
Controlling the optical properties of boron subphthalocyanines and their analogues
Mathias
Dowds
 and
Mogens Brøndsted
Nielsen
*
and
Mogens Brøndsted
Nielsen
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: mbn@chem.ku.dk
First published on 17th November 2020
Abstract
Boron subphthalocyanines (SubPcs) are cone shaped π-conjugated molecules comprised of three azomethine-bridged isoindole units and a central boron atom with an axial substituent. These molecules are particularly interesting for their optical properties offering potential applications for organic photovoltaics, organic light-emitting diodes, photodynamic therapy, and fluorescence imaging. In this review, we summarize how absorption and fluorescence properties can be finely tuned by substituent groups at either the periphery of the SubPc or at the axial position at the central boron atom. By suitable functionalization, fluorescence can for example be controlled by acid/base stimuli or by light/heat stimuli causing isomerization of an appended photo/thermoswitch. Moreover, optical properties can be tuned by contraction or expansion of the π-conjugated core. Key synthetic protocols for functionalization at peripheral and axial positions are also covered.
 Mathias Dowds | Mathias Dowds currently works as a postdoctoral researcher at the University of Copenhagen. He studied chemistry at the University of Kiel and received his PhD from ibidem in 2019 working with molecular switches and polymers under the supervision of Prof. Anne Staubitz. In 2020, he joined the group of Mogens Brøndsted Nielsen with a research scholarship of the German Research Foundation (DFG) and is now focussing on influencing the fluorescence properties of boron subphthalocyanines with external triggers. |
 Mogens Brøndsted Nielsen | Mogens Brøndsted Nielsen is Professor of organic chemistry at University of Copenhagen. He received his Ph.D. in synthetic and supramolecular chemistry in 1999 from University of Southern Denmark under supervision of Prof. Jan Becher. He spent one year during his Ph.D. studies with Prof. J. Fraser Stoddart at UCLA. After graduating, he spent two years as post doc in the field of acetylenic scaffolding with Prof. François Diederich at ETH in Zürich and returned in 2002 to University of Southern Denmark as Assistant Professor. In 2004 he moved to University of Copenhagen as Associate Professor and was promoted to Professor in organic chemistry in 2008. His research interests cover acetylenic scaffolding, tetrathiafulvalene chemistry, photochromic molecules such as dihydroazulenes and norbornadienes, and boron subphthalocyanine chromophores. Overall, he targets advanced functional molecules for molecular electronics and light-harvesting applications. |
Design, System, ApplicationBoron subphthalocyanines (SubPcs) are stable molecules that are easily synthesized in one step from phthalonitrile and boron trichloride and can be further functionalized at peripheral and axial positions. On account of their cone shaped structures and axial substituent, SubPcs do not strongly associate by stacking interactions, which conveniently facilitates their isolation by chromatographic means. Functional groups, such as electron donors or acceptors or photochromic units, in direct conjugation at the periphery or along the axial direction can be used to control absorption and fluorescence properties of SubPcs, in some cases as a function of external stimuli such as light or heat. These molecules have future application potential within photovoltaics, organic light-emitting diodes, photodynamic therapy, and fluorescence imaging. |
Introduction
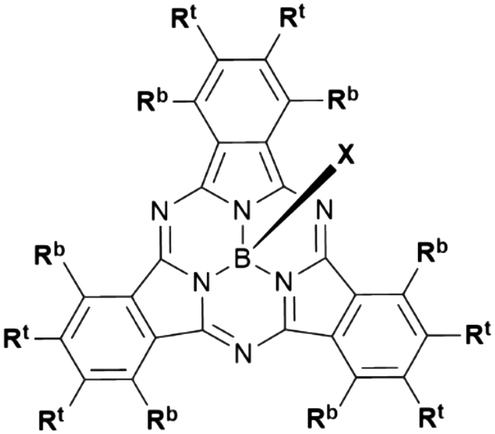
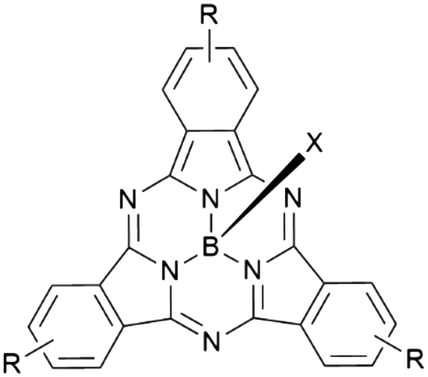
Boron subphthalocyanines (SubPcs) are a class of cone shaped macrocycles formed by three isoindole units connected by imine bridges that surround a central boron with an axial substituent (see Chart 1).1,2 The synthesis of SubPcs typically involves the cyclization of phthalonitriles in the presence of boron(III) salts (see Scheme 1).1,2 While other, larger metal ions facilitate the formation of four-membered macrocycles (phthalocyanines), the small boron core acts as template for three-membered rings. The atomic radius of boron slightly exceeds the size of the binding pocket of the SubPc macrocycle, forcing it to adapt a cone shape. The central boron ion coordinates to three pyrrole nitrogens and one axial ligand on the convex side of the macrocycle. Analogously, boron also facilitates the formation of three-membered analogues of porphyrazines and naphthalocyanines, SubPzs and SubNcs, respectively (see Chart 1).1,2 SubPzs are the smallest of these macrocycles that share a 14-electron π system surrounding the boron center. In SubPcs and SubNcs, the pyrrole groups of SubPz are fused to benzene and naphthalene.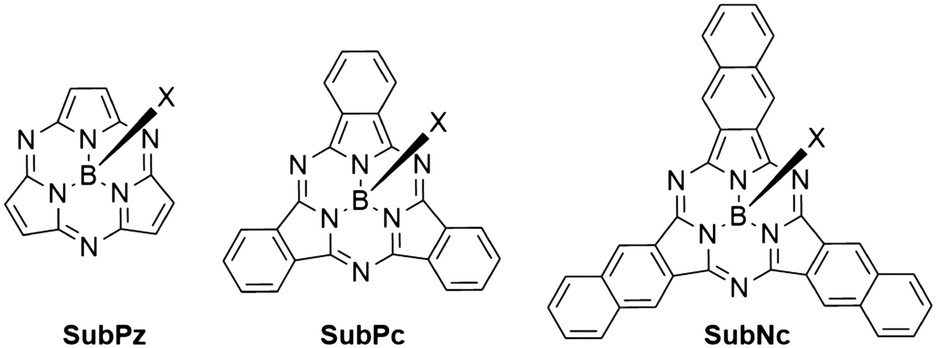 | ||
| Chart 1 Boron-centered macrocycles in this article. Subporphyrazine (SubPz), subphthalocyanine (SubPc) and subnaphthalocyanine (SubNc). | ||
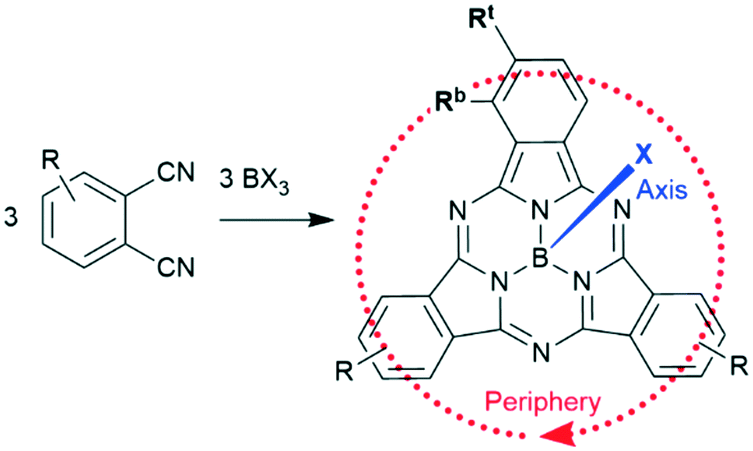 | ||
| Scheme 1 Synthesis and nomenclature of SubPcs: phthalonitriles and boron(III) halides form SubPc macrocycles. The axial ligand is highlighted in blue, the peripheral positions are surrounded by the red arrow. Rt represents a terminal substituent at the periphery and Rb a “bay” substituent at the periphery. | ||
Both, the cone shape and the axial ligand suppress aggregation in solution, increasing the solubility of SubPcs as compared to planar phthalocyanines. The absorption spectrum of SubPcs exhibits a Soret band at ca. 300 nm and a Q band at ca. 550–600 nm, depending on peripheral functionalization. Emission after photoexcitation occurs from the Q band with Stokes shifts ranging from 0–30 nm, typically mirroring its shape.1,2 Absorption and emission spectra of a SubPc with a 4-tert-butylphenoxy as the axial substituent is shown in Fig. 1.
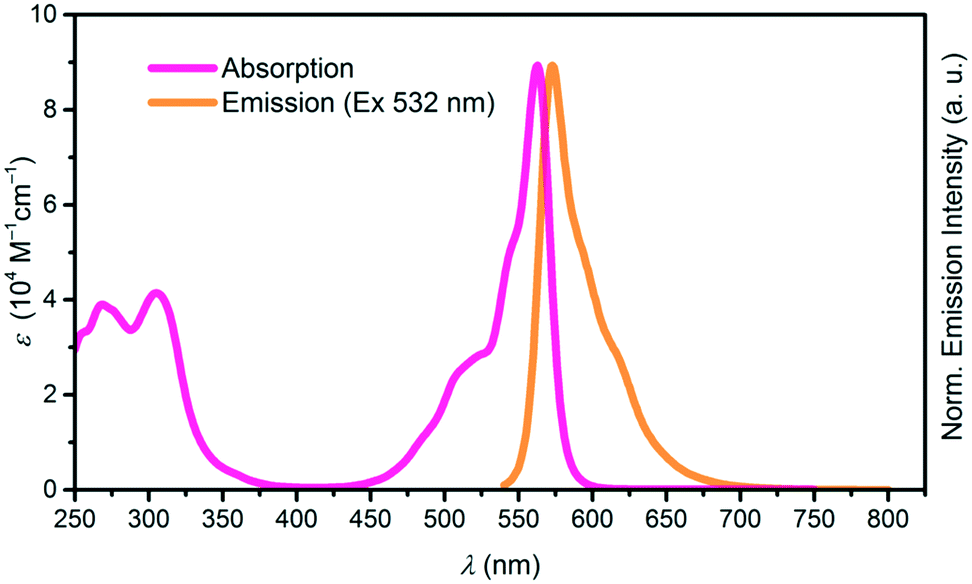 | ||
| Fig. 1 Absorption (pink) and emission (orange) spectra of SubPc with Rb, Rt = H and axial substituent X = p–tBuC6H4O. | ||
The absorption of SubPcs in the visible region combined with their capability to associate with C60 molecules and their adjustable redox properties led to the application of SubPcs as light-harvesting unit in organic photovoltaics.3–6 While the photoluminescence of SubPcs was applied in fluorescence imaging,7,8 the electroluminescent properties of SubPcs were used in organic light-emitting diodes.3 SubPcs show high triplet and singlet oxygen quantum yields and act as photosensitizer for singlet oxygen generation, which was used in photodynamic therapy.2,9,10
In this review, macrocycles with same peripheral substitution patterns share the same label, with sublabels for different axial ligands. If necessary, substituents and fragments are indexed by capital letters. The benzo groups of the SubPcs' isoindole units represent the periphery of the molecule and can be functionalized on two positions: the positions ortho to the pyrrole cycles are hereinafter labelled Rb (b for “bay” substituent) and the terminal positions meta to the pyrrole cycles are labelled Rt (see Scheme 1).
Reactivity at the axial position
Substitution reactions at the axial position
Typically, SubPcs are synthesized using boron(III) halides, leaving one halide as axial ligand (see Scheme 1).2 The stability of the B–X bonds corresponds to the halides' electronegativity in the order B–F > B–Cl > B–Br. Halide exchange reactions from chloride and bromide to fluoride can be induced by the Lewis acid BF3.11–13 SubPcs with chloride and bromide as axial substituents can undergo exchange reactions, while fluoride derivatives remain inert.11 In contrast to the reactivity of other boron halides, the axial ligand exchange of halides is faster for phenols and carboxylic acids than alcohols and amines, despite their lower nucleophilicity. This was attributed to the higher acidity of phenols and carboxylic acids.11 These substitution reactions followed second-order kinetics, with both SubPc and nucleophile concentration affecting the reaction rate. Guilleme et al.12 proposed a bimolecular mechanism that involves a weakening of the B–X bond by the hydroxy proton, while the new B–O bond was formed concertedly. Generally, the following parameters for the substitution of the axial ligand are acknowledged: i) the reactivity and reaction rate depend on the B–X bond strength, ii) the reaction rate increases with more acidic hydroxy groups, iii) electron donating substituents at the periphery enhance the reactivity, presumably due to stabilization of the positive charge at the boron atom in the transition state.12The ligand exchange of halides by carboxylic acids, hydroxy or amine ligands requires refluxing of SubPc 1 and a nucleophile in high boiling solvents (e.g. toluene) for hours and up to days (Scheme 2, reaction path (i)).2,14–17 To expand the scope of the ligand exchange beyond relatively acidic hydroxy groups, two pathways were developed: the introduction of the labile trifluoromethane sulfonate ligand18,19 as intermediate (Scheme 2, reaction path (ii)) and the activation of the B–X bond by Lewis acids20 (Scheme 2, reaction path (iii)).
