
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsequences of exchange-site heterogeneity and dynamics on the UV-visible spectrum of Cu-exchanged SSZ-13†
Hui
Li‡
 a,
Christopher
Paolucci‡
ab,
Ishant
Khurana
c,
Laura N.
Wilcox
c,
Florian
Göltl
d,
Jonatan D.
Albarracin-Caballero
c,
Arthur J.
Shih
c,
Fabio H.
Ribeiro
c,
Rajamani
Gounder
*c and
William F.
Schneider
*a
a,
Christopher
Paolucci‡
ab,
Ishant
Khurana
c,
Laura N.
Wilcox
c,
Florian
Göltl
d,
Jonatan D.
Albarracin-Caballero
c,
Arthur J.
Shih
c,
Fabio H.
Ribeiro
c,
Rajamani
Gounder
*c and
William F.
Schneider
*a
aDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, 182 Fitzpatrick Hall, Notre Dame, IN 46556, USA. E-mail: wschneider@nd.edu
bDepartment of Chemical Engineering, University of Virginia, 102 Engineer's Way, Charlottesville, VA 22904, USA
cCharles D. Davidson School of Chemical Engineering, Purdue University, 480 Stadium Mall Drive, West Lafayette, IN 47907, USA. E-mail: rgounder@purdue.edu
dDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
First published on 28th December 2018
Abstract
The speciation and structure of Cu ions and complexes in chabazite (SSZ-13) zeolites, which are relevant catalysts for nitrogen oxide reduction and partial methane oxidation, depend on material composition and reaction environment. Ultraviolet-visible (UV-Vis) spectra of Cu-SSZ-13 zeolites synthesized to contain specific Cu site motifs, together with ab initio molecular dynamics and time-dependent density functional theory calculations, were used to test the ability to relate specific spectroscopic signatures to specific site motifs. Geometrically distinct arrangements of two framework Al atoms in six-membered rings are found to exchange Cu2+ ions that become spectroscopically indistinguishable after accounting for the finite-temperature fluctuations of the Cu coordination environment. Nominally homogeneous single Al exchange sites are found to exchange a heterogeneous mixture of [CuOH]+ monomers, O- and OH-bridged Cu dimers, and larger polynuclear complexes. The UV-Vis spectra of the latter are sensitive to framework Al proximity, to precise ligand environment, and to finite-temperature structural fluctuations, precluding the precise assignment of spectroscopic features to specific Cu structures. In all Cu-SSZ-13 samples, these dimers and larger complexes are reduced by CO to Cu+ sites at 523 K, leaving behind isolated [CuOH]+ sites with a characteristic spectroscopic identity. The various mononuclear and polynuclear Cu2+ species are distinguishable by their different responses to reducing environments, with implications for their relevance to catalytic redox reactions.
1 Introduction
Copper ions exchanged onto zeolites are implicated as active sites for the selective catalytic reduction of nitrogen oxides,1–10 oxidation of NO to NO2,11,12 decomposition of NO and N2O,13–16 and partial methane oxidation to methanol.17–33 These Cu ions are associated with charge-compensating AlO4− tetrahedral sites that are distributed throughout the zeolite lattice. Because these Al substitutions are not ordered under typical zeolite synthesis conditions, a given framework and macroscopic Si/Al ratio will present a heterogeneous distribution of microscopic Al site ensembles, each of which provides a distinct exchange environment for a Cu ion. The precise Cu species and their relative densities in a given material are thus functions of framework topology,34–37 of the density and underlying distribution of framework Al (Si/Al), of the Cu loading (Cu/Al), and even of the protocols used to introduce Cu onto the zeolite supports.38–44 Further, multiple Cu ion site motifs may be of similar free energy at a given Al site or ensemble, and at finite temperatures these motifs may interchange at timescales relevant to observation or to catalytic turnover.45–47 Given these many sources of structural diversity, assignment of spectroscopic features to specific Cu motifs in a heterogeneous solid is a non-trivial endeavor, but a critical one to make connections between local structure and catalytic function.One strategy to reduce this complexity is to intentionally synthesize zeolites with framework Al distributions that present one or a few distinct Al site ensembles. This strategy is particularly promising for zeolites of relatively high symmetry, such as the chabazite (SSZ-13) framework that is composed of a single symmetry-distinct tetrahedral site. SSZ-13 samples synthesized using only organic N,N,N-trimethyl-1-adamantylammonium (TMAda+) structure-directing cations nominally contain only isolated framework Al sites,39 and are found to contain predominantly [CuOH]+ ions after aqueous Cu ion exchange and high temperature (>473 K) oxidation, based on titrimetric analysis and vibrational and X-ray spectroscopies.38 In contrast, SSZ-13 samples synthesized in the presence of TMAda+ and Na+ as co-structure-directing cations39 contain detectable fractions of paired Al sites in 6-membered rings (6MR). After Cu ion exchange and high temperature oxidative treatment, these paired Al sites are observed to preferentially host Cu2+ ions (Z2Cu) before isolated Al sites are occupied by [CuOH]+ ions (ZCuOH).38 Z2Cu and ZCuOH are expected to contribute differently to ultraviolet-visible (UV-Vis) absorption spectra based on ligand-field arguments and density functional theory (DFT) calculations, consistent with d–d transition and ligand-to-metal charge transfer (LMCT) features in experimentally-measured UV-Vis spectra that are observed to change with Cu content in Cu-SSZ-13 samples.6,48–50
These single Cu species may coexist with dimeric or higher nuclearity Cu clusters. Oxygen-bridged Cu dimers are well established to be present and quantifiable with CO temperature programmed reduction,15 to be plausible active sites for NO and N2O decomposition14,15,51–53 and partial CH4 oxidation in Cu-exchanged zeolites,17,30,33,54–62 including Raman63,64 and UV-Vis14,55 spectroscopic observations and CO temperature programmed reduction to quantify such sites. In Cu-SSZ-13 samples of certain composition (Si/Al = 5, Cu/Al = 0.3–1.6), oxygen-bridged Cu dimers and larger Cu oxide aggregates are detected by X-ray spectroscopy and are the dominant active sites for NO oxidation to NO2 under dry conditions.11 UV-Vis spectra of certain Cu-SSZ-13 samples (Si/Al = 13, Cu/Al = 0.45) following high temperature O2 treatment show features consistent with those for oxygen-bridged Cu dimers,48 and these features disappear upon reduction with CH4,18,50 implicating them as active sites for partial methane oxidation. The precise relationship between sample composition and treatment history, and the numbers and structures of Cu dimers or larger aggregates formed, however, remains less well resolved than such relationships for monomeric Cu sites in Cu-SSZ-13.
Here, we report UV-Vis spectra of model Cu-SSZ-13 zeolites prepared to contain predominantly Z2Cu or ZuOH sites, by virtue of their different framework Al arrangements and elemental compositions. We use supercell time-dependent DFT (TD-DFT) calculations to correlate observed UV-Vis spectral features with specific Cu motifs. We find that spectra computed for single, static, minimum energy structures are in poor agreement with experimental observation, because Cu ion dynamics have a significant impact on computed spectral features even at ambient temperature. We construct synthetic spectra by averaging over configurations visited during finite-temperature ab initio molecular dynamics (AIMD) simulations and show that these dynamically averaged spectra correspond closely with experimental observation. Further, experimental and simulated spectra are compared to identify features associated with a confounding subset of Cu dimers or larger aggregates in samples prepared to contain predominantly [CuOH]+ species. These results resolve several inconsistencies in spectral and site assignments reported in Cu-exchanged zeolites.
