
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed intramolecular decarbonylative coupling of acylindoles and diarylketones through the cleavage of C–C bonds†
Tian-Yang
Yu‡
,
Wen-Hua
Xu‡
 ,
Hong
Lu
and
Hao
Wei
*
,
Hong
Lu
and
Hao
Wei
*
Key Laboratory of Synthetic and Natural Functional Molecule of Ministry of the Education, College of Chemistry & Materials Science, Northwest University, Xi'an 710069, China. E-mail: haow@nwu.edu.cn
First published on 16th October 2020
Abstract
We report here cobalt–N-heterocyclic carbene catalytic systems for the intramolecular decarbonylative coupling through the chelation-assisted C–C bond cleavage of acylindoles and diarylketones. The reaction tolerates a wide range of functional groups such as alkyl, aryl, and heteroaryl groups, giving the decarbonylative products in moderate to excellent yields. This transformation involves the cleavage of two C–C bonds and formation of a new C–C bond without the use of noble metals, thus reinforcing the potential application of decarbonylation as an effective tool for C–C bond formation.
Introduction
Catalytic decarbonylation has attracted considerable attention in organic synthesis.1 In particular, intramolecular decarbonylative coupling emerged as a more efficient and higher atom-economic strategy for the efficient formation of chemical bonds. This method allows for a common carbonyl group to serve as a “traceless handle” for chemical bond formation. In the past few decades, advances in intramolecular decarbonylative coupling, which can be used to create C–C, C–P, C–N, C–O, C–S and C–Si bonds through transition-metal catalysis, have been made (Scheme 1A).2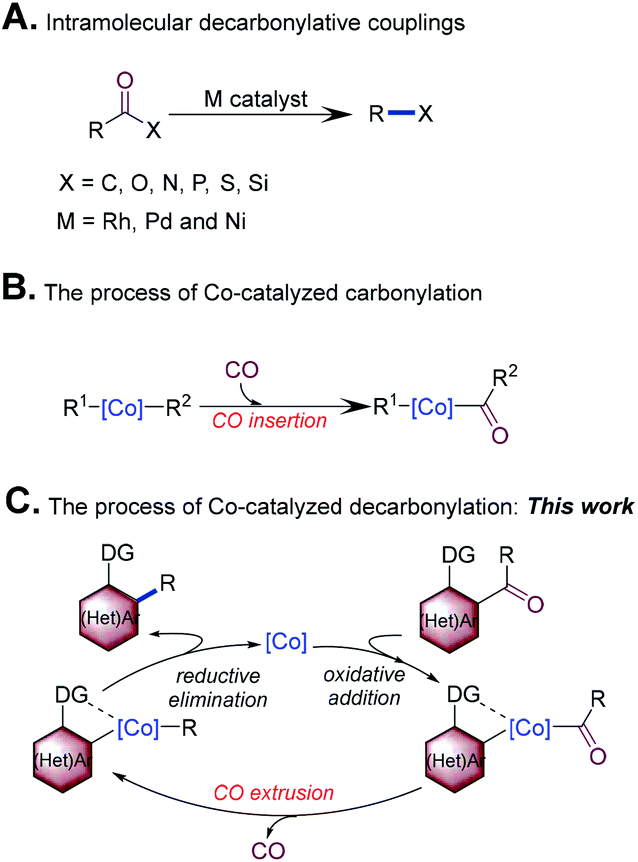 | ||
| Scheme 1 Inspiration for Co-catalyzed decarbonylation. | ||
Synthesis of bi(hetero)aryls very common in the pharmaceutical, agrochemical, and materials industries.3 From the view point of step and atom economy, transition-metal-catalyzed intramolecular decarbonylative coupling of ketones would offer a distinct strategy for the synthesis of bi(hetero)aryls, which has presented many attractive features. For example, ketones can be readily prepared, are stable, and generally have low toxicity. In addition, the use of this strategy helps to reduce the amount of harmful waste products (CO is the major side product). Finally, this reaction allows carbonyl of ketones to become a “traceless handle” for the construction of the C–C bond, which will improve the flexibility of synthetic design.1c,k Although highly attractive, the typical use of expensive and noble Rh complexes may hinder the development of this field.4 First row transition metals (iron, cobalt, nickel, and copper) are typically inexpensive and earth-abundant; the replacement of second row transition metals as catalysts with first row transition metals would be revolutionary. Pioneering work in this field has demonstrated that nickel enables the decarbonylation of ketones.5 In recent years, cobalt has received considerable attention in the activation of strong σ-bonds owing to its low price, ability to access multiple oxidation states, and its extensive, yet reactive, organometallic chemistry.6 Co carbonyl complexes are also known to catalyze several carbonylative processes (Scheme 1B).7 Decarbonylation involves C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–C bond oxidative addition, CO extrusion, and C–C bond forming reductive elimination, which can be regarded as the reverse process of carbonylation (Scheme 1C). The excellent affinity of Co with a carbonyl group inspired us to explore the use of this metal as the catalyst for decarbonylation. To our knowledge, Co-catalyzed decarbonylation is challenging and remains unsolved so far.8 Herein, we disclose the first Co-catalyzed decarbonylation of ketones with the assistance of an N-containing directing group,9 in which an intramolecular decarbonylative coupling of acylindoles and diarylketones was realized with an inexpensive cobalt precatalyst.
