
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of the RiPP trojan horse nucleotide antibiotic microcin C is directed by the N-formyl of the peptide precursor†
Shi-Hui
Dong
 ab,
Alexey
Kulikovsky
ade,
Inna
Zukher
d,
Paola
Estrada
a,
Svetlana
Dubiley
de,
Konstantin
Severinov
*def and
Satish K.
Nair
*abc
ab,
Alexey
Kulikovsky
ade,
Inna
Zukher
d,
Paola
Estrada
a,
Svetlana
Dubiley
de,
Konstantin
Severinov
*def and
Satish K.
Nair
*abc
aDepartment of Biochemistry, University of Illinois at Urbana-Champaign, Illinois, USA. E-mail: snair@illinois.edu
bCarl Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Illinois, USA
cCenter for Biophysics and Quantitative Biology, University of Illinois at Urbana-Champaign, Illinois, USA
dInstitute of Gene Biology, Russian Academy of Science, 34/5 Vavilo str., 11934 Moscow, Russia
eCenter for Life Sciences, Skolkov Institute of Science and Technology, 3 Nobel str., 143026 Moscow, Russia
fWaksman Institute for Microbiology, 190 Frelinghuysen Road, Piscataway, New Jersey, USA. E-mail: severik@waksman.rutgers.edu
First published on 26th December 2018
Abstract
Microcin C7 (McC) is a peptide antibiotic modified by a linkage of the terminal isoAsn amide to AMP via a phosphoramidate bond. Post-translational modification on this ribosomally produced heptapeptide precursor is carried out by MccB, which consumes two equivalents of ATP to generate the N–P linkage. We demonstrate that MccB only efficiently processes the precursor heptapeptide that retains the N-formylated initiator Met (fMet). Binding studies and kinetic measurements evidence the role of the N-formyl moiety. Structural data show that the N-formyl peptide binding results in an ordering of residues in the MccB “crossover loop”, which dictates specificity in homologous ubiquitin activating enzymes. The N-formyl peptide exhibits substrate inhibition, and cannot be displaced from MccB by the desformyl counterpart. Such substrate inhibition may be a strategy to avert unwanted McC buildup and avert toxicity in the cytoplasm of producing organisms.
Introduction
Microcin C7 (McC) is a modified heptapeptide, produced by some strains of E. coli. The terminal α-carboxylate of McC peptide is conjugated to 5′-phosphate of AMP via a phosphoramidate linkage to yield an aspartamide-adenylate that is linked through an N–P bond (Fig. 1).1,2 McC is a RiPP (ribosomally synthesized and post-translationally modified peptide),3 wherein the heptapeptide MRTGNAN (1 or 2), the product of the mccA gene, is modified by the enzyme MccB to yield the adenylate (3 or 4).4,5 The peptide component of McC facilitates both export from the producing strain through MccC major facilitator superfamily membrane transporter and import into susceptible cells via the inner membrane YejABEF transporter.6,7 Upon import into target cells, no-specific proteases cleave the peptide conjugate to release a “warhead” – an inert analog of aspartyl adenylate 5, with non-hydrolyzable N-acyl phosphoramidate link 6.8 The toxin inhibits aspartyl-tRNA synthetase to halt protein synthesis.9 | ||
| Fig. 1 (A) Biosynthetic gene cluster for the McC toxin from E. coli. (B) MccB-catalyzed production of the peptide-adenylate resulting in installation of an N–P linkage. (C) Chemical structures of aspartyl adenylate generated by aspartyl-tRNA. Synthetase (5) and the processed McC toxin (6). Mature McC toxin (6) has a tailoring aminopropyl modification that increases it affinity for the target. | ||
