DOI:
10.1039/C6RA06837E
(Paper)
RSC Adv., 2016,
6, 40225-40237
A comparative binding mechanism between human serum albumin and α-1-acid glycoprotein with corilagin: biophysical and computational approach†
Received
15th March 2016
, Accepted 6th April 2016
First published on 7th April 2016
Abstract
The binding of corilagin with plasma serum proteins like human serum albumin (HSA) and α-1-acid glycoprotein (AGP) was investigated under physiological conditions. To understand the pharmacological importance of the corilagin molecule, anti-inflammatory activity on mouse macrophages (RAW 264.7) cell lines was studied. This study reveals that corilagin caused an increase in inhibition growth of inflamed macrophages in concentration-dependent manner with an IC50 value of 66 μM. Further, intrinsic fluorescence of HSA and AGP was quenched upon titration of corilagin, and the binding constants obtained from fluorescence emission was found to be Kcorilagin 4.2 ± 0.02 × 105 M−1 which corresponds to the free energy of −7.6 kcal M−1 at 25 °C for a HSA–corilagin complex. Interestingly, corilagin showed binding with AGP, an acute phase protein, and the binding constant was found to be Kcorilagin = 1.5 ± 0.01 × 104 M−1 and its free energy was −5.6 kcal M−1 at 25 °C. Further, the average binding distance, r, between the donor (HSA) and acceptor (corilagin) was calculated and found to be 1.32 nm according to Förster's theory of non-radiation energy transfer. Later, circular dichroism studies emphasized that there are marginal changes in secondary structural conformation of HSA in the presence of corilagin. Corilagin is specifically bound to site I of HSA which was proved by site specific marker, phenylbutazone. Furthermore, the binding details between corilagin and HSA revealed that corilagin was bound to subdomain IIA through multiple interactions like hydrogen bonding and hydrophobic effects. Molecular dynamic studies (MD) also suggest that binding is very precise to site I (IIA domain) on HSA. Also, MD studies showed that HSA–corilagin complex reaches equilibration state at around 4 ns, which proves that the HSA–corilagin complex is stable in nature, hence the experimental and computational results are in agreement. Thus, examining the interaction mechanism of corilagin with plasma proteins may play a critical role in developing corilagin inspired drugs.
1 Introduction
Corilagin (beta-1-O-galloyl-3,6-(R)-hexahydroxydiphenoyl-D-glucose), a gallotannin identified in several plants, is a novel member of the phenolic tannin family with its molecular formula of C27H22O18 having molecular mass of 634.45 Da. It has been shown to exhibit versatile medicinal activities. There has been little research on the effect of corilagin on cancer; instead, most research has been focused on its use as an anti-viral, hypolipidemic, hypotensive, and anticoagulant agent.1 Some pharmacological activities of corilagin have already been described, such as antiatherogenic,2 antioxidant,3 hepatoprotective,4 and antitumor.5 Furthermore, corilagin was also able to inhibit the release of cytokines such as TNF-α, IL-1β and IL-6 as well as the production of nitric oxide, both of which are mediators of inflammation and pain. Corilagin has been reported as a TNF-α-releasing inhibitor in inflammatory scenarios.6 It's proven that corilagin has the potential to reduce HSV-1 induced inflammatory insult to the brain and anti-inflammatory activity in a cellular model.7 Thus, it's confirmed that corilagin is an inhibitor of TNF-α and can restrain radiation-induced microglia activation via suppression of the NF-κB pathway.6 Corilagin is protective against Gal/LPS-induced liver injury through suppression of oxidative stress and apoptosis. Several reports suggest that Phyllanthus amarus containing tannins including corilagin and geraniin exhibit a high degree of antiviral activity against HIV infection.8 In spite of the broad use of corilagin as mentioned, their binding with plasma protein is unclear. In this work we have described the detailed investigation on the binding mechanism of corilagin with plasma proteins.
Binding of various drugs to plasma proteins has a pivotal role for drug distribution and its action. Most drugs reversibly bind to plasma proteins and are transported in the blood circulatory system in a free state and a dissolved state in the plasma and may form a complex with plasma.9 So, among all the plasma proteins, drugs mostly bind with human serum albumin (HSA), which is the most predominant protein, a negative acute-phase protein, and to some extent with α-1-acid-glycoprotein (AGP) which is a positive acute phase protein.10,11 Absorption, distribution, metabolism, excretion, stability, and toxicity primarily depend on the binding of endogenous and exogenous molecules to HSA.12,13 Studies on the binding of drugs with HSA may provide information of structural features that determine the therapeutic effectiveness of drugs, and this has been studied for many years. HSA is a major circulatory protein and the most abundant protein in blood plasma and it is comprised of 585 amino acid residues. HSA is a non-glycosylated single chained polypeptide having 67 kDa mass, which organizes to form a heart-shaped protein with approximately 67% α-helical content.14–18 HSA binds mainly to acidic, neutral, and basic drugs, whereas basic drugs bind exclusively to AGP. HSA is a helical protein with turns and extended loops, and resembles a heart shape monomer, with approximate dimensions of 80 × 80 × 30 Å. It consists of three domains: I (residues 1–195), II (196–383), and III (384–585), plus seven water molecules and the overall structure is stabilized by 17 disulfide bridges.19,20 There are two primary binding sites on the protein, known as Sudlow's sites I and II. Aromatic and heterocyclic ligands have been found to bind within two hydrophobic pockets in sub-domains IIA and IIIA. Further, seven binding sites for fatty acids are localized in sub-domains IB, IIIA, and IIIB, and on the sub-domain interfaces. HSA also has a high-affinity metal binding site at the N-terminus.21 Usually, drugs bind to one or very few high-affinity sites with typical association constants in the range of 103 to 106 M−1.13 We have extensively studied the binding of natural and synthesized compounds with HSA, including trimethoxyflavone, coumarintyramine, β-sitosterol, coumarin derivatives, 7-hydroxy coumarin derivative, chito-oligomers, asiatic acid and lupeol. These results showed a strong binding towards HSA, which lead to the complexion of protein and ligand, and hence resulted in conformational changes of HSA.22–28
AGP, another plasma protein also known as Orosomucoid, is an acute phase plasma protein and is the principal extracellular lipocalin present in blood. AGP consists of 183 amino acid residues and five N-linked oligosaccharides, with a molecular weight of 44 kDa. The five carbohydrate chains account for about 40% of the total mass and render AGP very soluble and confer acidic (pI ∼ 2.8–3.8) properties with a net negative charge at physiological pH.29 AGP is mainly synthesized in the liver and is secreted into the blood circulatory system.30 It is reported that AGP is also synthesized and secreted by other organs which include the heart, lungs, and stomach.29 The major biological role of AGP is not well understood, though numerous in vitro and in vivo activities have been reported such as drug transport, inhibition of platelet aggregation, and modulation of lymphocyte proliferation.31–34 Similar to plasma proteins, the binding and distribution of wide range of exogenous and endogenous ligands is one among the major aspects of AGP.9 AGP–drug interactions are a focus of great importance in the pharmaceutical sciences as this interaction is a major factor in drug transport to tissue receptors, storage sites, and clearing organs35 in some chronic diseases. In most cases, ligand interactions at the protein binding level will significantly affect the efficacy, free concentration, distribution, rate of delivery, and elimination rate of drugs; thus, such studies will provide information on those structural features that determine the therapeutic affectivity of drugs, and have been an interesting research field for many years in life sciences, chemistry, industrial, biological and clinical medicine.36 Hence, it's important to understand the binding nature of protein with corilagin, a natural compound, and also pharmacokinetics of corilagin to understand the impact of ligand delivery and transport during acute (AGP) and normal phase (HSA) conditions. The present study emphasizes the molecular interactions of corilagin with plasma proteins and the role of cytotoxicity activities. Consequently, this helps in understanding the binding mechanism with plasma protein and drug delivery clues.
2 Results and discussion
2.1 Cell response assay
Biological and pharmacological importance, and its effect of corilagin on cell response of mouse macrophages (RAW 264.7), was examined by using the MTT assay. In order to induce the inflammation in mouse macrophages cell line lipopolysaccharide (LPS) (1 μg mL−1) was used. Earlier reports show that corilagin has anti-inflammatory and antioxidant properties.3,7,37 In a dose dependent manner corilagin has shown anti-inflammatory properties with an IC50 of 66 μM (Fig. SI 1†). The results obtained reveal that corilagin showed an increased inhibition in the growth of inflamed macrophages in a dose dependent manner. We have also tested corilagin with other cell lines including HT-29 (human colon adenocarcinoma cell line), HepG2 (hepatic carcinoma), and Mcf7 (breast cancer cell lines); however, corilagin is specific to macrophages, which indicates that it can act explicitly against inflammatory conditions. Thus, we can confidently project that corilagin is a potential therapeutic agent.
