of recent developments on process fundamentals, models and applications, Desalination, 2018, 434, 121–160 CrossRef CAS
- H. Strathmann, Electrodialysis, a mature technology with a multitude of new applications, Desalination, 2010, 264, 268–288 CrossRef CAS
- P. M. Biesheuvel, Y. Fu and M. Z. Bazant, Diffuse charge and Faradaic reactions in porous electrodes, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 061507 CrossRef CAS
- P. M. Biesheuvel, S. Porada, M. Levi and M. Z. Bazant, Attractive forces in microporous carbon electrodes for capacitive deionization, J. Solid State Electrochem., 2014, 18, 1365–1376 CrossRef CAS
- P. M. Biesheuvel, H. V. M. Hamelers and M. E. Suss, Theory of water desalination by porous electrodes with immobile chemical charge, Colloids Interface Sci. Commun., 2015, 9, 1–5 CrossRef CAS
- D. D. Caudle, Electrochemical demineralization of water with carbon electrodes, U.S. Department of the Interior, 1966 Search PubMed.
- Y. Oren and A. Soffer, Electrochemical parametric pumping, J. Electrochem. Soc., 1978, 125, 869–875 CrossRef CAS
- J. C. Farmer, D. V. Fix, G. V. Mack, R. W. Pekala and J. F. Poco, Capacitive deionization of NH4ClO4 solutions with carbon aerogel electrodes, J. Appl. Electrochem., 1996, 26, 1007–1018 CrossRef CAS
- S. Jeon,
et al. Desalination via a new membrane capacitive deionization process utilizing flow-electrodes, Energy Environ. Sci., 2013, 6, 1471 RSC
- J.-S. Kim and J.-H. Choi, Fabrication and characterization of a carbon electrode coated with cation-exchange polymer for the membrane capacitive deionization applications, J. Membr. Sci., 2010, 355, 85–90 CrossRef CAS
- J.-B. Lee, K.-K. Park, H.-M. Eum and C.-W. Lee, Desalination of a thermal power plant wastewater by membrane capacitive deionization, Desalination, 2006, 196, 125–134 CrossRef CAS
- E. Avraham, B. Yaniv, A. Soffer and D. Aurbach, Developing ion electroadsorption stereoselectivity, by pore size adjustment with chemical vapor deposition onto active carbon fiber electrodes. Case of Ca2+/Na+ separation in water capacitive desalination, J. Phys. Chem. C, 2008, 112, 7385–7389 CrossRef CAS
- S. A. Hawks,
et al. Using ultramicroporous carbon for the selective removal of nitrate with capacitive deionization, Environ. Sci. Technol., 2019, 53, 10863–10870 CrossRef CAS
- Y.-J. Kim, J.-H. Kim and J.-H. Choi, Selective removal of nitrate ions by controlling the applied current in membrane capacitive deionization (MCDI), J. Membr. Sci., 2013, 429, 52–57 CrossRef CAS
- J.-H. Yeo and J.-H. Choi, Enhancement of nitrate removal from a solution of mixed nitrate, chloride and sulfate ions using a nitrate-selective carbon electrode, Desalination, 2013, 320, 10–16 CrossRef CAS
- J. J. Lado,
et al. Removal of nitrate by asymmetric capacitive deionization, Sep. Purif. Technol., 2017, 183, 145–152 CrossRef CAS
- J. Lee, S. Kim, C. Kim and J. Yoon, Hybrid capacitive deionization to enhance the desalination performance of capacitive techniques, Energy Environ. Sci., 2014, 7, 3683–3689 RSC
- M. Pasta, C. D. Wessells, Y. Cui and F. La Mantia, A desalination battery, Nano Lett., 2012, 12, 839–843 CrossRef CAS
- K. C. Smith and R. Dmello, Na-ion desalination (NID) enabled by Na-blocking membranes and symmetric Na-intercalation: porous-electrode modeling, J. Electrochem. Soc., 2016, 163, A530–A539 CrossRef CAS
- D. Desai,
et al. Electrochemical desalination of seawater and hypersaline brines with coupled electricity storage, ACS Energy Lett., 2018, 3, 375–379 CrossRef CAS
- A. Mani and M. Z. Bazant, Deionization shocks in microstructures, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 061504 CrossRef
- D. I. Oyarzun, A. Hemmatifar, J. W. Palko, M. Stadermann and J. G. Santiago, Ion selectivity in capacitive deionization with functionalized electrode: theory and experimental validation, Water Res.: X, 2018, 1, 100008 Search PubMed
- D. I. Oyarzun, A. Hemmatifar, J. W. Palko, M. Stadermann and J. G. Santiago, Adsorption and capacitive regeneration of nitrate using inverted capacitive deionization with surfactant functionalized carbon electrodes, Sep. Purif. Technol., 2018, 194, 410–415 CrossRef CAS
- C. Liu,
et al. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater, Nat. Energy, 2017, 2, 17007 CrossRef CAS
- X. Su,
et al. Electrochemically-mediated selective capture of heavy metal chromium and arsenic oxyanions from water, Nat. Commun., 2018, 9, 4701 CrossRef
- M. E. Suss,
et al. Water desalination via capacitive deionization: what is it and what can we expect from it?, Energy Environ. Sci., 2015, 8, 2296–2319 RSC
- T. Kim and J. Yoon, CDI ragone plot as a functional tool to evaluate desalination performance in capacitive deionization, RSC Adv., 2014, 5, 1456–1461 RSC
- S. A. Hawks,
et al. Performance metrics for the objective assessment of capacitive deionization systems, Water Res., 2019, 152, 126–137 CrossRef CAS
- H. Wang,
et al. Three-dimensional macroporous graphene architectures as high performance electrodes for capacitive deionization, J. Mater. Chem. A, 2013, 1, 11778–11789 RSC
- R. Zhao, P. M. Biesheuvel and A. van der Wal, Energy consumption and constant current operation in membrane capacitive deionization, Energy Environ. Sci., 2012, 5, 9520–9527 RSC
