
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMinimalistic supramolecular proteoglycan mimics by co-assembly of aromatic peptide and carbohydrate amphiphiles†
Alexandra
Brito‡
 ab,
Yousef M.
Abul-Haija‡§
c,
Diana Soares
da Costa
ab,
Ramon
Novoa-Carballal
ab,
Rui L.
Reis
abd,
Rein V.
Ulijn
*efg,
Ricardo A.
Pires
*abd and
Iva
Pashkuleva
*ab
ab,
Yousef M.
Abul-Haija‡§
c,
Diana Soares
da Costa
ab,
Ramon
Novoa-Carballal
ab,
Rui L.
Reis
abd,
Rein V.
Ulijn
*efg,
Ricardo A.
Pires
*abd and
Iva
Pashkuleva
*ab
a3B's Research Group, I3Bs – Research Institute on Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, Parque de Ciência e Tecnologia, Zona Industrial da Gandra, 4805-017 Barco, Guimarães, Portugal. E-mail: pashkuleva@i3bs.uminho.pt; rpires@i3bs.uminho.pt
bICVS/3Bs–PT Government Associate Laboratory, Braga/Guimarães, Portugal
cDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK
dThe Discoveries Centre for Regenerative and Precision Medicine, Headquarters at University of Minho, Avepark, 4805-017 Barco, Guimarães, Portugal
eAdvanced Science Research Center (ASRC) at the Graduate Center, City University of New York (CUNY), 85 St Nicholas Terrace, New York, New York 10031, USA. E-mail: rein.ulijn@asrc.cuny.edu
fDepartment of Chemistry, Hunter College, City University of New York, 695 Park Avenue, New York 10065, USA
gPhD Programs in Biochemistry and Chemistry, The Graduate Center of the City University of New York, New York 10016, USA
First published on 21st December 2018
Abstract
We report the co-assembly of aromatic carbohydrate and dipeptide amphiphiles under physiological conditions as a strategy to generate minimalistic proteoglycan mimics. The resulting nanofibers present a structural, fluorenylmethoxycarbonyl-diphenylalanine (Fmoc-FF) core and a functional carbohydrate (Fmoc-glucosamine-6-sulfate or -phosphate) shell. The size, degree of bundling and mechanical properties of the assembled structures depend on the chemical nature of the carbohydrate amphiphile used. In cell culture medium, these nanofibers can further organize into supramolecular hydrogels. We demonstrate that, similar to proteoglycans, the assembled gels prolong the stability of growth factors and preserve the viability of cultured cells. Our results demonstrate that this approach can be applied to the design of extracellular matrix (ECM) substitutes for future regenerative therapies.
Introduction
Molecular self-assembly is a versatile bottom-up approach for the design and synthesis of functional biomaterials.1–5 The main advantage of this approach is the possibility to create structural and functional biomimetics with simple and systemically customizable compositions.3–5 As an example, bioactive peptide amphiphiles (PA) of varying complexity have been used as simple functional analogues of various proteins.2–4 These building blocks can code specific biochemical information and transfer it to the surrounding (biological) environment. Glycosylation, the process of functional decoration of proteins with carbohydrate chains, is the most common post-translational modification that diversifies the roles of proteins by instructing their folding and by activating specific recognition events and different signalling pathways.6 Attached carbohydrate chains can also have a protective role which contributes to the stability and activity of proteins.6,7 So far, these diverse roles of carbohydrates have received little attention, compared to peptides, in the design of molecular self-assembling blocks for the synthesis of functional supramolecular gels.8–14Aiming to develop extracellular matrix (ECM) mimics, recent studies introduced the use of peptide amphiphiles (PAs) end-on functionalized with carbohydrate moieties.8–11 Upon assembly of such amphiphiles, the carbohydrate units are exposed on the surface of the assembled fibre and are able to participate in multivalent biospecific interactions. While this approach seems straightforward for generating supramolecular mimics of proteoglycans, the introduction of the carbohydrate unit is challenging in terms of PA synthesis and assembly as it may disturb the delicate balance of forces governing the process of self-organization.15,16
Multicomponent co-assembly offers a possibility to expand the chemical diversity and functional complexity of supramolecular systems without compromising the simplicity of the building blocks.17 The feasibility of this approach has been demonstrated by a combination of different PAs.15–18 We hypothesized that a similar strategy can also be applied for co-assembly of simple carbohydrate amphiphiles (CAs, 1) and their peptide analogues (2) and the result will be a synergistic modular system mimicking proteoglycans (Scheme 1).
