
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTitanosilicate zeolite precursors for highly efficient oxidation reactions†
Risheng
Bai
 ab,
M. Teresa
Navarro
b,
Yue
Song
a,
Tianjun
Zhang
a,
Yongcun
Zou
a,
Zhaochi
Feng
c,
Peng
Zhang
d,
Avelino
Corma
*b and
Jihong
Yu
*ae
ab,
M. Teresa
Navarro
b,
Yue
Song
a,
Tianjun
Zhang
a,
Yongcun
Zou
a,
Zhaochi
Feng
c,
Peng
Zhang
d,
Avelino
Corma
*b and
Jihong
Yu
*ae
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: jihong@jlu.edu.cn
bInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, 46022 Valencia, Spain. E-mail: acorma@itq.upv.es
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
dDepartment of Chemistry, Dalhousie University, Halifax, Nova Scotia B3H 4R2, Canada
eInternational Center of Future Science, Jilin University, Changchun 130012, China
First published on 23rd October 2020
Abstract
Titanosilicate zeolites are catalysts of interest in the field of fine chemicals. However, the generation and accessibility of active sites in titanosilicate materials for catalyzing reactions with large molecules is still a challenge. Herein, we prepared titanosilicate zeolite precursors with open zeolitic structures, tunable pore sizes, and controllable Si/Ti ratios through a hydrothermal crystallization strategy by using quaternary ammonium templates. A series of quaternary ammonium ions are discovered as effective organic templates. The prepared amorphous titanosilicate zeolites with some zeolite framework structural order have extra-large micropores and abundant octahedrally coordinated isolated Ti species, which lead to a superior catalytic performance in the oxidative desulfurization of dibenzothiophene (DBT) and epoxidation of cyclohexene. It is anticipated that the amorphous prezeolitic titanosilicates will benefit the catalytic conversion of bulky molecules in a wide range of reaction processes.
Introduction
Zeolites are crystalline microporous silica-based materials with well-defined pores and cavities that play a fundamental role in ion-exchange, adsorption, and separation, as well as petrochemical and fine-chemical catalytic applications.1–7 The uniform micropores with sizes of molecular dimensions (ca. 0.25–1 nm) endow zeolites with unrivalled catalytic properties, especially shape selectivity.8,9 However, such microporous zeolites cannot process bulky molecules with kinetic diameters larger than 1.0 nm. Only the active sites located on the crystal surface or at the pore mouths are accessible for those bulky molecules. Besides, such confined environments in microporous zeolites also dramatically decrease the transport efficiency of reactants and products.10In recent years, extra-large pore zeolites have shown interesting potential as efficient catalysts in the field of fine chemicals and petrochemicals, but suffer from high product cost and low (hydro)thermostability.11,12 Nano-sized zeolites are expected to greatly lower the transport limitations, but are obstructed by the separation processes from the zeolite synthesis liquid or the catalytic reaction system.13,14 Creating hierarchical structures in the microporous zeolites has proven to be an efficient method for increasing the mass transport efficiency, but leads to the intrinsic limited ability to process bulky molecules as well as complicated and time/energy-consuming procedures.15–17 Seeking alternative materials that better combine the intrinsic shape selectivity of zeolites and the accessibility towards the active site is of great significance in zeolite catalysis.
With the deepening of research on the crystallization mechanism in recent years, in both clear synthesis gels and dense synthesis gels, it is generally accepted that the crystallization of zeolites proceeds via the following steps: (1) induction period, (2) nucleation, and (3) crystal growth.18–20 The mixed synthesis reactants, referred to as the primary amorphous phase, can transform into the secondary amorphous phase by equilibrating with the solution phase.18 The secondary amorphous phase is a pseudo-steady-state intermediate, exhibiting some short-range order of the zeolite framework structures, and can be converted to crystalline zeolites with long-range order zeolite frameworks. Tsuruta et al. presented that the short-range order of Si and Al atoms in the amorphous aluminosilicate is similar to that in crystalline zeolite crystals in the zeolite A system by using X-ray and electron diffraction.56 With extended X-ray absorption-fine structure (EXAFS) and X-ray absorption near edge structure (XANES) spectroscopies, Walton et al. found that the structure of the amorphous gallium silicates is similar to the structure of the finally crystallized gallium hydroxosodalite zeolite.57 Although the secondary amorphous phases demonstrate some spectroscopic and size-exclusive characteristics of zeolites, the lack of long-range periodicity of a crystalline zeolite lattice makes it amorphous and unsuitable for X-ray diffraction (XRD).21–24 Therefore, these zeolite precursors are also regarded as X-ray amorphous zeolites or embryonic zeolites.
Corma et al. constructed amorphous zeolite precursors of ITQ-7, ITQ-21, ITQ-30, ZSM-12, Beta, and NU-87, which exhibited pore dimensions and topologies very close to their corresponding crystalline zeolites, through the self-assembling of organic templates and silica.21 Inagaki et al. regulated the crystal growth kinetics of zeolite Y (FAU) and found that the X-ray amorphous zeolite phase, taken at the end of the induction period, showed the highest catalytic activity toward the Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate.25 Recently, Haw et al. prepared X-ray amorphous ZSM-5 precursors with tetrapropylammonium hydroxide (TPAOH) as the structure directing agent, which presented superior catalytic performances in the dealkylation of 1,3,5-triisopropylbenzene, which cannot penetrate through the micropores of zeolites.23 The above demonstrated high catalytic activities of the amorphous titanosilicate zeolites were ascribed to the more open structure of X-ray amorphous zeolites, which dramatically increased the accessibility towards the catalytic active sites and enhanced the mass transport efficiency.
Heteroatom-containing zeolites (e.g. Ti, Sn, B, etc.) with isolated active sites in the zeolite framework exhibit unique catalytic performances in numerous important processes.26–31 Titanosilicate zeolites, as a typical family of heteroatom-containing zeolites, have been widely used in oxidation reactions with H2O2 as the oxidant under mild conditions, such as alkene epoxidation, phenol hydroxylation, cyclohexanone ammoximation, and deep oxidative desulfurization.32–35 The catalytic activity of titanosilicate zeolites strongly correlates with the lattice Ti contents and the Ti coordination states.36,37 The length difference between the Ti–O bond (1.80 Å) and Si–O bond (1.61 Å) distorts the local structure around Ti in the titanosilicate zeolites, requiring high framework flexibility for the isomorphous substitution of Ti ions for Si.36,38,39
In addition, Guo et al. thoroughly investigated the active titanium species in TS-1 zeolite, and identified a new titanium species with a characteristic Raman band at 695 cm−1 when excited at the 266 nm laser line, confirmed by DFT calculations.40 The newly discovered octahedrally coordinated titanium species showed superior catalytic performance in the epoxidation reactions in comparison to the tetrahedrally coordinated one. Very recently, Xu et al. constructed a mononuclear TiO6 species in TS-1 zeolite via seed-assisted microwave irradiation.41 The octahedrally coordinated titanium species was identified with UV-Raman (excited at the 266 nm laser line) and X-ray absorption spectroscopy. The obtained TS-1 zeolite catalyst with mononuclear TiO6 species displayed outstanding catalytic performance and stability in the epoxidation of 1-hexene. Increasing the accessibility towards the Ti sites and the mass transport efficiency, improving the Ti contents, and constructing Ti catalytic species with higher activity have been widely considered to be key for improving the catalytic efficiency of titanosilicate catalysts.