2 Results
2.1 Z2Cu
First, UV-Vis spectra were collected on a Cu-SSZ-13 sample that contains only Z2Cu sites.38,65 This sample was prepared by starting from an SSZ-13 sample (Si/Al = 5) crystallized using a mixture of TMAda+ and Na+ cations to contain a finite and detectable fraction of paired Al sites,39 followed by aqueous ion exchange with a cupric nitrate solution to achieve a composition (Cu/Al = 0.21) demonstrated previously to contain only isolated Z2Cu sites through X-ray absorption spectroscopy, Brønsted acid site exchange stoichiometries with Cu2+ and Co2+ (2H+ replaced per Cu2+ or Co2+), and IR spectroscopy38 (sample preparation details in ESI Section S2.1 and elemental analysis in ESI Table S4†). This Cu-SSZ-13 sample was treated in flowing dry air to 673 K for 2 h, cooled to 300 K, and UV-Vis spectra were collected from 7000 to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1. Spectra are reported in quasi-absorption (K–M) units in Fig. 1a, and show a broad d–d transition feature from about 8000 to 16000 cm−1 characteristic of a d9 Cu2+ ion and a broad ligand-to-metal charge transfer (LMCT) feature from about 30000 to 50000 cm−1. The spectrum is consistent with that previously reported for a Cu-SSZ-13 sample after high temperature oxidative treatment.6
000 cm−1. Spectra are reported in quasi-absorption (K–M) units in Fig. 1a, and show a broad d–d transition feature from about 8000 to 16000 cm−1 characteristic of a d9 Cu2+ ion and a broad ligand-to-metal charge transfer (LMCT) feature from about 30000 to 50000 cm−1. The spectrum is consistent with that previously reported for a Cu-SSZ-13 sample after high temperature oxidative treatment.6
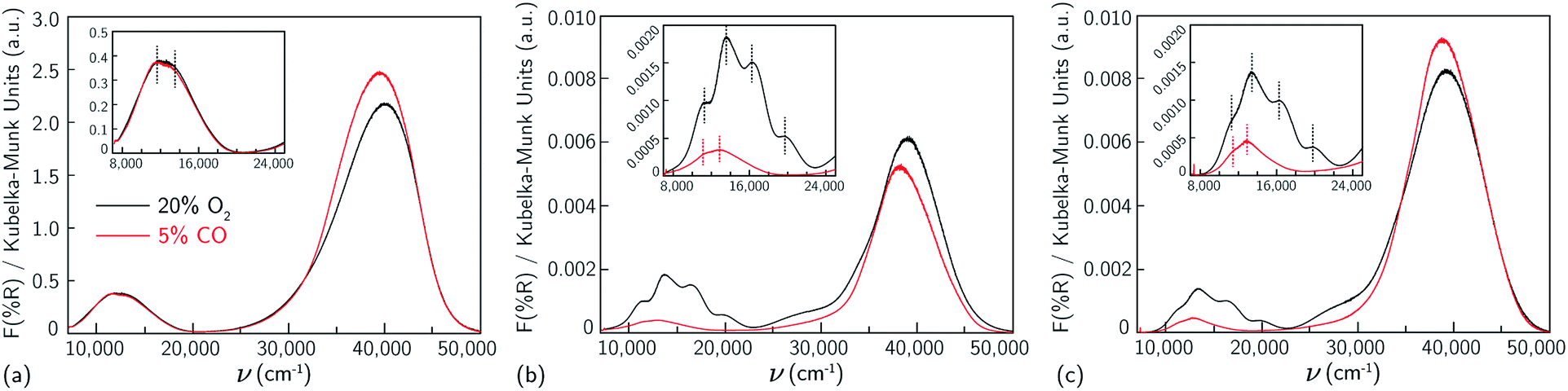 | ||
| Fig. 1 UV-visible spectra collected at 300 K of samples containing predominantly (a) Z2Cu (Si/Al = 5, Cu/Al = 0.21), (b) ZCuOH (Si/Al = 15, Cu/Al = 0.24) and (c) ZCuOH (Si/Al = 15, Cu/Al = 0.15), after 20% O2 treatment at 673 K (black), and 5% CO treatment at 523 K (red). Insets show magnification of the d–d transition region. | ||
The Cu2+ ions in this zeolite sample are associated with ensembles of two Al centers separated by two or one intervening Si tetrahedral (T-) sites in the same six-membered ring (6MR), which we term “para” and “meta” respectively. We used a triclinic 12 T-site SSZ-13 supercell described elsewhere11,66 to represent each ensemble (Fig. 2). In each case, a single Cu ion was placed within the 6MR and the structure annealed at 673 K for 150 ps using thermostated AIMD (computational details in ESI Section S1†). 400 equally spaced configurations were extracted from the trajectories and relaxed. In each Al ensemble, all configurations relaxed to one of three energy minima shown in Fig. 2a and e. These three minima differ in the combination of Al-adjacent and non-Al-adjacent framework oxygen (Of) that comprise the first Cu coordination sphere. The framework distorts to accommodate these different Cu-containing minima, behavior consistent with structural distortions reported previously in calculations of metal-exchanged MFI,67,68 MOR,67,69 and FER,70 and inferred from the appearance of two T–O–T deformation bands (900, 950 cm−1) in DRIFTS spectra after Cu exchange into SSZ-13.71 Despite these structural differences, the three para configurations differ in energy by less than 5 and meta by less than 20 kJ mol−1 (Fig. 2a, e, and also ESI Table S1†). The lowest-energy para and meta configurations differ in absolute energy by 21 kJ mol−1 (ESI Table S1†).
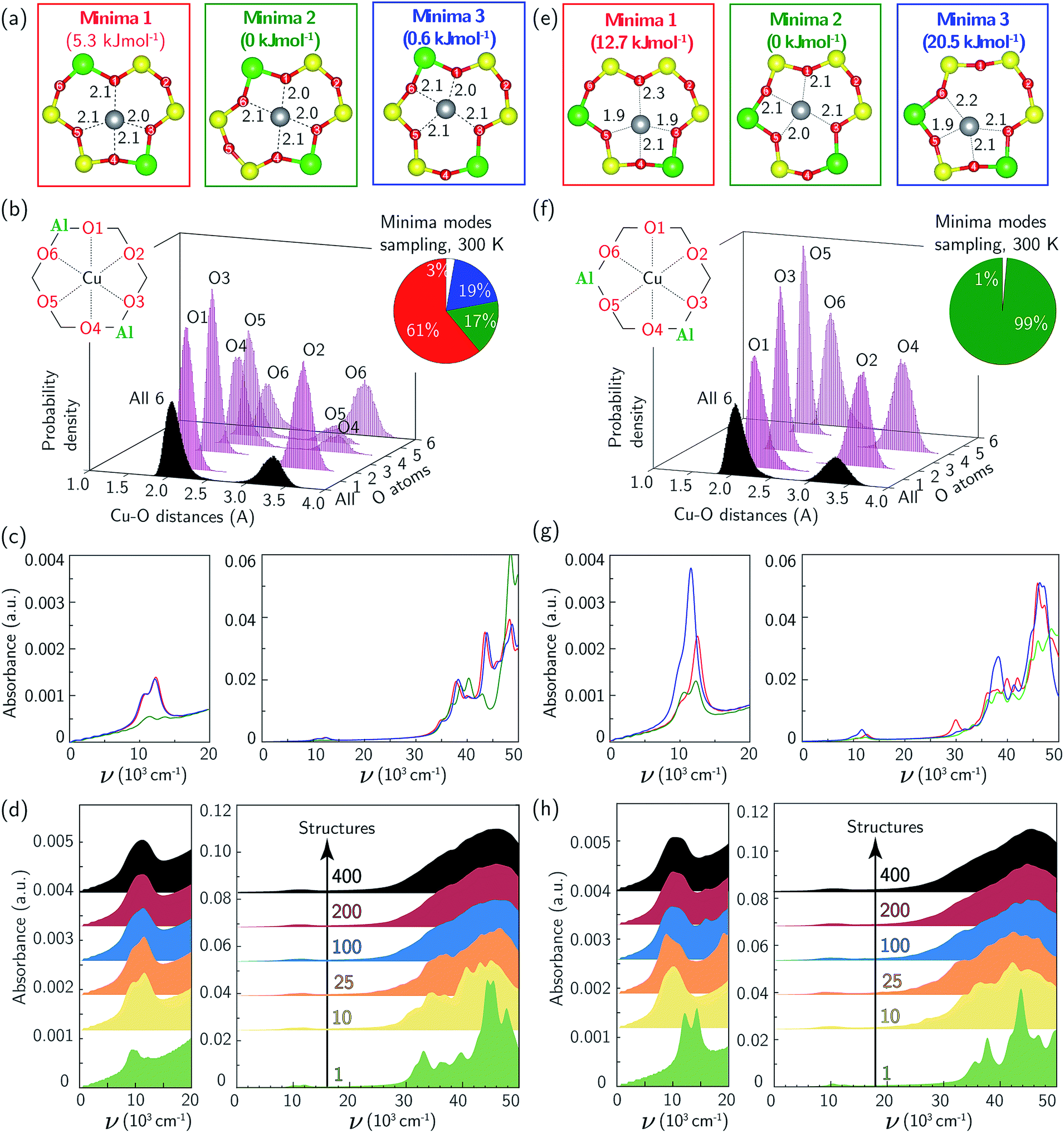 | ||
| Fig. 2 (a) DFT-computed local Cu environment in three Z2Cu para minima. (b) Individual and aggregate para Z2Cu Cu–Of distance histograms collected over 150 ps 300 K AIMD. Inset indicates percent time spent in each minimum. (c) TD-DFT-computed UV-Vis spectra of three Z2Cu para minima, color-coded by (a). (d) UV-Vis spectra averaged over 1 (green), 10 (yellow), 25 (orange), 100 (blue), 200 (red), and 400 (black) para Z2Cu snapshots equally spaced in time along the 150 ps trajectory. (e–h) Corresponding results for Z2Cu meta configuration. | ||
We performed additional AIMD at 300 K to gain insight into variations in Cu coordination environment at finite temperature38,72 that might influence the observed spectra. Fig. 2b reports histograms of distances between Cu and each of the six 6MR Of, collected at 0.6 fs intervals over the 150 ps simulation of para Z2Cu. Distances segregate into two groups centered at 2.1 and 3.3 Å, corresponding to Cu-coordinated and free Of, each group having widths >0.5 Å (Table S2†) that arise from finite temperature fluctuations of the lattice and Cu. The unimodal O1, O2, and O3 and bimodal O4, O5, and O6 distributions are due to transitions between the three different minimum energy configurations of Fig. 2a. Inspection of the trajectory shows that transitions occur by extension of the four Cu–Of bonds, distortion of the lattice, and relaxation into an adjacent minimum. We used these observations to categorize each AIMD frame into one of the three minima or into an intermediate state (example structure in ESI Fig. S2†), based on the identity and number of Of within 2.6 Å of Cu, chosen to represent the upper bound of a Cu–O coordination and shown in the pie chart inset to Fig. 2b. All three minima are visited, and the fractions of time spent in each is generally consistent with the relative energies of the corresponding minima. A non-negligible fraction of time is also spent outside any minimum.
