
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-triggered nitric oxide (NO) release from photoresponsive polymersomes for corneal wound healing†
Yutian
Duan
a,
Yong
Wang
b,
Xiaohu
Li
c,
Guozhen
Zhang
 d,
Guoying
Zhang
a and
Jinming
Hu
*a
d,
Guoying
Zhang
a and
Jinming
Hu
*a
aCAS Key Laboratory of Soft Matter Chemistry, Hefei National Laboratory for Physical Science at the Microscale, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: jmhu@ustc.edu.cn
bDepartment of Ophthalmology, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui 230022, China
cDepartment of Radiology, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui 230022, China
dHefei National Laboratory for Physical Sciences at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 5th November 2019
Abstract
Polymersomes have been extensively used in the delivery of both small and macromolecular payloads. However, the controlled delivery of gaseous therapeutics (e.g., nitric oxide, NO) remains a grand challenge due to its difficulty in loading of gaseous payloads into polymersomes without premature leakage. Herein, NO-releasing vesicles could be fabricated via the self-assembly of NO-releasing amphiphiles, which were synthesized by the direct polymerization of photoresponsive NO monomers (abbreviated as oNBN, pNBN, and BN). These monomers were rationally designed through the integration of the photoresponsive behavior of N-nitrosoamine moieties and the self-immolative chemistry of 4-aminobenzyl alcohol derivatives, which outperformed conventional NO donors such as diazeniumdiolates (NONOates) and S-nitrosothiols (SNOs) in terms of ease of preparation, stability of storage, and controllability of NO release. The unique design made it possible to selectively release NO by a light stimulus and to regulate the NO release rates. Importantly, the photo-mediated NO release could be manipulated in living cells and showed promising applications in the treatment of corneal wounds. In addition to delivering NO, the current design enabled the synergistic delivery of NO and other therapeutic payloads by taking advantage of NO release-mediated traceless crosslinking of the vesicles.
Introduction
Polymersomes (also known as polymeric vesicles) with morphological similarities to cellular membranes and viral capsids have been widely used in artificial cells, drug carriers, imaging agents, and so on, which are characterized by their unique nanostructures containing hydrophilic lumens enclosed with a hydrophobic bilayer membrane.1–6 The analogs of polymersomes, liposomes, have been clinically used (e.g., Doxil®) with the capability to optimize the biodistributions and attenuate the side effects of doxorubicin (DOX). Therefore, there is mounting interest in developing polymersome-based nanotherapeutic agents. In comparison with liposomes, polymersomes exhibited excellent stability yet insufficient permeability, which inhibited the release of encapsulated payloads. To resolve this problem, a plethora of methods such as the design of stimuli-responsive polymersomes,7–9 the introduction of channel proteins within bilayers,10 and post-modification of bilayer membranes11 have been proposed to regulate the permeability of polymersomes.Nitric oxide (NO), a diatom radical, has been recognized as an important gaseous transmitter in cardiovascular, immune, central nervous systems, and so on.12,13 Recently, the therapeutic potential of NO in the treatment of cancers, inflammations, and wounds has attracted much attention.14,15 Moreover, polymeric NO donors displayed increased half-lives, optimized biodistributions, and increased therapeutic outcomes.16–19 Although both hydrophilic and hydrophobic payloads could be successfully delivered by polymeric vesicles, the delivery of gaseous therapeutics (e.g., NO) using vesicular carriers remains elusive due to the difficulty in loading of gaseous NO into vesicles. To address this problem, NO donors were encapsulated into vesicular assemblies to develop NO-releasing vesicles. For example, a NO-releasing donor, diethylenetriamine diazeniumdiolate (DETA NONOate), and an anticancer drug, irinotecan (CPT-11), were loaded into the aqueous lumen of poly(D,L-lactic-co-glycolic acid) (PLGA) capsules and synergistic release of NO and CPT-11 could be achieved. However, premature NO release was observed due to the poor stability of NONOates even at physiological pH.20 Fortunately, the labile NONOates could be selectively caged by diverse protecting groups to minimize spontaneous hydrolysis of NONOates.21,22 In this regard, hybrid vesicles loaded with β-galactosidase (β-Gal) were fabricated and the extra addition of the β-galactosyl-pyrrolidinyl diazeniumdiolate (β-Gal-NONOate) donor into the hybrid vesicles led to enzyme-catalyzed NO release.23 However, the biomedical applications of this NO-releasing vesicle may be limited by the different in vivo distributions of the NO donors and the enzyme-loaded vesicles. Despite some achievements, NO-releasing vesicles were routinely fabricated by loading small molecule-based NO donors into either the hydrophilic (e.g., interior or exterior aqueous medium) or the hydrophobic (e.g., bilayer membrane) domains of vesicular assemblies. However, polymersomes that can directly release NO without the extra loading of small molecule NO donors have not been achieved, which were expected to overcome the drawbacks of previously developed NO-releasing vesicles such as low NO loading contents, premature NO leakage, and uncontrolled biodistributions of NO donors and catalysts.
On the other hand, we recently proposed a new strategy to concomitantly regulate the stability and permeability of polymersomes using traceless cross-linking chemistry.24–26 In this context, highly reactive primary amines could be in situ generated within the initially hydrophobic bilayer membranes under specific stimuli through the self-immolative degradation of carbamate linkages. These amine moieties further underwent inter/intrachain amidation reactions, synchronically permeabilizing and cross-linking bilayer membranes. As a result, the synergistic release of payloads from both the aqueous lumens and bilayer membranes could be obtained without the disintegration of vesicles. We surmised that if the traceless crosslinking process could be activated by the triggered NO release, the co-release of NO and other payloads could be possibly achieved. To this end, it is of crucial importance to develop stimuli-responsive NO donors that could be coupled with self-immolative chemistry. We hypothesized that light irradiation could be a potential trigger because of the spatiotemporal precision of the light stimulus. Moreover, light irradiation has been previously employed to regulate the NO release from photoresponsive NO donors.27–32 For example, N-nitrosoaniline-based NO releasers have been designed and photo-triggered NO release could be achieved by the homolysis of N–NO bonds.33–38 The resulting anilinyl radicals can be reduced in situ to form the corresponding aniline derivatives.33,39–41 Note that aniline derivatives (e.g., 4-aminobenzyl alcohol) have been extensively used to fabricate self-immolative linkers.42–44 Therefore, if the amine group of 4-aminobenzyl alcohol (ABA) could be caged by NO with the formation of photoresponsive N-nitrosoamine residues, the photo-triggered cleavage of the N–NO bond could likely give rise to the reformation of an intact ABA intermediate and the activation of traceless cross-linking reactions.
In this work, we synthesized three kinds of N-nitrosoaniline-based NO donors (abbreviated as oNBN, pNBN, and BN) by nitrosation of 4-aminobenzyl alcohol-based precursors. Unlike conventional NONOate- and SNO-based donors with poor thermo-stabilities, these NO donors can be directly polymerized into amphiphilic block copolymers via reversible addition–fragmentation chain transfer (RAFT) polymerization. The resulting amphiphiles self-assembled into diverse nanostructures (e.g., vesicles) with NO loading contents of ∼2 μmol mg−1. Importantly, the NO release rates could be drastically tuned and the photo-triggered NO release could spontaneously activate the traceless cross-linking reactions, cross-linking and permeabilizing the bilayer membranes (Scheme 1), which can be further used to modulate the release of encapsulated payloads.