In this work, with the understanding of zeolite formation processes, we prepared a series of amorphous titanosilicate zeolites (ATZ) with a short-range order structure as well as tunable Si/Ti ratio and pore sizes by using quaternary ammonium ions as the organic structure directing agents (OSDAs). The OSDA used in the zeolite synthesis system not only provides shape commensurate with the cages and channels in zeolites but also is in charge of the nucleus formation and the crystal growth process; the lower the charge density of the OSDA, the higher the requirement for nucleation.42,43 A series of commercialized and low charge density home-made quaternary ammonium cations have been employed to obtain amorphous titanosilicate zeolites, which are characterized by specific porosities, open structures, and abundant octahedrally coordinated Ti species. The obtained amorphous titanosilicate zeolite catalysts show enhanced catalytic performance in the oxidative desulfurization of dibenzothiophene (DBT) and the epoxidation of cyclohexene, with respect to the final zeolite structures.
Results and discussion
A series of amorphous titanosilicate zeolites (ATZ) with short-range order were synthesized with a molar composition of 1.0 SiO2: 0.025–0.2 TiO2: 0.25–0.50 OSDA: 10–30 H2O under hydrothermal conditions (170 °C for 6 h) by using TEA+, TMAda+, PhHE2+, DPhHE2+, and PhDC2+ as the structure directing agents (Scheme 1), and the obtained amorphous titanosilicates were named ATZ-TEA, ATZ-TMAda, ATZ-PhHE, ATZ-DPhHE, and ATZ-PhDC, respectively. The detailed synthesis conditions of ATZ samples are presented in Table 1. Notably, the Si/Ti ratio of the successfully prepared ATZ samples can be as low as 5, demonstrating the high plasticity of the ATZ samples. | ||
| Scheme 1 Organic cations utilized as structure directing agents (OSDA). | ||
| Sample | x | y | Si/Tic | S BET (m2 g−1) | S micro (m2 g−1) | S ext (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) | I 960/800 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Initial gel: SiO2: 0.025TiO2: xOSDA: yH2O. b Prepared with TiO2/SiO2 = 0.2. c Measured by inductively coupled plasma (ICP). d Specific surface area calculated from the nitrogen adsorption isotherm using the BET method. Smicro (micropore area), Sext (external surface area), and Vmicro (micropore volume) calculated using the t-plot method. e Total pore volume at P/P0 = 0.995. f V meso = Vtotal − Vmicro. g FT-IR relative intensity of the bands at 960 and 800 cm−1. | ||||||||||
| ATZ-TEA | 0.50 | 10 | 40.8 | 605 | 352 | 253 | 1.89 | 0.14 | 1.75 | 2.17 |
| ATZ-TEA-5b | 0.50 | 10 | 4.9 | 623 | 283 | 340 | 2.05 | 0.12 | 1.93 | 2.97 |
| ATZ-TMAda | 0.50 | 10 | 40.7 | 747 | 319 | 428 | 1.93 | 0.13 | 1.80 | 1.93 |
| ATZ-PhHE | 0.25 | 30 | 41.3 | 537 | 318 | 219 | 1.40 | 0.14 | 1.26 | 2.31 |
| ATZ-DPhHE | 0.25 | 30 | 41.2 | 730 | 628 | 102 | 0.41 | 0.25 | 0.16 | 2.13 |
| ATZ-PhDC | 0.25 | 30 | 40.5 | 498 | 299 | 199 | 1.76 | 0.13 | 1.63 | 1.89 |
All the harvested titanosilicate samples are X-ray amorphous (Fig. 1a). Infrared spectroscopy (IR) was used to characterize the short-range orders of the ATZ samples (Fig. 1b). The absorption bands in the IR spectra at around 1080 and 800 cm−1 correspond to the asymmetric and symmetric Si–O stretching motions; the band at 460 cm−1 is ascribed to a bending Si–O–Si mode.44 A small absorption band, that can be observed at 550 cm−1, is assigned to the bending vibrations of external Si–O, and also is generally attributed to the stretching vibration of double rings in the MFI structure.45 The absorption band at 960 cm−1 is attributed to the vibration of the Si–O–Ti or Si–O bond perturbed by Ti atoms in the framework.46,47 The intensity ratio between the absorption bands at 960 cm−1 and 800 cm−1 (I960/880) is usually used to evaluate the relative content of Ti in the framework.48 Generally, a larger I960/880 value means a higher framework titanium content. As summarized in Tables 1 and S1,† ATZ samples exhibited larger I960/880 values than the conventional TS-1 zeolite sample (1.16), which was added as a benchmark, indicating outstanding ability to incorporate framework titanium species.
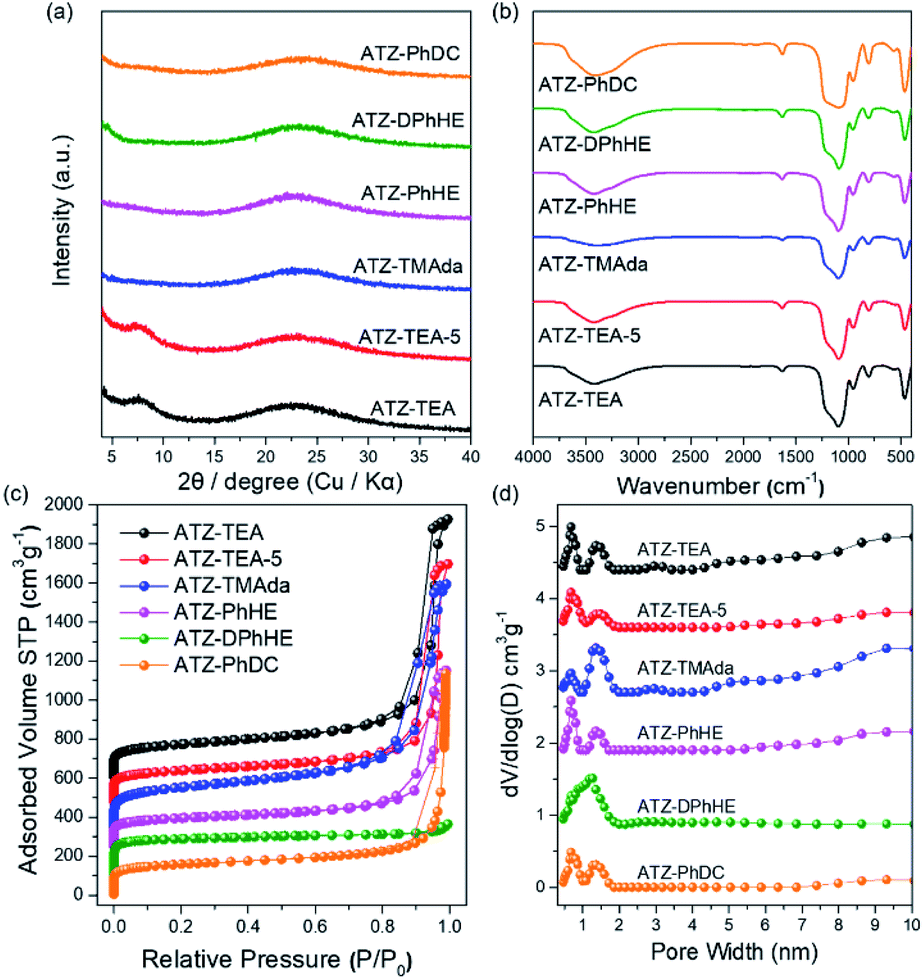 | ||
| Fig. 1 (a) XRD patterns, (b) FT-IR spectra, (c) N2 adsorption and desorption isotherms, and (d) pore size distributions of the ATZ samples. | ||
The nitrogen adsorption–desorption isotherms of the ATZ samples show combined type I–IV isotherms (Fig. 1c). The significant uptakes at low relative pressure demonstrate the existence of abundant microporous structures; and the hysteresis loops at a relative pressure of 0.8 < P/P0 < 0.99 are ascribed to the aggregation of small particles and the existence of mesopores. ATZ samples exhibit similar micropore volumes and higher total pore volumes in comparison to the crystallized TS-1 zeolite sample (Table S1 and Fig. S1†). Taking into account the comparison of the detailed data of the samples obtained from PhHE, and DPhHE that have been summarized in Table 1, it can be found that the Brunauer–Emmett–Teller (BET) area and micropore volume are positively correlated with the size of the utilized organic template (SBET: 537, and 730 m2 g−1, respectively; Vmicro: 0.14, and 0.25 cm3 g−1, respectively). It is worth noting that TEA+ usually directs the zeolite structure through