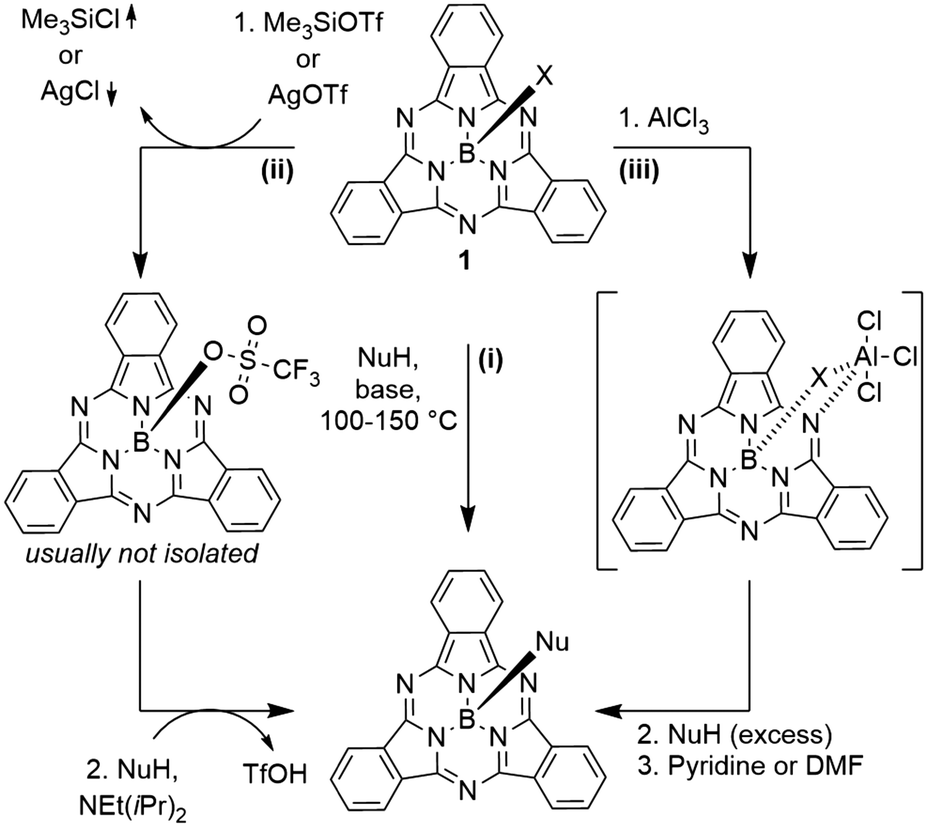 | ||
| Scheme 2 Axial ligand exchange reactions of halides by nucleophiles: i) direct exchange by nucleophiles, ii) via a triflate-SubPc intermediate and iii) via activation of the B–X bond with Lewis acids. | ||
Sulfonates can be introduced by nucleophilic substitution of axial hydroxy groups either by corresponding sulfonic anhydrides19,21 or ligand exchange reactions of SubPcs with axial chloride ligands with silver(I) sulfonate or trimethylsilyl sulfonate salts (Scheme 2, reaction path (ii)).18,22,23 Only methane sulfonate and trifluoromethane sulfonate showed a higher reactivity than chloride, with trifluoromethane sulfonate reacting faster than bromide.19 Both reactions, ligand exchange to triflate and a subsequent ligand exchange by oxygen, nitrogen or sulfur nucleophiles take place at comparably low temperatures (20–50 °C).8,15,18,22,24–26 Recently, SubPc 1 with trifluoromethane sulfonate as axial ligand was isolated and crystalized.22 Due to the high sensitivity of the axial triflate ligand towards hydrolysis, the corresponding SubPcs are typically not isolated, but subjected to the nucleophile directly.8,15,18,24,26,27
Morse and Bender20 were the first ones to describe an activation of the axial B–Cl bond in SubPcs by aluminum chloride (Scheme 2, reaction path (iii)). The formed SubPc(Cl)·AlCl3 complex reacts with phenol under condensation of HCl, before the resulting complex SubPc(OPh)·Al(OPh)3 is finally quenched by a Lewis base.20 This method allows the use of nucleophiles with lower acidity such as alcohols at temperatures below 60 °C.20 By this gentle method, it was shown possibly by Nielsen and co-workers to use TMS-protected alkynes26,28–31 or ferrocene28 as carbon nucleophiles in the ligand exchange at room temperature. Alternatively, when no additional electrophilic groups are present in the SubPc derivative, organometallic reagents such as Grignard acetylides or ferrocenyllithium compounds can be used to obtain axial B–C bonds.24
Subsequent reactions of axial substituents
Axial B–O and B–C bonds are stable enough to allow subsequent functionalization of the axial ligands. Prato reactions of aldehyde groups along the axial direction with fullerenes were widely employed successfully.32–38 Both axial alkynes and phenyl halides were used in Sonogashira coupling reactions.26,39,30 Alkyne groups can also be used as reactants in alkyne–azide click reactions.17,40–42 In a [2 + 2] cycloaddition of tetracyanoethylene followed by retro-electrocyclization, it was possible to introduce tetracyanobutadiene (TCBD) electron acceptors C, D in close proximity to the SubPc (see Scheme 3).29,43–45 | ||
| Scheme 3 SubPcs with axial (top left) and peripheral (top right) acetylene substituents, which were transferred into tetracyanobutadiene (TCBD) groups (bottom). i) Tetracyanoethylene, THF (anhydrous), room temp. ii) tetracyanoethylene, o-dichlorobenzene, room temp. | ||
Influence of the axial substituent on optical properties
Halides
Fulford et al.11 compared the chemical and physical properties of the three halide (F, Cl, Br) SubPcs 1e–g in depth (Table 1, entries 1–3). The solubility of 1e, 1f and 1g differs significantly, with Cl-SubPc 1f showing the lowest solubility in a range of nonpolar and polar aprotic solvents and F-SubPc 1e showing the highest solubility. Photophysically, the variation of halides results in only minor changes in the absorption maxima (λmax = 562–566 nm) and emission bands (λmax = 568–572 nm). The fluorescence quantum yield is lowest in Br-SubPc 1g, presumably due to a heavy atom effect. The quantum yields of F-SubPc 1e and Cl-SubPc 1f were found to be about five- and seven-fold larger. The reported quantum yields of Cl-SubPc 1f are in the range of 0.25–0.33.11,24,40,46,†‡ As SubPcs with axial halide ligands are often used as precursors for further functionalization, only few examples of Cl-SubPc and F-SubPc derivatives with identical, peripherally substituted SubPc cores were reported. In those examples, chloride causes a slight bathochromic shift of absorption and emission maxima as compared to fluoride with similar quantum yields (see e.g.Table 2, entries 2 and 3).13,46,47|
|
||||||
|---|---|---|---|---|---|---|
| # | Cmpd. | X | λ Abs (nm) | λ PL (nm) | Φ PL | Ref. |
| a In toluene. b In CH2Cl2. c In CHCl3. d In THF. e In DMF. f In DMF + H2SO4. g In CHCl3 + CF3COOH. | ||||||
| 1 | 1e | F | 562a | 568a | 0.22a | 11 |
| 2 | 1f | Cl | 565a | 571a | 0.31a | 11 |
| 564b | 581b | 0.33b | 24 | |||
| 565c | 571c | 0.25c | 40 | |||
| 563d | 574d | 0.29d | 46 | |||
| 3 | 1g | Br | 566a | 572a | 0.04a | 11 |
| 4 | 1h |
|
563a | n.r. | n.r. | 49 |
| 5 | 1i |
|
563c | 572c | 0.14c | 40, 50 |
| 6 | 1j |
|
563b | 592b | 0.18b | 52 |
| 7 | 1k |
|
562c | 571c | 0.0074c | 50 |
| 563e | 572e | 0.0065e | ||||
| 8 | 1l |
|
564f,g | 573f | 0.12f | 50 |
| 572g | 0.15g | |||||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| # | Cmpd. | Rt | Rb | X | λ Abs (nm) | λ PL (nm) | Φ PL | Ref. |
| a In toluene. b In CH2Cl2. c In THF. d Absolute ΦPL. e Solvent not specified.† Corrected values. | ||||||||
| 1 | 7a | F | H | F | 555a | 559a | 0.19a,† | 13 |
| 2 | 2c | F | F | F | 580a | 580a | 0.18a,† | 13 |
| 3 | 2d | F | F | Cl | 571b | 584b | 0.30b | 46 |
| 573c | 582c | 0.20c,d | 47 | |||||
| 4 | 8a | Cl | H | Cl | 580a | 580a | 0.12a,† | 13 |
| 574a | 584a | 0.27a,† | 53 | |||||
| 574a | 584a | 0.39a,c | 59 | |||||
| 5 | 9a | Cl | Cl | Cl | 600a | 600a | 0.12a,† | 13 |
| 6 | 7b | F | H |
|
553e | 565e | 0.16e | 16 |
| 7 | 8b | Cl | H |
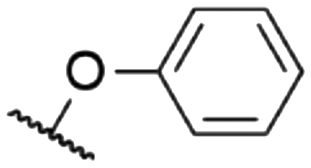
|
571e | 585e | 0.14e | 16 |
| 8 | 8c | Cl | H |
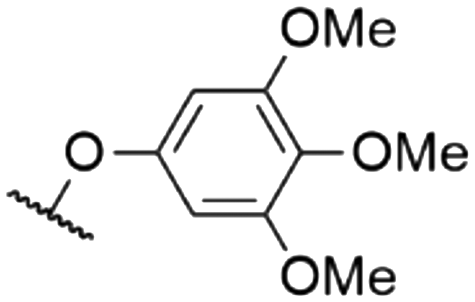
|
572a | 578a | 0.001a,† | 53 |
| 9 | 8d | Cl | H |
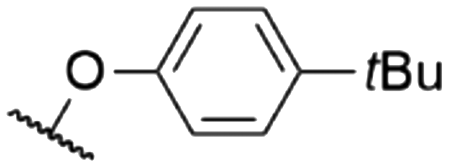
|
571a | 578a | 0.15a,† | 53 |
| 10 | 8e | Cl | H |
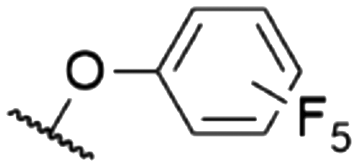
|
572a | 578a | 0.18a,† | 53 |
| 11 | 2e | F | F |
|
569a | 589a | 0.17a,† | 52 |
| 12 | 2f | F | F |
|
571a | 589a | 0.16a,† | 52 |
Phenolates
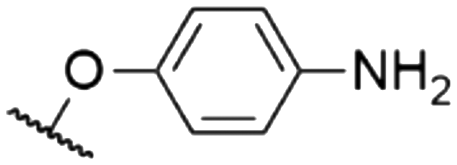
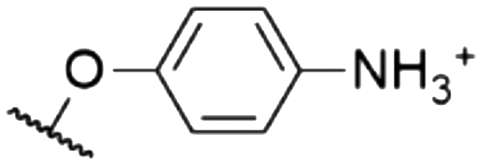
As mentioned above, axial halides can be substituted by oxygen nucleophiles. Often, phenolates are chosen as substituents due to the larger stability of the B–O bond as compared to B–Cl or B–Br bonds or for the higher solubility of the B–OAryl derivatives.2,11 Alkoxides can be introduced as convenient spacers between the SubPc core and other chromophores.17,24,26,48Typically, a substitution of axial halides by phenolates does not shift the absorption and emission bands significantly. The fully protonated H12SubPc core 1 was substituted with various phenolate groups (Table 1, entries 4–8, compounds 1h–l). The absorption of the Q-bands was reported in the range of 560–566 nm and the emission maxima were found between 570–571 nm.8,11,24,40,46,49–52 However, the axial substituent can drastically influence the emission quantum yields. Torres et al.53 compared SubPc systems bearing terminal chloride and different phenoxy substituents (Table 2, entries 8–10). They reported a fluorescence quenching by two orders of magnitude caused by the electron-rich 3,4,5-trimethoxyphenoxy substituent in SubPc 8c as compared to chloride in 8a, 4-tert-butylphenoxy in 8d and pentafluorophenoxy in 8e, respectively. This reduced fluorescence was ascribed to intramolecular electron transfer from the phenolate ligand to the boron core.†,53 Such a quenching of the fluorescence was also observed for aminophenolate substituents (Table 1, entries 7 and 8).40,50,51 Protonation of the amino nitrogen in SubPc 1k by acids reduced the electron transfer in 1l, and increases of the quantum yields by factors between 10 and 50 were reported.40,50,51
Alkoxides
In contrast to phenolates, alkoxides exert only minor influences on the fluorescence properties of SubPcs. Thus, alkyl chains are typically selected as spacers in connecting SubPcs to aromatic groups.8,17,24,26,41,54Boron–carbon bonds
SubPcs with various axial phenyl ligands show relatively unaffected Q absorption bands at 566 nm, and photoluminescence maxima at 583–590 nm with quantum yields of 0.17.55,† Terminal alkynes were used as rigid spacer for placing other aromatic systems in the proximity of SubPcs without affecting the absorption and fluorescence properties themselves.24,26,28–31,43,56 However, if alkynes are used to connect electron rich substituents such as anilines with electron poor SubPc rings, they allow an electron transfer, leading to quenching of the SubPc fluorescence (Scheme 3, compound 2a).43Variation of the periphery
Pre-synthetic: variation of the dinitrile precursor
The easiest way to introduce substituents and functional groups into the periphery of SubPcs is probably the use of functionalized phthalonitriles in the formation of SubPcs. This method has been widely employed to introduce halide substituents in the peripheral positions (see Table 2). Typically, symmetrically functionalized precursors are used, allowing the synthesis of SubPcs with two13,16,31,33–35 (7) or four13,31,51,57,58 (2) fluorine, two13,16,31,53,59,60 (8) or four13 (9) chlorine, and two iodine61,62 substituents per isoindole unit. Other substituents can also be incorporated by this method, as long as they withstand the reaction conditions during the SubPc formation.33,34,57Formation of asymmetrical SubPcs
