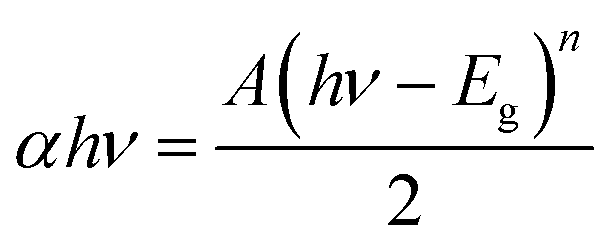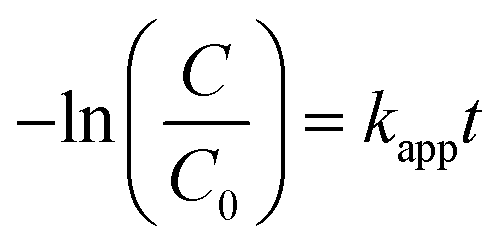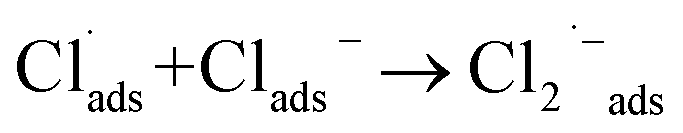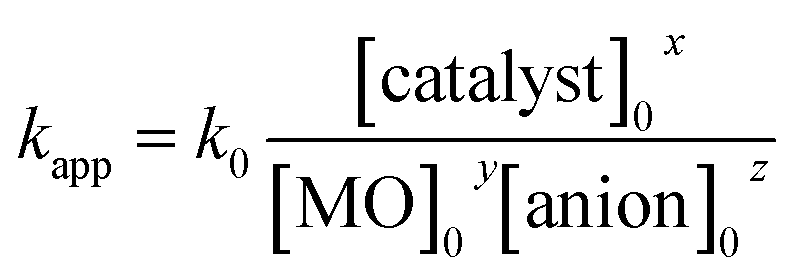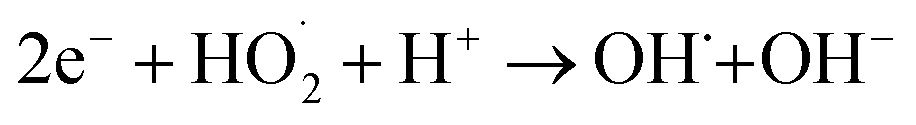DOI:
10.1039/C9RA00548J
(Paper)
RSC Adv., 2019,
9, 8977-8993
Photocatalytic activity of Ni0.5Zn0.5Fe2O4@polyaniline decorated BiOCl for azo dye degradation under visible light – integrated role and degradation kinetics interpretation†
Received
22nd January 2019
, Accepted 4th March 2019
First published on 19th March 2019
Abstract
Herein, we demonstrated the excellent improvement in photocatalytic degradation performance of BiOCl upon facile heterogeneous decoration with an integrated Ni0.5Zn0.5Fe2O4@polyaniline nanocomposite for an organic pollutant, methyl orange dye (MO), under visible light irradiation. The physico-chemical nature of the heterogeneous nanocomposite was characterized by XRD, FTIR, HRTEM-EDX and XPS analyses. The tuning of the band gap and optical sensitivity of BiOCl using Ni0.5Zn0.5Fe2O4@polyaniline were measured by DRS, PL and EIS techniques. To validate the transformation of the BiOCl photocatalyst to a visible light active photocatalyst due to the incorporation of Ni0.5Zn0.5Fe2O4@polyaniline and to gain insight into the origin of the synergistic effect for dye degradation by the heterostructured nanocomposite, we explored the effects of process parameters such as catalyst dosage, initial dye concentration, pH and the presence of inorganic anions on the extent of photo degradation. To get more details about reaction kinetics, a kinetic model using non-liner regression analysis was developed and the validity of the model was tested by comparing the experimental values with the calculated data. Based on the intermediate product formation, identified by GC-MS, a probable degradation pathway and a mechanism based on the electrochemical behaviour of the developed catalyst and trapping experiments were also proposed.
1. Introduction
The presence of toxic organic pollutants has had great repercussions on the environment and public health and become a global issue recently.1,2 The research in the field of effluent treatment has proclaimed that the use of semiconductor photocatalysts is expected to be a potential solution to degrade toxic organic pollutants and could solve many environmental problems. Among the various semiconductor photocatalysts, the integrated heterogeneous nanoscale photocatalysts appear to be one of the most promising technologies and a thriving strategy to treat wastewater as they use direct solar energy and generate no secondary pollutants.3,4 The search for an integrated nanoscale heterogeneous catalyst has been ongoing since the materialisation of TiO2 as a photocatalyst that is limited in its application as a visible light photocatalyst because of its large band gap, rapid recombination of photogenerated charge carriers and low photostability.5,6 Toward this goal, many researchers have successfully designed nanostructured heterogeneous photocatalysts to manifest visible light harvesting and photocatalytic performance of TiO2 marked by intrinsic fast collection and recombination of photoinduced charge carriers.7,8 In this plethora of heterostructured nanosize photocatalysts, in recent years, bismuth based photocatalysts have also attracted great attention in the design of heterogeneous photocatalysis due to their outstanding electrical, optical and photocatalytic properties.9 Among them, the ternary oxide semiconductor and member of the Sillen family of composites bismuth oxychloride (BiOCl) has drawn much interest due to its good biocompatibility, high photocorrosion stability, unique layered structure and dye photosensitization degradation capability.10,11 In this respect, many researchers have explored strategies to build heterostructured BiOCl photocatalysts with improved photocatalytic activity and a broader photoresponse by doping metal or non-metal elements or forming heterojunctions.12
With this particular interest, many research efforts have also been made to combine conjugated compounds/polymers such as graphene, carbon, polyaniline etc. with semiconductor photocatalysts to improve efficiency, especially in terms of cost, the catalytic degradation performance and photocorrosion stability.13,14 Conjugated conducting polyaniline, as an innovative new material, exhibits many extraordinary physical properties such as a chemically inert nature, excellent mobility of charge carriers and relatively good optical properties.15,16 Under visible light, polyaniline being an excellent photosensitizer acts as an efficient electron donor as well as a good hole transporter.17 For example, the role of polyaniline serving as an electron transfer medium in polyaniline–BiOCl photocatalysts has been demonstrated by Wang et al.18 On the basis of these properties, the incorporation of polyaniline into heterojunction photocatalysts not only enhances catalytic efficiency but also accelerates the degradation kinetics due to the integrated nanostructure and synergistic functionalities. Therefore, polyaniline (PAni) provides a potential platform for designing hybrid heterostructures to advance into the field of visible light driven catalysis for sustainable water treatment.
Lately, the PAni@transition metal ferrite based hetero-nanocomposites have attracted attention due to their reasonable catalytic activity under visible light with a narrow band gap and efficient charge transfer by the hopping process within different valence states.19 Additionally PAni@transition metal ferrites exhibit the integrated advantages of a narrow band gap energy corresponding well to the spectrum of sunlight, which makes them promising nanomaterials in the area of visible light driven photocatalysis. The integration of ferrites with other semiconductors or conductive materials adds many advantages such as photocorrosion stability, potential separation efficiency of photogenerated charge carriers, low toxicity, easy preparation and separation, superparamagnetic properties, cost effectiveness as well as overall enhancement in the photocatalytic activity and stability of composites.20–22 Among the classes of transition metal ferrites, nickel ferrite is significant because of its excellent chemical stability, remarkable mechanical hardness and ferromagnetic behaviour.23 What's more, in our previous work, we successfully fabricated dual phase nickel-zinc ferrite (Ni0.5Zn0.5Fe2O4) through a co-precipitation method24 and explored the enhancement of conductivity of polyaniline in the presence of multiferrite.25
Regarding morphological and architectural manipulation, tailoring of the heterojunction between two semiconductors also plays a crucial role in facilitating the efficiency of energy harvesting and enhances the separation of charge carriers for photoinitiated electro-chemical devices such as solar cells and catalytic reactors.26 Although heterostructured photocatalysts are not categorised based on the photocatalytic and photoelectrochemical reactions in which they are active as these technologies face common challenges for achieving immense efficiency, commercially, heterostructured photocatalysts are mainly established in the fields of water splitting into oxygen and hydrogen for energy conversion and depollution of organic contaminants in aqueous medium. The water splitting efficiency is now well controlled by tuning the valence and conduction band energy gap of electrochemical compounds to the level of the water redox couple. Oppositely, in the domain of the photocatalytic degradation of organic pollutants, it is generally believed that modulated photocatalysts are able to generate active radicals such as OH˙ (hydroxyl radicals) and O2˙− (superoxide radical ions) or the mechanism may only proceed through photogenerated holes and electrons produced from the catalyst surfaces leading to reaction intermediates by the basic three steps, i.e. the generation, transfer and consumption of charge. In efforts to connect each step in the charge kinetics, some fundamental models have been considered for the synthesis and manufacture of photocatalytic materials with the band-gap energy principles. The materialization of different models has moved the photocatalytic process forward from single component to multicomponent catalysts and the systems have developed more complicated kinetics as the generation and consumption of charge may occur simultaneously at different components as well as the transfer of charge taking place from one component to another one. Moreover, the surface chemistry, morphology and their correlation with the charge carrier transport ability in the photocatalysts have already been reported in several studies and it was reported that the reaction kinetics, mechanism and photonic reaction are complicated and difficult to determine, especially for reactions on heterostructure catalyst surfaces.27 Therefore, dye degradation is mainly unfavoured as a model reaction route for evaluating activities in different catalysts and the formation of intermediate products. Recently, several research works reported the excellent improvement and accelerated photocatalytic dye degradation of heterostructured photocatalysts28 but there are limited pertinent literature reports, to the best of our knowledge, that properly address the kinetics, mechanism and the intermediate product formation based on the surface activity of heterostructured nanoscale photocatalysts. In order to regulate reactive species and intermediate products kinetics, their studies may play very important task for the theoretical as well as practical applications of the heterostructured photocatalysts.
Considering the recent developments, in our previous work, we have reported heterojunction photocatalysts of BiOCl integrated with functionalised spinel ferrites (Ni0.5Zn0.5Fe2O4@PAni) and their excellent synergistically improved photocatalytic performance in the degradation of organic dye under visible light.29 The focus was put on the heterostructured photocatalyst synthesis, surfaces and the characteristics needed to prepare an efficient photocatalyst for degradation of an organic pollutant in aqueous solution driven by visible light. Motivated by the synergistically propitious photocatalytic performance of Ni0.5Zn0.5Fe2O4@polyaniline decorated BiOCl nanosheets prepared through a simple and facile mixing method to degrade an organic dye under visible light very effectively, in the present study, we have gained insight into the engineering aspects and mechanism for understanding the commendable photoactivity of the composite for the depollution of methyl orange in aqueous medium. The analysis of the physico-chemical characterisation and electro-chemical activity correlation of the composite suggested that the commending photoactivity predominantly benefits from the integrative synergistic modulation of the band gap, better photoredox capability and enhanced adsorption capacity. In contrast, the present work mainly focuses on the evaluation of degradation kinetics for methyl orange dye under visible light at multiple catalyst interfaces in the Ni0.5Zn0.5Fe2O4@polyaniline decorated BiOCl photocatalyst. Also, in studying the engineering aspects of the charge kinetics and gaining deeper understanding, the study unveils the influence of the key factors such as initial dye concentration, catalyst dose, pH and inorganic anions in the reaction kinetic rates along with modelling of these parameters using nonlinear regression analysis. Furthermore, based on the analysis of electrochemical characterization, intermediate products detected by GC/MS and experimental data analysis help to illuminate the underlying synergistic role of integrated Ni0.5Zn0.5Fe2O4@polyaniline for photocatalytic degradation and the mechanistic pathways for the degradation of organic pollutants in water, e.g. MO dye. Hopefully, such knowledge may be worthy, helpful and lay the groundwork for the development of heterostructured nanophotocatalysts to address the challenges of environmental issues and industrialization of the process technology.