a clustered mode, presenting a clustered/aggregated conformation, thus resulting in a sample with comparable surface areas and micropore volumes to PhHE2+ which exhibits a bulkier molecule size (605 vs. 537 m2 g−1, and 0.14 vs. 0.14 cm3 g−1, respectively).49 The micropore surface area of ATZ-TEA-5 is smaller than that of the ATZ-TEA (283 vs. 352 m2 g−1), indicating that the structural order decreases with increasing Ti content. The pore diameter of ATZ samples, determined according to nonlocal density functional theory, ranges from 0.5–2 nm, showing a strong correlation with the size of the OSDAs and verifying the existence of microporous structures (Fig. 1d). It is widely accepted that the quaternary ammonium cations could order the surrounding (alumino)silicate species by progressively displacing water molecules in the hydration sphere, thus forming the inorganic–organic composite species. Therefore, the utilized organic templates are closely related to the textural properties of the prepared ATZ samples.
The transmission Electron Microscopy (TEM) images of the obtained ATZ samples illustrate that all the samples exhibit a branched worm-like morphology, with sizes ranging from 10 to 20 nm (Fig. 2). The worm-like morphology is the typical morphological feature of the amorphous zeolite material. It is notable that the ATZ-DPhHE sample, prepared by using DPhHE2+ as the OSDA, exhibits a larger particle size (50–100 nm) than the others. The two phenyl groups in the DPhHE2+ provide stronger hydrophobicity to the template, which dramatically facilitates the overlap of the hydration sphere of the OSDA and the raw Si and Ti species, enhancing the polymerization and evolution processes. Notably, all these ATZ samples exhibit plentiful intra- and inter-particle mesopores, ascribed to the filling effect of the organic templates and the aggregation of the nanoparticles, respectively.
 | ||
| Fig. 2 TEM images of the obtained (a) ATZ-TEA, (b) ATZ-TEA-5, (c) ATZ-TMAda, (d) ATZ-PhHE, (e) ATZ-DPhHE, and (f) ATZ-PhDC samples. Scale bar, 50 nm. | ||
13C CP MAS NMR spectroscopy and thermal gravimetric analyses of the ATZ samples provide insights into the interaction of organic templates with the formed porous structures. As shown in Fig. S2,† the 13C CP MAS NMR spectra of the ATZ samples indicate that the organic templates remain intact in the obtained amorphous zeolite structures. The TG curves of all the ATZ samples show three stages of weight loss (Fig. S3†). In the case of the ATZ-TEA sample, the weight loss (2.6 wt%) below 170 °C is ascribed to the release of absorbed water; the second weight loss (6.8 wt%) in the higher temperature range (170–350 °C) is attributed to the loosely attached cations probably located on the surface of zeolitic units, and the third weight loss (3.7 wt%) is due to the decomposition of TEA+ that has been occluded inside the zeolitic units. As summarized in Table S2,† the weight loss of ATZ-TMAda and ATZ-PhDC samples (30.2 and 17.2 wt%, respectively) is higher than that of the rest of the samples, demonstrating the strong structure-directing ability of the corresponding OSDAs and the highly open structure of the obtained samples.
The 29Si MAS NMR spectra of ATZ samples show similar silicon microenvironments, and display resonance bands centered at −110 ∼ −112 ppm, ascribed to the tetrahedrally coordinated silicon. Notably, the resonance bands are also wider than those of the crystallized TS-1 zeolite, ascribed to the incompletely formed zeolite frameworks (Fig. S4†).
The electronic state of Ti in these amorphous titanosilicates was examined using an ultraviolet-visible (UV-vis) spectrometer. As shown in Fig. 3a, the adsorption bands of all the ATZ samples mainly appear at around 220 and 270 nm, which are attributed to the tetrahedrally coordinated titanium and octahedrally coordinated titanium species, respectively.37,50 It should be noted that the characteristic peak at about 330 nm for anatase species cannot be observed in these six amorphous samples, even for the ATZ-TEA-5 sample, with the highest Ti content, up to 14.4 wt%. However, the crystallized TS-1 zeolite sample shows significant characteristic bands of anatase TiO2 (Fig. S5a†). The results indicate that the open structure of the amorphous titanosilicates can protect the extra-framework titanium from aggregating into anatase species, and stabilize the titanium as catalytically active tetra- and hexa-coordinated species.
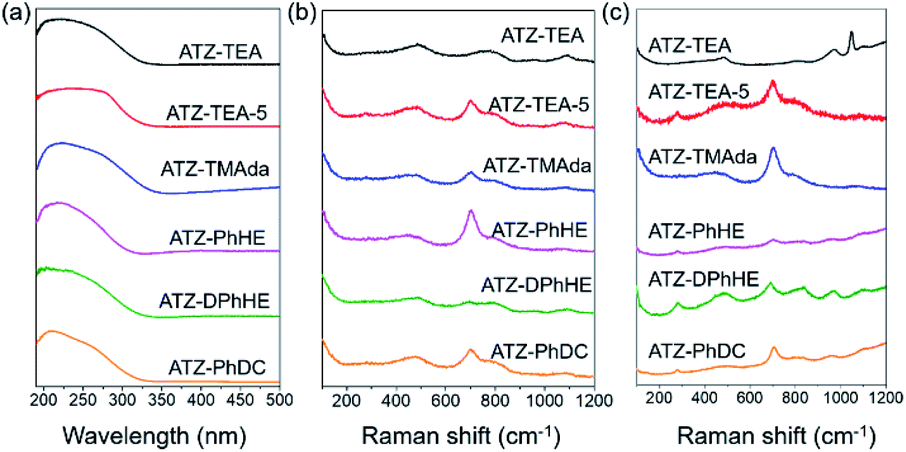 | ||
| Fig. 3 (a) UV-vis spectra, (b) UV-Raman spectra excited at 266 nm, and (c) UV-Raman spectra excited at 325 nm of the obtained ATZ samples. | ||
Ultraviolet resonance Raman (UV-Raman) spectroscopy with excitation lines at 266 and 325 nm is utilized to get a deeper insight into the Ti coordination states in the ATZ samples and the conventional TS-1 zeolite sample.51–53Fig. 3b shows the UV-Raman spectra of the titanosilicate samples, excited by a 266 nm laser line. The absorption band at around 695 cm−1 is attributed to the mononuclear octahedrally coordinated Ti species on the basis of DFT calculations (Scheme 2).40 All the ATZ samples exhibit such characteristic bands, suggesting that the octahedrally coordinated Ti species can be formed during the polymerization of the titanosilicates and can be well retained in the open structure.