Analogous results for meta Z2Cu are shown in Fig. 2f. Again, Cu–Of distances segregate into the same two groups, and the aggregate histogram of all Cu–Of distances have identical means and standard deviations in the para and meta cases. The individual Cu–Of histograms are all unimodal. The large energy difference between minima guarantees that meta Z2Cu remains in the lowest energy configuration throughout the simulation.
We used TD-DFT to compute frequency-dependent dielectric tensors and corresponding optical absorption spectra of the six minima. The computational details and codes for the VASP inputs and subsequent analysis are provided in ESI Section S1.2† and also on the external Zenodo repository (DOI: 10.5281/zenodo.1473128). Computed spectra are shown in Fig. 2c and g, reported in arbitrary K–M units. All six spectra exhibit features in the d–d and LMCT regions not evident in the experimental spectrum in Fig. 1a. Coordinatively similar (as measured from Cu–O distances and O–Cu–O angles) para minima 1 and 3 and meta minimum 2 exhibit equivalent spectra. Absolute intensities are greatest for lowest symmetry and least for highest symmetry minima.73 Neither any individual spectrum nor a Boltzmann weighting of all spectra at 300 K (shown in ESI Fig. S3†) recovers the experimental spectrum in Fig. 1a.
To simulate the effects of these geometric variations on observed spectra, we computed the absorption spectra of 400 equally spaced snapshots from the 300 K AIMD trajectories (ESI Fig. S4a (para) and b (meta)†). Individual spectra are sensitive to local structure. The d–d transitions shift generally to higher frequency with decreasing mean Cu–Of distance (ESI Fig. S5†), consistent with behavior expected from ligand field arguments. The LMCT features do not simply correlate with the Cu–Of distances. Fig. 2d and h show the results of averaging increasing numbers of spectra computed from configurations extracted with equal spacing along the AIMD trajectories. Sharp features in both the d–d and LMCT regions become broadened upon averaging an increasing number of spectra from 1 to 200 structures and converge at approximately 200 structures, as evidenced by the small changes resulting from further averaging up to 400 structures. In contrast to spectra computed from the minimum energy structures, the final averaged meta and para UV-Vis spectra are indistinguishable and consistent with experimental observations in Fig. 1a.
2.2 ZCuOH
For comparison to the sample containing predominantly Z2Cu sites, we synthesized different Cu-SSZ-13 samples that contain predominantly the ZCuOH site motif that results when Cu2+ ions are exchanged near isolated framework Al sites. We previously reported that crystallization of SSZ-13 zeolites in the presence of only TMAda+ cations produces a material with predominantly isolated framework Al, reflected in the inability to exchange divalent Co2+ cations.39,40 A sample of SSZ-13 (Si/Al = 15) was synthesized using this method and exchanged with different amounts of Cu2+ (Cu/Al = 0.15, 0.24) to generate two Cu-SSZ-13 samples of different ZCuOH density (sample preparation details and elemental analysis in ESI Section S2.1 and Table S4†).After Cu2+ ion exchange, each model ZCuOH sample was treated in flowing oxygen at 673 K (20% O2, balance He) for 120 min (details in ESI Section S2.2†). UV-Vis spectra of these Cu-SSZ-13 samples collected at 300 K are shown in Fig. 1b and c. Spectra of both samples show absorbance features in the 8000 to 22000 cm−1 region that are centered around ∼ 11059, 13593, 16379, and 20077 cm−1, and a shoulder in the 24000 to 30000 cm−1 range, and these features are higher in intensity for the sample with higher Cu content. These spectra are similar to those reported following similar pretreatments of Cu-SSZ-13 materials that are expected to contain predominantly ZCuOH species,18,48,50 by virtue of the Na+-free synthesis methods used to prepare the parent SSZ-13 sample. In contrast, these spectra are markedly different from those reported on materials expected to only contain Z2Cu species (Korhonen et al.6 and Fig. 1a). The four d–d transitions have different relative intensities in the two Cu-SSZ-13 samples shown in Fig. 1b and c. Literature Cu-SSZ-13 samples prepared to contain ZCuOH sites and exposed to the same O2 pre-treatment also share the same four d–d transitions but again with different relative intensities.18,48,50 We conclude that a mononuclear ZCuOH species cannot be solely responsible for the quadruplet feature. The sample-dependent variation suggests the presence of additional multinuclear ZCuOH-derived species with structures and populations that depend on synthesis, treatment and composition.
Da Costa et al.15 reported that CO reduces multinuclear Cu-oxo species in Cu-ZSM-5 to isolated Cu+ (d10) ions that do not exhibit d–d transitions. Similarly, we hypothesize that multinuclear Cu-oxo species present in Cu-SSZ-13 samples after treatment in O2 at 673 K will be reduced by CO at 523 K, leaving behind only isolated ZCuOH species and any residual Z2Cu sites. Model Z2Cu and ZCuOH samples were held in a flowing stream of 5% CO at 523 K until no further changes in UV-Vis spectra were observed (details in ESI Section S2.4†), prior to sealing the UV-Vis cell and cooling to 300 K to record the spectra shown in Fig. 1, an approach similar to that applied by Ipek et al. to Cu-SSZ-13 samples containing mixtures of Z2Cu and ZCuOH sites.18 As expected, no changes were observed to the d–d transition region in the spectrum of the Z2Cu sample upon CO exposure (Fig. 1a). In sharp contrast, the d–d features at 16379 and 20077 cm−1 and LMCT transition at 27000 cm−1 in the spectra of both ZCuOH samples (Fig. 1b and c) disappeared after exposure to CO, and features at 11059 and 13593 cm−1 were shifted to 11350 and 13000 cm−1 and decrease markedly in absorbance. Despite differences in the d–d quadruplet feature intensity that are detectable after high temperature O2 treatment, the d–d transition features of both ZCuOH samples become similar after CO treatment. These findings indicate that not all Cu2+ sites in Cu-SSZ-13 are reducible to Cu+ in the presence of CO, that the Cu2+ sites remaining after CO reduction are similar for both samples (Fig. 1b and c, red), and that these signatures are of isolated ZCuOH sites.
We used the same triclinic supercell to describe a [CuOH]+ ion-exchanged near an isolated Al. Each T-site in the chabazite lattice is common to two 8MR, one 6MR, and three 4MR. We used 473 K AIMD and geometry optimizations to compare the energies of the [CuOH]+ ion in each of these orientations. The two 8MR orientations are isoenergetic and the Cu–X (X = Si, Al, O) radial distribution function (RDF) computed from their AIMD trajectories are identical (ESI Fig. S6†). From a nudged elastic band calculation, the two 8MR minima are separated by a 63 kJ mol−1 barrier (ESI Fig. S7†). Similar calculations with the [CuOH]+ ion directed into a 6MR and 4MR result in configurations 15 and 45 kJ mol−1 higher in energy. We thus expect a [CuOH]+ ion to adopt and remain in one of the 8MR orientations at typical conditions of observation here.