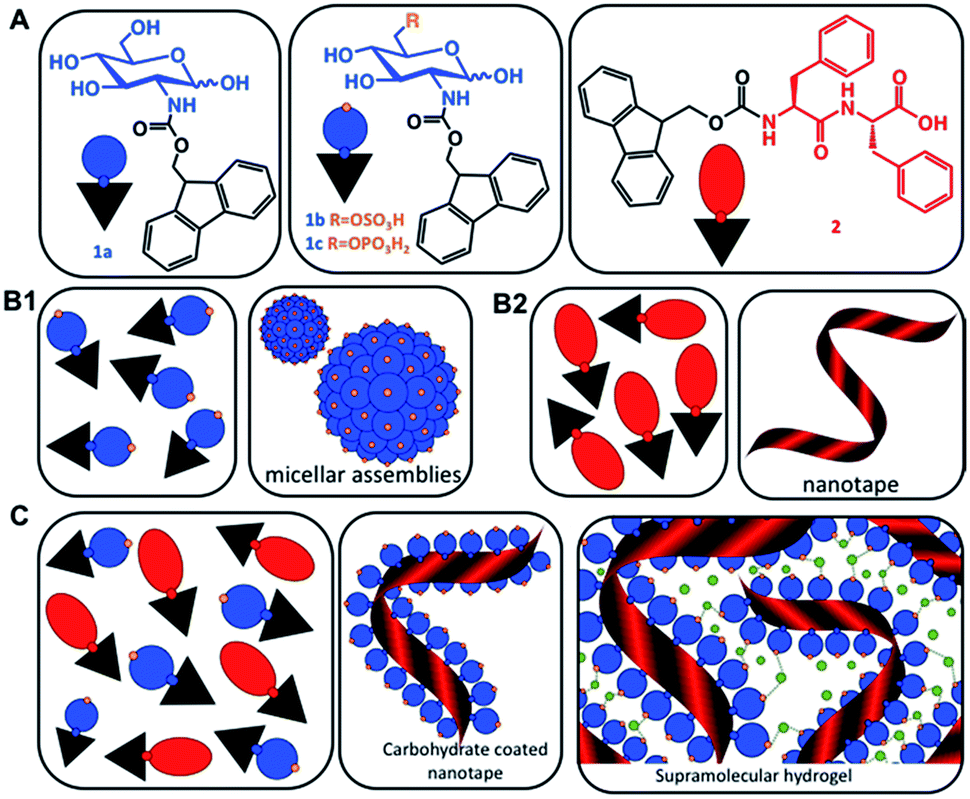 | ||
| Scheme 1 (A) Chemical structures of the carbohydrate amphiphiles (1) and the peptide analogue Fmoc-FF (2). (B) Schematic representation of the assembly of the individual components (1) and (2) in water resulting in the formation of micelles and nanotapes, respectively. (C) Co-assembly of (1) and (2) to form carbohydrate functionalized peptide nanotapes and supramolecular hydrogels. Green dots represent divalent cations. | ||
The range of carbohydrates found in biological systems provide a rich library of water soluble, chiral building blocks.19 Simple carbohydrate amphiphiles (CAs) functionalized with aromatic moieties such as naphthalene and fluorenylmethoxycarbonyl (Fmoc) have been recently developed and shown to form self-assembled structures.20–22 However, the amphiphiles bearing the naphthalene unit do not form gels and the ones functionalized with Fmoc require a heating–cooling cycle or a specific enzymatic stimulus to enable the formation of gels under physiological conditions.20,21
Herein, we describe bi-component systems, in which fluorenylmethoxycarbonyl-diphenylalanine (Fmoc-FF) provides the structural component, while Fmoc-carbohydrate is the solvent-exposed functional element, which can be systematically exchanged (Scheme 1). We demonstrate that these modular materials combine the simplicity of small molecules with the versatility and biofunctionality of glycans and proteins, respectively.
Results and discussion
Self-assembly of aromatic carbohydrate amphiphiles (1) and Fmoc-FF (2)
The amphiphiles Fmoc-glucosamine-6-sulfate (GlcN6S, 1b) and Fmoc-glucosamine-6-phosphate (Fmoc-GlcN6P, 1c) were obtained from the respective glucosamines in a single-step reaction using an excess of Fmoc chloride (details on the synthesis and chemical characterization of the amphiphiles are provided in the ESI, Fig. S1–S13†).Fmoc-GlcN (1a) is not soluble in water at room temperature. Upon temperature rising (80 °C), it dissolves and organizes spontaneously into nanofibers that, at room