The adenylyl transfer reaction catalyzed by MccB consumes two molar equivalents of ATP: the first is used for the attachment of AMP to the α-carboxylate of the MRTGNAN peptide, which subsequently undergoes an intermolecular reaction to form a peptide-succinimide intermediate.10 Attack of the succinimidyl nitrogen from this intermediate onto the α-phosphate of the second ATP equivalent forms the N–P bond. Hydrolysis of the succinimide ring yields the modified peptide-adenylate in which the terminal Asn encoded by the mccA gene is converted to an isoAsn (i.e. an aspartamide).10 Export of the prodrug from the producing organism is carried out by a member of the major facilitator superfamily of membrane transporters (MccC).4
Biosynthetic clusters that produce McC-like compounds universally contain only the genes encoding for an MccA-like precursor peptide, the MccB adenylyltransferase and the MccC transporter,11 but often possess genes that install additional decorations on the peptide aspartamide-adenylate, such as the 3-aminopropyl moiety found on McC 6.12,13 Producing strains have developed strategies to avert toxic effects of prematurely processed intact McC (3 or 4) in their cytoplasm. Specifically, MccE and MccF carry out either toxin modification or hydrolysis, respectively, to protect against accumulation inside of the host.14,15
All RiPP systems must utilize means to guard against the unregulated synthesis of the precursor peptide, which may result in build-up of the modified bioactive product within the cell. In most RiPP systems, this failsafe is ensured by the presence of the leader sequence in the processed substrate, as the bioactive natural product is only elaborated upon removal of the leader peptide by highly specific proteases, typically coupled with extracellular export.3 Seemingly, no such control point exists for McC, and the catalytic efficiencies of MccE (kcat/KM of 7.1 × 103 M−1 s−1)16 and MccF (kcat/KM of 3.95 × 104 M−1 s−1)17 for toxin inactivation may not be sufficient to overcome toxic buildup of 6 within the producing cell. Thus, McC biosynthesis may be regulated by other means.
Results and discussion
The initial characterization of intact McC purified from a producing strain showed that the molecule lacked a free α-amino group.18 Subsequent structural elucidation of the isolated product using 1H NMR was consistent with N-formylation of the initiator Met of the peptide conjugate 4.5 Moreover, systematic analysis of substrate scope of the YejABEF importer that internalizes intact McC into target cells demonstrated that 4 was a significantly better substrate than the peptide that lacked the N-formyl moiety 3.19 These data are consistent with two possibilities: (a) that only the N-formylated precursor 2 is used in the McC biosynthetic pathway, and/or (b) that N-formylation is necessary for toxin export out from producing cells.In order to distinguish between these two possibilities, we carried out HPLC analysis of the MccB adenylyltransferase-catalyzed reaction products using each 1 and 2 precursor peptides as substrates (Fig. 2A and B). Upon incubation of deformyl peptide 1 (50 μM) with ATP and sub-stoichiometric concentrations of MccB (1 μM), substantial amounts of the substrate remain even after 1 hour, consistent with observations in prior studies using the same desformyl substrate (Fig. 2A).10,20 In contrast, incubation of N-formylated peptide 2 (50 μM) with the enzyme resulted in the near complete conversion of the substrate after 10 minutes (Fig. 2B). To provide a quantitative assessment of the two substrates, we carried out Michaelis–Menten kinetics of MccB using either 1 or 2 as substrates (Fig. 2C and D).