O)–C bond oxidative addition, CO extrusion, and C–C bond forming reductive elimination, which can be regarded as the reverse process of carbonylation (Scheme 1C). The excellent affinity of Co with a carbonyl group inspired us to explore the use of this metal as the catalyst for decarbonylation. To our knowledge, Co-catalyzed decarbonylation is challenging and remains unsolved so far.8 Herein, we disclose the first Co-catalyzed decarbonylation of ketones with the assistance of an N-containing directing group,9 in which an intramolecular decarbonylative coupling of acylindoles and diarylketones was realized with an inexpensive cobalt precatalyst.
Results and discussion
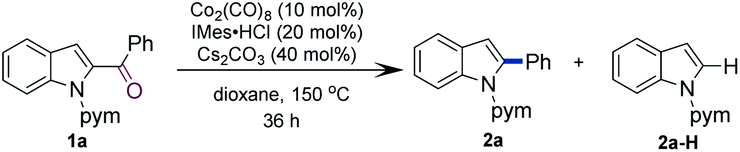
N-Pyrimidinyl 2-benzoyl indole 1a was employed as the model substrate. After a careful survey of the reaction parameters (see the ESI†), we discovered that the combined use of Co2(CO)8, IMes·HCl, and Cs2CO3 in dioxane afforded 2-phenyl indole 2a in 93% yield (Table 1, entry 1). Control experiments were carried out to understand the role of each reactant (see Table 1). In the absence of Co2(CO)8, the desired product was not observed (entry 2). The reaction without the ligand afforded the desired product, albeit in a lower yield (entry 3). When IMes·HCl (20 mol%) loading was reduced to IMes·HCl (10 mol%), the reaction produced 85% yield of 2a (entry 4). Other NHC ligands also promoted this reaction but gave lower yields as compared to that obtained with IMes (entries 5–7). Phosphine ligands such as P(n-Bu)3 or PCy3 also promoted this reaction but the yields were lower (entries 8 and 9). Dioxane as the solvent proved to be superior to toluene or iPrOH (entries 10 and 11). Co(II) complexes such as CoBr2 and salen Co complexes were not reactive for this transformation (entries 12 and 13). Next, several other coordinating groups were tested (Scheme 2). Comparable results were obtained when replacing the pyrimidyl group with a pyridyl group (1a-1), while no product was observed when the pyrimidyl group was removed (1a-2) or replaced with a weakly coordinating group such as carboxamide or urea (1a-3 and 1a-4).|
|
|||
|---|---|---|---|
| Entry | Deviations from above | Yieldb [%] | |
| 2a | 2a-H | ||
| a Standard conditions: 1a (0.1 mmol), Co2(CO)8 (10 mol%), IMes·HCl (20 mol%), Cs2CO3 (40 mol%), and dioxane (0.5 mL) at 150 °C in a sealed tube, 36 h. b Isolated yields. | |||