- L. Wang, J. E. Dykstra and S. Lin, Energy efficiency of capacitive deionization, Environ. Sci. Technol., 2019, 53, 3366–3378 CrossRef CAS
- L. Wang and S. Lin, Intrinsic tradeoff between kinetic and energetic efficiencies in membrane capacitive deionization, Water Res., 2018, 129, 394–401 CrossRef CAS
- S. Hand, X. Shang, J. S. Guest, K. C. Smith and R. D. Cusick, Global sensitivity analysis to characterize operational limits and prioritize performance goals of capacitive deionization technologies, Environ. Sci. Technol., 2019, 53, 3748–3756 CrossRef CAS
- T. Liu,
et al. Exceptional capacitive deionization rate and capacity by block copolymer–based porous carbon fibers, Sci. Adv., 2020, 6, eaaz0906 CrossRef CAS
- J. Oladunni,
et al. A comprehensive review on recently developed carbon based nanocomposites for capacitive deionization: From theory to practice, Sep. Purif. Technol., 2018, 207, 291–320 CrossRef CAS
- B. Jia and W. Zhang, Preparation and application of electrodes in capacitive deionization (CDI): a state-of-art review, Nanoscale Res. Lett., 2016, 11, 64 CrossRef
- C.-L. Yeh, H.-C. Hsi, K.-C. Li and C.-H. Hou, Improved performance in capacitive
deionization of activated carbon electrodes with a tunable mesopore and micropore ratio, Desalination, 2015, 367, 60–68 CrossRef CAS
- Y.-J. Kim and J.-H. Choi, Enhanced desalination efficiency in capacitive deionization with an ion-selective membrane, Sep. Purif. Technol., 2010, 71, 70–75 CrossRef CAS
- S. Nadakatti, M. Tendulkar and M. Kadam, Use of mesoporous conductive carbon black to enhance performance of activated carbon electrodes in capacitive deionization technology, Desalination, 2011, 268, 182–188 CrossRef CAS
- Z. Chen,
et al. A study of the effect of carbon characteristics on capacitive deionization (CDI) performance, Desalination, 2018, 433, 68–74 CrossRef CAS
- H. Li, L. Pan, C. Nie, Y. Liu and Z. Sun, Reduced graphene oxide and activated carbon composites for capacitive deionization, J. Mater. Chem., 2012, 22, 15556–15561 RSC
- R. L. Zornitta,
et al. High-performance activated carbon from polyaniline for capacitive deionization, Carbon, 2017, 123, 318–333 CrossRef CAS
- C.-C. Hsu, Y.-H. Tu, Y.-H. Yang, J.-A. Wang and C.-C. Hu, Improved performance and long-term stability of activated carbon doped with nitrogen for capacitive deionization, Desalination, 2020, 481, 114362 CrossRef CAS
- R. W. Pekala,
et al. Carbon aerogels for electrochemical applications, J. Non-Cryst. Solids, 1998, 225, 74–80 CrossRef CAS
- L. Li, L. Zou, H. Song and G. Morris, Ordered mesoporous carbons synthesized by a modified sol–gel process for electrosorptive removal of sodium chloride, Carbon, 2009, 47, 775–781 CrossRef CAS
- Y. Liu,
et al. Carbon aerogels electrode with reduced graphene oxide additive for capacitive deionization with enhanced performance, Inorg. Chem. Front., 2014, 1, 249–255 RSC
- G. Luo, Y. Wang, L. Gao, D. Zhang and T. Lin, Graphene bonded carbon nanofiber aerogels with high capacitive deionization capability, Electrochim. Acta, 2018, 260, 656–663 CrossRef CAS
- X. Liu,
et al. Nitrogen-doped hierarchical porous carbon aerogel for high-performance capacitive deionization, Sep. Purif. Technol., 2019, 224, 44–50 CrossRef CAS
- H. Li, T. Lu, L. Pan, Y. Zhang and Z. Sun, Electrosorption behavior of graphene in NaCl solutions, J. Mater. Chem., 2009, 19, 6773–6779 RSC
- H. Li, L. Zou, L. Pan and Z. Sun, Novel graphene-like electrodes for capacitive deionization, Environ. Sci. Technol., 2010, 44, 8692–8697 CrossRef CAS
- Z.-Y. Yang,
et al. Sponge-templated preparation of high surface area graphene with ultrahigh capacitive deionization performance, Adv. Funct. Mater., 2014, 24, 3917–3925 CrossRef CAS
- W. Dianbudiyanto and S.-H. Liu, Outstanding performance of capacitive deionization by a hierarchically porous 3D architectural graphene, Desalination, 2019, 468, 114069 CrossRef CAS
- D.-C. Han,
et al. High-performance capacitive deionization using nitrogen and phosphorus-doped three-dimensional graphene with tunable pore size, Electrochim. Acta, 2020, 336, 135639 CrossRef CAS
- Y. Wimalasiri and L. Zou, Carbon nanotube/graphene composite for enhanced capacitive deionization performance, Carbon, 2013, 59, 464–471 CrossRef CAS
- A. Amiri, Y. Chen, C. Bee Teng and M. Naraghi, Porous nitrogen-doped MXene-based electrodes for capacitive deionization, Energy Stor. Mater., 2020, 25, 731–739 CrossRef
- B. Chen,
et al. MXene as a Cation-Selective Cathode Material for Asymmetric Capacitive Deionization, ACS Appl. Mater. Interfaces, 2020, 12, 13750–13758 CrossRef CAS
- W. Peng, W. Wang, G. Han, Y. Huang and Y. Zhang, Fabrication of 3D flower-like MoS2/graphene composite as high-performance electrode for capacitive deionization, Desalination, 2020, 473, 114191 CrossRef CAS
- F. Xing,
et al. Chemically exfoliated MoS2 for capacitive deionization of saline water, Nano Energy, 2017, 31, 590–595 CrossRef CAS
- T. Gao,
et al. Mesoporous carbon derived from ZIF-8 for high efficient electrosorption, Desalination, 2019, 451, 133–138 CrossRef CAS
- S. Zhao,
et al. High capacity and high rate capability of nitrogen-doped porous hollow carbon spheres for capacitive deionization, Appl. Surf. Sci., 2016, 369, 460–469 CrossRef CAS
- C. Tsouris,
et al. Mesoporous Carbon for Capacitive Deionization of Saline Water, Environ. Sci. Technol., 2011, 45, 10243–10249 CrossRef CAS