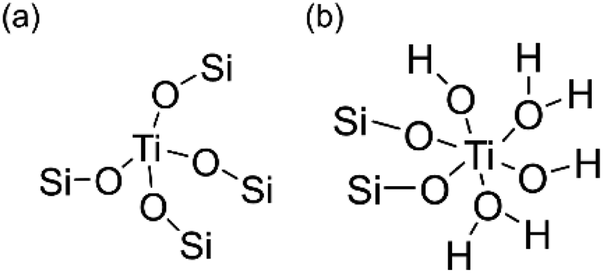 | ||
| Scheme 2 Proposed structures of (a) tetrahedrally coordinated and (b) octahedrally coordinated Ti species. | ||
The absorption bands sited at 144, 390, 516, and 637 cm−1 in the UV-Raman spectra with an excitation wavelength of 325 nm are ascribed to the anatase species. Fig. 3c demonstrates that ATZ samples exhibit no characteristic bands of anatase species, suggesting that these samples are free of anatase, while a band at 144 cm−1 exists in the microporous TS-1 zeolite sample, implying the presence of anatase species in the zeolite crystals, which is in accordance with the UV-vis spectra results (Fig. S5†).
XAFS is adequate to reveal the coordination environment and local structure of the Ti species in titanosilicates. XANES can determine the coordination geometry of Ti in a qualitative manner. Generally, the intensity of the pre-edge peaks positively correlates with the structural symmetry of the Ti species.54Fig. 4a shows the Ti K-edge XANES spectra of the calcined titanosilicate samples. The anatase sample displays three weak pre-edge peaks at 4967, 4970, and 4972 eV. A single pre-edge peak at 4968 eV, attributed to the symmetrical structure of tetrahedrally coordinated Ti, can be observed in the pure tetrahedral-coordinated TS-1 (TS-1-TiO4) zeolite sample. Compared to the anatase and TS-1-TiO4 samples, the ATZ-TEA sample only exhibits a single pre-edge peak at 4969 eV. The absence of the characteristic peaks of anatase in the ATZ-TEA sample indicates that it is free of anatase species. The decreased maximum intensity of the single pre-edge peak of the ATZ-TEA sample while also shifting towards lower energy should be attributed to the formation of the distorted octahedrally coordinated Ti species.
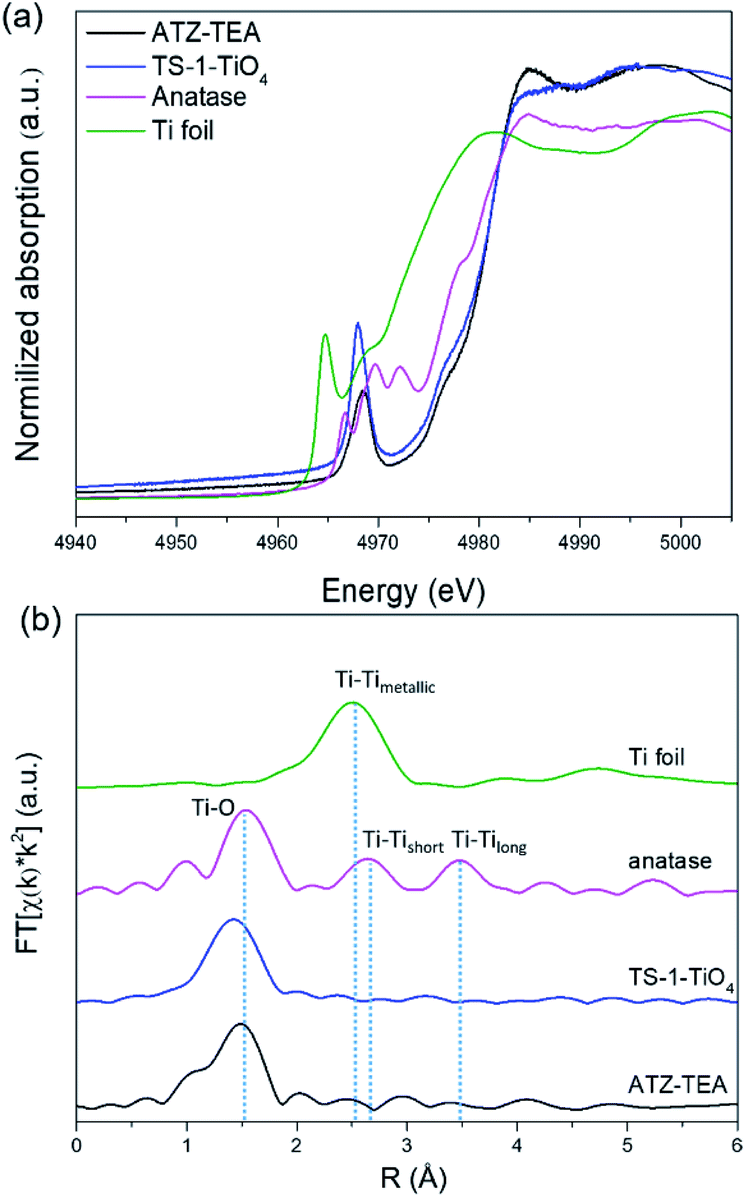 | ||
| Fig. 4 (a) Ti K-edge XANES spectra and (b) Fourier transform k2-weighted Ti EXAFS spectra in the R spacing of the ATZ-TEA sample, TS-1-TiO4 zeolite sample, anatase TiO2, and Ti foil reference. | ||
The Fourier-transformed extended X-ray absorption fine structure spectra (FT-EXAFS) of ATZ-TEA and anatase TiO2 are shown in Fig. 4b. The FT curve of anatase TiO2 shows characteristic peaks at 1.5, 2.6, and 3.5 Å, attributed to the Ti–O, Ti–Tishort, and Ti–Tilong scattering, respectively. The ATZ-TEA sample exhibits a major peak at 1.5 Å, ascribed to the Ti–O bond, and the absence of Ti–Ti peak at 2.5 Å indicates negligible Ti–O–Ti coordination, suggesting that all Ti atoms are isolated in the ATZ-TEA sample. That is, the local structure of newly detected TiO6 can be solidly confirmed as a kind of mononuclear Ti species. In addition, the thermal and static disorder in the absorber-scatterer distances of the ATZ-TEA sample is 0.0022, significantly higher than that of TS-1-TiO4 (0.0004), implying the high flexibility and distortion of the structure in the ATZ-TEA sample, leading to the slightly lower coordination number of the Ti–O shell than that of TS-1-TiO4 (3.6 vs. 4).55 Besides, the longer Ti–O distance in the ATZ-TEA sample than TS-1-TiO4 (1.84 vs. 1.82 Å) substantiates the existence of octahedrally coordinated Ti species (Table S3†).