2.2 Analysis of fluorescence emission spectra data
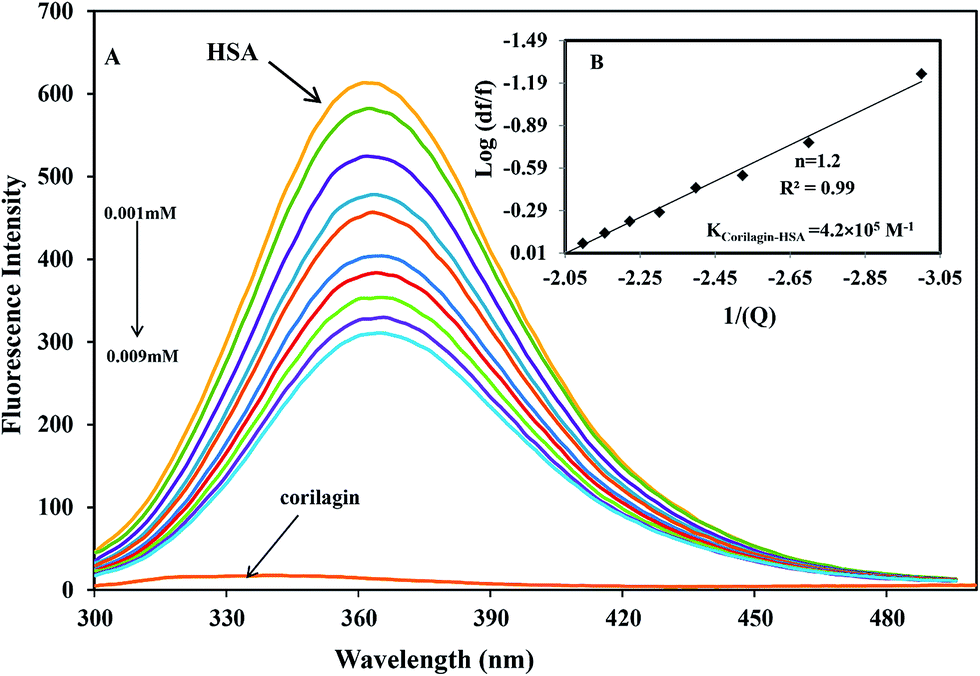
The binding affinity of protein-drug interaction was done by using fluorescence emission spectroscopy. The main emission spectra of HSA and AGP is due to presence of aromatic amino acid tryptophan, phenyl alanine, and tyrosine. Among all the amino acids tryptophan has a high quantum yield so the major fluorescence of HSA and AGP is dominated by tryptophan residue. The fluorescence emission spectrum of HSA is due to the presence of single residue Trp-214 which is located in subdomain IIA, one of the major binding sites. The process of fluorescence quenching is observed due to numerous molecular interactions such as energy transfer, reaction at excited state, static and dynamic quenching, and molecular rearrangements. Thus, intrinsic fluorescence protein quenching has been used to reveal the interaction mechanism with a ligand. When small molecules bind to HSA, it changes intrinsic fluorescence intensity due to the tryptophan residue.38 Fig. 1A shows the fluorescence emission spectra of HSA with various amounts of corilagin following an excitation at 285 nm. HSA exhibits a strong fluorescence emission band at 360 nm. Increasing concentrations of corilagin (0.001–0.009 mM) to HSA caused a quenching of fluorescence emission without any peak shift, hence this indicates that HSA–corilagin forms a complex. The fluorescence quenching is due to a decrease in the quantum yield of Trp-214 by variety of induced molecular interactions with corilagin. There are numerous reports that have showed the quenching of intrinsic fluorescence of HSA upon interacting with various drug molecule.25,26,39–43
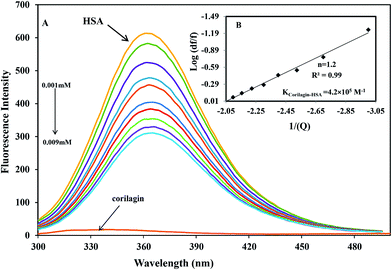 |
| | Fig. 1 Fluorescence emission spectra of HSA–corilagin in 0.1 M phosphate buffer with pH 7.2, λex = 285 nm, and temperature = 25 ± 1 °C. (A) Free HSA (0.001 mM) and free HSA with different concentrations corilagin of 0.001–0.009 mM. (B) Plot of log(dF/F) against log[Q]. λex = 285 nm and λem = 360 nm. | |
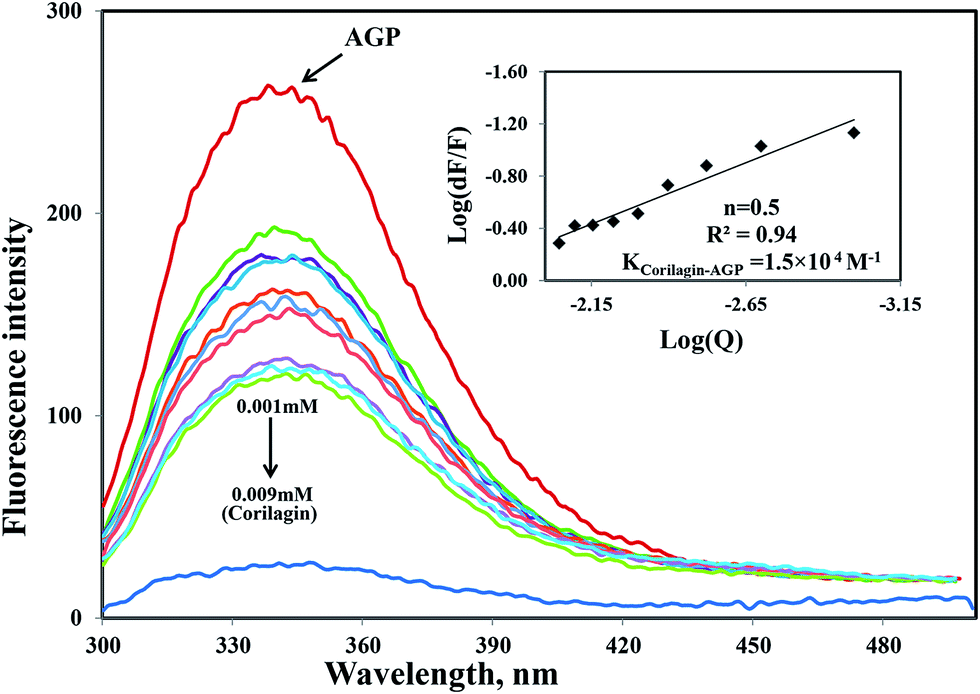
Similarly, the same concentration of corilagin was titrated against AGP which lead to a decrease in fluorescence emission maximum of AGP, indicating that corilagin was binding to AGP (Fig. 2). Thus, the plasma proteins HSA and AGP both bind to corilagin, as these two proteins are major carrier molecules for different endogenous and exogenous drugs in the blood circulatory system. Since HSA is a negative acute-phase protein and AGP is a positive acute-phase protein, binding studies with both HSA and AGP play a major role in deciding the pharmacokinetic role of corilagin in various biological conditions, especially during diseased and inflammatory conditions. Thus, to understand the pharmacokinetic behavior, the interaction of corilagin with AGP and HSA is of utmost importance in both normal and pathological conditions.
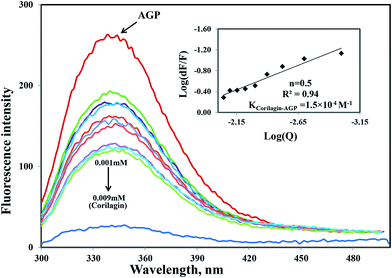 |
| | Fig. 2 Fluorescence emission spectra were measured for AGP along with corilagin in 0.1 M phosphate buffer with pH 7.2, λex = 285 nm, and temperature = 25 ± 1 °C. Free AGP (0.001 mM) and free AGP with different concentrations of corilagin, 0.001–0.009 mM. Insert: plot of log(dF/F) against log[Q]. λex = 285 nm and λem = 340 nm. | |
The quenching system is classified as static or dynamic quenching. These quenchings are well known by differences in the fluorescence temperature, life time, and viscosity.38 Dynamic quenching is a process in which the fluorophore and the quencher (drug) come into contact during the transient existence of an excited state. Static quenching is a process in which fluorophore (HSA)–quencher (corilagin) complex formation takes place. To verify whether it is static or dynamic quenching in HSA–corilagin complex, we plotted F0/F against Q. The resulting plot is linear for HSA–corilagin complexes indicating that the quenching is mainly static in these protein–drug complexes.41 The Kq was estimated according to the Stern–Volmer equation:
| | |
F0/F = 1 + Kqt0[Q] = 1 + KD[Q]
| (1) |
where
F and
F0 are the fluorescence intensities in presence and absence of quencher, [Q] is the quencher concentration, and
KD is the Stern–Volmer quenching constant (
Kq), which can be written as
KD =
Kqt0; where
Kq is the bimolecular quenching rate constant and
t0 is the lifetime of the fluorophore in the absence of quencher; lifetime of the fluorophore for HSA is 5.6 ns.
44–46 The quenching constant (
Kq) for corilagin is calculated (Fig. SI 2
†) to be 6.2 × 10
13 M
−1 s
−1. As this value is much greater than the maximum collisional quenching constant 2.0 × 10
10 M
−1 s
−1,
22,41,47–50 hence, the static quenching mechanism is prevalent in these HSA–corilagin complexes.
The decrease in fluorescence indicates that corilagin is binding to HSA, causing microenvironment changes in HSA due to formation of HSA–corilagin complexes.
| |
log[(F0 − F)/F] = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ks + n × log[Q] Ks + n × log[Q]
| (2) |
where
Q,
n, and
Ks are the quencher concentration, number of binding sites, and binding constant, respectively. From this equation the plotted results indicated a good linear relationship. The number of corilagin molecules binding to HSA was calculated to be 1.2, suggesting that HSA interacts with corilagin in a close relationship of one-to-one ratio (
Fig. 1B). From the slope of the plot, binding constants of corilagin were calculated from the intercept as 4.2 ± 0.02 × 10
5 M
−1 which indicates strong binding of corilagin to HSA. There is a good correlation and near to the computational calculated binding constant of 1.5 × 10
4 M
−1 obtained as lowest free energy. The binding constants and free energy of the best docking conformer are close and consistent with the binding constants and free energy determined by fluorescence studies. Thus, these results are corroborated with the experimental data. Interestingly, we have reported that natural compounds binding to HSA showed similar results on betulinic acid, feruloyl maslinic acid, trimethoxyflavone, and coumaroyl tyramine; and their binding constants were
KBA = 1.685 ± 0.01 × 10
6 M
−1,
KFMA = 1.42 ± 0.01 × 10
8 M
−1,
KTMF = 1.0 ± 0.01 × 10
3 M
−1, and
KCT = 4.5 ± 0.01 × 10
5 M
−1.
24,25,51,52
Interestingly, the binding of corilagin with AGP also was stronger and the binding constant was found to be 1.5 ± 0.01 × 104 M−1 which is in the range of Food and Drug Administration (FDA) data. Further, the computational calculated binding constant as 1.0 × 103 M−1 was obtained as lowest free energy. However, corilagin has good interactions with HSA and AGP; in fact, binding constants with AGP fell in the range of known FDA approved drugs like furosemide, imipramine, and benzyl penicillin which have binding constants of 2.6 × 104, 2.5 × 104, and 1.2 × 103 M−1, respectively with HSA13 and which are known to have significant pharmaceutical importance. These derivatives possess binding constants in the range of 103 to 104 thus suggesting that corilagin binds strongly with HSA and AGP, which has importance in the disposition of these molecules. It is known that in most of the normal cases HSA acts a carrier for various drug molecules; however, in pathological conditions, the AGP also plays a major role in transporting drug molecules. Recently, we also reported that chitosan oligomers were strongly bound to AGP, whereas other molecules like 7-hydroxycoumarin derivatives showed weak binding to AGP.22 Thus, corilagin is a potent phytochemical and can bind to both HSA and AGP which are pharmacologically important proteins.
Free energy calculations. The standard free energy is calculated according to the following equation:| | |
ΔG° = −RTlnK
| (3) |
where ΔG is a free energy, K is a binding constant at the corresponding temperature, which can be obtained from fluorescence data, and R is the gas constant. The calculated free energy change is −7.6 kcal M−1 at 25 °C. The calculated computational free energy value is −5.71 kcal M−1 and the results are in agreement with the experimental data. Here the lower free energy value is mainly due to a hydrophobic interaction of corilagin binding to HSA. In the case of AGP, the free energy calculated from experimental and computational means was −5.6 and −3.83 kcal M−1, respectively. The difference in free energy and binding constants from experimental and computational, may be due to differences in arrangement solution and crystal structural. Hence, it is validation of both the experimental and computationally calculated free energies. In addition, similar types of interactions, such as hydrophobic and hydrogen bonding, were observed with our recent studies on natural as well as synthesized compounds trimethoxyflavone, coumaroyl tyramine, asiatic acid, chitosan oligomers and 7-hydroxy coumarin derivatives with HSA.22,24–27,40
2.3 Energy transfer from HSA to corilagin
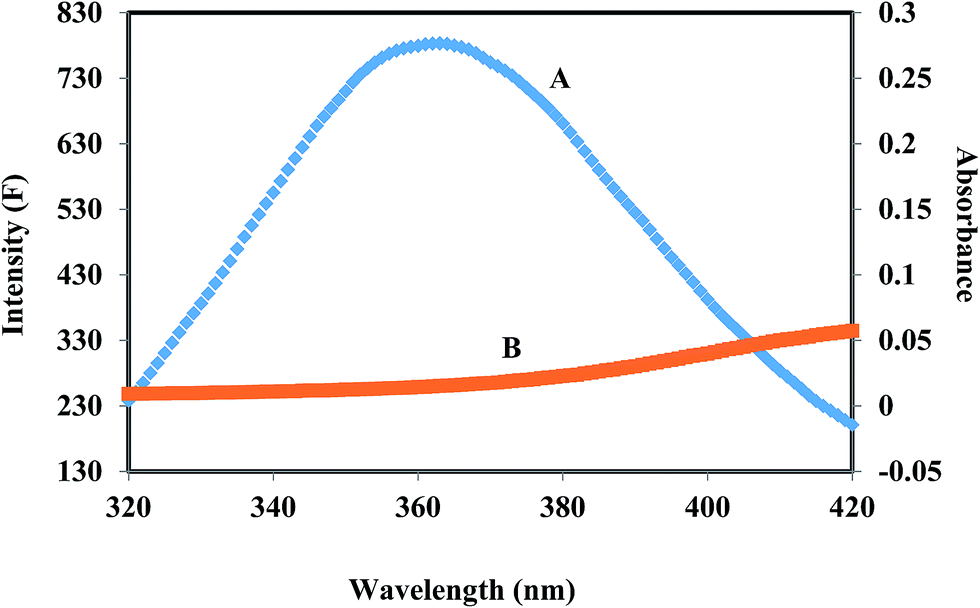
HSA has a single tryptophan residue (Trp-214). The distance r between the Trp-214 in HSA and the bound corilagin could be determined using fluorescence resonance energy transfer (FRET). FRET has been widely used to determine the molecular distance between acceptor and donor molecules. Generally, FRET occurs whenever the emission spectrum of a fluorophore (donor) overlaps with the absorption spectrum of another molecule (acceptor). The overlap of the UV absorption spectrum of corilagin with the fluorescence emission spectra of HSA are shown in Fig. 3. The distance between the donor and acceptor and extent of spectral overlaps determines the extent of energy transfer. The distance between the donor and acceptor can be calculated according to Fösters theory.53 The efficiency of energy transfer, E, can be calculated as| | |
E = 1 − F/F0 = R06/R06 + r6
| (4) |
where F and F0 are the fluorescence intensities of HSA in the presence and absence of corilagin, r the distance between acceptor and donor, and R0 is the critical distance when the transfer efficiency is 50%. R0 can be determined as| | |
R06 = 8.8 × 10−25 × K2 × N−4 × ϕ × J
| (5) |
where K2 is the spatial orientation factor of the dipole, N the refractive index of the medium, ϕ the fluorescence quantum yield of the donor, and J is the overlap integral of the fluorescence emission spectrum of the donor and the absorption spectrum of the acceptor. J can be calculated as| | |
J = ∑F(λ)ε(λ)λ4Δλ/∑F(λ)Δλ
| (6) |
where F(λ) is the fluorescence intensity of the fluorescent donor of wavelength, λ and ε(λ) is the molar absorption coefficient of the acceptor at wavelength, λ. In the present case, K2 = 2/3, N = 1.336 and ϕ = 0.118 for HSA.54 From the above mentioned equations we are able to calculate J = 9.41 × 10−15 cm3 L mol−1, R0 = 2.57 nm, E = 0.49, and r = 1.32 nm for HSA. The donor–acceptor distance, r < 8 nm (ref. 55 and 56) indicates that energy transfer from HSA to corilagin occurs with high possibility, thus the existence of static quenching due to complex formation between HSA (Trp-214) and corilagin.