temperature, form a highly hydrated scaffold (Fig. 1A).20 On the other hand, Fmoc-FF (2) dissolves in water only at basic pH (∼11) and self-assembles upon pH decrease.23–25 The assembly of 2 is controlled by peptidic hydrogen bonds that result in the formation of β-sheet-like structures that are further stabilized by aromatic π–π interactions, whilst the organization of 1a is driven by a combination of CH–π and π–π (T-shape) stacking interactions.20,24,26
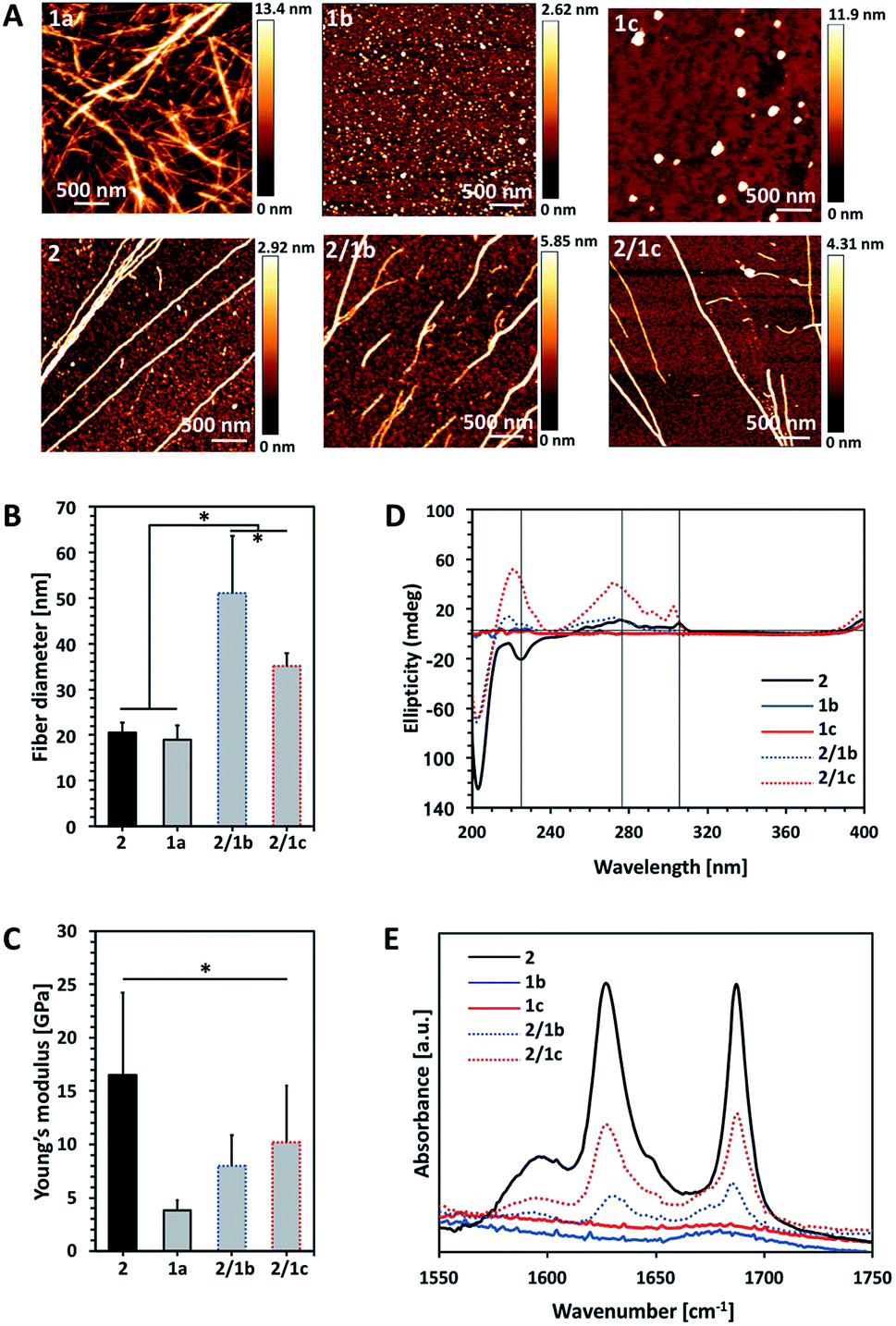 | ||
Fig. 1 Characterization of single (10 mM) and bi-component (ratio PA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CA 2:1) assemblies: (A) AFM images; (B) fibre diameter and (C) Young's modulus of the assembled fibres measured by AFM; (D) CD spectra of the PA Fmoc-FF (2), its CA analogues Fmoc-GlcN6S (1b) and Fmoc-GlcN6P (1c), and the respective co-assemblies; (E) FTIR spectra of the precursor amphiphiles and their co-assemblies. Statistically significant differences (p < 0.01) are marked with (*). Of note, Fmoc-GlcN (1a, 10 mM) was dissolved at 80 °C and the solution gels upon its cooling to room temperature and thus, AFM analysis was performed on the gel. :CA 2:1) assemblies: (A) AFM images; (B) fibre diameter and (C) Young's modulus of the assembled fibres measured by AFM; (D) CD spectra of the PA Fmoc-FF (2), its CA analogues Fmoc-GlcN6S (1b) and Fmoc-GlcN6P (1c), and the respective co-assemblies; (E) FTIR spectra of the precursor amphiphiles and their co-assemblies. Statistically significant differences (p < 0.01) are marked with (*). Of note, Fmoc-GlcN (1a, 10 mM) was dissolved at 80 °C and the solution gels upon its cooling to room temperature and thus, AFM analysis was performed on the gel. | ||
We compared the properties of the formed assemblies by AFM (Fig. 1B and C, black outlines) and found that the fibres have a similar size but their Young's moduli are significantly different: the PA 2 assembles in very rigid fibres (∼16 GPa) that have a 4-fold higher modulus than the ones obtained from the carbohydrate analogue 1a (∼4 GPa).