The catalytic activity of the ATZ catalysts for the oxidative desulfurization of DBT and the epoxidation of cyclohexene with tert-butyl hydroperoxide (TBHP) as the oxidizing agent was tested in comparison with that of the nano-sized TS-1 zeolite catalyst. The amorphous titanosilicate catalysts show much superior catalytic performance in the oxidative desulfurization of DBT compared to the conventional microporous TS-1 zeolite catalyst (micro-TS-1, crystallized for 24 h), and better performance than the intermediately crystallized TS-1 catalyst (TS-1-inter, crystallized for 6 h), which exhibits hierarchical structures and is free of anatase (Fig. 5a, S6–S7, and Tables S1 and S4†). The outstanding catalytic performance of ATZ catalysts can be ascribed to the open structure of the amorphous zeolites, the abundant active octahedrally coordinated titanium species, and being free of anatase TiO2 species, which significantly enhance the accessibility towards the active sites and the transport efficiency, and increase the intrinsic catalytic activity of the titanium catalytic sites. Fig. S8† displays good correlations between ln(c0/ct) and the reaction time over the ATZ-TEA catalyst, implying that, when fixing the amount of the catalyst, the DBT conversion rate has a first-order dependence on the concentration of the reactant.
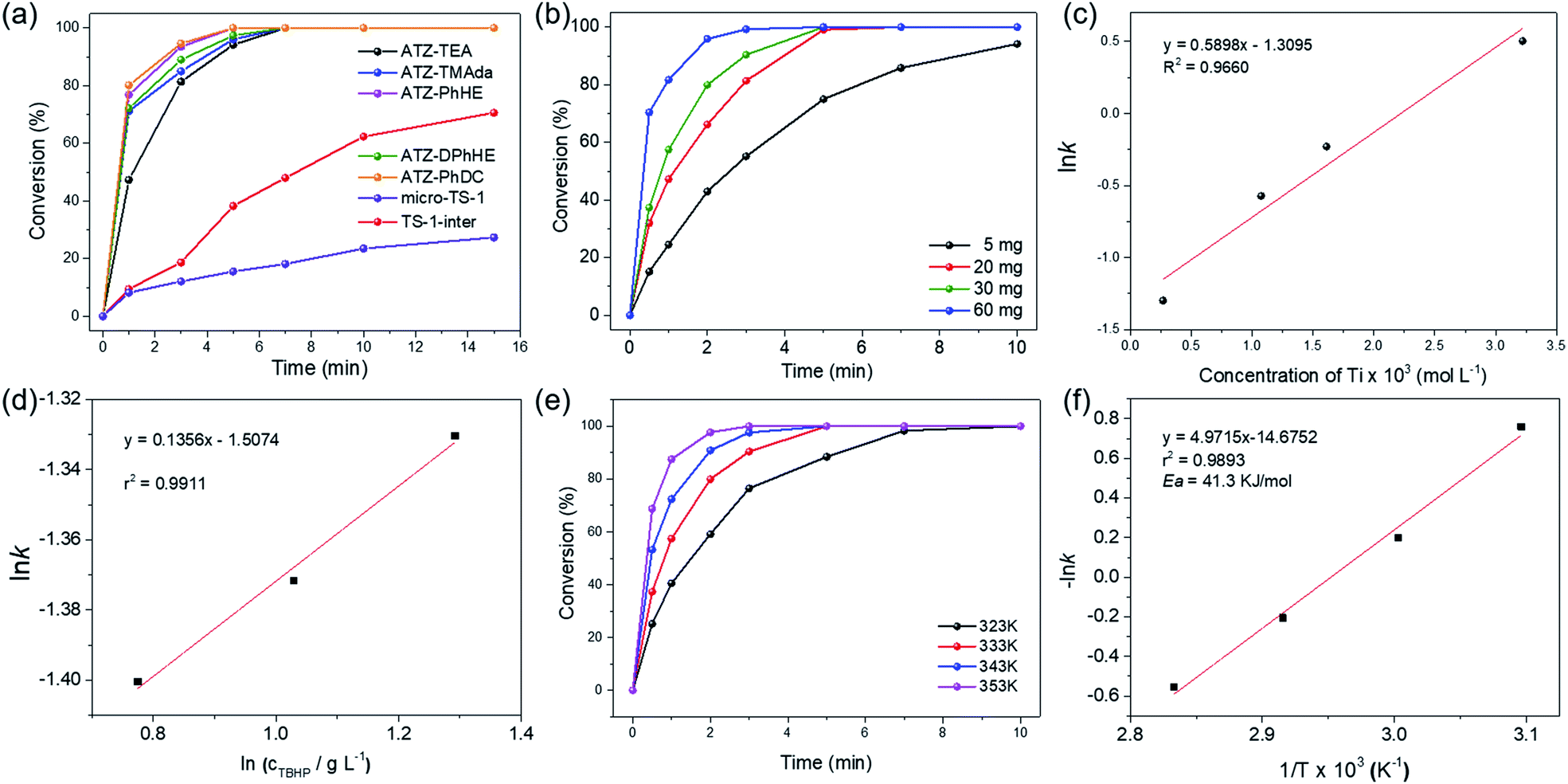 | ||
| Fig. 5 (a) Time-course variation of DBT conversion over 20 mg of the series of ATZ catalysts at 333 K; (b) time-course variation of DBT conversion with various Ti concentrations and (c) pseudo first-order rate constant for different concentrations of the ATZ-TEA catalyst at 333 K; (d) pseudo zero-order rate constant for different concentrations of TBHP; (e) time-course variation of DBT conversion at different reaction temperatures and (f) Arrhenius plot for the conversion of DBT over the 30 mg ATZ-TEA catalyst. Reaction conditions: 10 mL of 500 ppm model fuels, n(sulphide)/n(TBHP) = 0.5. | ||
Additionally, the detailed effects of the reaction conditions, such as the amount of the titanosilicate catalyst, and the molar ratio of TBHP to DBT, were investigated with the ATZ-TEA catalyst as a typical catalyst. As shown in Fig. 5b, the reaction rate increases when increasing the concentration of the titanosilicate catalyst. The linear logarithm plot for the apparent reaction rate constant versus catalyst concentration indicates that, when fixing the concentration of DBT and TBHP, the reaction rate of DBT fits a first-order dependence on the catalyst concentration (Fig. 5c and S9†). Fig. 5d demonstrates that the concentration of TBHP has a negligible effect on the reaction rate, indicating the zero-reaction order in TBHP under such performed reaction conditions. Hence, the reaction rate of oxidative desulfurization of DBT can be expressed as:
| r = k × [Ti] × [DBT] |
Fig. 5e shows the oxidation of DBT at different reaction temperatures over the ATZ-TEA catalyst. With the increase of the reaction temperature, the conversion rates of DBT are improved, demonstrating that the oxidation of DBT follows pseudo first-order kinetics (Fig. 5f and S10†). The apparent activation energy (Ea) of the ATZ-TEA catalyst is 41.3 kJ mol−1, which is calculated from the Arrhenius plot, and indicates that the reaction is kinetically controlled (Fig. 5f).