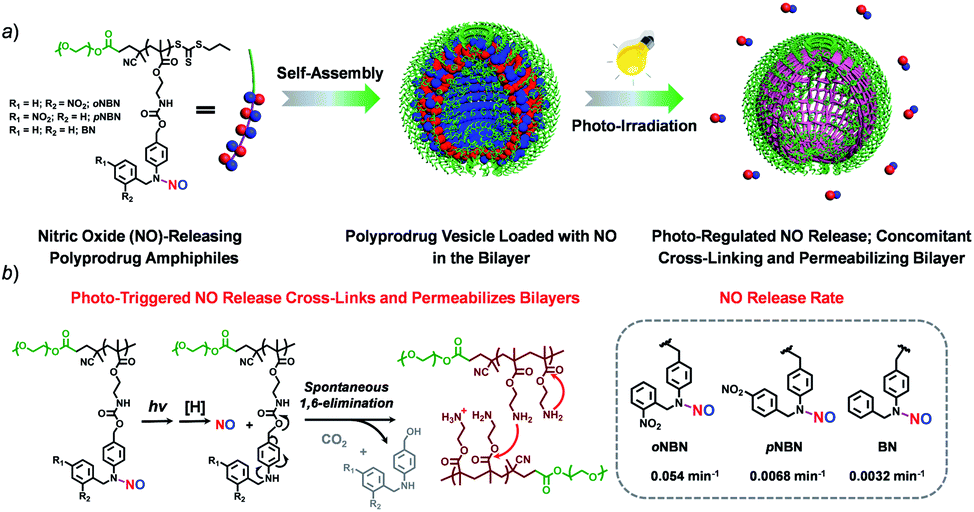 | ||
| Scheme 1 (a) Schematic illustration of the formation of vesicles assembled from NO-releasing amphiphiles. The resulting vesicles encompass photoresponsive N-nitrosoamine moieties within the bilayer membranes shielded by hydrophilic PEO coronas. (b) Under light irradiation, the cleavage of N–NO bonds induces the 1,6-elimination reaction with the release of primary amine groups that further undergo inter/intrachain amidation reactions, concomitantly cross-linking and permeabilizing the bilayers. | ||
Results and discussion
Synthesis and characterization of NO prodrug monomers
We proposed a facile route to synthesize NO-releasing monomers. First, Schiff bases were formed through equivalent mixing of the corresponding benzaldehydes and 4-aminobenzyl alcohol in ethanol, and the resultant Schiff bases can be readily separated by filtration and no further purification procedures were applied. The Schiff bases were subsequently reduced with sodium borohydride (NaBH4) and the generated aniline derivatives were treated with sodium nitrite (NaNO2) and glacial acetic acid at room temperature, giving rise to the corresponding nitrosated precursors. Finally, the hydroxyl groups were functionalized with 2-isocyanatoethyl methacrylate to afford the targeted monomers (oNBN, pNBN, and BN; Scheme 2a). Notably, the entire synthesis processes were implemented at room temperature and no harsh reaction conditions and tedious separation protocols were involved. In sharp contrast, the preparation of conventional NO donors (e.g., NONOates and SNOs) suffered from either tough reaction conditions (e.g., high pressure) or tedious purification procedures.12 The chemical structures of the intermediates and the named monomers were unequivocally identified by nuclear magnetic resonance (NMR) spectroscopy, electrospray ionization mass spectroscopy (ESI-MS), and high-performance liquid chromatography (HPLC) (Fig. S1–S8†).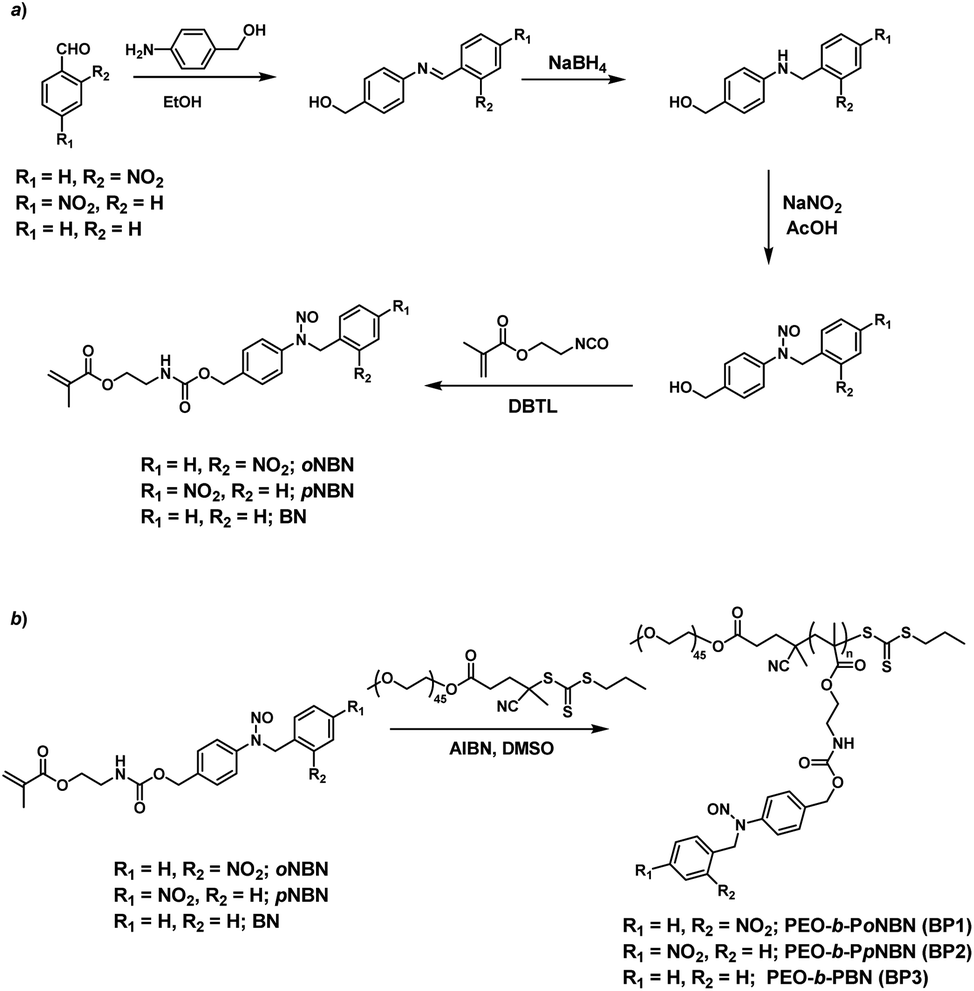 | ||
| Scheme 2 Synthetic routes of (a) NO-releasing monomers, oNBN, pNBN, and BN and (b) amphiphilic diblock copolymers (BP1–BP3) through RAFT polymerization. | ||
With oNBN, pNBN, and BN monomers in hand, we first verified the thermo-stabilities of the three monomers. To avoid thermo-induced polymerization of the monomers, the hydroxyl-terminated precursors were evaluated. Upon heating the three monomers at 70 °C (a typical reaction temperature for RAFT polymerization) for up to 24 h, we did not observe any changes in the HPLC elution profiles (Fig. S9†), suggesting excellent thermo-stabilities of the synthesized monomers; however, NONOate- and SNO-based NO donors have low thermo-stability and have to be stored at low temperature.12 Next, we verified whether the three monomers could release NO under light irradiation. Upon irradiating the three monomers with a hand-held UV lamp (365 nm, ∼4 mW cm−2; the irradiation intensity remained constant in all the experiments unless otherwise noted), progressive changes in the UV-vis absorbance spectra were observed for all the monomers. This result suggested that all three monomers were responsive to light irradiation (Fig. S10†). The evolution of absorbance intensities at 251 nm (for oNBN), 258 nm (for pNBN), and 254 nm (for BN) could be fitted by the first-order kinetics (R2 > 0.97); the apparent photolysis rate constants (kobs) were calculated to be ∼0.1 min−1, 0.013 min−1, and 0.0023 min−1, respectively. The kobs of two control samples, N-(2-cyanobenzyl)-N-(4-(hydroxymethyl)phenyl)nitrous amide with an electron-withdrawing group and N-(4-(hydroxymethyl)phenyl)-N-(2-methoxybenzyl)nitrous amide with an electron-donating group, was determined to be 0.0017 min−1 and 0.0016 min−1, respectively (Fig. S11†), which were quite similar to that of the BN monomer (0.0023 min−1) without any substituent group (Fig. S10e and f†). These results likely ruled out the effect of the substituent groups on modulation of the NO release rates and suggested that the photoresponsive nature of o-nitrobenzyl amine played a crucial role in the faster NO release behavior. In addition, we also measured the NO release rate of a previously reported N-nitrosoamine-based NO donor, N,N′-di-secbutyl-N,N′-dinitroso-1,4-phenylenediamine (BNN6), and found that the photolysis process of BNN6 exhibited an apparent rate constant of 0.005 min−1 (Fig. S12†), twenty times slower than that of the oNBN donor.35 Note that bis-N,N-nitroso-p-phenylenediamine (BNN) derivatives have been recognized as high performance caged NO donors34 and have been used in biological systems.35,45 Thus, our design by the introduction of photoresponsive o-nitrobenzyl moieties adjacent to the N-nitrosoamine moieties provided a feasible way to sensitize N-nitrosoamine-based NO donors, which may reduce light irradiation exposure when used in biological systems.