The co-assembly of 2 and 1a is challenging as both compounds have limited solubility in water and dissolve under different conditions. We functionalized 1a with polar groups aiming to improve the solubility and to impart relevant functionality. Sulphate groups (1b) were chosen because of their biological relevance: most ECM glycosaminoglycans are sulphated and they interact specifically with other biological entities (involved in cell–cell and cell–ECM interactions) depending on their sulphation patterns.6 On the other hand, carbohydrates internalized by cells are activated via their functionalization with phosphate groups and thus, we have also chosen this functionality (1c). As expected, incorporation of these functional groups resulted in improved solubility and affected the assembly of Fmoc-GlcN6S (1b) and Fmoc-GlcN6P (1c): the introduced polar, negatively charged groups cause electrostatic repulsion between the modified CA, thus disturbing the unidirectional assembly observed for 1a. Such Coulomb forces favour self-assembled morphologies with a high surface to volume ratio. Indeed, 1b and 1c form micellar assemblies with negative zeta potentials (−112.0 ± 23.7 mV for 1b and −74.4 ± 5.3 mV for 1c), indicating that the sulphate group of 1b and the phosphate group of 1c are displayed on the surface of the micelles formed in water (Fig. 1A). Of note, the micelles of 1c are bigger than the ones assembled from 1b due to the different Coulomb forces (Fig. S17†). The circular dichroism (CD) analysis supports the formation of micellar assemblies: no signals were observed in the CD spectra of 1b and 1c (Fig. 1D).
Co-assembly of 2 with either 1b or 1c was performed in water at room temperature. The respective amphiphiles were dissolved at basic pH (∼11) at different ratios (i.e. PA:CA of 2:0.5, 2:1, 2:2) but transparent solutions were observed only for the ratios 2:0.5 and 2:1 (Fig. S14†). The pH was then adjusted to 8 by drop-wise addition of HCl. CD spectra of these solutions were recorded 24 h after the pH adjustment and indicated the formation of assemblies that clearly differ from 2 (Fig. 1D and S18†). The peak at 204 nm is associated with the π–π* transition and is clearly observed for all systems that contain 2. Formation of a superhelical arrangement (n–π* transition) is shown by the signal at 225 nm in the spectrum of 2.26,27 A blue shift of this peak, observed in the mixed systems (217 nm for 2/1b and 220 nm for 2/1c), indicates changes in the superhelical structure.28,29 This shift was concomitant with polarity changes upon addition of CAs and increased intensity especially for 2/1c, indicating co-assembly of the CAs with 2. Signals associated with the π–π* transition of the Fmoc groups (the signal with the maximum at 278 nm and the peak at 306 nm in the spectrum of 2, Fig. 1D) are also blue-shifted in the spectra of the mixed systems confirming co-assembly.27
TEM and AFM characterization corroborated the CD and fluorescence data. The co-assembled fibres differ from the single component fibres in several aspects: both 2/1b and 2/1c fibres are bundled (and appear branched), thicker and less stiff than the Fmoc-FF fibres (Fig. 1A–C and S16, S20†). Furthermore, the co-assembled fibres have higher Young's moduli compared to the single component CA (1a) fibres, i.e. the co-assemblies have intermediate mechanical properties when compared with the PA and CA single component fibres. FTIR spectroscopy was also performed to characterize the co-assemblies (Fig. 1E).30,31 Absorptions at 1625 and 1687 cm−1 are associated with the formation of H-bonding by Fmoc-peptide amide and carbamate groups.32 As expected, these peaks are missing in the spectra of CA (1b and 1c) solutions. On the other hand, the amide I peaks are clearly observed for the co-assembled systems (2/1b and 2/1c) demonstrating that the β-sheet structure remains intact in the presence of the CA although the lower intensity of these peaks suggests that a degree of intercalation might have occurred.
Previously, different models for assembly of two-component systems have been proposed.16,17,31 Our results (AFM; CD and FTIR spectra) indicate that 2 co-assembles with 1b and 1c into core–shell nanofibers with the carbohydrate moieties exposed on the surface. Such co-assembly would involve intercalation between the Fmoc moieties of the CA and PA on the formed fibre surface (Scheme 1B), thus shielding the hydrophobic Fmoc while exposing the carbohydrate unit.31 We studied the surface charge of the formed nanofibers in order to confirm this organization (Table S1†). The value determined for 2 was −0.04 mV, which agrees with our previous work showing that the apparent pKa of the terminal carboxylic acid in 2 is substantially shifted due to the favoured self-assembly of the uncharged form.23 The co-assembled systems 2/1b and 2/1c have negative zeta potentials, −56.0 ± 1.5 mV and −55.4 ± 6.1 mV respectively. As expected, the absolute values are smaller than the ones of the precursor CAs due to the lower density of the functional groups on the co-assembled fibres as compared with the single component micelles. These results further confirm our hypothesis for co-assembly resulting in the formation of systems with a peptide-based core and carbohydrate shell.