We have measured external and internal diffusion and have found that the reaction is not controlled by diffusion. The potential external diffusion control was investigated by performing the oxidative desulfurization of DBT at different stirring speeds (400 rpm, 600 rpm, and 800 rpm). When the stirring speed was over 400 rpm, there was no significant enhancement in the rate of the reactions (Fig. S11†). We measured the rate of the reaction for two reactants with different molecular sizes: thiophene (Th) and dibenzothiophene (DBT). Th could penetrate, at least some, within TS-1, while DBT cannot penetrate within the pores of MFI. Then we measured the initial rate for the oxidative desulfurization of the two molecules with the microporous TS-1 and the amorphous titanosilicate zeolite precursor (ATZ-TEA) that contains extra-large pores. The results show that with the larger DBT molecule, the ratio of the initial reaction rate, with respect to thiophene, is much larger with the amorphous titanosilicate zeolite precursor with extra-large pores than with TS-1 (21.7 vs. 8.9 at the reaction time of 1 min). This indicates the benefit of the zeolite precursor with extra-large pores, also from the point of view of reactant and product diffusion.
Furthermore, to get a deeper insight into the nature of the active Ti sites, we treated the ATZ-TEA catalyst with acid leaching, which removed most of the octahedrally coordinated Ti species, as evinced by the UV-vis spectrum and the UV-Raman spectrum excited at a wavelength of 266 nm (Fig. S13†). ICP suggested that the acid treatment removed ∼30% titanium species (Si/Ti increased from 40.8 to 58.0). The catalytic activity of the acid treated ATZ-TEA catalyst was also evaluated with the oxidation of DBT. Fig. S14† shows that the acid treatment dramatically deactivated the catalyst, implying that the octahedrally coordinated Ti species was responsible for the catalytic activity.
The durability tests for the oxidation of DBT over the ATZ-TEA catalyst were also conducted. The used catalyst was regenerated by calcination in air. As presented in Fig. S15,† 91% of the initial catalytic activity remained after 10 runs. UV-vis spectroscopy, N2 adsorption–desorption isotherms, and TEM imply that the microenvironment of Ti and microporosity of the open structure are well maintained, showing high catalytic stability (Fig. S16, S17 and Table S1†).
Moreover, the catalytic activity of ATZ-TEA was also evaluated by the epoxidation of cyclohexene with TBHP as the oxidant and with nano-sized TS-1 and Ti-Beta (Si/Ti = 25) as the reference catalysts, which were proved to mainly exhibit tetrahedrally coordinated Ti by UV-vis spectroscopy (Fig. S5 and S18–S20†). The conversion of cyclohexene over the ATZ-TEA catalyst is 42% of the maximum theoretical value, which is much higher than that over the TS-1 zeolite catalyst (around 16% of the maximum theoretical value) and Ti-Beta zeolite catalyst (around 28% of the maximum theoretical value) (Fig. 6 and Tables S5–S7†). The efficiency of TBHP is higher than that for the Ti-Beta catalyst (95 vs. 92%). The epoxide selectivity over ATZ-TEA is also higher than that of the Ti-Beta catalyst (69 vs. 44%). The superior catalytic performance of the ATZ catalyst can be ascribed to the open structure and the active octahedrally coordinated Ti species. Due to the wettability of titanosilicate catalysts influencing the activity and selectivity largely, we silylated Ti-Beta and ATZ-TEA catalysts with dimethoxydimethylsilane to increase the hydrophobicity. Fig. S20 and S21† indicated that the silylation treatment transformed most of the octahedrally coordinated Ti species into a tetrahedrally coordinated form. As summarized in Fig. 6 and Table S8,† the silylated ATZ-TEA sample (named ATZ-TEA-Sil) showed dramatically enhanced catalytic activity and epoxide selectivity in comparison to the non-silylated ATZ-TEA (71% and 86% vs. 42% and 69%, respectively). But the silylated Ti-Beta (Ti-Beta-Sil) exhibited slightly improved catalytic performance in comparison to the fresh one (with the conversion of cyclohexene: 30% vs. 28%) (Fig. 6 and Table S9†). The briefly increased activity over the Ti-Beta-Sil catalyst can be ascribed to the hindrance effect aroused by the silylated methyl groups, which is almost negligible in the case of the ATZ-TEA sample due to the large external surface area.
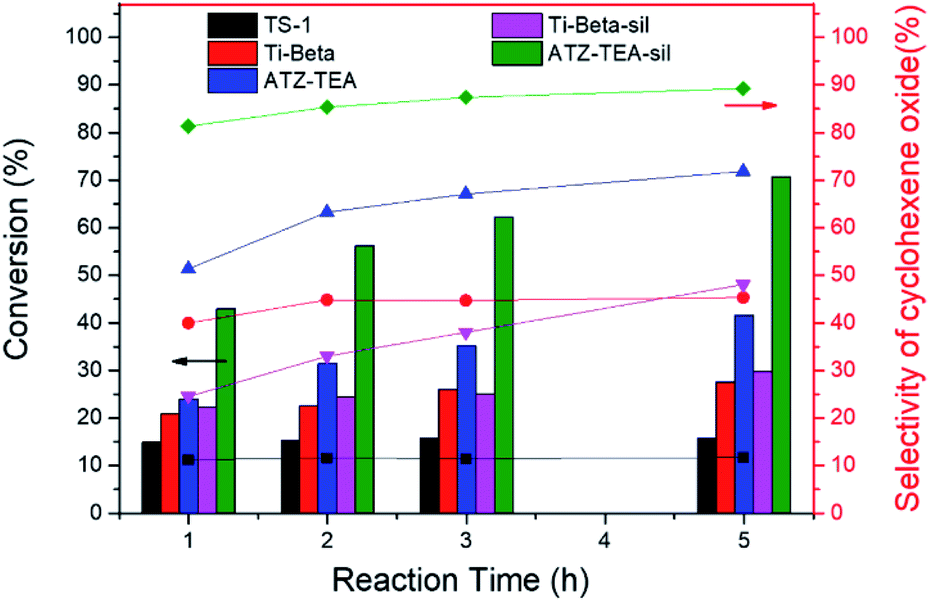 | ||
| Fig. 6 Epoxidation of cyclohexene over TS-1, Ti-Beta, Ti-Beta-Sil, ATZ-TEA, and ATZ-TEA-Sil catalysts. Reaction conditions: cat., 30 mg; cyclohexene, 56 mmol; TBHP (80 wt%), 14 mmol; temp., 333 K, and 3 bar of N2. | ||
Conclusions
In summary, a series of prezeolitic long-range amorphous titanosilicates but with an open structure of zeolite units and high amounts of catalytically active titanium species were prepared by using quaternary ammonium cations as the organic structure directing agents under hydrothermal conditions. Comprehensive characterization indicated that the amorphous titanosilicates exhibited some open structure of the zeolite units with pore dimensions ranging from 0.5 to 2 nm. Besides, the textural properties of ATZ samples strongly correlated with the utilized organic template. UV-vis, UV-Raman, and X-ray absorption spectra evidenced the presence of octahedrally coordinated isolated titanium species, which remained stable upon calcination. The outstanding catalytic performance over ATZ catalysts in the oxidative desulfurization of dibenzothiophene and the epoxidation of cyclohexene could be attributed to the highly active octahedrally coordinated isolated titanium sites and the abundant open structure of zeolite units, which dramatically enhanced the transport efficiency and active site accessibility. This work opens a new avenue for expanding the catalytic applications of current microporous titanosilicate zeolites for processing bulky molecules.Experimental methods