To confirm the NO release under light irradiation, a spin trapping agent, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), was used to in situ capture the generated NO radical with the formation of 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl (PTI).46 This transition can be easily monitored by electron paramagnetic resonance (EPR) spectroscopy. In the absence of NO-releasing monomers, there were no EPR signal changes after UV light irradiation, suggesting the inert nature of PTIO under light irradiation (Fig. 1a). Moreover, direct mixing of the monomers and PTIO did not change the EPR signals of PTIO either, indicative of no premature NO leakage. However, once shining light on the mixtures of PTIO and NO-releasing monomers, the characteristic EPR signals of PTI were observed and typical EPR spectra were recorded after 20 min (Fig. 1b–d). The above result confirmed that the formation of NO under light irradiation, which was ascribed to the homolysis rather than the heterolysis of N–NO bonds in the NO-releasing monomers, in accordance with a previous conclusion that homolytic cleavage of N-nitrosoamine was more energetically favorable.47 Density functional theory (DFT) calculation revealed that the N–N bond dissociation energy (BDE) of oNBN, pNBN, and BN was 20.8 kcal mol−1, 22.0 kcal mol−1, and 32.5 kcal mol−1, respectively (Fig. S13†), revealing that the release of NO from the oNBN monomer was easier than from the other two monomers, in good agreement with the kinetic data (Fig. S10†).
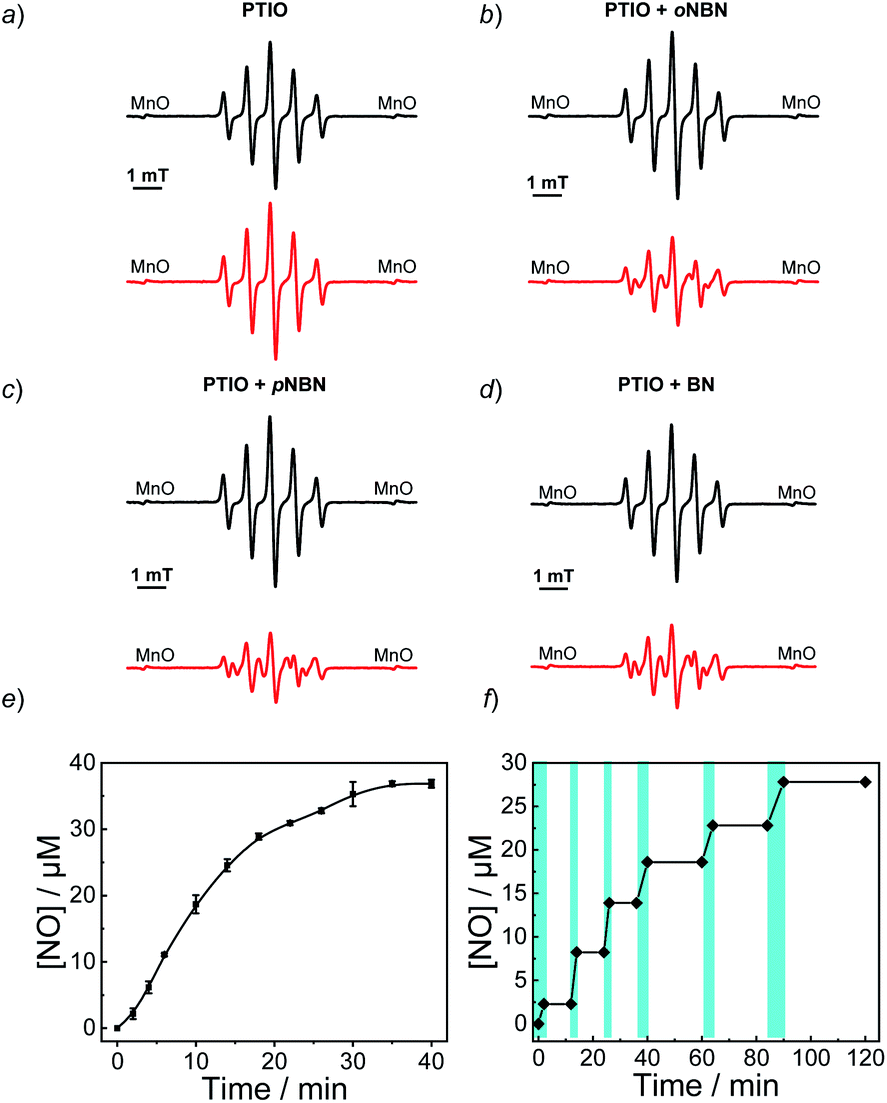 | ||
| Fig. 1 EPR spectroscopy of aqueous solutions (20 mM PBS buffer containing 20% DMSO, pH 7.4) for (a) PTIO, (b) oNBN with PTIO, (c) pNBN with PTIO, and (d) BN with PTIO before (black lines) and after (red lines) UV 365 nm irradiation (4 mW cm−2) for 20 min. In all cases, the concentrations of PTIO and NO-releasing monomers were 20 μM. (e and f) NO release profiles of the oNBN monomer (40 μM) in an aqueous medium (20 mM PBS buffer containing 5% DMSO) under (e) continuous (mean ± s.d.; n = 3) or (f) intermittent (the cyan areas indicate the light on) UV 365 nm irradiation (4 mW cm−2). | ||
Besides the EPR technique, the photo-triggered NO release was observed by the remarkable fluorescence turn-on of a previously reported NO-selective fluorescent probe based on pyronine chromophores (Fig. S14†).48 The NO yield was ∼92% for the oNBN monomer using the standard Griess assay (Fig. 1e), representing a relatively high NO yield as compared to that of previously reported N-nitrosamine-based NO donors.33 We assumed that the high NO yield was likely ascribed to the enhanced photo-reactivity of the oNBN monomer that may avoid adverse side reactions. Moreover, the NO release process could be alternately activated and deactivated by switching on/off the incident light (Fig. 1f), making the spatiotemporal regulation of NO release possible.