| 1 | “Standard conditions” with 1a | 93 | <5 |
| 2 | Without Co2(CO)8 | 0 | 0 |
| 3 | Without IMes·HCl | 18 | 0 |
| 4 | IMes·HCl (10 mol%) | 85 | <5 |
| 5 | ICy·HCl instead of IMes·HCl | 68 | 10 |
| 6 | SIPr·HCl instead of IMes·HCl | 14 | 17 |
| 7 | IPr·HCl instead of IMes·HCl | 47 | 20 |
| 8 | P(n-Bu)3 instead of IMes·HCl | 72 | <5 |
| 9 | PCy3 instead of IMes·HCl | 80 | <5 |
| 10 | Toluene instead of dioxane | 86 | <5 |
| 11 | i PrOH instead of dioxane | 0 | 70 |
| 12 | CoBr2 instead of Co2(CO)8 | 0 | 10 |
| 13 | Salen Co(II) instead of Co2(CO)8 | 0 | 19 |

|
|||
 | ||
| Scheme 2 Screening of directing groups. | ||
With the optimized reaction conditions in hand, we next investigated the scope of indole substrates. As shown in Table 2, electron-donating and -withdrawing substituents at the 3-, 4-, 5-, and 6-positions were compatible with the reaction conditions. Common functional groups such as aryl fluoride (2b), cyanide (2c), acetyl (2d), ether (2e and 2f), trifluoromethyl (2h) and ester (2i) were unaffected by the reaction conditions, thus highlighting the versatility of the transformation. The reaction efficiency was unaffected when a 3-methyl group (2k) was introduced, thus indicating tolerance of steric hindrance. A wide variety of electron-donating (2r) or electron-withdrawing (2m, 2s and 2t) substituents on the benzene ring were also well tolerated. Polyaromatic ketones (2u and 2v) reacted smoothly to give the corresponding decarbonylation products. The substrates could be changed from indole to pyrrole, with pyrimidine as a directing group, to furnish the desired product 2x in moderate yield. In addition, aryl alkyl ketones were suitable for this transformation, and the corresponding 2-alkyl indoles (2ya–2yf) were obtained in moderate yields.10 Note that aryl alkyl ketones had failed in the Ni system. However, under the standard conditions, no products were detected when secondary alkyl aryl ketones were used (2yg, 2yh and 2yi), affording byproduct 2a-H.

We also investigated the decarbonylation of diaryl ketones. For these substrates, ICy instead of IMes in the Co catalysis gave better yields (see the ESI†). A variety of diaryl ketones bearing a 2-pyridyl directing group also underwent decarbonylation to deliver the corresponding products in moderate to good yields. Further, a wide variety of functional groups, including ethers, trifluoromethyl, fluorides, and esters, were tolerated in the reaction. The directing group was also extended to a pyrimidine (4m) and a pyrazole (4o); however, no product was observed when the directing group was replaced with an oxime (4p).
