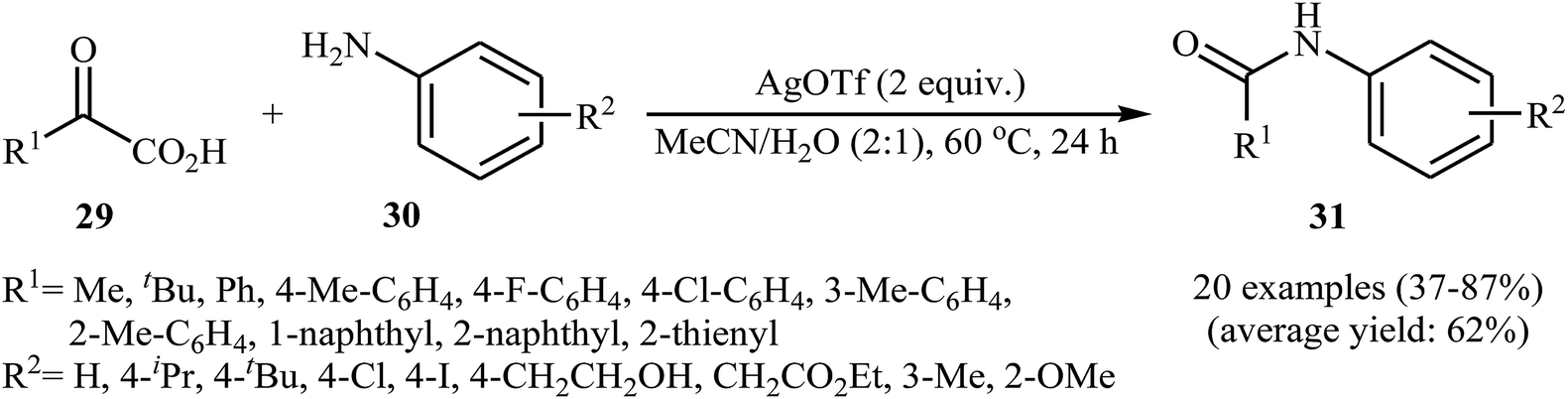DOI:
10.1039/C9RA00929A
(Review Article)
RSC Adv., 2019,
9, 8964-8976
Recent developments in decarboxylative cross-coupling reactions between carboxylic acids and N–H compounds
Received
3rd February 2019
, Accepted 4th March 2019
First published on 18th March 2019
Abstract
Carboxylic acids and their derivatives are ubiquitous compounds in organic chemistry, and are widely commercially available in a large structural variety. Recently, carboxylic acids have been frequently used as non-toxic and environmentally benign alternatives to traditional organohalide coupling partners in various carbon–carbon and carbon–heteroatom cross-coupling reactions. Along this line, several methods have been reported for the synthesis of nitrogen-containing organic compounds through decarboxylative cross-coupling reactions between carboxylic acids and N–H compounds. This review focuses on recent advances and discoveries on these reactions with special attention on the mechanistic aspects of the reactions.
 Sattar Arshadi | Sattar Arshadi was born in Miandoab, west Azerbaijan Province, Iran, in 1973. He received his B.Sc. degree in chemistry in the University of Kermanshah (1997) and his M.Sc. (2000) under supervision of Professor Issa Yavari and PhD (2004) in organic chemistry under supervision of Professor M. Z. Kassaee Both in Tarbiat Modarres University, Tehran, Iran. Then, he went to the University of Payame Noor as an Assistant Professor (2005). His main research interest is computational chemistry (especially on rearrangements and interactions in nano systems), organic synthesis and spectral studies of organic compounds. |
 Saeideh Ebrahimiasl | Saeideh Ebrahimiasl was born in Tabriz, Iran, in 1973. She received her PhD degree in nanomaterials and nanotechnology from the Institute of Advanced Materials, University Putra Malaysia in 2010. Professor Majid Monajjemi, Professor Wan Zin Wan Yunus, Professor Zulkarnain Zainal, Professor Mohd Zobir in physical chemistry, nanotechnology and electrochemistry were her most famous supervisors and examiners. Now she is Head of the Industrial nanotechnology Research Center in Tabriz and is an academic member of the Islamic Azad University of Ahar. Her research interests include synthesis and application of nanomaterials in different sciences. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her PhD degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Associate Professor. Her research interests include inorganic and organic synthesis, new methodologies in nanomaterial synthesis. |
 Aazam Monfared | Aazam Monfared was born in Tehran, Iran, in 1965. She received her B.S. degree in Pure Chemistry from University of Shahid Beheshti, Tehran, Iran, and her M.S. degree in Organic chemistry from Shahid Beheshti University, Tehran, Iran, in 1991 under the supervision of Prof. A. Rustaiyan. She received her PhD degree in 1999 under the supervision of Prof. A. Rustaiyan in Shahid Beheshti University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as an Associate Professor. Her research interests include organic synthesis, phytochemistry, drug synthesis, nanochemistry, methodologies and theoretical chemistry. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Carbon–nitrogen bond formation is the key strategy in the synthesis of nitrogen-containing organic molecules that have widespread application in many fields such as medicinal chemistry,1 agrochemistry,2 and organic synthesis.3 Over the past decades, several protocols have been developed to construct various C–N bonds. General synthetic methods toward the construction of C(sp3)–N bonds involve the nucleophilic substitution reactions between nitrogen nucleophiles and alkyl halides,4 N-alkylation of nitrogen nucleophiles with alcohols under Mitsunobu conditions,5 and hydroamination reactions of alkenes with nitrogen nucleophiles.6 The most popular methods for the formation of C(sp2)–N bonds include the SNAr reaction of electron-poor aryl halides with nitrogen nucleophiles,7 Buchwald–Hartwig reaction,8 Ullmann coupling,9 and Chan–Lam amination.10 The construction of C(sp)–N bonds mainly relies on the coupling of haloalkynes with N-nucleophiles.11 However, most of these methods, if not all, suffer from use of high cost, toxic or hazardous starting materials, limited substrate scope, prolonged reaction times and harsh reaction conditions. Thus, the development of a general, convenient, and truly efficient protocol for the formation of all the three kinds of C–N bonds (Calkyl–N, Caryl–N, Calkynyl–N) remains a challenge.
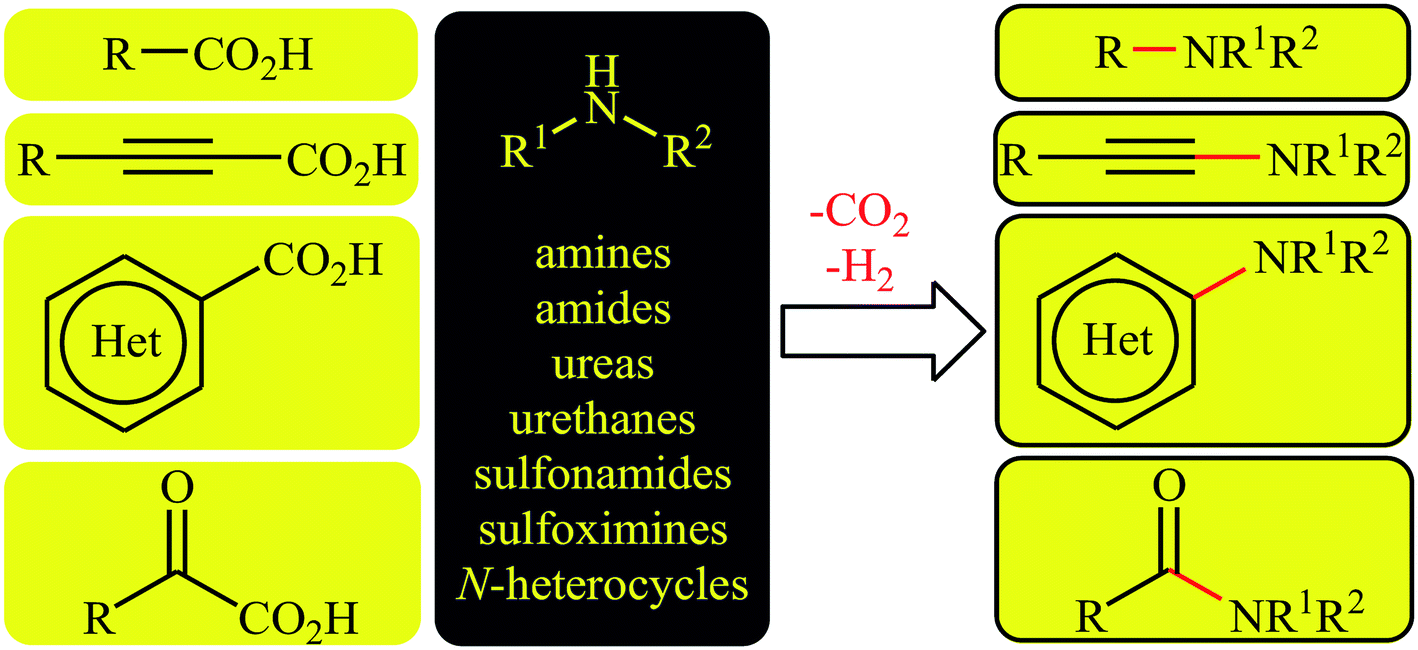
In recent years, decarboxylative cross-coupling reactions has emerged as a novel, selective, and powerful strategy to carbon–carbon12–15 and carbon–heteroatom16–18 bonds formations. This synthetic strategy utilizes readily available, high stable, very soluble, and low toxic carboxylic acids as alternative to traditional coupling partners containing unfavorable (pseudo)halide-leaving groups and extrudes carbon dioxide as an innocuous by-product. Thus, these reactions meet the criteria of green chemistry. Since a number of discoveries and developments in the decarboxylative C–N cross-coupling reactions have occurred during the current decade, a comprehensive review in this interesting and novel research field seems to be timely. As a part of our continuing reviews on cross-coupling reactions19 and new methodologies in organic synthesis,20 in this focus-review we will highlight recent progress in the decarboxylative cross-coupling reactions between carboxylic acids and N–H compounds (Fig. 1), with special emphasis on the mechanistic aspects of the reactions. It is noted that the reactions were classified based on the type of starting carboxylic acids (e.g. alkyl, aryl, and alkynyl carboxylic acids).