- X. Xu,
et al. Capacitive deionization using nitrogen-doped mesostructured carbons for highly efficient brackish water desalination, Chem. Eng. J., 2019, 362, 887–896 CrossRef CAS
- P. Zhang, J. Li and M. B. Chan-Park, Hierarchical Porous Carbon for High-Performance Capacitive Desalination of Brackish Water, ACS Sustainable Chem. Eng., 2020, 8, 9291–9300 CrossRef CAS
- J.-B. Lee, K.-K. Park, H.-M. Eum and C.-W. Lee, Desalination of a thermal power plant wastewater by membrane capacitive deionization, Desalination, 2006, 196, 125–134 CrossRef CAS
- A. Hassanvand, K. Wei, S. Talebi, G. Q. Chen and S. E. Kentish, The role of ion exchange membranes in membrane capacitive deionisation, Membranes, 2017, 7, 54 CrossRef
- Y.-J. Kim and J.-H. Choi, Improvement of desalination efficiency in capacitive deionization using a carbon electrode coated with an ion-exchange polymer, Water Res., 2010, 44, 990–996 CrossRef CAS
- L. Wang and S. Lin, Mechanism of selective ion removal in membrane capacitive deionization for water softening, Environ. Sci. Technol., 2019, 53, 5797–5804 CrossRef CAS
- J. Ma,
et al. Carbon black flow electrode enhanced electrochemical desalination using single-cycle operation, Environ. Sci. Technol., 2020, 54, 1177–1185 CrossRef CAS
- Y. Oren, Capacitive deionization (CDI) for desalination and water treatment — past, present and future (a review), Desalination, 2008, 228, 10–29 CrossRef CAS
- M. E. Suss,
et al. Capacitive desalination with flow-through electrodes, Energy Environ. Sci., 2012, 5, 9511–9519 RSC
- S. Jeon,
et al. Desalination via a new membrane capacitive deionization process utilizing flow-electrodes, Energy Environ. Sci., 2013, 6, 1471 RSC
- C. He, J. Ma, C. Zhang, J. Song and T. D. Waite, Short-circuited closed-cycle operation of flow-electrode CDI for brackish water softening, Environ. Sci. Technol., 2018, 52, 9350–9360 CrossRef CAS
- J. Ma,
et al. Water recovery rate in short-circuited closed-cycle operation of flow-electrode capacitive deionization (FCDI), Environ. Sci. Technol., 2019, 53, 13859–13867 CrossRef CAS
- K. Tang, S. Yiacoumi, Y. Li and C. Tsouris, Enhanced water desalination by increasing the electroconductivity of carbon powders for high-performance flow-electrode capacitive deionization, ACS Sustainable Chem. Eng., 2019, 7, 1085–1094 CrossRef CAS
- S. Liu, V. Q. Do and K. C. Smith, Modeling of electrochemical deionization across length scales: Recent accomplishments and new opportunities, Curr. Opin. Electrochem., 2020, 22, 72–79 CrossRef CAS
- M. Z. Bazant, K. Thornton and A. Ajdari, Diffuse-charge dynamics in electrochemical systems, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 021506 CrossRef
- P. M. Biesheuvel and M. Z. Bazant, Nonlinear dynamics of capacitive charging and desalination by porous electrodes, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 031502 CrossRef CAS
- A. M. Johnson and J. Newman, Desalting by Means of Porous Carbon Electrodes, J. Electrochem. Soc., 1971, 118, 510 CrossRef CAS
- P. M. Biesheuvel, Y. Fu and M. Z. Bazant, Diffuse charge and Faradaic reactions in porous electrodes, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 061507 CrossRef CAS
- M. A. Murad and C. Moyne, A dual-porosity model for ionic solute transport in expansive clays, Comput. Geosci., 2007, 12, 47 CrossRef
- A. Revil and N. Linde, Chemico-electromechanical coupling in microporous media, J. Colloid Interface Sci., 2006, 302, 682–694 CrossRef CAS
- A. Hemmatifar, M. Stadermann and J. G. Santiago, Two-dimensional porous electrode model for capacitive deionization, J. Phys. Chem. C, 2015, 119, 24681–24694 CrossRef CAS
- M. Simoncelli,
et al. Blue Energy and Desalination with Nanoporous Carbon Electrodes: Capacitance from Molecular Simulations to Continuous Models, Phys. Rev. X, 2018, 8, 021024 CAS
- A. N. Shocron and M. E. Suss, The effect of surface transport on water desalination by porous electrodes undergoing capacitive charging, J. Phys.: Condens. Matter, 2017, 29, 084003 CrossRef
- X. Shang, R. D. Cusick and K. C. Smith, A combined modeling and experimental study assessing the impact of fluid pulsation on charge and energy efficiency in capacitive deionization, J. Electrochem. Soc., 2017, 164, E536–E547 CrossRef CAS
- S. Porada,
et al. Direct prediction of the desalination performance of porous carbon electrodes for capacitive deionization, Energy Environ. Sci., 2013, 6, 3700 RSC
- S. Porada,
et al. Water desalination using capacitive deionization with microporous carbon electrodes, ACS Appl. Mater. Interfaces, 2012, 4, 1194–1199 CrossRef CAS
- J. Nordstrand and J. Dutta, Dynamic Langmuir Model: A Simpler Approach to Modeling Capacitive Deionization, J. Phys. Chem. C, 2019, 123, 16479–16485 CrossRef CAS
- J. Nordstrand and J. Dutta, Simplified Prediction of Ion Removal in Capacitive Deionization of Multi-Ion Solutions, Langmuir, 2020, 36, 1338–1344 CrossRef CAS
- D.-H. Lee,
et al. Selective lithium recovery from aqueous solution using a modified membrane capacitive deionization system, Hydrometallurgy, 2017, 173, 283–288 CrossRef CAS
- A. Siekierka and M. Bryjak, Hybrid capacitive deionization with anion-exchange membranes for lithium extraction, E3S Web Conf., 2017, vol. 22, p. 00157 Search PubMed.
