
Electrochemically mediated deionization: a review†
 *a
*a
Abstract
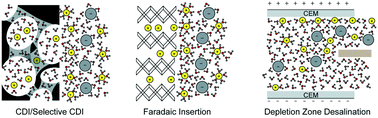
Electrochemical deionization technologies allow generation of potable water from contaminated sources and extraction of valuable resources from seawater, brines and industrial wastewater—all in an environmentally friendly and energy efficient manner. In this review, we detail existing electrode materials, cell architectures, and charge transfer mechanisms related to electrochemically driven desalination and selective element extraction in aqueous environments. More specifically, we address capacitive and faradaic charge-transfer processes—ion electrosorption and ion intercalation, respectively. We also address selective electrosorption/electrodeposition at functionalized electrode surfaces. The selectivity is driven by ion specific interactions at the electrode, such as chelation and redox activity. Electrochemically mediated deionization strategies can help address the global need for potable water. Their widespread adoption hinges on a thorough understanding of the range of available deionization methods—which we attempt to provide in this review.
- This article is part of the themed collections: Emerging Investigator Series, 2021 MSDE Symposium Collection and Molecular Engineering for Water Technologies
 Please wait while we load your content...
Please wait while we load your content...