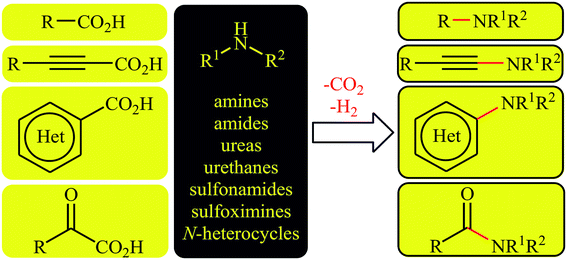 |
| | Fig. 1 Decarboxylative C–N cross-coupling reactions. | |
2. Cross-coupling reactions between Calkyl–CO2H and N–H bonds
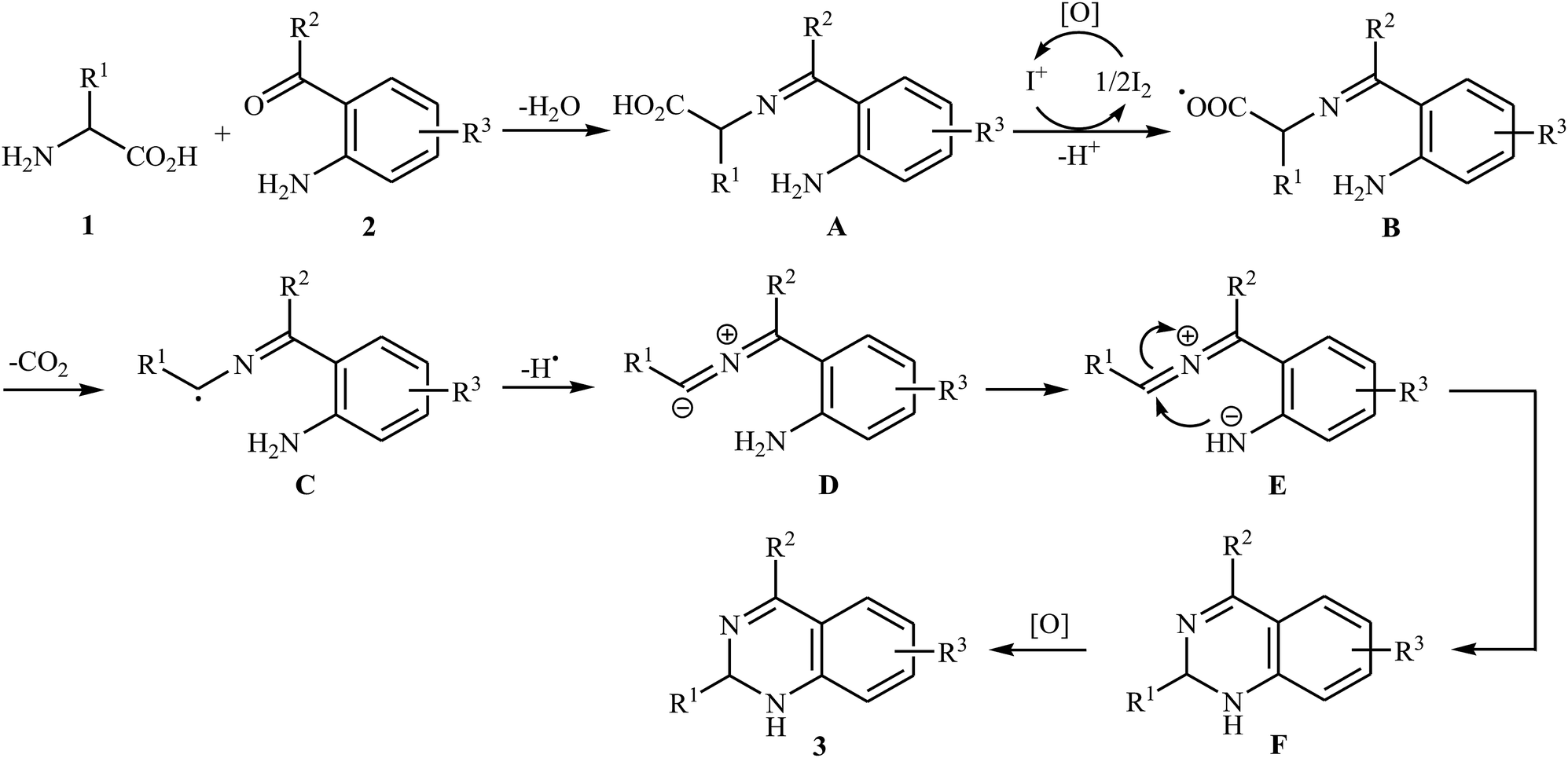
Decarboxylative cross-coupling reactions between aliphatic carboxylic acids and N–H compounds has scarcely been studied. In fact, only four examples of such reactions were reported in literature thus far. In 2011, Yan and Wang reported a promising metal-free protocol for the synthesis of biologically important quinazoline derivatives 3 through the hitherto unknown intramolecular oxidative decarboxylative coupling reaction between primary α-amino acids 1 and 2-aminobenzoketones 2 using molecular iodine as catalyst and TBHP (tert-butyl hydroperoxide) as the terminal oxidant.21 The reaction was carried out in an atmosphere of air at 80 °C, tolerated various electron-rich and electron-poor substrates, and provided functionalized quinazolines 3 in good to quantitative yields (Scheme 1a). However, the steric hindrance of the substrates greatly affected the efficiency of the transformation. For example, trimethyl substituted 2-aminobenzaldehyde failed to undergo the cyclization and 2-aminobenzaldehyde bearing 2,5-dimethyl substituents led to poor yield of the desired product. As shown in Scheme 2, this reaction is believed to proceed through a condensation/oxidization/CO2 elimination/H+ elimination/1,6-H transfer/cyclization/aromatization sequential process. In a related investigation, the same authors showed that 2-arylquinazolines 6 were formed from oxidative decarboxylative amination of carboxylic acids 4 with 2-aminobenzoketones 5 and ammonium acetate in a similar process employing Cu(OAc)2 as a catalyst and molecular oxygen as the sole oxidant in NMP at 120 °C (Scheme 1b).22
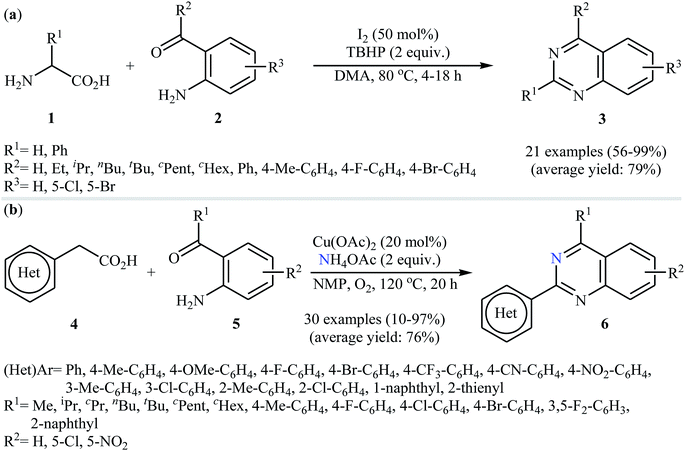 |
| | Scheme 1 (a) I2-catalyzed decarboxylative C–N coupling reactions of primary α-amino acids 1 and 2-aminobenzoketones 2 developed by Wang; (b) synthesis of 2-arylquinazolines 6 through decarboxylative amination of arylacetic acids 4 with 2-aminobenzoketones 5 and NH4OAc. | |
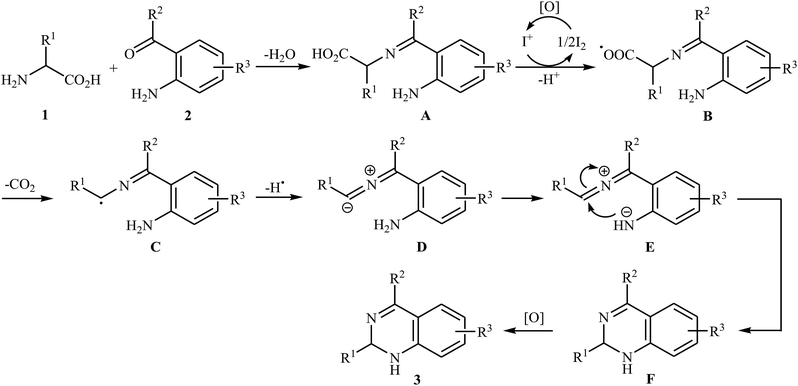 |
| | Scheme 2 Mechanistic proposal for the formation of quinazoline derivatives 3. | |
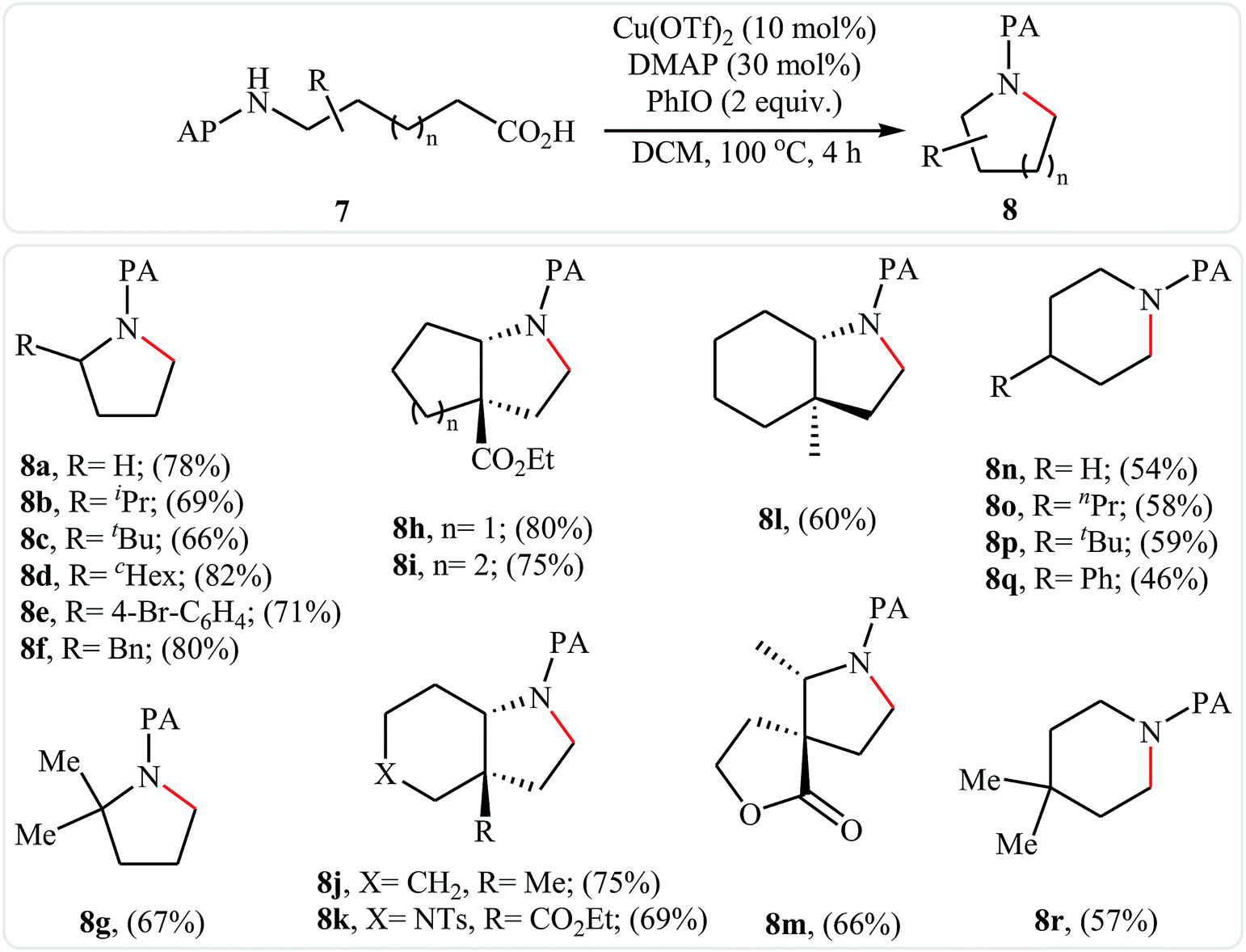
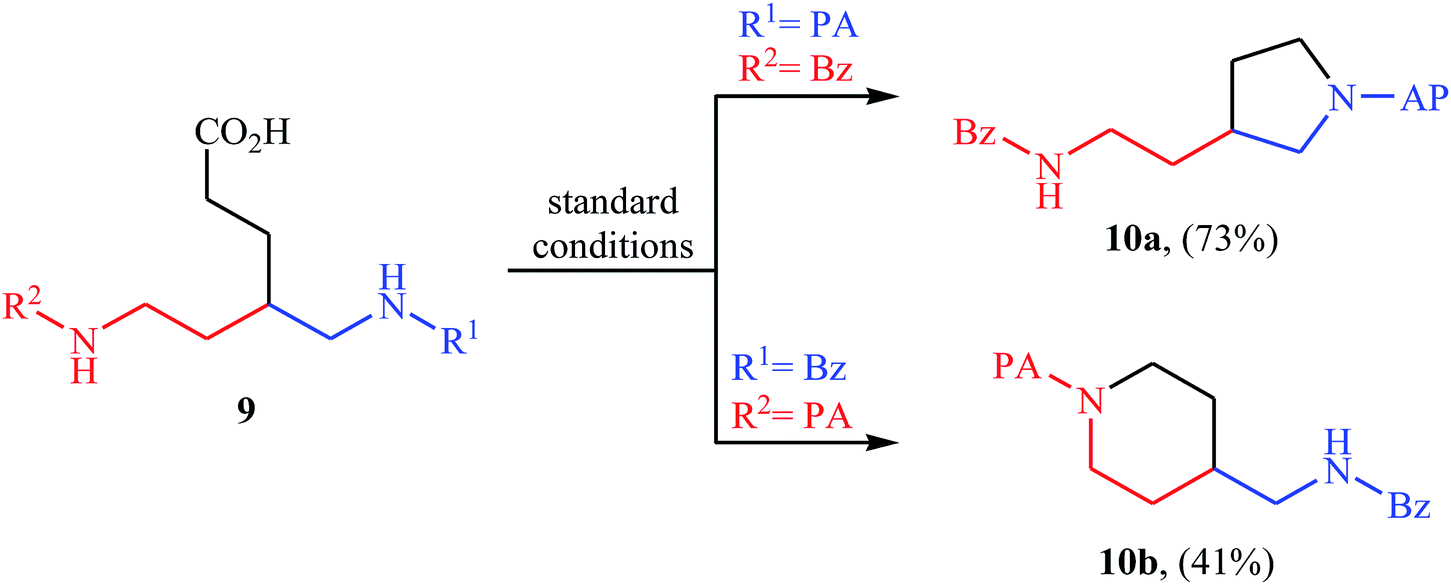
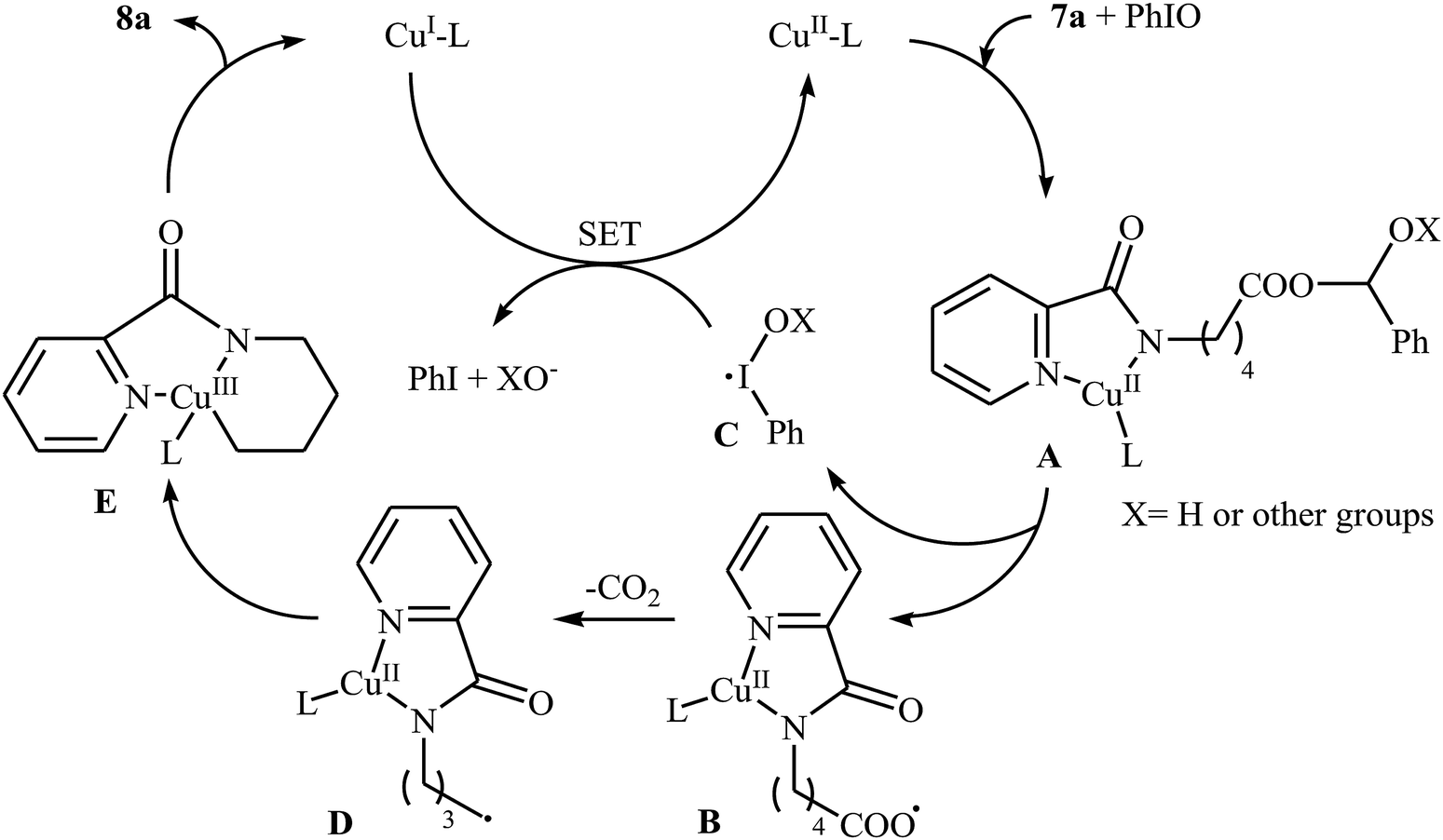
In 2016, primary aliphatic carboxylic acids 7 were found by Xiao-Fu research team to undergo intramolecular decarboxylative C–N coupling reaction in the presence of 10 mol% of Cu(OTf)2 as a low-cost commercially available catalyst, 30 mol% of DMAP as additive and 2 equiv. of PhIO as an oxidant, using dichloromethane as solvent at 100 °C, to afford the corresponding 5- and 6-membered cyclic amines 8 in moderate to high yields (Scheme 3).23 The reaction was also tolerated secondary aliphatic carboxylic acids and provided the expected products with yield range from 42 to 48%. However, tertiary carboxylic acids failed to participate in this transformation. The results revealed that the cyclization of chiral primary carboxylic acids under the standard condition gave the products with retention of configuration