Characterization of the supramolecular gels
Upon contact with cell culture medium (pH 7.4), the PA 2 and its co-assemblies with the CAs form self-sustained macroscopic gels (Fig. 2A).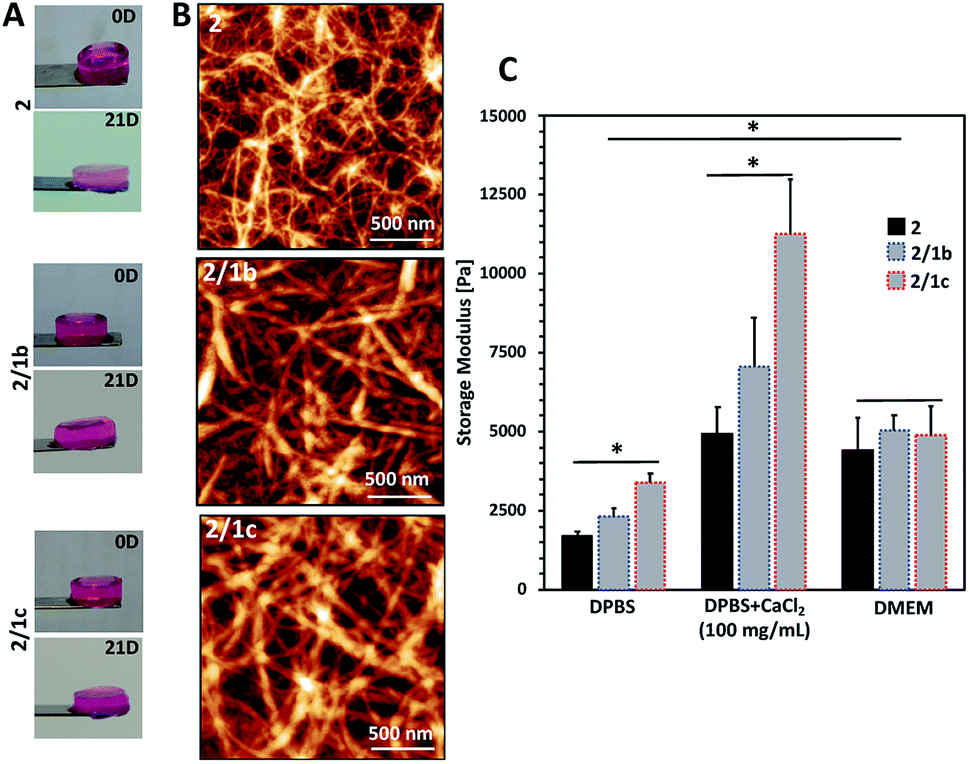 | ||
| Fig. 2 Characterization of the formed gels: (A) optical images showing the macroscopic integrity of the gels after formation in cell culture medium (DMEM) at 37 °C (0D) and after immersion for 21 days under the same conditions (21D); (B) tangled nanofibrous structures observed using an AFM; (C) mechanical properties of the gels determined by rheology. DPBS: Dulbecco's Phosphate-Buffered Saline (with no Ca2+ and Mg2+); DMEM: Dulbecco's Modified Eagle's Medium; DMEM was supplemented with 10% fetal bovine serum. | ||
We used an AFM to analyse the gels and observed a typical entangled fibrous network for all studied compositions (Fig. 2B and S21†). However, two major differences can be observed in the AFM images: fibres of 2 are thinner and more entangled compared to the ones generated by co-assembly (2/1b and 2/1c). These differences are also reflected in the viscoelastic properties of the gels: mixed systems form more robust gels in all studied media (Fig. 2C and S23†). Previous studies have demonstrated that gelation of similar PA systems can be either pH- or salt-triggered via the formation of salt bridges between the anions exposed on the fibre surface (Scheme 1C).33,34 Our rheological studies confirmed that these stabilizing interactions are also at play in our current system: upon reduction of the pH by adding Dulbecco's Phosphate-Buffered Saline (DPBS, pH 7.4, only monovalent cations), we observed the formation of soft gels. The moduli of the formed gels increased significantly in the presence of divalent Ca2+ cations (Fig. 2C, DPBS + CaCl2). This difference depends on the anions exposed on the fibre surface and we observed the following order HPO42−/H2PO4− (2/1c 3.3-fold increase) > SO42− (2/1b, 3-fold increase) > COO− (2, 2.7-fold increase). These results confirmed that the carbohydrate units are exposed on the fibre surface.
After confirming the exposure of the carbohydrate units on the fibre surface and their ability to form gels under physiological conditions, we evaluated the bioactivity of the generated co-assemblies by their ability to interact specifically with proteins. We designed two experimental setups to evaluate two fundamental roles of proteoglycans in the ECM: the ability of the assembled gels to protect growth factors and to host cells.