- D. I. Oyarzun, A. Hemmatifar, J. W. Palko, M. Stadermann and J. G. Santiago, Adsorption and capacitive regeneration of nitrate using inverted capacitive deionization with surfactant functionalized carbon electrodes, Sep. Purif. Technol., 2018, 194, 410–415 CrossRef CAS
- W. Tang, P. Kovalsky, B. Cao, D. He and T. D. Waite, Fluoride removal from brackish groundwaters by constant current capacitive deionization (CDI), Environ. Sci. Technol., 2016, 50, 10570–10579 CrossRef CAS
- C. J. Gabelich, T. D. Tran and I. H. Suffet, Electrosorption of inorganic salts from aqueous solution using carbon aerogels, Environ. Sci. Technol., 2002, 36, 3010–3019 CrossRef CAS
- S.-J. Seo,
et al. Investigation on removal of hardness ions by capacitive deionization (CDI) for water softening applications, Water Res., 2010, 44, 2267–2275 CrossRef CAS
- B. E. Conway, Transition from “supercapacitor” to “battery” behavior in electrochemical energy storage, J. Electrochem. Soc., 1991, 138, 1539 CrossRef CAS
- S. Porada, A. Shrivastava, P. Bukowska, P. M. Biesheuvel and K. C. Smith, Nickel hexacyanoferrate electrodes for continuous cation intercalation desalination of brackish water, Electrochim. Acta, 2017, 255, 369–378 CrossRef CAS
- H. Kim,
et al. Aqueous rechargeable Li and Na ion batteries, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS
- K. Singh, S. Porada, H. D. de Gier, P. M. Biesheuvel and L. C. P. M. de Smet, Timeline on the application of intercalation materials in Capacitive Deionization, Desalination, 2019, 455, 115–134 CrossRef CAS
- H. Yoon, J. Lee, S. Kim and J. Yoon, Review of concepts and applications of electrochemical ion separation (EIONS) process, Sep. Purif. Technol., 2019, 215, 190–207 CrossRef CAS
- W. Li, J. R. Dahn and D. S. Wainwright, Rechargeable lithium batteries with aqueous electrolytes, Science, 1994, 264, 1115–1118 CrossRef CAS
- J.-Y. Luo, W.-J. Cui, P. He and Y.-Y. Xia, Raising the cycling stability of aqueous lithium-ion batteries by eliminating oxygen in the electrolyte, Nat. Chem., 2010, 2, 760–765 CrossRef CAS
- H. Pan, Y.-S. Hu and L. Chen, Room-temperature stationary sodium-ion batteries for large-scale electric energy storage, Energy Environ. Sci., 2013, 6, 2338 RSC
- H. Kim,
et al. New iron-based mixed-polyanion cathodes for lithium and sodium rechargeable batteries: combined first principles calculations and
experimental study, J. Am. Chem. Soc., 2012, 134, 10369–10372 CrossRef CAS
- Y.-U. Park,
et al. A new high-energy cathode for a Na-ion battery with ultrahigh stability, J. Am. Chem. Soc., 2013, 135, 13870–13878 CrossRef CAS
- F. Sauvage, E. Baudrin and J.-M. Tarascon, Study of the potentiometric response towards sodium ions of Na0.44−xMnO2 for the development of selective sodium ion sensors, Sens. Actuators, B, 2007, 120, 638–644 CrossRef CAS
- J. F. Whitacre, A. Tevar and S. Sharma, Na4Mn9O18 as a positive electrode material for an aqueous electrolyte sodium-ion energy storage device, Electrochem. Commun., 2010, 12, 463–466 CrossRef CAS
- J. Lee, S. Kim, C. Kim and J. Yoon, Hybrid capacitive deionization to enhance the desalination performance of capacitive techniques, Energy Environ. Sci., 2014, 7, 3683–3689 RSC
- Y. H. Jung, C. H. Lim, J.-H. Kim and D. K. Kim, Na2FeP2O7 as a positive electrode material for rechargeable aqueous sodium-ion batteries, RSC Adv., 2014, 4, 9799 RSC
- S. Kim, J. Lee, C. Kim and J. Yoon, Na2FeP2O7 as a novel material for hybrid capacitive deionization, Electrochim. Acta, 2016, 203, 265–271 CrossRef CAS
- W. Song,
et al. Aqueous sodium-ion battery using a Na3V2(PO 4)3electrode, ChemElectroChem, 2014, 1, 871–876 CrossRef CAS
- W. Zhao, L. Guo, M. Ding, Y. Huang and H. Y. Yang, Ultrahigh-desalination-capacity dual-ion electrochemical deionization device based on Na3V2(PO 4)3@C–AgCl electrodes, ACS Appl. Mater. Interfaces, 2018, 10, 40540–40548 CrossRef CAS
- D.-H. Nam and K.-S. Choi, Bismuth as a new chloride-storage electrode enabling the construction of a practical high capacity desalination battery, J. Am. Chem. Soc., 2017, 139, 11055–11063 CrossRef CAS