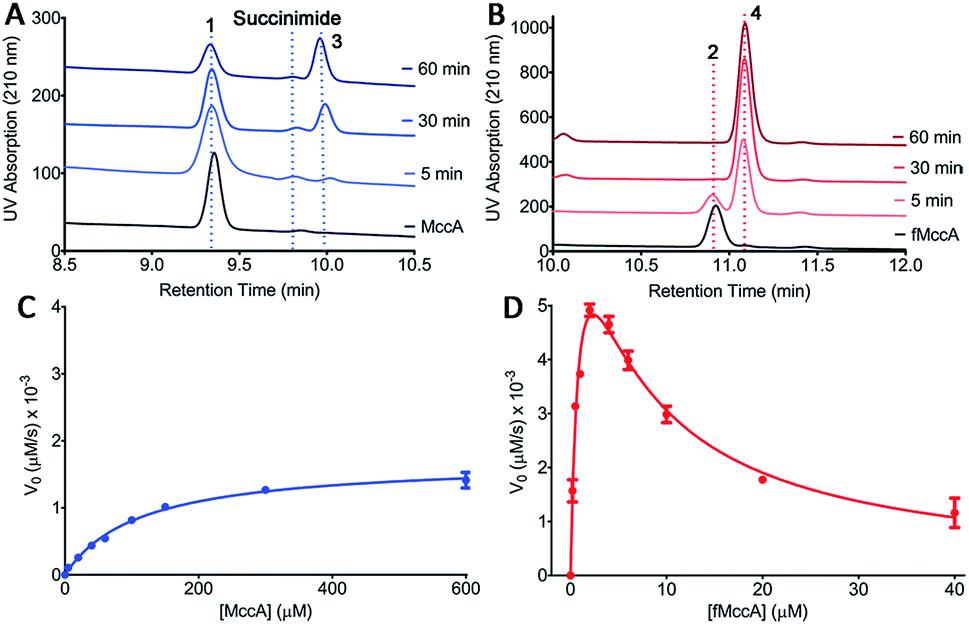 | ||
| Fig. 2 (A and B) HPLC analysis of the time-course of the MccB reaction progress using either (A) desformyl MccA 1 or (B) N-formylated MccA 2 as a substrate. (C and D) Michaelis–Menten kinetics curves for MccB with either (C) 1 or (D) 2 as a substrate. Error bars represent standard deviation of the mean (n = 3). | ||
Consistent with the chromatographic analysis, the catalytic efficiency of MccB using N-formylated 2 (kcat/KM of 3.6 × 104 M−1 s−1) was nearly 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times greater than that with the desformyl 1 peptide (kcat/KM of 3.0 M−1 s−1). Most notably, the kinetic data with N-formylated peptide 2 are consistent with strong substrate inhibition (Ki of ∼5.5 μM), as discussed below.
000 times greater than that with the desformyl 1 peptide (kcat/KM of 3.0 M−1 s−1). Most notably, the kinetic data with N-formylated peptide 2 are consistent with strong substrate inhibition (Ki of ∼5.5 μM), as discussed below.
We next sought to understand the molecular rationale underlying the observed four orders of magnitude difference in catalytic efficiency observed with the N-formylated 2 relative to desformyl 1 peptide. To this end, we determined the 1.6 Å resolution co-crystal structure of E. coli MccB in complex with ATP, MgCl2 and N-formylated MccA (2) (Fig. 3A; ESI Table S1†). In prior studies,20 the substrate peptide was directed away from the ATP-binding site. In contrast, our structure presents the MccA substrate bound to MccB in a productive manner consistent with the reaction scheme. Clear and continuous electron density can be observed for the peptide aspartamide-adenylate 4 (generated in situ) within the active site (Fig. 3B; ESI Fig. S1†).