Interactions of the supramolecular gels with basic fibroblast growth factors (FGF-2)
Basic fibroblast growth factor (FGF-2) is a glycoprotein that has a pleiotropic effect in different tissues and organs: it is crucial for processes such as angiogenesis, development, wound healing, cell survival and differentiation, among others.35,36 It is also known for its thermal instability: it loses most of its activity after 24 h of incubation at 37 °C (Fig. S24†).37 In its native environment – the pericellular space – FGF-2 interacts specifically with cell surface proteoglycans, which protect it from degradation and aggregation.6 Electrostatic interactions and hydrogen bonding are the main driving forces for the formation of proteoglycan-FGF-2 complexes.36,38 Proteoglycan charge density is crucial for these interactions as basic amino acid residues in the growth factor, usually ordered in Cardin–Weintraub sequences, are the main contributors to the formation of the complexes.6,38 Indeed, addition of heparin (the most negatively charged natural biomolecule) or heparan sulphate to the FGF-2 incubation media prolongs its activity.37,39,40 We therefore tested the developed systems for their capacity to protect FGF-2 and preserve its bioactivity under physiological conditions (Fig. 3).9 We added the growth factor into the gels (details are provided in the ESI†) and the successful encapsulation was confirmed by immunostaining and SDS-PAGE (Fig. 3, 0D). Of note, immunostaining allows visualisation only of active FGF-2 as it is based on the recognition of the growth factor by a specific antibody with a fluorescent tag. The gels with the encapsulated growth factor were maintained under standard cell culture conditions (DMEM at 37 °C) for 7 days and then characterised again (Fig. 3, 7D). Our results demonstrated that mixed systems (2/1b and 2/1c) protect better the FGF-2 from degradation when compared to the non-functionalized peptide nanofibers (2): stronger co-localization (Fig. 3A) and more protein (Fig. 3B) are visible for the FGF-2 loaded in the co-assembled gels after 7 days. Moreover, ELISA tests did not show any release of the growth factor to the surrounding media for this period of time. These results indicate that FGF-2 is simply entrapped in the Fmoc-FF gel and thus, attainable for degradation (although slower as compared with free FGF-2). On the other hand, in the mixed systems, FGF-2 interacts with the carbohydrates exposed on the fiber surface, thus copycatting the natural proteoglycan protection mechanism.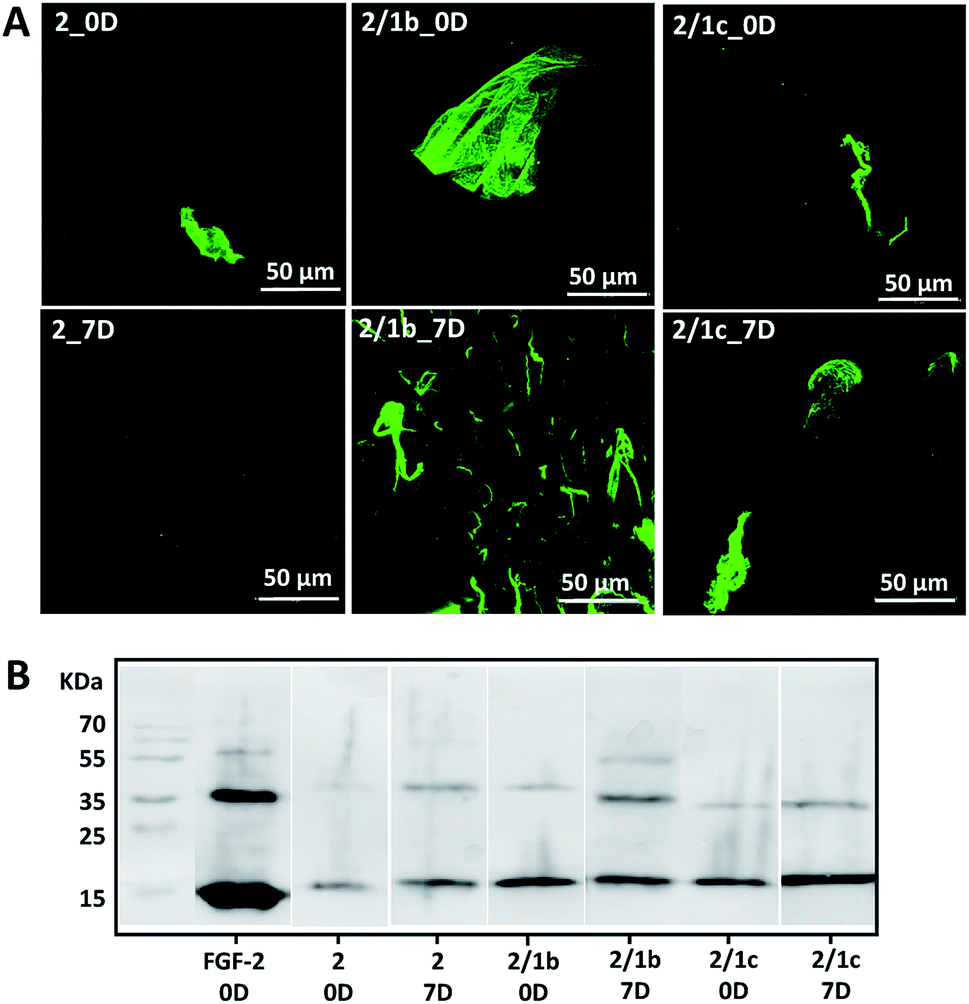 | ||
| Fig. 3 Interaction of one- and bi-component supramolecular hydrogels with FGF-2: (A) bioactive FGF-2 (green) distribution and stability within the gels demonstrated for 0 and 7 days by confocal imaging and immunostaining. Of note, FGF-2 was not homogenised intentionally to facilitate the visualization of the growth factor by fluorescent microscopy; (B) SDS-PAGE analysis of FGF-2 entrapped (0D) and preserved in the hydrogel after 7 days. | ||
Cytotoxicity of the supramolecular gels