2. Experimental section
Chemicals
All chemicals were of analytical grade and used as received. The details of all chemicals used are given in the ESI.†
Preparation of the photocatalysts
Synthesis of Ni0.5Zn0.5Fe2O4@polyaniline nanofibers (N@PAni). The Ni0.5Zn0.5Fe2O4@polyaniline (N@PAni) nanofibers were prepared by an in situ polymerization method. The detailed synthetic procedure is given in the ESI.†
Synthesis of BiOCl. BiOCl powder was prepared by a facile hydrolysis method. The detailed synthetic procedure is given in the ESI.†
Synthesis of Ni0.5Zn0.5Fe2O4@polyaniline modified BiOCl photocatalyst (N@P/B). The Ni0.5Zn0.5Fe2O4@polyaniline/BiOCl photocatalysts were synthesized by dispersive mixing of N@PAni nanofibers and prepared BiOCl in tetrahydrofuran (THF). First, a stock solution of N@PAni was prepared by dispersing ferrite nanoparticles in tetrahydrofuran with 0.45 g l−1 concentration. A measured amount of as-prepared BiOCl powder was added to 200 ml of the above N@PAni solution, which was then sonicated for 15 min and finally stirred for 24 h. The filtered precipitates were washed with water and ethanol several times and dried in a vacuum oven at 80 °C for 24 h.
Characterization techniques
The prepared composite was characterized using XRD, FTIR spectroscopy, HRTEM-EDX, XPS, DRS, PL and EIS. The instrumental details and the techniques are given in the ESI.†
Adsorption and photocatalytic experiments. Methyl orange dye was used for the dye removal study using photocatalysts. The detailed experimental procedure is explained in the ESI.†
Trapping study. To confirm the main reactive oxygen species involved in the catalytic reaction, trapping studies were carried out. The analysis procedure is described in the ESI.†
3. Results and discussion
Physicochemical characterization
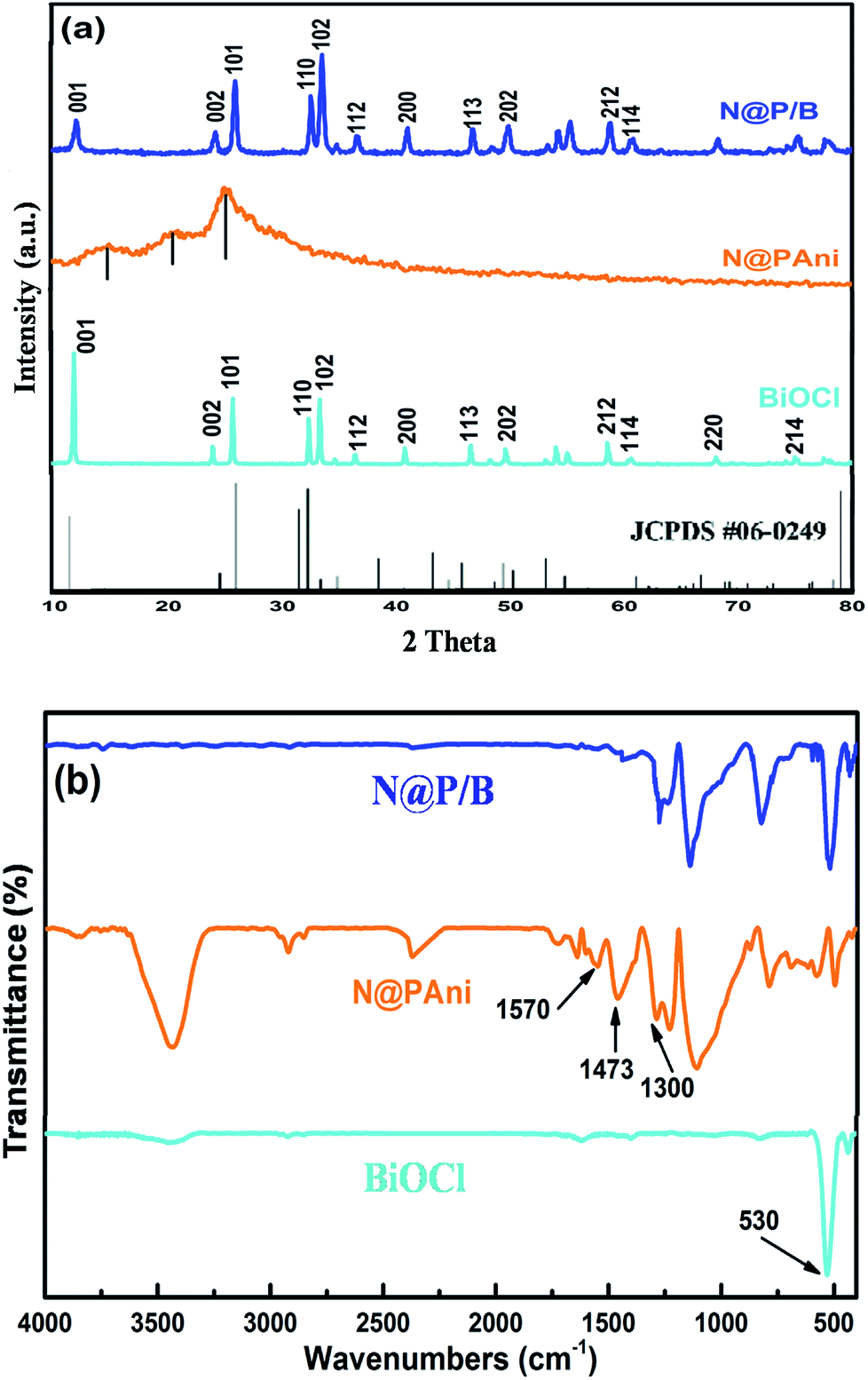
The phase composition and crystallographic structures of pure BiOCl, N@PAni nanofibers and the N@P/B composite were confirmed by XRD patterns (Fig. 1a). The strong and narrow diffraction peaks of pure BiOCl at 11.98°, 24.10° and 33.4° match the (001), (101) and (102) planes of tetragonal BiOCl (a = b= 3.891 Å, c = 7.369 Å, JCPDS no. 06-0249), respectively, which primarily confirmed that the as-synthesized BiOCl was crystalline.18 The XRD pattern of the N@PAni nanofibers revealed broad diffraction peaks at 13.30°, 20.60° and 25.40° attributed to the semi crystalline phases of the HCl doped polyaniline chain periodicity30 but the characteristic crystalline peaks of nickel zinc ferrite were absent, probably due to the presence of a very small fraction of nanocrystals. The XRD pattern of the as-prepared N@P/B composite shows strong and sharp peaks similar to the BiOCl diffraction pattern but of lesser intensity. The intensity ratio of I001/I102 for the N@P/B composite is 0.804, which is much smaller than that of pure BiOCl (I001/I102 = 1.7), hence the (001) peak is no longer dominant for the as-synthesized catalyst. This confirms that the presence of heterophase N@PAni nanofibers in BiOCl lowered the crystallinity of the N@P/B composite and also indicates the stacking of small-size N@PAni nanofibers in the layer of BiOCl.
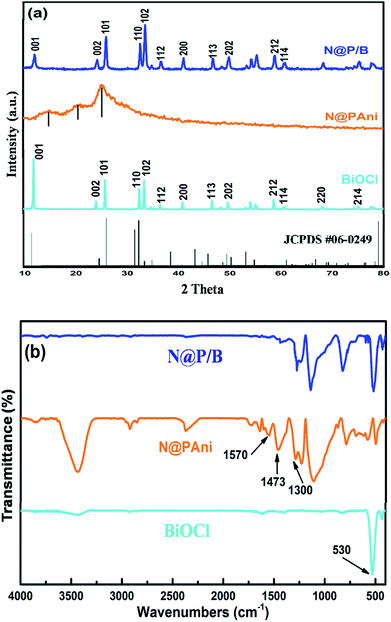 |
| | Fig. 1 (a) XRD pattern and (b) FTIR spectra of pure BiOCl, N@PAni and the N@P/B composite. | |
Fig. 1b shows the FTIR spectra of the pure BiOCl, N@PAni nanofibers and N@P/B composite. The IR spectrum of BiOCl displays diffused bands at 3730 cm−1 and 1398 cm−1 that may be due to stretching from water molecules and an intensive peak at 530 cm−1 that is assigned to the Bi–O bond.18 The FTIR spectrum of the prepared N@PAni reveals a broad weak band around 3444 cm−1 attributed to O–H stretching vibrations from water molecules, and bands at 1570 and 1473 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching deformation of quinonoid and benzenoid units of polyaniline, respectively. The weak bands appearing at low wave numbers (580 and 420 cm−1) are characteristic peaks of the tetrahedral and octahedral sites of NZF nanocrystals, respectively.24 However, the FTIR spectrum of the N@P/B composite displayed the characteristic peaks of polyaniline (1577 and 1490 cm−1) as well as the absorption peak of BiOCl (532 cm−1) shifted to higher wave numbers. In the figure, the characteristic peaks of NZF do not appear due to the low concentration of magnetic nanocrystals. The above results confirm some interactions between polyaniline functional groups and BiOCl.
C stretching deformation of quinonoid and benzenoid units of polyaniline, respectively. The weak bands appearing at low wave numbers (580 and 420 cm−1) are characteristic peaks of the tetrahedral and octahedral sites of NZF nanocrystals, respectively.24 However, the FTIR spectrum of the N@P/B composite displayed the characteristic peaks of polyaniline (1577 and 1490 cm−1) as well as the absorption peak of BiOCl (532 cm−1) shifted to higher wave numbers. In the figure, the characteristic peaks of NZF do not appear due to the low concentration of magnetic nanocrystals. The above results confirm some interactions between polyaniline functional groups and BiOCl.