but in the cases of secondary carboxylic acids diastereocontrol of this reaction was not good and mixture of diastereoisomers were obtained. Interestingly, this reaction also showed outstanding site-selectivity in the C–N bond formation process. The selectivity was found to be dictated by the N-protecting groups on the substrates. An example of a site-selective reaction is shown in Scheme 4, where the substrates having both the Bz- and PA-protected amino groups, the products cyclized at the PANH side were obtained as the sole products. Noteworthy, the size of newly forming rings would not affect the site-selectivity. Based on literature reports, a possible mechanism was proposed by the authors (Scheme 5), whereby the reaction is initiated by reaction of starting carboxylic acid 7a with PhIO in the presence of CuII–L to give hypervalent iodine(III) intermediate A, which undergoes a homolytic cleavage of one I–O bond affording two radical intermediates B and C. The intermediate B is then convert to an alkyl radical intermediate D through a decarboxylation process. Then, oxidative addition of the alkyl radical to the CuII which is chelated by the directing group affords the intermediate E, which after reductive elimination delivers the final product 8a and the intermediate CuI–L. Finally, the whole catalytic cycle is accomplished with the oxidation of CuI–L to CuII–L in the presence of intermediate C or decomposition products thereof. Shortly thereafter, Yang, Jiang, and Shi theoretically investigated the detailed mechanism of this reaction using density functional theory (DFT) calculations.24 Some important information of this study is listed below: (i) the reaction proceeds through a IIII–O bond heterolysis/single electron transfer/hydrogen atom transfer/decarboxylation/proton transfer/reductive elimination sequential process; (ii) The heterolytic cleavage of the IIII–O bond is much easier with the help of +HDMAP, whereas the homolytic pathways need to overcome higher enthalpy barriers and thus show less feasibility; and (iii) the chelation of the PA directing group to the Cu(III) center significantly facilitates the proton transfer process, which is the rate-determining step.
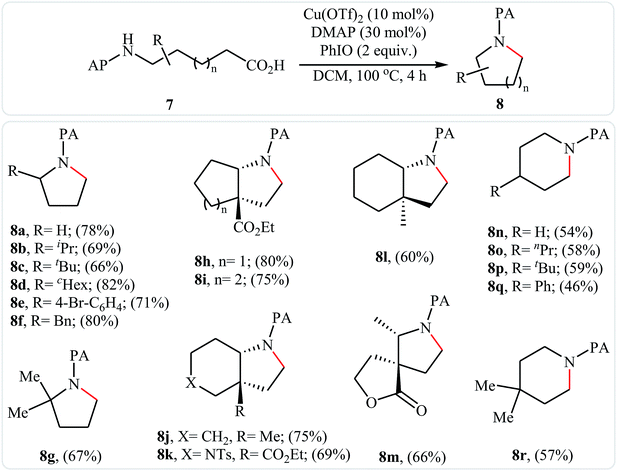 |
| | Scheme 3 Cu(II)-catalyzed intramolecular decarboxylative C–N coupling reaction of primary aliphatic carboxylic acids 7. | |
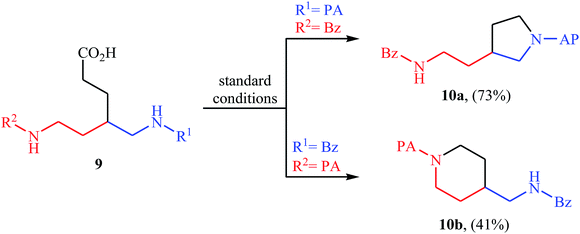 |
| | Scheme 4 Site-selectivity controlled by a directing group in the intramolecular decarboxylative C–N coupling process. | |
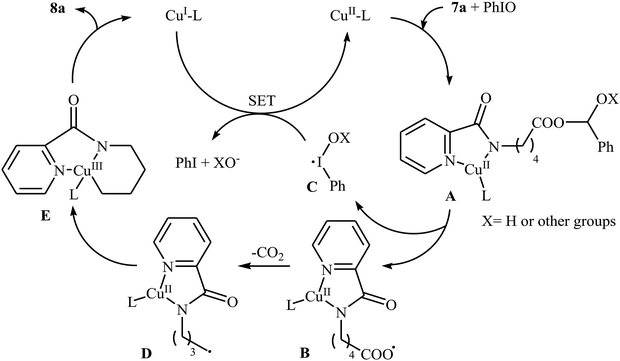 |
| | Scheme 5 Mechanism proposed to explain the formation of cyclic amines 8. | |
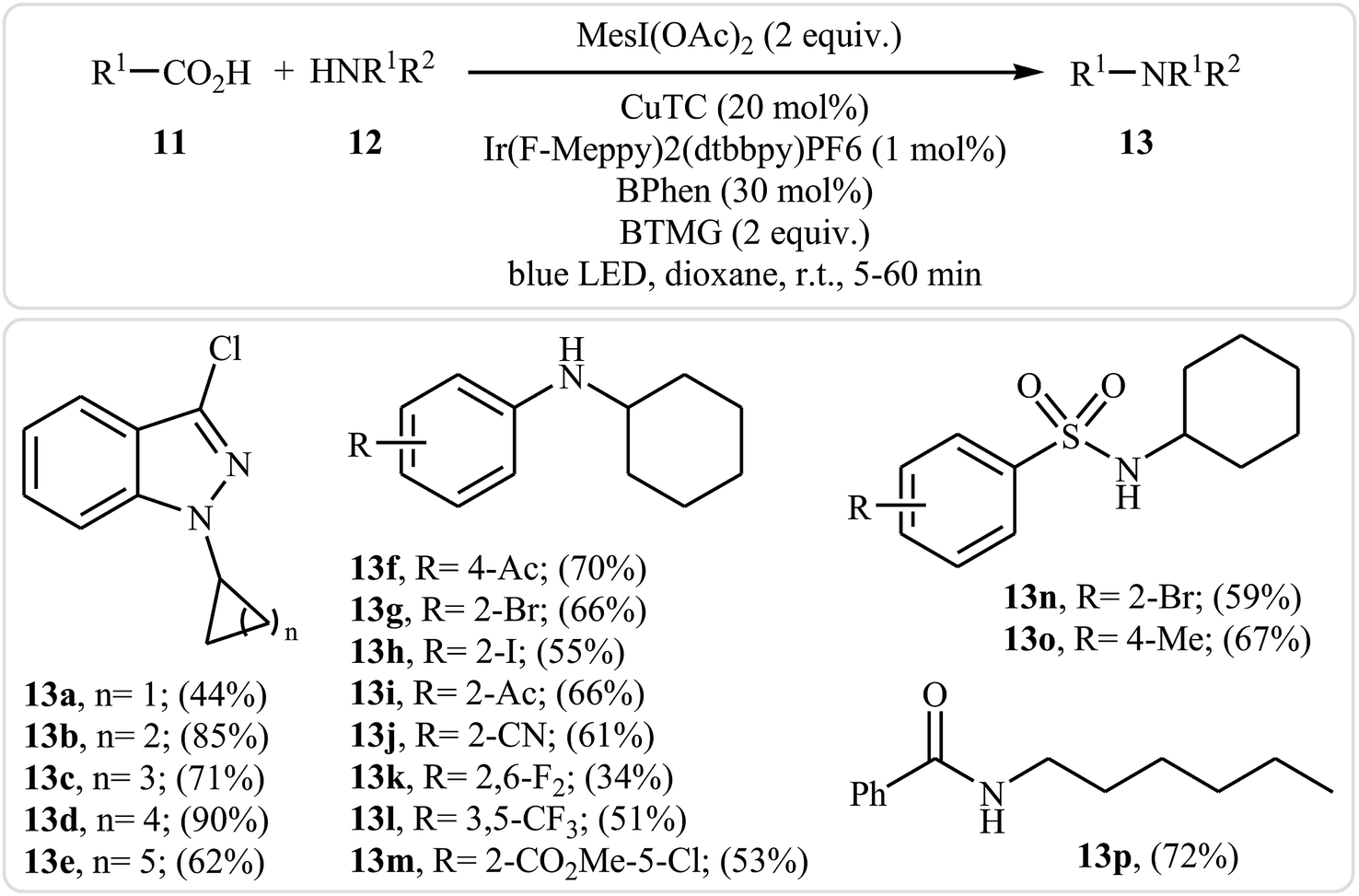
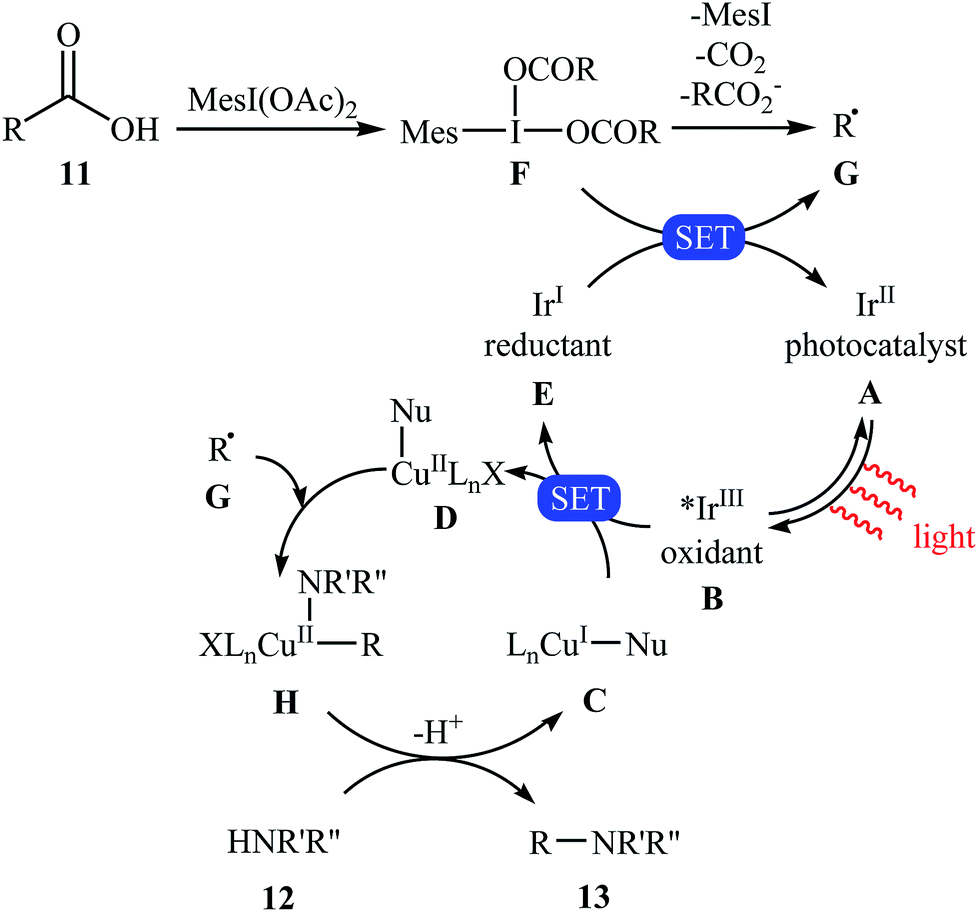