Next, we tested the ability of the gels to support viable cells. Initially, we used L929 cells that are recommended for standard in vitro cytotoxicity tests. Our results demonstrate that cells remain viable either when they are seeded on top of the gels (Fig. 4A and S27†) or when they are encapsulated inside the gels (Fig. 4B and S27†). Chondrogenic cell line ATDC5 (Fig. S25†) and epithelial HeLa cells (Fig. S26†) were also tested with the assembled materials to prove their versatility. In all the cases, we observed a majority of viable cells (Fig. S27†). The culture of these cell lines requires the use of media supplemented with proteins (10% fetal bovine serum, FBS) that can affect the mechanical properties of the gels.41 Our results demonstrated that indeed the presence of proteins affects significantly the storage moduli of the gels: when DPBS supplemented with CaCl2 was replaced with DMEM (the same Ca2+ concentration) supplemented with FBS we observed a significant decrease of the storage moduli, especially for the co-assembled systems (Fig. 2C and S23†). Of note, the substitution of DMEM with DMEM/F12 does not induce any significant change in the mechanical properties of the gels (Fig. S23†). These data suggest that the proteins from the medium interact with the mixed systems and disrupt partially the ionic crosslinking bridges between different nanofiber segments. As the mechanical properties of the assembled gels under cell culture conditions are very similar, we suggest that the observed different number of adherent/encapsulated cells is due to the different chemistry of the generated supramolecular systems and their specific interactions with the cultured cells.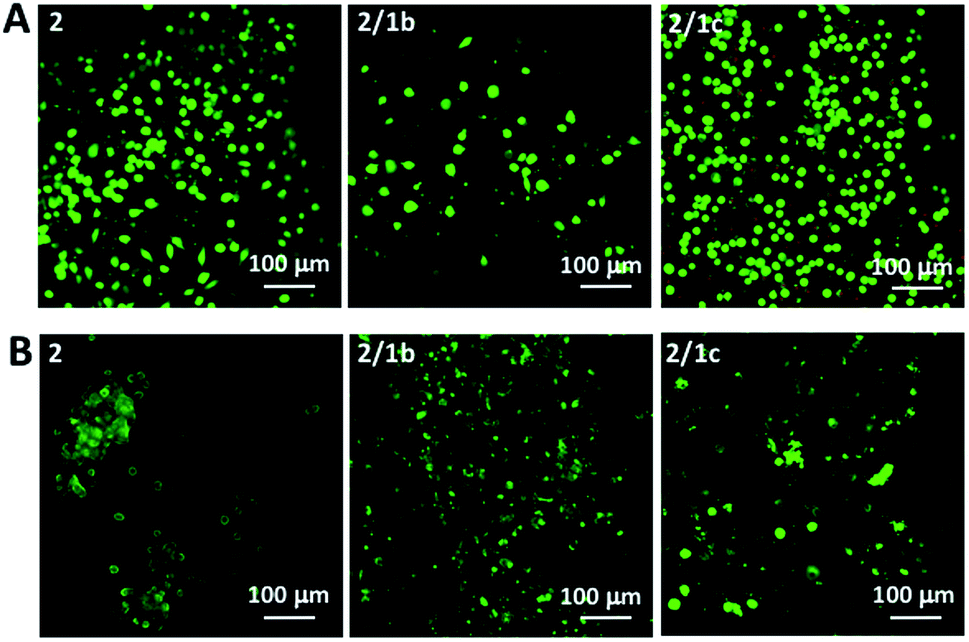 | ||
| Fig. 4 Live/dead assays for L929 cultured (A) on top of the gels or (B) encapsulated within the gels for 48 h. Dulbecco's Modified Eagle's Medium (DMEM) was used for these tests. | ||
Conclusions
We have successfully designed and synthesized supramolecular, functional glycoprotein mimics by co-assembly of simple building blocks. In addition to the simplicity of the building blocks, the proposed approach has several other advantages: the assembly is carried out under physiological conditions, and the formed hydrogels are stable, non-cytotoxic and biofunctional. We also demonstrated that by changing the chemical design in a systematic way we are able to control the biofunctionality and mechanical properties of the supramolecular gels – features that are of key importance in the design of materials for biomedical and tissue engineering applications.Conflicts of interest
Rein V. Ulijn is a director of Biogelx, Ltd, an organization with a financial interest in the subject matter and materials (Fmoc-FF) discussed in this publication.Acknowledgements
We acknowledge the EU's H2020 and FP7 framework programmes (Forecast 668983; CHEM2NATURE 692333; THE DISCOVERIES CTR 739572; ERC AdG ComplexiTE 321266) and the Portuguese FCT (IF/00032/2013; BD/113794/2015; BPD/85790/2012; M-ERA-NET2/0001/2016 – INCIPIT; ENMed/001/2015 – CytoNanoHeal). We thank P. Frederix for his help in the FTIR measurements and M. Mullin for her help in the TEM imaging.References
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed.