- S. I. Park, I. Gocheva, S. Okada and J. Yamaki, Electrochemical properties of NaTi2(PO4)3 anode for rechargeable aqueous sodium-ion batteries, J. Electrochem. Soc., 2011, 158, A1067 CrossRef CAS
- M. Pasta,
et al. Full open-framework batteries for stationary energy storage, Nat. Commun., 2014, 5, 3007 CrossRef
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Highly reversible open framework nanoscale electrodes for divalent ion batteries, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS
- L. Guo,
et al. A Prussian blue anode for high performance electrochemical deionization promoted by the faradaic mechanism, Nanoscale, 2017, 9, 13305–13312 RSC
- S. Choi,
et al. Battery electrode materials with omnivalent cation storage for fast and charge-efficient ion removal of asymmetric capacitive deionization, Adv. Funct. Mater., 2018, 28, 1802665 CrossRef
- F. Chen,
et al. Dual-ions electrochemical deionization: a desalination generator, Energy Environ. Sci., 2017, 10, 2081–2089 RSC
- S. Kim,
et al. Hybrid electrochemical desalination system combined with an oxidation process, ACS Sustainable Chem. Eng., 2018, 6, 1620–1626 CrossRef CAS
- P. Srimuk,
et al. Two-dimensional molybdenum carbide (MXene) with divacancy ordering for brackish and seawater desalination via cation and anion intercalation, ACS Sustainable Chem. Eng., 2018, 6, 3739–3747 CrossRef CAS
- P. Srimuk,
et al. MXene as a novel intercalation-type pseudocapacitive cathode and anode for capacitive deionization, J. Mater. Chem. A, 2016, 4, 18265–18271 RSC
- M. R. Lukatskaya,
et al. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide, Science, 2013, 341, 1502–1505 CrossRef CAS
- S. C. Anderson, Mineral Industry Surveys, 2017 Search PubMed.
- S. M. Bennett, Mineral Industry Surveys, 2017 Search PubMed.
- D.-H. Nam, M. A. Lumley and K.-S. Choi, A desalination battery Combining Cu3[Fe(CN)6]2 as a Na-storage electrode and Bi as a Cl-storage electrode enabling membrane-free desalination, Chem. Mater., 2019, 31, 1460–1468 CrossRef CAS
- J. Lee,
et al. Enhancement in desalination performance of battery electrodes via improved mass transport using a multichannel flow system, ACS Appl. Mater. Interfaces, 2019, 11, 36580–36588 CrossRef CAS
- B. W. Byles, D. A. Cullen, K. L. More and E. Pomerantseva, Tunnel structured manganese oxide nanowires as redox active electrodes for hybrid capacitive deionization, Nano Energy, 2018, 44, 476–488 CrossRef CAS
- T. Wu,
et al. Highly stable hybrid capacitive deionization with a MnO2 anode and a positively charged cathode, Environ. Sci. Technol. Lett., 2018, 5, 98–102 CrossRef CAS
- B. W. Byles, B. Hayes-Oberst and E. Pomerantseva, Ion removal performance, structural/compositional dynamics, and electrochemical stability of layered manganese oxide electrodes in hybrid capacitive deionization, ACS Appl. Mater. Interfaces, 2018, 10, 32313–32322 CrossRef CAS
- K. Wang,
et al. Metal–organic-frameworks-derived NaTi 2 (PO 4 ) 3 /carbon composites for efficient hybrid capacitive deionization, J. Mater. Chem. A, 2019, 7, 12126–12133 RSC
- T. F. Fuller, Simulation and optimization of the dual lithium ion insertion cell, J. Electrochem. Soc., 1994, 141, 1 CrossRef CAS
- K. C. Smith, Theoretical evaluation of electrochemical cell architectures using cation intercalation electrodes for desalination, Electrochim. Acta, 2017, 230, 333–341 CrossRef CAS
- S. Liu and K. C. Smith, Quantifying the trade-offs between energy consumption and salt removal rate in membrane-free cation intercalation desalination, Electrochim. Acta, 2018, 271, 652–665 CrossRef CAS
- K. Singh, H. J. M. Bouwmeester, L. C. P. M. de Smet, M. Z. Bazant and P. M. Biesheuvel, Theory of water desalination with intercalation materials, Phys. Rev. Appl., 2018, 9, 064036 CrossRef CAS
- J. Lee, S. Kim and J. Yoon, Rocking chair desalination battery based on prussian blue electrodes, ACS Omega, 2017, 2, 1653–1659 CrossRef CAS