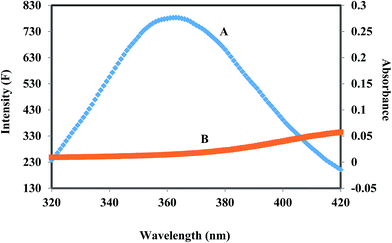 |
| | Fig. 3 The overlap of fluorescence spectrum of HSA (A) and absorbance spectrum of corilagin (B) [λex = 285 nm, λem = 360 nm c(HSA)/c(corilagin) = 1:1]. | |
2.4 Displacement experiment using site specific marker for human serum albumin with corilagin
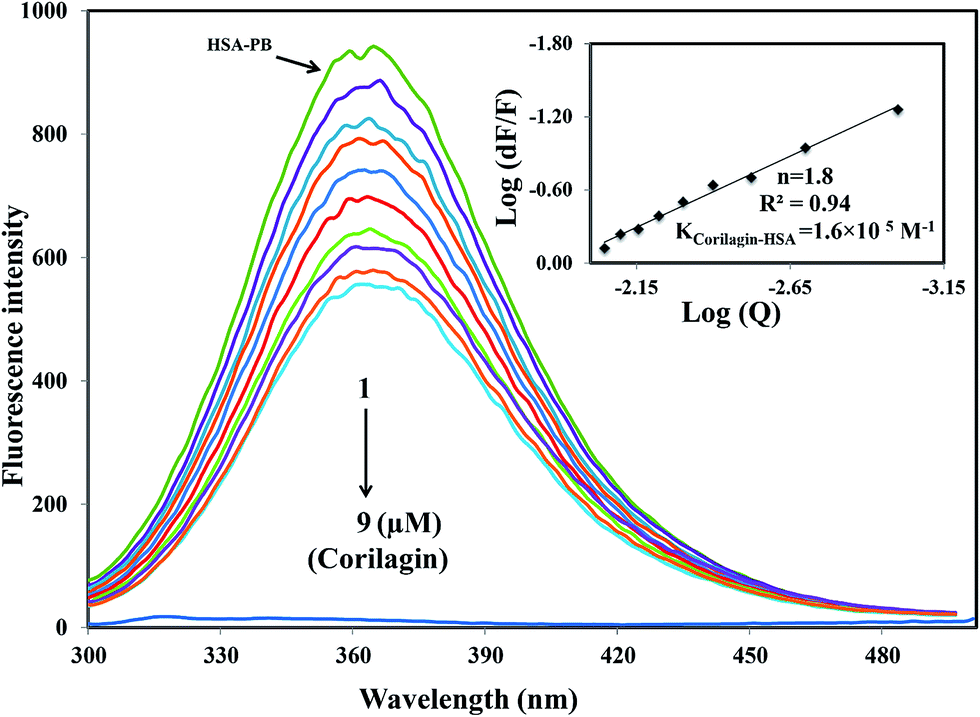
A majority of ligands bind to HSA at primary sites known as sudlow site I and II, which are located deep inside the subdomain IIA and IIIA, respectively. Site-specific competitive experiments were conducted at room temperature in which lidocaine (subdomain IB marker), phenylbutazone (site I marker), and ibuprofen (site II marker) were used as marker drug molecules that exclusively bind HSA. In the site-specific displacement experiments, a solution of corilagin was gradually titrated with HSA and the concentration of HSA and site marker is 1:1. Then, the fluorescence intensities are measured and analyzed; Fig. 4 shows that the addition of phenylbutazone (site I marker) slightly changes the fluorescence spectra of HSA which indicates that it has bound to HSA. Later on, corilagin was titrated continuously with various concentrations where HSA and site markers are kept at constant concentration. Corilagin has to compete with lidocaine (subdomain IB marker), phenylbutazone (site I marker), and ibuprofen (site II marker) to get the chance to bind to HSA if they bind to HSA in the same site. This was proved by the gradual decrease in fluorescence intensity, but the intensity was much lower in the absence of site-specific markers. The binding constant of the corilagin–HSA system is ∼3.9 ± 0.02 × 104 (lidocaine), 1.6 ± 0.01 × 105 (phenylbutazone), and 3.7 ± 0.02 × 104 M−1 (ibuprofen), respectively. The original binding constant of HSA–corilagin is 4.2 ± 0.02 × 105 M−1. The binding constants in the presence of phenylbutazone, and in its absence, are close to each other whereas binding constants in the presence of other site-specific markers like lidocaine and ibuprofen are less which indicates that corilagin competed and displaced phenylbutazone from the binding site. Thus, the displacement measurement confirms that corilagin is bound to Sudlow's site I of HSA (Fig. 4). Similar kinds of competitive displacement studies were reported in our pervious papers.22,48
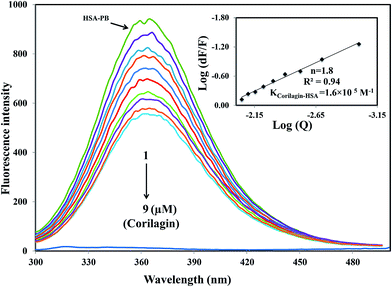 |
| | Fig. 4 Displacement of phenylbutazone from HSA–phenylbutazone complex by corilagin. Fluorescence emission spectra were recorded on Perkin-Elmer LS55 fluorescence spectrometer for corilagin with HSA in presence of site probe markers. Phenylbutazone is referred as site-I probe. The concentrations of HSA and phenylbutazone were maintained constant at a concentration of 1 μM whereas the corilagin was varied from 0 to 9 μM. Inserts: modified Stern–Volmer plot. Plot of log(dF/F) against log[Q] λex = 285 nm, λem = 360 nm. | |
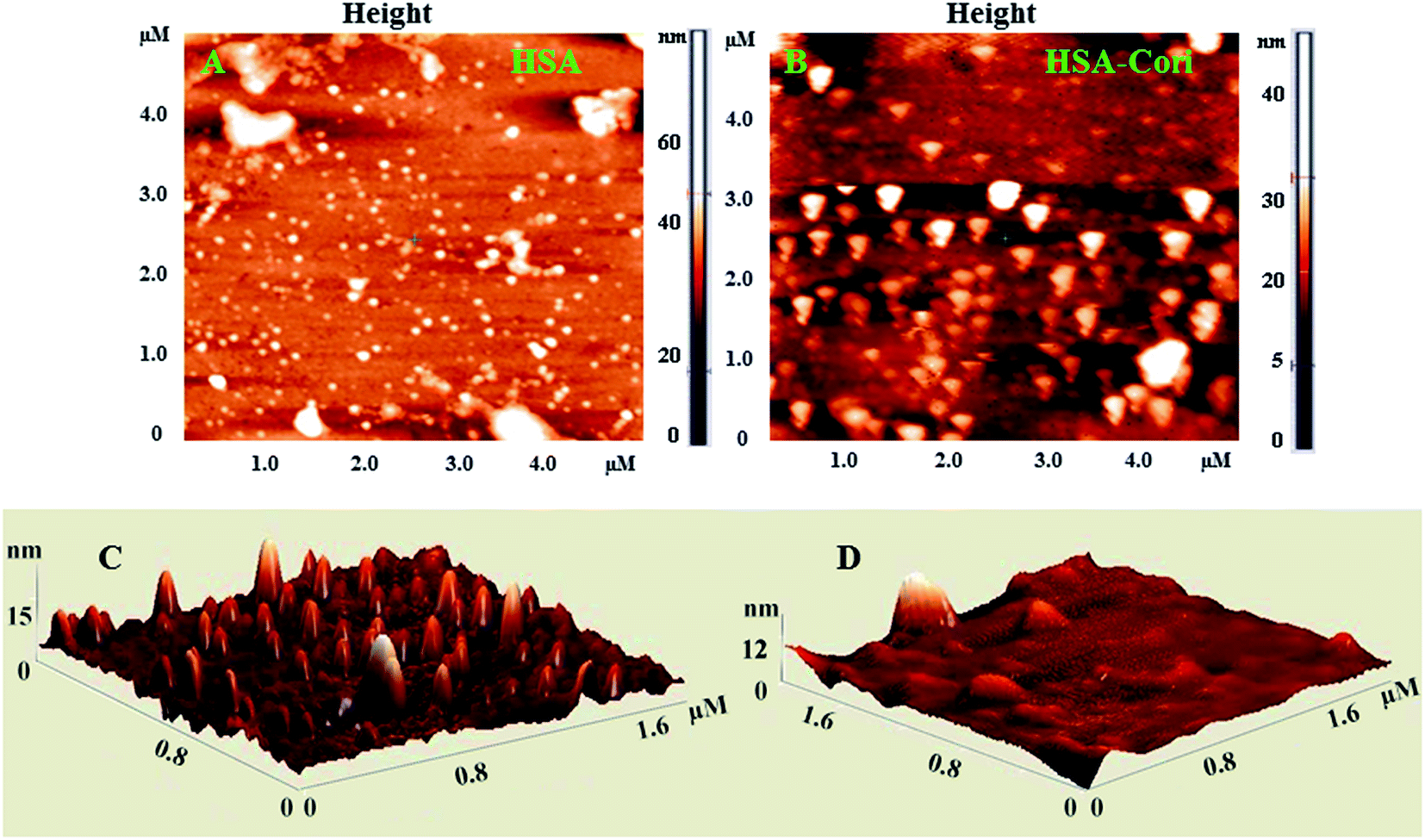
2.5 Atomic force microscopy (AFM)
To investigate topography changes in HSA upon addition of corilagin, the un-liganded HSA and HSA–corilagin complexes were clearly visualized by using AFM in triplicate. Fig. 5A and B shows the results obtained from AFM for the un-liganded HSA and its HSA–corilagin complexes. The data shows that HSA was adsorbed evenly on the mica glass surface. The mean height of the different HSA molecules was 137.3 ± 2.47 nm. These dimensions are nearer to a previous report of HSA.57 After addition of corilagin, the HSA molecule became swollen, and the mean height of HSA reached to 337.5 ± 3.05 nm which indicates the aggregation or HSA–corilagin complexation. Thus, the microenvironment surrounding HSA became more hydrophobic after interacting with corilagin. Therefore, minimizing some factors unfavorably affecting the formation of a stable complex structure, the HSA molecule reduced its surface area of contact with water by molecular aggregation. Mostly proteins are aggregated under certain conditions like a special composition ion, appropriate pH value, and concentration of a protein solution. Moreover, protein–protein hydrophobic interaction is an important factor to cause proteins aggregation.58 The different shapes and size distributions point towards distinctly different forms of morphology of free HSA and HSA–corilagin complex. These results again support the formation of a protein ligand complex and, as a result, the morphology is different from the free protein (Fig. 5C and D). Hence, the results exhibit that a hydrophobic interaction between HSA and corilagin may exist.