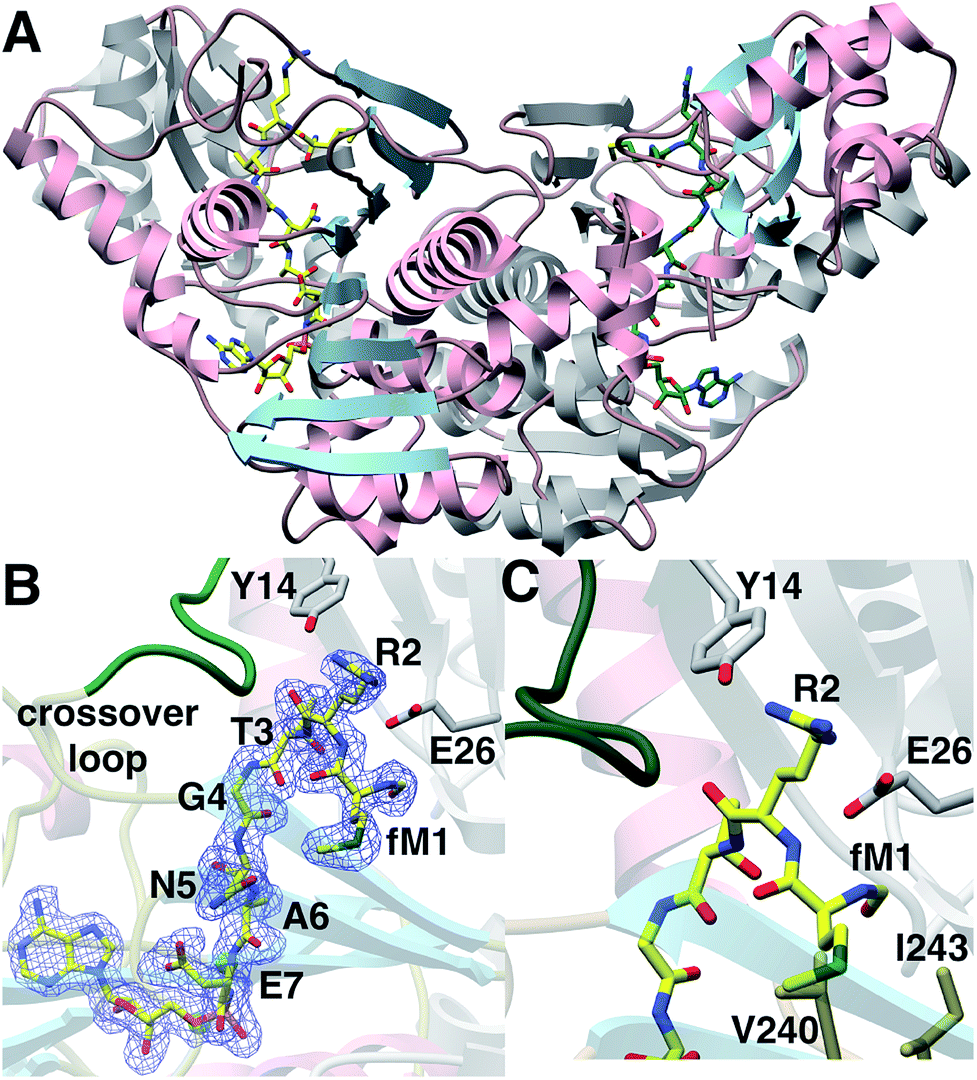 | ||
| Fig. 3 (A) Structure of MccB homodimer bound to product 4. One monomer is colored in gray and the other in pink and blue. (B) Coordinates of the MccB-4 complex superimposed on a difference Fourier map (Fobs − Fcalc) contoured at 2.5σ using phases from the final model with the coordinate of 4 removed prior to a round of simulated annealing refinement. (C) Close-up view of the N-formyl binding pocket of MccB. | ||
The N-formyl moiety is situated in a pocket formed, along one side, by Val240, and Ile243 of one MccB monomer and, on the other side, by Gly23 and Glu26 from the other monomer (Fig. 3C). The formyl oxygen is engaged with the backbone of Gly23 and the side chain of Glu26. Perhaps as a result of the productive binding mode for 4, electron density can also be observed of residues Lys262 through Lys271, which correspond to the “crossover loop” that connects the two active sites in homologous ubiquitin-like protein activating enzymes (E1) (ESI Fig. S2†). These newly observed contacts reveal that a number of residues located within the “crossover loop” engage in interactions with the MccA peptide.