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nickel hexacyanoferrate nanoparticle electrodes for aqueous sodium and potassium ion batteries, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS
- J. Ahn,
et al. High performance electrochemical saline water desalination using silver and silver-chloride electrodes, Desalination, 2020, 476, 114216 CrossRef CAS
- T. Kim, C. A. Gorski and B. E. Logan, Low energy desalination using battery electrode deionization, Environ. Sci. Technol. Lett., 2017, 4, 444–449 CrossRef CAS
- X. Hou,
et al. Coupling desalination and energy storage with redox flow electrodes, Nanoscale, 2018, 10, 12308–12314 RSC
- E. S. Beh, M. A. Benedict, D. Desai and J. B. Rivest, A redox-shuttled electrochemical method for energy-efficient separation of salt from water, ACS Sustainable Chem. Eng., 2019, 7, 13411–13417 CrossRef CAS
- S. J. Kim, Y.-C. Wang, J. H. Lee, H. Jang and J. Han, Concentration polarization and nonlinear electrokinetic flow near a nanofluidic channel, Phys. Rev. Lett., 2007, 99, 044501 CrossRef
- A. Mani, T. A. Zangle and J. G. Santiago, On the propagation of concentration polarization from microchannel−nanochannel interfaces part I: analytical model and characteristic analysis, Langmuir, 2009, 25, 3898–3908 CrossRef CAS
- T. A. Zangle, A. Mani and J. G. Santiago, On the propagation of concentration polarization from microchannel−nanochannel interfaces part II: numerical and experimental study, Langmuir, 2009, 25, 3909–3916 CrossRef CAS
- E. V. Dydek,
et al. Overlimiting current in a microchannel, Phys. Rev. Lett., 2011, 107, 118301 CrossRef
- E. V. Dydek and M. Z. Bazant, Nonlinear dynamics of ion concentration polarization in porous media: The leaky membrane model, AIChE J., 2013, 59, 3539–3555 CrossRef CAS
- D. Deng,
et al. Overlimiting current and shock electrodialysis in porous media, Langmuir, 2013, 29, 16167–16177 CrossRef CAS
- Q. Pu, J. Yun, H. Temkin and S. Liu, Ion-enrichment and ion-depletion effect of nanochannel structures, Nano Lett., 2004, 4, 1099–1103 CrossRef CAS
- Y.-C. Wang, A. L. Stevens and J. Han, Million-fold preconcentration of proteins and peptides by nanofluidic
filter, Anal. Chem., 2005, 77, 4293–4299 CrossRef CAS
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Direct seawater desalination by ion concentration polarization, Nat. Nanotechnol., 2010, 5, 297–301 CrossRef CAS
- S. Schlumpberger, N. B. Lu, M. E. Suss and M. Z. Bazant, Scalable and continuous water deionization by shock electrodialysis, Environ. Sci. Technol. Lett., 2015, 2, 367–372 CrossRef CAS
- D. Deng,
et al. Water purification by shock electrodialysis: deionization, filtration, separation, and disinfection, Desalination, 2015, 357, 77–83 CrossRef CAS
- M. A. Alkhadra, T. Gao, K. M. Conforti, H. Tian and M. Z. Bazant, Small-scale desalination of seawater by shock electrodialysis, Desalination, 2020, 476, 114219 CrossRef CAS
- K. M. Conforti and M. Z. Bazant, Continuous ion-selective separations by shock electrodialysis, AIChE J., 2020, 66, e16751 CrossRef CAS
- M. A. Alkhadra, K. M. Conforti, T. Gao, H. Tian and M. Z. Bazant, Continuous separation of radionuclides from contaminated water by shock electrodialysis, Environ. Sci. Technol., 2019, 54(1), 527–536 Search PubMed
- K. N. Knust, D. Hlushkou, R. K. Anand, U. Tallarek and R. M. Crooks, Electrochemically mediated seawater desalination, Angew. Chem., Int. Ed., 2013, 52, 8107–8110 CrossRef CAS
- R. Dhopeshwarkar, D. Hlushkou, M. Nguyen, U. Tallarek and R. M. Crooks, Electrokinetics in Microfluidic Channels Containing a Floating Electrode, J. Am. Chem. Soc., 2008, 130, 10480–10481 CrossRef CAS
- Y. Cerci, Y. A. Cengel and B. Wood, The minimum separation work for desalination processes, Proc. ASME Adv. Energy Syst. Div., 1999, vol. 39, pp. 545–552 Search PubMed.
- Y. A. Cengel, Y. Cerci and B. Wood, Second law analysis of separation processes of mixtures, Proc. ASME Adv. Energy Syst. Div., 1999, vol. 39, pp. 537–543 Search PubMed.