Asymmetrical functionalization of the SubPc periphery leads to regioisomers and enantiomers. This asymmetry can derive from phthalonitriles without C2v symmetry and/or using a mixture of phthalonitriles in the synthesis. In case of 4-iodophthalonitrile, two different geometries of the formed SubPc 10 are feasible that display C3 and C1 symmetry. These two regioisomers are intrinsically chiral, leading to a pair of enantiomers of each as shown in Fig. 2. The C1 and C3 regioisomers can be separated by chromatography, and the resulting triiodo-SubPc 10 was widely used as versatile precursor for further functionalization of SubPcs.36,45,62–71meta-Substituted phthalonitriles were furthermore used to integrate other functional groups in the SubPc periphery.7,69,72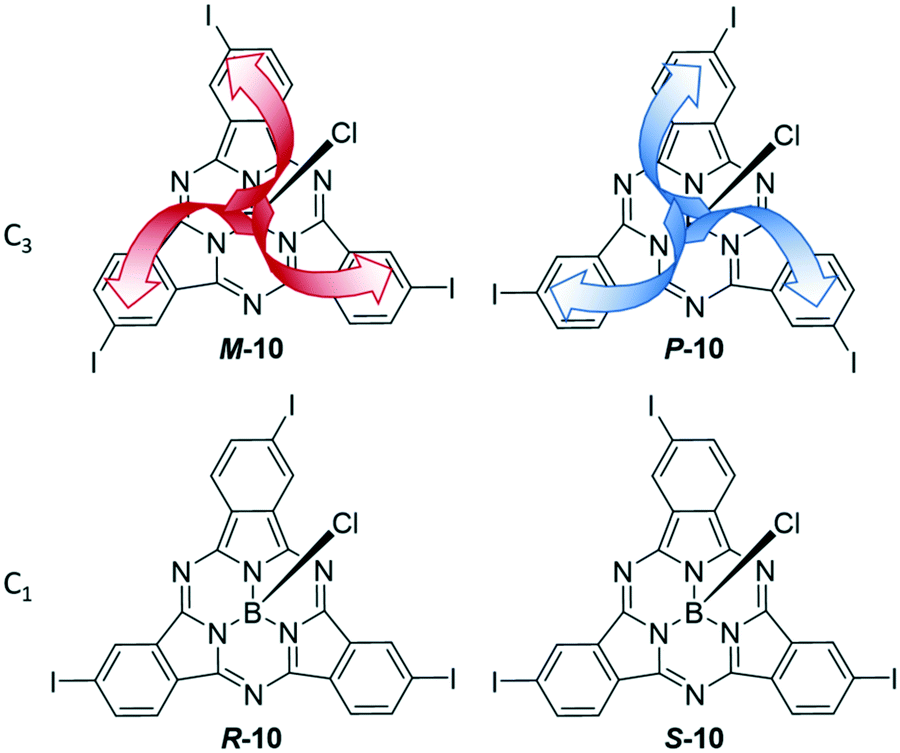 | ||
| Fig. 2 Regio- and stereoisomers of SubPc 10 with one peripheral iodo substituent per isoindole unit. Top row: Pair of enantiomers with C3 symmetry, M-10 (top left) and P-10 (top right) orientation of the iodide substituents. Bottom row: C1 enantiomers R-10 (left) and S-10 (right). | ||
It is also possible to synthesize SubPcs with different substituents at the individual isoindole units by using mixtures of phthalonitriles in the SubPc synthesis. Thus, SubPcs with one peripheral iodo substituent are accessible after separation of the other possible products.29,45,54,69,73 By this method, isoindole units with different properties can be combined, e.g. electron deficient tetrafluoro isoindoles,46,47,74,75 3,4-diiodo phthalonitriles for further functionalization,70,74,76,77 solubilizing substituents,70,74,77,78 fused aromatic systems,46,47,74,75,78 and dinitriles yielding SubPc dimers and trimers.59,75,79,80
Post-synthetic: reactions at the periphery
The aromatic isoindole units can be employed in various reactions, especially iodo-substituted SubPcs were broadly utilized for further functionalization. They were applied in palladium-catalyzed cross-coupling reactions, such as Sonogashira reactions,26,39,44,45,64–67,73,76 Buchwald–Hartwig couplings of amides,63 Stille reactions,69 Suzuki reactions,62,68 and palladium-catalyzed cyanations.70,77 The iodo substituents were furthermore transferred to boronic esters that were subsequently employed in Suzuki–Miyaura coupling reactions.68 The condensation of hydroxy groups and boronic acids also yielded boronic esters.81 Iodo SubPcs were used in nucleophilic substitutions with phenothiazene;36 similarly, toluene sulfonate groups were substituted by thiophenols.35It was possible to transfer alkynes B and D into tetracyanobutadienes C and E by [2 + 2] cycloaddition of tetracyanoethylene (TCNE) and subsequent retro-electrocyclization (Scheme 3).43,45
When two neighboring terminal positions bear nitrile groups, the SubPc could be used as precursor for the formation of new SubPc macrocycles.59,75,79,80
Terminal 1,3-dithiol-2-one motifs were successfully transferred into tetrathiafulvalene and tetrathiafulvalene-vinylogue.47,82
Influence of the periphery on optical properties
Electron-withdrawing and electron-donating groups
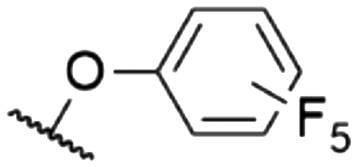
The effect of decreasing electron density in the SubPc macrocycle can be studied by comparing SubPcs with axial chloride ligands and different halide substituents in the periphery (Table 2). Unsubstituted Cl-SubPc 1f possesses an absorption maximum at 564 nm and shows photoluminescence at 571–574 nm with reported quantum yields of 0.25–0.33† (Table 1, entry 2). Substituting the terminal positions with fluorine results in SubPc 7a with a hypsochromic shift of the Q-band's absorption to 555 nm and the emission to 559 nm, while the quantum yield was reduced to 0.19† (Table 2, entry 1).13 Substitution of the axial fluoride ligand to pentafluorophenolate increased the observed Stokes shift of SubPc 7b to 12 nm, with a similar quantum yield of 0.16 (Table 2, entry 6).16The introduction of six additional fluorine substituents at the bay positions (SubPc 2c, Table 2, entry 2) induced a significant bathochromic shift of 25 nm (absorption) and 21 nm (emission) respective to the F6SubPc 7a with only six fluorine substituents, with a comparable quantum yield. SubPc 8a with chlorine substituents (Table 2, entry 4) at the terminal positions showed a bathochromic shift of 10 nm (absorption) and 13 nm (emission).53,59 Notably, the reported Stokes shifts of the emission differ between 10 nm (ref. 53 and 59) and 0 nm.13 The quantum yields were reported to be between 0.39 (ref. 59)§ and 0.12.13 Substitution of all terminal and bay positions with chlorines resulted in SubPc 9a and further bathochromic shifts of the absorption and emission bands to 600 nm, with no observable Stokes shift (Table 2, entry 5), while the quantum yield did not change as compared to SubPc 8a with terminal chlorine substituents (Table 2, entry 4).13 A SubPc 11 with four trifluoroethoxy substituents per isoindole unit showed an absorption maximum of the Q band at 600 nm in dioxane and the corresponding emission appeared at 621 nm with an extraordinarily high quantum yield of 0.85 (Table 3, entry 1).25,80
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| # | Cmpd. |
|
X | λ Abs (nm) | λ PL (nm) | Φ PL | Ref. | |
| a In dioxane. b In CHCl3. c In DMSO. d In acetone. e In toluene. f In CH2Cl2. | ||||||||
| 1 | 11 |
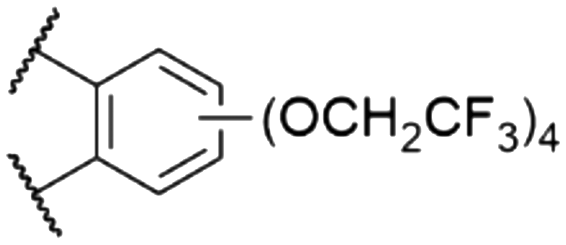
|
Cl | 600a | 621a | 0.85a | 25, 80 | |
| 2 | 12a |
|
|
568b | 601c | n.r. | 7 | |
| 3 | 13a |
|
R = H | Cl | 563d | 615d | n.r. | 81 |
| 4 | 13b | R = H | O-Ph | 562d | 616d | n.r. | 81 | |
| 5 | 13c | R = H | O–C6F5 | 567d | 581d | n.r. | 81 | |
| 6 | 14a | R = TIPS | Cl | 573e | 588e | n.r. | 81 | |
| 7 | 14b | R = TIPS | O-Ph | 524e | 602e | n.r. | 81 | |
| 8 | 14c | R = TIPS | O–C6F5 | 576e | 589e | n.r. | 81 | |
| 9 | 15 |
|
|
O-Ph | 570f | 579f | 0.19f | 78 |
The introduction of electron donating substituents such as tert-butyl or hydroxy groups, or o-diisopropylphenol ether in terminal positions leads to small bathochromic shifts of the Q band absorption maxima (Table 3, entries 2–5, 9, compounds 12a, 13a–c, 15).7,78,81 The reported emission maxima strongly depend on the axial substituent and showed Stokes shifts between 9 and 54 nm.7,78,81 Substitution of the hydroxy groups to triisopropylsilyl (TIPS) ethers in SubPcs 14a,c resulted in bathochromic shifts of 10 and 9 nm for axial chloride and pentafluorophenol ligands, respectively (Table 3, entries 6 and 8).81 In contrast, introduction of the TIPS group leads to a dramatic hypsochromic shift of 38 nm in the Q band absorption with phenol as axial ligand in SubPc 14b (Table 3, entry 7). SubPcs 14a–c with terminal silyl ethers also showed dramatic changes of the absorption and emission wavelengths, depending on the axial substituent (Table 3, entries 6–8). In this series, an axial phenol ligand (Table 3, entry 7) induced a hypsochromic shift in 14b of 59 nm as compared to the SubPc 14a with an axial chloride (Table 3, entry 6). The emission bands were also affected by the change from hydroxy groups to TIPS ethers and were reported to be 588 nm, 602 nm, and 589 nm for the SubPcs 14a,b,c with axial chloride, phenol and pentafluorophenol ligands, respectively (Table 3, entries 6–8). Notably, the Stokes shift of the TIPS-SubPc 14b with an axial phenolate ligand was observed to be 78 nm (Table 3, entry 7).81
Contraction of the peripheral π-system
Subporphyrazines (SubPzs, see Chart 1) are analogues of SubPcs with a reduced peripheral π-system: formed from dicyanoethylene precursors, they miss the peripheral benzo groups. However, they also possess an aromatic 14 π-electron macrocycle and are capable of fluorescence.2 The fluorescence of a SubPz 16a with fused 1,3-dithiol-2-one rings (Scheme 4) is almost fully quenched, which was attributed to a heavy atom effect.82 The 1,3-dithiol-2-one fused ethylene building block was also used to obtain mixed SubPcs 17 and 18 with tetrafluoro isoindole units (Scheme 4).47 Here, the heavy atom effect could also be observed: compared to the fully fluorinated SubPc 2d (Table 2, entry 3), the quantum yield of the fluorescence decreased by a factor of 5 upon exchanging one isoindole by the ethylene unit in 18 and was further reduced by a factor of 3 upon introducing a second ethylene unit in 17. The Q bands of these systems undergo blue shifts and line broadening from three tetrafluoro isoindole units (2d, 573 nm) to one (18, 570 nm), two (17, 556 nm) and three (16a, 530 nm) ethylene units.47,82 The SubPc 18 with one dithiolone unit and two tetrafluorobenzene units was further reacted to form tetrathiafulvalene-vinylogues 19 and 20 (TTFV, Scheme 4).47 This substitution pattern resulted in significant splitting of the Q band's absorption into two bands (622 nm and 832 nm). The resulting TTFV-SubPcs 19 and 20 were found to be nonfluorescent, which was ascribed to quenching of the excited state by an intramolecular charge transfer.47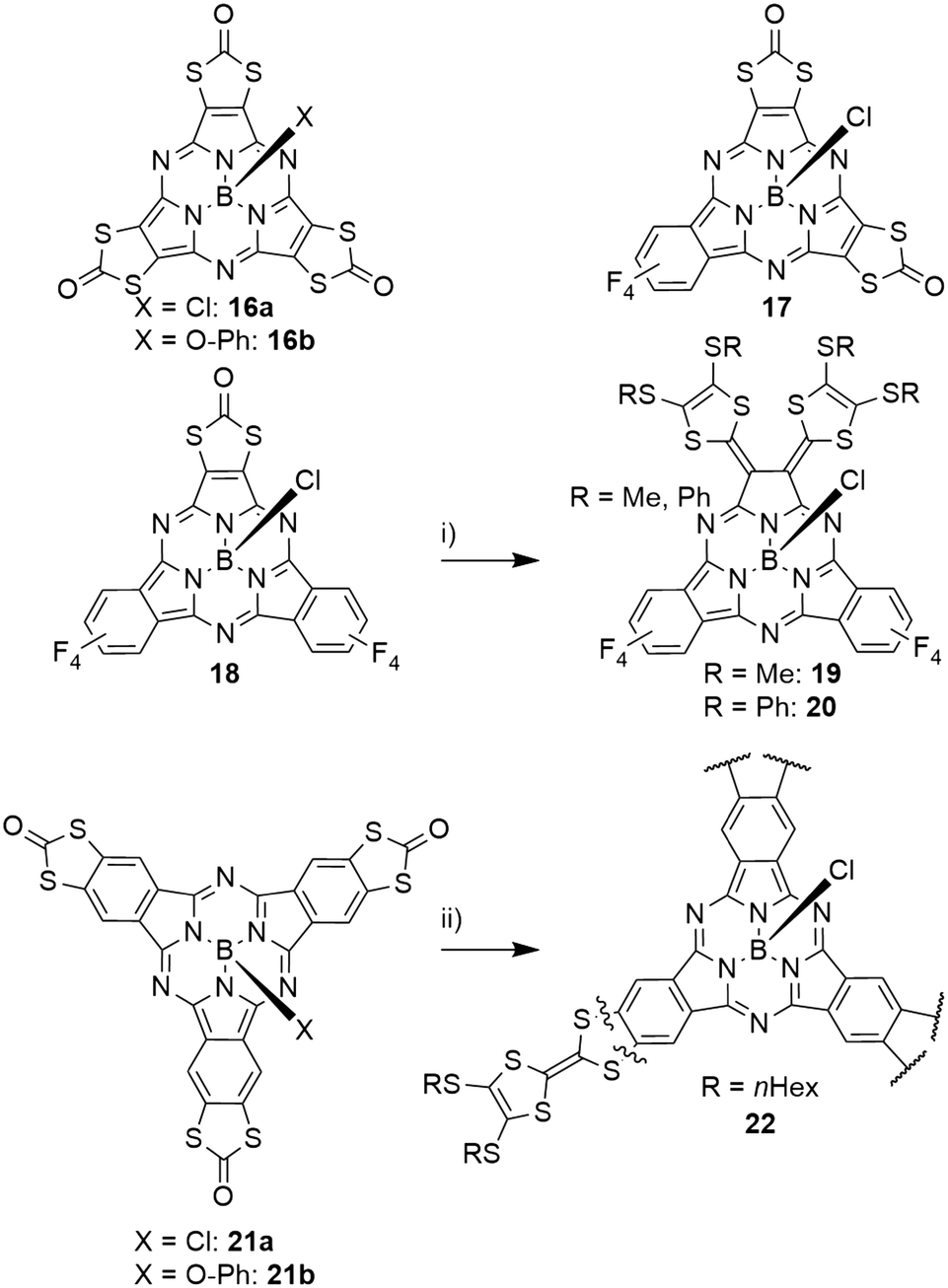 | ||