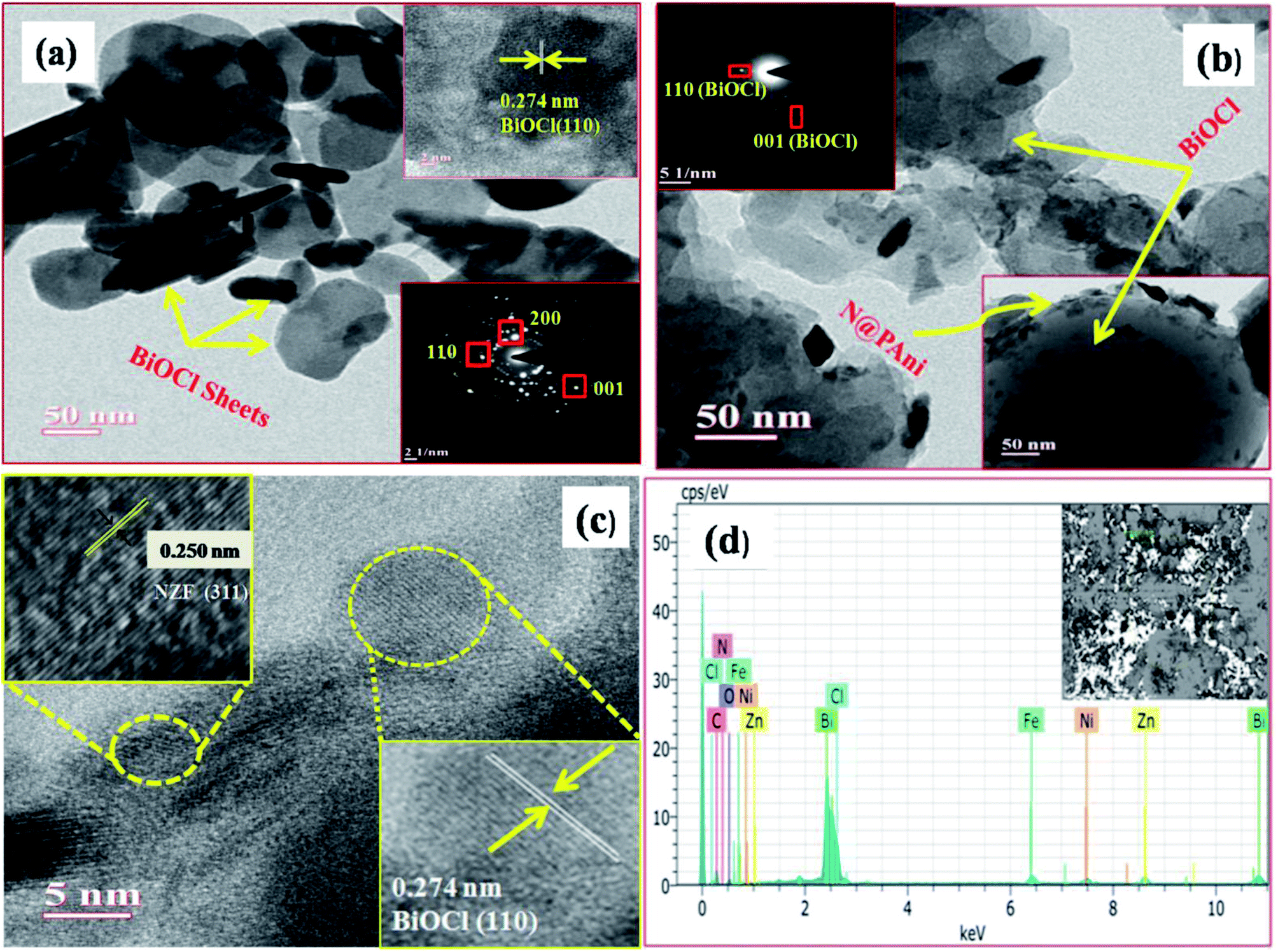
Fig. 2a–d displays the surface morphologies as well as inner structure of BiOCl and the N@P/B composite examined by TEM and HRTEM. The TEM image of pure BiOCl (Fig. 2a) showed two dimensional sheet like nanostructures with a regular thickness of about 30–40 nm and a relatively smooth surface. The low resolution TEM image of N@P/B evidenced that the composite has a morphology like the BiOCl sheets with even distributions of N@PAni throughout the BiOCl surfaces (Fig. 2b). In the respective HRTEM image (Fig. 2c), two distinct lattice fringes were found with d-spaces of 0.250 nm and 0.274 nm, which accord well with the (311) plane of NZF nanocrystals and (110) plane of tetragonal BiOCl, respectively.20 This implies the heterogeneous decoration of N@PAni composite particles over BiOCl sheets and confirms the formation of a heterojunction structure. The selected area electron diffraction pattern (inset Fig. 2c) and HRTEM image indicate that the exposed facet of the composite is the (110) facet of BiOCl. In addition, the elemental content of N@P/B was further investigated by EDS. As shown in Fig. 2d, the peaks of N, H, Ni, Zn and Fe originating from the nanofibers certify that N@PAni exits in the as-prepared samples.
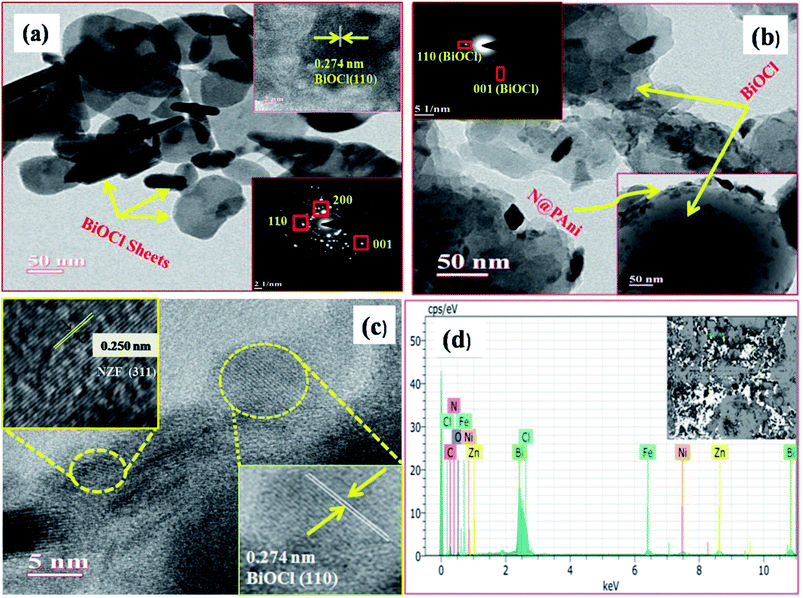 |
| | Fig. 2 TEM images of BiOCl (a) and N@P/B (b), HRTEM image and SAED diffraction pattern of N@P/B (c) and EDX spectra of the N@P/B (d) composite. | |
The Raman spectrum (Fig. 3) of BiOCl shows two strong peaks at 142.8 cm−1 and 198.4 cm−1, which are in agreement with the published values.31 The bands at 142.8 cm−1and 198.4 cm−1 are ascribed to the A1g and the Eg internal Bi–Cl stretching modes in BiOCl. For the N@P/B composite, the bands and vibrations are similar to those of the pure BiOCl sample. In the composite, the bands at 141.5 cm−1 and 200.3 cm−1 are assigned to the A1g and the Eg internal Bi–Cl stretching modes, which show a small blue and red shift, respectively, compared with that of pure BiOCl. A broad diffused band peak at 411.2 cm−1 is related to the C–H deformation and another band at 577.4 cm−1 is attributed to the amine deformation of the polyaniline.32 In contrast, the Raman signals for NZF cannot be clearly observed in the hybrid as the NZF nanoparticles have been enveloped by the polyaniline, although a very weak symmetric stretching of oxygen atom in Fe–O at 297.3 cm−1 is observed. The Raman spectral study confirms the successful formation of the N@P/B heterostructure. The band shifting corresponding to 142.8 cm−1 and 198.4 cm−1 suggests that an intensive interaction exists between BiOCl and N@PAni.
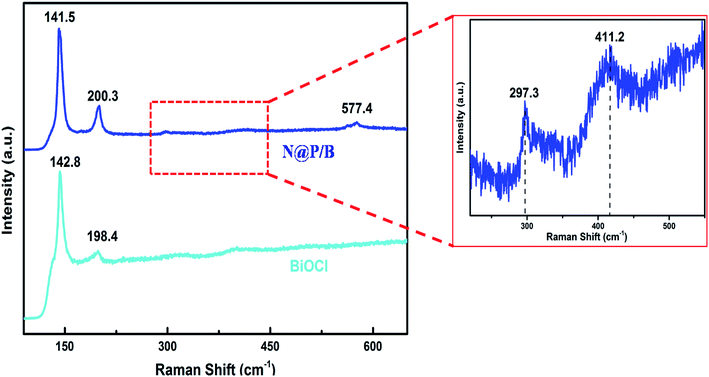 |
| | Fig. 3 Raman spectra of BiOCl, NZF/BiOCl, PAni/BiOCl and N@P/B. | |
Band-gap and visible light harvesting
The UV-vis-DRS technique was employed to find the band gap value of the prepared composite and the light harvesting ability of the developed catalyst. To investigate the contribution of BiOCl and N@PAni to the light absorption properties of the as-prepared composite, the UV-vis DRS spectra of BiOCl, N@PAni and the N@P/B nanocomposite were collected in the wavelength range from 200 nm to 800 nm, as shown in Fig. 4a. The DRS spectrum of BiOCl displays a strong absorption in the ultraviolet region of 200–400 nm, indicating more photocatalytic activity in the UV region of light (λ < 390 nm). BiOCl shows an absorption edge at 320 nm, which corresponds to a band gap of about 3.2 eV. In the absorption spectrum of the N@PAni nanofibers, there is a broad absorption region from 200 to 800 nm. This result indicates that the nanofibers show significant absorption of visible light. The absorption spectrum of the N@P/B nanocomposite displays an absorption profile similar to that of BiOCl with a visible absorption region from 380 nm to 420 nm. It is interesting to note that the DRS spectral response of the composite was greatly extended and enhanced. This observation clearly reveals that the loading of N@PAni nanofibers enhances the absorption of BiOCl in the visible region due to the high absorption coefficient of polyaniline.33 Thus, improved absorption is expected to contribute to the improvement of the overall activity of the composite. This observation suggests the coexistence of N@PAni and BiOCl components, which is consistent with the results of XRD and HRTEM images. Moreover, the colour of the composite also changes from white to grey (inset of Fig. 4a) with the loading of N@PAni nanofibers. The band gap energies of BiOCl, N@PAni and N@P/B were determined to be 3.2 eV, 1.2 eV and 2.1 eV (Fig. 4b), as measured from the Tauc plot followed by the equation:| |
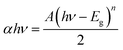 | (1) |
where A, α, Eg and ν are the proportional constant, absorption coefficient, band gap and light frequency, respectively. Among them, n depends on the characteristic transition in the semiconductor (i.e., n = 1 for direct transition or n = 4 for indirect transition).
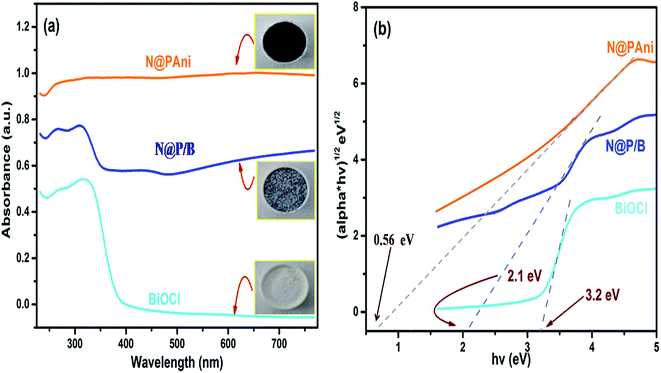 |
| | Fig. 4 (a) UV-vis diffuse reflectance spectra and (b) plot of (αhν)1/2 versus hν for the band gap energies of BiOCl, N@PAni and the N@P/B composite along with colour change images as the inset of (a). | |
Surface chemistry and interfacial interaction
XPS analysis was carried out to determine the surface electronic states and chemical composition of the N@P/B composite (Fig. 5). The full XPS spectrum (Fig. S1†) indicated that the as-prepared N@P/B contained Bi, O, Cl, N, Fe, Ni and Zn elements, which is in complete agreement with the EDX analysis. The two intense peaks at 159.1 and 164.9 eV were assigned to Bi 4f7/2 and Bi 4f5/2, respectively, indicating bismuth existed as Bi3+ in the composite.34 The broad peak of Cl 2p around 198.6 eV can be resolved into Cl 2p1/2 (198.3 eV) and Cl 2p3/2 (199.1 eV) energy levels, which are characteristic of the Cl− ion. The overlapping between Cl 2p1/2 and Cl 2p3/2 regions caused the appearance of a single peak of chloride ion for BiOCl.35 The diffused XPS signal for O 1s was asymmetric and could be fitted to three peaks situated at 529.1, 530.4 and 532.2 eV, which were ascribable to O2− in the NZF system, Bi–O bonds and chemisorbed oxygen, respectively, revealing three different kinds of oxygen species in the composite.36 The peaks at 284.7 and 286.3 eV can be assigned to carbon from XPS analysis. The N 1s core spectrum shows diffuse peaks at around 399 eV with a binding energy centred at 398.2 eV, 399.1 eV and 400.5 eV resulting from the quinonoid imine (–N–), benzenoid amine (–NH–) and imine (–NH+) groups, respectively. Fig. 5 shows the Fe 2p peak at 710.9 eV, which is consistent with the photoelectron peaks of Fe3+ in the composite.37 Similarly, weak satellite peaks appeared at 854.5 eV and 1022.1 eV that correspond to characteristic peaks of nickel and zinc, Ni 2p3/2 and Zn 2p3/2, respectively.38 Therefore, the decoration of N@PAni nanofibers might change the charge distribution of the BiOCl surface and chemical bonds could be formed at the interface of BiOCl and the N@PAni nanofibers. This indicates the presence of N@PAni, which is consistent with the results of XRD.