Preparation of titanosilicate zeolite precursors
Titanosilicate zeolite precursors were prepared with the following molar composition: 1.0 SiO2: 0.025–0.200 TiO2: 0.25–0.50 OSDA: 10–30 H2O under hydrothermal conditions (170 °C for 6 h), in which OSDA can be tetraethylammonium (TEA+), trimethyladamantylammonium (TMAda+), N,N,N,N′,N′,N′-hexaethyl-1,4-benzenedimethanaminium, (PhHE2+), N,N,N,N′,N′,N′-hexaethyl-[1,1′-biphenyl]-4,4′-dimethanaminium (DPhHE2+), and N,N′-dicyclohexyl-N,N,N′,N′-tetramethyl-1,4-benzenedimethanaminium, (PhDC2+). The typical synthesis procedure is as follows: the organic template was mixed with distilled water and stirred for 15 min. Afterward, tetrabutyl titanate (TBOT) and tetraethyl orthosilicate (TEOS) were added into the system and stirred for 6 h to achieve complete hydrolysis. Then the synthesis gel was introduced into a teflon-lined stainless-steel autoclave. Crystallization was carried out at 170 °C under static conditions for 6 h. After centrifugation, washing, and drying, the products were calcined at 550 °C for 6 h under an air atmosphere to remove the organic template.Preparation of conventional TS-1 zeolites TS-1-inter and micro-TS-1
With tetrapropylammonium hydroxide (TPAOH) as the organic template and TBOT as the titanium source, the conventional TS-1 zeolite samples were synthesized with a molar composition of 1.0 SiO2: 0.025 TiO2: 0.3 TPAOH: 30 H2O under hydrothermal conditions at 170 °C for 6–24 h. The samples crystallized for 6 h and 24 h were named TS-1-inter and micro-TS-1, respectively. A typical preparation procedure is similar to that of the ATZ samples.Preparation of the nano-sized Ti-Beta zeolite
The Ti-Beta zeolite was synthesized in basic medium from a gel of the following molar composition: SiO2: 0.067 TiO2: 0.56 TEAOH: 0.35 H2O2: 7 H2O: 5 wt% Seed. The gel preparation involved the hydrolysis under stirring of TEOS in an aqueous solution containing TEA+ and H2O2. Later, tetraethyl orthotitanate (TEOT) was added and the solution was kept under stirring until the ethanol formed in the hydrolysis of TEOS was evaporated. Dealuminated zeolite Beta seeds were also added and the mixture was loaded into teflon-lined stainless-steel autoclaves and heated at 140 °C under rotation (60 rpm) for 20 days. The solid was recovered by centrifugation, washed with distilled water and calcined in static air at 580 °C for 3 h.Acid treatment
Typically, 1 g ATZ-TEA sample was treated with 50 mL 2 M HNO3 water solution at 60 °C for 2 h, and then thoroughly washed with distilled water.Silylation conditions
The silylation process was performed using a standard procedure under a N2 flow. Calcined ATZ-TEA or Ti-Beta zeolite (1 g) previously dehydrated at 300 °C under vacuum was brought into contact with a solution of the silylating agent (dimethoxydimethylsilane, 2.8 mmol) in dry toluene (10 mL) at 80 °C under reflux for 1 hour. Then, a solution of triethylamine (2.8 mmol) in toluene (5 mL) was also added and the mixture was refluxed for an additional hour at 80 °C. The zeolite was washed with toluene and dried at 100 °C. The silylated ATZ-TEA and Ti-Beta samples were named ATZ-TEA-Sil and Ti-Beta-Sil, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research and Development Program of China (Grant 2016YFB0701100), the National Natural Science Foundation of China (Grant 21621001, 21920102005 and 21835002), the 111 Project (B17020), the European Union through the European Research Council (grant ERC-AdG-2014-671093, SynCatMatch), and the Spanish Government through “Severo Ochoa” (SEV-2016-0683, MINECO) for supporting this work. The APS was operated for the U.S. DOE Office of Science by the Argonne National Laboratory, and the CLS@APS facilities (Sector 20) were supported by the U.S. DOE under contract no. DEAC02-06CH11357, and the Canadian Light Source and its funding partners. R. Bai acknowledges the China Scholarship Council for the financial support. José Gaona Miguélez is also acknowledged for technical help.Notes and references
- Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268–7316 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS.
- E. M. Gallego, M. T. Portilla, C. Paris, A. León-Escamilla, M. Boronat, M. Moliner and A. Corma, Science, 2017, 355, 1051–1054 CrossRef CAS.
- J. Shin, D. Jo and S. B. Hong, Acc. Chem. Res., 2019, 52, 1419–1427 CAS.
- M. G. Clerici, Top. Catal., 2000, 13, 373–386 CrossRef CAS.
- G. Bellussi, R. Millini, P. Pollesel and C. Perego, New J. Chem., 2016, 40, 4061–4077 RSC.
- B. Smit and T. L. M. Maesen, Nature, 2008, 451, 671–678 CrossRef CAS.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- J. C. Groen, W. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef CAS.
- R. Bai, Q. Sun, N. Wang, Y. Zou, G. Guo, S. Iborra, A. Corma and J. Yu, Chem. Mater., 2016, 28, 6455–6458 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, J. L. Jordá, C. Martínez and M. Moliner, Nature, 2006, 443, 842–845 CrossRef CAS.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494–2513 CrossRef CAS.
- H. Awala, J.-P. Gilson, R. Retoux, P. Boullay, J.-M. Goupil, V. Valtchev and S. Mintova, Nat. Mater., 2015, 14, 447–451 CrossRef CAS.
- R. Bai, Y. Song, Y. Li and J. Yu, Trends Chem., 2019, 1, 601–611 CrossRef.
- K. Li, J. Valla and J. Garcia-Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS.
- D. Schneider, D. Mehlhorn, P. Zeigermann, J. Kaerger and R. Valiullin, Chem. Soc. Rev., 2016, 45, 3439–3467 RSC.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- G. Feng, P. Cheng, W. Yan, M. Boronat, X. Li, J.-H. Su, J. Wang, Y. Li, A. Corma, R. Xu and J. Yu, Science, 2016, 351, 1188–1191 CrossRef CAS.
- A. Chawla, N. Linares, R. Li, J. García-Martínez and J. D. Rimer, Chem. Mater., 2020, 32, 3278–3287 CrossRef CAS.