| Scheme 4 SubPzs 16–20 (top and middle left) and a SubPc (bottom left) with fused 1,3-dithiol-2-one rings. If fused to SubPz, these rings can be transferred into tetrathiafulvalene vinylogue 19, 20 (middle row). In contrast, dithiol-2-ones fused to a SubPc core 21a,b are transferred into tetrathiafulvalene (22) (bottom row). i) Dithiomethyl-1,3-dithol-2-thione or dithiophenyl-1,3-dithiol-2-thione, 1-chloronaphthalene, triethyl phosphite, 120 °C, 2 h. ii) dithiohexyl-1,3-dithol-2-thione or, 1-chloronaphthalene, triethyl phosphite, reflux, 5 h. | ||
Expansion of the peripheral π-system
The effect of incremental expansion of the peripheral benzo groups was studied by comparing SubPc 2d with three tetrafluorobenzene moieties with SubPcs 23, 24 and 25 in which one, two, or all three moieties were exchanged with naphthalene (Table 4, entries 1–3).46 Upon introduction of the first naphthalene unit, both absorption and emission of the Q band of 23 were red shifted by ca. 40 nm. Introducing the second and third naphthalene unit resulted in further bathochromic shifts of the absorption band by 24 (24) and 14 nm (25), respectively, to reach 651 nm in SubNc 25.46,49 Notably, the emission band was shifted by 12 nm with the second naphthalene and by 24 nm in the substitution of the third naphthalene to 674 nm in 25. The SubNc 24 also exhibited the largest Stokes shift in the mentioned series, being 23 nm. The observed quantum yields were for all systems in the range of 0.16 and 0.3.46 Introducing fluorine and chlorine substituents in the peripheral positions of SubNcs 26a and 27a did not change the photophysical properties drastically compared to the parent SubNc 25.83,84 Bromine substituents in 28a,b reduce the quantum yield of the emission by ca. 50%, iodine substituents in 29a,b lead to further decrease of the quantum yields to ca. one-fourth of the quantum yield of SubNc with proton (25), fluorine (26a,b), or chlorine (27a,b) substituents.83,84
1,3-Dithiol-2-one units were also fused to the peripheral benzene ring of the SubPcs 21a,b (Scheme 4).82 Compared to S-alkyl substituted SubPcs, the Q band absorption of 21a was found to have a hypsochromic shift of ca. 20 nm, due to the electron-withdrawing effect of the S2CO rings.82 Following the same strategy as described for SubNcs above, tetrafluorobenzene was gradually exchanged by tetrathiafulvalene-fused benzene units (TTF, Scheme 4).85 This resulted in small bathochromic shifts of ca. 7 nm with each introduced TTF unit. Introducing the first TTF unit reduced the emission quantum yield to 0.06, a reduction by a factor of five compared to that of SubPc 2d with three tetrafluorobenzene units.46,85 The fluorescence was further reduced to a quantum yield of 6 × 10−4 upon introduction of the second TTF unit and the symmetrical SubPc 22 with three TTF units did not exhibit any detectable fluorescence.82,85 This quenching can be explained by electron-transfer processes from the TTF moieties to the SubPc.85
Heterocycles in the peripheral π system
Subphthalocyanines with triimide rings fused to the peripheral benzo groups showed absorption maxima of the Q band at approx. 590 nm, which were independent from the axial ligand and substituents at the imide nitrogen.86 The series of triimide SubPcs emitted at ca. 600 nm, with quantum yields between 0.09 and 0.15.86The 3,4-thieno-fused SubPzs 30 and 31 display multiple Q-band absorptions at 605, 577, 520 nm and 636, 598, 544 nm, respectively (Chart 2 and Fig. 3). They exhibit very small fluorescence quantum yields of 0.06 and 0.04, respectively, in CH2Cl2, with emission maxima at 616 and 658 nm.87 Fusion of three benzo[b]thiophene units to the SubPc periphery did not result in quenched fluorescence in 32a,b. Here, the absorption and emission characteristics are influenced by the geometry. The C3 regioisomer 32a shows one absorption maximum of the Q band at 558 nm and emits at 572 nm with a quantum yield of 0.11. On the other hand, the Q band absorption of the C1 isomer 32b is split and broadened, with a maximum at 562 nm. The emission of the C1 isomer appears at 578 nm with a quantum yield of 0.12.88
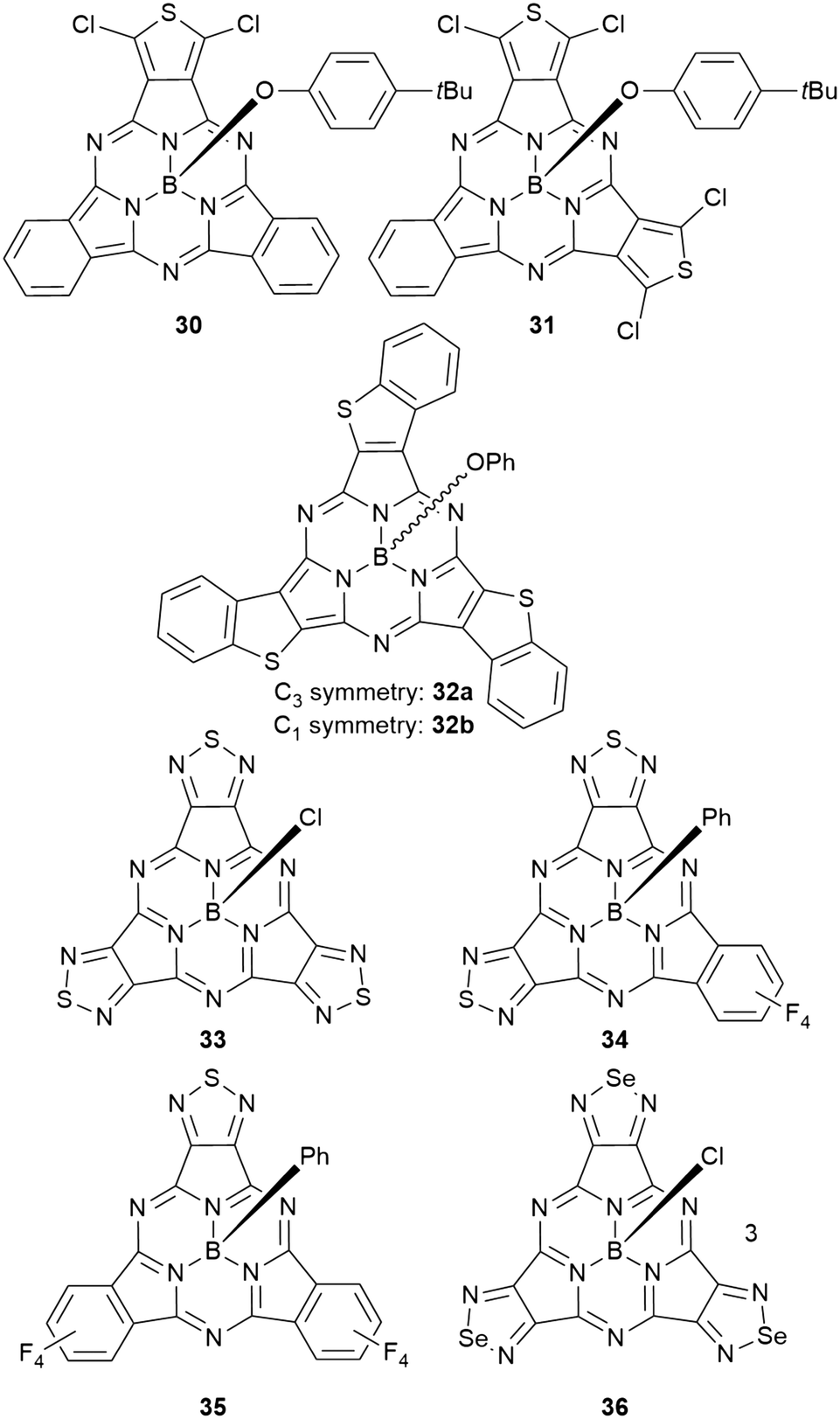 | ||
| Chart 2 SubPcs 30–36 with benzene rings substituted by 5-membered heterocycles. | ||
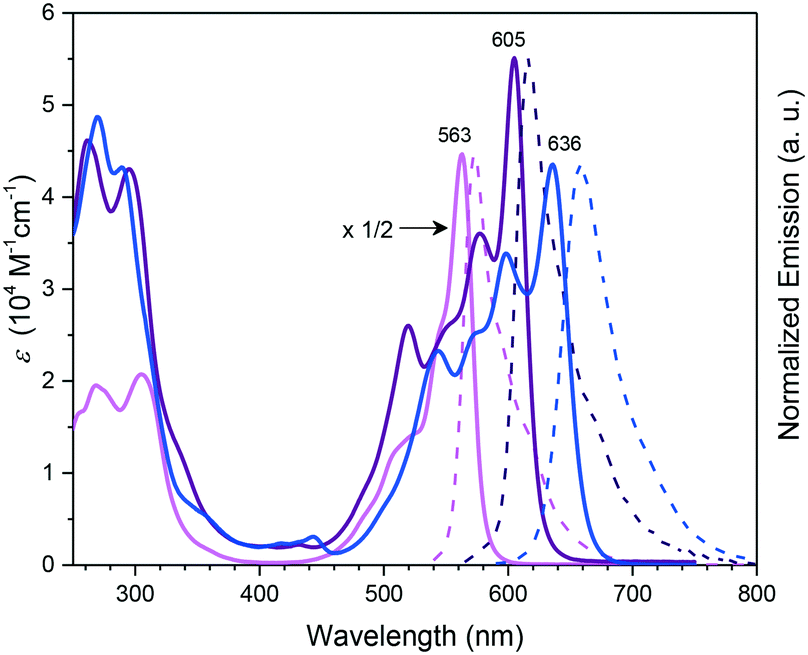 | ||
| Fig. 3 UV-vis absorption (full line) and normalized emission (dashed line) spectra of SubPc 1h (pink), and the thieno-fused SubPzs 30 (purple) and 31 (blue) recorded in CHCl3. The spectrum of 1h was multiplied by a factor of 1/2 for clarity. Reproduced from ref. 87 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Peripheral benzo groups of SubPcs can also be exchanged for other aromatic heterocycles, such as 1,2,5-thiadiazole (33–35, Chart 2). The Q band's absorption of the resulting SubPc 33 with three thiadiazole units showed a hypsochromic shift of 25 nm as compared to unsubstituted SubPc 1f (Table 1, entry 2). This shift is less pronounced than the hypsochromic shift of a SubPc 41a with three phenanthroquinoxaline (PQ) units (Chart 3, vide infra). Being mixed with tetrafluorobenzene moieties (Chart 2), the Q bands of the resulting SubPcs 34 and 35 showed distinct shoulders representing Q bands for thiadiazole- and tetrafluorobenzene-dinitrile units.89 However, these absorption bands exhibited bathochromic shifts compared to corresponding absorptions of the SubPcs 2d and 33 with three identical peripheral moieties.89 Within this series, the SubPc 33 with three thiadiazole units showed the highest emission quantum yield of 0.24, while the fluorescence of the mixed SubPcs 34 and 35 was reduced by a factor of 2.89,90 Substituting the sulfur of thiadiazoles by selenium resulted in SubPc 36 with three selenadiazole rings in the periphery (Chart 2).90 This substitution resulted in bathochromic shifts of absorption and emission not only in respect to thiadiazole–SubPc 33, but also in respect to unsubstituted SubPc 1f. The fluorescence quantum yield was reduced to 0.03 by the heavy atom effect of selenium.90
 | ||
| Chart 3 SubPc analogues 37–41 with pyrazine or phenantro[4,5-fgh]quinoxaline (PQ) motifs in the periphery. | ||
By substituting two carbon atoms in the peripheral benzo groups, SubPc analogues 37a,b and 38a,b with pyrazine moieties were described (Chart 3).90,91 These electron deficient peripheral aromatic rings induced a hypsochromic shift of 30 nm (37a, R = H) and 13 nm (38a, R = Ph), compared to unsubstituted SubPc 1f (Table 1, entry 2).90,91 The reported emission bands were 542 nm (37a) and 560 nm (38a), with Stokes shifts of 10 nm and 9 nm, respectively. The terminal substitution with phenyl rings increased the quantum yield from 0.15 to 0.31.91
Phenanthro[4,5-fgh]quinoxalines (PQ) moieties were introduced in the periphery of SubPcs 39–41 and mixed with 5,6-diisopropylphenoxy moieties (Chart 3).78 Compared to the SubPc 14a with three diisopropylphenoxy moieties (Table 3, entry 5), introducing one PQ unit in 39b resulted in a bathochromic shift of 13 nm, and a second PQ unit in 40b redshifted the Q band absorption by another 5 nm.78 The introduction of a third PQ unit in 41b did not result in further bathochromic shifts, leaving the absorption maximum at 588 nm. The emission band is also redshifted upon exchange of one PQ unit by 24 nm to 603 nm in 39b.78 Introducing further PQ moieties resulted in small hypsochromic shifts of the emission to 600 and 598 nm for two (40b) and three (41b) PQ units, respectively. The reported quantum yields of the PQ SubPcs were 0.19 and 0.18 for 41b and 41b, respectively.78 The SubPc 39b with only one PQ unit, however, exhibited a smaller quantum yield of 0.11.78
Acetylene units were used to arrange SubPc multimers in various scaffolds (vide infra), and to connect SubPcs with other functional units. This allows maintaining a conjugation between the SubPc core and the aromatic substituents while guaranteeing a certain distance between the units. If connecting 4-aniline derivatives at the periphery of SubPcs 3a,b (Scheme 3),44,45 three of such electron donating groups (EDGs) induce a bathochromic shift of the absorption to 601 nm in toluene and chloroform (C3 and C1 isomer, respectively).44,45 The observed emission spectra revealed Stokes shifts of 12 nm (ref. 44) (toluene, C3 isomer) and 26 nm (ref. 45) (chloroform, C1 isomer). The emission quantum yield was only reported for the C3 isomer to be 0.27 in toluene.44 After transformation of those acetylenes into TCBD groups (4a,b, Scheme 3, vide supra), the Q band absorption is redshifted to 622 nm in toluene44 (C3 isomer) and to 605 nm with an intense shoulder at 636 nm in chloroform45 (C1 isomer). Simultaneously, the introduction of TCBD groups resulted in a new absorption band at ca. 460 nm, assigned to a charge-transfer transition.44,45 The Q band fluorescence of the C3 isomer was reported with a maximum of 650 nm in toluene, while charge transfer reduced the quantum yield to 0.02.44