- D. Hlushkou, K. N. Knust, R. M. Crooks and U. Tallarek, Numerical simulation of electrochemical desalination, J. Phys.: Condens. Matter, 2016, 28, 194001 CrossRef CAS
- C. D. Davies, S. E. Johnson and R. M. Crooks, Effect of Chloride Oxidation on Local Electric Fields in Microelectrochemical Systems, ChemElectroChem, 2019, 6, 4867–4876 CrossRef CAS
- C. D. Davies and R. M. Crooks, Focusing, sorting, and separating microplastics by serial faradaic ion concentration polarization, Chem. Sci., 2020, 11, 5547–5558 RSC
- I. Tabushi, Y. Kobuke and T. Nishiya, Extraction of uranium from seawater by polymer-bound macrocyclic hexaketone, Nature, 1979, 280, 665–666 CrossRef CAS
- C. Liu,
et al. Direct/alternating current electrochemical method for removing and recovering heavy metal from water using graphene oxide electrode, ACS Nano, 2019, 13, 6431–6437 CrossRef CAS
- J. Xu,
et al. Remediation of heavy metal contaminated soil by asymmetrical alternating current electrochemistry, Nat. Commun., 2019, 10, 2440 CrossRef
- G. V. Myasoedova, I. I. Antokol'skaya and S. B. Savvin, New chelating sorbents for noble metals, Talanta, 1985, 32, 1105–1112 CrossRef CAS
- G. V. Myasoedova, S. B. Savvin and E. Blasius, Chelating sorbents in analytical chemistry, Crit. Rev. Anal. Chem., 1986, 17, 1–63 CrossRef CAS
- G. V. Myasoedova, POLYORGS as complexing sorbents for preconcentration of trace metals, Fresenius' J. Anal. Chem., 1991, 341, 586–591 CrossRef CAS
- C. Kantipuly, S. Katragadda, A. Chow and H. Gesser, Chelating polymers and related supports for separation and preconcentration of trace metals, Talanta, 1990, 37, 491–517 CrossRef CAS
- T. Wu,
et al. Amidoxime-functionalized macroporous carbon self-refreshed electrode materials for rapid and high-capacity removal of heavy metal from water, ACS Cent. Sci., 2019, 5, 719–726 CrossRef CAS
- P.-O. Morin, T. Bura and M. Leclerc, Realizing the full potential of conjugated polymers: innovation in polymer synthesis, Mater. Horiz., 2016, 3, 11–20 RSC
- T. M. Swager, 50th anniversary perspective : conducting/semiconducting conjugated polymers, A personal perspective on the past and the future, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS
- G. R. Whittell and I. Manners, Metallopolymers: new multifunctional materials, Adv. Mater., 2007, 19, 3439–3468 CrossRef CAS
- X. Su and T. A. Hatton, Redox-electrodes for selective electrochemical separations, Adv. Colloid Interface Sci., 2017, 244, 6–20 CrossRef CAS
- X. Su, H. J. Kulik, T. F. Jamison and T. A. Hatton, Anion-Selective Redox Electrodes: Electrochemically Mediated Separation with Heterogeneous Organometallic Interfaces, Adv. Funct. Mater., 2016, 26, 3394–3404 CrossRef CAS
- J. Kim, J. H. Kim and K. Ariga, Redox-active polymers for energy storage nanoarchitectonics, Joule, 2017, 1, 739–768 CrossRef CAS
- X. Su and T. A. Hatton, Electrosorption at functional interfaces: from molecular-level interactions to electrochemical cell design, Phys. Chem. Chem. Phys., 2017, 19, 23570–23584 RSC
- M. A. Vorotyntsev and S. V. Vasilyeva, Metallocene-containing conjugated polymers, Adv. Colloid Interface Sci., 2008, 139, 97–149 CrossRef CAS
- M. Burgess, J. S. Moore and J. Rodríguez-López, Redox active polymers as soluble nanomaterials for energy storage, Acc. Chem. Res., 2016, 49, 2649–2657 CrossRef CAS
- P. Sengodu and A. D. Deshmukh, Conducting polymers and their inorganic composites for advanced Li-ion batteries: a review, RSC Adv., 2015, 5, 42109–42130 RSC
- J. Xing,
et al. Electrically switched ion exchange based on polypyrrole and carbon nanotube nanocomposite for the removal of chromium(VI) from aqueous solution, Ind. Eng. Chem. Res., 2018, 57, 768–774 CrossRef CAS
- A. G. MacDiarmid,
et al. The concept of ‘doping’ of conducting polymers: the role of reduction potentials [and discussion], Philos. Trans. R. Soc., A, 1985, 314, 3–15 CrossRef
- H. J. Ahonen, J. Lukkari and J. Kankare, n- and p-doped poly(3,4-ethylenedioxythiophene): two electronically conducting states of the polymer, Macromolecules, 2000, 33, 6787–6793 CrossRef CAS
- D. Kiefer,
et al. Double doping of conjugated polymers with monomer molecular dopants, Nat. Mater., 2019, 18, 149–155 CrossRef CAS
- P. Burgmayer and R. W. Murray, Ion gate electrodes, Polypyrrole as a switchable ion conductor membrane, J. Phys. Chem., 1984, 88, 2515–2521 CrossRef CAS
- P. Burgmayer and R. W. Murray, An ion gate membrane: electrochemical control of ion permeability through a membrane with an embedded electrode, J. Am. Chem. Soc., 1982, 104, 6139–6140 CrossRef CAS
- W. E. Price, C. O. Too, G. G. Wallace and D. Zhou, Development of membrane systems based on conducting polymers, Synth. Met., 1999, 102, 1338–1341 CrossRef CAS
- H. Zhao, W. E. Price and G. G. Wallace, Synthesis, characterisation and transport properties of layered conducting electroactive polypyrrole membranes, J. Membr. Sci., 1998, 148, 161–172 CrossRef CAS
- J. M. Davey, S. F. Ralph, C. O. Too, G. G. Wallace and A. C. Partridge, Electrochemically controlled transport of metal ions across polypyrrole membranes using a flow-through cell, React. Funct. Polym., 2001, 49, 87–98 CrossRef CAS
- H. N. M. E. Mahmud, A. K. O. Huq and Y. R. Binti, The removal of heavy metal ions from wastewater/aqueous solution using polypyrrole-based adsorbents: a review, RSC Adv., 2016, 6, 14778–14791 RSC