Very recently, MacMillan and colleagues reported an elegant intermolecular version of this reaction using a synergistic combination of copper(I) and photoredox catalysis.25 With the mesl(OAc)2/CuTC/Ir(F-Meppy)2(dtbbpy)PF6/BPhen/BTMG system, direct C–N coupling of a variety of primary, secondary and tertiary alkyl carboxylic acids 11 with a range of nitrogen nucleophiles (N-heterocycles, amides, sulfonamides and anilines) 12 proceeded efficiently to produce N-alkyl products 13 in moderate to almost quantitative yields. Some reported examples are shown in Scheme 6. It is noted that the reaction took place in dioxane at room temperature under irradiation of blue LED light (34 W) and tolerated a library of important functional groups, including fluoro, chloro, bromo, iodo, amide, ether, and ester functionalities. Thus this procedure offers scope for further elaboration of products. The authors assume that the mechanistic pathway of this transformation involves the initial formation of triplet-excited-state *IrIII complex B via the excitation of photocatalyst A under light irradiation. Meanwhile, coordination of the nitrogen nucleophile 12 with a copper(I) precatalyst followed by deprotonation forms the copper(I)-amido species C, which after oxidation by complex B gives the copper(II)-amido system D and the iridium(II) complex E. Next, reduction of iodomesitylene dicarboxylate F (which is generated from carboxylic acid 11 and iodomesitylene diacetate) by IrII species E leads to the desired alkyl radical G, while reconstituting the ground-state photocatalyst A. Finally, the capture of alkyl radical G by copper(II)-amido complex D yields copper(III) complex H, which upon reductive elimination affords C(sp3)–N bearing adduct 13 and regenerate copper(I) catalyst C (Scheme 7).
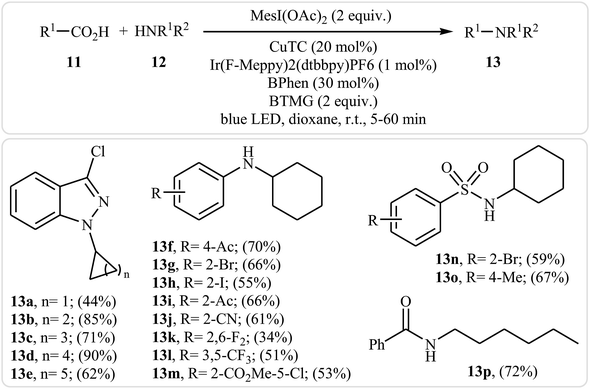 |
| | Scheme 6 Decarboxylative cross-coupling of alkyl carboxylic acids 11 with nitrogen nucleophiles 12 developed by MacMillan research team. | |
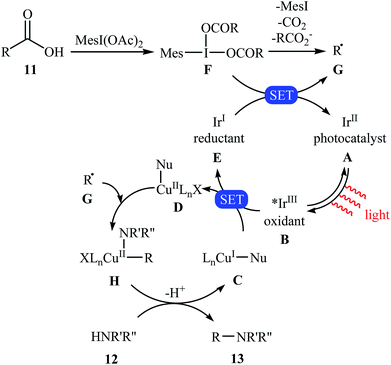 |
| | Scheme 7 Plausible mechanism for reaction in Scheme 6. | |
3. Cross-coupling reactions between Caryl–CO2H and N–H bonds
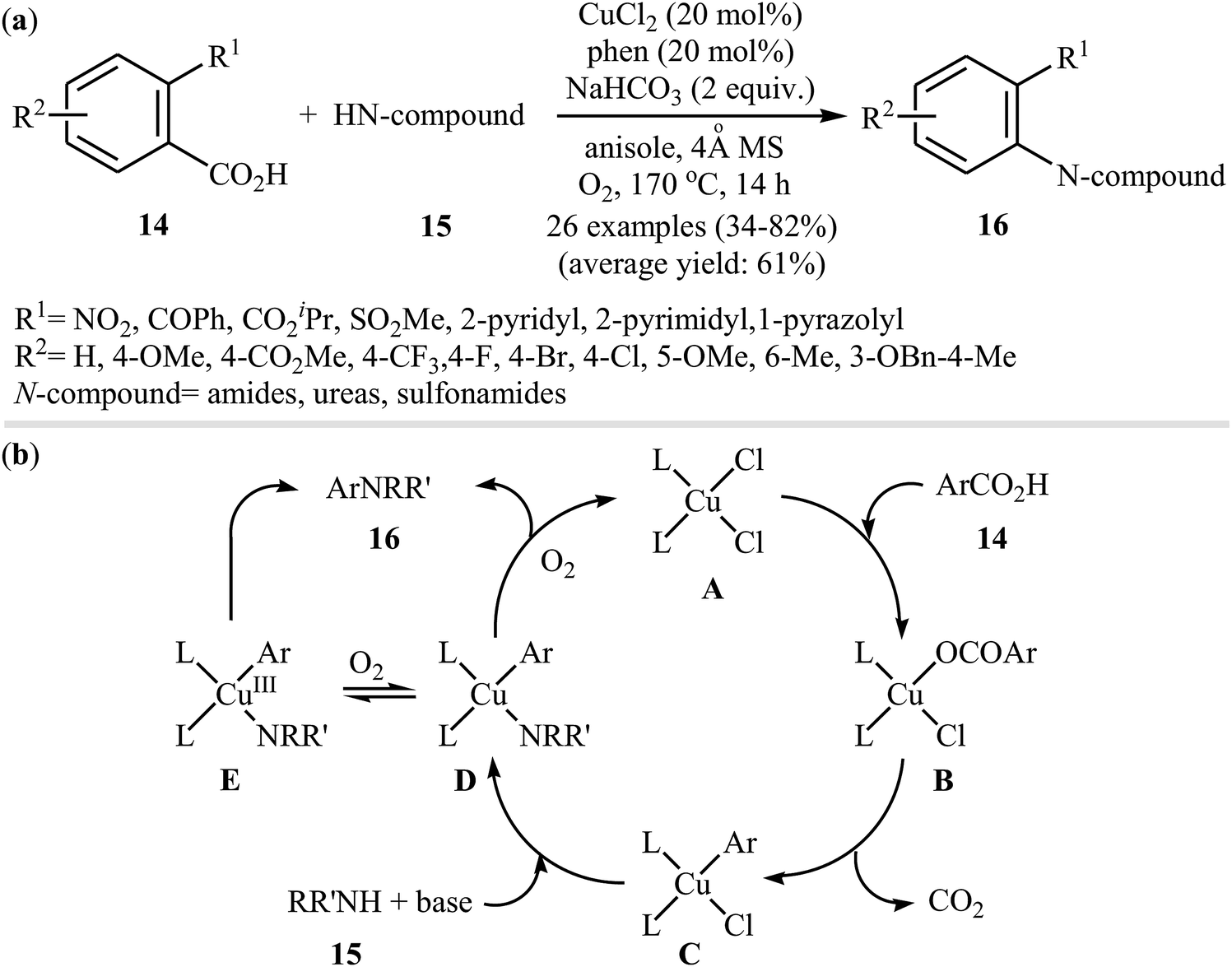
In 2012, Zhang, Patel, and Mainolfi demonstrated a simple and highly efficient method for the construction C(aryl)–N bonds from aromatic benzoic acids and amide derivatives via Cu-catalyzed decarboxylative cross-coupling reaction using molecular oxygen as the sole oxidant.26 After studying a number of catalysts, such as CuI, CuCl, CuCl2, Cu(OAc)2, and Cu(OTf)2, ligands, such as bpy and phen, and solvents, such as diglyme, tAmyOH, toluene, and anisole, a combination of CuCl2/phen/anisole with 2.0 equiv. of sodium bicarbonate as an inexpensive base at 170 °C was found to be optimum with respect to the yield of product isolated. Under the optimized conditions, several ortho-substituted benzoic acid derivatives 14 underwent coupling with various N-nucleophiles 15 and gave the expected N-arylated products 16 in modestly to high yields (Scheme 8a). The plausible mechanism for this decarboxylative C–N coupling transformation is shown in Scheme 8b and starts with the generation of a copper(II) carboxylate intermediate B from the reaction of benzoic acid 14 with copper catalyst A through a deprotonation process, which is followed by its decarboxylation to furnish intermediate C. Subsequently, the reaction of this intermediate with N-nucleophile 15 in the presence of a base gives complex D that is in equilibrium with the complex E. Finally, reductive elimination of these complexes affords the final product 16.