NO-releasing mechanism of the monomers
To unravel the mechanism of photo-induced NO release from the NO-releasing monomers, especially for the oNBN monomer containing both photoresponsive 2-nitrobenzyl amine and N-nitrosoamine moieties bonded to the same nitrogen atom, N-(2-nitrobenzyl)-N-phenylnitrous amide (M1, Fig. S15†) and N-benzyl-N-phenylnitrous amide control molecules (M3, Fig. S15†) were synthesized. Under identical UV light irradiation (4 mW cm−2), the progressive consumption of M1 and M3 and the simultaneous formation of N-(2-nitrobenzyl)aniline (M2) and N-benzylaniline (M4) were detected by HPLC, which were identified by comparison with the corresponding authentic samples. Moreover, the reaction rate of M1 was faster than that of M3, in good agreement with the fact that oNBN released NO more quickly than the BN monomer (Fig. S10†). In addition, we found that the M2 intermediate was subjected to a further photolysis reaction with the formation of o-nitrosobenzaldehyde (M5) and aniline (M6), which was, however, not observed for M4. Notably, if the cleavage of 2-nitrobenzyl amine moieties proceeded prior to that of N-nitrosoamine moieties, nitrogen gas rather than NO would be released.49 Although the simultaneous cleavage of N-nitrosoamine and 2-nitrobenzyl amine moieties in M1 cannot be completely ruled out, the primary photolysis process likely followed two steps, namely, the fast cleavage of N-nitrosoamine moieties with the release of NO and the relatively slow photo-degradation of 2-nitrobenzyl amine residues, as evidenced by the formation of an M2 intermediate (Fig. S15†). We, therefore, proposed the following reaction mechanism: under light irradiation, N-nitrosoamines underwent photolysis and a NO radical was released. The resultant anilinyl radical was then reduced to anilines in aqueous solutions.41 It is worth noting that the formation of aniline derivatives led to spontaneous 1,4- or 1,6-elimination reactions if esters, carbonates, or carbamate linkages were present in the ortho- or para-positions.44To further elucidate the NO release mechanism of the monomers, a counterpart compound, NB(NO)A, was synthesized (Fig. S16†) and the photolysis process was investigated (Fig. 2a and b). As expected, akin to the M1 donor, the photo-mediated NO release generated an anilinyl radical that could be reduced with the formation of a secondary aniline intermediate (i.e., NBA), followed by a spontaneous 1,6-elimination reaction with the release of N-(2-nitrobenzyl)-4-aminobenzyl alcohol (A) and 4-nitroaniline (B).50 Note the existence of the A intermediate even after the complete consumption of the NB(NO)A donor. This result, once again, confirmed that the photo-degradation of NB(NO)A was composed of a fast NO release process and a relatively slow photolysis process of 2-nitrobenzyl amine derivatives. Indeed, we examined the photolysis process of an authentic A compound and the result revealed an apparent photolysis rate constant of 0.024 min−1 (Fig. S17†), whereas the NO release of oNBN had an apparent rate constant of 0.1 min−1 (Fig. S8a and b†). The further photolysis of the A compound likely gave rise to the elution peak close to A in Fig. 2b, which was assigned to o-nitrosobenzene according to HRMS analysis.51
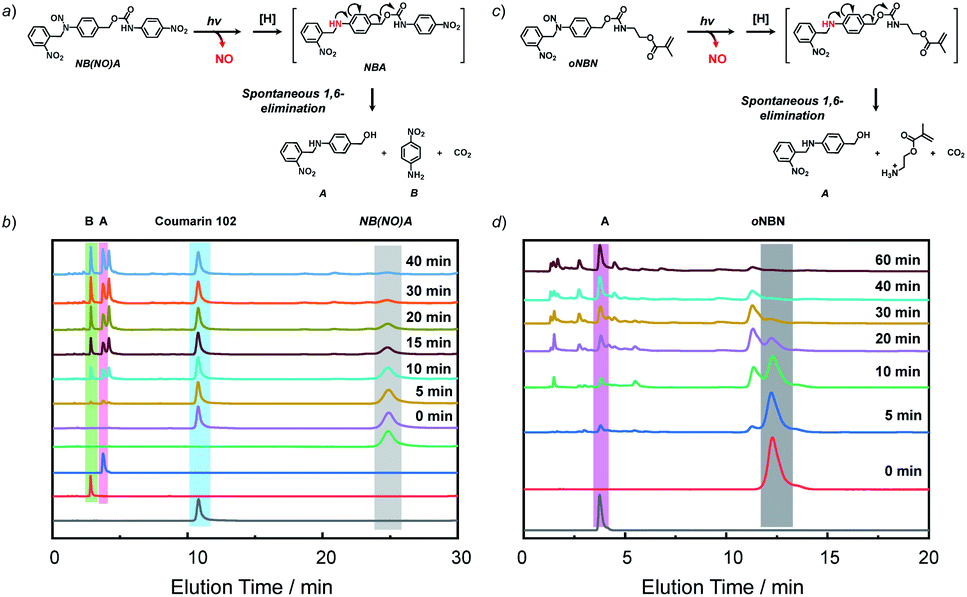 | ||
| Fig. 2 (a and c) Proposed degradation mechanism of the (a) NB(NO)A model compound and (c) oNBN monomer under light irradiation. (b and d) HPLC profiles recorded at 254 nm for (b) NB(NO)A and (d) oNBN (0.25 mM; CH3CN/H2O, v/v = 1/1 as the eluent) under UV 365 nm irradiation (4 mW cm−2) for varying durations. Coumarin 102 was used as the internal standard in (b). | ||
Because the oNBN monomer had a comparable chemical structure to the NB(NO)A model compound, the photo-mediated NO release of the oNBN monomer was proposed to follow a similar reaction mechanism. Indeed, the photo-triggered NO release of the oNBN monomer concurred well with the NB(NO)A compound and the formation of an A intermediate was observed (Fig. 2c and d). Notably, after the initial increase, the absorbance at 251 nm exhibited a gradual decrease when subjected to a longer irradiation period (Fig. S10a and b†), which agreed well with the absorbance changes of the authentic A compound (Fig. S17†). In contrast, the decrease in absorbance intensity was not observed for the pNBN and BN monomers (Fig. S10c–f†). On the other hand, due to the generation of more reactive aliphatic primary amine residues that can further induce several side reactions including, but not limited to, amidation of ester bonds and reaction with p-benzoquinone imine and 2-nitrosobenzaldehyde intermediates,25 some unidentified products were observed in the HPLC elution profiles (Fig. 2d). The detailed photolysis reaction mechanism should be further elucidated. Taken together, we tentatively assumed that the photo-mediated NO release mechanism was as follows: (1) photo-mediated homolytic cleavage of N–NO bonds with the release of a NO radical, (2) in situ reduction of the anilinyl radical with the formation of secondary aniline derivatives, and (3) spontaneous 1,6-elimination with the formation of primary amines (the A intermediate from the oNBN monomer underwent further photo-degradation).50,52 Of note, the in situ generation of primary amines further induced inter/intrachain amidation reactions within the vesicles that synergistically cross-linked and permeabilized the resulting vesicles (Scheme 1), in accordance with our previous reports.25,35,53
Synthesis of NO-releasing amphiphiles and photo-mediated NO release
Knowing that N-nitrosoamine-based monomers could selectively release NO under light irradiation, subsequently, we synthesized PEO-b-PoNBN, PEO-b-PpNBN, and PEO-b-PBN amphiphilic diblock copolymers via reversible addition–fragmentation chain transfer (RAFT) polymerization using a poly(ethylene oxide) (PEO)-based macroRAFT agent (Scheme 2b). The unique thermo-stability of these NO-releasing monomers made them compatible with RAFT polymerization conditions. The NMR results of the as-synthesized amphiphiles, PEO45-b-PoNBN25 (BP1), PEO45-b-PpNBN30 (BP2), and PEO45-b-PBN46 (BP3), are shown in Fig. S18.† The structural parameters of the as-synthesized amphiphiles are summarized in Table S1.† All the amphiphiles (BP1–BP3) exhibited relatively narrow polydispersities (Mw/Mn < 1.2) and the NO loading contents were calculated to be 1.88 μmol mg−1 for BP1, 1.94 μmol mg−1 for BP2, and 2.24 μmol mg−1 for BP3, respectively. Note that the NO loading contents were quite comparable to those of previously reported NONOate-based polymeric NO donors.54Using 1,4-dioxane as a cosolvent, BP1 and BP2 self-assembled into vesicles while BP3 with the longest hydrophobic block length self-assembled into spherical nanoparticles, as confirmed by transmission electron microscopy (TEM) (Fig. 3a and S19†). This result was somewhat surprising considering that a longer hydrophobic chain led to a higher packing parameter, which may suggest that the dipole–dipole interactions of nitrobenzene moieties within BP1 and BP2 copolymers played a crucial role in the self-assembly process.55 Dynamic light scattering (DLS) analysis revealed the as-assembled BP1 vesicles, BP2 vesicles, and BP3 nanoparticles having intensity-average hydrodynamic diameters, 〈Dh〉, of 168 nm, 159 nm, and 90 nm, respectively (Table S1†).