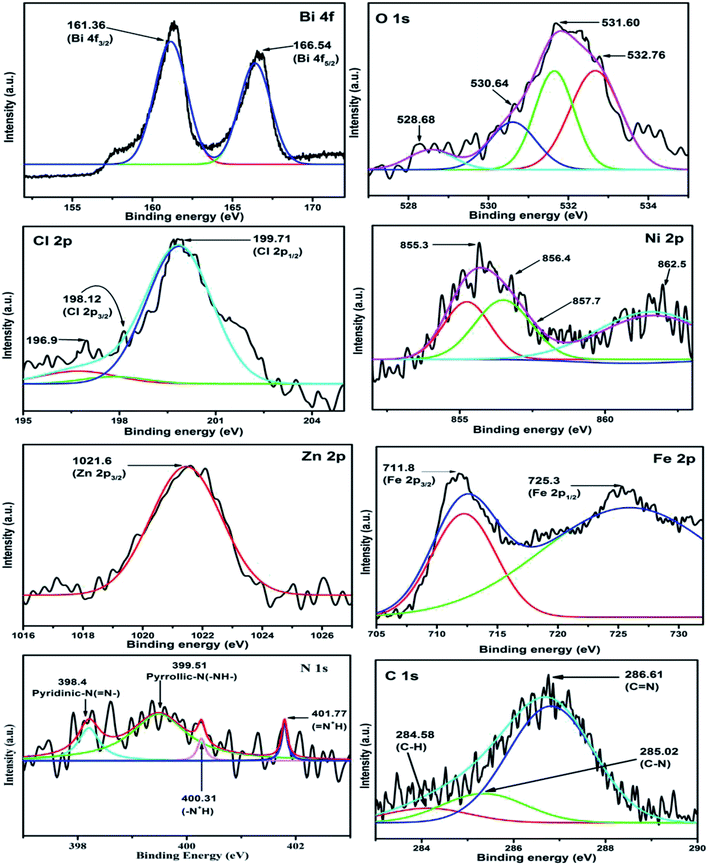 |
| | Fig. 5 Individual XPS spectra of Bi 4f, O 1s, Cl 2p, Ni 2p, Zn 2p, Fe 2p, N 1s and N 1s. | |
Photocatalytic activity and degradation kinetics
In our previous publication,29 we reported that a Ni0.5Zn0.5Fe2O4@polyaniline/BiOCl heterostructured photocatalyst with 20 wt% Ni0.5Zn0.5Fe2O4@polyaniline showed commendable catalytic performance in the degradation of various organic pollutants of dye wastewater driven by visible light. In continuation of this study, to evaluate the role of integrated Ni0.5Zn0.5Fe2O4@polyaniline in BiOCl for advantageous catalytic activity through heterojunctions, we investigated the photo-degradation kinetics of different physically mixed photocatalysts such as BiOCl/Ni0.5Zn0.5Fe2O4, BiOCl/PAni, BiOCl/Ni0.5Zn0.5Fe2O4/PAni, Ni0.5Zn0.5Fe2O4@PAni, Ni0.5Zn0.5Fe2O4@PAni/BiOCl as well as bare PAni, Ni0.5Zn0.5Fe2O4, and BiOCl. The photocatalytic performances of the synthesized composites were evaluated by using MO degradation under visible light irradiation. Fig. 6a shows the photocatalytic degradation efficiency of MO under different conditions. During photolysis, negligible degradation of dye (only 5%) was observed even after 60 min. The developed Ni0.5Zn0.5Fe2O4@PAni/BiOCl (N@P/B) multi-component hybrid nanostructure with heterogeneous interfacial junctions showed promising photocatalytic performance and accelerated degradation kinetics over other catalysts. Amongst all, N@P/B showed the highest photoreactivity and degraded the methyl orange dye almost completely within 60 min of visible light irradiation. The enhancement in photocatalytic activity is attributed to the integrated form of Ni0.5Zn0.5Fe2O4@PAni enabling a high rate of surface oxidation–reduction reactions, photon absorption and generation of electron–hole and separation-migration of active charge carriers.39 To observe the excellent photocatalytic activity of the N@P/B heterostructured samples, we have compared the photocatalytic performances of some heterojunction catalysts already reported earlier (Table 1).
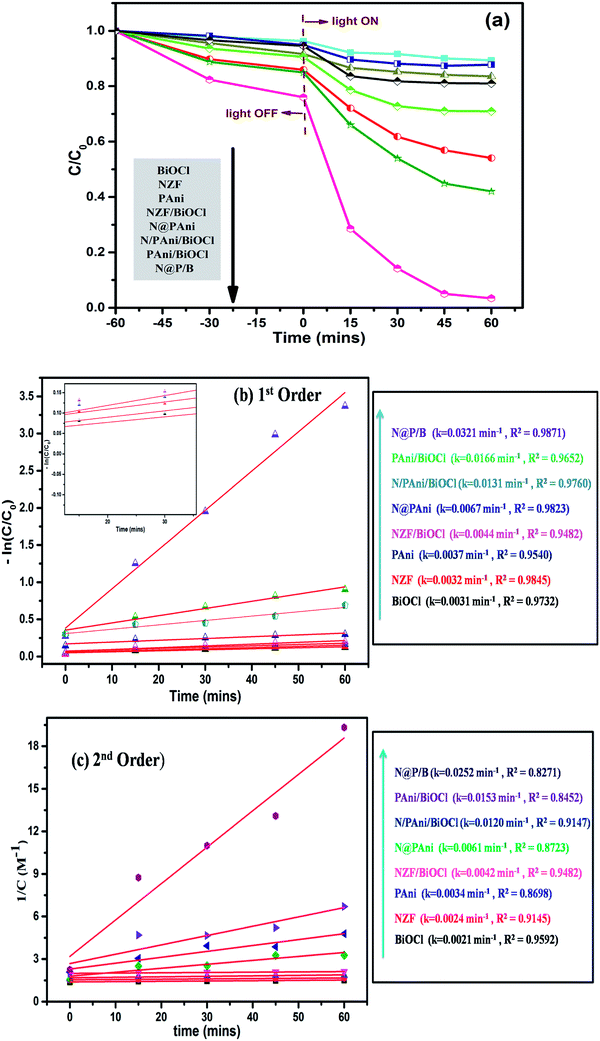 |
| | Fig. 6 (a) Photocatalytic degradation of MO dye, (b) 1st order kinetic plot and (c) 2nd order kinetic plot for photocatalytic degradation of MO over BiOCl, NZF, PAni, NZF/BiOCl, N@PAni, PAni/BiOCl, N/PAni/BiOCl and N@P/B. | |
Table 1 Comparison of various photocatalyst systems for dye removal in the literature
| Photocatalyst |
Dye (conc.) |
Irradiation source |
Decolourisation time (min) |
Catalyst dose |
Degradation efficiency |
Cyclic stability (runs/efficiency) |
Ref. |
| BiOCl/PANI |
MO (10 mg l−1) |
Visible |
210 min |
1 g l−1 |
∼67% |
3 runs/∼67% |
18 |
| CoFe2O4/BiOCl |
RhB (20 mg l−1) |
Visible |
6 h |
1 g l−1 |
∼95% |
3 runs/∼80% |
20 |
| PAn/TiO2 |
MB (10 mg l−1) |
Sunlight |
90 min |
2 g l−1 |
∼80% |
4 runs/∼55% |
27 |
| BiOCl-PT |
RhB (10 mg l−1) |
UV |
180 min |
0.3 g l−1 |
∼86% |
Not studied |
32 |
| BiOCl/BiVO4 |
MO (20 mg l−1) |
Visible |
11 h |
1 g l−1 |
∼85% |
6 runs/(not mentioned) |
34 |
| BiOCl–TiO2 |
MB (20 mg l−1) |
Visible |
120 min |
1 g l−1 |
∼97% |
5 runs/(not mentioned) |
35 |
| N@P/B |
MO (10 mg l−1) |
Visible |
60 min |
1 g l−1 |
∼96% |
4 runs/∼90% |
This study |
Further, the degradation reaction kinetics was studied for the different composites by using pseudo first (eqn (2)) and second order (eqn (3)) kinetic models.
| |
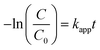 | (2) |
| |
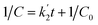 | (3) |
where
kapp is the apparent rate constant (min
−1), and
C0 and
C are the concentration of dye initially and at time
t (min), respectively.
Fig. 6b and c present the pseudo first and second order kinetics of the degradation reaction. It can be inferred that the degradation reaction clearly follows pseudo first order kinetics rather than second order kinetics. The analysis of the degradation constant reveals that the N@P/B heterostructure exhibits the highest rate as
kapp is about 7.5 times higher than that of BiOCl.
Degradation kinetics control parameters
pH is an important factor in the photodegradation process as it has a significant effect on the adsorption–desorption properties at the catalyst surface. To study the effect of pH on the performance of the catalyst, photocatalytic degradation of MO dye was carried out in the pH range 3–11. Photocatalytic efficiency of the N@P/B composite at pH 3, 5, 6, 7, 8, 9 and 11 was found to be 48.34%, 66.02%, 87.30%, 96.56%, 91.54%, 78.43% and 72.57%, respectively (Fig. 7a). The results indicate that at low and high pH values, photodegradation rates are slow. The reason for this is that at high pH, a high OH ion content enhances electron–hole separation. However, photogenerated CO2 will be trapped in the solution and carbonate and bicarbonate are formed in an alkaline system. Both carbonates are efficient scavengers of hydroxyl radicals due to their very high rate constants with hydroxyl radicals (k = 3.9 × 108 M−1 s−1 for carbonate and k = 8.5 × 106 M−1 s−1 for bicarbonate).40 On the other hand, at low pH, the acidic medium hinders the formation of hydroxyl radicals and subsequently reduces the degradation rate.41 The optimum pH value for photocatalytic degradation is 7 and the first order rate constant at pH 7 is 0.0301 min−1.
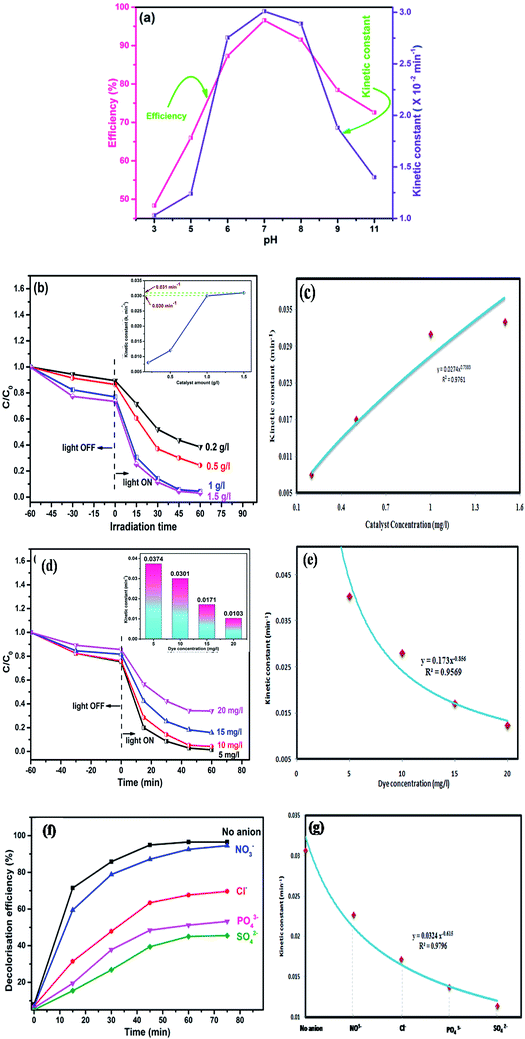 |
| | Fig. 7 Effect of (a) pH, (b) photocatalyst dose, (d) dye concentration and (f) presence of anions on photocatalytic activity of N@P/B and variation of apparent kinetic constant for dye degradation at different (c) photocatalyst dose, (e) dye concentration and (g) anions. | |
Another crucial kinetic parameter in the photo degradation process is the catalyst concentration. The effect of catalyst dose was found by varying the amounts of N@P/B from 0.2 to 1.5 g l−1 for 10 mg l−1 MO dye solution. It can be seen that the photodegradation rate increases with increasing amounts of photocatalyst (Fig. 7b). Decolorization of MO dye with different amounts of N@B/P composite follows pseudo first order kinetics (inset of Fig. 7c). It was observed that kapp significantly increases with an increase in the dose of photocatalyst from 0.2 to 1 g l−1. But, the rate constant (kapp) values for the 1 and 1.5 g l−1 catalyst dose are approximately the same and hence 1 g l−1 can be taken as the optimum catalyst loading. Also, reaction rate constants were found to be 0.008, 0.012, 0.0301 and 0.031 min−1 for catalyst doses of 0.2, 0.5, 1 and 1.5 g l−1, respectively. The enhanced photodegradation is attributed to the increased number of photoactive sites on the photocatalyst surface, which increases the number of adsorbed photons and dye molecule adsorption. However, upon a further increase of the photocatalyst dosage beyond the optimum, the penetration depth of the photons reduced, which reduces catalyst efficiency.42
Additionally, the effect of initial dye concentration was also studied for the N@P/B catalyst dose of 1 g l−1 at four different dye concentrations of 5, 10, 15 and 20 mg l−1. The results displayed in Fig. 7d show that the photocatalytic degradation strongly depends on the initial concentration of methyl orange. As the concentration of dye increases from 5 to 20 mg l−1, the photocatalytic decolorization decreases from 98.58% to 52.05% within 60 min of visible light irradiation. At a low dye concentration, MO dye molecules adsorb on the active surface of the catalyst and react with photogenerated holes and hydroxyl radicals on the catalyst surface. These reactive species effectively degrade MO. But, at high concentrations (≥15 mg l−1), shielding of light by the MO solution would lead to poor interaction with the catalyst and finally resulting in insufficient generation of electron–hole pairs.43,44 This causes an insufficient hydroxyl radical concentration for the removal of a high dosage of dye. Another reason for the lower photocatalytic efficiency at high dye concentration may be that crowding of molecules over the catalyst surface might block the active sites and eventually decrease hydroxyl radical production.45 Also, the reaction rate constants for the different dye concentrations are included in the inset of Fig. 7d. The rate constants for the dye concentration of 5 and 10 mg l−1 are approximately the same and hence 10 mg l−1 was taken as the concentration for catalytic degradation study.
Besides catalyst dose and dye concentration, the presence of inorganic anions such as chloride, sulphate, carbonate, nitrate, phosphate etc. also affects the photocatalytic degradation rate. Generally, anions have an inhibitory effect on decolourisation efficiency due to the occurrence of competitive adsorption and the formation of less active inorganic radical species. The effect of inorganic anions on the photocatalytic degradation of MO solution by the N@P/B composite was studied using the corresponding sodium salts. Fig. 7f shows the influence of various inorganic anions (i.e., Cl−, NO3−, PO43− and SO42−) at the same concentration (0.1 M) on the decolorization activity of the N@P/B composite. Compared to the control test in the aqueous MO dye solution without anions, the existence of all the anions reduced the MO degradation to a certain degree except nitrate. It was observed that the SO42− anions have the strongest inhibition effect on decolourisation efficiency. The decolourisation inhibition effect due to anions is in the following decreasing order: SO42− > PO43− > Cl− > NO3− (Fig. 7g). The inhibition effect is due to the h+ and ˙OH scavenging properties of the adsorbed anions (eqn (4)–(9)) and the results agree with a published report.46
| |
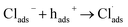 | (4) |
| |
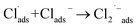 | (5) |
| | |
SO42−ads + hads+ → SO4˙−ads
| (6) |
| | |
SO42−ads + ˙OHads → SO4˙−ads + OHads−
| (7) |
| | |
PO43−ads + hads+ → PO4˙2−ads
| (8) |
| | |
PO43−ads + ˙OHads → PO4˙2−ads
| (9) |
From the above reactions, it can be concluded that the catalyst surface active sites are blocked by the radical anions, which are not easily oxidizable, and subsequently the photocatalytic efficiency decreases.
Degradation rates and modelling
A non-linear regression analysis was used to evaluate the pseudo first order rate constant (kapp) for the given photocatalytic system. This method was used to find the relationship between rate constant and operational parameters including initial dye concentration, catalyst dosage and presence of anions. The apparent reaction rate constant for the photocatalytic degradation of dye was evaluated by using an empirical power law, as47
| kapp = a(operational parameter)b |
where a and b are model parameters. The model parameters (a and b) were calculated for each operational parameter with non-linear regression and the results are shown in Fig. 7c, e and g. kapp increases with increasing catalyst concentration (Fig. 7c). On the other hand, kapp reduces with increasing initial concentration of dye (Fig. 7e) and in the presence of anions (Fig. 6g). As previously discussed, degradation of MO follows pseudo first order kinetics, therefore it is inferred that kapp is a function of the operational parameters as follows:
With non-linear regression analysis, the values of x, y and z were estimated and the average value of k0 was calculated as 0.077. Finally,
A comparison between experimentally and theoretically calculated kapp values for the decolorization of MO dye is shown in Fig. 8. The results from the plot reveal that the developed kinetic rate equation is in good agreement with the observed experimental data.
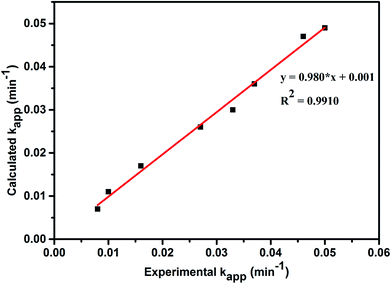 |
| | Fig. 8 Comparison between experimental and calculated kapp values obtained from new kinetic model equation. | |
Degradation intermediate formation and reaction pathways
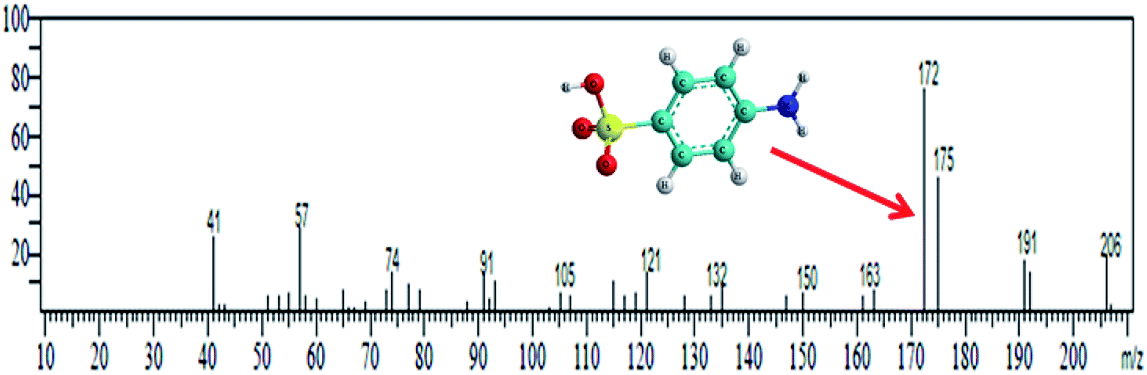
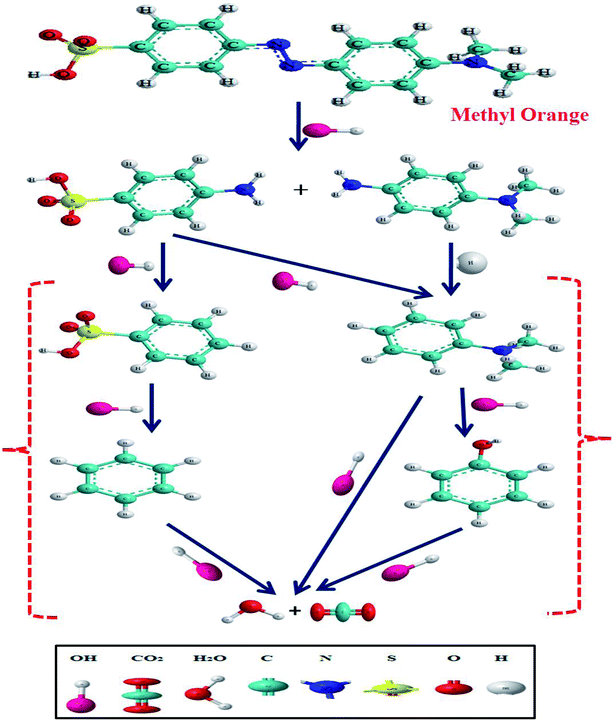
Methyl orange dye contains a –NN– double bond and a sulfonyl group located on the p-benzene ring. GC-MS analysis was conducted for the detection of different degraded fragmental products of MO dye, which were obtained after degradation under visible light in the presence of the N@P/B composite. Generally, hydroxyl radicals and photogenerated holes are responsible for the degradation of dye. Reactive species may attack different chemical bonds of the dye or directly oxidize MO to form different intermediates. The sample after 45 min of irradiation showed two m/z peaks of medium intensity at 136 and 172 and a lower intensity m/z peak at 121 in the GCMS analysis (Table 2). These peaks can be attributed to the substituted aromatic amines. This indicates that the degradation proceeded through the symmetric cleavage of the azo group, which results in the formation of N,N-dimethylbenzenamine, N,N-dimethyl-p-phenylenediamine and sulfanilic acid.47,48 After 1 h of visible light irradiation, the degradation products of MO disappear without the formation of new by-products. It seems likely that the intermediates were further degraded into smaller molecules during the degradation experiments.49,50 Eventually, it is assumed that complete mineralization of MO dye leads to CO2, H2O, SO42−, NH4+ and NO3− as end products.51 Based on the findings reported in this paper and previous literature data,52,53 the expected dye intermediates and their possible chemical structures are suggested in Fig. 9 and the pathways for formation of the intermediates are schematically presented in Scheme S1.†
Table 2 GC-MS spectral data of intermediate products of MO
| Degradation intermediates |
Retention time (min) |
Main ions (m/z) |
Mass spectrum |
| N,N-Dimethylbenzenamine & N,N-dimethyl-p-phenylenediamine |
10.327 |
121 & 136 |
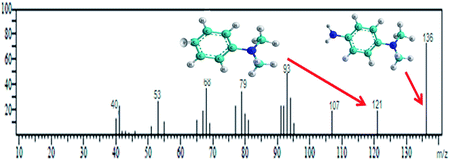 |
| Sulfanilic acid |
15.223 |
172 |
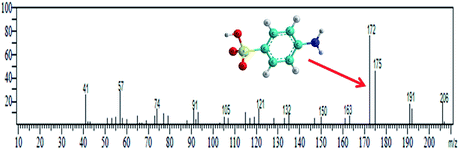 |
 |
| | Fig. 9 GCMS-expected dye intermediates with their possible chemical structures. | |
To further investigate the fate of MO dye and its degradation intermediates, the extent of mineralization was quantified by TOC analysis. It was observed that ∼75% of the carbon content of the dye solution was mineralized. It can be concluded that MO can be completely mineralized by the hydroxyl radicals/photo holes, and degradation intermediates can be converted into CO2, H2O and inorganic ions.
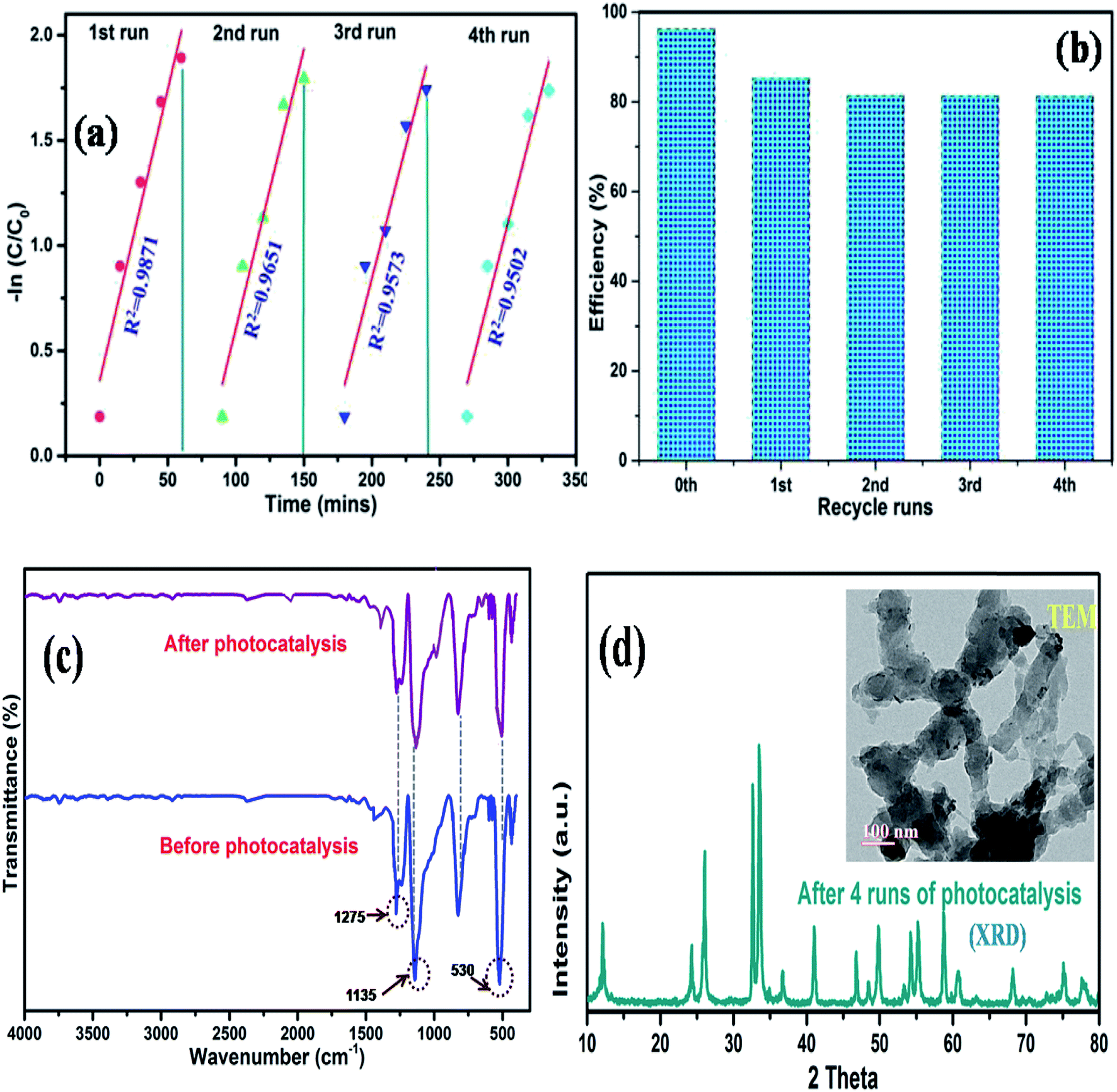
Reusability and photocorrosion stability
The photocorrosion stability is another criterion to determine the service life of a catalyst. Therefore, to further understand the superiority of the synthesized composite, we measured the cycling performance of N@P/B during 4 runs of photo degradation experiments. In between each cycle, the used catalyst was centrifuged, washed and dried in an oven at 60 °C after each run. Fig. 10a and b shows that the decolorization efficiency of the photocatalyst decreased after each run. This reduction in photo activity is due to the adsorption of some organic molecules on the surface of the catalyst, which permanently block the active sites of the catalyst. Moreover, the physico-chemical stability of the as-prepared composite was further inspected by FTIR spectroscopy. Fig. 10c displays that the FTIR spectra of the recycled composite did not change after the repeated use compared with the fresh sample. XRD and TEM results of the catalyst obtained after four recycles (Fig. 10d and inset figure) also confirmed that the phase and morphology of the catalysts remained unchanged. It is clear from the spectra that the chemical structure of the composite remains the same after photocatalysis, which demonstrates the outstanding recyclability and stability of the as-synthesized N@P/B composite.
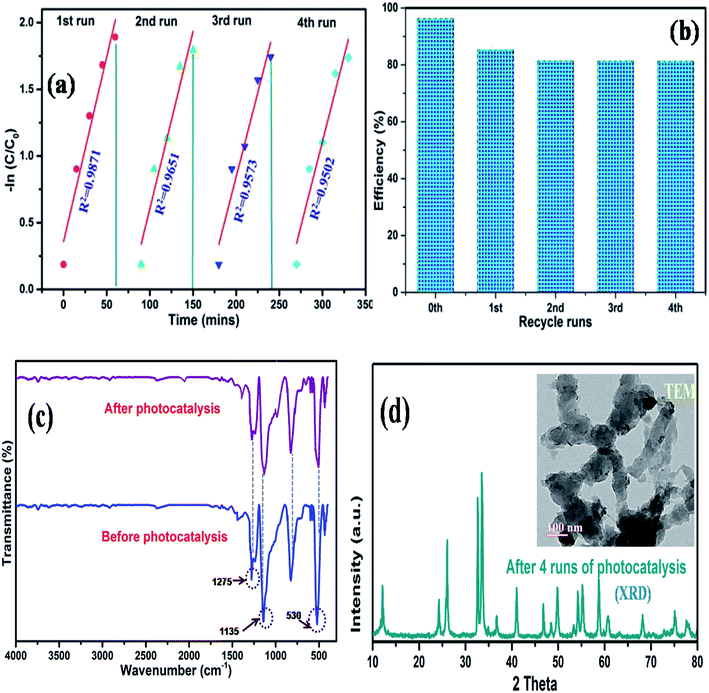 |
| | Fig. 10 Recycling runs of (a) photo degradation kinetics of MO dye, (b) degradation efficiency of N@P/B, (c) FTIR patterns of the N@P/B photocatalyst before and after the 4th run under visible light and (d) XRD pattern and TEM image of the recycled N@P/B after the 4th cycle. | |
Electrochemical properties and efficient charge transfer kinetics
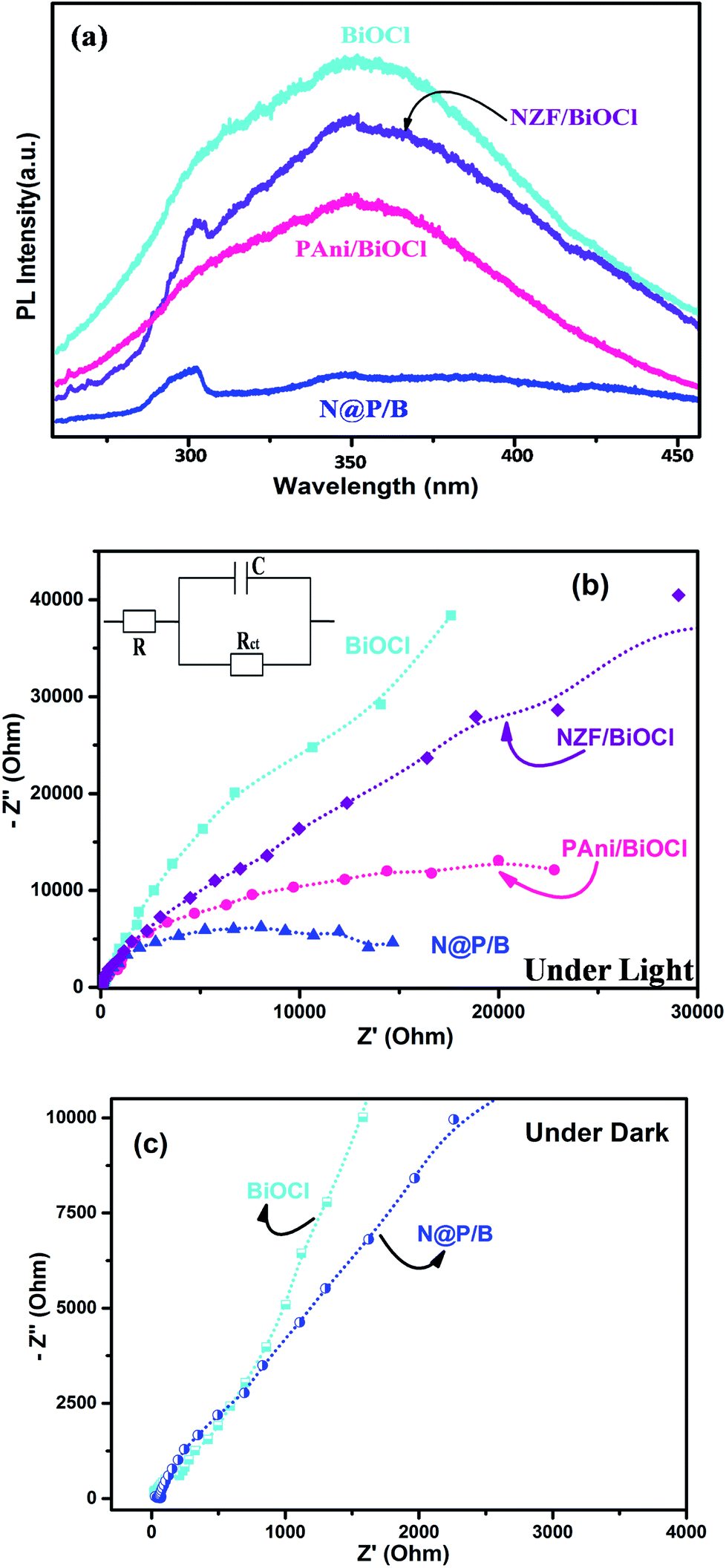
The enhanced rate of photocatalytic degradation of BiOCl decorated with N@PAni from a simple solution mixing method may be due to some intrinsic role of the in situ synthesized NZF and PAni composite in band tuning, charge separation efficiency and accelerated charge carrier photogeneration. To ascertain the enhancement in the degradation efficiency, we tried to figure out the rate of recombination of the photogenerated charge carriers and participation of reactive radical species in the catalytic reaction system. The photoluminescence technique is commonly used to study the recombination rate, life span and behaviour of photogenerated electron–hole pairs.54 Here, to check the role of N@PAni in the BiOCl composite, the PL spectra of pure BiOCl, NZF/BiOCl, PAni/BiOCl and the N@P/B composite were studied and are presented in Fig. 11a. The excitation light source was a 280 nm He–Cd laser. The emission peak for BiOCl was observed at around 320 nm under visible light whereas the N@P/B composite showed a weaker emission peak. This drastic quenching of photoluminescence intensity was observed after the introduction of the N@PAni nano-scale composite, implying that the photogenerated electrons and holes experience better separation in the heterojunction. The above results also demonstrate that the efficient transfer of photogenerated electrons from NZF to BiOCl via polyaniline occurs, which reduces the direct recombination of electrons and holes, leading to efficient charge separation.55 In an earlier report, it was shown that magnetic photocatalysts transport holes predominantly via surface states and not directly from the valence band.56 The coating of PAni on transition metal ferrites favoured the diffusion of holes to surface states of the composite particles and slowed down the charge separation. As a result, this enhances the interfacial charge transfer kinetics and ensures more photon utilization during the photocatalytic reaction.57
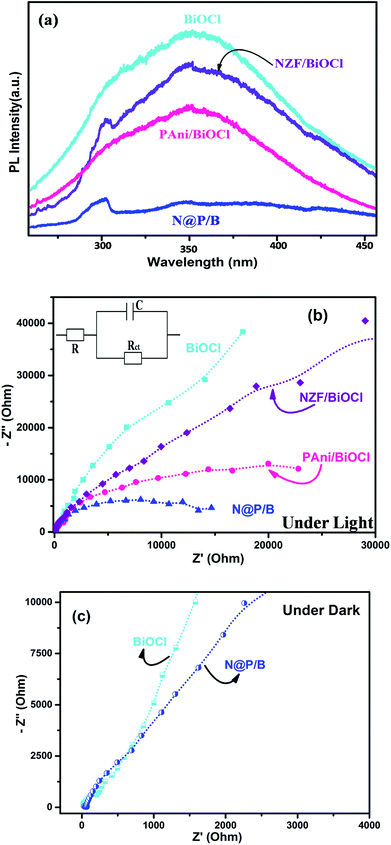 |
| | Fig. 11 (a) Photoluminescence analysis, (b) EIS analysis of BiOCl, NZF/BiOCl, PAni/BiOCl and N@P/B composite, inset: the equivalent circuit diagram, and (c) EIS analysis of BiOCl and N@P/B under dark conditions. | |
To further ascertain the interfacial charge transfer property, the EIS response was compared in terms of Nyquist plots of the BiOCl, NZF/BiOCl, PAni/BiOCl and N@P/B electrodes and fitted using an equivalent circuit (inset in Fig. 11b). The circuit is composed of R, which is the intrinsic bulk solution resistance, Rct, which is the interfacial charge transfer resistance, and C, the constant for the electrode/electrolyte interface, respectively.58 Under light, all electrodes show a semicircle arc, suggesting charge transfer controls the kinetics at the electrode interface. Normally, the smaller the radius, the better the charge transfer ability.25 It was found that the N@P/B electrode has the smallest radius among all electrodes of BiOCl, which suggested that the composite possesses a lower charge transfer impedance. A significant decrease in charge-transfer resistance in the N@P/B composite indicates accelerated ion diffusion and electron transfer at cycling rates. The surface area of N@P/B in contact with the electrolyte solution itself facilitates electron transfer from N@PAni nanofibers within the whole electrode. The multiple cations through strong interaction with PAni chains in N@P/B are the centres of redox sites, enable electron transport and enhance the separation efficiency of electron–hole pairs.59 This implied that the composite catalyst provides appropriate electronic channels and enhances the separation of photoelectrons and holes, as already evidenced by the PL studies. Very recently, it was reported that the presence of PAni in heterostructure composite electrodes of magnetic materials enabled good electrochemical performance due to its unique hierarchical structure, short pathway for ion penetration and the connection between the PAni chains and multiferrite oxides.19,60 In the dark, the Nyquist plots (Fig. 11c) of BiOCl and N@P/B are almost the same, including a minor semicircle part at high frequencies corresponding to charge transfer resistance and a major straight line part at low frequencies corresponding to mass transfer resistance. The pronounced straight line part at low frequencies indicated that mass transfer was a dominant process at this stage compared to the charge transfer.
Interfacial charge kinetics and mechanistic insights
The generation and transport of photogenerated reactive charge carriers including active oxygen groups, singlet oxygen, superoxide radicals and hydroxyl radicals have been proposed as the key parameter for the enhanced catalytic activity of semiconductor nanomaterials.61 It is emphasized that the generated electron–hole pairs (eCB−/hVB+) on the catalyst surface react with the adsorbed species and generate active radicals such as hydroxyl radicals (OH˙) and superoxide radical ions (O2˙−) through redox reactions. These radicals participate in the oxidative degradation of the absorbed dye molecules on the catalyst surface. Therefore, to better understand the mechanism, it is imperative to identify the responsible reactive species in the photocatalytic degradation of organic dye over N@P/B. Fig. 12 shows different experimental kinetics in the presence of specific reactive species scavenging chemicals. As shown in the figure, when EDTA-2Na and IPA were added into the degradation system, the degradation efficiency was remarkably decreased.62 This indicates that the holes and hydroxyl radicals are the main active species in the dye degradation. This suggests that holes directly or indirectly, via adsorbed water to hydroxyl radicals, degrade MO dye. However, when any one of benzoquinone or AgNO3 was added into the degradation system, photodegradation activity was hardly affected. This indicates that e˙ and O2− do not play an important role in the degradation process.
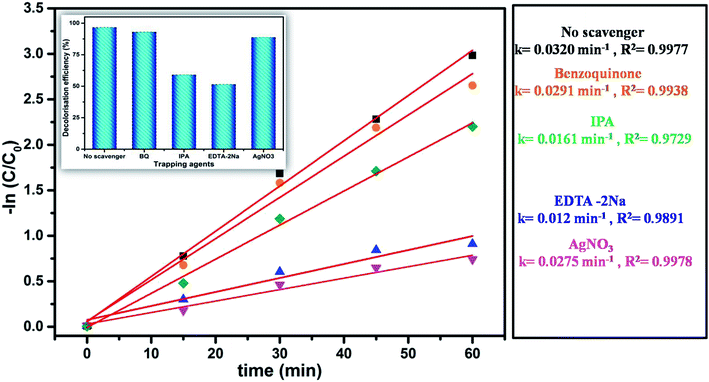 |
| | Fig. 12 Effect of scavengers on degradation kinetics of N@P/B composite. | |
To gain deeper insight into the rate of charge transfer in the heterojunction architecture, as illustrated in the PL, EIS and trapping analysis, it was observed that the rate of hydroxyl radical formation and transport of holes on the catalyst surface are the main cause for the high rate of oxidation of MO dye under visible light. It is assumed that the generated photoholes may react with absorbed water (eqn (13)) on the catalyst surface to yield h+ and OH˙, which play an indirect role in the degradation of the dye. The produced hydroxyl radical may now directly react with the adsorbed dye molecules, forming an intermediate, and finally degrade the dye (eqn (15)–(17)).
| | |
N@P/B + hν → N@P/B (e− + h+)
| (12) |
| | |
N@P/B (h+) + H2O/OH− → OH˙ + H+
| (13) |
| | |
N@P/B (e−) + O2 → O2˙−
| (14) |
| |
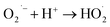 | (15) |
| |
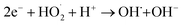 | (16) |
| |
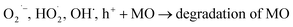 | (17) |
Based on the results of our experiments as well previous literature, a probable schematic mechanism of carrier transfer and intermediate product generation during dye degradation over N@P/B is represented in Scheme 1. When N@PAni and BiOCl contact and form a heterojunction, the band structure of BiOCl may be tuned and reconfigured. In the first stage, the energy bands of NZF and PAni are combined and form an equilibrated Fermi level along the path of the diffusion carriers. In the second stage, the energy bands of N@PAni and BiOCl shift upward and downwards carrying the diffused charges till the Fermi levels equilibrate. When light irradiates the heterojunction, generated holes and electrons appear in the valence band and conduction band at a higher rate and there is low charge separation efficiency due to the small energy gap in the heterostructured composite. NZF in the present work may act as a co-catalyst and play a combined synergistic role with PAni, similar to the case of carbon-doped Fe2O3 nano-scale photocatalysts.63,64 Due to the band alignment and low potential difference25 in N@PAni, electrons easily transfer from the conduction band of N@PAni to the conduction band of BiOCl and simultaneously the holes in the valence band of BiOCl move to the valence band of N@PAni. NZF mainly may plays two roles in the enhancement of catalytic performance. One is to support the charge separation and transport through the formation of junctions/interfaces with the light sensitizing PAni chains and the other is possibly to serve as a reaction site to accept the separated charges for surface reaction on N@PAni.65 As a result, the photogenerated electron–hole pairs are effectively separated at a higher rate, which is crucial for the higher rate of degradation. Further, as the generated holes very quickly move to PAni and easily transport to acceptors like adsorbed dye molecules (eqn (17)) or water (eqn (7)), the formation of ˙OH and dye degradation are consequently accelerated, which may support the scavenger study results and kinetic analysis.
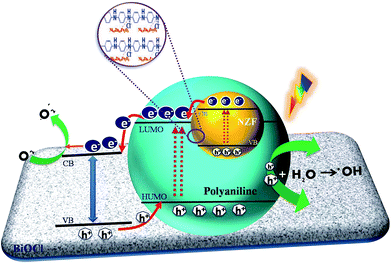 |
| | Scheme 1 Suggested charge carrier transport on N@P/B surfaces in aqueous solution. | |
4. Conclusion
The present work provides a tangible platform to design a multi-component heterostructured photocatalyst for visible light harvesting that restricts the recombination rates of the photogenerated electrons and holes in BiOCl by generating interfacial interaction with a Ni0.5Zn0.5Fe2O4@polyaniline nanocomposite. The enhanced kinetics and propitious catalytic performance of the N@P/B composite could be credited to the strong interfacial interaction in N@PAni that maximizes the interfacial contact between Ni0.5Zn0.5Fe2O4 and polyaniline as well as synergistically accelerating charge transfer at different interfaces in BiOCl. The PL spectral analysis and the EIS studies also successfully demonstrated that N@PAni on BiOCl can effectively transfer the surface electrons and prolong the lifetime of the photogenerated charge carriers. Furthermore, due to the high adsorption capacity of the dye molecules, the deposited N@PAni can also remarkably increase the surface adsorption of the N@P/B composite. Also, it was found that the reaction of MO degradation follows the first order kinetic model and the presence of inorganic ions has an inhibitory effect on dye degradation in a sequence of SO42− > PO43− > Cl− > NO3−. The structural information about the degradation intermediates obtained from GC-MS analysis provided the degradation route, which followed demethylation and hydroxylation processes. It is believed that the combined effect of BiOCl and the formation of the N@PAni heterojunction contribute to the accelerated degradation efficiency of dye molecules driven under visible light.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors highly thank the Guru Gobind Singh Indraprastha University, Delhi for providing the financial assistance through Indraprastha Research Fellowship (IPRF No. GGSIPU/DRC/Ph.D./Adm./IPRF/2012/94) to carry out the research program. We gratefully acknowledge the instrumentation support by AIRF-JNU Delhi, DST-SAIF Kochi and IIT Bombay. Central characterization facilities at MRC MNIT Jaipur are gratefully acknowledged. We also express our special thanks to all technical staff for their support.
References
- C.-C. Wang, X.-D. Du, J. Li, X.-X. Guo, P. Wang and J. Zhang, Appl. Catal., B, 2016, 193, 198–216 CrossRef CAS.
- E. M. Dias and C. Petit, J. Mater. Chem. A, 2015, 3, 22484–22506 RSC.
- J. Kou, C. Lu, J. Wang, Y. Chen, Z. Xu and R. S. Verma, Chem. Rev., 2017, 117, 1445–1514 CrossRef CAS PubMed.
- X. J. Wen, C. G. Niu, L. Zhang, C. Liang and G. M. Zeng, J. Catal., 2017, 356, 283–299 CrossRef CAS.
- J. W. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- Y. F. Li, D. H. Xu, J. I. Oh, W. Z. Shen, X. Li and Y. Yu, ACS Catal., 2012, 2, 391–398 CrossRef CAS.
- D. Pan, J. Jiao, Z. Li, Y. Guo, C. Feng, Y. Liu, L. Wang and M. Wu, ACS Sustainable Chem. Eng., 2015, 10, 2405–2413 CrossRef.
- Y. Tang, W. Di, X. Zhai, R. Yang and W. Qin, ACS Catal., 2013, 3, 405–412 CrossRef CAS.
- L. Sun, L. Xiang, X. Zhao, C. J. Jia, J. Yang, Z. Jin, X. Cheng and W. Fan, ACS Catal., 2015, 6, 3540–3551 CrossRef.
- M. Li, J. Zhang, H. Gao, F. Li, S. E. Lindquist, N. Wu and R. Wang, ACS Appl. Mater. Interfaces, 2016, 10, 6662–6668 CrossRef PubMed.
- Q. Li, Z. Guan, D. Wu, X. Zhao, S. Bao, B. Tian and J. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 6958–6968 CrossRef CAS.
- C. W. Tan, G. Q. Zhu, M. Hojamberiev, K. Okada, J. C. Liang, X. Luo and Y. Liu, Appl. Catal., B, 2014, 152, 425–436 CrossRef.
- B. K. Gupta, G. Kedawat, Y. Aggarwal, P. Kumar, J. Dwivedi and S. K. Dhawan, RSC Adv., 2015, 5, 10623–10631 RSC.
- Z. Wu, X. Yuan, J. Zhang, H. Wang, L. Jiang and G. Zeng, ChemCatChem, 2017, 9, 41–64 CrossRef CAS.
- D. W. Hatchett and M. Josowicz, Chem. Rev., 2008, 108, 746–769 CrossRef CAS PubMed.
- H. Zhang, J. Zong and Y. Zhu, Environ. Sci. Technol., 2008, 42, 3803–3807 CrossRef CAS PubMed.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
- Q. Wang, J. Hui, J. Li, Y. Cai, S. Yin, F. Wang and B. Su, Appl. Surf. Sci., 2013, 283, 577–583 CrossRef CAS.
- X. Chen, Y. Dai, J. Guo, T. Liu and X. Wang, Ind. Eng. Chem. Res., 2012, 55, 568–578 CrossRef.
- I. Y. Choi, I. Y. Kim, W. D. Cho, S. J. Kang, T. K. Leung and Y. Sohn, RSC Adv., 2015, 5, 79624–79634 RSC.
- Y. H. Zhu, R. Jiang, Q. Y. Fu, R. R. Li, J. Yao and T. S. Jiang, Appl. Surf. Sci., 2016, 369, 01–10 CrossRef.
- D. Yang, J. Feng, L. Jiang, X. Wu, L. Sheng, Y. Jiang, T. Wei and Z. Fan, Adv. Funct. Mater., 2015, 25, 7080–7087 CrossRef CAS.
- K. N. Harish, N. Bhojya, P. N. Prashanthkumar and R. Viswanath, ACS Sustainable Chem. Eng., 2013, 1, 1143–1153 CrossRef CAS.
- S. Kumar, V. Singh, S. Aggarwal, U. K. Mandal and R. K. Kotnala, J. Phys. Chem. C, 2010, 114, 6272–6280 CrossRef CAS.
- S. Kumar, V. Singh, S. Aggarwal, U. K. Mandal and R. K. Kotnala, Compos. Sci. Technol., 2010, 70, 249–254 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- B. Liu, X. Zhao, C. Terashima, A. Fujishima and K. Nakata, Phys. Chem. Chem. Phys., 2014, 16, 8751–8760 RSC.
- S. Kumar, K. Ojha and A. K. Ganguli, Adv. Mater. Interfaces, 2017, 4, 1600981 CrossRef.
- R. Tanwar, B. Kaur and U. K. Mandal, Appl. Catal., B, 2017, 211, 305–322 CrossRef CAS.
- S. Kumar, V. Singh, S. Aggarwal and U. K. Mandal, Colloid Polym. Sci., 2009, 287, 1107–1110 CrossRef CAS.
- Y. Tian, C. F. Guo, Q. Wang and Q. Liu, Appl. Surf. Sci., 2012, 258, 1949–1954 CrossRef CAS.
- L. Zhang and M. Wan, J. Phys. Chem. B, 2003, 107, 6748–6753 CrossRef CAS.
- S. Min, F. Wang and Y. Han, J. Mater. Sci., 2007, 42, 9966–9972 CrossRef CAS.
- H. Gnayem and Y. Sasson, J. Phys. Chem. C, 2015, 119, 19201–19209 CrossRef CAS.
- J. Xia, L. Xu, J. Zhang, S. Yin, H. Li, H. Xu and J. Dia, CrystEngComm, 2013, 15, 10132–10141 RSC.
- S. H. Piao, M. Bhaumik, A. Maity and H. J. Choi, J. Mater. Chem. C, 2015, 3, 1861–1868 RSC.
- Ta-I. Yang, R. N. C. Brown, L. C. Kempel and P. Kofinas, J. Nanopart. Res., 2010, 12, 2967–2978 CrossRef CAS.
- M. Guerrero, A. Altube, E. Garcia-Lecina, E. Rossinyol, M. D. Baro, E. Pellicer and J. Sort, ACS Appl. Mater. Interfaces, 2014, 6, 13994–14000 CrossRef CAS PubMed.
- L. Ye, J. Liu, C. Gong, L. Tian, T. Peng and L. Zan, ACS Catal., 2012, 2, 1677–1683 CrossRef CAS.
- S. Borji, S. Nasseri, A. H. Mahvi, R. Nabizadeh and A. H. Javadi, J. Environ. Health Sci. Eng., 2014, 12, 101–110 CrossRef PubMed.
- B. K. Avasarala, S. R. Tirukkovalluri and S. Bojja, J. Hazard. Mater., 2011, 186, 1234–1240 CrossRef CAS PubMed.
- Z. He, Y. Shi, C. Gao, L. Wen, J. Chen and S. Song, J. Phys. Chem. C, 2014, 118, 389–396 CrossRef CAS.
- D. Sun, J. Li, L. He, B. Zhao, T. Wang, R. Li, S. Yin, Z. Feng and T. Sato, CrystEngComm, 2014, 16, 7564–7574 RSC.
- B. Neppolian, H. C. Choi, S. Sakthivel, B. Arabindoo and V. Murugesan, J. Hazard. Mater., 2002, 89, 303–317 CrossRef CAS PubMed.
- M. Asilturk, F. Sayilkan and E. Arpac, J. Photochem. Photobiol., A, 2009, 203, 64–71 CrossRef.
- M. Abdullah, K. C. Gary and R. W. Matthews, J. Phys. Chem., 1990, 94, 6820–6825 CrossRef CAS.
- L. G. Devi and K. E. Rajashekhar, J. Mol. Catal. A: Chem., 2011, 334, 65–76 CrossRef CAS.
- S. Xie, P. Huang, J. J. Kruzic, X. Zeng and H. Qian, Sci. Rep., 2016, 6, 21947–21957 CrossRef CAS PubMed.
- A. Mohammad, K. Kapoor and S. M. Mobin, ChemistrySelect, 2016, 1, 3483–3490 CrossRef CAS.
- J. T. Spadaro, L. Isabelle and V. Renganathn, Environ. Sci. Technol., 1994, 28, 1389–1393 CrossRef CAS PubMed.
- L. G. Devi, S. G. Kumar, K. M. Reddy and C. Munikrishnappa, J. Hazard. Mater., 2009, 164, 459–467 CrossRef PubMed.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS.
- C. Baiocchi, C. M. Brussino, E. Pramauro, A. Bianco-Prevot, L. Palmisao and G. Marci, Int. J. Mass Spectrom., 2002, 214, 247–256 CrossRef CAS.
- K. Dai, H. Chen, T. Peng, D. Ke and H. Yi, Chemosphere, 2007, 69, 1361–1367 CrossRef CAS PubMed.
- Y. H. Zheng, C. Q. Chen, Y. Y. Zhan, X. Y. Lin, Q. Zheng, K. M. Wei and J. F. Zhu, Inorg. Chem., 2007, 46, 6675–6682 CrossRef CAS PubMed.
- G. Colon, S. Murcia Lopez, M. C. Hidalgo and J. A. Navio, Chem. Commun., 2010, 46, 4809–4811 RSC.
- B. Klahr, S. Gimenez, F. Faabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294 CrossRef CAS PubMed.
- B. Li, C. Zhenhua, W. Shixuan, W. Qiang and S. Zhurui, Dalton Trans., 2018, 47, 10288–10298 RSC.
- S. Khilari, S. Pandit, J. L. Varanasi, D. Das and D. Pradhan, ACS Appl. Mater. Interfaces, 2015, 7, 20657–22066 CrossRef CAS PubMed.
- X. Gao, X. Liu, Z. Zhu, X. Wang and Z. Xie, Sci. Rep., 2016, 6, 01–10 CrossRef PubMed.
- Y. Xia, Z. He, J. Su, Y. Liu, B. Tang and X. Li, Nano, 2018, 13(06), 1850070–1850082 CrossRef CAS.
- M. Liang, Z. Yang, Y. Yang, Y. Mei, H. Zhou and S. Yang, J. Mater. Sci.: Mater. Electron., 2019, 30(2), 1310–1321 CrossRef.
- H. Yu, J. Tian, F. Chen, P. Wang and X. Wang, Sci. Rep., 2015, 5, 13083 CrossRef CAS PubMed.
- Y. Hou, F. Zou, A. Dagg and P. Feng, Nano Lett., 2012, 12, 6464–6473 CrossRef CAS PubMed.
- N. Guo, Y. Liang, S. Lan, L. Liu, J. Zhang, G. Ji and S. Gan, J. Phys. Chem., 2014, 118, 18343–18355 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00548j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*