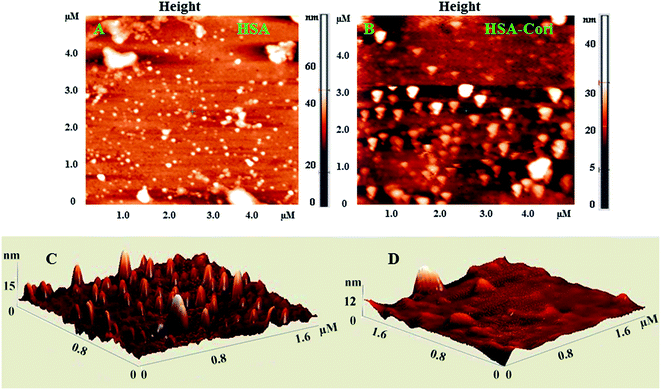 |
| | Fig. 5 2D AFM images of (A) free HSA (B) HSA–corilagin complex. The corresponding 3D images are given by (C) and (D) respectively, scan areas are given in brackets. | |
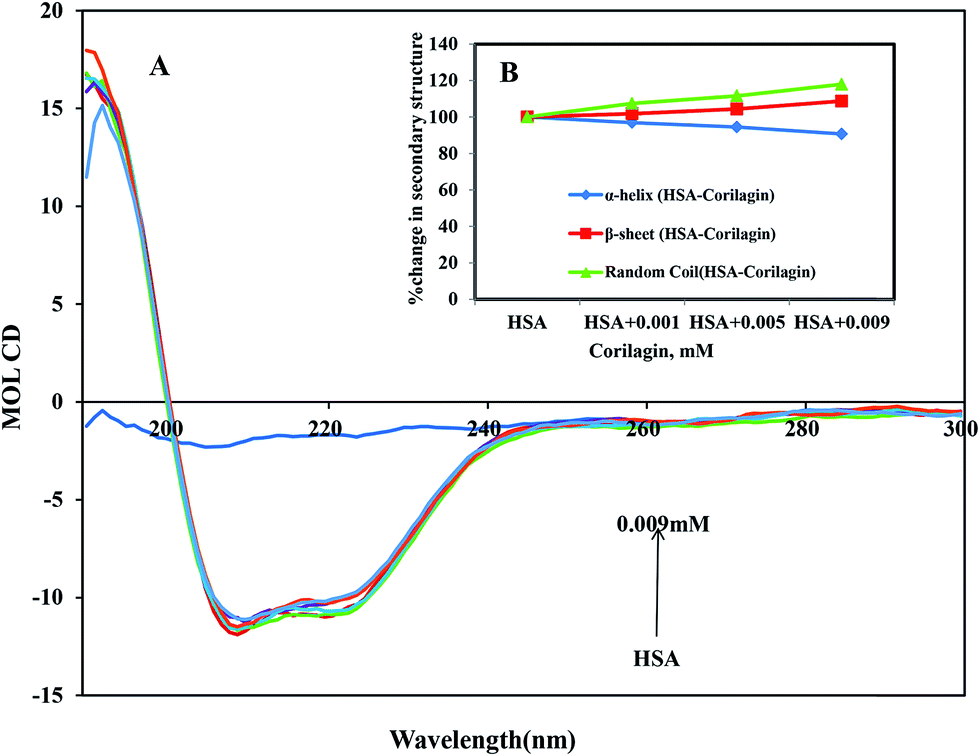
2.6 Circular dichroism (CD) spectroscopy
CD is the most vigorous analytical spectroscopy technique which helps in understanding the changes in the secondary and tertiary structure of proteins in the far-UV region. The possible effect on the overall structure of HSA upon binding with corilagin was monitored by CD spectroscopy (Fig. 6). The secondary structure of HSA can be determined by CD spectroscopy in the “far-UV” spectral region (190–250 nm). HSA has two absorption peaks at 208 and 222 nm and these peaks originate from α-helix in the protein. Addition of corilagin (0.001–0.009 mM) to HSA (0.001 mM) resulted in a decrease in absorbance intensity at 208 nm and a significant change at 222 nm, which indicated a decrease in the α-helix with an increase in the β-sheets and random coils induced by a specific interaction between corilagin and HSA. Also, it indicates conformational changes induced by corilagin on the overall structure of HSA and resultant formation of HSA–corilagin complexes. Free HSA has 58% α-helix, 23% β-sheets, and 19% random coils, which is in agreement with the previous reports.25,52,59 The original crystal structure, however, was reported to be 67% of the α-helical content;14 in our study it was 58%. The differences in α-helical contents could originate from different structural arrangements of the protein in a solid state (X-ray structure) and in aqueous solution (CD measurements). However, there was no change in tertiary structure of HSA when corilagin binds to it. The percentages of the secondary structural elements of HSA and the corilagin–HSA complex are shown in Table 1. A slight decrease in the α-helical content from 58 ± 2.5 to 52.6 ± 1.8 and an increase in the β-turns from 23 ± 0.73 to 25 ± 0.1, and random coils from 19 ± 1.0 to 22.4 ± 0.8 indicates that the addition of corilagin induced a partial unfolding of the secondary structure of HSA. The partial unfolding may be due to microenvironment changes within the proximity of tryptophan residue while binding to corilagin, which was also revealed by a decrease in fluorescence emission quenching and AFM data. The slight decrease of α-helical content with an increase in the β-strand and random coils may be due to the formation of HSA–corilagin complex with hydrogen and hydrophobic interactions. Our results are in agreement with previous studies that advocate partial loss and conformational changes in overall structure of HSA upon complexion with ligand binding.22,51,52,60–63
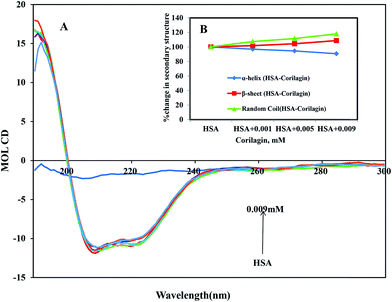 |
| | Fig. 6 (A) Circular dichroism spectra of free HSA and its corilagin complexes in aqueous solution with a protein concentration of 0.001 mM and corilagin concentrations were 0.001, 0.005, and 0.009 mM. Spectra were recorded with a JASCO J-810 CD spectropolarimeter. A quartz cell with a path length of 0.02 cm was used. (B) The secondary structural changes of HSA and HSA–corilagin; the plot represents the concentration-dependent secondary structural changes of free HSA and HSA–corilagin. | |
Table 1 Secondary structural analysis of HSA and HSA plus different concentrations (0.003, 0.005 and 0.009 mM) of corilagin. On the basis of Fig. 6, the data was analyzed by web-based software CDNN 2.2
| Corilagin |
HSA |
HSA + 0.001 mM |
HSA + 0.005 mM |
HSA + 0.009 mM |
| α-Helix% |
58 ± 2.5 |
56.2 ± 2.03 |
54.8 ± 2.04 |
52.6 ± 1.8 |
| β-Sheet% |
23 ± 0.73 |
23.4 ± 0.6 |
24 ± 0.8 |
25 ± 0.1 |
| Random coils% |
19 ± 1.0 |
20.4 ± 0.6 |
21.2 ± 0.4 |
22.4 ± 0.8 |
2.7 Thermal stability of HSA and HSA–corilagin complexes
In order to determine the stability of HSA–corilagin complexes, temperature-dependent CD was carried out for HSA and HSA with 0.009 mM of corilagin, from 25–85 °C. The secondary structural conformation of protein is not significantly changed up to 60 °C in HSA–corilagin complexes (Fig. SI 3†). Above 65 °C the α-helical content decreased dramatically due to thermal denaturation, while the β-sheets and random coil content increased in both HSA–corilagin complexes. An earlier report showed that the Tm of HSA alone was around 65 °C, which shows that the unfolding of protein occurs only after this point.25 In the secondary structural conformation it was noticed in corilagin plus HSA complexation (0.009 mM) that the α-helical contents were 52.6 ± 1.8% and 33.7 ± 2.0%, β-sheets 25 ± 0.73% and 38.2 ± 1.0%, and random coils 22.4 ± 0.8% and 28.1 ± 2.0%, respectively, which indicates that there is no release of corilagin from its complexation. Thus, this result indicated that even at 60 °C the HSA–corilagin complex is stable, thus the protein complex is thermodynamically and conformationally stable.
2.8 Molecular docking studies
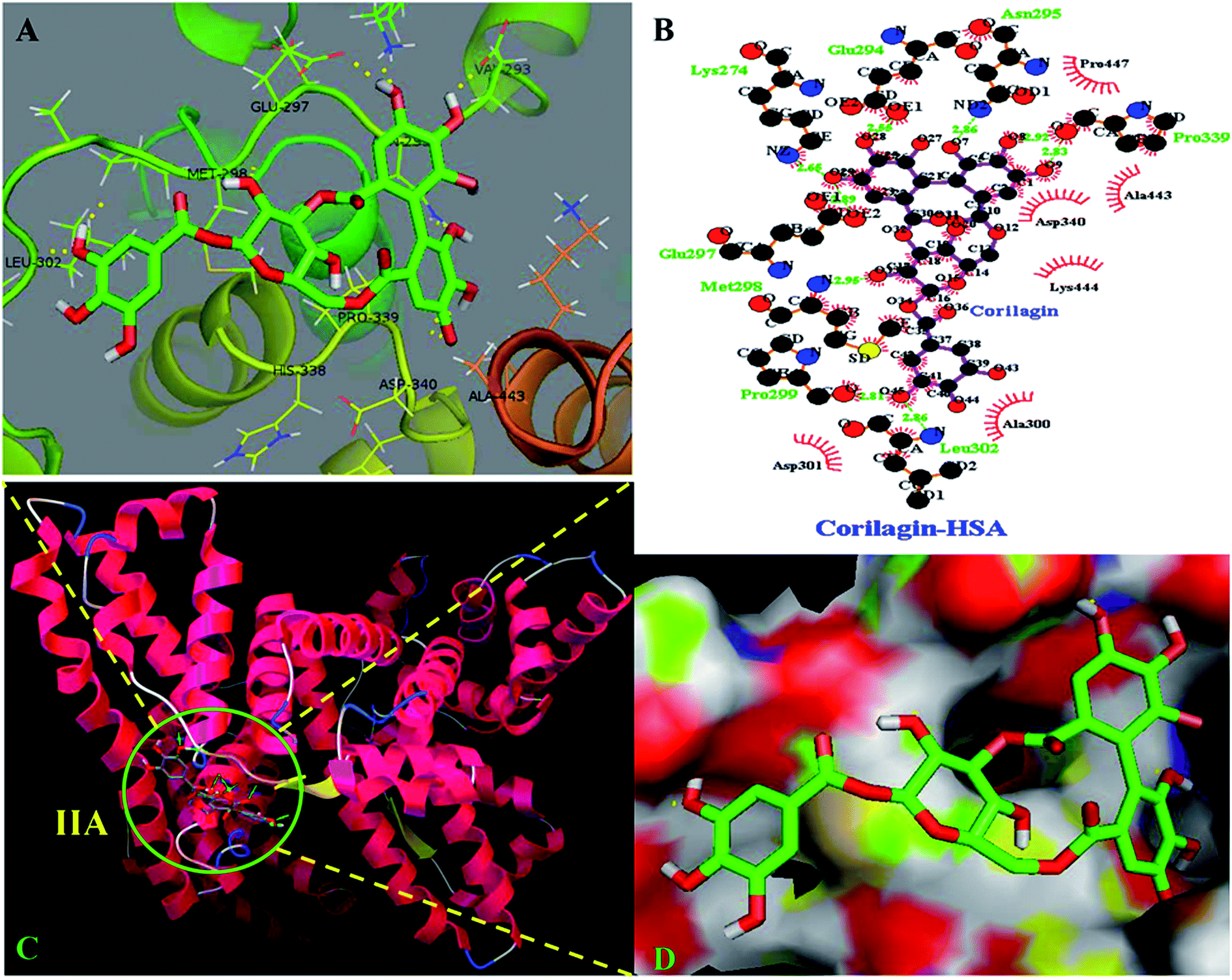
The site marker competitive experiments indicated that corilagin binding site on HSA was site I (subdomain IIA). To further describe the binding site and residues involved in the interaction of corilagin with HSA, molecular docking was done using Autodock (4.2.3) software to generate possible conformations upon binding of corilagin with HSA and AGP. The crystal structure of HSA and AGP was taken from the Protein Data Bank (entry PDB code 1AO6 and 3kQ0). The crystal structure of three homologous domains in HSA can be further divided into six-helix and four-helix subdomains (A and B).64 Due to presence of multiple binding sites, HSA has an extraordinary ability to bind various small molecules.19 Based on the site-specific markers used in our experiment, we determined that corilagin binds specifically to the site I (subdomain IIA). Further, we have defined the binding site using a molecule docking technique to determine the primary binding site of corilagin on HSA. By using Auto Dock software 4.2.3 about 30 conformers were generated from docking simulation and we chose the conformer on the basis of least free energy of binding and score ranking, which also matches the free energy obtained from fluorescence emission.65 The corilagin complex is stabilized by 8 hydrogen bonds between corilagin to LYS274, GLU294, ASN295, GLU297, MET298, PRO299, LEU302, and PRO339 of the protein with lengths of 2.65, 2.56, 2.86, 2.89, 2.98, 2.81, 2.86, and 2.98 Å. The minimum binding energy conformer was observed as −5.71 kcal M−1. We even validated the docking results by using X-score of Auto-doc conformer, wherein we obtained a reasonably good binding energy value of −8.73 kcal mol−1 and binding affinity of docked corilagin with HSA was 6.40 μM. That fact gives a moderate correlation with the experimentally determined values −7.6 kcal M−1 having an inhibition constant of 65.16 μM, and the binding constant was 1.5 × 104 M−1 (Fig. 7A). Docking results determined that corilagin was binding to HSA at subdomain IIA which is Sudlow's site I with the most stable docking conformer (Fig. 7B and C). It was also observed that the corilagin binding site in HSA was fully covered by hydrophobic interactions (Fig. 7D). Thus, corilagin is surrounded by hydrophobic and hydrophilic amino acids such as ALA300, GLY328, ASP340, and ALA443. Therefore, corilagin binds with HSA mainly by hydrophobic interactions with above mentioned amino acids involved in the interactions, along with few hydrophilic interactions that are shown in Fig. 7C. These results are in accordance with the free energy calculations obtained from the binding constant which was derived from fluorescence quenching data.
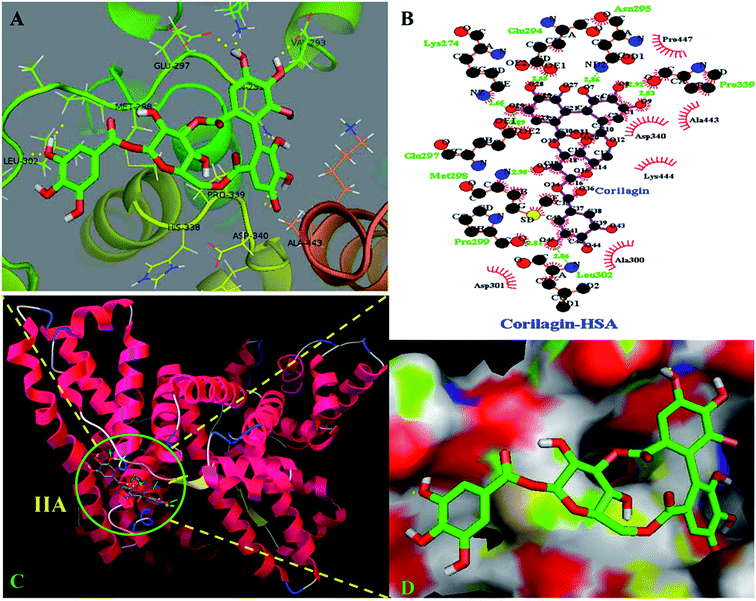 |
| | Fig. 7 Schematic representation of least binding energy docked conformation obtained from docking simulation. (A) Pymol stereo view of corilagin binding site to HSA IIA domain in which corilagin is rendered as capped sticks and surrounding residues as lines (LYS274, GLU294, ASN295, GLU297, MET298, PRO299, LEU302, and PRO339). (B) A graphical representation of Ligplot data to show the hydrophobic interactions of HSA with corilagin. (C) Corilagin bound to IIA domain on HSA (protein and ligand colored blue, red, yellow, and green, respectively). (D) The hydrophobic and hydrophilic amino acid residues surrounding corilagin. | |
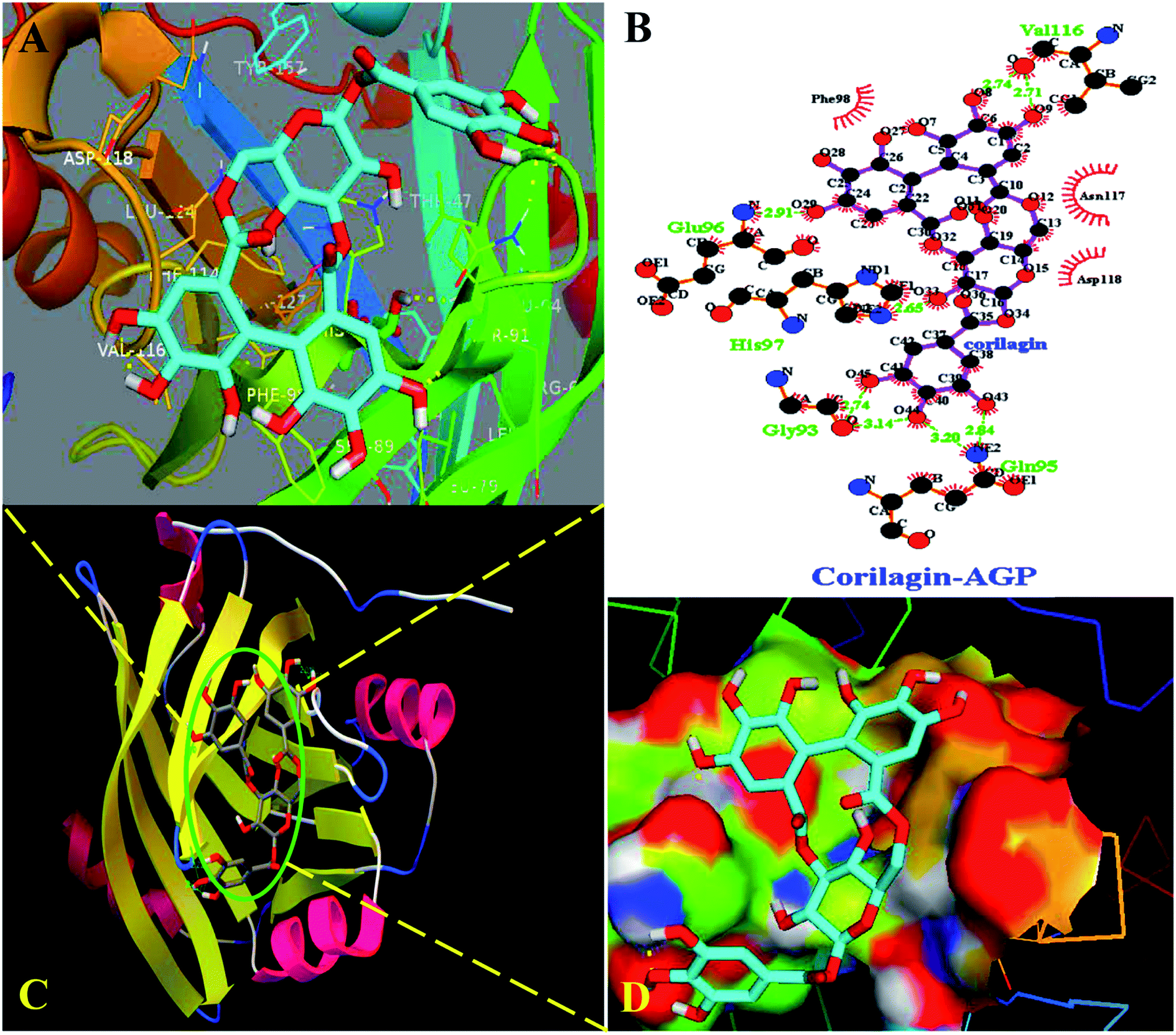
Interestingly, docking studies revealed that corilagin interacts with AGP with binding constants of 1.0 × 103 M−1 and free energy was found to be −3.83 kcal M−1 which is nearer to the experimental 1.5 ± 0.01 × 104 M−1 and −5.6 kcal M−1 values (Fig. 8). The corilagin–AGP complex is stabilized by hydrophobic amino acids and with 7 hydrogen bond between the compound and GLY93, GLU96, VAL116, HIS97 and GLN95 with bond length of 2.74, 2.91, 2.71, 2.14, 2.65, 2.84 and 3.20 Å. Overall the interactions of corilagin are varied between HSA and AGP, but the binding constants and free energy of experimental and computational are comparatively less than has, suggesting that HSA–corilagin complex is more stable than AGP–corilagin complex. The results of docking studies indicate that the interactions between corilagin and HSA, AGP are dominated by hydrogen and hydrophobic interactions, which is in excellent agreement with free energies that are obtained from fluorescence data. However, the pharmacological behavior could change differently in diseased conditions and thus, corilagin binding to plasma protein of AGP would certainly be an important process. Particularly, AGP is an acute phase protein; hence, differential interactions were observed for the corilagin molecule with HSA and AGP.
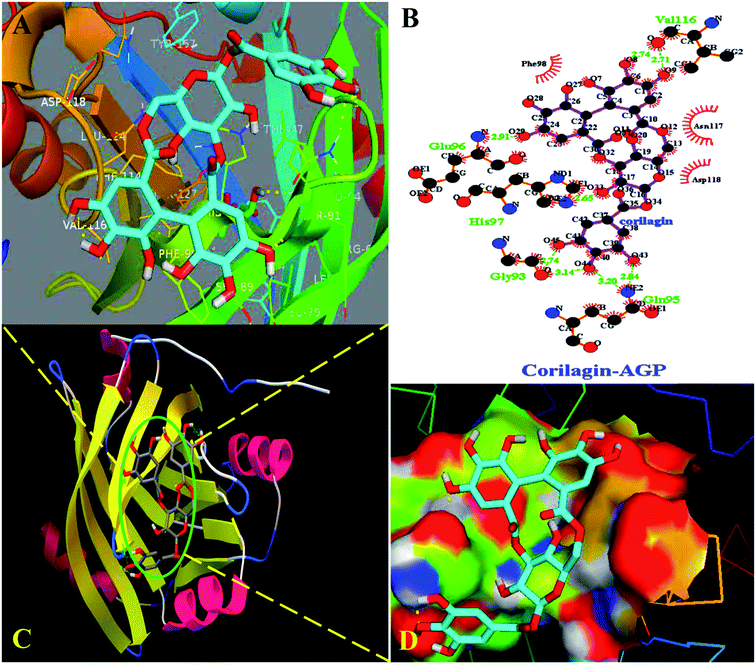 |
| | Fig. 8 Docking conformation of AGP–corilagin complex obtained from Auto dock v 4.2.3. (A) Pymol Stereo view of corilagin bound to AGP in which corilagin is rendered as capped sticks and surrounding residues as lines (GLY93, GLU96, VAL116, HIS97, and GLN95). (B) Ligplot analysis of AGP with corilagin to show the hydrophobic interactions and the binding pocket showing hydrophobic and hydrophilic amino acid residues surrounding the probe corilagin. (C) Corilagin bound to IIA domain on AGP (protein and ligand colored blue, red, yellow, and green, respectively). (D) The hydrophobic and hydrophilic amino acid residues surrounding the corilagin. | |
2.9 Molecular dynamics simulations
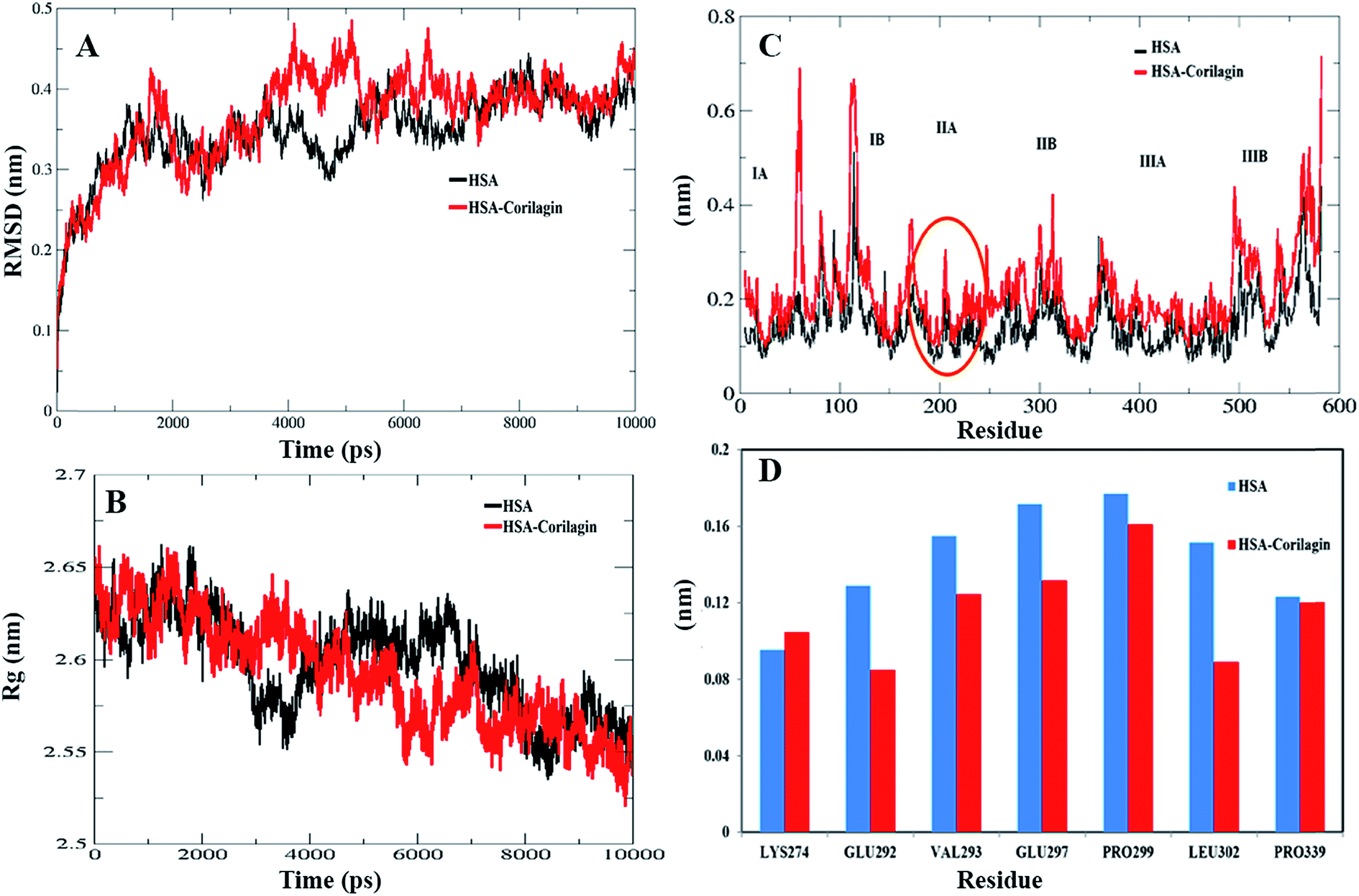
The least binding energy conformer complex was chosen on the basis of the docking results and its stability properties were obtained by calculating the root mean square deviation (RMSD), root mean square fluctuation (RMSF) and radius of gyration (Rg) during MD. The stability of the HSA–corilagin complex gives credibility of the docked results. The mentioned parameters i.e. RMSD, Rg, and RMSF values of atoms in un-liganded HSA and liganded HSA–corilagin with respect to the initial structures were calculated along 10000 ps trajectories (Fig. 9A and B). Free HSA and HSA–corilagin complex RMSD values steadily increased until 1000 ps followed by a slow increase up to 4000 ps. After this, there was no further increment of RMSD values and it reached equilibrium. After reaching equilibrium, the RMSD values of the C–Cα–N backbone for both the HSA and HSA–corilagin complex were calculated for the 1–10000 ps time scale (Fig. 9A). For HSA, the data point fluctuations are 0.45 ± 0.053 nm whereas for the complex they are 0.40 ± 0.036 nm. The decrease in RMSD value of the complex from that of the free HSA indicates that the ligand, upon binding with protein, showed slight structural change, increased rigidity, and stability. Thus, the MD simulations studies showed that these complexes remain in a stable binding position with slight RMSD fluctuations, confirming the integrity of docked conformer forecast by AutoDock 4.2.3. Remarkably, our data is in agreement with the earlier reports from our group.23,26,48,66 The protein reliability is analyzed by plotting Rg values against the function of time. Rg was used to assess the stability of the back bone atoms of HSA and HSA–corilagin complex (Fig. 9B). With the free HSA alone and HSA–corilagin, the Rg value is stabilized at about 2000 ps, which implies that the MD achieved equilibrium at 4000 ps. First, the Rg value of both free HSA and HSA–corilagin complex was 2.64 nm. The free HSA and HSA–corilagin complex was stabilized at 2.57 ± 0.03 nm (Fig. 9B). The Rg value of HSA, which is shown experimentally by neutron scattering in aqueous solution, was 2.74 ± 0.35 nm which showed that it synchronized with the experimental data. Previous studies showed that the Rg of HSA determined experimentally from neutron scattering in aqueous solution was 2.74 ± 0.035 nm, which indicates that the simulations performed are identical to the experimental values.67 Also, the present result is closely related with our previous studies where β-sitosterol, asiatic acid, and piperidine stabilized from 2.59 ± 0.03 nm to 2.40 ± 0.031 nm; 2.42 ± 0.03 nm to 2.45 ± 0.01 nm; and 2.64 ± 0.021 nm to 2.57 ± 0.03 nm for free HSA and HSA-with complexes, respectively.23,26,48 MD simulations provided insights of stability of the complexes of docked conformers. Coming to the RMSD figure, it gives the overall stability of the docked conformer specifically, based on fluctuation of the Cα-backbone relative to native HSA alone. Also, MD data showed that HSA plus corilagin reaches equilibration state at around 4000 ps, which indicated a stable HSA and corilagin complex. We further studied the RMSD and time evolution of the radius of gyration in the course of 20 ns of MD simulations of free HSA and the HSA–corilagin complex results are shown in Fig. SI 4; † as seen, the initial RMSD and Rg values are stabilized as mentioned above. Thus, based on the RMSD and Rg values that are obtained in this work, it can be concluded that HSA exhibits a slight conformational change when it combines with corilagin. RMSD provided the evaluation of the structural drift values of Cα atoms from the initial structure as a function of time. From 0–20 ns trajectory data the RMSD value for free HSA is 0.45 ± 0.04 nm, while the HSA–corilagin complex fluctuated initially and stabilized at around 4 ns simulation time with data point 0.43 ± 0.03 nm. The HSA–corilagin complex showed less fluctuation which indicates that only a few atomic fluctuations are in the magnitude of both RMSD and Rg where the protein–ligand complex reaches an equilibrium state. Thus, both experimental and computational results suggest a slight conformational change probably around the binding pocket of the HSA. So, during simulation the change of Rg value from HSA to HSA–corilagin over simulation time indicates stabilization and slight conformational changes in the secondary structure of HSA when bound to the corilagin. This observation again supports a partial change of CD spectral results as shown and discussed earlier. The local protein mobility was analyzed by calculating the RMSF values of HSA alone and from the HSA–corilagin complex. The RMSF values were plotted against residue numbers based on the 10000 ps trajectory (Fig. 9C). The profile atomic fluctuations were found to be very similar to those of HSA and HSA–corilagin complex. Here, the results show that, except subdomain IIA, all other domains IA, IB, IIB, and IIIA of HSA showed high fluctuation which indicated that corilagin is more rigid at site I, particularly IIA domain (Fig. 9C). This rigidity was located in individual residues of LYS351, GLU354, ALA213, VAL216, and ASP324 of HSA–corilagin (Fig. 9D). The specific binding was also confirmed from the site specific probes which showed that corilagin binds to IIA domain (Fig. 4). Thus, in site I the fluctuations are less when compared with the other drug binding sites IB and site II. Therefore, this study provides evidence that HSA binds to sites I (IIA subdomain) and interacts specifically with corilagin through conformational adjustments of the protein structure, in aggregation with ligand conformational variation at these sites. Based on the RMSD, Rg, and RMSF values obtained in this work, it can be concluded that the HSA molecule exhibits a slight conformational change when it combines with corilagin.
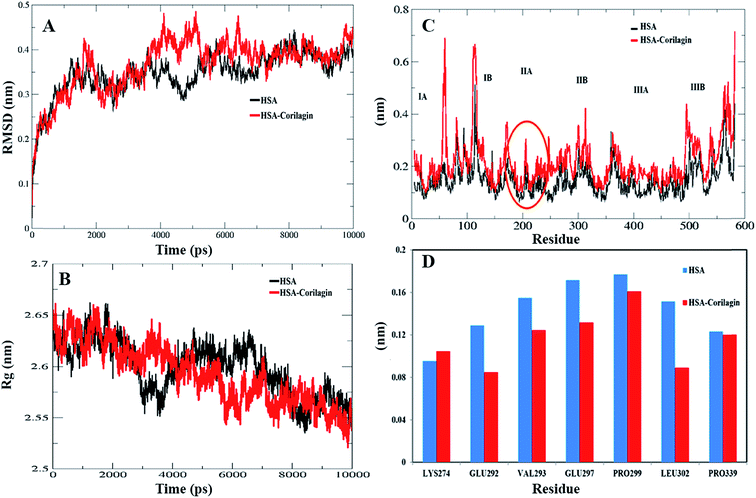 |
| | Fig. 9 (A) Root mean square deviation (nm) of un-ligand HSA and ligand HSA (HSA–corilagin). (B) Time dependence of the radius of gyration (Rg) for the backbone atoms of un-ligand HSA and ligand HSA (HSA–corilagin). (C) RMSF values against residue numbers. The RMSF values of un-ligand HSA and HSA–corilagin complex were plotted against residue numbers. (D) The profile of atomic fluctuations. Atomic fluctuations of un-liganded HSA and HSA–corilagin complex to the active site amino acid residues present in the IIB subdomain of HSA which is site I. | |
3 Materials and methods
3.1 Preparation of stock solutions
Fat free HSA was purchased from Sigma-Aldrich (USA), and dissolved in physiological aqueous solution of 0.1 M phosphate buffer at pH 7.2, at a concentration of 1.5 mM. Corilagin was purchased from Natural remedies Pvt., Ltd, (Bengaluru, India). Stock solutions were prepared by dissolving (1.5 mM) in 20:80% ethanol/water mixture. It is known that 20% ethanol does not affect the absorption of HSA and AGP and their structures. HSA and AGP solutions were prepared on basis of their molecular weights, respectively. All other materials were of analytical grade and double distilled water was used to prepare solutions. The chemicals used were purchased from Sigma-Aldrich.
We also optimized the incubation time of corilagin binding to HSA and AGP by fluorescence emission and found 5 min was the maximum binding time. Thus, we fixed the incubation of corilagin with HSA and AGP at 5 min and followed the same time for further experiments. Upon titration of corilagin with HSA and AGP, there was no precipitation which confirms that the mixture is clear.
3.2 Cell response assay (MTT assay)
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)tetrazolium reduction assay was the first homogeneous cell viability assay developed for a 96-well format that was suitable for high throughput screening (HTS).68 Cell response was carried out by the MTT assay, the cell lines taken are the mouse macrophages (RAW 264.7) which were sub cultured and were seeded in 96 well plates at a density of 5 × 103 cells. To induce inflammation, cells were pretreated with lipopolysaccharide (LPS) at 1 μg mL−1 for 4 h. After a while the cells were treated with corilagin in increasing concentrations of 10, 20, 40, 60, 80, and 100 μM for 48 h in a volume of 100 μL. The cells were grown in same media without corilagin as a control. At the end, 20 μL of MTT (5 mg mL−1 in PBS) was added and cells were incubated for 4 h. About 100 μL of DMSO was added to each well and mixed with repeated pipetting to dissolve the MTT crystals. Finally, cell response was measured at absorbance of 570 nm by using a micro plate reader (μ Quant Biotek Instrument, Inc.). Experiments were carried out in triplicates. Taking the control as 100%, reference cell response was calculated. The mean ± SE was calculated and reported as the cell response (%) vs. concentration (μM).
3.3 Fluorescence spectroscopy
A LS-55 spectrofluorometer (PerkinElmer Co., USA) equipped with 1.0 cm quartz cells was used to carry out the fluorescence emission spectra. Fluorescence emission spectra were recorded at room temperature with wavelength range from 300–500 nm, excitation wavelength of 285 nm, and band width of 5 nm for both excitation and emission. The experimental sample (protein and drug) was maintained at room temperature. HSA and AGP concentrations were 1 μM, and the different concentrations of corilagin were 1 to 9 μM in 0.1 M phosphate buffer at pH 7.2. Three independent experiments were performed and for each time three independent identical spectra were obtained.
Increasing concentration of corilagin with HSA and AGP and increasing absorbance of excitation or emission radiation introduces an inner filter effect that may decrease the fluorescence intensity and result in a nonlinear relationship between the observed fluorescence intensity and the concentration [Q] of the corilagin. Such an effect can be corrected using the following equation:
| | |
Fcor = Fobs10(Aexc + Aemi)/2
| (7) |
where
Fobs is the observed fluorescence and
Fcor is the corrected fluorescence intensity,
Aexc and
Aemi represent the absorbance at the fluorescence excitation (285 nm) and emission wavelengths for HSA (360 nm) and AGP (340 nm), respectively.
3.4 Effect of site marker on corilagin binding with HSA
To identify the binding site of corilagin in HSA structure, an equilibrium dialysis was carried out. In the experiment lidocaine (IB specific marker), phenylbutazone (site I specific marker) and ibuprofen (site II specific marker) were employed as site-specific markers. The concentration of HSA and the site specific probe was maintained at constant concentration of 0.001 mM, whereas corilagin was titrated with increased concentrations from 0.001 to 0.009 mM. The excitation wavelength for site-specific markers lidocaine, phenylbutazone, and ibuprofen with HSA was 285 nm. The modified Stern–Volmer equation was used to measure the fluorescence quenching data.
3.5 Atomic force microscopy (AFM)
To observe the morphological changes of free HSA and HSA–corilagin, an AFM experiment was carried out by NT-MDT solver scanning probe microscopy (Germany) equipment. The cantilever (0.3 mm) have an Au high reflectivity coating, tip height 14–16 μm, force constant (5, 5 N m−1) and the typical imaging resonance frequency was 140 kHz. The samples were imaged by AFM in noncontact mode. All of the samples were prepared as follows: (1) before washing with water, free HSA with 30 μL of 1.5 μmol L−1 HSA was added to a glass slide and incubated for 15 min at 288 K; (2) HSA–corilagin complexes with free HSA samples were prepared as defined in step (1) before adding 20 μL of a 15 mol L−1 corilagin solution, incubated for 15 min, washed with water, dried under N2 for 5 min, and then later the samples were dried overnight for taking an AFM image in air.
3.6 Circular dichroism spectroscopy
CD measurements were carried out with a Jasco J-810 spectropolarimeter using a quartz cell with a path length of 0.2 cm. Far UV-CD (190–300 nm) spectra were collected at room temperature. The concentration of HSA was 0.001 mM and the concentrations of corilagin were 0.001, 0.005, and 0.009 mM. Three scans were accumulated at a speed of 100 nm min−1 and data was collected. The CD data were used to determine the relative amounts of changes in the secondary structural elements of protein. CDNN 2.1 software was used to calculate the secondary structural elements of protein.51,52
3.7 Molecular docking
Molecular docking is an important computational procedure performed to find out the exact binding site on a protein which fits geometrically and energetically by using Auto Dock 4.2.3 software; this has been used widely because it shows acceptable fee energy values relative to experimentally observed docking data.69 The HSA (PDB Id: 1A06) and AGP (PDB Id: 3KQ0) crystal structures were obtained from the Brookhaven Protein Data Bank. A three-dimensional structure of corilagin was built from a 2D structure and geometry was optimized by using Discovery Studio 3.5 software. Molecular docking was performed using the AutoDock (4.2.3) program. The PDB structures were optimized and used as input for AutoDock Tools. To find the binding site and types of interactions involved in the formation of HSA–corilagin complexes, docking was performed using the Lamarckian genetic algorithm implemented in AutoDock 4.2.3. We found that this is the best performing docking method in terms of its ability to find the lowest energy and its structure prediction accuracy; it also incorporates ligand flexibility. AutoDock 4.2.3 takes water as solvent by default, and polar hydrogens were added using the MGL tools interface.70,71 For each docking simulation 30 different conformers were obtained from AutoDock, the conformer with the least binding free energy must match with experimental data for further analysis as reported earlier.22,23,40,66 For post docking analysis to compare and validate our docking results we used X-score v1.2.1, a consensus scoring function wherein it calculates the negative logarithm of the dissociation constant of the ligand to protein, and predicts the binding energy (kcal mol−1) and binding affinity of the ligand. X-score was reported to have an accuracy of ±2.2 kcal mol−1 relative to the actual binding energy.72.
3.8 Molecular dynamics simulation
Molecular dynamics simulation is an important parameter to find out the stability of protein-drug complex. A 10000 ps MD simulation of the complex was carried out with a Gromacs v4.6.3 package with force field GROMOS96 43a1, and was used for un-liganded HSA and HSA–corilagin complexes.73,74 The initial conformation was chosen as the least binding energy docking conformation. The topology parameters of HSA were created by using the Gromacs program. The topology parameters of HSA–corilagin were built by the Dundee PRODRG2.5 server.75 Then the complex was immersed in a cubic box (7.335 × 6.135 × 8.119 nm3) of extended simple point charge (SPC) water molecules, 15Na+ counter ions, and 43623 solvent atoms were added to each simulation box to maintain electro-neutrality and to release conflicting contacts. Simulations were performed in the NPT (i.e., constant number of molecules, constant pressure, and constant temperature) ensemble, at a temperature of 300 K and 1 bar, respectively maintained using a Berendsen thermostat76 with a coupling constant of 1.0 ps. Protein and water/ions were coupled independently. Pressure coupling used the Berendsen barostat with a coupling constant of 1.0 ps. Long-range electrostatic interactions were calculated using the particle mesh Ewald method77 with a 10 Å cut-off. The LINCS algorithm,78 was used to restrain bond lengths. Each system was energy minimized followed by a short 200 ps simulation during which the protein (and if present, ligand) non-hydrogen atoms were harmonically restrained with a force constant of 1000 kJ mol−1 Å−2. For further information see previous articles.22,23,48,66,79
4 Conclusions
In this work, the interaction between corilagin and serum proteins was studied in vitro and in silico by biophysical and molecular dynamics techniques which, in turn, provide valuable information. Corilagin showed a clear decrease in the percentage growth with increase in inhibition of inflamed mouse macrophages (RAW 264.7 cell line). Hence, it indicates that corilagin is a potent anti-inflammatory agent. Later, fluorescence studies showed that corilagin quenched the intrinsic fluorescence of HSA and AGP through the static quenching mode and hydrogen and hydrophobic interactions played a major role in the interaction. Further, CD studies revealed that the secondary structural content of protein was found to decrease upon binding of corilagin. These changes in the secondary structure of protein are well supported by the fractional fluctuation in the RMSD and Rg values of MD studies. Displacement experiments showed that corilagin binds specifically to sub-domain IIA, and molecular docking offered a molecular level understanding with ability to find the participation of particular chemical groups. The study provided accurate and quantifiable data on the binding mechanism of corilagin with drug carrier proteins of HSA and AGP and also helped in understanding its effect on serum protein function during distribution in blood.
Acknowledgements
This work was supported by Science and Engineering Research Board, No-SB/EMEQ-064/2014, Department of Science and Technology (Nos SR/SO/BB-0123/2010 and DST-FIST), UPE-2 University of Hyderabad and DBT-CREBB, India. We thank CIL and BIF, University of Hyderabad, for CD and Bioinformatics facilities. We thank Dr Naidu Subbarao, School of Computational and Integrative Sciences, JNU, New Delhi, India for helping us in X-score programme. We acknowledge the AFM facility provided by the School of Chemistry, University of Hyderabad. We thank Mr M. Druga Prasad for helping with AFM imaging.
References
- K. C. C. Liu, M.-T. Lin, S.-S. Lee, J.-F. Chiou, S. Ren and E. J. Lien, Planta Med., 1999, 65, 043–046 CrossRef CAS PubMed.
- W. Duan, Y. Yu and L. Zhang, Yakugaku Zasshi, 2005, 125, 587–591 CrossRef CAS.
- Y. Chen and C. Chen, Neurochem. Int., 2011, 59, 290–296 CrossRef CAS PubMed.
- S. Kinoshita, Y. Inoue, S. Nakama, T. Ichiba and Y. Aniya, Phytomedicine, 2007, 14, 755–762 CrossRef CAS PubMed.
- D. K.-P. Hau, G.-Y. Zhu, A. K.-M. Leung, R. S.-M. Wong, G. Y.-M. Cheng, P. B.-S. Lai, S.-W. Tong, F.-Y. Lau, K.-W. Chan and W.-Y. Wong, Phytomedicine, 2010, 18, 11–15 CrossRef CAS PubMed.
- S. Okabe, M. Suganuma, Y. Imayoshi, S. Taniguchi, T. Yoshida and H. Fujiki, Biol. Pharm. Bull., 2001, 24, 1145–1148 CAS.
- L. Zhao, S.-L. Zhang, J.-Y. Tao, R. Pang, F. Jin, Y.-J. Guo, J.-H. Dong, P. Ye, H.-Y. Zhao and G.-H. Zheng, Int. Immunopharmacol., 2008, 8, 1059–1064 CrossRef CAS PubMed.
- F. Notka, G. Meier and R. Wagner, Antiviral Res., 2004, 64, 93–102 CAS.
- M. Otagiri, Yakugaku Zasshi, 2009, 129, 413 CrossRef CAS.
- Z. Israili and P. Dayton, Drug Metab. Rev., 2001, 33, 161–235 CrossRef CAS PubMed.
- D. N. Bailey and J. R. Briggs, Ther. Drug Monit., 2004, 26, 40–43 CrossRef CAS PubMed.
- K. Yamasaki, V. T. Chuang, T. Maruyama and M. Otagiri, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5435–5443 CrossRef CAS PubMed.
- A. Varshney, P. Sen, E. Ahmad, M. Rehan, N. Subbarao and R. H. Khan, Chirality, 2010, 22, 77–87 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Mol. Biol., 1998, 5, 827–835 CAS.
- S. Curry, P. Brick and N. P. Franks, Biochim. Biophys. Acta, 1999, 1441, 131–140 CrossRef CAS.
- A. A. Bhattacharya, S. Curry and N. P. Franks, J. Biol. Chem., 2000, 275, 38731–38738 CrossRef CAS PubMed.
- D. Carter, X.-M. He, S. H. Munson, P. D. Twigg, K. M. Gernert, M. B. Broom and T. Y. Miller, Science, 1989, 244, 1195–1198 CAS.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
- T. Peters Jr, All about albumin: biochemistry, genetics, and medical applications, Academic Press, San Diego, 1996 Search PubMed.
- D. P. Yeggoni, M. Gokara, D. Mark Manidhar, A. Rachamallu, S. Nakka, C. S. Reddy and R. Subramanyam, Mol. Pharmacol., 2014, 11, 1117–1131 CrossRef CAS PubMed.
- B. Sudhamalla, M. Gokara, N. Ahalawat, D. G. Amooru and R. Subramanyam, J. Phys. Chem. B, 2010, 114, 9054–9062 CrossRef CAS PubMed.
- S. Neelam, M. Gokara, B. Sudhamalla, D. G. Amooru and R. Subramanyam, J. Phys. Chem. B, 2010, 114, 3005–3012 CrossRef CAS PubMed.
- M. Gokara, B. Sudhamalla, D. G. Amooru and R. Subramanyam, PLoS One, 2010, 5, e8834 Search PubMed.
- M. Gokara, T. Malavath, S. K. Kalangi, P. Reddana and R. Subramanyam, J. Biomol. Struct. Dyn., 2014, 32, 1290–1302 CAS.
- M. Gokara, G. B. Kimavath, A. R. Podile and R. Subramanyam, J. Biomol. Struct. Dyn., 2015, 33, 196–210 CAS.
- M. Kallubai, A. Rachamallu, D. P. Yeggoni and R. Subramanyam, Mol. BioSyst., 2015, 11, 1172–1183 RSC.
- T. Fournier, N. Medjoubi-N and D. Porquet, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1482, 157–171 CrossRef CAS.
- E. Athineos, J. C. Kukral and R. J. Winzler, Arch. Biochem. Biophys., 1964, 106, 338–342 CrossRef CAS PubMed.
- O. Pos, R. A. Oostendorp, M. E. Van Der Stelt, R. J. Scheper and W. Van Dijk, Inflammation, 1990, 14, 133–141 CrossRef CAS PubMed.
- B. F. T. Hochepied, H. Baumann and C. Libert, Cytokine Growth Factor Rev., 2003, 14, 25–34 CrossRef PubMed.
- C. Gambacorti-Passerini, M. Zucchetti, D. Russo, R. Frapolli, M. Verga, S. Bungaro, L. Tornaghi, F. Rossi, P. Pioltelli and E. Pogliani, Clin. Cancer Res., 2003, 9, 625–632 CAS.
- P. N. Bories, J. Feger, N. Benbernou, J.-D. Rouzeau, J. Agneray and G. Durand, Inflammation, 1990, 14, 315–323 CrossRef CAS PubMed.
- J. M. Kremer, J. Wilting and L. Janssen, Pharmacol. Rev., 1988, 40, 1–47 CAS.
- G. Lambrinidis, T. Vallianatou and A. Tsantili-Kakoulidou, Adv. Drug Delivery Rev., 2015, 5, 583 Search PubMed.
- X.-R. Dong, M. Luo, L. Fan, T. Zhang, L. Liu, J.-H. Dong and G. Wu, Int. J. Mol. Med., 2010, 25, 531–536 CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2009 Search PubMed.
- P. Kandagal, S. Kalanur, D. Manjunatha and J. Seetharamappa, J. Pharm. Biomed. Anal., 2008, 47, 260–267 CrossRef CAS PubMed.
- A. Garg, D. M. Manidhar, M. Gokara, C. Malleda, C. S. Reddy and R. Subramanyam, PLoS One, 2013, 8, e63805 CAS.
- D. Agudelo, P. Bourassa, J. Bruneau, G. Berube, E. Asselin and H.-A. Tajmir-Riahi, PLoS One, 2012, 7, e43814 CAS.
- P. Bourassa, S. Dubeau, G. M. Maharvi, A. H. Fauq, T. J. Thomas and H. A. Tajmir-Riahi, Biochimie, 2011, 93, 1089–1101 CrossRef CAS PubMed.
- S. Tayyab, M. S. Zaroog, S. R. Feroz, S. B. Mohamad and S. N. A. Malek, Int. J. Pharm., 2015, 491, 352–358 CrossRef CAS PubMed.
- M. Zolfagharzadeh, M. Pirouzi, A. Asoodeh, M. R. Saberi and J. Chamani, J. Biomol. Struct. Dyn., 2014, 32, 1936–1952 CAS.
- N. Tayeh, T. Rungassamy and J. R. Albani, J. Pharm. Biomed. Anal., 2009, 50, 107–116 CrossRef CAS PubMed.
- E. Rahnama, M. Mahmoodian-Moghaddam, S. Khorsand-Ahmadi, M. R. Saberi and J. Chamani, J. Biomol. Struct. Dyn., 2015, 33, 513–533 CAS.
- M. F. Zhang, Z. Q. Xu, Yu-Shu Ge, F. L. Jiang and Y. Liu, J. Photochem. Photobiol., B, 2012, 108, 34–43 CrossRef CAS PubMed.
- D. P. Yeggoni, A. Rachamallu and R. Subramanyam, J. Biomol. Struct. Dyn., 2015, 33, 1336–1351 CAS.
- O. A. A. Hamdi, S. R. Feroz, J. A. Shilpi, E. H. Anouar, A. K. Mukarram, S. B. Mohamad, S. Tayyab and K. Awang, Int. J. Mol. Sci., 2015, 16, 5180–5193 CrossRef CAS PubMed.
- D. P. Yeggoni and R. Subramanyam, Mol. BioSyst., 2014, 10, 3101–3110 RSC.
- R. Subramanyam, M. Goud, B. Sudhamalla, E. Reddeem, A. Gollapudi, S. Nellaepalli, V. Yadavalli, M. Chinnaboina and D. G. Amooru, J. Photochem. Photobiol., B, 2009, 95, 81–88 CrossRef CAS PubMed.
- R. Subramanyam, A. Gollapudi, P. Bonigala, M. Chinnaboina and D. G. Amooru, J. Photochem. Photobiol., B, 2009, 94, 8–12 CrossRef CAS PubMed.
- T. Förster, Mod. Quantum Chem., 1996, vol. 3, 93 Search PubMed.
- D. Epps, T. Raub, V. Caiolfa, A. Chiari and M. Zamai, J. Pharm. Pharmacol., 1998, 51, 41–48 CrossRef.
- Y.-J. Hu, Y. Liu, J.-B. Wang, X.-H. Xiao and S.-S. Qu, J. Pharm. Biomed. Anal., 2004, 36, 915–919 CrossRef CAS PubMed.
- B. Valeur and J.-C. Brochon, New trends in fluorescence spectroscopy: applications to chemical and life sciences, Springer Science & Business Media, 2012 Search PubMed.
- D. Kowalczyk, J.-P. Marsault and S. Slomkowski, Colloid Polym. Sci., 1996, 274, 513–519 CAS.
- P. L. Privalov, Adv. Protein Chem., 1979, 33, 167–241 CrossRef CAS PubMed.
- D. P. R. Yeggoni, M. M. Darla, C. S. Reddy and R. Subramanyam, J. Biomol. Struct. Dyn., 2015, 1–46 Search PubMed.
- F. Zsila, Z. Bikádi and M. Simonyi, Biochem. Pharmacol., 2003, 65, 447–456 CrossRef CAS PubMed.
- C. D. Kanakis, P. A. Tarantilis, H. A. Tajmir-Riahi and M. G. Polissiou, J. Agric. Food Chem., 2007, 55, 970–977 CrossRef CAS PubMed.
- Y. L. Jiang, Bioorg. Med. Chem., 2008, 16, 6406–6414 CrossRef CAS PubMed.
- R. Beauchemin, C. N'Soukpoe-Kossi, T. Thomas, T. Thomas, R. Carpentier and H. Tajmir-Riahi, Biomacromolecules, 2007, 8, 3177–3183 CrossRef CAS PubMed.
- N. A. Kratochwil, W. Huber, F. Müller, M. Kansy and P. R. Gerber, Biochem. Pharmacol., 2002, 64, 1355–1374 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- C. Malleda, N. Ahalawat, M. Gokara and R. Subramanyam, J. Mol. Model., 2012, 18, 2589–2597 CrossRef CAS PubMed.
- M. Kiselev, G. IuA, G. Dobretsov and M. Komarova, Biofizika, 2001, 46, 423 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- B. Gorelik and A. Goldblum, Proteins: Struct., Funct., Bioinf., 2008, 71, 1373–1386 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- R. Wang, L. Lai and S. Wang, J. Comput.-Aided Mol. Des., 2002, 16, 11–26 CrossRef CAS PubMed.
- H. J. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun, 1995, 91, 43–56 CrossRef CAS.
- E. Lindahl, B. Hess and D. Van Der Spoel, J. Mol. Model., 2001, 7, 306–317 CAS.
- A. W. Schuttelkopf and D. M. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- H. J. Berendsen, J. v. Postma, W. F. van Gunsteren, A. DiNola and J. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- W. R. Scott, P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Krüger and W. F. van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596–3607 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06837e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.