In order to validate the interactions with product observed in the crystal structure, we analyzed the binding of wild-type MccB to fluorescein isothiocyanate (FITC) labeled peptide (2) that is not a substrate, yielding a Kd ∼ 3.6 μM (ESI Table S2 and Fig. S3 and S4†). Competition fluorescence polarization assays using unlabeled 2 yields a Ki ∼ 9 μM. We next tested a panel of variants of MccB with amino acid substitutions at both the N-formyl site, and at the crossover loop (Table 1; ESI Table S3 and Fig. S5–S7†). Removal of part of the N-formyl binding pocket in the Ile243 → Ala variant results in near complete loss of binding to 2. Likewise, Ala mutation at residues along the Lys262–Lys271 crossover loop severely compromises binding, as is seen in Val264Ala (Kd ∼ 79 μM), and Tyr268Ala (Kd ∼ 18 μM) MccB derivatives, with substitutions of residues that line the peptide-binding pocket (ESI Fig. S2†). These data affirm the role of the crossover loop in reinforcing productive binding of the MccA substrate. The specificity of complementary interactions between a given E1 activating enzyme and its cognate ubiquitin-like protein (Ubl) substrate is a hallmark of the Ubl activation.21–24
| Variant | K d (μM) | IC50 (μM) | K i (μM) | [Protein] (μM) |
|---|---|---|---|---|
| Y14A | 143.40 ± 35.90 | NA | NA | NA |
| E26A | 91.11 ± 4.70 | NA | NA | NA |
| E26D | 539.40 ± 358.40 | NA | NA | NA |
| E26Q | 16.93 ± 3.11 | 59.34 ± 8.75 | 13.46 | 30 |
| S212A | 2.06 ± 0.05 | 6.99 ± 1.02 | 2.87 | 2 |
| F217A | 8.85 ± 0.88 | 32.07 ± 7.36 | 11.86 | 10 |
| I243A | >800 | NA | NA | NA |
| V264A | 79.17 ± 12.81 | 682.50 ± 61.55 | 399.29 | 50 |
| Y268A | 17.57 ± 1.80 | 93.35 ± 15.65 | 37.28 | 20 |
| Y268F | 4.15 ± 0.37 | 36.83 ± 3.18 | 15.14 | 5 |
| H333A | 5.01 ± 0.37 | 26.55 ± 2.98 | 11.62 | 5 |
In order to gain additional inferences regarding MccB substrate specificity based on homologous Ubl conjugation systems, we carried out comparative analysis with the structure of eukaryotic E1s, namely those of Uba1 in complex with ubiquitin, Sae2 in complex with SUMO, and Uba3 in complex with NEDD8 (ESI Fig. S2†).21–24 In each of these structures, the residue corresponding to Arg72 in ubiquitin affects substrate recognition via complementary interactions with residues in the crossover loop of the cognate E1.24 In our co-crystal structure, Arg2* (the asterisk designates the numbering of residues in the substrate) from the N-formylated precursor 4 is directed towards the peptide binding clamp in the adjacent monomer, where Tyr14 forms platform along one edge, and Glu26 wedges in between the N-formyl oxygen of fMet1* and the guanidino group of Arg2* (Fig. 3C). The importance of these residues is highlighted by the nearly 40-fold increase in Kd (∼143 μM) for Tyr14 → Ala, and 25-fold increase in Kd (∼91 μM) for Glu26 → Ala MccB (Table 1; ESI Fig. S5†).
As our kinetic measurements of MccB suggested substrate inhibition (Ki of ∼5.5 μM) by the N-formylated 2, but not with the desformyl 1 peptide, we analyzed differences in binding to MccB in greater detail using competition fluorescence polarization (FP) assays with FITC-labeled precursor peptides. Label-free N-formylated 2 was able to efficiently compete with the labeled peptide (Ki ∼ 9 μM) (Fig. 4A). In contrast, FITC-labeled desformyl 1 only weakly bound to MccB with a Kd of ∼378.7 μM (ESI Fig. S4 and S8†). Notably, desformyl 1 could not displace bound FITC-labeled N-formylated 2, even at concentrations greater than 5 mM (Fig. 4A), while N-formylated 2 was able to displace FITC-labeled desformyl 1 with a Ki ∼ 52 μM (ESI Fig. S9†). Hence, N-formylation of the MccA precursor enables substrate inhibition of MccB.
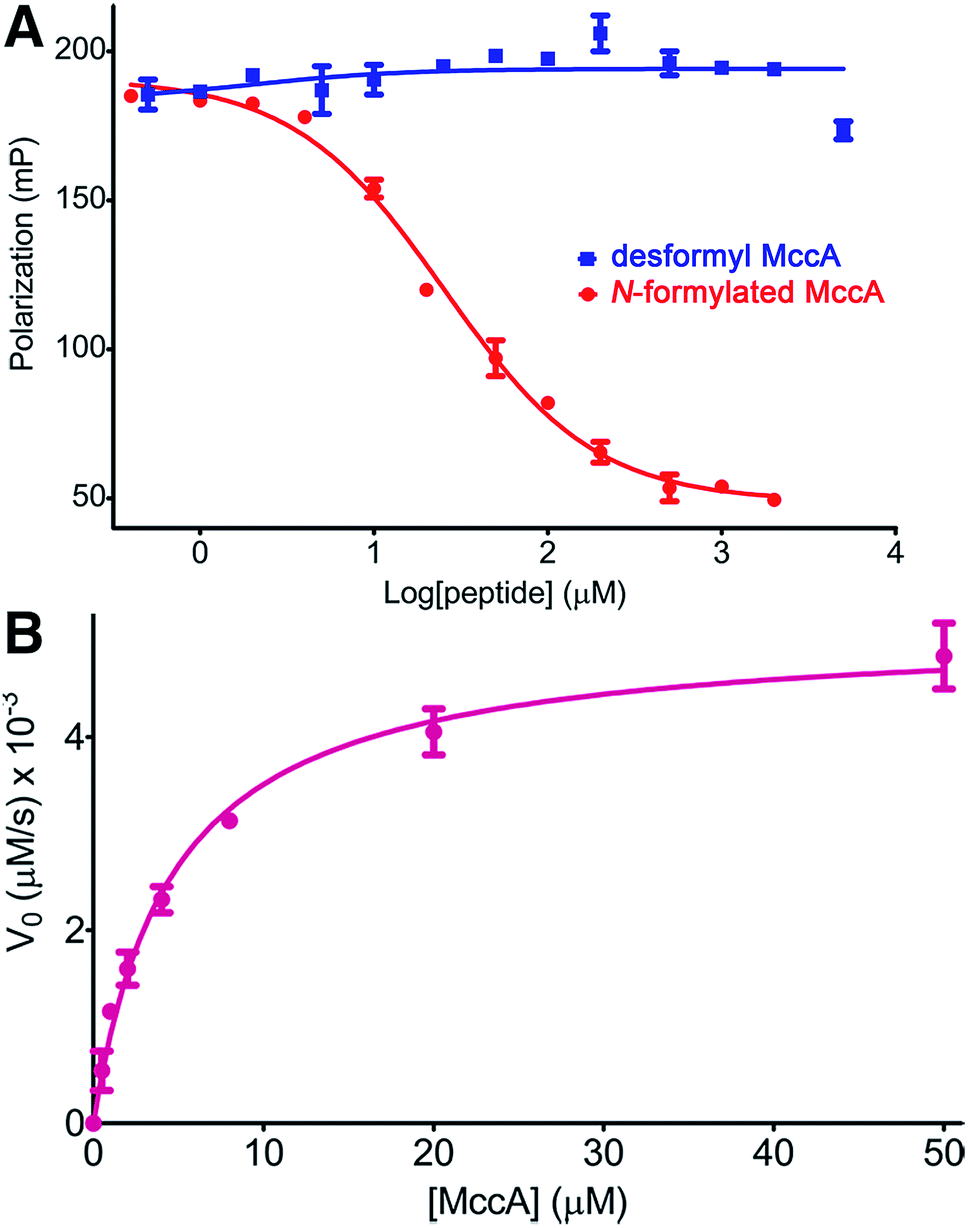 | ||
| Fig. 4 (A) Competition FP binding curves of MccB using FITC labeled N-formylated 2 as probe. (B) Michaelis–Menten kinetics curve for Glu26 → Ala MccB with N-formylated 2 as substrate. Error bars represent standard deviation of the mean (n = 3). | ||
Based on our structural data, we hypothesized that the interactions established between Glu26 in MccB and the fMet1* and Arg2* of 2 may contribute to substrate inhibition. In order to test this theory, we carried out Michaelis–Menten kinetics of Glu26 → Ala MccB using N-formylated 2 as a substrate. This variant demonstrated a 33-fold decrease in catalytic efficiency (kcat/KM of 1.1 × 103 M−1 s−1) (Fig. 4B). More importantly, no substrate inhibition by 2 was observed with Glu26 → Ala MccB, confirming that the interactions between Glu26 of MccB, and fMet1* and Arg2* of 2 play a role in mediating substrate inhibition (Fig. 3C). Hence, Arg2* plays a role analogous to that of Arg72 in ubiquitin in establishing the specificity of MccB with the N-formylated MccA precursor peptide.
Conclusions
The inhibition of enzymatic turnover at high concentrations of substrate is prevalent throughout primary metabolism, and occurs across various classes of catalysts.25 Our studies highlight a rare example of the use of substrate inhibition in a natural product biosynthetic pathway. We have previously demonstrated that an intrinsic transcription terminator located between the mccA and mccB, coupled with a ribosome-mediated stabilization of the monocistronic mccA transcript results in a 20-fold excess of the precursor peptide, relative to the MccB adenylase.26 The role of the interaction between Glu26 and fMet1* and Arg2* of 2 in mediating substrate inhibition is notable as similar interactions are conserved across various classes of E1-like activating enzyme and cognate substrates. When the MccC-mediated efflux becomes saturated, production of excess product 4 may result in processing and cellular toxicity, given that the immunity determinants have catalytic efficiencies in the order of ∼104 M−1 s−1. Substrate inhibition limits this unwanted situation, and excess production with most RiPP natural products cellular toxicity is minimal, as the leader peptide, required for post-translational modifications, must be removed for bioactivity to appear. However, this is not the case with McC as intracellular processing of endogenous 4 will liberate active toxin 6 within the producing organism. Thus, inhibition of MccB by substrate 2 may restrict overproduction of 4 providing an expedient means to avert the unwanted buildup of a toxic metabolite within the producing organism at conditions of excess precursor peptide synthesis or decreased export of mature compound.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Keith Brister and colleagues for facilitating data collection at LS-CAT (Argonne National Labs, IL). This work was Supported by NIH AI117210 (to K. S. and S. K. N.), and from Skoltech and the Russian Science Foundation grant 16-14-10356 (to S. D.).References
- K. Severinov and S. K. Nair, Future Microbiol., 2012, 7, 281–289 CrossRef CAS PubMed.
- K. Severinov, E. Semenova, A. Kazakov, T. Kazakov and M. S. Gelfand, Mol. Microbiol., 2007, 65, 1380–1394 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- J. E. Gonzalez-Pastor, J. L. San Millan, M. A. Castilla and F. Moreno, J. Bacteriol., 1995, 177, 7131–7140 CrossRef CAS PubMed.
- J. I. Guijarro, J. E. Gonzalez-Pastor, F. Baleux, J. L. San Millan, M. A. Castilla, M. Rico, F. Moreno and M. Delepierre, J. Biol. Chem., 1995, 270, 23520–23532 CrossRef CAS PubMed.
- T. Kazakov, A. Metlitskaya and K. Severinov, J. Bacteriol., 2007, 189, 2114–2118 CrossRef CAS PubMed.
- M. Novikova, A. Metlitskaya, K. Datsenko, T. Kazakov, A. Kazakov, B. Wanner and K. Severinov, J. Bacteriol., 2007, 189, 8361–8365 CrossRef CAS PubMed.
- T. Kazakov, G. H. Vondenhoff, K. A. Datsenko, M. Novikova, A. Metlitskaya, B. L. Wanner and K. Severinov, J. Bacteriol., 2008, 190, 2607–2610 CrossRef CAS PubMed.
- A. Metlitskaya, T. Kazakov, A. Kommer, O. Pavlova, M. Praetorius-Ibba, M. Ibba, I. Krasheninnikov, V. Kolb, I. Khmel and K. Severinov, J. Biol. Chem., 2006, 281, 18033–18042 CrossRef CAS PubMed.
- R. F. Roush, E. M. Nolan, F. Lohr and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 3603–3609 CrossRef CAS PubMed.
- O. Bantysh, M. Serebryakova, K. S. Makarova, S. Dubiley, K. A. Datsenko and K. Severinov, mBio, 2014, 5, e01059-14 CrossRef PubMed.
- A. Kulikovsky, M. Serebryakova, O. Bantysh, A. Metlitskaya, S. Borukhov, K. Severinov and S. Dubiley, J. Am. Chem. Soc., 2014, 136, 11168–11175 CrossRef CAS PubMed.
- A. Metlitskaya, T. Kazakov, G. H. Vondenhoff, M. Novikova, A. Shashkov, T. Zatsepin, E. Semenova, N. Zaitseva, V. Ramensky, A. Van Aerschot and K. Severinov, J. Bacteriol., 2009, 191, 2380–2387 CrossRef CAS PubMed.
- M. Novikova, T. Kazakov, G. H. Vondenhoff, E. Semenova, J. Rozenski, A. Metlytskaya, I. Zukher, A. Tikhonov, A. Van Aerschot and K. Severinov, J. Biol. Chem., 2010, 285, 12662–12669 CrossRef CAS PubMed.
- A. Tikhonov, T. Kazakov, E. Semenova, M. Serebryakova, G. Vondenhoff, A. Van Aerschot, J. S. Reader, V. M. Govorun and K. Severinov, J. Biol. Chem., 2010, 285, 37944–37952 CrossRef CAS PubMed.
- V. Agarwal, A. Metlitskaya, K. Severinov and S. K. Nair, J. Biol. Chem., 2011, 286, 21295–21303 CrossRef CAS PubMed.
- V. Agarwal, A. Tikhonov, A. Metlitskaya, K. Severinov and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4425–4430 CrossRef CAS PubMed.
- J. F. Garcia-Bustos, N. Pezzi and C. Asensio, Biochem. Biophys. Res. Commun., 1984, 119, 779–785 CrossRef CAS PubMed.
- G. H. Vondenhoff, B. Blanchaert, S. Geboers, T. Kazakov, K. A. Datsenko, B. L. Wanner, J. Rozenski, K. Severinov and A. Van Aerschot, J. Bacteriol., 2011, 193, 3618–3623 CrossRef CAS PubMed.
- C. A. Regni, R. F. Roush, D. J. Miller, A. Nourse, C. T. Walsh and B. A. Schulman, EMBO J., 2009, 28, 1953–1964 CrossRef CAS PubMed.
- D. T. Huang, H. W. Hunt, M. Zhuang, M. D. Ohi, J. M. Holton and B. A. Schulman, Nature, 2007, 445, 394–398 CrossRef CAS PubMed.
- I. Lee and H. Schindelin, Cell, 2008, 134, 268–278 CrossRef CAS PubMed.
- L. M. Lois and C. D. Lima, EMBO J., 2005, 24, 439–451 CrossRef CAS PubMed.
- B. A. Schulman and J. W. Harper, Nat. Rev. Mol. Cell Biol., 2009, 10, 319–331 CrossRef CAS PubMed.
- M. C. Reed, A. Lieb and H. F. Nijhout, BioEssays, 2010, 32, 422–429 CrossRef CAS PubMed.
- I. Zukher, M. Novikova, A. Tikhonov, M. V. Nesterchuk, I. A. Osterman, M. Djordjevic, P. V. Sergiev, C. M. Sharma and K. Severinov, Nucleic Acids Res., 2014, 42, 11891–11902 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, X-ray crystallographic data, and FP biding curves. See DOI: 10.1039/c8sc03173h |
| This journal is © The Royal Society of Chemistry 2019 |