The benzothiadiazole electron acceptor was additionally introduced, replacing one of the three electron donating aniline groups (Chart 4).45 The resulting SubPc 42 with two electron donating and one electron withdrawing substituent showed similar absorption and emission properties like the SubPc 3a with three aniline substituents. Density functional theory (DFT) calculations reveal, however, significant charge-transfer character of the HOMO–LUMO transition, moving electron density from the aniline donor groups to the benzothiadiazole acceptor.45 The benzoic acid moiety allows anchoring of the molecule to TiO2 for dye-sensitized solar cell fabrication; one such cell (solid-state device) exhibited a power conversion efficiency (PCE) of 1.54%,45 being significantly smaller than related systems with porphyrin as the sensitizer (with PCEs of 4–13%).92–94
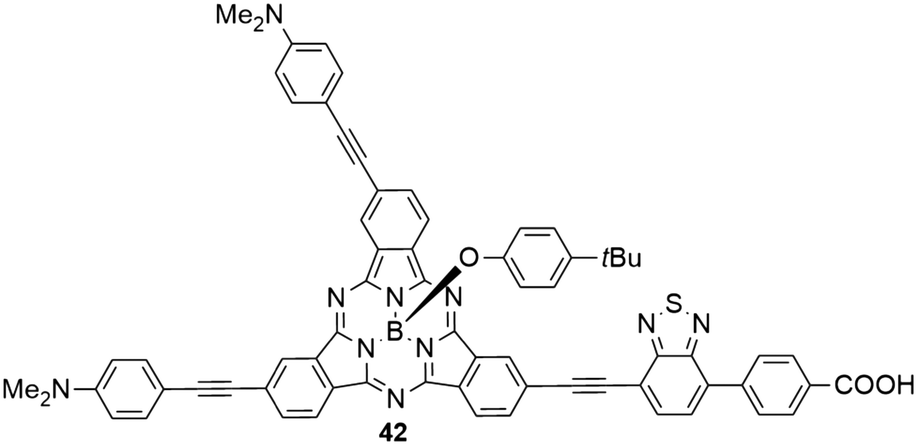 | ||
| Chart 4 SubPc 42 with two electron donating (4-dimethylaminophenyl-acetylene) substituents and one electron withdrawing benzothiadiazole–benzoic acid substituent. | ||
Combination of SubPcs with other chromophores
Ferrocenes
SubPcs were functionalized with ferrocene (FC) groups on both axial24,28,29,95,96 and peripheral29,62,97 positions. In axial positions, FC was connected to the SubPc via acetylene,24,28,29 methoxy,24 phenoxy,95 and carboxylate24,96 linkers, as well as linked directly to the central boron atom.24 It was also possible to form a SubPc dimer 1m linked by ferrocene–diacetylene (Chart 9).28 In all mentioned examples, the introduction of an axial ferrocene moiety did not alter the absorption properties significantly. The absorption maxima shifted by less than 5 nm compared to their respective analogue with an axial chloride substituent.24,29,95,96 However, the FC groups did have dramatic effects on the SubPcs' fluorescence, with quenching rates between 80 (ref. 95) and 99 (ref. 24) percent being reported. Time-resolved transient spectroscopy showed similar excited states for SubPcs with axial ferrocene units as compared to chloride.24 While the excited state of the chloro-SubPc persisted for >900 ps without significant spectral changes, the initial excited state of SubPcs with FC substituents was quenched within 2 ps and the systems returned to the initial ground state within 1 ns.24 This fast deactivation was attributed to electron transfer from the ferrocene ligands to the SubPc core, where the life time of the charge transfer state depends on the bridge unit.24 DFT calculations supported this finding.24,97In contrast to axial substitution, ferrocene units do affect the absorption properties when attached to the periphery of SubPcs. In SubPcs with axial 4-t-butylphenoxy ligands, the substitution of three peripheral protons by ferrocene lead to a bathochromic shift of 44 nm in chloroform; substituting a second set of three peripheral protons shifted the Q band's absorption maximum by another 26 nm from 605 and 631 nm, respectively.62 Similar to FC in axial ligands, an efficient electron transfer from ferrocene to the SubPc core leads to rapid decay of excited states and thus prevents fluorescence.62 These findings were also supported by DFT calculations.62,97 Substitution of only one peripheral proton by acetylene–ferrocene lead to a bathochromic shift of 6 nm in toluene, corresponding to an absorption maximum at 569 nm.29
Perylenes
When perylene diimide (PDI) units are placed in axial positions of SubPcs 2g,h (Chart 5), the individual absorption properties of both chromophores are maintained.58 The absorption maxima corresponding to the Q band appear at ca. 530 nm in CH2Cl2 and are split in case of electronic conjugation between PDI and SubPc. The SubPc's Q band absorption was not influenced by electronic conjugation with maxima at ca. 572 nm in CH2Cl2.58 As suggested by the absorption spectra, the individual emission properties of PDI and SubPc are only maintained if the linking unit prevents electron transfer. E.g., a PDI–SubPc dyad 2g with a meta-phenylenebisoxy linker showed two emission bands, corresponding to the emissions of the individual fluorophores PDI and SubPc at 530 and 580 nm, respectively.58 In contrast, the PDI–SubPc dyad 2h with para-phenylenebisoxy linker showed only one emission band corresponding to the Q band emission of SubPc at 580 nm with increased intensity.58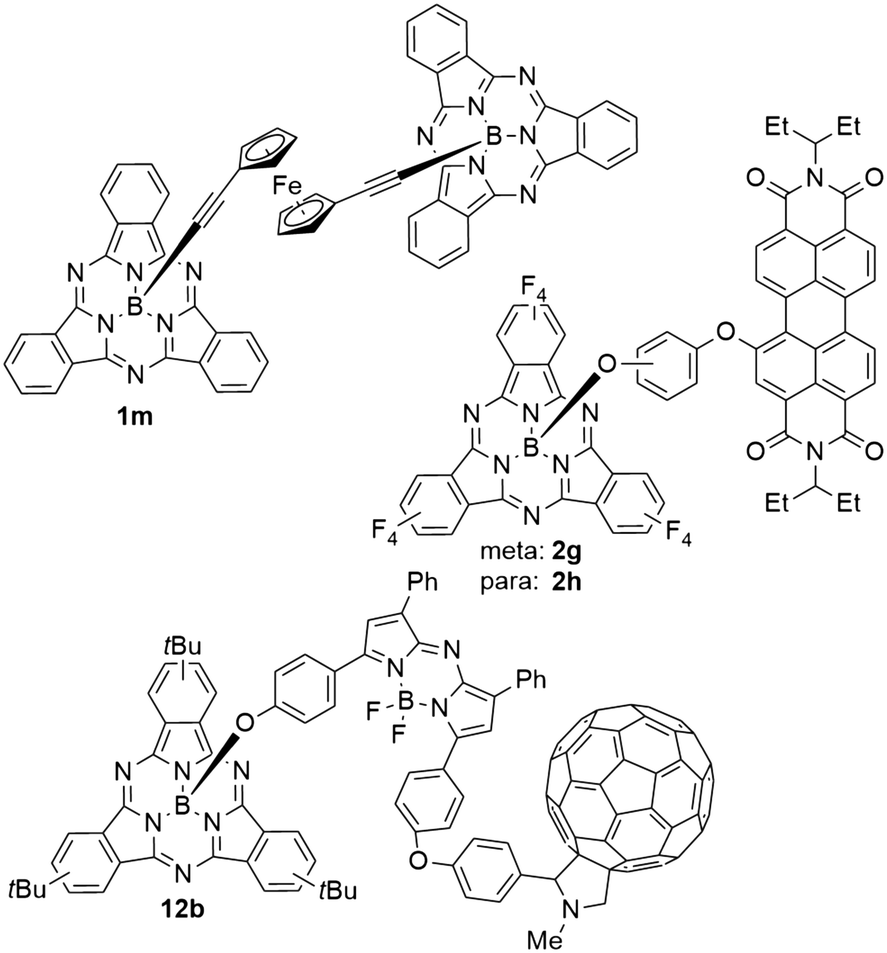 | ||
| Chart 5 SubPcs axially connected to other chromophores. Top: Ferrocene-bisacetylene links two SubPc units in the dimer 1m. Middle: A SubPc core and a PDI unit are linked axially by 1,3-phenylenebisoxy (2g) or 1,4-phenylenebisoxy (2h) units. Bottom: A BODIPY–fullerene dyad was introduced as axial ligand in SubPc 12b. | ||
Whereas axial substitution of SubPc with PDI units did not change the absorption maximum of the SubPc Q band significantly, substituting each isoindole unit with PDI substituents in the peripheral positions lead to bathochromic shift of the SubPc's Q band absorption.67 If connected directly, the absorption maximum was found to be at 590 nm in chloroform. The connection of PDI and SubPc via an acetylene bridge seemed to increase the electron transfer and induced a bathochromic shift of the SubPc's Q band absorption to 615 nm.67
BODIPY
SubPcs with unsubstituted periphery were also axially connected to dipyrrometheneboron difluoride (BODIPY) fluorophores.17,38,41,48 The absorption spectra of the resulting dyads showed characteristic bands of both SubPc and BODIPY. The absorption maxima of the SubPcs' Q band did not vary with the used linker unit and were found to be 567 nm in benzene in case of phenolate linkers48 and alkoxy triazoles17,41 after linking the SubPc with BODIPY by click reactions. The unaffected absorption spectra of the SubPc and BODIPY units indicate no ground state interaction, i.e. conjugation between the two dyes.17,41,48 Emission spectroscopy of the dyad systems revealed interaction within the excited states, again independent from the linking unit. If the BODIPY's π–π* transition absorbs at higher wavelengths than the SubPc's Q band absorption, the emission intensity of the SubPcs' Q bands was reduced in the dyads as compared to chloro SubPc.17,41,48 Simultaneously, the emission intensity of the π–π* transition of BODIPY increased.17,41,48 The reversed effect was observed when the BODIPY's π–π* transition absorbs at lower wavelengths than the SubPc's Q band.17,41 The size of this energy transfer from the high energy emission to the low energy emission depended on the overlap between the fluorescence spectrum of SubPc and the absorption spectrum of BODIPY.17,41,48 The energy transfer was confirmed as Förster resonance energy transfer (FRET) by time-resolved fluorescence measurements and DFT calculations.17,41,56 The quantum yield of the energy transfer was determined to be as high as 0.80.41In a triad 12b of aza-BODIPY that was functionalized with SubPc and C60 on opposed sites of the central methylene unit (Chart 5), competitive electron transfer pathways are feasible.38 The absorption spectrum of the triad resembles features of the parent structural motifs, without shifting the absorption maximum of the SubPc Q band. In contrast, the fluorescence of the system is quenched by more than 98% when excited at the SubPc Q band.38 In a SubPc–aza-BODIPY dyad, this excitation induced emission from the aza-BODIPY π–π* transition.38 The fluorescence after excitation of the aza-BODIPY unit showed only one third of the emission intensity of the unsubstituted aza-BODIPY unit and was quenched by another 75% when incorporated in the triad.38 These effects result from energy transfer from the SubPc to the aza-BODIPY unit and further to the C60. Thus, this triad architecture formed charge separated states with a positive charge at the SubPc unit and a negative charge on the aza-BODIPY or the C60 moiety.38
Fullerenes
When SubPc is axially connected to fullerenes, the absorption of the SubPc Q band is not affected.26,32–37,98 Similar to BODIPY units, fullerenes act as electron acceptor in SubPc–C60 dyads, quenching the SubPc fluorescence by >98%.26,32–37,98A direct binding of fullerenes to the SubPc core is not required for this energy transfer. It was shown that coordination of fullerenes and SubPc's is sufficient to quench the SubPc fluorescence.49,73,99 This complexation can be enhanced by preorganization of the two SubPc units in dimers linked peripherally by o-(diacetylene)-benzene73 and prevented by bulky substituents in the periphery of SubPc.100
As described above, connecting SubPc axially to a fullerene via a BODIPY linker, the triad 12b showed energy transfer from the SubPc unit to the fullerene (Chart 5).38
Switches
Cyanovinyl-terpyridines can act as sensor for cyanide ions by addition to the vinyl double bond, interrupting the conjugation.101 Attached to the periphery of a SubPc, the overall emission was quenched. Upon addition of cyanide, the SubPc fluorescence was switched on.101Combining SubPcs with photoswitchable moieties bears the potential of influencing and changing the fluorescence properties of SubPcs by irradiation with light. So far, SubPcs were linked to azobenzene (AB), dithienylethene (DTE), and dihydroazulene (DHA) photoswitches (Chart 6(A)).