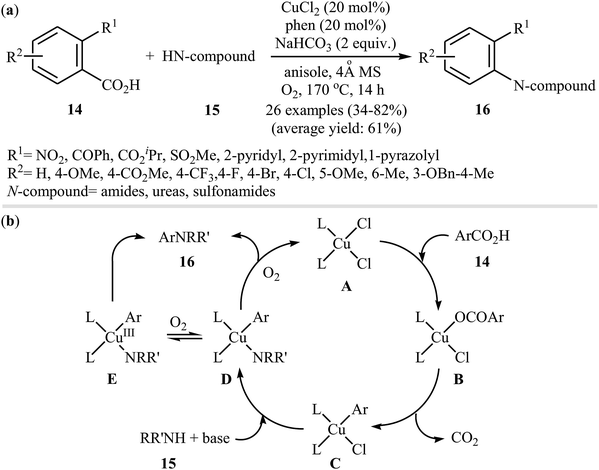 |
| | Scheme 8 (a) Cu(II)-catalyzed decarboxylative C–N coupling reaction between ortho-substituted benzoic acid derivatives 14 and nitrogen nucleophiles 15; (b) mechanism proposed to explain the N-arylated products 16 synthesis. | |
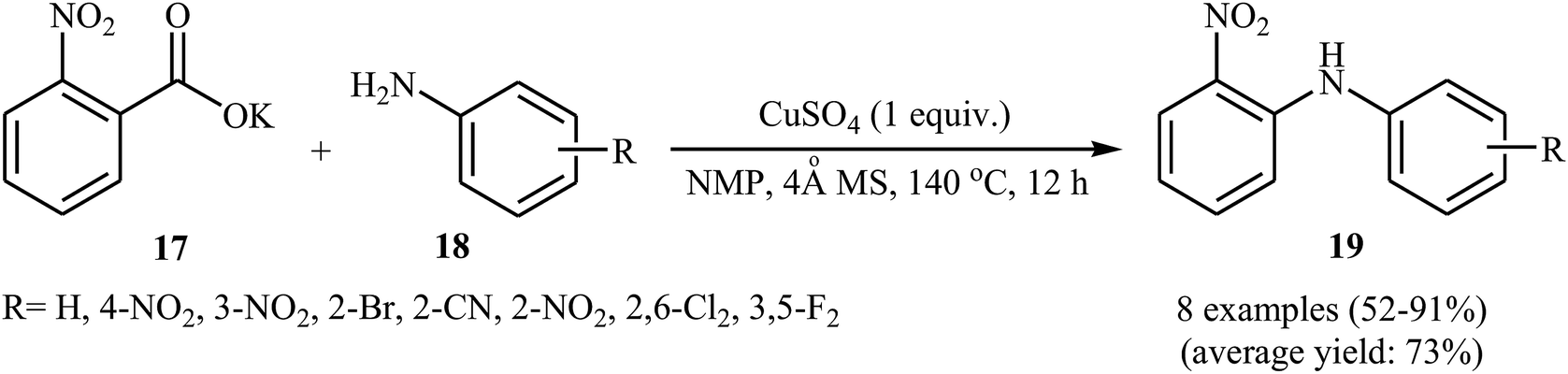
Not long after this report, Jia and co-workers extended the above cross-coupling to anilines.27 They showed that the reaction of potassium 2-nitrobenzoate 17 with primary anilines 18 in the presence of 1.0 equiv. of CuSO4 as mediator in NMP afforded the corresponding diarylamines 19 in moderate to excellent yields (Scheme 9). Satisfactorily, a series of important functional groups such as F, Cl, Br, NO2, and CN were well tolerated by the reaction conditions employed, thus providing a useful approach to functionalized diarylamines. Noteworthy, the reaction works also for amides if the Ag2CO3 additive is used. However, just like previous work, this protocol requires harsh reaction conditions (140 °C). In 2017, Sarkate and colleagues improved the efficiency of this reaction in the terms of reaction time, temperature, and yield by performing the process under microwave irradiation.28
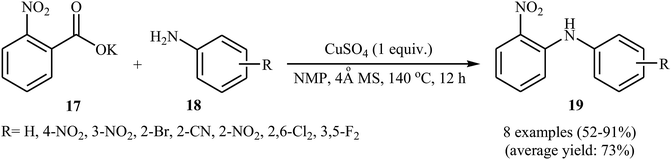 |
| | Scheme 9 Jia's synthesis of diarylamines 19. | |
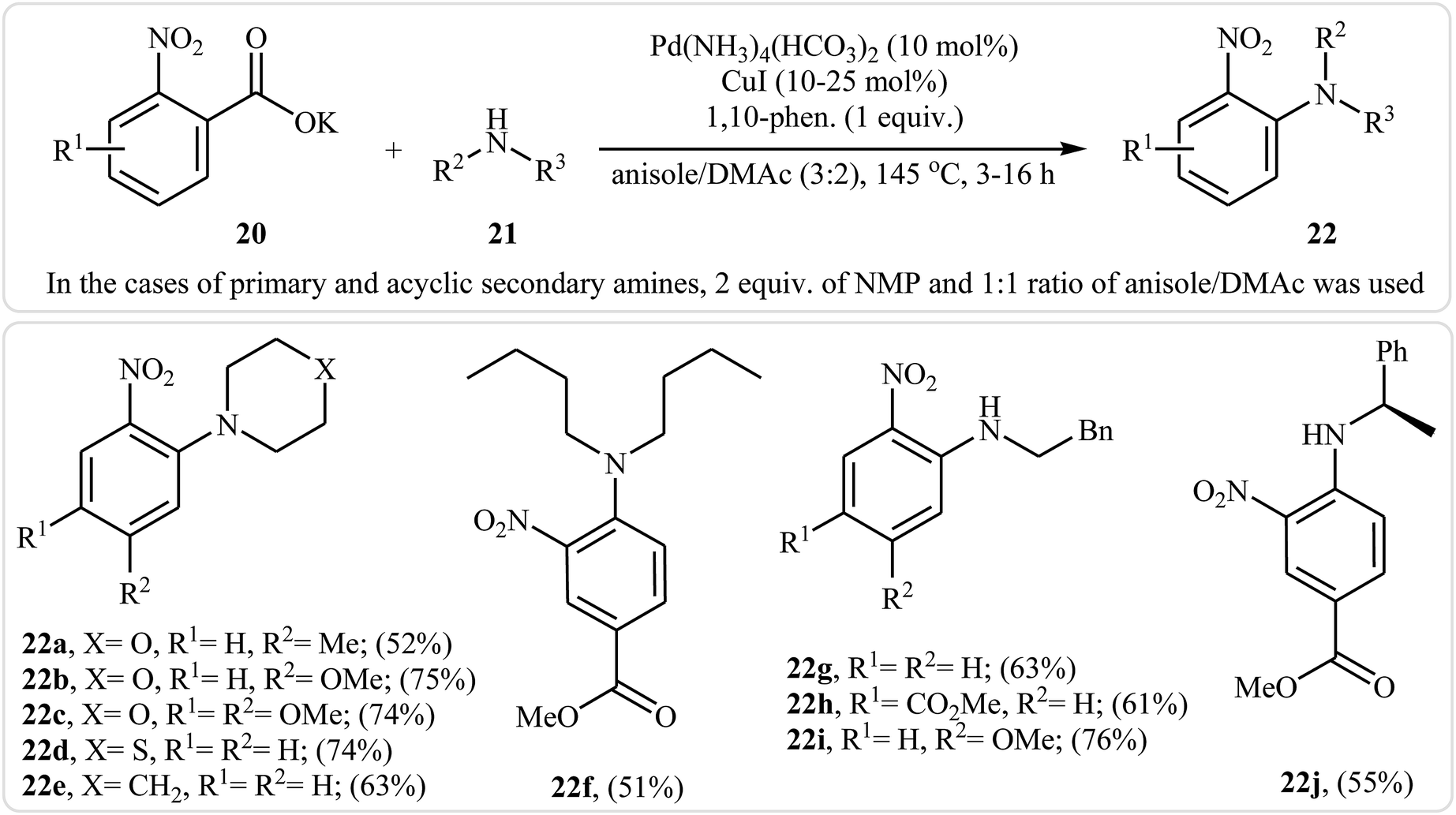
Very recently, Goossen and co-workers extended the substrate scope of this reaction system and reported the decarboxylative ipso-amination of potassium salt of electron-deficient benzoic acids 20 with aliphatic amines 21.29 A variety of N-arylated amines 22 were obtained in up to 81% yields in a solvent mixture of anisole and dimethylacetamide (DMAc) employing Pd(NH3)4(HCO3)2/CuI/1,10-phen combination as a catalytic system at 145 °C. Some reported examples are shown in Scheme 10. It should be mentioned that, in the cases of cyclic secondary amines, the best results were obtained under an air atmosphere without any additional oxidant, while for primary and acyclic secondary amines, the use of over-stoichiometric amounts of N-methylmorpholine N-oxide (NMO) as an oxidant gave the best results. Beside aliphatic amines, electron-rich anilines have also been successfully applied in this methodology.
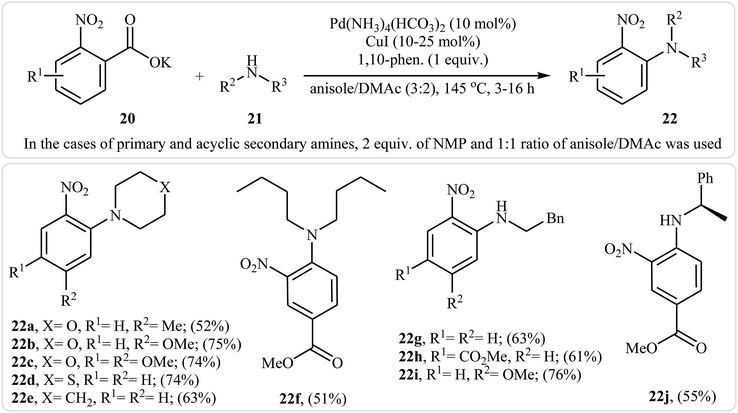 |
| | Scheme 10 Decarboxylative ipso-amination of benzoic acids 20 with aliphatic amines 21 reported by Gooßen. | |
4. Cross-coupling reactions between Ccarbonyl–CO2H and N–H bonds
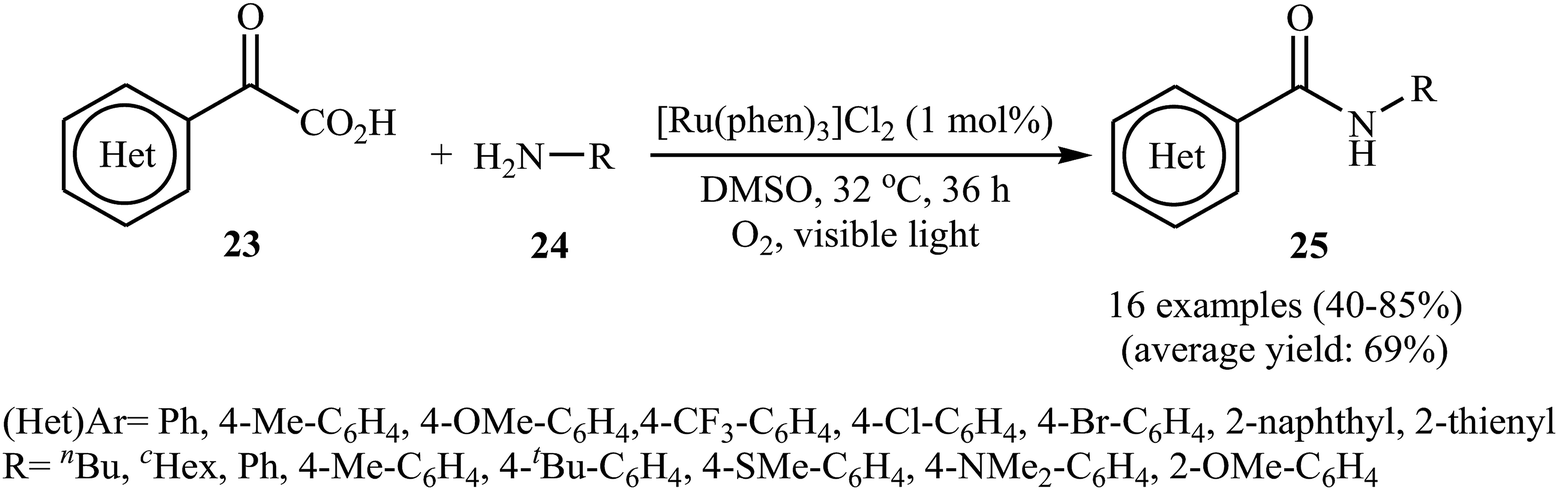
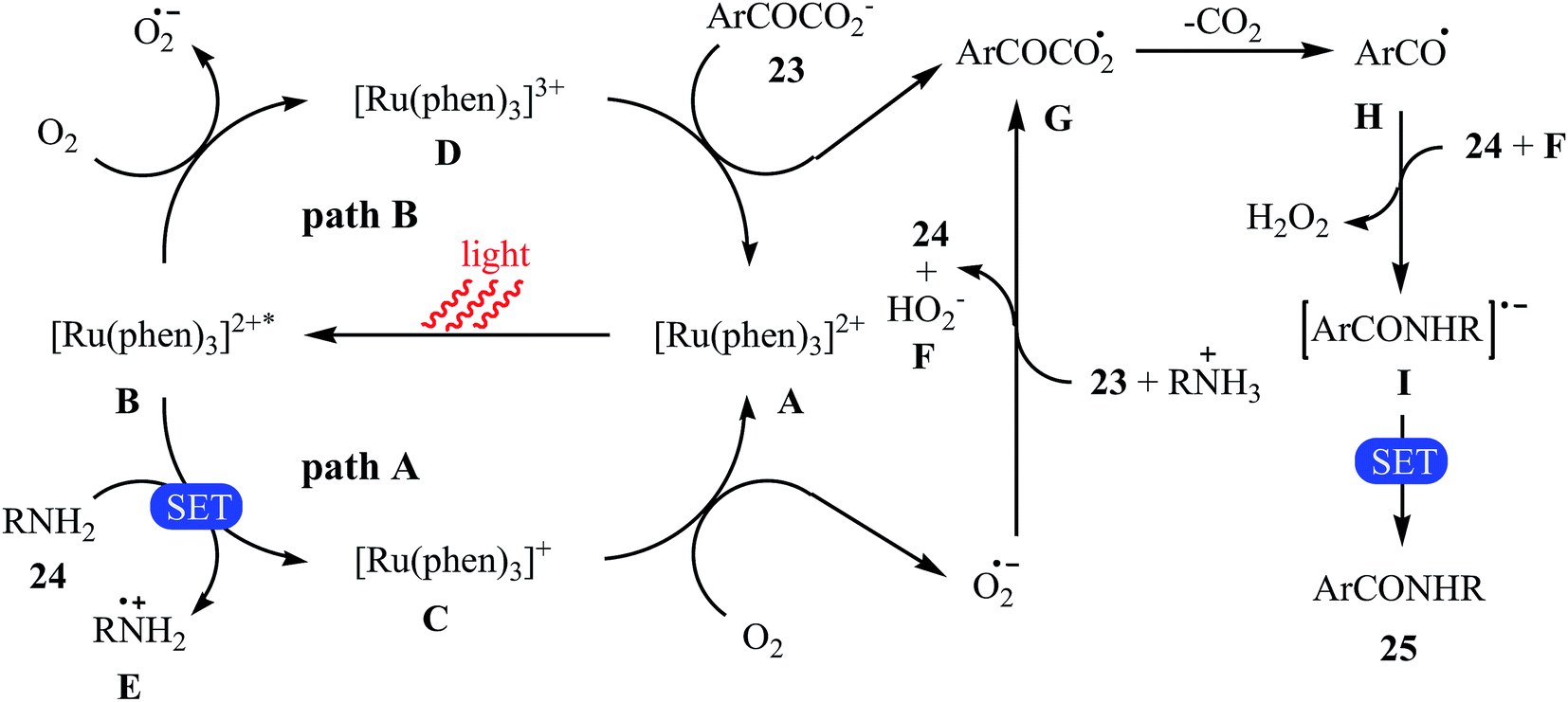
The first example of decarboxylative amidation of α-keto acids with amines was reported by Lan–Lei's research team in 2014; a broad range of 2-oxo-2-arylacetic acids 23 were reacted with various aliphatic and aromatic primary amines 24 in the presence of [Ru(phen)3]Cl2 as a photocatalyst under irradiation by visible light in the presence of molecular oxygen and afforded the corresponding secondary amides 25 in moderate to high yields (Scheme 11).30 2-Oxo-2-methylacetic acid did not work well in the reaction and therefore no other 2-oxo-2-alkylacetic acids were examined in the protocol. Furthermore, the reaction did not give satisfactory yield with N-propargylamine. Interestingly, when 2-phenylenediamine or analogously oxygen- or mercapto-substituted anilines are used, the corresponding 2-substituted benzazoles can be prepared through a tandem cross-coupling and condensation reaction. The authors proposed mechanism of this visible-light-mediated decarboxylative amidation reaction is depicted in Scheme 12. Similar amidation work was also reported by Xu and co-workers.31 They showed that the reaction of similar substrates in a mixed solvent of dioxane and water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under the irradiation of a 23 W fluorescent light bulb and catalyst-free conditions afforded the expected amides in 48 hours.