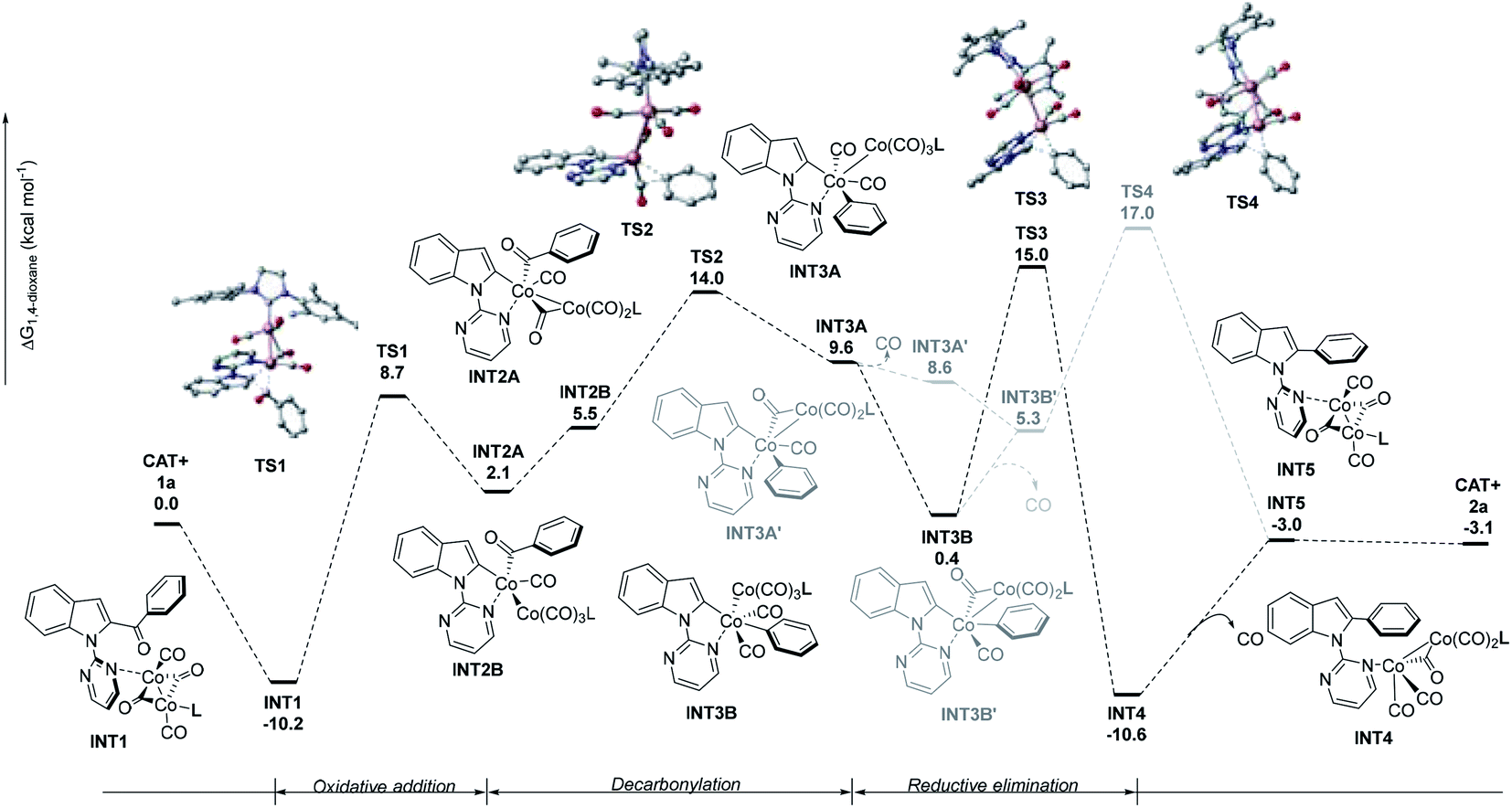
A preliminary density functional theory (DFT) study was conducted to explore the reaction mechanism (Fig. 1). The geometries were optimized at the B3LYP/DZ level of theory11 and the electronic energies were further improved by single-point calculations at the M06/TZ level.12 Solvation effects in 1,4-dioxane were treated by the continuum implicit solvation model SMD.13a In the simulation, we chose Co2(CO)4(NHC) to be the catalyst as can be seen from Fig. 2 (The justifications are described in the ESI†). The dissociation of CO ligands is necessary to initiate catalysis and create coordination sites (Co2(CO)8 + L = Co2(CO)4L (CAT) + 4CO (ΔG = 25.2 kcal mol−1)). The catalytic cycle commences upon coordination between the Co(0) complex (CAT) and substrate 1a, giving the catalyst–substrate complex INT1.
 | ||
| Fig. 1 Density functional theory (DFT) calculated pathways for the intramolecular decarbonylative coupling. | ||
Then, INT1 undergoes oxidative addition to form INT2AviaTS1, requiring an activation free energy of 18.9 kcal mol−1; this step is endergonic by 12.3 kcal mol−1. INT2A is converted to INT2Bvia the transfer of carbon monoxide. Next, INT2B is converted to INT3Avia decarbonylation, requiring an activation free energy of 8.5 kcal mol−1. The decarbonylation step was endergonic by 4.1 kcal mol−1. INT3A can rearrange into intermediate INT3B, which then undergoes reductive elimination to form INT4 (viaTS3). Finally, INT4 is converted to INT5 with the release of CO.14 The dissociation of CO from INT3A or INT3B was also considered but deemed unlikely as the energy barrier (TS4) is 2 kcal mol−1 higher than that of TS3. The overall free energy for the reaction is −3.1 kcal mol−1. Considering the activation free energy of each step, evidently, the highest energy barrier is the transition from INT1 to TS1 (18.9 kcal mol−1). According to the energetic span approximation,13b,c the rate-determining transition state (RDTS) and the rate-determining intermediate (RDI) are TS3 and INT1, respectively, thus, the energetic span δE = 25.2 kcal mol−1.