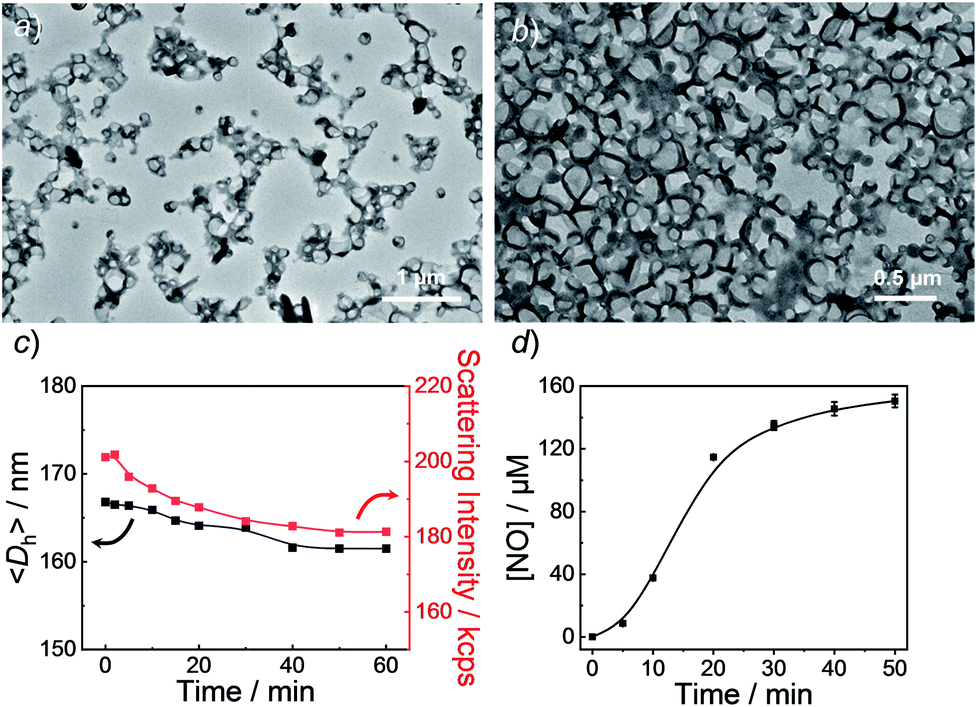 | ||
| Fig. 3 TEM images of aqueous dispersions (0.1 g L−1) of BP1 vesicles (a) before and (b) after UV 365 nm irradiation. (c) Evolution of hydrodynamic diameters, 〈Dh〉, and corresponding scattering intensities and (d) NO release profile of BP1 vesicles under UV 365 nm irradiation (4 mW cm−2). Data represent mean ± s.d. (n = 3). | ||
Next, we sought to verify the NO release profiles of the resulting nanoassemblies by monitoring the UV-vis spectra (Fig. 4). The UV-vis spectra displayed very similar changes to the monomers under UV 365 nm irradiation (Fig. S10†). The absorbance changes can be well-fitted by a mono-exponential function (R2 > 0.98) and the apparent photolysis rate constants (kobs) of BP1, BP2, and BP3 assemblies were calculated to be 0.054 min−1, 0.0068 min−1, and 0.0032 min−1, respectively. Note that the NO release rates of the nanoparticles were generally slower than that of the corresponding monomers. We tentatively ascribed this result to the encapsulation of NO-releasing monomers in the hydrophobic domains of vesicles/micelles. Although slower, the NO release rate of BP1 vesicles was still ∼7.9-fold faster than that of BP2 vesicles and ∼16.9-fold faster than that of BP3 nanoparticles (Fig. 4), substantiating that the NO release rates could be effectively tuned by the introduction of photoresponsive o-nitrobenzyl moieties. Although UV-vis results revealed that a photolysis reaction occurred (Fig. 4a and b), there were only slight decreases in the 〈Dh〉 and corresponding scattering intensities (Fig. 3c) within 60 min irradiation. After 60 min light irradiation, we re-examined the morphology of the BP1 vesicles and TEM results revealed that the vesicular nanostructure was retained (Fig. 3b), in accordance with the DLS result that precluded the possibility of vesicle disassembly. This phenomenon can be interpreted by the photo-mediated NO release that induced the spontaneous 1,6-elimination reaction, in situ producing highly reactive primary amine moieties and cross-linking the bilayer membrane through inter/intrachain cross-linking reactions (Scheme 1).25,35,53 Because BP1 released NO faster than BP2 and BP3 which could shorten the irradiation period in biological applications, we further investigated the in vitro and in vivo applications of these NO-releasing materials using BP1 as a representative example.
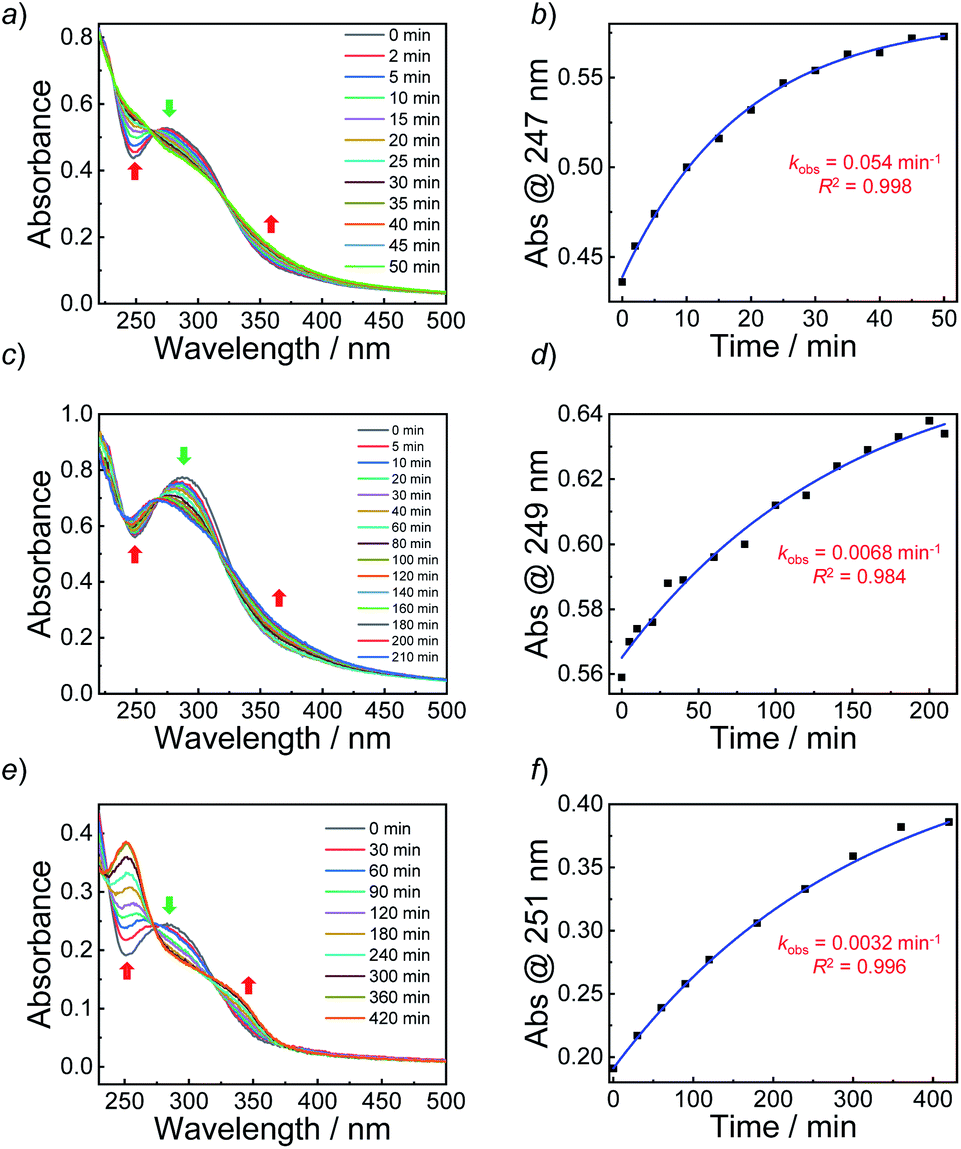 | ||
| Fig. 4 (a, c and e) Absorbance spectra and (b, d and f) absorbance intensity changes together with a mono-exponential fit of aqueous dispersions (0.1 g L−1, 25 °C) of (a and b) BP1 vesicles, (c and d) BP2 vesicles, and (e and f) BP3 micelles under UV 365 nm irradiation (4 mW cm−2). | ||
In vitro and in vivo NO release
It is known that NONOate-based NO donors are highly susceptible to acidic pH, while ultrasound could lead to NO release from N-nitrosoamine-based BNN6 donors.45 In contrast, we found that neither acidic pH nor sonication could trigger the NO release from BP1 vesicles, as evidenced by negligible changes in the fluorescence intensity of the pyronine probe (Fig. S20†). Moreover, BP1 vesicles were relatively stable in the presence of an esterase (0.2 U L−1), fetal bovine serum (FBS), DMEM, and cell lysate, and in a relatively broad pH range (4–8), as evidenced