Again to explore the consequence of ion dynamics on spectroscopy, we performed additional AIMD simulations at 300 K for 150 ps on a [CuOH]+ ion in one of the 8MR orientations. During the course of the simulation the Cu ion remained coordinated to the same two Of, bond distances fluctuated, and the OH ligand rotated between Cu–OH rotational conformers twice. Fig. 3b reports histograms of the two Cu–Of and Cu–OH distances. The Cu–OH bond is shorter and has a narrower distribution than the Cu–Of bond. The Cu–Of mean distances are slightly shorter than those from the Z2Cu simulations while the standard deviations are the same as the Z2Cu trajectories (ESI Table S2†). Thus, the coordination environment around ZCuOH varies less than Z2Cu. The inset to Fig. 3b reports the fraction of the trajectory spent in each of the two rotational conformations.
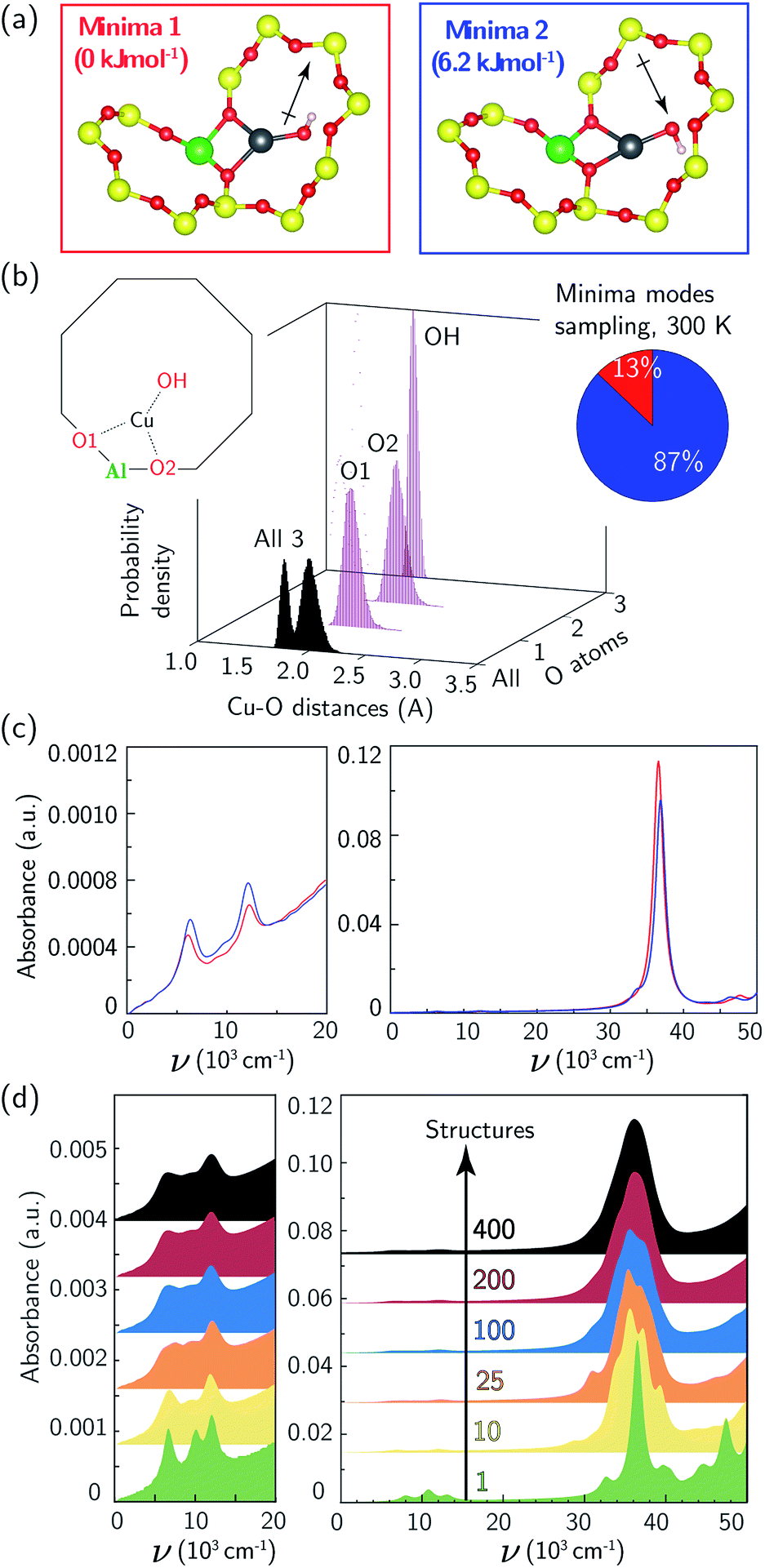 | ||
| Fig. 3 (a) DFT-computed local Cu environment in two 8MR ZCuOH minima. (b) Individual and aggregate Cu–O distance histograms collected over 150 ps of 300 K AIMD. Inset indicates percent time spent in each minimum. (c) TD-DFT-computed UV-Vis spectra of two ZCuOH minima, color-coded by (a). (d) UV-Vis spectra averaged over 1 (green), 10 (yellow), 25 (orange), 100 (blue), 200 (red), and 400 (black) snapshots equally spaced in time along the 150 ps trajectory. | ||
The 8MR [CuOH]+ ion can exist in one of two rotational conformers that differ in energy by 6 kJ mol−1 and are distinguished by whether the OH ligand points into or out of the 8MR (Fig. 3a). Fig. 3c reports the computed UV-Vis spectra of a relaxed 8MR [CuOH]+ ion; each conformer yields a spectrum with two equivalent sharp features in the d–d transition region and a single sharp LMCT band. Predicted ZCuOH absorption intensities are less than either the Z2Cu para and meta spectra in Fig. 2c and g, consistent with the higher symmetry of ZCuOH and prior predictions that ZCuOH may have small or unobservable d–d transitions.49,74 However the two spectra in Fig. 3c for the two ZCuOH isomers are only in rough correspondence with the observed spectrum of the ZCuOH samples.
We computed the absorption spectra of 400 equally spaced structures chosen from the 300 K AIMD simulation; all computed spectra are overlaid in Fig. S4c.† Significant variations are present in the d–d (7000 to 14000 cm−1) and LMCT (30000 to 50000 cm−1) regions (ESI Fig. S6c†), with shorter mean Cu–O distances again correlating with shifts to higher frequency d–d transitions (ESI Fig. S5†). Fig. 3d reports spectra averaged over 1, 10, 25, 100, 200, and 400 structures. The two sharp d–d features at 8000 and 13000 cm−1 broaden and begin to merge, while the LMCT region converges to a peak spanning 30000 to 45000 cm−1. The averaged d–d and LMCT regions ≈1000 cm−1 are red-shifted but similar in shape to those observed after CO reduction of the ZCuOH samples (Fig. 1b and c). Further, the decrease in computed intensity of the d–d relative to LMCT bands in ZCuOH compared to Z2Cu models corresponds with experimental observation. These observations support both the assignment of the Fig. 1b and c spectra following CO treatment (red) to isolated ZCuOH and the conclusion that the quadruplet features after O2 treatment (black) cannot be solely assigned to ZCuOH but rather have contributions from multinuclear Cu complexes.