Finally, the utility of this reaction was demonstrated for a variety of valuable target motifs (Scheme 3A). The N-pyrimidyl directing group could be smoothly removed from 2a to obtain the corresponding N–H indole 5a in 82% yield. In the presence of IBr, 5a reacted with acetophenone to give the corresponding carbazole derivative 6a.15 Such scaffolds are prevalent in natural products and drug candidates with intriguing bioactivities,16 thus highlighting the synthetic applicability of the present method. The protocol was additionally applied to the synthesis of bazedoxifene, a third-generation selective estrogen receptor modulator (SERM).17 As shown in Scheme 3B, the synthesis was initiated by the conversion of ketones 7 to 8 under standard conditions. The removal of the directing group from 8 gave indole derivative 9. The conversion of 9 to bazedoxifene has been reported.17c
 | ||
| Scheme 3 Synthetic utility. | ||
Conclusions
In summary, we have discovered a simple Co system that can catalyze the decarbonylation of ketones via the cleavage of two C–C bonds. This work showcases the unique ability of the Co catalyst to trigger C–C bond disconnections, and further reinforces the potential of using decarbonylation as a tool for chemical bond formation and cleavage. However, this coupling reaction is still associated with significant drawbacks such as the requirement of high temperatures and directing groups. The use of directing groups aids the decarbonylation, but it complicates the whole synthesis and limits the application of this strategy in synthesis.18 Studies aimed at expanding this decarbonylative strategy to simple ketones without directing groups are ongoing in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (NSFC 21901202 and 21971205) and Natural Science Basic Research Program of Shaanxi (2020JQ-576). We thank the chemical HPC center of NWU for computational resources. Dedicated to Professor Peng-Fei Xu on the occasion of his 56th birthday.Notes and references
- For recent reviews and highlights, see: (a) A. Dermenci and G. Dong, Sci. China: Chem., 2013, 56, 685–701 CrossRef CAS; (b) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS; (c) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567–581 RSC; (d) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS; (e) Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS; (f) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS; (g) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC; (h) R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 6708–6710 CrossRef CAS; (i) C. Liu and M. Szostak, Org. Biomol. Chem., 2018, 16, 7998–8010 RSC; (j) L. Guo and M. Rueping, Chem. – Eur. J., 2018, 24, 7794–7809 CrossRef CAS; (k) Q. Zhao and M. Szostak, ChemSusChem, 2019, 12, 2983–2987 CrossRef CAS; (l) H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00153.
- For selected recent examples for intramolecular decarbonylative coupling, see: (a) M. Murakami, H. Amii and Y. Ito, Nature, 1994, 370, 540–541 CrossRef CAS; (b) T. Kondo, A. Nakamura, T. Okada, N. Suzuki, K. Wada and T. A. Mitsudo, J. Am. Chem. Soc., 2000, 122, 6319–6320 CrossRef CAS; (c) T. Kondo, Y. Taguchi, Y. Kaneko, M. Niimi and T. A. Mitsudo, Angew. Chem., Int. Ed., 2004, 43, 5369–5372 CrossRef CAS; (d) T. Matsuda, M. Shigeno and M. Murakami, Chem. Lett., 2006, 35, 288–289 CrossRef CAS; (e) A. Dermenci, R. E. Whittaker and G. Dong, Org. Lett., 2013, 15, 2242–2245 CrossRef CAS; (f) A. Dermenci, R. E. Whittaker, Y. Gao, F. Cruz, Z. Yu and G. Dong, Chem. Sci., 2015, 6, 3201–3210 RSC; (g) R. E. Whittaker and G. Dong, Org. Lett., 2015, 17, 5504–5507 CrossRef CAS; (h) R. Yu, X. Chen, S. F. Martin and Z. Wang, Org. Lett., 2017, 19, 1808–1811 CrossRef CAS; (i) X. Liu, H. Yue, J. Jia, L. Guo and M. Rueping, Chem. – Eur. J., 2017, 23, 11771–11775 CrossRef CAS; (j) R. Takise, R. Isshiki, K. Muto, I. Kenichiro and J. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 3340–3343 CrossRef CAS; (k) N. Ichiishi, C. A. Malapit, Ł. Woźniak and M. S. Sanford, Org. Lett., 2018, 20, 44–47 CrossRef CAS; (l) C. Liu and M. Szostak, Chem. Commun., 2018, 54, 2130–2133 RSC; (m) K. Ishitobi, R. Isshiki, K. K. Asahara, C. Lim, K. Muto and J. Yamaguchi, Chem. Lett., 2018, 47, 756–759 CrossRef CAS; (n) Z.-J. Zheng, C. Jiang, P.-C. Shao, W.-F. Liu, T.-T. Zhao, P.-F. Xu and H. Wei, Chem. Commun., 2019, 55, 1907–1910 RSC; (o) T. Morioka, S. Nakatani, T. Sakamato, T. Kodama, S. Ogoshi, N. Chatani and M. Tobisu, Chem. Sci., 2019, 10, 6666–6671 RSC; (p) W. Srimontree, W. Lakornwong and M. Rueping, Org. Lett., 2019, 21, 9330–9333 CrossRef CAS; (q) C. E. Brigham, C. A. Malapit, N. Lalloo and M. S. Sanford, ACS Catal., 2020, 10, 8315–8320 CrossRef CAS.