- A. Corma and M. J. Díaz-Cabañas, Microporous Mesoporous Mater., 2006, 89, 39–46 CrossRef CAS.
- R. Li, A. Chawla, N. Linares, J. G. Sutjianto, K. W. Chapman, J. G. Martínez and J. D. Rimer, Ind. Eng. Chem. Res., 2018, 57, 8460–8471 CrossRef CAS.
- K.-G. Haw, J.-P. Gilson, N. Nesterenko, M. Akouche, H. El Siblani, J.-M. Goupil, B. Rigaud, D. Minoux, J.-P. Dath and V. Valtchev, ACS Catal., 2018, 8, 8199–8212 CrossRef CAS.
- M. Akouche, J.-P. Gilson, N. Nesterenko, S. Moldovan, D. Chateigner, H. E. Siblani, D. Minoux, J.-P. Dath and V. Valtchev, Chem. Mater., 2020, 32, 2123–2132 CrossRef CAS.
- S. Inagaki, K. Thomas, V. Ruaux, G. Clet, T. Wakihara, S. Shinoda, S. Okamura, Y. Kubota and V. Valtchev, ACS Catal., 2014, 4, 2333–2341 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef CAS.
- J. Grand, S. N. Talapaneni, A. Vicente, C. Fernandez, E. Dib, H. A. Aleksandrov, G. N. Vayssilov, R. Retoux, P. Boullay, J.-P. Gilson, V. Valtchev and S. Mintova, Nat. Mater., 2017, 16, 1010–1015 CrossRef CAS.
- F. Dubray, S. Moldovan, C. Kouvatas, J. Grand, C. Aquino, N. Barrier, J.-P. Gilson, N. Nesterenko, D. Minoux and S. Mintova, J. Am. Chem. Soc., 2019, 141, 8689–8693 CrossRef CAS.
- A. Carati, C. Flego, D. Berti, R. Millini, B. Stocchi and C. Perego, Stud. Surf. Sci. Catal., 1999, 125, 45–52 CrossRef CAS.
- C. Perego, A. Carati, P. Ingallina, M. A. Mantegazza and G. Bellussi, Appl. Catal., A, 2001, 221, 63–72 CrossRef CAS.
- W. O. Parker and R. Millini, J. Am. Chem. Soc., 2006, 128, 1450–1451 CrossRef CAS.
- N. A. Grosso-Giordano, A. S. Hoffman, A. Boubnov, D. W. Small, S. R. Bare, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2019, 141, 7090–7106 CrossRef CAS.
- R. Bai, Q. Sun, Y. Song, N. Wang, T. Zhang, F. Wang, Y. Zou, Z. Feng, S. Miao and J. Yu, J. Mater. Chem. A, 2018, 6, 8757–8762 RSC.
- J. A. Martens, P. Buskens, P. A. Jacobs, A. van der Pol, J. H. C. van Hooff, C. Ferrini, H. W. Kouwenhoven, P. J. Kooyman and H. van Bekkum, Appl. Catal., A, 1993, 99, 71–84 CrossRef CAS.
- X. Zhang, Y. Wang and F. Xin, Appl. Catal., A, 2006, 307, 222–230 CrossRef CAS.
- W. Fan, R.-G. Duan, T. Yokoi, P. Wu, Y. Kubota and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 10150–10164 CrossRef CAS.
- L. Xu, D. D. Huang, C. G. Li, X. Ji, S. Jin, Z. Feng, F. Xia, X. Li, F. Fan, C. Li and P. Wu, Chem. Commun., 2015, 51, 9010–9013 RSC.
- S. Bordiga, F. Bonino, A. Damin and C. Lamberti, Phys. Chem. Chem. Phys., 2007, 9, 4854–4878 RSC.
- D. Gleeson, G. Sankar, C. R. A. Catlow, J. M. Thomas, G. Spanó, S. Bordiga, A. Zecchina and C. Lamberti, Phys. Chem. Chem. Phys., 2000, 2, 4812–4817 RSC.
- Q. Guo, K. Sun, Z. Feng, G. Li, M. Guo, F. Fan and C. Li, Chem.–Eur. J., 2012, 18, 13854–13860 CrossRef CAS.
- W. Xu, T. Zhang, R. Bai, P. Zhang and J. Yu, J. Mater. Chem. A, 2020, 8, 9677–9683 RSC.
- T. Moteki and T. Okubo, Chem. Mater., 2013, 25, 2603–2609 CrossRef CAS.
- D. Zhu, L. Wang, D. Fan, N. Yan, S. Huang, S. Xu, P. Guo, M. Yang, J. Zhang, P. Tian and Z. Liu, Adv. Mater., 2020, 32, 2000272 CrossRef CAS.
- P. Pichat, C. Franco-Parra and D. Barthomeuf, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 991–996 RSC.
- X. Wang, G. Li, W. Wang, C. Jin and Y. Chen, Microporous Mesoporous Mater., 2011, 142, 494–502 CrossRef CAS.
- P. Ratnasamy, D. Srinivas and H. Knozinger, Adv. Catal., 2004, 48, 1–169 CAS.
- M. Liu, Z. Chang, H. Wei, B. Li, X. Wang and Y. Wen, Appl. Catal., A, 2016, 525, 59–67 CrossRef CAS.
- P. Kumar, J. K. Gupta, G. Muralidhar and T. S. R. P. Rao, Stud. Surf. Sci. Catal., 1998, 113, 463–472 CrossRef CAS.
- T. Ikuno, W. Chaikittisilp, Z. Liu, T. Iida, Y. Yanaba, T. Yoshikawa, S. Kohara, T. Wakihara and T. Okubo, J. Am. Chem. Soc., 2015, 137, 14533–14544 CrossRef CAS.
- A. Zecchina, S. Bordiga, C. Lamberti, G. Ricchiardi, C. Lamberti, G. Ricchiardi, D. Scarano, G. Petrini, G. Leofanti and M. Mantegazza, Catal. Today, 1996, 32, 97–106 CrossRef CAS.
- C. Li, G. Xiong, Q. Xin, J. k. Liu, P. l. Ying, Z. c. Feng, J. Li, W. b. Yang, Y. z. Wang and G. r. J. A. C. I. E. Wang, Angew. Chem., Int. Ed., 1999, 38, 2220–2222 CrossRef CAS.
- C. Li, G. Xiong, J. Liu, P. Ying, Q. Xin and Z. Feng, J. Phys. Chem. B, 2001, 105, 2993–2997 CrossRef CAS.
- T. Zhang, Y. Zuo, M. Liu, C. Song and X. Guo, ACS Omega, 2016, 1, 1034–1040 CrossRef CAS.
- H. K. Nguyen, G. Sankar and R. A. Catlow, J. Porous Mater., 2017, 24, 421–428 CrossRef CAS.
- R. Anderson, G. Mountjoy, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 1998, 232, 72–79 CrossRef.
- Y. Tsuruta, T. Satoh, T. Yoshida, O. Okumura and S. Ueda, Stud. Surf. Sci. Catal., 1986, 28, 1001–1007 CrossRef CAS.
- R. Walton and D. O’Hare, J. Phys. Chem. Solids, 2001, 62, 1469–1479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04603e |
| This journal is © The Royal Society of Chemistry 2020 |