2.3 Cu dimers
The additional features in Fig. 1b and c following 673 K 20% O2 treatment are from CO-reducible Cu species. A discrete, exchanged [CuO]+ ion is an unlikely candidate because it has a unfavorable computed formation free energy and its computed spectrum (ESI Fig. S8†) is inconsistent with experimental observation. More likely are dimeric or larger Cu clusters. To test the potential for isolated ZCuOH to condense into dimers, we considered two [CuOH]+ ions located at Al separated by four T-sites in the same 8-MR using a 36-T-site supercell. Fig. 4a, structures C and E show the ZCu(OH)2CuZ condensation product and of its ZCuOCuZ dehydration product, respectively (details in ESI Section S1.12 and Fig. S9†). Within the generalized gradient and harmonic approximations, the free energies to form ZCu(OH)2CuZ and dehydrate to ZCuOCuZ are computed to be −108 and −116 kJ mol−1, respectively (details in ESI Section S1.12†). These energetics are consistent with the formation of dimers from [CuOH]+ ions of suitable proximity.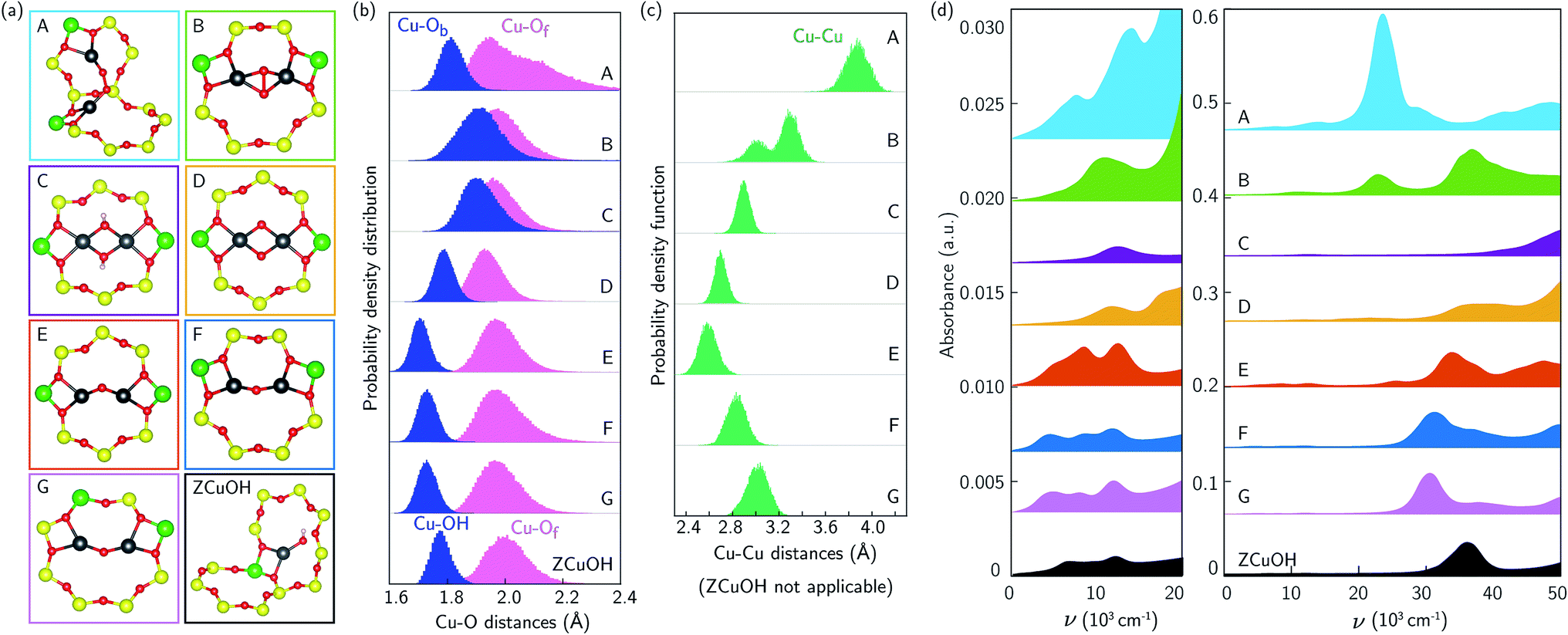 | ||
| Fig. 4 (a) Optimized geometries of Cu dimers A-G, and ZCuOH for reference. (b)/(c) Aggregate Cu–O distance (b) and Cu–Cu distance (c) histograms for dimers A–G and ZCuOH collected over 150 ps 300 K AIMD. Bridging Ob and OH distances are presented in blue, Of distances are in pink, Cu–Cu distances are in green. (d) Averaged spectra of Cu dimers A–G (left panel is from 0–20000 cm−1, right panel is from 0 to 50000 cm−1, each spectra is offset on the y-axis). Colors of spectra are consistent with those in (a). Averaged ZCuOH spectrum is plotted at the bottom in black for reference. | ||
We performed 150 ps AIMD at 300 K on these two dimer structures in the 12-T-site supercell. In both trajectories the dimers remain roughly in the plane of the 8MR and retain coordination to the same bridging and framework O, unlike the more dynamic Z2Cu behavior described above. Both dimers vibrate internally and against the framework. Histograms of the Cu–O and Cu–Cu distances are shown in Fig. 4b and c. In ZCuOCuZ, Cu–Ob (bridging O) distances are systematically shorter and fluctuate less than Cu–Of. The Cu–Cu separation oscillates around 2.7 Å, and Cu–O–Cu angle varies from 90 to 115°. The Cu(OH)2 core of ZCu(OH)2CuZ remains essentially planar and tilted at an angle of ≈35° with respect to the 8MR plane during the AIMD (structure in ESI Fig. S12†). The Cu–Ob(H) and Cu–Of distances cover a similar range, and the Cu–Cu separation oscillates around 3 Å.
We used TD-DFT and spectral averaging methods identical to those above to simulate UV-Vis spectra of both dimers at 300 K. We observed computed spectra to be sensitive to the geometries of the dimers, similar to the monomer Z2Cu and ZCuOH. Fig. S10† shows spectra averaged over various numbers of snapshots; spectral averaging converge after ≈200 structures. Fig. 4d shows the spectra averaged over 400 snapshots. The spectrum of ZCu(OH)2CuZ structure (C) exhibits a broad and low intensity d–d feature around 12400 cm−1 and a LMCT band blue-shifted to beyond 50000 cm−1. In contrast, the spectrum of ZCuOCuZ structure (E) has two distinct d–d features around 8500 and 12400 cm−1 and an LMCT band edge that begins near 30000 cm−1. These two are clearly distinct from one another and from the computed spectrum of ZCuOH.
The spectroscopy of Cu dimers may be sensitive to Al proximity, through its influence on geometric and electronic structures. To test this effect, we constructed two additional ZCuOCuZ models with two Al placed third- and second-nearest-neighbor in an 8MR and introduced a Cu–O–Cu dimer so as to maintain Cu–O distances and a Cu–O–Cu angle similar to previous reports.11,18Fig. 4a shows the optimized structures F and G used to initiate subsequent dynamics. At 3NN, the ZCuOCuZ dimer is symmetrically coordinated to Of associated with Al; at 2NN, this symmetry is broken, although both Cu remain bound to two Of. During subsequent 300 K AIMD simulations the Cu ions retain their coordination; as shown in the histograms, Cu–O distances vary across the same ranges at all Al placements while Cu–Cu distances and Cu–O–Cu angles vary considerably with Al separation. While the computed spectra of the optimized structures are different, these differences largely disappear during averaging. As shown in Fig. 4d, averaged spectra have similar d–d features and differ only in the LMCT band edge position.
The Cu dimer spectroscopy could also be sensitive to geometric isomerism. To test this effect, we considered several examples of Cu dimers bridged by two O, a well known motif that exhibits several geometric isomers that are sensitive to Al separation.34,74–76Fig. 4a structures A, B, and D correspond to three different Al placements and three different isomers, all of which were obtained by geometry relaxations beginning from literature structures.11,18 A and B have triplet and D has a singlet ground states, consistent with earlier results.34,75 Computed spectra at the optimized geometries (ESI Fig. S10†) exhibit sharp and distinct peaks in both the d–d and LMCT regions. We performed AIMD on all three isomers; distance histograms collected during the simulations are shown in Fig. 4b and c. The dioxo dimer D is the least variable across the trajectory; dimers A and B sample much larger Cu–Of and Cu–Cu distances, respectively. During the finite temperature simulation, dimer A moves from a cis to a trans μ-peroxo orientation whereas the optimized geometry has a slightly twisted O–O linkage, resulting in smaller Cu–O–O angles (geometry comparison in ESI Fig. S13†). Fig. 4d reports computed spectra averaged over 400 snapshots. Spectra differ significantly in band location and intensity both in the d–d and LMCT regions.
UV-Vis spectra are thus sensitive to Cu dimer composition and structure and dynamics. Comparisons with the experimental spectra collected after 20% O2 treatment at 673 K in Fig. 1b and c are complicated by the ill-defined number and nuclearity of Cu species present in the samples. Nevertheless, we can make some useful connections. The relative intensities of the d–d transitions for some dimeric Cu species (in particular A, B, and E) are computed to be greater than that of monomeric ZCuOH, consistent with the observation of decreases in d–d peak intensity following CO reduction. Structures B, E, and F have features that roughly correspond with those observed at 11059, 13593, 16379, and 20077 cm−1 (Fig. 1b and c), but red-shifted by an amount similar to that found in the comparison of computed and observed ZCuOH spectra. Structures C and D contain features that are plausible candidates for the features observed at 16000 and 19000 cm−1. Structures A and B contain features that could account for the broad low energy LMCT shoulder from ≈22000 to 27000 cm−1 that disappears after the CO reduction treatment. All these results imply that the variations in the quadruplet feature in the d–d region of nominally ZCuOH samples shown in Fig. 3b and c are associated at least in part with the contributions of different numbers and/or types of dimers from sample to sample.