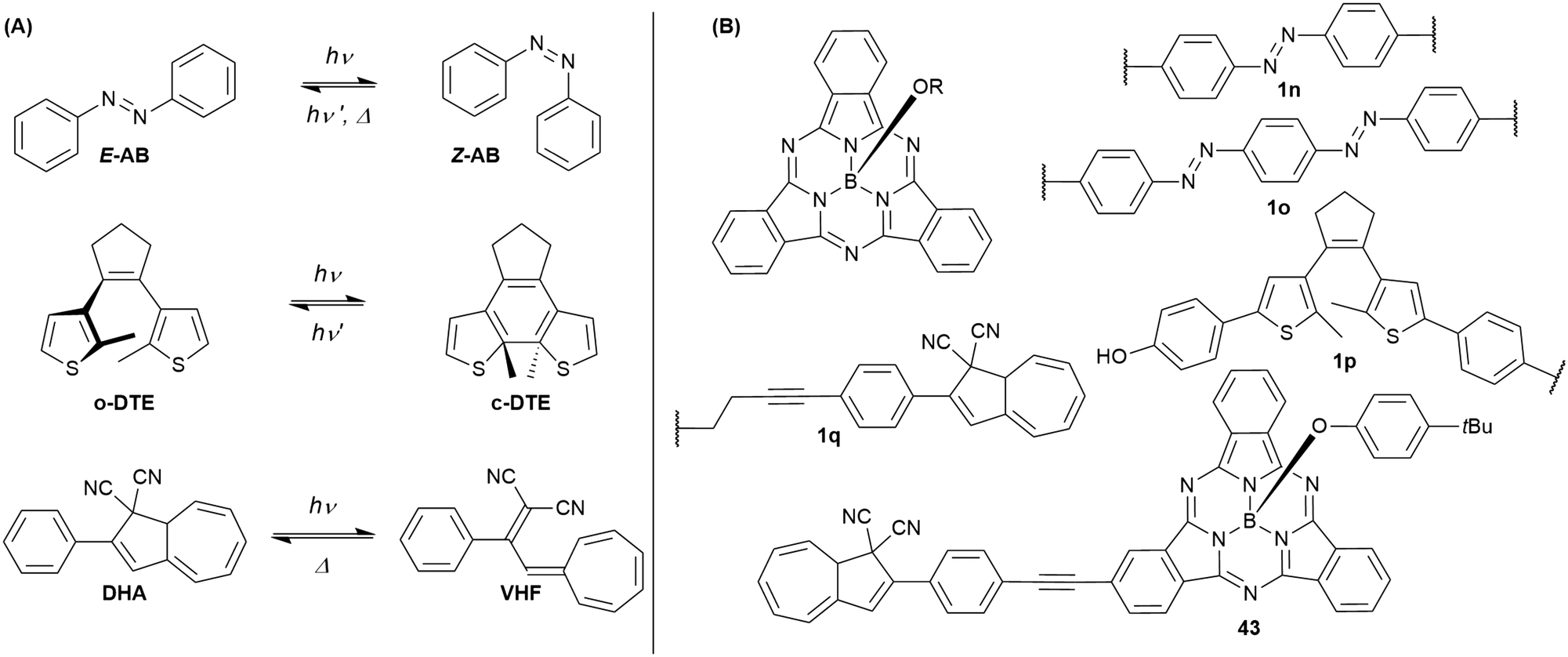 | ||
| Chart 6 (A) Photoswitchable systems that were connected to SubPcs: E/Z-azobenzene (AB), open/closed dithienylethene (DTE); dihydroazulene/vinylheptafulvalene (DHA/VHF). (B) Structures of SubPcs with photoswitchable substituents in the axial (1n–q) and peripheral (43) position. | ||
By substitution of their axial chlorides by (4,4′)-dihydroxyazobenzene, two SubPc cores were linked.102 The resulting triad 1n (Chart 6(B)) is still capable to undergo E/Z-photoisomerization as described in Chart 6(A). Upon irradiation with 365 nm light, the azobenzene (AB) unit switches from its E to its Z configuration. The SubPc–AB–SubPc triad 1n shows absorption bands that are characteristic for the two molecule classes, namely the SubPcs' Q band at ca. 560 nm and the AB's π–π* band at ca. 360 nm. Photoisomerization with UV light leads to a decrease of the E-AB's π–π* band. This effect can be reversed by irradiation with 570 nm light and was repeated for nine cycles without signs of photodegradation. The AB's photoisomerization is accompanied by changes in the fluorescence of 1o. Upon irradiation with 365 nm light, the emission intensity at the emission maximum (587 nm in DMF) dropped to ca. 50% of its initial value. Irradiation with 570 nm light restored the initial emission behavior. The reduced fluorescence intensity for the two SubPc fluorophores linked by E-azobenzene was attributed to a change in the rate of non-radiative decay.102
Similarly, bisazobenzene units can be used to connect two SubPc units at their axial positions (1o, Chart 6).103 Due to the close proximity and electronical coupling of the two azo groups, the Z,Z isomer is not accessible by photoisomerization. Thus, the observed changes in the absorption spectra were rather small as compared to 1n. Nevertheless, the photoswitching with 385 nm light could be reversed by irradiation with 570 nm light and was repeated for nine cycles without any signs of photodegradation. Upon irradiation with 385 nm light, the fluorescence of the SubPcs' Q band was reduced by one third, which was also ascribed to changes in the rate of non-radiative decay.103
In contrast to the dihydroxy azobenzene photoswitches, a dihydroxy dithienylethene (DTE) could only be linked to one SubPc 1p (Chart 6).104 The SubPc–DTE dyad 1p showed an absorption maximum at 565 nm and did undergo ring closure upon irradiation with 312 nm light to form a photostationary state with ca. 75% c-DTE. This corresponded to increased absorption in the region of the Q band of SubPc, as c-DTE also has a broad absorbance band between 400 and 600 nm. The photoisomerization was reversed by irradiation with 570 nm light. The dyad 1o exhibited fluorescence with a maximum at 587 nm and a quantum yield of 0.09 in DMSO. The emission intensity was reduced to ca. 50% by irradiation with UV light, which was fully reversible upon irradiation with visible light. The reduced fluorescence of the closed-ring isomer was ascribed to resonant energy transfer or electron transfer.104
SubPc has also been functionalized at either axial (1q) or peripheral positions (43) with the dihydroazulene photoswitch (DHA) (Chart 6).54 DHA undergoes a photoisomerization to a vinylheptafulvene (VHF) that thermally returns to DHA. Due to the stereocenter at C8a of DHA, compound 1q exists as a racemic mixture, while the peripherally substituted compound 43 exists as a mixture of diastereoisomers. These compounds are interesting for fluorescence tuning using light and heat stimuli. The axially substituted compound 1q has a fluorescence quantum yield of 0.30 in toluene, and the DHA thus has no significant influence of the SubPc fluorescence. By irradiating the compound with 420 nm light, the DHA was photoisomerized to VHF. This isomer exhibited almost no SubPc fluorescence. Heating the sample in toluene at 60 °C induced VHF-to-DHA back-conversion with restoration of the SubPc fluorescence. The SubPc fluorescence could in this way be turned on and off over several irradiation-heating cycles as shown in Fig. 4.
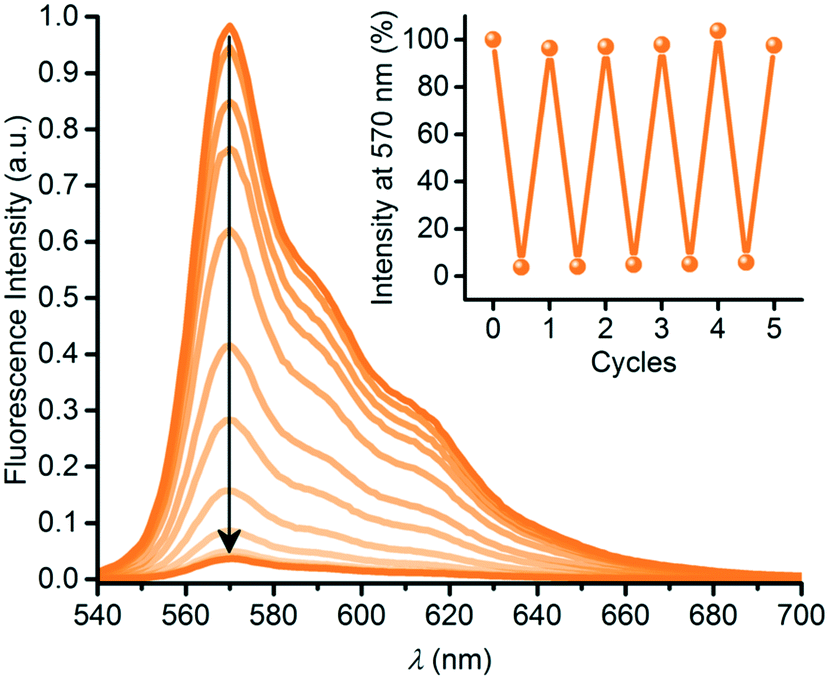 | ||
| Fig. 4 Reversible fluorescence quenching during photoisomerization of SubPc-DHA 1q into SubPc-VHF by irradiation at 420 nm for 0 to 14 minutes. Inset: Fluorescence switching over five cycles. For each cycle, fluorescence was regained after heating (60 °C) the sample in the dark for 14–26 h. Reproduced from ref. 54 with permission from The Royal Society of Chemistry. | ||
Also, compound 43 showed reduced SubPc emission after DHA-to-VHF conversion, but not to the same degree as for 1q, and in this case photoisomerization also caused some degradation, and the original fluorescence was not fully restored after one cycle.
Advanced architectures: SubPc dimers, trimers and polymers
Fused periphery
In dimers (44 and 45) and trimers (46) of SubPcs with fused peripheral benzo groups (Chart 7), the absorption properties change drastically with increasing molecule size.59 The Q band absorption is shifted bathochromically and splits into several bands.59,80 The transitions at lowest energies showed the highest extinction coefficients.59,80 With two peripheral chlorine substituents per isoindole unit, the dimer 44 showed an absorption maximum of 709 nm in toluene, corresponding to a bathochromic shift of 135 nm compared to the monomeric SubPc 8a. Expanding the structure to a trimer 46 shifted the absorption maximum by another 68 nm to 777 nm.59 The same trend was observed for a pair of SubPc monomer 11 (Table 3, entry 1) and dimer 45 with trifluoroethoxy substituents in the periphery (Chart 7).80 Here, the Q band absorption was shifted from 621 nm to 730 nm in 1,4-dioxane when going from one to two SubPc units.80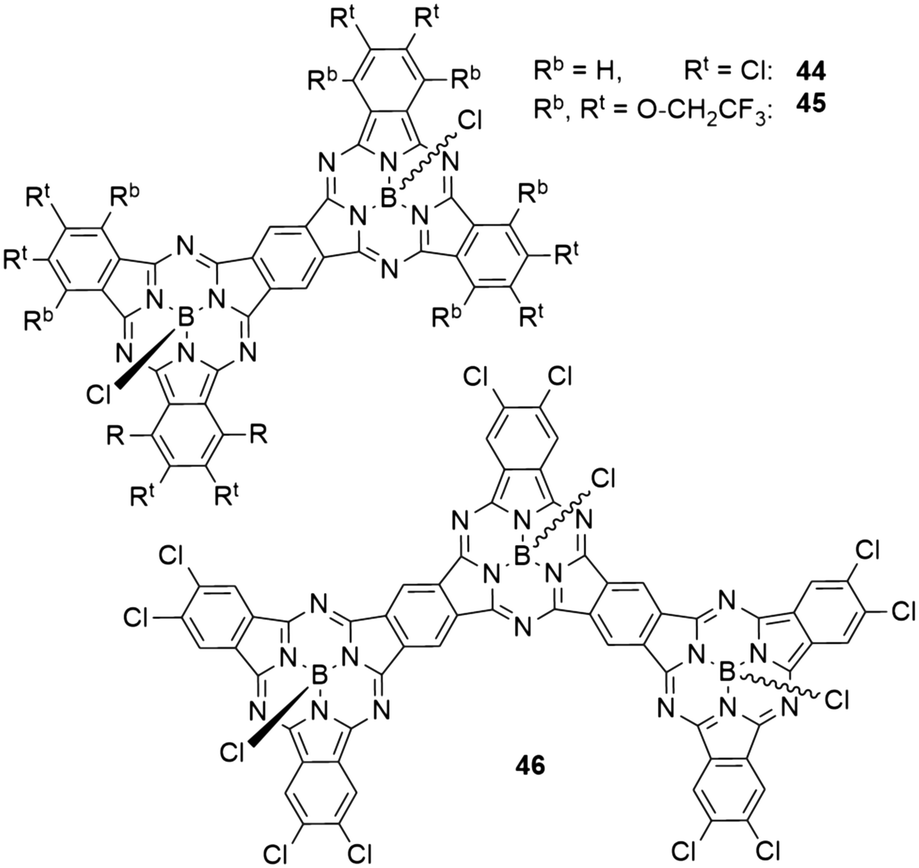 | ||
| Chart 7 Fused SubPc dimers 44, 45 and trimer 46. | ||
The emission maxima also exhibited bathochromic shifting upon increasing molecule size. With peripheral chlorides, the observed emission maxima in toluene were 584, 712, and 784 nm with quantum yields of 0.39, 0.26, and 0.19 for monomer 8a, dimer 44, and trimer 46, respectively.59 For the trifluoroethoxy substituted monomer 11 and dimer 43, the longest wavelength maxima were reported to be 621 and 744 nm in 1,4-dioxane, respectively.80 Notably, the dimer 45 exhibited a second emission band at 622 nm.80 The emission quantum yields were determined to be 0.85 for the monomer 11 and 0.31 for the dimer 45.80
Acetylenic scaffolds
A variety of cyclic and acyclic acetylenic scaffolds of SubPc has been prepared and studied (Chart 8).26,30,76,105,106 For example, three and two SubPcs have been fused to dehydro[18]annulene (47)105 and radiaannulene (48)76 cores, respectively, and two SubPcs have been linked axially by tetraethynylethene mono- and dimers (1r and 1s).26,30 While the axial anchoring of SubPcs in dimers 1r and 1s do not cause red shifted longest-wavelength absorptions, the longest-wavelength absorption maximum of the radiaannulene scaffold 48 is at 621 nm, and this compound absorbs light with a molar absorptivity >20.000 M−1 cm−1 in the entire region from 300 nm to ca. 640 nm.76 It has an emission maximum at 621 nm and exhibits a relatively small quantum yield of 0.15 in CH2Cl2, which may be explained by the electron-accepting radiaannulene core.76 Interestingly, this scaffold underwent four reversible reductions at low potentials, ultimately generating a tetraanion.76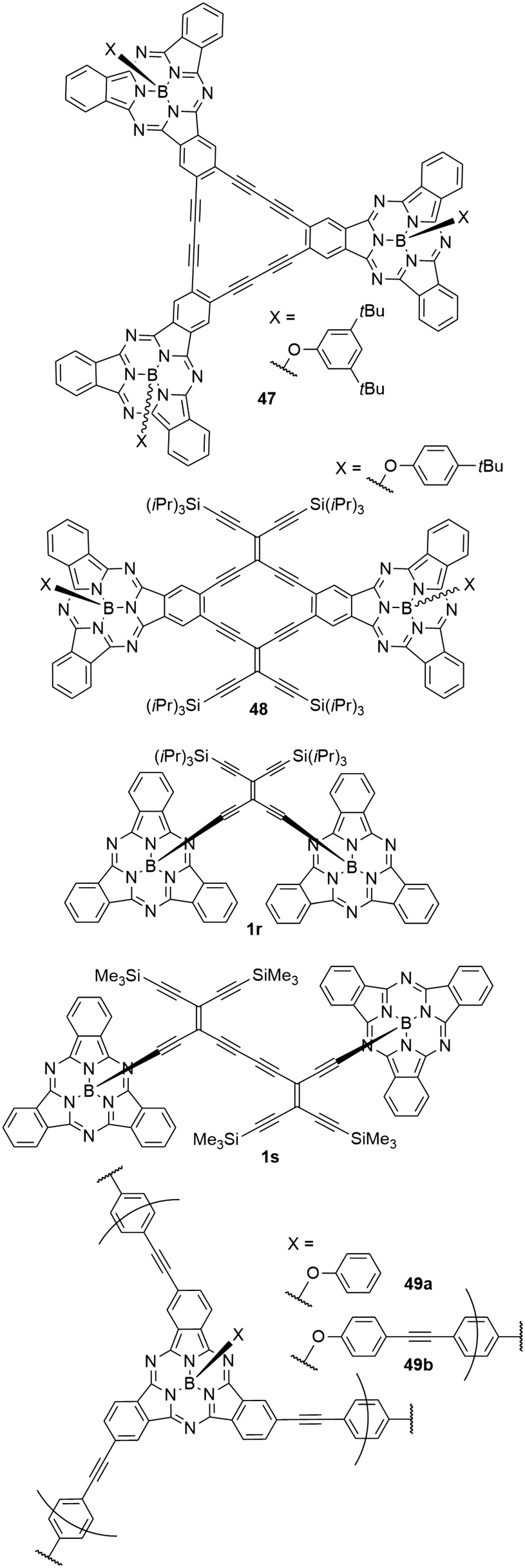 | ||
| Chart 8 Various scaffolds of SubPc multimers that were obtained by acetylenic scaffolding. | ||
It was also possible to form highly cross-linked conjugated polymers by linking SubPc with p-diethynylbenzene (49a,b, Chart 8).39 These amorphous polymers showed diffuse reflectance peaks of the Q bands in the solid state at 720 (49a) and 698 nm (49b).39 The diffuse reflectance of the corresponding monomers was found at 616 nm.39 The smaller bathochromic shift of the polymer 49b was explained by a decreased π conjugation caused by the axial linking units.39
Other connections
1,3,5-Phenylenetrioxy linker was used to connect three SubPc units (50) with different peripheral substituents (–H, –F, –OCH2CF3, Chart 9).25 The UV-vis spectrum of the trimer 50 showed a broadened Q band with a maximum at 566 nm in 1,4-dioxane, with a smaller peak at 600 nm. These peaks correspond to the Q band absorptions of the SubPcs 2d and 11 with axial chloride and peripheral fluoride or trifluoroethoxy substituents, respectively (Table 2, entry 3, and Table 3, entry 1). Upon excitation of the Q band by irradiation at 530 nm, the trimer 50 showed two emission peaks at 582 and 614 nm in 1,4-dioxane. The low energy emission originated from the trifluoroethoxy SubPc unit, while the maximum of the high energy emission corresponds to fluoro SubPc.25 The observed quantum yield of the trimer was 0.02.25 While the absorption characteristics suggest electronically independent SubPc units, the fluorescence behavior and DFT calculations indicated energy transfer from the unsubstituted SubPc core to the fluoro SubPc unit, either directly or via the trifluoroethoxy SubPc unit.25 | ||
| Chart 9 Top: Trimer 50 of three SubPc units with different electron density axially connected via 1,3,5-phenylenetrioxy. Bottom: Dimer 51 of two SubPc units connected via a [2.2]-paracyclophane macrocycle. | ||