- X. W. Du, J. Zhou, J. F. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- K. E. Inostroza-Brito, E. Collin, O. Siton-Mendelson, K. H. Smith, A. Monge-Marcet, D. S. Ferreira, R. P. Rodriguez, M. Alonso, J. C. Rodriguez-Cabello, R. L. Reis, F. Sagues, L. Botto, R. Bitton, H. S. Azevedo and A. Mata, Nat. Chem., 2015, 7, 897–904 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- D. S. Costa, R. L. Reis and I. Pashkuleva, Annu. Rev. Biomed. Eng., 2017, 19, 1–26 CrossRef PubMed.
- H. S. Azevedo and I. Pashkuleva, Adv. Drug Delivery Rev., 2015, 94, 63–76 CrossRef CAS PubMed.
- X. W. Du, J. Zhou, O. Guvench, F. O. Sangiorgi, X. M. Li, N. Zhou and B. Xu, Bioconjugate Chem., 2014, 25, 1031–1035 CrossRef CAS PubMed.
- S. S. Lee, T. Fyrner, F. Chen, Z. Álvarez, E. Sleep, D. S. Chun, J. A. Weiner, R. W. Cook, R. D. Freshman, M. S. Schallmo, K. M. Katchko, A. D. Schneider, J. T. Smith, C. Yun, G. Singh, S. Z. Hashmi, M. T. McClendon, Z. Yu, S. R. Stock, W. K. Hsu, E. L. Hsu and S. I. Stupp, Nat. Nanotechnol., 2017, 12, 821–829 CrossRef CAS PubMed.
- B. S. Kim, D. J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333–16337 CrossRef CAS PubMed.
- J. Zhou, X. W. Du, X. Y. Chen and B. Xu, Biochemistry, 2018, 57, 4867–4879 CrossRef CAS PubMed.
- Y. B. Lim, S. Park, E. Lee, H. Jeong, J. H. Ryu and M. S. Lee, Biomacromolecules, 2007, 8, 1404–1408 CrossRef CAS PubMed.
- J. Liu, Z. L. Sun, Y. Q. Yuan, X. Tian, X. Liu, G. X. Duan, Y. G. Yang, L. Yuan, H. C. Lin and X. M. Li, ACS Appl. Mater. Interfaces, 2016, 8, 6917–6924 CrossRef CAS PubMed.
- D. Yuan, J. Shi, X. Du, N. Zhou and B. Xu, J. Am. Chem. Soc., 2015, 137, 10092–10095 CrossRef CAS PubMed.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- Y. M. Abul-Haija, G. G. Scott, J. K. Sahoo, T. Tuttle and R. V. Ulijn, Chem. Commun., 2017, 53, 9562–9565 RSC.
- M. Delbianco, P. Bharate, S. Varela-Aramburu and P. H. Seeberger, Chem. Rev., 2016, 116, 1693–1752 CrossRef CAS PubMed.
- L. S. Birchall, S. Roy, V. Jayawarna, M. Hughes, E. Irvine, G. T. Okorogheye, N. Saudi, E. De Santis, T. Tuttle, A. A. Edwards and R. V. Ulijn, Chem. Sci., 2011, 2, 1349–1355 RSC.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- Z. M. Yang, G. L. Liang, M. L. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843–845, 10.1039/b616563.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- Y. Zhang, H. W. Gu, Z. M. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS PubMed.
- D. H. Chin, R. W. Woody, C. A. Rohl and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15416–15421 CrossRef CAS PubMed.
- S. Mondal, L. Adler-Abramovich, A. Lampel, Y. Bram, S. Lipstman and E. Gazit, Nat. Commun., 2015, 6, 8615 CrossRef CAS PubMed.
- Y. M. Abul-Haija, S. Roy, P. W. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973–979 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed.
- S. Fleming, P. W. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- S. Roy, N. Javid, P. W. J. M. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Chem.–Eur. J., 2012, 18, 11723–11731 CrossRef CAS PubMed.
- C. M. R. Perez, Z. Alvarez, F. Chen, T. Aytun and S. I. Stupp, ACS Biomater. Sci. Eng., 2017, 3, 2166–2175 CrossRef PubMed.
- S. Amorim, R. A. Pires, D. S. da Costa, R. L. Reis and I. Pashkuleva, Langmuir, 2013, 29, 7983–7992 CrossRef CAS PubMed.
- G. Chen, D. R. Gulbranson, P. Z. Yu, Z. G. Hou and J. A. Thomson, Stem Cells, 2012, 30, 623–630 CrossRef CAS PubMed.
- D. M. Ornitz, BioEssays, 2000, 22, 108–112 CrossRef CAS PubMed.
- D. Gospodarowicz and J. Cheng, J. Cell. Physiol., 1986, 128, 475–484 CrossRef CAS PubMed.
- O. Saksela, D. Moscatelli, A. Sommer and D. B. Rifkin, J. Cell Biol., 1988, 107, 743–751 CrossRef CAS PubMed.
- N. Javid, S. Roy, M. Zelzer, Z. M. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data. See DOI: 10.1039/c8sc04361b |
| ‡ These authors contributed equally. |
| § Present address: WestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK |
| This journal is © The Royal Society of Chemistry 2019 |