by negligible changes in UV-vis spectra (Fig. S21†). However, photo-mediated NO release could be readily detected using a pyronine probe (Fig. S22†). The generated NO concentration of BP1 vesicles at a concentration of 0.1 g L−1 was determined to be 154 μM (relative to ∼82% N-nitrosoaniline moieties; Fig. 3d). Moreover, in addition to UV light, we found that visible light (410 nm, 30 mW cm−2) can also activate the NO release process (Fig. S23†). Although there were no significant changes in NO concentrations after irradiation of 365 nm or 410 nm for 30 min, the NO release rates can be tuned by the irradiation wavelengths and intensities. The selective NO release under light irradiation thus made it possible to spatiotemporally control the NO release process, which could be potentially used in localized NO delivery in biological systems. To verify this idea, we monitored the photo-mediated NO release of BP1 vesicles in HeLa cells. Upon incubating BP1 vesicles and a pyronine probe with HeLa cells, we can readily observe red fluorescence of the activated pyronine probe after irradiating the cells with UV light for 20 min, suggesting photo-mediated intracellular NO release. In contrast, the control groups without BP1 vesicles with 20 min irradiation or with BP1 vesicles without irradiation had no detectable red fluorescence under otherwise identical conditions, revealing no spontaneous NO release (Fig. 5).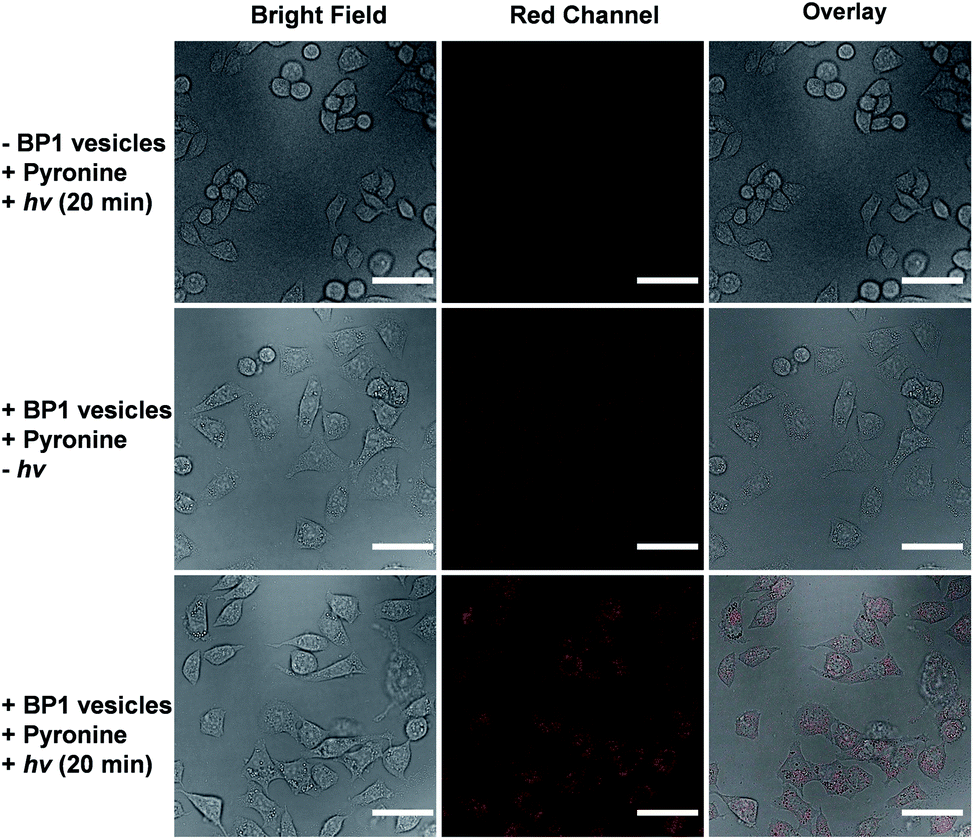 | ||
| Fig. 5 Confocal laser scanning microscopy (CLSM) images of HeLa cells co-incubated with BP1 vesicles (0.4 g L−1) and a pyronine probe (0.1 mM) without or with irradiation for 20 min. The scale bars are 50 μm. | ||
Given that NO has broad therapeutic functions, we further evaluated the potential use of BP1 vesicles in corneal wound healing by taking advantage of NO in mediating cell proliferation and migration.56 A corneal wound model was fabricated by scratching mouse corneas using a surgical blade and the wounded corneas could be stained with fluorescein sodium. We then used the BP1 vesicles as an eye drop for the treatment of the corneal wound. At predetermined time points (every 8 h), an irradiated BP1 dispersion was topically applied to the wounded corneas. Phosphate buffer solution (PBS), non-irradiated BP1 vesicles, and S-nitrosoglutathione (GSNO, a commercially available NO donor) were also used for the treatment of the corneal wound. We found that the irradiated BP1-based eye drop with light-controlled NO release gave rise to a faster corneal healing rate than that of PBS and non-irradiated BP1 vesicles. Specifically, after 88 h, the BP1-treated group with light irradiation was completely recovered (Fig. 6). Importantly, we did not observe obvious side effects of the BP1-based eye drop because the wounded areas were not adversely affected by the non-irradiated BP1 vesicles, suggesting good biocompatibility of the BP1 vesicles. Moreover, the irradiated BP1 vesicles exhibited negligible cytotoxicity as well (Fig. S24d†). Notably, GSNO was known to spontaneously release NO under biological conditions and the GSNO-treated group also exhibited a faster healing performance than the PBS-treated and non-irradiated control group, suggesting that it was the released NO that exerted a positive effect on the corneal wound healing process.