It was possible to incorporate [2.2]paracyclophane into the periphery of SubPc and use it as linker of two SubPc macrocycles.79 The SubPc units in the resulting anti-[2.2](1,4)-subphthalocyaninophane 51 (Chart 9) were obtained as three isomers. Depending on the orientation of the SubPcs in the π stacked paracyclophane, concave–concave (CC), concave–convex (CV) and convex–convex (VV) isomers were isolated. Due to the short distance of 2.9 Å between the parallel cyclophane benzene rings, the orientation of SubPc macrocycles influences the absorption and emission properties. During the synthesis, two regioisomers of monomeric SubPc macrocycles with one cyclophane as peripheral group were obtained. The endo and exo isomers show absorption maxima of the Q band at 586 and 588 nm in chloroform, respectively.79 The CC SubPc dimer 51 showed Q band absorption with a maximum at 594 nm and regular band structure.79 Flipping one SubPc to obtain the CV isomer resulted in broadening of the absorption signal accompanied by a decreased extinction coefficient.79 The Q band absorption of the VV isomer is split into two bands at 593 and 617 nm, both with similar intensity as the bands of the CV isomer. The CC, CV, and VV isomer of the dimer exhibited fluorescence with emission maxima at 607, 626, and 641 nm, and quantum yields of 0.15, 0.16, and 0.16 respectively.79
By condensation of SubPcs 13a,b with 2 peripheral hydroxy groups per isoindole unit (Table 3, entries 2 and 3) with 4-phenylenediboronic acid, two porous organic polymers with phenolate and pentafluorophenolate as axial linkers were formed.81 The polymers were amorphous solids with pore sizes of 1.7 nm (X = OPh) and 1.5 nm (X = OC6F5). The resulting polymers were characterized in solid state, and diffuse reflectance spectra showed the absorption peaks in the Q band region at 583 and 581 nm, accompanied by weaker bands extending into the near IR region.81 Notably, these peaks are shifted by ca. 50 and 60 nm compared to the powders of the monomers 13a and 13b, respectively.81 These shifts were explained by the formation of disordered J aggregates in the solid state. The photoemission was fully quenched in the polymers.81
Summary and outlook
Subphthalocyanines and their analogues with reduced or expanded peripheries are macrocycles whose absorption and fluorescence properties can be tuned to match the requirements for various applications. The narrow emission bands of SubPcs with tunable wavelengths are beneficial for the application in organic light-emitting diodes.3 On the other hand, organic photovoltaics depend upon small fluorescence quantum yields and broad absorption bands in the visible region.3,4 The application as sensitizer for the formation of singlet oxygen requires long-lived excited triplet states.2Oxygen- and carbon nucleophiles can be introduced as axial ligand via three different routes, with possible subsequent functionalization. The peripheral positions can be altered by means of cross-coupling methods. Axial substitution exhibited smaller influences on the photophysical properties of SubPcs than alteration of the periphery. Thus, axial substituents are mainly used to enhance solubility or photostability, or act as linker for other functional building blocks, such as ferrocenes, other fluorophores, photoswitches, or fullerenes. Nevertheless, electron rich axial ligands can induce charge transfers to electron poor macrocycles and thereby quench the fluorescence. Moreover, it was recently shown from electrochemical studies that the ability of the axial substituent to stabilize a radical at the boron enhances the first reduction of the SubPc, rendering it a stronger electron acceptor.31
Charge transfer processes can also occur from electron rich peripheral substituents or associated fullerene acceptors, also quenching the SubPc fluorescence. Fluorescence quenching was also observed in mixed systems with SubPcs and other chromophores due to Förster resonance energy transfer (FRET).
Decreasing the electron density in the periphery of SubPcs by electron withdrawing groups results in hypsochromic shifting of both absorption and emission from the Q band. Analogously, electron donating groups induce bathochromic shifts of the Q band. While reducing the size of the periphery shifted absorption and emission hypsochromically, SubNcs with expanded periphery show bathochromic shifts of the Q band in the NIR region (>600 nm).
The combination of SubPc with structural motifs that can undergo isomerization upon an external stimulus such as light may find use for optical memory devices, bioimaging, or a as controllable biological markers.107–109 The introduction of azobenzene, dithienylethene, and dihydroazulene photoswitches present successful examples of such systems exhibiting light-induced fluorescence switching by virtue of photoisomerization of the photoswitch appendage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. D. thanks the German Research Foundation for a Research Fellowship (#441212186). M. B. N. thanks the Independent Research Fund Denmark | Natural Sciences (#8021-00009B) for financial support.Notes and references
- N. Kobayashi, T. Ishizaki, K. Ishii and H. Konami, J. Am. Chem. Soc., 1999, 121, 9096–9110 CrossRef CAS.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS.
- G. E. Morse and T. P. Bender, ACS Appl. Mater. Interfaces, 2012, 4, 5055–5068 CrossRef CAS.
- G. de la Torre, G. Bottari and T. Torres, Adv. Energy Mater., 2017, 7, 1601700 CrossRef.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Coord. Chem. Rev., 2021, 428, 213605 CrossRef CAS.
- T. M. Grant, D. S. Josey, K. L. Sampson, T. Mudigonda, T. P. Bender and B. H. Lessard, Chem. Rec., 2019, 19, 1–21 CrossRef.
- F. Yurt, M. Ince, O. Er, H. Melis Soylu, K. Ocakoglu and O. Yilmaz, Photodiagn. Photodyn. Ther., 2019, 26, 361–365 CrossRef CAS.
- E. Winckel, M. Mascaraque, A. Zamarrón, Á. Juarranz de la Fuente, T. Torres and A. Escosura, Adv. Funct. Mater., 2018, 28, 1705938 CrossRef.
- P. Dharmaratne, D. N. Sapugahawatte, B. Wang, C. L. Chan, K.-M. Lau, C. B. Lau, K. P. Fung, D. K. P. Ng and M. Ip, Eur. J. Med. Chem., 2020, 200, 112341 CrossRef CAS.
- F. Yurt, T. Arslan, Z. Biyiklioglu, A. Tunçel, D. Ozel and K. Ocakoglu, J. Drug Delivery Sci. Technol., 2020, 56, 101567 CrossRef CAS.
- M. V. Fulford, D. Jaidka, A. S. Paton, G. E. Morse, E. R. L. Brisson, A. J. Lough and T. P. Bender, J. Chem. Eng. Data, 2012, 57, 2756–2765 CrossRef CAS.
- J. Guilleme, L. Martínez-Fernández, D. González-Rodríguez, I. Corral, M. Yáñez and T. Torres, J. Am. Chem. Soc., 2014, 136, 14289–14298 CrossRef CAS.
- E. Bukuroshi, J. Vestfrid, Z. Gross and T. P. Bender, New J. Chem., 2019, 43, 16730–16737 RSC.
- C. G. Claessens, D. González-Rodríguez, B. del Rey, T. Torres, G. Mark, H.-P. Schuchmann, C. von Sonntag, J. G. MacDonald and R. S. Nohr, Eur. J. Org. Chem., 2003, 2003, 2547–2551 CrossRef.
- P. V. Solntsev, K. L. Spurgin, J. R. Sabin, A. A. Heikal and V. N. Nemykin, Inorg. Chem., 2012, 51, 6537–6547 CrossRef CAS.
- B. Ebenhoch, N. B. A. Prasetya, V. M. Rotello, G. Cooke and I. D. W. Samuel, J. Mater. Chem. A, 2015, 3, 7345–7352 RSC.
- E. T. Eçik, E. Özcan, H. Kandemir, I. F. Sengul and B. Çoşut, Dyes Pigm., 2017, 136, 441–449 CrossRef.
- J. Guilleme, D. González-Rodríguez and T. Torres, Angew. Chem., Int. Ed., 2011, 50, 3506–3509 CrossRef CAS.
- A. S. Paton, G. E. Morse, D. Castelino and T. P. Bender, J. Org. Chem., 2012, 77, 2531–2536 CrossRef CAS.
- G. E. Morse and T. P. Bender, Inorg. Chem., 2012, 51, 6460–6467 CrossRef CAS.
- A. S. Paton, A. J. Lough and T. P. Bender, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o459–o464 CrossRef CAS.
- Z. Wang and X. Fu, Organometallics, 2017, 36, 285–290 CrossRef CAS.
- J. Guilleme, L. Martínez-Fernández, I. Corral, M. Yáñez, D. González-Rodríguez and T. Torres, Org. Lett., 2015, 17, 4722–4725 CrossRef CAS.
- E. Maligaspe, M. R. Hauwiller, Y. V. Zatsikha, J. A. Hinke, P. V. Solntsev, D. A. Blank and V. N. Nemykin, Inorg. Chem., 2014, 53, 9336–9347 CrossRef CAS.
- S. Mori, N. Ogawa, E. Tokunaga, S. Tsuzuki and N. Shibata, Dalton Trans., 2016, 45, 908–912 RSC.
- H. Gotfredsen, L. Broløs, T. Holmstrøm, J. Sørensen, A. Viñas Muñoz, M. D. Kilde, A. B. Skov, M. Santella, O. Hammerich and M. B. Nielsen, Org. Biomol. Chem., 2017, 15, 9809–9823 RSC.
- Y. Kage, S. Shimizu, G. Kociok-Köhn, H. Furuta and G. D. Pantoş, Org. Lett., 2020, 22, 1096–1101 CrossRef CAS.
- H. Gotfredsen, M. Jevric, S. L. Broman, A. U. Petersen and M. B. Nielsen, J. Org. Chem., 2016, 81, 1–5 CrossRef CAS.
- A. V. Muñoz, H. Gotfredsen, M. Jevric, A. Kadziola, O. Hammerich and M. B. Nielsen, J. Org. Chem., 2018, 83, 2227–2234 CrossRef.
- L. Broløs, M. D. Kilde and M. B. Nielsen, Org. Biomol. Chem., 2020, 18, 6077–6085 RSC.
- E. Bukuroshi, A. U. Petersen, L. Broløs, T. P. Bender and M. B. Nielsen, Eur. J. Inorg. Chem., 2020, 2020, 3481–3495 CAS.
- C. B. Kc, G. N. Lim and F. D'Souza, Angew. Chem., Int. Ed., 2015, 54, 5088–5092 CrossRef CAS.
- M. Rudolf, O. Trukhina, J. Perles, L. Feng, T. Akasaka, T. Torres and D. M. Guldi, Chem. Sci., 2015, 6, 4141–4147 RSC.
- L. Feng, M. Rudolf, O. Trukhina, Z. Slanina, F. Uhlik, X. Lu, T. Torres, D. M. Guldi and T. Akasaka, Chem. Commun., 2015, 51, 330–333 RSC.
- C. B. Kc, G. N. Lim and F. D'Souza, Chem. – Eur. J., 2016, 22, 13301–13311 CrossRef CAS.
- C. B. Kc, G. N. Lim and F. D'Souza, Chem. – Asian J., 2016, 11, 1246–1256 CrossRef CAS.
- R. Cantu, H. B. Gobeze and F. D'Souza, J. Porphyrins Phthalocyanines, 2016, 20, 987–996 CrossRef CAS.