3 Discussion
Cu-SSZ-13 samples used here and reported in the literature are now well understood to be intrinsically heterogeneous at the microscopic scale, as a result of variations in composition and location of Al77 and charge-compensating Cu ions, among other factors. Some of these differences are readily observed spectroscopically; for instance ZCuOH sites are clearly indicated by their distinctive O–H stretch vibration at 3660 cm−1,38,78,79 while others are more subtle to infer. The UV-Vis results reported here provide some guidance for distinguishing three types of Cu species. Z2Cu species are distinguished by relatively intense d–d transitions with maxima near 12000 and 14000 cm−1 (Fig. 1a), and these features persist upon exposure to CO. Isolated ZCuOH species are indicated by lower intensity d–d features near 11000 and 13000 cm−1, which also persist during exposure to CO. We find that samples intentionally prepared to contain exclusively ZCuOH always contain a confounding set of species that have relatively intense but irregular d–d features, which we assign to a mixture of higher nuclearity Cu oxo and hydroxo species. These species contribute four relatively prominent features in the d–d region, but they do not persist following CO reduction treatments.
Discrimination within these sets is more challenging. Samples prepared to contain only Z2Cu species potentially contain two geometrically distinct sites distinguished by the location of the charge-compensating Al,80 and such sites are predicted here to have distinct UV-Vis spectra if computed at one minimum energy structure. Accounting for the finite temperature fluctuations in Cu location between local minima and associated fluctuations in Cu coordination environment, however, attenuates these differences, such that the two sites become spectroscopically indistinguishable. Similar factors affect the interpretation of XAS,79,81–84 EPR49,83,85 spectroscopies, and X-ray diffraction5,82,85,86 patterns, and caution should be applied in inferring Z2Cu geometric information by comparison of observations to predictions from single, minimum energy structures.
In samples in which Cu exchange is predominantly associated with isolated Al T-sites and contain a majority of ZCuOH sites, observed UV-Vis spectra contain features in the d–d and LMCT regions that cannot be accounted for by the spectrum computed of these sites.18,48,49,87 Strategies to directly prepare and characterize Cu-SSZ-13 samples that contain exclusively ZCuOH sites, either by exchanging dilute amounts of Cu or using higher silica-content SSZ-13 supports, are unlikely to be successful because of the difficulties of observing dilute ZCuOH and the presence of a confounding set of framework Al sites that can stabilize dimeric forms of Cu even at low Al density. Therefore, a strategy that combines synthetic efforts to bias formation of predominantly one Cu site type with treatments that selectively remove minority Cu species, is more likely to allow access to individual Cu site types.
The exact shapes and positions of the features depend on the zeolite composition and the precise pretreatment conditions, including temperature, pressure and duration of O2 exposure, all suggestive of additional Cu sites produced dynamically. Calculations here show that [CuOH]+ ions can move between adjacent 8MRs with an activation barrier of 63 kJ mol−1 (ESI Fig. S7†) and that suitably proximal ions can condense into dimers. This proposal is consistent with experimental observations of a decrease in the approximately 3660 cm−1 vibrational feature associated with the ZCuO–H stretch with increasing temperature,41 corresponding Raman shifts for multiple Cu2Ox motifs.18,19,55 It is also consistent with the observation of dry NO oxidation to NO2 on nominally ZCuOH-containing samples, ascribed to dimeric Cu sites.11,88 Because computed UV-Vis spectra of Cu dimers (Fig. 4) are sensitive to Al proximity, extra-lattice ligands, and to finite temperature structural fluctuations, assignment of specific spectroscopic features to individual types of dimeric Cu species is not possible based on results reported here.
The samples prepared to contain Z2Cu sites have spectral features that are invariant to reduction in CO. Literature results on similar samples find that they are insensitive to exposure to He, O2, or CH4.6,19 The four features observed in the d–d region of the ZCuOH samples following oxidizing treatment are similar to those reported previously in similar samples,18,48,50 but these bands do respond differently to subsequent reducing treatments. We find that 5% CO exposure at 523 K reproducibly preserves a portion of the d–d and LMCT features, which we assign to isolated ZCuOH sites. In contrast, exposure to CH4 at 473 K results only in a decrease in the lower energy portion (25000 to 38000 cm−1) of the LMCT band and disappearance of the 29000 cm−1 band.18,50,87 These results suggest that at the same temperature CH4 reduces a different population of CuxOyHz moieties to Cu+ than exposure to inert (He), and exposure to CO reduces all of the CuxOyHz observed to reduce in either CH4 or He.18 Spectral features we assign here to isolated ZCuOH moieties are observed in similar materials to persist after CH4 exposure.18,50 Precise identification of the Cu dimer sites responsible for CH4 activation under different conditions remains an important challenge for experiment and computation.
4 Conclusions
Cu-exchanged zeolites remain a topic of great scientific interest because of their intriguing performance in catalyzing difficult transformations involving the nitrogen oxides and methane. Identification of active sites is complicated by the sensitivity of Cu exchange to zeolite framework types, the number and distribution of framework Al atoms, Cu content, and pretreatment conditions. The SSZ-13 zeolite framework is constructed of a single symmetry-distinct type of T-site, in principle reducing the number of possible distinct ion exchange sites and thus simplifying spectroscopic interrogation of those sites. Here we use directed synthetic approaches to emphasize different types of exchange sites, and DFT evaluations of site structure and spectral signatures to test this principle. We find that in SSZ-13 samples prepared to contain only isolated Al T-sites, and thus in principle a homogeneous array of [CuOH]+ ion exchange sites, always contain a confounding subset of O- or OH-bridged Cu dimers and/or larger aggregates in the Cu/Al exchange regime explored here. These latter species are intrinsically heterogeneous due to heterogeneity in bridging ligands and/or in arrangements of framework Al substituents, which likely involve more than one Al T-site. The underlying [CuOH]+ UV-Vis spectrum can be revealed by selective CO reduction of the polynuclear Cu species. The UV-Vis spectrum, even of isolated [CuOH]+ ions, is found to be influenced by the intrinsic, finite-temperature dynamics of the site, as revealed through AIMD and TD-DFT calculations. In SSZ-13 samples prepared to contain 2NN (meta) and 3NN (para) Al pairs in the 6MR, the same dynamical factors serve to obscure spectroscopic differences between these two distinct types of Z2Cu sites. While calculations performed at the optimized geometries predict that the meta and para Cu-exchange sites are spectroscopically distinct, those differences are indistinguishable after accounting for the finite-temperature fluctuations in Cu ion coordination environment.These results highlight the potential and the practical challenges of developing correlations between observed spectroscopy and the contributions of various Cu ion exchange sites and motifs to observed chemical reactivity. They highlight that precise characterization of active sites in this and similar systems demands a careful integration of chemical and spectroscopic interrogation with computational models that account for the structural and dynamical complexities of the materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The experimental research at Purdue on zeolite synthesis and characterization was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0019026. The computational work at Notre Dame was supported by the National Science Foundation GOALI program under award number CBET-1258690. We thank the Center for Research Computing at Notre Dame, and EMSL, a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory, for support of computational resources. We thank John R. Di Iorio (Purdue) for assistance with zeolite synthesis and helpful technical discussions. Florian Göltl contributions were supported in part through NSF grant number CHE-1800284.References
- C. Paolucci, J. Di Iorio, F. Ribeiro, R. Gounder and W. Schneider, Adv. Catal., 2016, 59, 1–107 Search PubMed.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. Peden and J. Szanyi, Recent advances in automotive catalysis for NOx emission control by small-pore microporous materials, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- F. Gao, J. H. Kwak, J. Szanyi and C. H. Peden, Current understanding of Cu-exchanged chabazite molecular sieves for use as commercial diesel engine DeNOx catalysts, Top. Catal., 2013, 56, 1441–1459 CrossRef CAS.
- I. Nova and E. Tronconi, Urea-SCR technology for deNOx after treatment of diesel exhausts, Springer, 2014 Search PubMed.