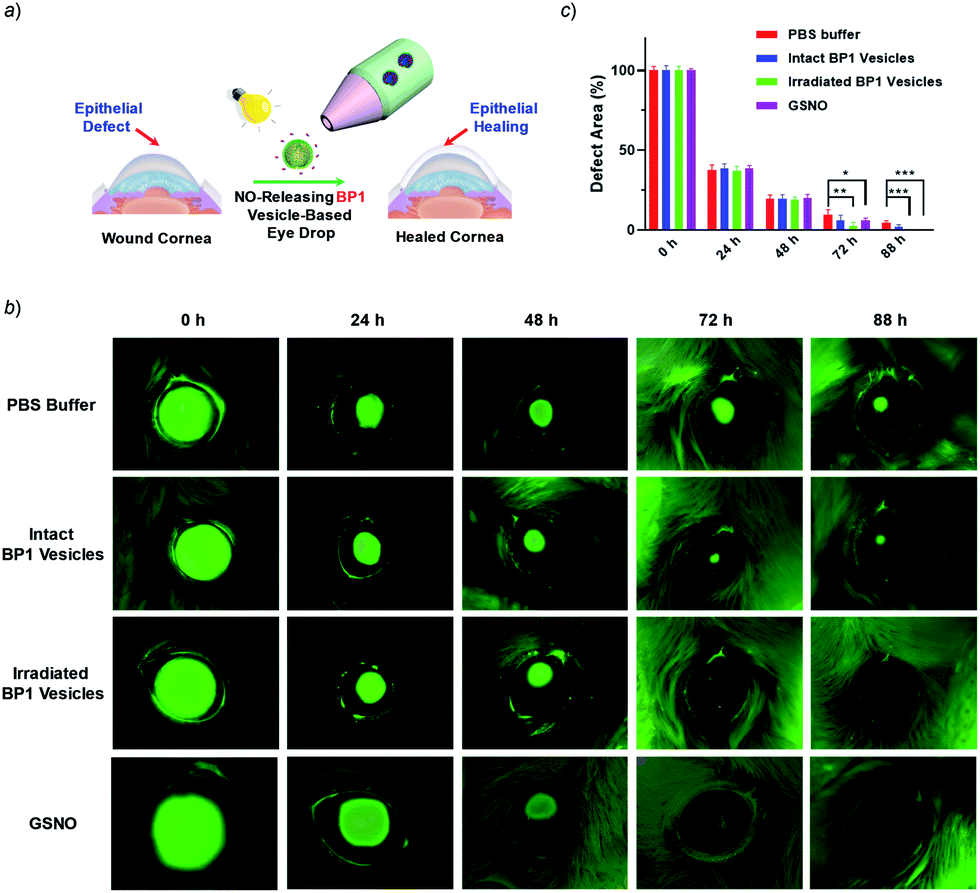 | ||
| Fig. 6 (a) Schematic illustration of the in vivo corneal healing process using NO-releasing BP1 vesicle-based eye drops. (b) Representative photographs of the wounded corneas treated with PBS buffer, untreated BP1 vesicles, irradiated BP1 vesicles, and GSNO, respectively. After each treatment, the corneas were stained with 0.5% sodium fluorescein solution and were imaged by using a Dino-lite hand-held microscope. (c) Quantitative analysis of the wounded areas in (b) using ImageJ software. Values are presented as mean ± s.d. (n = 4). p values were calculated in comparison with the group treated with PBS, **p < 0.01; ***p < 0.001. | ||
To understand the faster healing processes and explore the potential of the NO-releasable vesicles in the treatment of human corneal wounds, we monitored the migration of human corneal epithelial cells (HCECs) to mimic the corneal healing process using the wound healing assay.57 Intriguingly, we observed that the scratch recovery process was highly dependent on the irradiation time (Fig. S24a and b†) and a 10 min irradiation led to a faster healing of the scratch. Quantitative analysis by Griess assay revealed that the nitrite concentrations in the supernatant of cell lysates were positively correlated with the irradiation time. Similarly, a noticeable increase of the nitrite level was observed for cells after GSNO treatment, which confirmed that the light-triggered NO release accounted for the faster wound healing (Fig. S24c†). Notably, there were obvious increases in the cell viability after 20 min irradiation at the tested BP1 concentrations of 0.05–0.4 g L−1, which became more pronounced upon increasing the incubation time, in agreement with the accelerated wound healing performance (Fig. S25†). Moreover, the irradiated BP1 vesicles were nontoxic to HCECs as well at a concentration up to 0.4 g L−1 (Fig. S26†). Taken together, the photo-triggered NO release exerted a positive effect on the increased cell migration and viability which may synergistically contribute to the faster corneal healing process (Fig. 6).58
Finally, BP1 self-assembled into vesicular assemblies and the photo-triggered NO release resulted in the formation of cross-linked vesicles with enhanced permeability (Scheme 1). We envisioned that the NO-releasing vesicles can be further employed to encapsulate other therapeutic agents, exerting a synergistic therapeutic effect.1 Subsequently, we used a polarity-sensitive probe, Nile red (NR), as a model drug to mimic the controlled release behavior of encapsulated payloads. The fluorescence of NR-loaded BP1 vesicles continuously dropped when subjected to light irradiation, indicative of photo-triggered NR release from BP1 vesicles. However, there were no changes in NR fluorescence without light irradiation; we did not observe remarkable photobleaching of NR fluorophores either (Fig. S27†). This result encouraged us to fabricate a nanovector for synergistic delivery of NO and chemotherapy drugs. Previous results revealed that NO can reverse the multidrug resistance (MDR) effect of cancer cells, improving the therapeutic performance.59 After loading doxorubicin into the aqueous lumens of BP1 vesicles, we found that the DOX release was highly dependent on the irradiation time. Specifically, less than 15% DOX was released without light irradiation but over 70% DOX was released after 10 min light irradiation (Fig. S28†). Thus, the NO-releasable BP1 vesicles could be employed for synergistic delivery of NO and DOX, which may exhibit better performance in treating cancers.
Conclusions
In conclusion, we have proposed a direct polymerization approach to fabricate polymeric NO donors through the polymerization of N-nitrosoamine-based monomers. These monomers exhibited excellent thermo-stability and were compatible with RAFT polymerization conditions, and outperformed widely used NONOate and SNO donors. Moreover, NO release rates could be efficiently modulated. The RAFT polymerization of the NO monomers led to the formation of well-defined NO-releasing amphiphiles. Intracellular NO release could be achieved by taking advantage of the spatiotemporal precision of light stimuli. Preliminary results demonstrated that the light-mediated NO release from the vesicular assemblies could stimulate corneal wound healing. Moreover, the co-release of NO and chemotherapy drugs can be achieved by means of photo-mediated cross-linking and permeabilization of vesicle bilayers. The screening of other photoresponsive moieties capable of shifting the irradiation wavelengths to the near-infrared (NIR) region without compromising the NO release performance is currently underway.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The financial support from the Natural Science Foundation of China (NNSFC) project (51722307, 51673179, and 51773190), the Fundamental Research Funds for the Central Universities (WK3450000003), the China Postdoctoral Science Foundation (2017M612090), and the Inter-Collegiate Key Projects of Nature Science of Anhui Province (KJ2018A0197) is gratefully acknowledged.References
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS.
- X. L. Hu, Y. G. Zhang, Z. G. Xie, X. B. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Y. Q. Zhu, B. Yang, S. Chen and J. Z. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- L. G. Wang, L. Chierico, D. Little, N. Patikarnmonthon, Z. Yang, M. Azzouz, J. Madsen, S. P. Armes and G. Battaglia, Angew. Chem., Int. Ed., 2012, 51, 11122–11125 CrossRef CAS.
- H. L. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS.
- F. H. Meng, Z. Y. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- T. Einfalt, D. Witzigmann, C. Edlinger, S. Sieber, R. Goers, A. Najer, M. Spulber, O. Onaca-Fischer, J. Huwyler and C. G. Palivan, Nat. Commun., 2018, 9, 1127 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- P. G. Wang, M. Xian, X. P. Tang, X. J. Wu, Z. Wen, T. W. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- S. P. Nichols, W. L. Storm, A. Koh and M. H. Schoenfisch, Adv. Drug Delivery Rev., 2012, 64, 1177–1188 CrossRef CAS.
- W. P. Fan, B. C. Yung and X. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 8383–8394 CrossRef CAS.
- Z. Sadrearhami, T. K. Nguyen, R. Namivandi-Zangeneh, K. Jung, E. H. H. Wong and C. Boyer, J. Mater. Chem. B, 2018, 6, 2945–2959 RSC.
- Y. Kang, J. Kim, Y. M. Lee, S. Im, H. Park and W. J. Kim, J. Controlled Release, 2015, 220, 624–630 CrossRef CAS.
- J. Kim, G. Saravanakumar, H. W. Choi, D. Park and W. J. Kim, J. Mater. Chem. B, 2014, 2, 341–356 RSC.
- M. C. Jen, M. C. Serrano, R. van Lith and G. A. Ameer, Adv. Funct. Mater., 2012, 22, 239–260 CrossRef CAS.
- M. F. Chung, H. Y. Liu, K. J. Lin, W. T. Chia and H. W. Sung, Angew. Chem., Int. Ed., 2015, 54, 9890–9893 CrossRef CAS.
- P. H. Ruane, K. M. Bushan, C. M. Pavlos, R. A. D'Sa and J. P. Toscano, J. Am. Chem. Soc., 2002, 124, 9806–9811 CrossRef CAS.
- K. Sharma and H. Chakrapani, Nitric Oxide, 2014, 43, 8–16 CrossRef CAS.
- Y. Zhang, N. Gal, F. Itel, I. Westensee, E. Brodszkij, D. Mayer, S. Stenger, M. Castellote-Borrell, T. Boesen, S. R. Tabaei, F. Höök and B. Städler, Nanoscale, 2019, 11, 11530–11541 RSC.
- X. R. Wang, G. H. Liu, J. M. Hu, G. Y. Zhang and S. Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 3138–3142 CrossRef CAS PubMed.
- Z. Y. Deng, Y. F. Qian, Y. Q. Yu, G. H. Liu, J. M. Hu, G. Y. Zhang and S. Y. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS.
- Y. M. Li, G. H. Liu, X. R. Wang, J. M. Hu and S. Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 1760–1764 CrossRef CAS.
- K. Hishikawa, H. Nakagawa, T. Furuta, K. Fukuhara, H. Tsumoto, T. Suzuki and N. Miyata, J. Am. Chem. Soc., 2009, 131, 7488–7489 CrossRef CAS.
- T. Suzuki, O. Nagae, Y. Kato, H. Nakagawa, K. Fukuhara and N. Miyata, J. Am. Chem. Soc., 2005, 127, 11720–11726 CrossRef CAS.
- S. Diring, D. O. Wang, C. Kim, M. Kondo, Y. Chen, S. Kitagawa, K. Kamei and S. Furukawa, Nat. Commun., 2013, 4, 2684 CrossRef.
- E. B. Caruso, S. Petralia, S. Conoci, S. Giuffrida and S. Sortino, J. Am. Chem. Soc., 2007, 129, 480–481 CrossRef CAS.
- S. Sortino, S. Petralia, G. Compagnini, S. Conoci and G. Condorelli, Angew. Chem., Int. Ed., 2002, 41, 1914–1917 CrossRef CAS.
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC.
- N. Ieda, Y. Hotta, N. Miyata, K. Kimura and H. Nakagawa, J. Am. Chem. Soc., 2014, 136, 7085–7091 CrossRef CAS.
- S. Namiki, T. Arai and K. Fujimori, J. Am. Chem. Soc., 1997, 119, 3840–3841 CrossRef CAS.
- J. Fan, Q. J. He, Y. Liu, F. W. Zhang, X. Y. Yang, Z. Wang, N. Lu, W. P. Fan, L. S. Lin, G. Niu, N. Y. He, J. B. Song and X. Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13804–13811 CrossRef CAS.
- T. Ohwada, M. Miura, H. Tanaka, S. Sakamoto, K. Yamaguchi, H. Ikeda and S. Inagaki, J. Am. Chem. Soc., 2001, 123, 10164–10172 CrossRef CAS.
- K. Piech, T. Bally, A. Sikora and A. Marcinek, J. Am. Chem. Soc., 2007, 129, 3211–3217 CrossRef CAS.
- E. Y. Zhou, H. J. Knox, C. J. Reinhardt, G. Partipilo, M. J. Nilges and J. Chan, J. Am. Chem. Soc., 2018, 140, 11686–11697 CrossRef CAS.
- H. H. He, Y. Xia, Y. X. Qi, H. Y. Wang, Z. Wang, J. M. Bao, Z. Q. Zhang, F. G. Wu, H. L. Wang, D. J. Chen, D. H. Yang, X. W. Liang, J. Q. Chen, S. Zhou, X. Liang, X. H. Qian and Y. J. Yang, Bioconjugate Chem., 2018, 29, 1194–1198 CrossRef CAS.
- H. Okuno, N. Ieda, Y. Hotta, M. Kawaguchi, K. Kimura and H. Nakagawa, Org. Biomol. Chem., 2017, 15, 2791–2796 RSC.
- Z. Q. Zhang, J. Y. Wu, Z. H. Shang, C. Wang, J. G. Cheng, X. H. Qian, Y. Xiao, Z. P. Xu and Y. J. Yang, Anal. Chem., 2016, 88, 7274–7280 CrossRef CAS.
- G. H. Liu, X. R. Wang, J. M. Hu, G. Y. Zhang and S. Y. Liu, J. Am. Chem. Soc., 2014, 136, 7492–7497 CrossRef CAS.
- G. H. Liu, G. F. Zhang, J. M. Hu, X. R. Wang, M. Q. Zhu and S. Y. Liu, J. Am. Chem. Soc., 2015, 137, 11645–11655 CrossRef CAS.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970–2984 CrossRef CAS PubMed.
- J. Fan, N. Y. He, Q. J. He, Y. Liu, Y. Ma, X. Fu, Y. J. Liu, P. Huang and X. Y. Chen, Nanoscale, 2015, 7, 20055–20062 RSC.
- T. Akaike, M. Yoshida, Y. Miyamoto, K. Sato, M. Kohno, K. Sasamoto, K. Miyazaki, S. Ueda and H. Maeda, Biochemistry, 1993, 32, 827–832 CrossRef CAS.
- X. Q. Zhu, J. Q. He, Q. Li, M. Xian, J. M. Lu and J. P. Cheng, J. Org. Chem., 2000, 65, 6729–6735 CrossRef CAS PubMed.
- Y. Q. Sun, J. Liu, H. X. Zhang, Y. Y. Huo, X. Lv, Y. W. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS PubMed.
- J. M. Hu, M. R. Whittaker, J. F. Quinn and T. P. Davis, Macromolecules, 2016, 49, 2741–2749 CrossRef CAS.
- S. U. Hettiarachchi, B. Prasai and R. L. McCarley, J. Am. Chem. Soc., 2014, 136, 7575–7578 CrossRef CAS.
- Y. V. Il'ichev, M. A. Schworer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef.
- R. Erez and D. Shabat, Org. Biomol. Chem., 2008, 6, 2669–2672 RSC.
- Z. Y. Deng, S. Yuan, R. P. Xu, H. J. Liang and S. Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 8896–8900 CrossRef CAS.
- H. B. Jin, L. Yang, M. J. R. Ahonen and M. H. Schoenfisch, J. Am. Chem. Soc., 2018, 140, 14178–14184 CrossRef CAS.
- X. R. Wang, J. M. Hu, G. H. Liu, J. Tian, H. J. Wang, M. Gong and S. Y. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS.
- H. W. Choi, J. Kim, J. Kim, Y. Kim, H. B. Song, J. H. Kim, K. Kim and W. J. Kim, ACS Nano, 2016, 10, 4199–4208 CrossRef CAS.
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS.
- J. H. Park, J. Y. Kim, D. J. Kim, M. Kim, M. Chang, R. S. Chuck and C. Y. Park, Sci. Rep., 2017, 7, 8093 CrossRef.
- X. Zhang, G. Tian, W. Y. Yin, L. M. Wang, X. P. Zheng, L. Yan, J. X. Li, H. R. Su, C. Y. Chen, Z. J. Gu and Y. L. Zhao, Adv. Funct. Mater., 2015, 25, 3049–3056 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04039k |
| This journal is © The Royal Society of Chemistry 2020 |