- V. Bandi, F. P. D'Souza, H. B. Gobeze and F. D'Souza, Chem. Commun., 2016, 52, 579–581 RSC.
- Q. Xu, Y. Gao, X. Wu, H. Hang, H. Li, Y. Chen, W. Wang and H. Tong, New J. Chem., 2019, 43, 16385–16390 RSC.
- Y. Bernhard, E. Gigot, V. Goncalves, M. Moreau, N. Sok, P. Richard and R. A. Decréau, Org. Biomol. Chem., 2016, 14, 4511–4518 RSC.
- E. Özcan, G. Keşan, B. Topaloğlu, E. Tanrıverdi Eçik, A. Dere, F. Yakuphanoglu and B. Çoşut, New J. Chem., 2018, 42, 4972–4980 RSC.
- A. Şenocak, T. Basova, E. Demirbas and M. Durmuş, Electroanalysis, 2019, 31, 1697–1707 CrossRef.
- K. A. Winterfeld, G. Lavarda, J. Guilleme, M. Sekita, D. M. Guldi, T. Torres and G. Bottari, J. Am. Chem. Soc., 2017, 139, 5520–5529 CrossRef CAS.
- K. A. Winterfeld, G. Lavarda, J. Guilleme, D. M. Guldi, T. Torres and G. Bottari, Chem. Sci., 2019, 10, 10997–11005 RSC.
- H. Gotfredsen, T. Neumann, F. E. Storm, A. V. Muñoz, M. Jevric, O. Hammerich, K. V. Mikkelsen, M. Freitag, G. Boschloo and M. B. Nielsen, ChemPhotoChem, 2018, 2, 976–985 CrossRef CAS.
- K. McAuliffe, M. Kaster, R. Szlag and E. Trivedi, Int. J. Mol. Sci., 2017, 18, 1177 CrossRef.
- Y. Wang, S. Mori, H. Furuta and S. Shimizu, Angew. Chem., Int. Ed., 2019, 58, 10975–10979 CrossRef CAS.
- S. Çetindere, B. Çoşut, S. Yeşilot, M. Durmuş and A. Kılıç, Dyes Pigm., 2014, 101, 234–239 CrossRef.
- H. M. Rhoda, M. P. Kayser, Y. Wang, A. Y. Nazarenko, R. V. Belosludov, P. Kiprof, D. A. Blank and V. N. Nemykin, Inorg. Chem., 2016, 55, 9549–9563 CrossRef CAS.
- Y. Bernhard, P. Winckler, R. Chassagnon, P. Richard, É. Gigot, J.-M. Perrier-Cornet and R. A. Decréau, Chem. Commun., 2014, 50, 13975–13978 RSC.
- Y. Bernhard, P. Richard and R. A. Decréau, Tetrahedron, 2018, 74, 1047–1052 CrossRef CAS.
- G. E. Morse, M. G. Helander, J. F. Maka, Z.-H. Lu and T. P. Bender, ACS Appl. Mater. Interfaces, 2010, 2, 1934–1944 CrossRef CAS.
- C. Duan, G. Zango, M. García Iglesias, F. J. M. Colberts, M. M. Wienk, M. V. Martínez-Díaz, R. A. J. Janssen and T. Torres, Angew. Chem., Int. Ed., 2017, 56, 148–152 CrossRef CAS.
- H. Gotfredsen, M. D. Kilde, M. Santella, A. Kadziola and M. B. Nielsen, Mol. Syst. Des. Eng., 2019, 4, 199–205 RSC.
- C. Bonnier, D. S. Josey and T. P. Bender, Aust. J. Chem., 2015, 68, 1750 CrossRef CAS.
- C. Azarias, L. Cupellini, A. Belhboub, B. Mennucci and D. Jacquemin, Phys. Chem. Chem. Phys., 2018, 20, 1993–2008 RSC.
- I. Sánchez-Molina, C. G. Claessens, B. Grimm, D. M. Guldi and T. Torres, Chem. Sci., 2013, 4, 1338 RSC.
- L. C. Kasi Viswanath, L. D. Shirtcliff, S. Krishnan, N. V. Handa and K. Darrell Berlin, Tetrahedron Lett., 2014, 55, 4199–4202 CrossRef CAS.
- R. Fujishiro, H. Sonoyama, Y. Ide, T. Fujimura, R. Sasai, N. E. M. Kaufman, Z. Zhou, M. G. H. Vicente and T. Ikeue, J. Porphyrins Phthalocyanines, 2020, 24, 211–219 CrossRef CAS.
- K. L. Sampson, X. Jiang, E. Bukuroshi, A. Dovijarski, H. Raboui, T. P. Bender and K. M. Kadish, J. Phys. Chem. A, 2018, 122, 4414–4424 CrossRef CAS.
- M. Obłoza, Ł. Łapok, J. Solarski, T. Pędziński and M. Nowakowska, Chem. – Eur. J., 2018, 24, 17080–17090 CrossRef.
- J. Fernández-Ariza, R. M. Krick Calderón, J. Perles, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, Chem. Commun., 2017, 53, 8525–8528 RSC.
- J. Guilleme, E. Cavero, T. Sierra, J. Ortega, C. L. Folcia, J. Etxebarria, T. Torres and D. González-Rodríguez, Adv. Mater., 2015, 27, 4280–4284 CrossRef CAS.
- J. Guilleme, M. J. Mayoral, J. Calbo, J. Aragó, P. M. Viruela, E. Ortí, T. Torres and D. González-Rodríguez, Angew. Chem., Int. Ed., 2015, 54, 2543–2547 CrossRef CAS.
- J. Guilleme, J. Aragó, E. Ortí, E. Cavero, T. Sierra, J. Ortega, C. L. Folcia, J. Etxebarria, D. González-Rodríguez and T. Torres, J. Mater. Chem. C, 2015, 3, 985–989 RSC.
- H. Chan, H.-L. Wong, M. Ng, C.-T. Poon and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 7256–7263 CrossRef CAS.
- H. Hang, Z. Zhang, X. Wu, Y. Chen, H. Li, W. Wang, H. Tong and L. Wang, J. Mater. Chem. C, 2018, 6, 7141–7148 RSC.
- A. J. Beneto, J. Y. Jeong and J. S. Park, Dalton Trans., 2018, 47, 16036–16039 RSC.
- T. Kawata, Y. Chino, N. Kobayashi and M. Kimura, Langmuir, 2018, 34, 7294–7300 CrossRef CAS.
- G. Zango, T. Sakurai, B. Urones, H. Saeki, W. Matsuda, M. V. Martínez-Díaz, S. Seki and T. Torres, Chem. – Eur. J., 2018, 24, 8331–8342 CrossRef CAS.
- J. Dubbert, A. Höing, N. Riek, S. K. Knauer and J. Voskuhl, Chem. Commun., 2020, 56, 7653–7656 RSC.
- F. Yurt, F. A. Sarı, M. Ince, S. G. Colak, O. Er, H. M. Soylu, C. C. Kurt, C. B. Avci, C. Gunduz and K. Ocakoglu, J. Photochem. Photobiol., A, 2018, 367, 45–55 CrossRef CAS.
- H. Gotfredsen, T. Holmstrøm, A. V. Muñoz, F. E. Storm, C. G. Tortzen, A. Kadziola, K. V. Mikkelsen, O. Hammerich and M. B. Nielsen, Org. Lett., 2018, 20, 5821–5825 CrossRef CAS.
- L. Tejerina, M. V. Martínez-Díaz and T. Torres, Org. Lett., 2019, 21, 2908–2912 CrossRef CAS.
- S. Nakano, Y. Kage, H. Furuta, N. Kobayashi and S. Shimizu, Chem. – Eur. J., 2016, 22, 7706–7710 CrossRef CAS.
- H. Lissau, C. L. Andersen, F. E. Storm, M. Santella, O. Hammerich, T. Hansen, K. V. Mikkelsen and M. B. Nielsen, Chem. Commun., 2018, 54, 2763–2766 RSC.
- G. Zango, J. Zirzlmeier, C. G. Claessens, T. Clark, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, Chem. Sci., 2015, 6, 5571–5577 RSC.
- H. Pan, W. Liu, C. Wang, K. Wang and J. Jiang, Chem. – Eur. J., 2016, 22, 9488–9492 CrossRef CAS.
- Q. Liu, S. Shimizu and N. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 5187–5191 CrossRef CAS.
- N. Shibata, S. Mori, M. Hayashi, M. Umeda, E. Tokunaga, M. Shiro, H. Sato, T. Hoshi and N. Kobayashi, Chem. Commun., 2014, 50, 3040–3043 RSC.
- G. M. Eder, B. R. Walker and P. L. McGrier, RSC Adv., 2017, 7, 29271–29274 RSC.
- Y. Wang, K. Uchihara, S. Mori, H. Furuta and S. Shimizu, Org. Lett., 2019, 21, 3103–3107 CrossRef CAS.
- K. Yamamoto, A. Takagi, M. Hada, R. Taniwaki, T. Mizutani, Y. Kimura, Y. Takao, K. Moriwaki, F. Matsumoto, T. Ito, T. Iwai, K. Hida, T. Mizuno and T. Ohno, Tetrahedron, 2016, 72, 4918–4924 CrossRef CAS.
- K. Shirai, A. Takagi, R. Taniwaki, M. Kurata, K. Kinugasa, K. Yamamoto, T. Mizutani, Y. Takao, K. Moriwaki, T. Ito, T. Iwai, F. Matsumoto and T. Ohno, Tetrahedron, 2018, 74, 4220–4225 CrossRef CAS.
- S. Shimizu, Y. Yamazaki and N. Kobayashi, Chem. – Eur. J., 2013, 19, 7324–7327 CrossRef CAS.
- X. Huang, M. Hu, X. Zhao, C. Li, Z. Yuan, X. Liu, C. Cai, Y. Zhang, Y. Hu and Y. Chen, Org. Lett., 2019, 21, 3382–3386 CrossRef CAS.
- H. Gotfredsen, F. E. Storm, A. V. Muñoz, M. Santella, A. Kadziola, O. Hammerich, K. V. Mikkelsen and M. B. Nielsen, Chem. – Eur. J., 2017, 23, 16194–16198 CrossRef CAS.
- H. Shang, L. Zhao, D. Qi, C. Chen and J. Jiang, Chem. – Eur. J., 2014, 20, 16266–16272 CrossRef CAS.
- M. Hamdoush, I. A. Skvortsov, M. S. Mikhailov, G. Pakhomov and P. A. Stuzhin, J. Fluorine Chem., 2017, 204, 31–36 CrossRef CAS.
- M. Hamdoush, K. Nikitin, I. Skvortsov, N. Somov, Y. Zhabanov and P. A. Stuzhin, Dyes Pigm., 2019, 170, 107584 CrossRef CAS.
- P. A. Stuzhin, I. A. Skvortsov, Y. A. Zhabanov, N. V. Somov, O. V. Razgonyaev, I. A. Nikitin and O. I. Koifman, Dyes Pigm., 2019, 162, 888–897 CrossRef CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Gratzel, Science, 2011, 334, 629–634 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- M. E. El-Khouly, M. A. El-Kemary, A. El-Refaey, K.-Y. Kay and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2016, 20, 1148–1155 CrossRef CAS.
- P. J. Swarts and J. Conradie, Inorg. Chem., 2020, 59, 7444–7452 CrossRef CAS.
- M. H. Rasmussen, A. L. Vishart, F. E. Storm and K. V. Mikkelsen, J. Phys. Chem. A, 2018, 122, 7620–7627 CrossRef CAS.
- C. B. Kc, G. N. Lim, M. E. Zandler and F. D'Souza, Org. Lett., 2013, 15, 4612–4615 CrossRef CAS.
- I. Sánchez-Molina, B. Grimm, R. M. Krick Calderon, C. G. Claessens, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 10503–10511 CrossRef.
- L. A. Burt, C. G. Bezzu, C. J. McMonagle, S. A. Moggach and N. B. McKeown, J. Porphyrins Phthalocyanines, 2016, 20, 1034–1040 CrossRef CAS.
- J. B. Arockiam and J. S. Park, Spectrochim. Acta, Part A, 2019, 207, 112–117 CrossRef CAS.
- M. Shi, Y. Zhao, H. Xu, J. Mack, L. Yin, X. Wang and Z. Shen, RSC Adv., 2016, 6, 71199–71205 RSC.
- M. Shi, J. Mack, L. Yin, X. Wang and Z. Shen, J. Mater. Chem. C, 2016, 4, 7783–7789 RSC.
- M. Shi, J. Chen and Z. Shen, J. Porphyrins Phthalocyanines, 2016, 20, 1082–1089 CrossRef CAS.
- R. S. Iglesias, C. G. Claessens, M. Á. Herranz and T. Torres, Org. Lett., 2007, 9, 5381–5384 CrossRef CAS.
- H. Gotfredsen, M. Jevric, A. Kadziola and M. B. Nielsen, Eur. J. Org. Chem., 2016, 2016, 17–21 CrossRef CAS.
- F. M. Raymo and M. Tomasulo, J. Phys. Chem. A, 2005, 109, 7343–7352 CrossRef CAS.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- T. Fukaminato, J. Photochem. Photobiol., C, 2011, 12, 177–208 CrossRef CAS.
- J. D. Dang, M. V. Fulford, B. A. Kamino, A. S. Paton and T. P. Bender, Dalton Trans., 2015, 44, 4280–4288 RSC.
- C. Bonnier and T. Bender, Molecules, 2015, 20, 18237–18245 CrossRef CAS.
- D. González-Rodríguez, T. Torres, D. M. Guldi, J. Rivera, M. Á. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 6301–6313 CrossRef.
- H. Kietaibl, Monatsh. Chem., 1974, 105, 405–418 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
Footnotes |
| † In the ref. 11, 55, 110 and 111, the quantum yields were determined using (phenoxy)-F12-SubPc 2e as reference with a reported quantum yield of 0.40,112 which was later determined to be 0.17.33,43 For the sake of clarity, we corrected the respective quantum yields by the resulting factor of 0.425. The quantum yields in the ref. 13 and 53 were determined using reference values that refer to ref. 11 and were also corrected. |
| ‡ The graphical abstract shows a perspective of the crystal structure of SubPc 1f, as reported in ref. 113. Image processing and rendering was performed using UCSF chimera.114 |
| § The quantum yield was determined using an absolute method. |
| This journal is © The Royal Society of Chemistry 2021 |