:1) under the irradiation of a 23 W fluorescent light bulb and catalyst-free conditions afforded the expected amides in 48 hours.
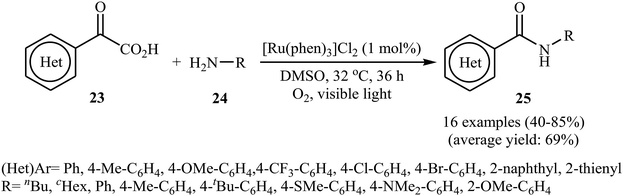 |
| | Scheme 11 Ru-catalyzed decarboxylative amidation of α-keto acids 23 with amines 24 reported by Lei. | |
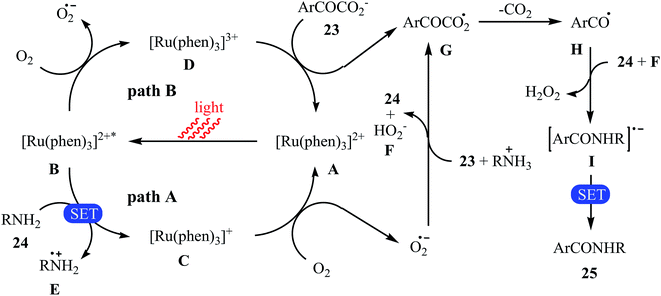 |
| | Scheme 12 Mechanism proposed to explain the secondary amides 25 synthesis. | |
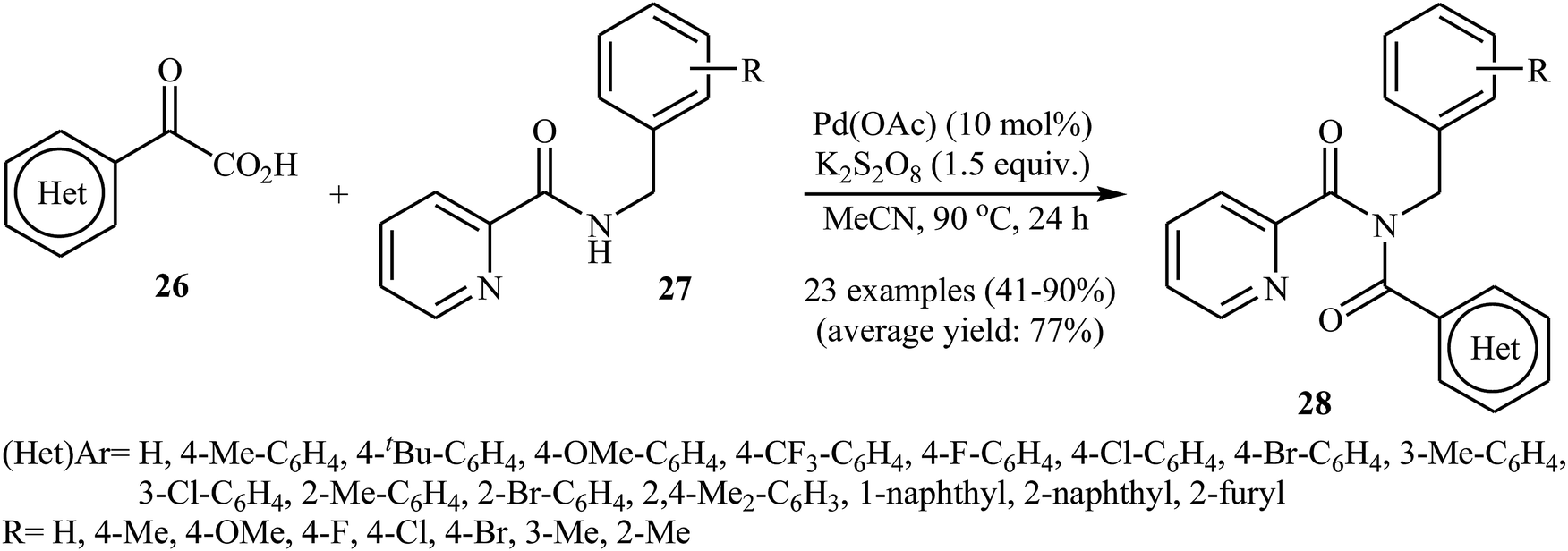
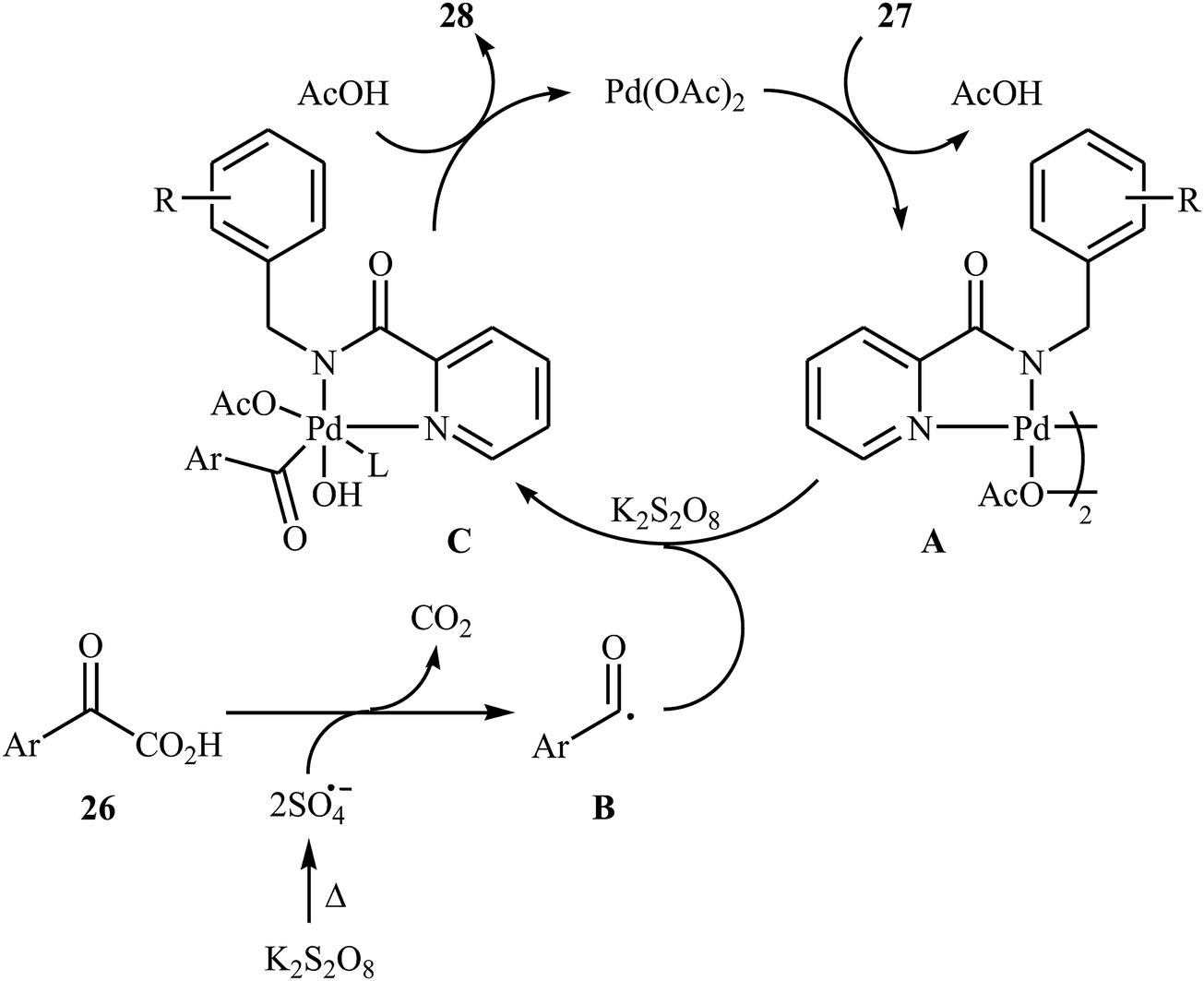
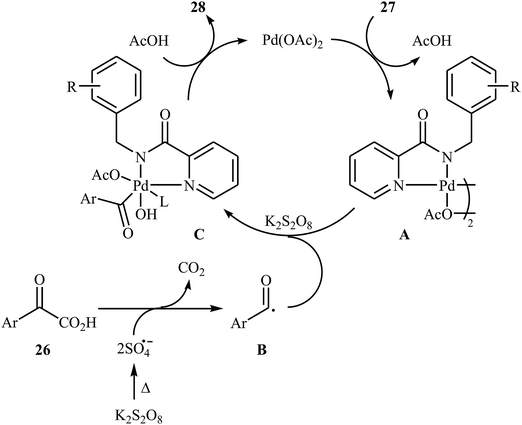
In 2016, Liu, Wang, and co-workers demonstrated an interesting amidation of α-keto acids 26 with N-benzylpicolinamide derivatives 27 through a palladium-catalyzed decarboxylative process.32 This new methodology provides a concise and efficient synthesis of various substituted imides 28 in fair to excellent yields. The results proved that the reaction was not dependent on the electronic and steric effects of substituents in the phenyl ring of N-benzylpicolinamides. Thus, the system tolerated both electron-withdrawing and electron-donating substituents on the para, ortho, and meta positions of aromatic ring of amides according to Scheme 13. However, the outcome of reaction was strongly dependent on the electronic character of the substituents on the phenyl ring periphery of 2-oxo-2-arylacetic acids. Generally, electron-rich carboxylic acids afforded better yields compared to electron-poor ones. It is noted that the strongly electron-poor 2-oxo-2-(p-NO2-phenyl)acetic acid failed to afford the product. To probe the mechanistic pathway of this reaction, a free-radical trap test was investigated. The reaction was completely inhibited in the presence of 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO), a radical scavenger, which suggested that a radical process is probably involved in this reaction. The authors mentioned that the reaction mechanism involves three steps (Scheme 14): (i) chelation of picolinamide 27 with Pd(OAc)2 to generate the five-membered intermediate A; (ii) reaction of A with the acyl radical B (generated by decarboxylation of α-keto acid 26 in the presence of K2S2O8) to form the Pd(IV)-intermediate C; and (iii) reductive elimination of intermediate C to afford the final product 28 along with the release of Pd(II) species.
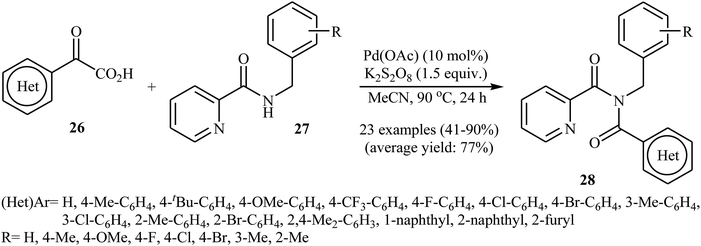 |
| | Scheme 13 Synthesis of imides 28 via Pd-catalyzed decarboxylative amidation of α-keto acids 26 with N-benzylpicolinamides 27. | |
 |
| | Scheme 14 Plausible mechanism for Liu–Wang's synthesis of imides 28. | |
In a related investigation, He and Xu along with their co-workers reported an Ag-catalyzed synthesis of secondary amides 31 from α-keto acids 29 and anilines 30 under an air atmosphere and mild conditions.33 In this study, various silver catalysts (e.g., AgF, AgOTf, AgBF4, AgNO3, Ag2CO3, Ag2O), and binary solvents (e.g., MeCN/H2O, DMSO/H2O, DMF/H2O, NMP/H2O, DCE/H2O) were examined and 2 equiv. of AgOTf in MeCN/H2O (2:1) was found to be optimal for this transformation. This reaction tolerates a wide range of functional groups such as fluoro, chloro, iodo, methoxy, hydroxyl and ester functionalities and could be applied to both aromatic and aliphatic α-keto acids (Scheme 15). Recently, the methodology was extended to a series of NH-free sulfoximines, using CuBr/K2S2O8 combination as an effective catalytic system, and the N-aroylsulfoximines were obtained with yield range from 14 to 91%.34
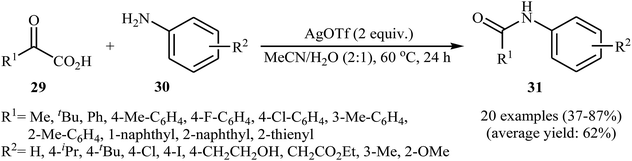 |
| | Scheme 15 Ag-catalyzed synthesis of secondary amides 31 from α-keto acids 29 and anilines 30 via a decarboxylative process. | |
5. Cross-coupling reactions between Calkynyl–CO2H and N–H bonds
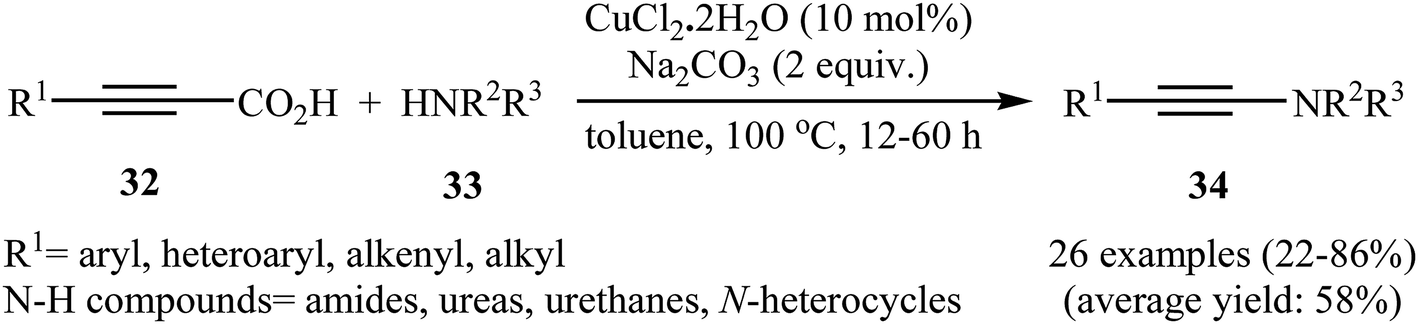
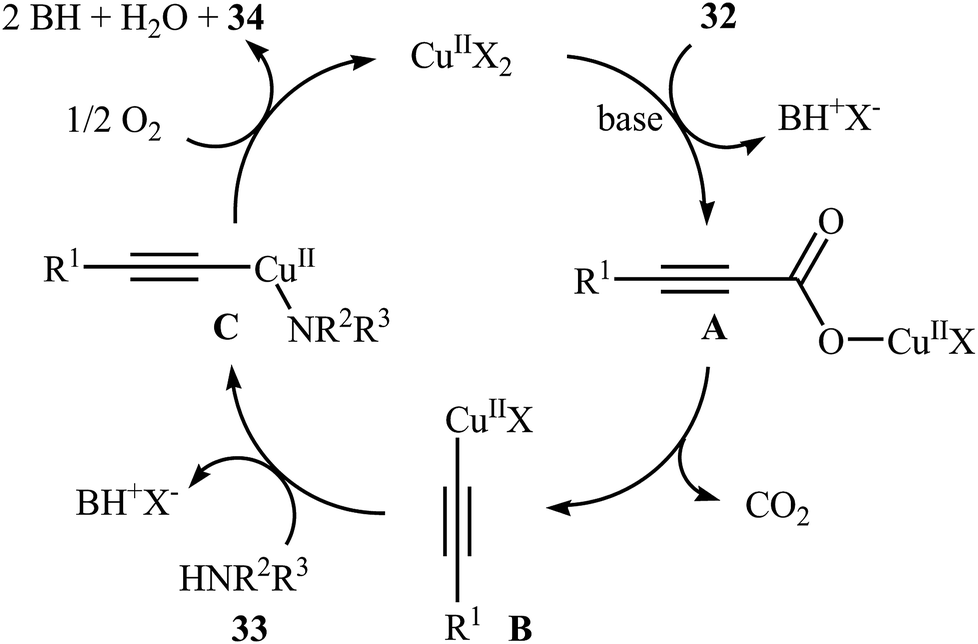
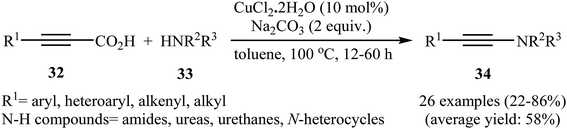
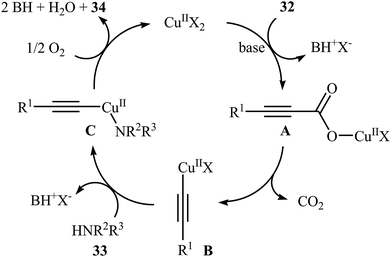
Although direct decarboxylative cross-coupling of alkynyl carboxylic acids with C–H bonds has been well known,35 the decarboxylative hetero-coupling reactions between C(sp)–CO2H and heteroatom–H (especially N–H) bonds were less developed. One of the earliest reports of the C–N bond forming reactions through decarboxylative cross-coupling of alkynyl carboxylic acids with N-nucleophiles has been reported by Jia and Jiao in 2010, when a range of aryl-, heteroaryl-, alkenyl-, and alkyl-substituted propiolic acids 32 underwent coupling reaction with N-nucleophiles 33 in the presence of 10 mol% of CuCl2 + 2H2O as a catalyst and 2.0 equiv. of Na2CO3 as a base in toluene to form the corresponding C(sp)–N bearing adducts 34 within 12–60 h (Scheme 16).36 The results showed that the reactivity order for the carboxylic acids under these reaction conditions was aryl-substituted propiolic acids > alkyl-substituted propiolic acids > alkenyl-substituted propiolic acids > heteroaryl-substituted propiolic acids. It is noteworthy that other commercially available Cu catalysts such as CuBr2, Cu2O, Cu(OAc)2 + H2O and CuSO4 + 5H2O were also found to promote this coupling reaction; albeit with reduced efficiencies. A proposed mechanistic possibility is depicted in Scheme 17. This transformation start with the formation of copper(II) intermediate A by reaction between the Cu(II) catalyst and carboxylic acid 32 in the presence of a base. Its decarboxylation leads to the alkynyl copper(II) intermediate B, which after nucleophilic attack by N-nucleophile 33 affords the CuII(alkynyl)(amidate) intermediate C. Finally, reductive elimination of this intermediate provides the expected product 34 and the copper catalyst is recycled.
 |
| | Scheme 16 Cu(II)-catalyzed amidation of propiolic acids 32 with nitrogen nucleophiles 33. | |
 |
| | Scheme 17 Plausible mechanism for the reaction in Scheme 16. | |
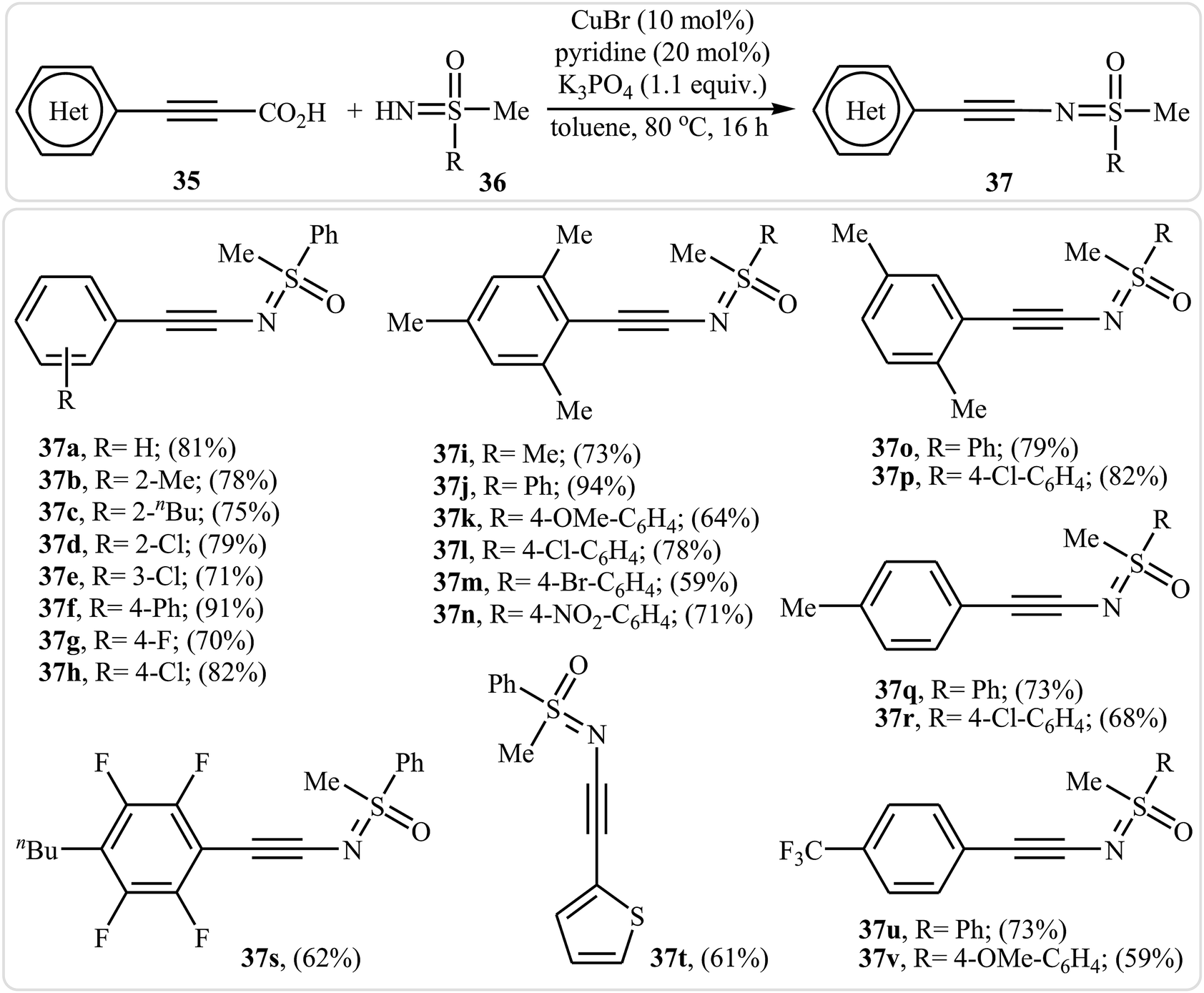
Inspired by this work, in 2013, the group of Bolm developed an interesting N-alkynylated sulfoximines synthetic strategy via Cu(I)-catalyzed decarboxylative coupling of aryl propiolic acids with sulfoximines under air atmosphere.37 Different variables affecting the reaction (e.g., such as catalyst, base, additive, solvent, and temperature) were carefully studied and optimized. It was found that using CuBr/K3PO4/pyridine combination as a catalytic system resulted in the best yields. Among the various solvents like DMF, DMSO, DCM, toluene, and 1,4-dioxane, toluene was the most efficient for this reaction. It should be mentioned that the presence of pyridine was crucial for the success of the reaction since it minimize the occurrence of the Glaser–Hay homocoupling. Various aryl propiolic acids 35 reacted well with N–H sulfoximines 36 under the standard conditions to produce the corresponding sulfoximidoyl-functionalized alkynes 37 in good to excellent yields (Scheme 18). It is observed that several functional groups, such as F, Cl, Br, OMe, and NO2 groups, on the aromatic ring of both substrates were well-tolerated. This transformation provides many opportunities for applications to organic synthesis. For example, the synthesized N-alkynylated sulfoximines could be used to prepare alkenylated sulfoximines by reduction of their alkyne moieties, and cyclobutenone sulfoximines by reaction with isobutyryl chloride.
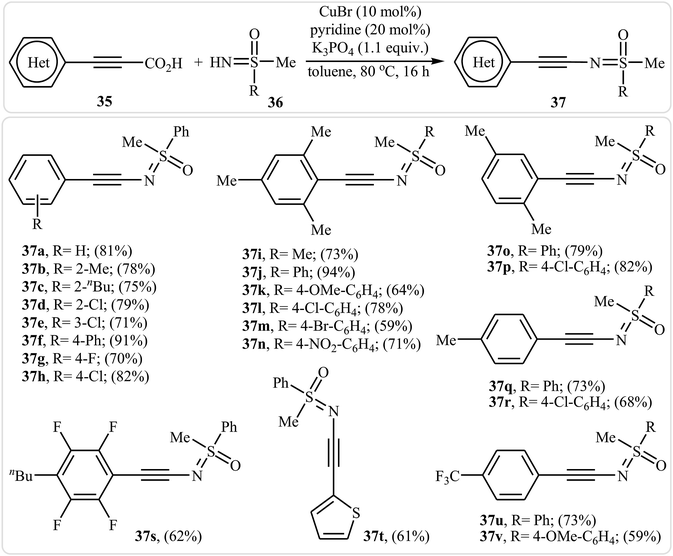 |
| | Scheme 18 Bolm's synthesis of sulfoximidoyl-functionalized alkynes 37. | |
6. Conclusion
In summary, this focus-review highlights the recent discoveries and developments in decarboxylative cross-coupling reactions between carboxylic acids and N–H compounds. This strategy enables the facile construction of carbon–nitrogen bonds by avoiding the use of environmentally hazardous organohalide or organometallic reagents, thereby providing an efficient and green approach to various biologically and synthetically important nitrogen-containing organic molecules. As illustrated, all the three kinds of C–N bonds [C(sp3)–N, C(sp2)–N, C(sp)–N] could be effectively formed by this methodology demonstrating the general applicability of the procedure. Despite stellar achievements during the past nine years in this research arena, many challenges still remain to be overcome: (i) the number of reported examples are narrow and there is further need to study the scope and limitations of these reactions; (ii) most of the reactions covered in this review are limited to the use of bulk metal catalysts. Therefore, exploration of organocatalysts and nano-sized metal catalysts are highly desirable from the view point of green chemistry; and (iii) other reactions such as coupling of alkenyl carboxylic acids with N–H compounds should be explored. We conclude this review by hoping that it will stimulate researchers to further thinking and research in this domain.
Conflicts of interest
There are no conflicts to declare.
References
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
-
(a) S. A. Lawrence, Amines: synthesis, properties and applications, Cambridge University Press, Cambridge, UK, 2004 Search PubMed;
(b) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS;
(c) P. Devendar and G.-F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed;
(d) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed.
-
(a) F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159–2232 CrossRef CAS PubMed;
(b) A. El Kadib, ChemSusChem, 2015, 8, 217–244 CrossRef CAS PubMed;
(c) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed;
(d) S. Arshadi, E. Vessally, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, RSC Adv., 2017, 7, 13198–13211 RSC;
(e) N. Bhattacharyya, S. Jha, S. Jha, T. Y. Bhutia and G. Adhikary, Int. J. Appl. Pharm., 2012, 4, 295–304 Search PubMed.
-
(a) R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS;
(b) H. Saeidian and M. Abdoli, J. Sulfur Chem., 2015, 36, 463–470 CrossRef CAS.
- K. K. Swamy, N. B. Kumar, E. Balaraman and K. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed.
- T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
-
(a) H. Bader, A. R. Hansen and F. McCarty, J. Org. Chem., 1966, 31, 2319–2321 CrossRef CAS;
(b) P. Magdolen, M. Mečiarová and Š. Toma, Tetrahedron, 2001, 57, 4781–4785 CrossRef CAS;
(c) J. L. Bolliger and C. M. Frech, Tetrahedron, 2009, 65, 1180–1187 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- J. X. Qiao and P. Y. Lam, Synthesis, 2011, 829–856 CrossRef CAS.
-
(a) M. O. Frederick, J. A. Mulder, M. R. Tracey, R. P. Hsung, J. Huang, K. C. Kurtz, L. Shen and C. J. Douglas, J. Am. Chem. Soc., 2003, 125, 2368–2369 CrossRef CAS PubMed;
(b) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742–7743 CrossRef CAS PubMed.
- G. J. Perry and I. Larrosa, Eur. J. Org. Chem., 2017, 3517–3527 CrossRef CAS PubMed.
-
(a) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380–7391 RSC;
(b) P. Liu, G. Zhang and P. Sun, Org. Biomol. Chem., 2016, 14, 10763–10777 RSC.
- T. Zhang, N.-X. Wang and Y. Xing, J. Org. Chem., 2018, 83, 7559–7565 CrossRef CAS PubMed.
- N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC.
- A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS.
-
(a) Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS;
(b) M. Satish Kumar, K. C. Rajanna, M. Venkateswarlu, P. Venkanna and P. K. Saiprakash, Synth. Commun., 2015, 45, 2251–2258 CrossRef CAS.
-
(a) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef CAS;
(b) E. Vessally, R. Mohammadi, A. Hosseinian, K. Didehban and L. Edjlali, J. Sulfur Chem., 2018, 39, 443–463 CrossRef CAS;
(c) A. Hosseinian, L. Zare Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur Chem., 2018, 39, 674–698 CrossRef CAS;
(d) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(e) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(f) K. Nejati, S. Ahmadi, M. Nikpassand, P. D. K. Nezhad and E. Vessally, RSC Adv., 2018, 8, 19125–19143 RSC;
(g) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(h) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(i) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(j) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(k) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(l) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed.
-
(a) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS;
(b) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef CAS;
(c) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(d) E. Vessally, K. Didehban, M. Babazadeh, A. Hosseinian and L. Edjlali, J. CO2 Util., 2017, 21, 480–490 CrossRef CAS;
(e) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2017, 21, 342–352 CrossRef CAS;
(f) K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef CAS;
(g) E. Vessally, R. Mohammadi, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 24, 361–368 CrossRef CAS;
(h) S. Farshbaf, L. Z. Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(i) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(j) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC;
(k) E. Vessally, A. Hosseinian, L. Edjlali, M. Babazadeh and K. Didehban, Mini-Reviews Org. Chem., 2018, 15, 315–323 CrossRef CAS;
(l) E. Vessally, A. Hosseinian, M. Babazadeh, L. Edjlali and R. Hosseinzadeh-Khanmiri, Curr. Org. Chem., 2018, 22, 315–322 CrossRef CAS;
(m) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 71662–71675 RSC;
(n) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 99781–99793 RSC;
(o) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, RSC Adv., 2017, 7, 7079–7091 RSC;
(p) S. Arshadi, E. Vessally, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, RSC Adv., 2017, 7, 13198–13211 RSC;
(q) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D Esrafili, Curr. Org. Synth., 2017, 14, 557–567 CrossRef CAS;
(r) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–637 CrossRef CAS PubMed;
(s) E. Vessally, R. Hosseinzadeh-Khanmiri, E. Ghorbani-Kalhor, M. Es' haghi and A. Bekhradnia, RSC Adv., 2017, 7, 19061–19072 RSC;
(t) S. Soleimani-Amiri, E. Vessally, M. Babazadeh, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 28407–28418 RSC;
(u) E. Vessally, A. Hosseinian, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, J. Iran. Chem. Soc., 2017, 14, 2339–2353 CrossRef CAS;
(v) M. Babazadeh, S. Soleimani-Amiri, E. Vessally, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 43716–43736 RSC;
(w) E. Vessally, M. Babazadeh, K. Didehban, A. Hosseinian and L. Edjlali, Curr. Org. Chem., 2017, 21, 2561–2572 CrossRef CAS;
(x) E. Vessally, M. Babazadeh, A. Hosseinian, L. Edjlali and L. Sreerama, Curr.
Org. Chem., 2018, 22, 199–205 CrossRef CAS;
(y) E. Vessally, M. Babazadeh, K. Didehban, A. Hosseinian and L. Edjlali, Curr. Org. Chem., 2018, 22, 286–297 CrossRef CAS;
(z) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed.
- Y. Yan and Z. Wang, Chem. Commun., 2011, 47, 9513–9515 RSC.
- Y. Yan, M. Shi, B. Niu, X. Meng, C. Zhu, G. Liu, T. Chen and Y. Liu, RSC Adv., 2016, 6, 36192–36197 RSC.
- Z.-J. Liu, X. Lu, G. Wang, L. Li, W.-T. Jiang, Y.-D. Wang, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2016, 138, 9714–9719 CrossRef CAS PubMed.
- Y.-N. Yang, J.-L. Jiang and J. Shi, Organometallics, 2017, 36, 2081–2087 CrossRef CAS.
- Y. Liang, X. Zhang and D. W. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed.
- Y. Zhang, S. Patel and N. Mainolfi, Chem. Sci., 2012, 3, 3196–3199 RSC.
- W.-J. Sheng, Q. Ye, W.-B. Yu, R.-R. Liu, M. Xu, J.-R. Gao and Y.-X. Jia, Tetrahedron Lett., 2015, 56, 599–601 CrossRef CAS.
- A. P Sarkate, D. N. Pansare, K. S Karnik, I. A Kale, S. S Bahekar and D. B Shinde, Curr. Microwave Chem., 2017, 4, 163–167 CrossRef.
- M. Pichette Drapeau, J. Bahri, D. Lichte and L. J. Goossen, Angew. Chem., Int. Ed., 2018, 131, 902–906 CrossRef.
- J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef CAS PubMed.
- W.-T. Xu, B. Huang, J.-J. Dai, J. Xu and H.-J. Xu, Org. Lett., 2016, 18, 3114–3117 CrossRef CAS PubMed.
- N. Xu, J. Liu, D. Li and L. Wang, Org. Biomol. Chem., 2016, 14, 4749–4757 RSC.
- X.-L. Xu, W.-T. Xu, J.-W. Wu, J.-B. He and H.-J. Xu, Org. Biomol. Chem., 2016, 14, 9970–9973 RSC.
- C. Pimpasri, L. Sumunnee and S. Yotphan, Org. Biomol. Chem., 2017, 15, 4320–4327 RSC.
- K. Park and S. Lee, RSC Adv., 2013, 3, 14165–14182 RSC.
- W. Jia and N. Jiao, Org. Lett., 2010, 12, 2000–2003 CrossRef CAS PubMed.
- D. L. Priebbenow, P. Becker and C. Bolm, Org. Lett., 2013, 15, 6155–6157 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Aazam Monfared
*a and
Esmail Vessally
d,
Aazam Monfared
*a and
Esmail Vessally