- F. J. Rodríguez, S. Gutiérrez, J. G. Ibanez, J. L. Bravo and N. Batina, The efficiency of toxic chromate reduction by a conducting polymer (polypyrrole): influence of electropolymerization conditions, Environ. Sci. Technol., 2000, 34, 2018–2023 CrossRef
- Y. Lin, X. Cui and J. Bontha, Electrically controlled anion exchange based on polypyrrole and carbon nanotubes nanocomposite for perchlorate removal, Environ. Sci. Technol., 2006, 40, 4004–4009 CrossRef CAS
- L. Wen and N. M. Kocherginsky, Doping-dependent ion selectivity of polyaniline membranes, Synth. Met., 1999, 106, 19–27 CrossRef CAS
- A. Mirmohseni, W. E. Price, G. G. Wallace and H. Zhao, Adaptive membrane systems based on conductive electroactive polymers, J. Intell. Mater. Syst. Struct., 1993, 4, 43–49 CrossRef
- J. Wang, B. Deng, H. Chen, X. Wang and J. Zheng, Removal of aqueous Hg(II) by polyaniline: sorption characteristics and mechanisms, Environ. Sci. Technol., 2009, 43, 5223–5228 CrossRef CAS
- D. Shao,
et al. PANI/GO as a super adsorbent for the selective adsorption of uranium(VI), Chem. Eng. J., 2014, 255, 604–612 CrossRef CAS
- S. J. Higgins, Conjugated polymers incorporating pendant functional groups—synthesis and characterisation, Chem. Soc. Rev., 1997, 26, 247–257 RSC
- R. Agarwal and M. K. Sharma, Selective electrochemical separation and recovery of uranium from mixture of uranium(VI) and lanthanide(III) ions in aqueous medium, Inorg. Chem., 2018, 57, 10984–10992 CrossRef CAS
- D. Shao,
et al. PANI/GO as a super adsorbent for the selective adsorption of uranium(VI), Chem. Eng. J., 2014, 255, 604–612 CrossRef CAS
- Y. Wang, D. Astruc and A. S. Abd-El-Aziz, Metallopolymers for advanced sustainable applications, Chem. Soc. Rev., 2019, 48, 558–636 RSC
- J.-C. Eloi, L. Chabanne, G. R. Whittell and I. Manners, Metallopolymers with emerging applications, Mater. Today, 2008, 11, 28–36 CrossRef CAS
- M. Gallei and C. Rüttiger, Recent Trends in Metallopolymer Design: Redox-Controlled Surfaces, Porous Membranes, and Switchable Optical Materials Using Ferrocene-Containing Polymers, Chem. – Eur. J., 2018, 24(40), 10006–10021 CrossRef CAS
- W. Tian, X. Mao, P. Brown, G. C. Rutledge and T. A. Hatton, Electrochemically nanostructured polyvinylferrocene/polypyrrole hybrids with synergy for energy storage, Adv. Funct. Mater., 2015, 25, 4803–4813 CrossRef CAS
- M. W. Espenscheid and C. R. Martin, Ion exchange voltammetry with electroactive ionomers, Electroanalysis, 1989, 1, 93–95 CrossRef CAS
- M. W. Espenscheid and C. R. Martin, Electroactive ion exchange polymers, J. Electroanal. Chem. Interfacial Electrochem., 1985, 188, 73–84 CrossRef CAS
- D. S. Achilleos and T. A. Hatton, Selective molecularly mediated pseudocapacitive separation of ionic species in solution, ACS Appl. Mater. Interfaces, 2016, 8, 32743–32753 CrossRef CAS
- Y. Ren, X. Mao and T. A. Hatton, An asymmetric electrochemical system with complementary tunability in hydrophobicity for selective separations of organics, ACS Cent. Sci., 2019, 5, 1396–1406 CrossRef CAS
- F. He, A. Hemmatifar, M. Z. Bazant and T. A. Hatton, Selective adsorption of organic anions in a flow cell with asymmetric redox active electrodes, Water Res., 2020, 115963 CrossRef CAS
- X. Su,
et al. Electrochemically-mediated selective capture of heavy metal chromium and arsenic oxyanions from water, Nat. Commun., 2018, 9, 4701 CrossRef
- J. C. Farmer,
et al. Electrosorption of chromium ions on carbon aerogel electrodes as a means of remediating ground water, Energy Fuels, 1997, 11, 337–347 CrossRef CAS
- C. Nie,
et al. Electrophoretic deposition of carbon nanotubes–polyacrylic acid composite film electrode for capacitive deionization, Electrochim. Acta, 2012, 66, 106–109 CrossRef CAS
- Y. Jung,
et al. Enhanced electrochemical stability of a zwitterionic-polymer-functionalized electrode for capacitive deionization, ACS Appl. Mater. Interfaces, 2018, 10, 6207–6217 CrossRef CAS
- M. S. Gaikwad and C. Balomajumder, Polymer coated capacitive deionization electrode for desalination: a mini review, Electrochem. Energy Technol., 2016, 2, 1–5 Search PubMed
- Z. Wang,
et al. Enhancement of heavy metals removal efficiency from liquid wastes by using potential-triggered proton self-exchange effects, Electrochim. Acta, 2014, 130, 40–45 CrossRef CAS
- V. Pascal, D. Laetitia, L. Joël, S. Marc and P. Serge, New concept to remove heavy metals from liquid waste based on electrochemical pH-switchable immobilized ligands, Appl. Surf. Sci., 2007, 253, 3263–3269 CrossRef CAS
- P. Viel,
et al. Electropolymerized poly-4-vinylpyridine for removal of copper from wastewater, Appl. Surf. Sci., 2003, 212–213, 792–796 CrossRef CAS
- T. Luo, S. Abdu and M. Wessling, Selectivity of ion exchange membranes: A review, J. Membr. Sci., 2018, 555, 429–454 CrossRef CAS
- E. Bakker, R. K. Meruva, E. Pretsch and M. E. Meyerhoff, Selectivity of polymer membrane-based ion-selective electrodes: self-consistent model describing the potentiometric response in mixed ion solutions of different charge, Anal. Chem., 1994, 66, 3021–3030 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0me00090f |
| ‡ These authors contributed equally. The sequence appears as a result of flip coins outcomes. |
| This journal is © The Royal Society of Chemistry 2021 |