- D. W. Fickel and R. F. Lobo, Copper coordination in Cu-SSZ-13 and Cu-SSZ-16 investigated by variable-temperature XRD, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Isolated Cu2+ ions: active sites for selective catalytic reduction of NO, Chem. Commun., 2011, 47, 800–802 RSC.
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Time-resolved copper speciation during selective catalytic reduction of NO on Cu-SSZ-13, Nat. Catal., 2018, 1, 221–227 CrossRef.
- T. V. Janssens, H. Falsig, L. F. Lundegaard, P. N. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko and C. Lamberti, A consistent reaction scheme for the selective catalytic reduction of nitrogen oxides with ammonia, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, The Cu–CHA deNOx Catalyst in Action: Temperature-Dependent NH3-Assisted Selective Catalytic Reduction Monitored by Operando XAS and XES, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. Peden, Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- A. A. Verma, S. A. Bates, T. Anggara, C. Paolucci, A. A. Parekh, K. Kamasamudram, A. Yezerets, J. T. Miller, W. N. Delgass, W. F. Schneider and F. H. Ribeiro, NO oxidation: A probe reaction on Cu-SSZ-13, J. Catal., 2014, 312, 179–190 CrossRef CAS.
- M. P. Ruggeri, I. Nova, E. Tronconi, J. A. Pihl, T. J. Toops and W. P. Partridge, In situ DRIFTS measurements for the mechanistic study of NO oxidation over a commercial Cu–CHA catalyst, Appl. Catal., B, 2015, 166, 181–192 CrossRef.
- G. Centi and S. Perathoner, Nature of active species in copper-based catalysts and their chemistry of transformation of nitrogen oxides, Appl. Catal., A, 1995, 132, 179–259 CrossRef CAS.
- M. H. Groothaert, J. A. Van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, Bis(μ-oxo)dicopper in Cu-ZSM-5 and its role in the decomposition of NO: A combined in situ XAFS, UV-vis-near-IR, and kinetic study, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS PubMed.
- P. Da Costa, B. Moden, G. D. Meitzner, D. K. Lee and E. Iglesia, Spectroscopic and chemical characterization of active and inactive Cu species in NO decomposition catalysts based on Cu-ZSM5, Phys. Chem. Chem. Phys., 2002, 4, 4590–4601 RSC.
- P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Coordination chemistry and reactivity of copper in zeolites, Coord. Chem. Rev., 2013, 257, 483–494 CrossRef CAS.
- M. J. Wulfers, R. F. Lobo, B. Ipek and S. Teketel, Conversion of Methane to Methanol on Copper-Containing Small-Pore Zeolites and Zeotypes, Chem. Commun., 2015, 51, 4447–4450 RSC.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- D. K. Pappas, et al., Methane to Methanol: Structure-Activity Relationships for Cu–CHA, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- A. R. Kulkarni, Z. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Cation-Exchanged Zeolites for the Selective Oxidation of Methane to Methanol, Catal. Sci. Technol., 2017, 8, 114–123 RSC.
- P. Tomkins, A. Mansouri, S. E. Bozbag, F. Krumeich, M. B. Park, E. M. C. Alayon, M. Ranocchiari and J. A. van Bokhoven, Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature, Angew. Chem., Int. Ed., 2016, 55, 5467–5471 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Cu–CHA – a model system for applied selective redox catalysis, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- M. Dusselier and M. E. Davis, Small-Pore Zeolites: Synthesis and Catalysis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS PubMed.
- B. E. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes, Chem. Rev., 2017, 118, 2718–2768 CrossRef PubMed.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- A. A. Latimer, A. R. Kulkarni, H. Aljama, J. H. Montoya, J. S. Yoo, C. Tsai, F. Abild-Pedersen, F. Studt and J. K. Nørskov, Understanding trends in C–H bond activation in heterogeneous catalysis, Nat. Mater., 2017, 16, 225 CrossRef CAS PubMed.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Monocopper active site for partial methane oxidation in Cu-exchanged 8MR zeolites, ACS Catal., 2016, 6, 6531–6536 CrossRef CAS.
- D. K. Pappas, et al., The nuclearity of the active site for methane to methanol conversion in Cu-mordenite: a quantitative assessment, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Selective anaerobic oxidation of methane enables direct synthesis of methanol, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. Hensen and E. A. Pidko, Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- Z. Lan and S. M. Sharada, Computational strategies to probe CH activation in dioxo–dicopper complexes, Phys. Chem. Chem. Phys., 2018, 20, 25602–25614 RSC.
- B. R. Goodman, W. F. Schneider, K. C. Hass and J. B. Adams, Theoretical analysis of oxygen-bridged Cu pairs in Cu-exchanged zeolites, Catal. Lett., 1998, 56, 183–188 CrossRef CAS.
- B. L. Trout, A. K. Chakraborty and A. T. Bell, Local spin density functional theory study of copper ion-exchanged ZSM-5, J. Phys. Chem., 1996, 100, 4173–4179 CrossRef CAS.
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, Local environment and nature of Cu active sites in zeolite-based catalysts for the selective catalytic reduction of NOx, ACS Catal., 2013, 3, 413–427 CrossRef CAS.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Siting and distribution of framework aluminium atoms in silicon-rich zeolites and impact on catalysis, Catal. Rev., 2012, 54, 135–223 CrossRef.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, Catalysis in a cage: Condition-Dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- J. R. Di Iorio and R. Gounder, Controlling the Isolation and Pairing of Aluminum in Chabazite Zeolites Using Mixtures of Organic and Inorganic Structure-Directing Agents, Chem. Mater., 2016, 28, 2236–2247 CrossRef CAS.
- J. R. Di Iorio, C. T. Nimlos and R. Gounder, Introducing catalytic diversity into single-site chabazite zeolites of fixed composition via synthetic control of active site proximity, ACS Catal., 2017, 7, 6663–6674 CrossRef CAS.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Revisiting the nature of Cu sites in the activated Cu-SSZ-13 catalyst for SCR reaction, Chem. Sci., 2014, 8, 548–563 Search PubMed.
- J. D. Albarracin-Caballero, I. Khurana, J. R. Di Iorio, A. J. Shih, J. E. Schmidt, M. Dusselier, M. E. Davis, A. Yezerets, J. T. Miller, F. H. Ribeiro and R. Gounder, Structural and kinetic changes to small-pore Cu-zeolites after hydrothermal aging treatments and selective catalytic reduction of NOx with ammonia, React. Chem. Eng., 2017, 2, 168–179 RSC.
- J. E. Schmidt, R. Oord, W. Guo, J. D. Poplawsky and B. M. Weckhuysen, Nanoscale tomography reveals the deactivation of automotive copper-exchanged zeolite catalysts, Nat. Commun., 2017, 8, 1666 CrossRef PubMed.
- F. Göltl, P. Sautet and I. Hermans, The impact of finite temperature on the coordination of Cu cations in the zeolite SSZ-13, Catal. Today, 2016, 267, 41–46 CrossRef.
- P. Chen, A. Khetan, M. Jabłońska, J. Simböck, M. Muhler, R. Palkovits, H. Pitsch and U. Simon, Local dynamics of copper active sites in zeolite catalysts for selective catalytic reduction of NOx with NH3, Appl. Catal., B, 2018, 237, 263–272 CrossRef CAS.
- H. Li, C. Paolucci and W. F. Schneider, Zeolite Adsorption Free Energies from Ab Initio Potentials of Mean Force, J. Chem. Theory Comput., 2018, 14, 929–938 CrossRef CAS PubMed.
- L. Chen, H. Falsig, T. V. Janssens, J. Jansson, M. Skoglundh and H. Grönbeck, Effect of Al-distribution on oxygen activation over Cu–CHA, Catal. Sci. Technol., 2018, 8, 2131–2136 RSC.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Characterization of Cu-exchanged SSZ-13: a comparative FTIR, UV-Vis, and EPR study with Cu-ZSM-5 and Cu-β with similar Si/Al and Cu/Al ratios, Dalton Trans., 2013, 42, 12741–12761 RSC.
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. Birk Rasmussen, L. F. Lundegaard and S. Mossin, Coordination Environment of Copper Sites in Cu–CHA Zeolite Investigated by Electron Paramagnetic Resonance, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- R. Oord, J. E. Schmidt and B. M. Weckhuysen, Methane-to-methanol conversion over zeolite Cu-SSZ-13, and its comparison with the selective catalytic reduction of NOx with NH3, Catal. Sci. Technol., 2018, 8, 1028–1038 RSC.