- (a) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS; (b) C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS; (c) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- (a) O. Daugulis and M. Brookhart, Organometallics, 2004, 23, 527–534 CrossRef CAS; (b) Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690–2694 CrossRef CAS.
- (a) T. Morioka, A. Nishizawa, T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2017, 139, 1416–1419 CrossRef CAS; (b) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586–589 CrossRef CAS.
- (a) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251–8254 CrossRef CAS; (b) D.-G. Yu, X. Wang, R. Zhu, S. Luo, X.-B. Zhang, B.-Q. Wang, L. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2012, 134, 14638–14641 CrossRef CAS; (c) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS; (d) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (e) F. H. Lutter, L. Grokenberger, M. S. Hofmayer and P. Knochel, Chem. Sci., 2019, 10, 8241–8245 RSC.
- (a) Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS; (b) Y. Li, Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC.
- (a) L. F. Halle, W. E. Crowe, P. B. Armentrout and J. L. Beauchamp, Organometallics, 1984, 3, 1694–1706 CrossRef CAS; (b) Z. Zhu, X. Li, S. Chen, P. Chen, B. A. Billett, Z. Huang and G. Dong, ACS Catal., 2018, 8, 845–849 CrossRef CAS; (c) X.-T. Min, D.-W. Ji, H. Zheng, B.-Z. Chen, Y.-C. Hu, B. Wan and Q.-A. Chen, Org. Lett., 2020, 22, 3386–3391 CrossRef CAS.
- (a) N. Chatani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 1999, 121, 8645–8646 CrossRef CAS; (b) R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408–1411 CrossRef CAS; (c) Z.-Q. Lei, F. Pan, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, Y.-X. Li, J. Sun and Z.-J. Shi, J. Am. Chem. Soc., 2015, 137, 5012–5020 CrossRef CAS; (d) Y. Hibe, Y. Ebe, T. Nishimura and H. Yorimitsu, Chem. Lett., 2017, 46, 953–955 CrossRef CAS; (e) Y. Lyori, K. Takahashi, K. Yamazaki, Y. Ano and N. Chatani, Chem. Commun., 2019, 55, 13610–13613 RSC.
- See the ESI† for details (Table S9†).
- (a) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS; (b) P. Jeffrey Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef; (c) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (d) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (e) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (b) A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. JonaS, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS; (c) L. E. Roy, P. Jeffrey Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS; (d) Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS; (b) S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS; (c) S. Kozuch and J. M. L. Martin, ChemPhysChem, 2011, 12, 1413–1418 CrossRef CAS.
- (a) M. A. Ortuñ, B. Dereli and C. J. Cramer, Inorg. Chem., 2016, 55, 4124–4131 CrossRef; (b) S. H. H. Eliasson and V. R. Jensen, Faraday Discuss., 2019, 220, 231–248 RSC.
- P. Ni, J. Tan, W. Zhao, H. Huang, F. Xiao and G. J. Deng, Org. Lett., 2019, 21, 3687–3691 CrossRef CAS.
- (a) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef; (b) H.-J. Knölker, Top. Curr. Chem., 2005, 244, 115–148 CrossRef; (c) I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2011, 309, 203–253 CrossRef; (d) H. Jiang, J. Sun and J. Zhang, Curr. Org. Chem., 2012, 16, 2014–2025 CrossRef CAS.
- (a) C. P. Miller, H. A. Harris and B. S. Komm, Drugs Future, 2002, 27, 117–121 CrossRef CAS; (b) B. S. Komm, Y. P. Kharode, P. V. N. Bodine, H. A. Harris, C. P. Miller and C. R. Lyttle, Endocrinology, 2005, 146, 3999–4008 CrossRef CAS; (c) S. Chen, X. K. Liu, X. Yu and Z. D. Yuan, Chin. J. New Drugs, 2013, 13, 1571–1573 Search PubMed.
- (a) J. Li and L. Ackermann, Chem. – Eur. J., 2015, 21, 5718–5722 CrossRef CAS; (b) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 57, 10378–10382 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and experimental details. CCDC 1965066. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04326e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |