DOI:
10.1039/C0PY00296H
(Paper)
Polym. Chem., 2011,
2, 625-631
N-Heterocyclic carbene-borane radicals as efficient initiating species of photopolymerization reactions under air
Received
9th September 2010
, Accepted 27th October 2010
First published on 16th November 2010
Abstract
Eight N-heterocyclic carbene-boranes (NHC-boranes) are proposed as new efficient co-initiators for acrylate photopolymerization reactions. They are particularly interesting in aerated conditions, where they help overcome the classical oxygen inhibition. The carbene boryl radicals that are the initiating species have been characterized by their transient absorption spectra obtained in laser flash photolysis (LFP) experiments. Rate constants for the generation of the carbene boryl radicals by hydrogen abstraction with t-butoxyl radical and triplet benzophenone as well as the reactions with oxygen, electron rich and electron poor alkenes, two alkyl halides (CHCl3 and iodopropane) and diphenyliodonium hexafluorophosphate have been measured. The reactivity of N-heterocyclic carbene borane radicals is clearly affected by the NHC substituent.
Introduction
In photopolymerization reactions that are involved in the radiation curing and laser imaging areas,1 the photoinitiating system (PIS) plays a pivotal role. Among PISs, Type II PISs are based on a hydrogen transfer process between a suitable photoinitiator (PI) (e.g.benzophenones, thioxanthones, ketocoumarins, camphorquinone or dyes…) and a co-initiator DH (e.g.amine, thiol, silane…).1 The quest for new and efficient co-initiators remains a big challenge in this area and the discovery of radicals exhibiting an enhanced reactivity should provide new opportunities for the development of new materials.
In recent work, we have been especially interested in the photopolymerization of low viscosity monomers (LVMs) under air. In that case, the ratio between the rate of polymerization Rp in air and in laminate can drop down to 10% when considering usual Type II systems. Some novel co-initiator structures such as the silanes1s–1t provide a noticeably better ratio. Recently, we checked the polymerization initiating ability of newly developed2–4N-heterocyclic carbene boranes (NHC-boranes)5 as well as classical amine-boranes6 in laminate. To extend this study, we selected eight new NHC-borane derivatives (Scheme 1) as suitable co-initiators for free radical polymerization (FRP) of LVMs under air. The co-initiator ability and the NHC substituent effect of these structures will be presented. Little is known about the kinetics of the NHC boryl radicals, so we measured rate constants for their formation and interaction with alkenes, oxygen, CHCl3, iodopropane and diphenyliodonium hexafluorophosphate by laser flash photolysis (LFP) experiments. The results will be discussed within the context of the small molecule chemistry6a,7 and will help to suggest structure/reactivity relationships for these N-heterocyclic carbene boranes as co-initiators of photopolymerization.
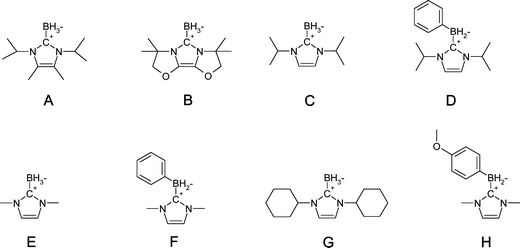 |
| | Scheme 1 Investigated compounds. | |
Experimental
(i) Investigated compounds
The borane complexes investigated are depicted in Scheme 1. NHC-boranes can be easily prepared according to established procedures.8 All NHCBs were prepared with analytical purity up to accepted standards for new organic compounds (>98%) which was checked by high field NMR analysis.8 The benzophenone model photoinitiator (BP), methyl acrylate (MA), ethyl vinyl ether (EVE), diphenyliodonium hexafluorophosphate (Ph2I+) were obtained from Aldrich. Ethyl dimethylaminobenzoate (EDB; Esacure EDB from Lamberti) was chosen as a reference amine co-initiator. The alkene stabilizers were removed by column purification (Aldrich AL-154) before the LFP experiments. 2,2′-di(ortho-chlorophenyl)-4,4′,5,5′-tetraphenylbiimidazole (o-Cl-HABI) was obtained from Tokyo Chemical Industry (TCI-Europe).
Nanosecond laser flash photolysis (LFP) experiments were carried out with a Q-switched nanosecond Nd/YAG laser (λexc = 355 nm, 9 ns pulses; energy reduced down to 10 mJ, from Powerlite 9010 Continuum) and an analyzing system consisting of a pulsed xenon lamp, a monochromator, a fast photomultiplier and a transient digitizer.9 The ketyl radical quantum yield (ΦK.) in the 3BP/NHC borane reaction was determined by a classical procedure.9,10 The t-BuO˙ radicals were generated at 355 nm by the direct cleavage of di-tert-butylperoxide as in ref. 6a and 9b.
(iii)
ESR spin trapping experiments
This ESR technique (ESR-ST) is powerful for the identification of the radical centers.11 The radicals were generated under the polychromatic light exposure of a Xe–Hg lamp (Hamamatsu, L8252, 150 W) of NHC-boranes in di-tert-butylperoxide/tert-butylbenzene (50/50); 6 mm quarts cylindrical ESR tubes were used and the samples were argon purged 15 min prior to measurements. The irradiation was carried out inside the cavity (TE102) of the spectrometer through a filter to cut off the light below 310 nm. The generated radicals were trapped by phenyl-N-tbutylnitrone (PBN). The ESR spectra simulations were carried out with the WINSIM software.12
(iv) DFT calculations
All the calculations were performed by using the hybrid functional B3LYP from the Gaussian 03 suite of program.13 Reactants and products were fully optimized at the B3LYP/6-31+G* level (checked for imaginary frequencies). The electronic absorption spectra were calculated with the time-dependent density functional theory at MPW1PW91/6-311++G** level on the relaxed geometry determined at UB3LYP/6-31+G* level.
(v)
Photopolymerization experiments
For film polymerization experiments, a given PI was dissolved into a bulk formulation based on ethoxylated pentaerythritol tetraacrylate (EPT from Cray Valey; viscosity = 150 cP).14 The films (20 μm thick) were deposited on a BaF2 pellet either under air or in laminate as in14c and irradiated with a polychromatic light (Xe–Hg lamp, Hamamatsu, L8252, 150 W). The evolution of the double bond content was followed by real-time FTIR spectroscopy (Nexus 870, Nicolet) at RT.14 In this paper, the Rp quantities refer to the maximum rates of the polymerization reaction and were determined from the maximum of the first derivative of the conversion vs. time curves.
Results and discussion
(1) Co-initiator photopolymerization ability of NHC-borane
The results for the acrylate FRP kinetics of ethoxylated pentaerythritol tetraacrylate (EPT) using benzophenone as the photoinitiator in both the absence and presence of the NHC-borane co-initiators are shown in Fig. 1 and Table 1; o-Cl-HABI was also studied as a PI (Fig. 2).
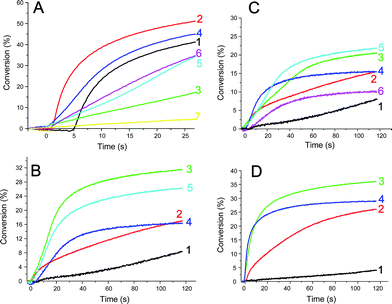 |
| | Fig. 1 (A) Radical photopolymerization ability of various BP/co-initiator couples (1%/1% w/w; in EPT, in laminate; λ > 300 nm): EDB (2); A (3); B (4); C (5); D (6); without co-initiator (1); without BP and B alone (7). (B) Radical photopolymerization ability of various BP/co-initiator couples (1%/1% w/w; in EPT, under air; λ > 300 nm): EDB (2); E (3); F (4); G (5); without co-initiator (1). (C) Radical photopolymerization ability of various BP/co-initiator couples (1%/1% w/w; in EPT, under air; λ > 300 nm): EDB (2); A (3); B (4); C (5); D (6); without co-initiator (1). (D) Radical photopolymerization ability of various BP/Ph2I+/co-initiator initiating systems (1%/1%/1% w/w; in EPT, under air; λ > 300 nm): EDB (2); C (3); D (4); without co-initiator (1). | |
Table 1 Radical photopolymerization. Polymerization rates of EPT using the BP/NHC-borane photoinitiating system (1%/1%, w/w) under UV light irradiation (Xe–Hg lamp)
| |
Laminated conditions |
Under air |
| Co–I |
R
p/[M0] × 100a |
R
p/[M0] × 100a |
|
R
p/[M0] (s−1) where [M0] is the initial monomer concentration.
Conversion reached at t = 120 s.
|
| — |
5.9 (56b) |
0.06 (7b) |
|
EDB
|
9.5 (60b) |
0.5 (16b) |
|
A
|
0.6 (56b) |
0.3 (20b) |
|
B
|
3 (60b) |
0.8 (16b) |
|
C
|
1.1 (59b) |
0.5 (22b) |
|
D
|
1.5 (58b) |
0.2 (10b) |
|
E
|
1.4 (61b) |
1.0 (31b) |
|
F
|
1.2 (58b) |
0.5 (16b) |
|
G
|
1.4 (61b) |
0.9 (26b) |
|
H
|
0.8 (53b) |
0.1 (9b) |
 |
| | Fig. 2 (A) Radical photopolymerization ability of various o-Cl-HABI/co-initiator couples (1%/1% w/w; in EPT, in laminate; λ > 300 nm): without co-initiator (1); without o-Cl-HABI and G alone (2); o-Cl-HABI/G (3). (B) Radical photopolymerization ability of various o-Cl-HABI/co-initiator couples (1%/1% w/w; in EPT, under air; λ > 300 nm): without co-initiator (1); without o-Cl-HABI and G alone (2); o-Cl-HABI/G (3). | |
In laminate (Fig. 1A, black line or line 1), the polymerization using BP alone is not very efficient and leads to an inhibition period of about 5 s. As previously,5 this is ascribed to the abstraction of a labile hydrogen from the monomer by the BP triplet state (3BP). In contrast, the addition of the NHC-borane co-initiators (1% w/w) suppresses the inhibition period; the polymerization process starts immediately under light irradiation (Fig. 1A). Because the inhibition period corresponds to the required time for the consumption of the oxygen dissolved in the formulation, this suggests that the NHC-boranes are good candidates to overcome the oxygen inhibition of the FRP. A noticeable structure effect on the polymerization profiles in laminate is found: the co-initiators ability decreases in the order B > C, D, E, F, G > H > A. Even though an inhibition period is avoided with NHC-boranes, the maximum Rps are usually lower than for BP alone. Only BP/B can be considered as competitive with the BP/EDB reference system.
In contrast, a completely different behavior for the NHC-borane based systems is observed under air (Fig. 1B–C show the different BP/co-initiator couples). Indeed, the Rps associated with all BP/NHC-borane systems are drastically improved compared to BP alone (where the polymerization does not occur at all). The co-initiator ability decreases in the order E > G > B > C, F > A > D > H and is better for B, E and G (and similar for C and F) than for the EDB. In addition to the decrease of the inhibition period previously observed in laminate, this shows the better ability of these compounds compared to EDB to suppress the oxygen inhibition. The use of o-Cl-HABI as a photoinitiator leads to similar trends (Fig. 2).
Some typical polymerization profiles of EPT upon addition of diphenyliodonium hexafluorophosphate (Ph2I+PF6−) to BP/co-initiator under air are shown in Fig. 1D. The Rps are strongly increased when using the BP/NHC-borane/Ph2I+PF6− three-component photoinitiating system (Fig. 1D, lines 3,4) instead of BP/EDB/Ph2I+PF6− (Fig. 1D, curve 2). This unambiguously shows the high reactivity of the NHC-boranes in combination with Ph2I+PF6− for a photopolymerization under air.
(2) Rate constants for reactions of boryl radical with small molecules
(a) Kinetic data on the formation of the NHC-boryl radicals.
The formation of the NHC-boryl radicals is well evidenced by ESR-spin trapping experiments (Fig. 3). For example, the spin adduct of the boryl radical derived from C is characterized by aN = 15.4 aH = 2.1 and aB = 4.4 G in excellent agreement with previous data on boryl radicals.3a,15
NHC-boryl radicals can be formed in LFP through the quenching of the tert-butoxyl radical t-BuO˙ (reaction (1)) or the benzophenone triplet state 3BP (reaction (2)); t-BuO˙ is easily obtained by the direct cleavage of di-tert-butylperoxide at 355 nm6,9b and 3BP is directly observed at 525 nm (intersystem crossing quantum yield: 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10). Following the rising time of the NHC-boryls (Fig. 4 and 5) and the 3BP lifetime provides a direct access to the rate constant kH1 and kH2, respectively (Table 2). The ketyl radical quantum yields (ΦK˙) were also measured in the usual way10 (ΦK˙s are equal to the boryl radical quantum yields).
10). Following the rising time of the NHC-boryls (Fig. 4 and 5) and the 3BP lifetime provides a direct access to the rate constant kH1 and kH2, respectively (Table 2). The ketyl radical quantum yields (ΦK˙) were also measured in the usual way10 (ΦK˙s are equal to the boryl radical quantum yields).
| | | t-BuO˙ + NHC–BH2R → t-BuOH + NHC–B˙HR (kH1) | (1) |
| | | 3BP + NHC– BH2R → BPH˙ + NHC– B˙HR (kH2) with R = H or Ph | (2) |
![(A) The kinetics of the formation of the NHC-boryl derived from A at λ = 400 nm in acetonitrile/di-tert-butylperoxide; the AA concentrations are 0; 0.002 and 0.0057 M. (B) The kinetics at 550 nm corresponding to the NHC-boryl derived from D for different CHCl3 concentrations from: [CHCl3] = 0 M to [CHCl3] = 0.084 M.](/image/article/2011/PY/c0py00296h/c0py00296h-f4.gif) |
| | Fig. 4 (A) The kinetics of the formation of the NHC-boryl derived from A at λ = 400 nm in acetonitrile/di-tert-butylperoxide; the AA concentrations are 0; 0.002 and 0.0057 M. (B) The kinetics at 550 nm corresponding to the NHC-boryl derived from D for different CHCl3 concentrations from: [CHCl3] = 0 M to [CHCl3] = 0.084 M. | |
Table 2 Rate constants (k1H, k2H) for the formation of the NHC-boryl radicals
| |
k
1
H
c (tBu-O˙) 107M−1s−1 |
k
2
H (3BP) 107M−1s−1 |
BDE (B–H)b kcal mol−1 |
|
Ketyl radical quantum yield (ΦK˙) in acetonitrile.
UB3LYP/6-31+G* level.
Acetonitrile/di-tert-butylperoxide.
|
|
AA
|
24 |
120 (0.7a) |
83.3 |
|
BB
|
11 |
250 (<0.15a) |
80.7 |
|
CC
|
26 |
45.5 (1a) |
82.6 |
|
DD
|
17 |
68.7 |
84.1 |
|
EE
|
26 |
96 |
81.9 |
|
FF
|
14 |
91.1 |
80.2 |
|
GG
|
21 |
100 (1a) |
82.8 |
|
HH
|
26.2 |
|
79.0 |
The transient absorption spectra for the NHC-boryl derived from A, B, C, E and G exhibit a maximum absorption at about 360 nm while those derived from D, F and H exhibit a visible light absorption at about 550 nm. This shows the influence of the phenyl Ph group on the NHC– B˙HR absorption spectra, which leads to a new red-shifted transition (Fig. 5B). The spectral features predicted by time-dependent density functional theory modeling (TD-DFT) fit quite well this experimental finding. A red shift of 129 nm is calculated between the maximum absorption spectra of the boryl derived from C (376 nm) and D (505 nm), respectively (MPW1PW91/6-311++G** level).
From this work, good to excellent hydrogen donating properties of N-heterocyclic carbene-borane complexes is noted as revealed by the high kH1 and kH2 rate constants (>107 M−1s−1). Interestingly, for the phenyl substitution, a slight decrease of kH1 is observed i.e. kH1(D) < kH1(C) and kH1(F) < kH1(E). This can be ascribed to a steric hindrance for the hydrogen abstraction process. The ΦK˙s are also strongly affected by the NHC structure (< 0.2 for B to 1 for C). 3BP being quite bulky, the change of ΦK˙ can probably be partly correlated with the steric hindrance in the NHC borane. 3BP reacts like an alkoxy radical. A similar behavior was recently found for other NHC-boranes.5
(b) Structure of the NHC-boryl radicals.
The different bond dissociation energy BDE(B–H) evaluated at UB3LYP/6-31+G* level for NHC-boranes are gathered in Table 2. Compared to amine (or phosphine)-boranes, the BDEs are strongly reduced (i.e. for triethylamine-borane, BDE(B–H) = 101 kcal mol−16a). This is in line with the excellent hydrogen donating properties of the NHC-boranes found here. Interestingly, BDEs(B–H) are quite similar for NHC–BH3 and NHC–BH2Ph showing a weak influence of the phenyl substitution. This can be rationalized by molecular orbital calculations. In previous work, a planar π-type structure for various NHC-boryl radicals was found.4 This was ascribed to a significant spin delocalization from the boron atom into the NHC ligand due to the empty p-orbital on the central carbene carbon atom. This holds true here in A, B, C, E and G. In the NHC–B˙HAr structure (D, F and H), however, the aryl is almost orthogonal to the NHC plane ruling out an important delocalization on the aryl ring (Fig. 6). This behavior is also exemplified by the spin density on the boron atom which is quite similar for NHC–B˙HAr and NHC–B˙H2 (Table 3).
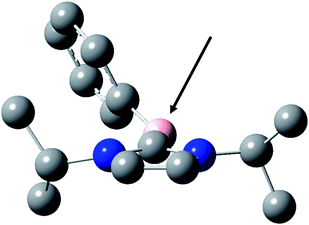 |
| | Fig. 6 Structure of the NHC-boryl radical derived from D (geometry optimized at UB3LYP/6-31+G* level). | |
| |
k
add (R˙/MA) 106 M−1s−1 |
k′add (R˙/O2) 108 M−1s−1 |
k
ox
(R˙/Ph2I+) 109 M−1s−1 |
k
Cl
(R˙/CHCl3) 107 M−1s−1 |
k
I (R˙/C3H7I) 107 M−1s−1 |
Spin Ba |
|
UB3LYP/6-31+G*.
For the addition to ethyl vinylether, a rate constant < 105 M−1s−1 is found.
|
|
AA
|
35 |
>5 |
1.4 |
|
|
0.57 |
|
BB
|
9.5 |
>5 |
1.1 |
|
|
0.51 |
|
CC
|
31b |
>6 |
0.7 |
1.3 |
8.3 |
0.56 |
|
DD
|
1.9 |
>6 |
0.6 |
0.5 |
2.3 |
0.53 |
|
EE
|
38 |
>6 |
0.8 |
0.6 |
18 |
0.55 |
|
FF
|
11 |
>6 |
0.4 |
0.15 |
2.8 |
0.50 |
|
GG
|
21 |
>6 |
|
|
|
0.56 |
|
HH
|
10.5 |
>6 |
|
|
|
0.48 |
(c) Reactivity of the NHC-boryl radicals.
Through the direct observation of the NHC-boryl radicals offered by LFP, important absolute radical/molecule and radical/ion reaction rate constants are accessible. All these kinetic data are compiled in Table 3. Because preliminary experiments showed that the NHC-boryl radicals react very rapidly with oxygen (kO2 > 5 × 108 M−1s−1, Table 3), all the kinetic experiments were carried out under argon.
(c-1) Addition of NHC–boryl radicals to double bonds.
The addition rate constants (kadd) to methyl acrylate (MA) and ethyl vinyl ether (EVE) are compiled in column 2 of Table 3. The addition to an electron deficient alkene (MA) is very fast (>1.9 × 106 M−1s−1). The back reaction can be probably ruled out (or is slow) because no fragmentation (with a concomitant boryl radical re-formation) is observed on a 200 μs time scale. Interestingly, NHC–B˙HPh radicals are characterized by lower kadd than NHC–B˙H2 (i.e. 31 × 106vs. 1.9 × 106 M−1s−1 for the boryl radicals derived from C and D, respectively). This can be ascribed to steric hindrance by the phenyl group. The addition rate constant to the electron rich alkene (EVE) is lower (< 105 M−1s−1 for the boryl radical derived from C which is one of the most reactive for the addition to MA). This combination of a slow addition to an electron rich alkene (EVE) and a fast addition to an electron poor alkene (MA) suggests a nucleophilic behavior for the boryl radicals investigated here. This is similar to that noted for other recently studied NHC-boryls as well as for classical amine-boryls.5
(c-2) Reactivity of NHC-boryl radicals towards alkyl halides.
The reaction of boryl radicals with alkyl halides is an important process in small molecule radical chemistry. The rate constants for the reactions of the NHC boryls with an alkylchloride (CHCl3) (kCl) and iodopropane (kI), gathered in columns 5–6 of Table 3 (Fig. 4B), are quite high (1.5 × 106 to 1.3 × 107 M−1s−1 for the reaction with CHCl3 and 2.3 × 107 to 8.3 × 107 M−1s−1 for iodopropane). Interestingly, as for the addition process (see above), NHC–B˙HPh radicals have a lower reactivity than NHC–B˙H2. This can be still ascribed to the crowded character of the NHC–B˙HPh radical center. NHC-boryl radicals exhibit low ionization potentials IP.5 Therefore, the charge transfer from the NHC-boryl radical to the alkyl halide might explain in part their high reactivity towards CHCl3 and iodopropane.
(c-3)
Oxidation of NHC-boryl radicals by Ph2I+PF6−.
Due to its very low reduction potential (∼−0.2 V), diphenyliodonium hexafluorophosphate is a classical compound used to oxidize radicals.16 The rate constants for the chemical oxidation of NHC-boryl radicals by Ph2I+PF6− were found to be quite close to the diffusion limit (k > 4 × 108 M−1s−1, Table 3), in line with the low IPs (previously reported5) and the high nucleophilic character (see above) of the NHC-boryls.
(3) The boryl radical reactivity and the polymerization initiation mechanism
The high intrinsic reactivity of the NHC-boryls towards the acrylate unit found above is in agreement with their good polymerization initiating ability. However, the Rps (Fig. 1–2) do not correlate with the radical reactivity towards MA (kadd), i.e. the boryl radical derived from E which is characterized by the highest kadd to MA does not lead to the highest Rp in laminate. This can be ascribed to the large difference of ΦK˙ in the NHC-borane series (from <0.15 for B to 1 for G). It was found previously that E exhibits the highest reactivity for radical processes.2
Interestingly, the high reactivity of NHC-boryl radicals with O2 is in full agreement with the ability of these structures to overcome the oxygen inhibition as previously described in amine-boranes in.6b They consume oxygen and re-generate new boryls (reactions (3a) and (3b)). The reactivity of ligated-boranes with the peroxyls generated by the addition of initiating or propagating radicals with O2 (which converts stable peroxyls into new initiating structures, reaction 3c) can also probably be invoked to explain the good ability of NHC-boranes to initiate the polymerization under air.
| | | NHC–B(˙)HR + O2 → NHC–BOO(˙)HR | (3a) |
| | | NHC–BOO(˙)HR + NHC–BH2R → NHC–B(OOH)HR + NHC–B(˙)HR | (3b) |
| | | ROO˙ + NHC–BH2R → ROO–H + NHC–B(˙)HR | (3c) |
For the three-component photoinitiating systems BP/NHC-BH2R/Ph2I+PF6−, the drastic increase of the polymerization rates compared to BP/NHC-BH2R systems stems from the high rate constants for the oxidation of the boryl radicals to borenium cations (4).3b,5 The release of a phenyl radical (Ph˙), which exhibits a higher polymerization initiating property than NHC-boryls, is also expected to increase Rp. Indeed, Ph˙ has a higher kadd (MA) [1.9 × 108 M−1s−1].17
| | | NHC–B˙HR + Ph2I+ → NHC–B+HR + PhI + Ph˙ | (4) |
Conclusions
NHC-boranes appear to be an excellent class of co-initiators for FRP. A remarkable behavior in the acrylate photopolymerization in aerated conditions is noted i.e.polymerization profiles better than for the reference system are obtained. The characterization of the derived NHC-boryl radicals by LFP has allowed the determination of the key rate constants for the elementary events. The NHC substitution affects the different processes. More interestingly, the phenyl substitution on the radical center is found to decrease both the formation and reaction rate constants of the boryls. The present work allows a better knowledge of the structure/reactivity relationship for the NHC-boryl radicals. The design of optimized co-initiators with better hydrogen donating properties and higher reaction rate constants with monomers, O2 and ROO˙ should be investigated in forthcoming works to further improve the photopolymerization process in aerated conditions.
References
-
(a)
Radiation Curing of Polymeric Materials, ed. C. E. Hoyle and J. F. Kinstle, ACS Symposium series, vol. 417, 1990 Search PubMed;
(b)
Lasers in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, CRC Press, Boca Raton, 1990 Search PubMed;
(c)
Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, Elsevier Science Publishers LTD, London, 1993 Search PubMed;
(d)
Chemistry and Technology of UV and EB Formulation for Coatings, Inks and Paints, ed. P K T Oldring, Sita Technology, London, 1994 Search PubMed;
(e)
Current Trends in Polymer Photochemistry, ed. N. S Allen, M. Edge, I. R. Bellobono and E. Selli, Ellis Horwood, New York, 1995 Search PubMed;
(f)
J. P. Fouassier
Photoinitiation, Photopolymerization and Photocuring: Fundamental and Applications, Hanser Publishers, New-York, 1995 Search PubMed;
(g)
Photopolymerization: Fundamentals and Applications, ed. A. B. Scranton, A. Bowman and R. W. Peiffer, ACS Symposium series, Washington DC, vol. 673, 1997 Search PubMed;
(h)
J. V. Crivello in Photoinitiators for Free Radical, Cationic and Anionic Photopolymerization, ed. G. Bradley, New York, 2nd edn, 1998 Search PubMed;
(i)
S. Davidson in Exploring the Science, Technology and Application of UV and EB Curing, Sita Technology Ltd, London, 1999 Search PubMed;
(j)
D. C. Neckers, W. Jager, in UV and EB at the Millenium, Sita Technology, London, 1999 Search PubMed;
(k)
K. Dietliker, A Compilation of Photoinitiators Commercially Available for UV Today, Sita Technology Ltd, Edinburgh/London, 2002 Search PubMed;
(l)
Photoinitiated Polymerization, ed. K. D. Belfied and J. V. Crivello , ACS Symposium series, Washington DC, vol. 847, 2003 Search PubMed;
(m)
Photochemistry and UV Curing, ed. J. P. Fouassier, Research Signpost, Trivandrum, India, 2006 Search PubMed;
(n)
R. Schwalm in UV Coatings: Basics, Recent Developments and New Applications, Elsevier, Oxford UK, 2007 Search PubMed;
(o)
W. Schnabel, Polymers and Light: Fundamentals and Technical Applications, Wiley-VCH, Weinheim, 2007 Search PubMed;
(p)
Macromolecular Engineering: From Precise Macromolecular Synthesis to Macroscopic Materials Properties and Applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, 2007 Search PubMed;
(q)
Lasers in Chemistry, ed. M. Lackner, Wiley-VCH., Weinheim, 2008 Search PubMed;
(r)
Handbook on Photochemistry and Photophysics of Polymer Materials, ed. N. S. Allen, Wiley, USA, in press Search PubMed;
(s) M.-A. Tehfe, J. Lalevée, D. Gigmes and J.-P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS;
(t) R. Souane, M. A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromol. Chem. Phys., 2010, 211, 1441–1445 CrossRef CAS.
-
(a) S. H. Ueng, M. Makhlouf Brahmi, E. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS;
(b) S.-H. Ueng, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, Org. Lett., 2010, 12, 3002–3005 CrossRef CAS;
(c) A. Solovyev, S.-H. Ueng, J. Monot, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, Org. Lett., 2010, 12, 2998–3001 CrossRef CAS.
-
(a) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS;
(b) T. Matsumoto and F. P. Gabbaï, Organometallics, 2009, 28, 4252–4253 CrossRef CAS.
-
(a) J. C. Walton, Angew. Chem., Int. Ed., 2009, 48, 2–5 CrossRef;
(b) J. C. Walton, M. Makhlouf Brahmi, L. Fensterbank, E. Lacôte, M. Malacria, Q. Chu, S.-H. Ueng, A. Solovyev and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 2350–2358 CrossRef CAS.
- M.-A. Tehfe, M. Makhlouf Brahmi, J.-P. Fouassier, D. P. Curran, M. Malacria, L. Fensterbank, E. Lacôte and Jacques Lalevée, Macromolecules, 2010, 43, 2261–2267 CrossRef CAS.
-
(a) J. Lalevée, N. Blanchard, A.-C. Chany, M. A. Tehfe, X. Allonas and J. P. Fouassier, J. Phys. Org. Chem., 2009, 22, 986–993 CrossRef CAS;
(b) J. Lalevée, M. A. Tehfe, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 9057–9062 CrossRef CAS.
-
(a) B. Sheeller and K. U. Ingold, J. Chem. Soc., Perkin Trans., 2001, 2, 480–486 Search PubMed;
(b) M. Lucarini, G. F. Pedulli and L. Valgimigli, J. Org. Chem., 1996, 61, 1161–1164 CrossRef CAS.
- The NHC-boranes were prepared according to established procedures (see ref. 2 and 3a) and also K. N. Henkel, G. Kratz, T. Kreutzberg, J. Boeseb, R. Maulitzb and A.H., Chem. Ber., 1993, 126, 2041–2045 Search PubMed; M. M. Brahmi, J. Monot, M. Desage-El Murr, D. P. Curran, L. Fensterbank, E. Lacôte and M. Malacria, J. Org. Chem., 2010, 75, 6983–6985 CAS.
-
(a) J. Lalevée, X. Allonas and J. P. Fouassier, J. Am. Chem. Soc., 2002, 124, 9613–9621 CrossRef CAS;
(b) J. Lalevée, B. Graff, X. Allonas and J. P. Fouassier, J. Phys. Chem. A, 2007, 111, 6991–6998 CrossRef CAS.
-
S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel Dekker, Inc, New-York, 1993 Search PubMed.
-
(a)
P. Tordo, Spin-trapping: recent developments and applications in Electron Spin Resonance, ed. N. M. Atherton, M. J. Davies and B. C. Gilbert, vol. 16. Cambridge, The Royal Society Of Chemistry; 1998 Search PubMed;
(b) C. F. Chignell, Pure Appl. Chem., 1990, 62, 301–308 CrossRef CAS;
(c) Y. Kotake and K. Kuwata, Bull. Chem. Soc. Jpn., 1981, 54, 394–399 CrossRef CAS;
(d) A. Criqui, J. Lalevée, X. Allonas and J.-P. Fouassier, Macromol. Chem. Phys., 2008, 209, 2223–2231 CrossRef CAS.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–112 CrossRef CAS.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision B.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed;
(b)
J. B. Foresman and A. Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian. Inc., 2nd edn, 1996 Search PubMed.
-
(a) J. Lalevée, X. Allonas, S. Jradi and J. P. Fouassier, Macromolecules, 2006, 39, 1872–1879 CrossRef CAS;
(b) J. Lalevée, L. Zadoina, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2494–2502 CrossRef CAS;
(c) M. El-Roz, J. Lalevee, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 8725–8732 CrossRef CAS.
-
(a) J. A. Baban, V. P. J. Marti and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1985, 1723–1733 RSC;
(b) J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1986, 1607–1611 RSC.
-
(a)
J. V. Crivello in Ring-Opening Polymerization, ed. D. J. Brunelle, Hanser, Munich, 1993 Search PubMed;
(b) J. V. Crivello and J. H. W. Lam, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 1059–1065 CrossRef CAS.
- J. Lalevée, X. Allonas and J. P. Fouassier, J. Phys. Chem. A, 2004, 108, 4326–4334 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 