- G. D. Lei, B. J. Adelman, J. Sárkány and W. M. H. Sachtler, Identification of copper(II) and copper(I) and their interconversion in Cu/ZSM-5 De-NOx catalysts, Appl. Catal., B, 1995, 5, 245–256 CrossRef CAS.
- B. Modén, P. Da Costa, B. Fonfé, D. K. Lee and E. Iglesia, Kinetics and Mechanism of Steady-State Catalytic NO Decomposition Reactions on Cu-ZSM5, J. Catal., 2002, 209, 75–86 CrossRef.
- B. Modén, P. Da Costa, D. K. Lee and E. Iglesia, Transient studies of oxygen removal pathways and catalytic redox cycles during NO decomposition on Cu-ZSM5, J. Phys. Chem. B, 2002, 106, 9633–9641 CrossRef.
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Oxygen precursor to the reactive intermediate in methanol synthesis by Cu-ZSM-5, J. Am. Chem. Soc., 2010, 132, 14736–14738 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, Selective oxidation of methane by the bis(μ-oxo)dicopper core stabilized on ZSM-5 and mordenite zeolites, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- S. Grundner, M. A. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Effect of Active Sites Structure on Activity of Copper Mordenite in Aerobic and Anaerobic Conversion of Methane to Methanol, Angew. Chem., Int. Ed., 2018, 57, 8906–8910 CrossRef CAS PubMed.
- J. A. van Bokhoven and C. Lamberti, in XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, ed. Y. Iwasawa, K. Asakura and M. Tada, Springer International Publishing, Cham, 2017; pp. 299–316 Search PubMed.
- N. Kosinov, C. Liu, E. J. Hensen and E. A. Pidko, Engineering of transition metal catalysts confined in zeolites, Chem. Mater., 2018, 30, 3177–3198 CrossRef CAS PubMed.
- D. Palagin, A. J. Knorpp, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Assessing the relative stability of copper oxide clusters as active sites of a CuMOR zeolite for methane to methanol conversion: size matters?, Nanoscale, 2017, 9, 1144–1153 RSC.
- E. Borfecchia, D. K. Pappas, M. Dyballa, K. A. Lomachenko, C. Negri, M. Signorile and G. Berlier, Evolution of active sites during selective oxidation of methane to methanol over Cu–CHA and Cu–MOR zeolites as monitored by operando XAS, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.07.028.
- M. J. Henson, M. a. Vance, C. X. Zhang, H.-C. Liang, K. D. Karlin and E. I. Solomon, Resonance Raman investigation of equatorial ligand donor effects on the Cu2O22+ core in end-on and side-on μ-peroxo-dicopper(II) and bis-μ-oxo-dicopper(III) complexes, J. Am. Chem. Soc., 2003, 125, 5186–5192 CrossRef CAS PubMed.
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, Cu-ZSM-5: A biomimetic inorganic model for methane oxidation, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. D. Iorio, J. D. Albarracin-caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- J. S. McEwen, T. Anggara, W. F. Schneider, V. F. Kispersky, J. T. Miller, W. N. Delgass and F. H. Ribeiro, Integrated operando X-ray absorption and DFT characterization of Cu-SSZ-13 exchange sites during the selective catalytic reduction of NOx with NH3, Catal. Today, 2012, 184, 129–144 CrossRef CAS.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Transition-metal ions in zeolites: Coordination and activation of oxygen, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- A. Godiksen, P. N. R. Vennestrøm, S. B. Rasmussen and S. Mossin, Identification and quantification of copper sites in zeolites by electron paramagnetic resonance spectroscopy, Top. Catal., 2017, 60, 13–29 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- S. Sklenak, P. C. Andrikopoulos, S. R. Whittleton, H. Jirglova, P. Sazama, L. Benco, T. Bucko, J. Hafner and Z. Sobalik, Effect of the Al siting on the structure of Co(II) and Cu(II) cationic sites in ferrierite. A periodic DFT molecular dynamics and FTIR study, J. Phys. Chem. C, 2013, 117, 3958–3968 CrossRef CAS.
- J. Luo, D. Wang, A. Kumar, J. Li, K. Kamasamudram, N. Currier and A. Yezerets, Identification of two types of Cu sites in Cu/SSZ-13 and their unique responses to hydrothermal aging and sulfur poisoning, Catal. Today, 2016, 267, 3–9 CrossRef CAS.
- F. Goeltl, P. Muller, P. Uchupalanun, P. Sautet and I. Hermans, Developing a Descriptor-Based Approach for CO and NO Adsorption Strength to Transition Metal Sites in Zeolites, Chem. Mater., 2017, 29, 6434–6444 CrossRef.
- D. Harris and M. Bertolucci, Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy, Dover Books on Chemistry Series, Dover Publications, 1989 Search PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper active sites in biology, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- B. R. Goodman, K. C. Hass, W. F. Schneider and J. B. Adams, Cluster Model Studies of Oxygen-Bridged Cu Pairs in Cu-ZSM-5 Catalysts, J. Phys. Chem. B, 1999, 103, 10452–10460 CrossRef CAS.
- M. H. Mahyuddin, T. Tanaka, Y. Shiota, A. Staykov and K. Yoshizawa, Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)]2+ Active Species in Large-Pore Zeolites, ACS Catal., 2018, 8, 1500–1509 CrossRef CAS.
- B. C. Knott, C. T. Nimlos, D. J. Robichaud, M. R. Nimlos, S. Kim and R. Gounder, Consideration of the Aluminum Distribution in Zeolites in Theoretical and Experimental Catalysis Research, ACS Catal., 2018, 8, 770–784 CrossRef CAS.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale, Determining the storage, availability and reactivity of NH3 within Cu–Chabazite-based Ammonia Selective Catalytic Reduction systems, Phys. Chem. Chem. Phys., 2014, 16, 1639–1650 RSC.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, Interaction of NH3 with Cu-SSZ-13 catalyst: A complementary FTIR, XANES, and XES study, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- S. Li, H. Li, R. Gounder, A. Debellis, I. B. Müller, S. Prasad, A. Moini and W. F. Schneider, First-Principles Comparison of Proton and Divalent Copper Cation Exchange Energy Landscapes in SSZ-13 Zeolite, J. Phys. Chem. C, 2018, 122, 23564–23573 CrossRef CAS.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga, C. Lamberti and I. A. Pankin, Composition-driven Cu-speciation and reducibility in Cu–CHA zeolite catalysts: a multivariate XAS/FTIR approach to complexity, Chem. Sci., 2017, 8, 6836–6851 RSC.
- C. W. Andersen, E. Borfecchia, M. Bremholm, M. R. V. Jørgensen, P. N. R. Vennestrøm, C. Lamberti, L. F. Lundegaard and B. B. Iversen, Redox-Driven Migration of Copper Ions in the Cu–CHA Zeolite as Shown by the In Situ PXRD/XANES Technique, Angew. Chem., Int. Ed., 2017, 56, 10367–10372 CrossRef CAS PubMed.
- G. Berlier, V. Crocellà, M. Signorile, E. Borfecchia, F. Bonino and S. Bordiga, in Struct. Bond., ed. M. P. F.Mingos, Springer, Berlin, Heidelberg, 2018, ch. 1, pp. 1–64 Search PubMed.
- A. Martini, E. Alladio and E. Borfecchia, Determining Cu–Speciation in the Cu–CHA Zeolite Catalyst: The Potential of Multivariate Curve Resolution Analysis of In Situ XAS Data, Top. Catal., 2018, 61, 1396–1407 CrossRef CAS.
- A. Godiksen, O. L. Isaksen, S. B. Rasmussen, P. N. Vennestrøm and S. Mossin, Site-Specific Reactivity of Copper Chabazite Zeolites with Nitric Oxide, Ammonia, and Oxygen, ChemCatChem, 2018, 10, 366–370 CrossRef CAS.
- C. W. Andersen, M. Bremholm, P. N. R. Vennestrom, A. B. Blichfeld, L. F. Lundegaard and B. B. Iversen, Location of Cu2+ in CHA zeolite investigated by X-ray diffraction using the Rietveld/maximum entropy method, IUCrJ, 2014, 1, 382–386 CrossRef CAS PubMed.
- R. Oord, Spectroscopic Insights into Copper-Based Microporous Zeolites for NH3-SCR of NOx and Methane-to-Methanol Activation, Ph.D. thesis, Utrecht University, 2017.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. Peden, Effects of Si/Al ratio on Cu/SSZ-13 NH 3-SCR catalysts: Implications for the active Cu species and the roles of Brønsted acidity, J. Catal., 2015, 331, 25–38 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05056b |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |