Detailed study of the reversible addition–fragmentation chain transfer polymerization and co-polymerization of 2-methacryloyloxyethyl phosphorylcholine†
Neha
Bhuchar
a,
Zhicheng
Deng
a,
Kazuhiko
Ishihara
b and
Ravin
Narain
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, AB T6G 2G6, Canada. E-mail: narain@ualberta.ca; Fax: +1-780-492 2881; Tel: +1-780-492-1736
bDepartment of Materials Engineering and Department of Bioengineering, School of Engineering, The University of Tokyo, 7-3-1, Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
First published on 11th November 2010
Abstract
In this paper, we report on a detailed study of the reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-methacryloyloxyethyl phosphorylcholine (MPC). The RAFT polymerization conditions were carefully monitored and it was noted that well-defined homopolymers with ideal low polydispersity (Mw/Mn < 1.2) could be achieved in methanol using 4-cyanopentanoic acid dithiobenzoate (CTP) as a chain transfer agent and 4,4′-azobis(4-cyanovaleric acid) (ACVA) as an initiator. A series of well-defined block copolymers having a range of compositions and molecular weight were prepared using poly(MPC) as the macroCTA. Statistical MPC-based copolymers of biological relevance were also synthesized and characterized.
Introduction
Zwitterionic compounds have drawn considerable attention in both industry and academia. Due to their polar nature, they are soluble in water. The presence of positive and negative charges on these ions can be easily exploited. We have studied the polymerization of 2-methacryloyloxyethyl phosphorylcholine (MPC), a zwitterionic monomer.1 The MPC is a methacrylate with a phosphorylcholine group, which is one of the representative phospholipid polar groups on the cell membrane.2,3 The MPC polymers showed excellent blood compatibility due to reduction of protein adsorption even when they contact with whole blood without anticoagulant.4–8 Based on the functionality of the MPC polymers, they have been used in various blood contacting medical devices such as oxygenator, catheter, cardiovascular stent, and implantable blood pump to prevent blood coagulation at these surfaces.9–18 Also, the grafting of the poly(MPC) on artificial hip joints improves lubrication and reduces wear of polyethylene liner of the hip joint.19 A range of MPC polymers (di-block, tri-block, random, graft copolymers) and hydrogels have been synthesized for surface modifications.14,20,21Due to the significant interest of MPC polymers, different controlled radical polymerization techniques have been used for the synthesis of well-defined MPC polymers. Controlled polymerization of MPC by an atom transfer radical polymerization (ATRP) was reported by Armes et al.22 The ATRP is a type of living radical polymerization (LRP) which is carried out using transition metal catalysts.23 The advantage of aqueous ATRP for MPC is the small time frame in which the reaction is completed. More than 96% yields were obtained within 10 min of the start of reaction. However, the copolymers with other vinyl compounds were obtained with extremely high polydispersities.22 Also, metal catalyst residues make ATRP ineffective for biomedical applications.23 Very recently, poly(MPC) copolymers have been prepared using soap free heterogeneous polymerization.24 With the development of reversible addition–fragmentation chain transfer (RAFT) polymerization technique,25–27 an increasing number of polymers for biomedical devices were prepared via RAFT due to its compatibility with various solvents, ideal tolerance to a wide range of conditions and monomers, as well as no metal catalyst involvement.27 The RAFT was employed in the synthesis of well-defined MPC polymers. Firstly, Yusa et al.28 reported RAFT polymerization of MPC in water using a combination of 4,4′-azobis(4-cyanopentanoic acid) (ACVA) and 4-cyanopentanoic acid dithiobenzoate (CTP). The polymerization of MPC in water was found to be very fast. The reaction time was reported to be around 2 h. However, the polydispersity of resulting MPC polymer was relatively high (Mw/Mn ≈ 1.27). Another factor is that the dissolution of solid phase ACVA and CTP in pure water was difficult, which may have directly affected the initial stages of the polymerization. Yu et al.29 improved the protocol by adding 5 wt% NaHCO3. However, at least 4 h stirring in an ice-bath was still required for the dissolution of ACVA and CTP.
Considering the above reasons as well as the significance of MPC polymers in the biomedical field, a detailed study of the RAFT polymerization of MPC in methanol has been carried out and its copolymerization with primary amine and sugar-based monomers is described.
The diblock copolymers of MPC unit with other monomer unit can be used to increase the specificity of a surface towards cell interaction. We have synthesized diblock copolymers of MPC with 2-aminoethyl methacrylamide hydrochloride (AEMA), 2-gluconamidoethyl methacrylamide (GAEMA), D-gluconaminoethyl methacrylate (GAMA) and 2-lactobionamidoethyl methacrylamide (LAEMA). AEMA is a cationic polymer and hence its copolymer with MPC can be used for DNA complexation.30GAEMA, GAMA and LAEMA are synthetic glycopolymers. Glycopolymers containing pendant saccharide groups are known to interact with proteinsvia multivalent interactions.31 Depending upon the type of pendant saccharide groups, these glycopolymers may be specific to particular biomolecules.32
Experimental
Materials
The MPC with 7.0 ppm inhibitor was obtained from NOF, Co. (Tokyo, Japan), which was synthesized by the method reported previously.12-Aminoethyl methacrylamide hydrochloride (AEMA),332-gluconamidoethyl methacrylamide (GAEMA),32D-gluconaminoethyl methacrylate (GAMA),34 and 2-lactobionamidoethyl methacrylamide (LAEMA)35 were synthesized according to previous reports. The CTA, cyanopentanoic acid dithiobenzoate (CTP), was synthesized as previously described.36,374,4′-Azobis(4-cyanovaleric acid) (ACVA) was purchased from Sigma Aldrich (Canada) and used as received. Methanol and HPLC grade water were purchased from Caledon Laboratory Chemicals (Canada).Methods
The number average molecular weight (Mn) and polydispersity (Mw/Mn) of the polymer samples were determined at a flow rate of 1.0 mL min−1 using a Viscotek conventional GPC system equipped with two Waters Ultrahydrogel linear WAT011545 columns (pore size: blend, exclusion limit = 7.0 × 106) and Viscotek model 250 dual detector. 0.50 M sodium acetate/0.50 M acetic acid buffer was used as eluent. The GPC was calibrated by six near-monodispersed poly(ethylene oxide) (PEO) standards (Mp—1.01 × 103 to 1.01 × 105 g mol−1). 1H-NMR spectra of the polymers were recorded on a Varian 400 or 500 MHz instrument.Homopolymerization of 2-methacryloyloxyethyl phosphorylcholine
Homopolymerization of MPC was conducted using RAFT technique in methanol, at 60 °C, ACVA as the initiator and CTP as the CTA. In a typical protocol, in a 10 mL Schlenk tube, MPC (2.0 g, 6.8 mmol), CTP (0.030 g, 0.10 mmol, target DPn = 63) and ACVA (1.5 × 10−2 g, 0.052 mmol) were dissolved in 6 mL methanol. The solution was then degassed via four freeze–vacuum–thaw cycles and placed in the 60 °C oil bath for 11 h. The conversion was determined by 1H-NMR spectroscopy using D2O as a solvent and by comparing the vinyl resonance of the monomer (appearing between 5.3 and 5.8 ppm) to the methyl resonance of the polymer (appearing between 0.5 and 1.4 ppm). The polymer was obtained by precipitating in a large quantity of acetone. The molecular weight and polydispersity were obtained from aqueous GPC after the methanol solution of polymer samples was dried in air. The resulting polymer molecular weight Mn was 1.4 × 104 g mol−1, indicating more than 80% conversion. The polydispersity (Mw/Mn) was around 1.1.Synthesis of diblock copolymer with poly(MPC) segment
The chain extension reaction was carried out using various monomers initiated from poly(MPC)-based macro-CTA. The synthesis of poly(MPC)-based macro-CTAs was carried out using the above mentioned procedure, employing 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of CTP/ACVA and quenched at ∼60% conversion. A typical procedure for block copolymerization of MPC with GAEMA is as follows: the poly(MPC)-based macro-CTA (0.20 g, 1.2 × 10−2 mmol, Mn = 1.6 × 104 g mol−1, Mw/Mn = 1.1), GAEMA (0.22 g, 0.70 mmol) and ACVA (1.7 × 10−3 g, 5.8 × 10−3 mmol) were dissolved in 1.5 mL water. After degassing via four freeze–vacuum–thaw cycles, the solution was kept in 70 °C oil bath for polymerization. The polymers were obtained by precipitating in acetone. The residual GAEMA was removed by washing with N,N′-dimethyl formaldehyde (DMF). (In the case of AEMA and GAMA, the residual monomer was removed by washing with methanol.) Molecular weight from the typical polymerization reaction for the synthesis of poly(MPC54-b-GAEMA55) was 3.3 × 104 g mol−1 with a polydispersity of 1.3.
:1 ratio of CTP/ACVA and quenched at ∼60% conversion. A typical procedure for block copolymerization of MPC with GAEMA is as follows: the poly(MPC)-based macro-CTA (0.20 g, 1.2 × 10−2 mmol, Mn = 1.6 × 104 g mol−1, Mw/Mn = 1.1), GAEMA (0.22 g, 0.70 mmol) and ACVA (1.7 × 10−3 g, 5.8 × 10−3 mmol) were dissolved in 1.5 mL water. After degassing via four freeze–vacuum–thaw cycles, the solution was kept in 70 °C oil bath for polymerization. The polymers were obtained by precipitating in acetone. The residual GAEMA was removed by washing with N,N′-dimethyl formaldehyde (DMF). (In the case of AEMA and GAMA, the residual monomer was removed by washing with methanol.) Molecular weight from the typical polymerization reaction for the synthesis of poly(MPC54-b-GAEMA55) was 3.3 × 104 g mol−1 with a polydispersity of 1.3.
Note: in the synthesis of poly(MPC54-b-AEMA54), the polymerization can be also conducted at 60 °C in 1.5 mL methanol.
Random copolymerization of 2-methacryloyloxyethyl phosphorylcholine
The random polymers of MPC with LAEMA, GAEMA, GAMA or AEMA were made by adding the monomers in 50:50 molar ratio. A typical polymerization process for the copolymerization of GAEMA and MPC is given, MPC (0.19 g, 0.65 mmol) and GAEMA (0.20g, 0.65 mmol) were dissolved in 2 mL water, followed by the addition of 0.30 mL 2-propanol stock solution of CTP (2.0 × 10−3 g, 8.0 × 10−3 mmol) and ACVA (1.0 × 10−3 g, 4.0 × 10−3 mmol). Acetic acid (0.2 mL) was added to prevent the hydrolysis of CTP in the aqueous solution.38 The solution was kept in a 70 °C oil bath for polymerization. The reaction was stopped after 20 h by quenching in liquid nitrogen. The molecular weight was analysed in GPC and the composition was confirmed using 1H-NMR. The random copolymerization of poly(MPC-stat-AEMA) was carried out in methanol at 60 °C, as the AEMA is soluble in methanol. The molecular weight (Mn) of the resulting polymer, poly(MPC50-stat-GAEMA45), for the above procedure was 2.8 × 104 and the PDI was 1.19.
Results and discussion
Detailed kinetic studies for the homopolymerization of 2-methacryloyloxyethyl phosphorylcholine
The RAFT technique was used for the homopolymerization of MPC. RAFT polymerization enables access to “site specific functionality” concurrently providing a control over the molecular weight and molecular weight distribution.27 As mentioned already, the polymerization of MPC using living polymerization techniques such as ATRP and RAFT has been reported. ATRP of MPC was extensively studied in methanol as solvent. However, a detailed study for the RAFT polymerization of MPC in methanol was still lacking. Therefore, in this work, the RAFT polymerization of MPC was conducted in methanol instead of water as solvent. Methanol provides a low viscosity solution for the preparation of high molecular weight polymers.39Synthesis of controlled molecular weight and low polydispersity polymers depends on several parameters. The choice of the chain transfer agent in the RAFT polymerization largely affects these parameters. The use of CTP for this reaction was determined by comparing the polymer samples prepared without chain transfer agent and in the presence of two different CTAs', S-1-dodecyl-S-(α,α′-dimethyl-α″-acetic acid) trithiocarbonate (CTAm) and CTP.Table 1 shows a comparison of the results of various polymerization reactions carried out using CTP, CTAm or without CTA.
It was observed that without the addition of chain transfer agent, the molecular weight was about double than the target molecular weight. The molecular weight was found to be controlled when either the dithio-based CTP or trithio-based CTAm was employed. The polydispersity, however, was better controlled by using CTP as a chain transfer agent. Similar results were also observed in the previous study of the RAFT polymerizations of LAEMA.35 Moreover, CTP, as the chain transfer reagent, is compatible with a wide variety of monomers. Therefore, CTP was selected as the chain transfer agent for the RAFT polymerizations of the MPC.
Compared to the previous reports28,29 in which the polymerization of the MPC was conducted in water, methanol was chosen as the solvent in this work to firstly eliminate the requirement for a long time for the dissolution of CTP and ACVA or any addition of salt. Moreover, water (if not acidified) can cause unwanted side reactions for example, hydrolysis of CTP, which can reduce the control of the polymerization.38 The rate of hydrolysis of CTP is found to be strongly temperature dependent. As reported by Levesque et al.,40 for a 24 h polymerization reaction in water, around 5.0–25% hydrolysis occurs at 20 °C. With the increase in temperature, this rate increases exponentially. For example, at 35 °C, 40–60% hydrolysis occurs over 24 h in the pH range 7.5–8.5.40 To avoid this and to facilitate ease of dissolution of CTP in the solvent, methanol was preferred. Methanol being a volatile solvent, the polymerizations were conducted at 60 °C and Schlenk tube was used to reduce the loss of solvent during the polymerization process.
Another important aspect in RAFT is the ratio of chain transfer agent to initiator. This ratio may affect the control of the polymerization. It turns out, in a fixed amount of CTA and lower initiator, fewer radicals are generated and hence termination reactions can be minimized. However, no obvious difference of the polydispersities was observed in our case (Table S1, Trail 1 (ESI†)). On the other hand, using less initiator usually slows down the pace of polymerization reaction. Considering the lower decomposition rate of ACVA in methanol,39 a CTP/ACVA ratio of 2.0 has been used in the polymerization unless otherwise mentioned. This was done to override the effect of organic solvent on the speed of polymerization reaction.41
Fig. 1 illustrates the kinetics of homopolymerization of MPC at the conditions described above. In the GPC trace (Fig. 1(A)), the shift in peaks to a shorter elution time indicates a gradual increase in the molar mass of the polymer with reaction time. Fig. 1(B) and (C) show the increase in conversion with time and the linear increase in molecular weight with conversion, respectively. ln ([M0]/[M]) vs. reaction time is a straight line indicating a first order reaction with respect to the monomer concentration. Low polydispersities were also observed even for the high molecular weight polymers (e.g.Mn = 1.26 × 104 g mol−1, Mw/Mn = 1.04, Mn = 2.18 × 104 g mol−1, Mw/Mn = 1.13). Comparing to previous reports (Mn = 1.21 × 104 g mol−1, Mw/Mn = 1.12,28Mn = 2.18 × 104 g mol−1, Mw/Mn = 1.27 ref. 28), lower polydispersity may be due to the well dissolution of CTP and ACVA at the very beginning of the polymerization. All these factors showed that the polymerization of the MPC in methanol was carried out in a controlled manner. For a target degree of polymerization (DPn) of 60, a high monomer conversion (>80%) was achieved after 10 h of reaction. Table S1 (ESI†) shows the synthetic parameters, molecular weights, and molecular weight distributions for the RAFT homopolymerization of the MPC.
 | ||
| Fig. 1 (A) Shifts in GPC peaks with the RAFT homopolymerization of MPC in methanol, using ACVA as the initiator and CTP as the chain transfer agent at 60 °C with a target DPn of 63 and CTP/ACVA = 2, (B) semi-logarithmic and conversion plot vs. reaction time for RAFT polymerization of poly(MPC) for the above mentioned reaction conditions and (C) evolution of molecular weight of poly(MPC) with conversion, confirming living polymerization. | ||
Molecular weight was found to increase up to ∼80%. The slow increase of the molecular weight after 80% conversion in the kinetic study experiment may be due to the high viscosity of the solution. It should be noted that poly(MPC) with different (higher) DPn as well as narrow-molecular weight distribution was also successfully synthesized (Fig. S1 in ESI†).
A summary of the polymerization kinetics is shown in Table 2. The results show that for higher CTP/ACVA ratio (5:1), the reaction is slower and takes almost twice longer to complete the reaction as compared to a CTP/ACVA ratio of 2. We noted that there was no significant difference between the polydispersities of the two polymers (with different CTP/ACVA ratios).
:1a
| Code | Reaction time/h | Conv.% (NMR) | M n (theory) (×104)/g mol−1 | M n (GPC) (×104)/g mol−1 | M w/Mn (GPC) |
|---|---|---|---|---|---|
| a The polymerization reaction was carried out at 60 °C, target DPn 63 and monomer concentration of 1.12 M. | |||||
| 3.1 | 7 | 43 | 1.03 | 8.4 | 1.08 |
| 3.2 | 9 | 56 | 1.33 | 1.01 | 1.11 |
| 3.3 | 11 | 65 | 1.54 | 1.12 | 1.10 |
| 3.4 | 23 | 78 | 1.84 | 1.34 | 1.13 |
With a fixed mole ratio of monomer to chain transfer agent, the solution concentrations and amount of initiator were varied for the kinetic studies. The results showed that the rate of the polymerizations strongly depended on the concentration of initiator (Fig. 2). For the same CTP/ACVA ratio with higher concentration of monomer (1.12 M), a faster rate of polymerization was observed (Table 3).
 | ||
| Fig. 2 Kinetic plot of the homopolymerization reaction of poly(MPC) in methanol at 70 °C, with varying concentrations and CTP/ACVA ratio: (1) CTP/ACVA = 2, (2) CTP/ACVA = 2 and (3) CTP/ACVA = 5, and varying concentrations, (1) 1.12 M, (2) 0.85 M, and (3) 1.12 M. | ||
All kinetics also brought out a similar inhibition time of 2.0 h in the polymerization process. This may be due to the fact that the monomer contains around 7 ppm of inhibitor. MPC monomer is highly hygroscopic, attempt to remove the inhibitor prior to the polymerization was unsuccessful.
Self-chain extension experiment of MPC
Poly(MPC)-based macro-CTA was obtained/purified by precipitating the polymer in acetone. Compared to the dialysis method, precipitation can effectively prevent the hydrolysis of the macroCTA.42 The accessibility of poly(MPC)-based macro-CTA was investigated in the self-chain extension experiments. The shift in the GPC peak after the sequential monomer addition confirmed the success of self-chain extension experiments. This formed the basis for the di-block copolymerization of MPC with other monomers.From previous discussions, for the synthesis of the first block, the monomer concentration of 1.1 M proved to be the most effective. Subsequently, the effect of monomer concentration on the synthesis of the second block was studied (Fig. S3 in ESI†). The results are summarized in Table S2 (ESI†). The results indicated that higher monomer concentration helps in the better control of molecular weight distribution (Table S1, Trial 2 (ESI†)). Also, the lower monomer concentration was used in order to prevent an overly high viscosity of the solution during polymerization reaction. Another factor that was studied for the self-chain extension of MPC was the CTP/ACVA ratio. Interestingly, when comparing the polymerizations with various ratios of CTP/ACVA (Table S2, Trial 2 and 3 (ESI†)), no obvious difference of polydispersities was observed in the homopolymerizations (1.07 against 1.09), but the difference was significant in their corresponding self-chain experiments (1.16 against 1.21). Fig. S2 (ESI†) shows the self-chain extension of MPC using a CTP/ACVA ratio of 5.0.
Similar results were also found in the self-chain experiments of LAEMA.35 Probing into the classic RAFT mechanism the answer may be found in the main equilibrium stage. The chains generated from initiator ACVA and the chains generated from the CTP leaving group have similar or even the same activity because of the same chemical structures. This maintained the equilibrium during the reaction. However, when the reaction was quenched, the “extra” (compared to the number of CTP mercapto groups) polymeric radicals became dead chains which could not be activated again. Therefore the difference in polydispersity was higher in the self-chain experiments.
Synthesis of diblock and random copolymers of MPC with other monomers
The MPC was copolymerized with various monomers. The AEMA bearing primary amino group has been copolymerized with MPC in the past by Sakaki et al.30 The AEMA unit in the polymer has been used for the DNA complexation while, the second block, the poly(MPC) may increase the water solubility and biocompatibility of the copolymer. It was observed that the biocompatibility, water solubility and nuclear resistance of the poly(MPC-co-AEMA) were better than those of the conventional cationic DNA carrier poly(L-lysine). Similarly, the motivation behind copolymerization of MPC with the synthetic glycopolymers is that these polymers contain saccharide moieties and thus entail various biological functions such as cell growth regulation, adhesion to specific biomolecules, etc. The biological applications of these polymers have been well studied by Narain et al.32Generally, the copolymerizations were conducted in a mixture of H2O and water miscible organic solvent because of the poor solubility of the second monomers and corresponding polymers in pure organic solvents. H2O was acidified by the addition of small amount of acetic acid to reduce the hydrolysis of CTA.38 As shown in Fig. 3(A) and (B), 4(A) and (B), S4(a) and (b), S5(a) and (b) (ESI†) in the diblock copolymerization experiments, a clear shift to high molecular weight in the GPC traces was observed in all cases indicating the success in blocking of these monomers on MPC.
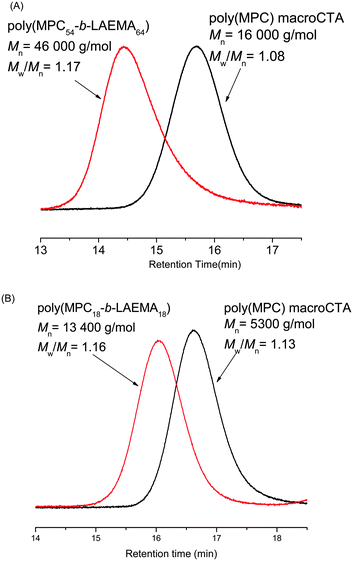 | ||
| Fig. 3 Diblock copolymerization of LAEMA with MPC using sequential monomer addition, forming (A) poly(MPC54-b-LAEMA64) and (B) poly(MPC18-b-LAEMA18). | ||
 | ||
| Fig. 4 Diblock copolymerization of GAEMA with MPC using sequential monomer addition, forming (A) poly(MPC54-b-GAEMA55) and (B) poly(MPC18-b-GAEMA18). | ||
Different DPn were also targeted in the diblock copolymerizations. A slight broadening of GPC traces was found in the samples with higher DPn (Fig. 3(A), 4(A), S4(a) and S5(a) (ESI†)) which may be due to the lower reactivity of the leaving group for the longer polymer chains (see also Table S3 in the ESI†). Another interesting observation was that poly(MPC)-based macroCTA, which is an ester, was compatible with all amide monomers used in this work (Fig. 5–7). For instance, copolymers with low PDI were successfully synthesized in the diblock copolymerization of MPC with GAEMA (amide) and GAMA (ester). The DP of the resulting copolymer was found to be slightly higher than what was actually targeted. This may have happened due to the termination of primary radicals before their addition to the monomer. This results in lower initiator efficiency.23Random copolymerization of MPC, on the other hand, yielded 80% conversion (Table 4). NMR spectroscopy was used to determine the monomer composition in the statistical copolymers (Fig. S6 and S7 (ESI†)).
 | ||
| Fig. 5 Structures of the monomers, initiator and chain transfer agent. | ||
 | ||
| Fig. 6 Homopolymerization of poly(MPC) by RAFT technique in methanol at 60 °C, using ACVA as the thermally degradable initiator and CTP as the chain transfer agent. | ||
 | ||
| Fig. 7 Di-block copolymerization of poly(MPC) macroCTA with GAEMA in water at 70 °C in the presence of ACVA as the initiator. | ||
| Code | Statistical copolymers of MPC | M n (theory) (×104)/g mol−1 | M n (GPC) (×104)/g mol−1 | M w/Mn (GPC) |
|---|---|---|---|---|
| a Polymerization was carried out in water, in the presence of acetic acid at 70 °C, using CTP as the macroCTA. | ||||
| 4.1 | Poly(MPC42-stat-LAEMA42) | 4.5 | 3.2 | 1.25 |
| 4.2 | Poly(MPC50-stat-GAEMA45) | 3.6 | 2.8 | 1.19 |
| 4.3 | Poly(MPC58-stat-GAMA48) | 3.61 | 3.2 | 1.20 |
| 4.4 | Poly(MPC52-stat-AEMA43) | 2.7 | 2.2 | 1.25 |
Diblock copolymerization of MPC with AEMA can be carried out in pure methanol as solvent. AEMA concentration of 0.25 M was recommended because of the high viscosity of the reaction solution at the end of the polymerization. The polydispersity of the resulting diblock copolymer was observed as low as 1.2 while similar experiment conducted in H2O resulted in a copolymerMw/Mn = 1.3. This result also indicated the advantage of using methanol as solvent. But higher target molecular weights of poly(MPC-stat-AEMA) cannot be synthesized in methanol. It has been observed that the copolymer formed precipitates out before the reaction can complete, hence, giving low molecular weight polymers. This can be explained by the fact that poly(MPC) segment increases the solubility in methanol. But as the reaction proceeds and the poly(MPC-b-AEMA) chain length increases, the solubility of the resulting co-polymer decreases due to incompatibility with the solvent.
Conclusion
We have hereby presented a detailed study of the RAFT polymerization of MPC. Methanol proves to be a better solvent for dissolution of CTP and ACVA. An ideal narrow polydispersity (Mw/Mn < 1.2) of poly(MPC) can be obtained up to Mn = 2.9 × 104 g mol−1 using methanol as solvent. The rate of RAFT polymerizations of MPC in methanol strongly depended on the initiator concentration (for a constant CTP concentration). Under reasonable viscosity of the reaction mixture, copolymerizations were found to proceed with good control a higher monomer concentration and the polydispersity of resulting self-blocking MPC polymer was slightly lower when higher ratio of CTP to ACVA was used in the preparation of macro-CTA. The poly(MPC)-based macro-CTP was successfully applied in the synthesis of block-type copolymer with various monomers. The diblock copolymers prepared via sequential monomer addition were synthesized with high yields and low polydispersities. Ongoing work in our laboratory will focus on the cytotoxicity studies of statistical and diblock copolymers composed of the poly(MPC) segment and the cationic polymer segment. These functionalized polymers may be used as carriers of drugs and genes to the cells.References
- K. Ishihara, T. Ueda and N. Nakabayashi, Polym. J. (Tokyo, Jpn.), 1990, 22, 355–360 CrossRef CAS.
- J. Yuan, S. Armes, Y. Takabayashi, K. Prassides, C. Leite, F. Galembeck and A. Lewis, Langmuir, 2006, 22, 10989–10993 CrossRef CAS.
- Y. Iwasaki and K. Ishihara, Anal. Bioanal. Chem., 2005, 381, 534–546 CrossRef CAS.
- K. Ishihara, R. Aragaki, T. Ueda, A. Watenabe and N. Nakabayashi, J. Biomed. Mater. Res., 1990, 24, 1069–1077 CrossRef CAS.
- K. Ishihara, N. Ziats, B. Tierney, N. Nakabayashi and J. Anderson, J. Biomed. Mater. Res., 1991, 25, 1397–1407 CrossRef CAS.
- K. Ishihara, K. Arai, N. Nakabayashi, S. Morita and K. Furuya, J. Biomed. Mater. Res., 1992, 26, 937–945 CrossRef CAS.
- K. Ishihara, H. Nomura, T. Mihara, K. Kurita, Y. Iwasaki and N. Nakabayashi, J. Biomed. Mater. Res., 1998, 39, 323–330 CrossRef CAS.
- L. Yan and K. Ishihara, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3306–3313 CrossRef CAS.
- K. Enomoto, T. Hasebe, R. Asakawa, A. Kamijo, Y. Yoshimoto, T. Suzuki, K. Takahashi and A. Hotta, Diamond Relat. Mater., 2010, 19, 806–813 CrossRef CAS.
- H. Lomas, J. Du, I. Canton, J. Madsen, N. Warren, S. Armes, A. Lewis and G. Battaglia, Macromol. Biosci., 2010, 10, 513–530 CrossRef CAS.
- K. Nam, T. Kimura, S. Funamoto and A. Kishida, Acta Biomater., 2010, 6, 403–408 CrossRef CAS.
- Y. Xu, M. Takai and K. Ishihara, Ann. Biomed. Eng., 2010, 38, 1938–1953 CrossRef.
- M. Kyomoto, T. Moro, Y. Takatori, H. Kawaguchi, K. Nakamura and K. Ishihara, Biomaterials, 2010, 31, 1017–1024 CrossRef CAS.
- T. Shimizu, T. Goda, N. Minoura, M. Takai and K. Ishihara, Biomaterials, 2010, 31, 3274–3280 CrossRef CAS.
- L. Lewis, Colloids Surf., B, 2000, 18, 261–275 CrossRef CAS.
- L. Lewis, P. Hughes, L. Kirkwood, S. Leppard, R. Redman, L. Tolhurst and P. Stratford, Biomaterials, 2000, 21, 1847–1859 CrossRef CAS.
- T. Moro, Y. Takatori, K. Ishihara, T. Konno, Y. Takigawa, T. Matsushita, U. I. Chung, K. Nakamura and H. Kawaguchi, Nat. Mater., 2004, 3, 829–836 CrossRef CAS.
- O. Katakura, N. Morimoto, Y. Iwasaki, K. Akiyoshi and S. Kasugai, J. Med. Dent. Sci., 2005, 52, 115–121 Search PubMed.
- M. Kyomoto, T. Moro, K. Saiga, F. Miyaji, H. Kawaguchi, Y. Takatori, K. Nakamura and K. Ishihara, Biomaterials, 2010, 31, 658–668 CrossRef CAS.
- C. Wang, A. Javadi, M. Ghaffari and S. Gong, Biomaterials, 2010, 31, 4944–4951 CrossRef CAS.
- Y. Iwasaki, M. Takamiya, R. Iwata, S. Yusa and K. Akiyoshi, Colloids Surf., B, 2007, 57, 226–236 CrossRef CAS.
- E. Lobb, Y. Ma, N. Billingham and S. Armes, J. Am. Chem. Soc., 2001, 123, 7913–7914 CrossRef CAS.
- K. Matyjaszewski and T. P. Davis, Handbook of Radical Polymerization, Wiley Interscience, 2002 Search PubMed.
- R. Kojima, M. C. Z. Kasuya, K. Ishihara and K. Hatanaka, Polym. J. (Tokyo, Jpn.), 2009, 41, 370–373 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- S. Yusa, K. Fukuda, T. Yamamoto, K. Ishihara and Y. Morishima, Biomacromolecules, 2005, 6, 663–670 CrossRef CAS.
- B. Yu, A. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS.
- S. Sakaki, M. Tsuchida, Y. Iwasaki and K. Ishihara, Bull. Chem. Soc. Jpn., 2004, 77, 2283–2288 CrossRef CAS.
- Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5031–5036 CrossRef CAS.
- Z. Deng, M. Ahmed and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 614–627 CrossRef CAS.
- Z. Deng, H. Bouchékif, K. Babooram, A. Housni, N. Choytun and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4984–4996 CrossRef CAS.
- R. Narain and S. Armes, Biomacromolecules, 2003, 4, 1746–1758 CrossRef CAS.
- Z. Deng, S. Li, X. Jiang and R. Narain, Macromolecules, 2009, 42, 6393–6405 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- D. Thomas, A. Convertine, R. Hester, A. Lowe and C. McCormick, Macromolecules, 2004, 37, 1735–1741 CrossRef CAS.
- W. Lyoo, D. Song, W. Lee, S. Han and S. Noh, J. Appl. Polym. Sci., 2006, 102, 4831–4834 CrossRef CAS.
- G. Levesque, P. Arsene, V. Fanneau-Bellenger and T. Pham, Biomacromolecules, 2000, 1, 400–406 CrossRef CAS.
- Y. Ma, Y. Tang, N. C. Billingham, S. P. Armes, A. L. Lewis, A. W. Lloyd and J. P. Salvage, Macromolecules, 2003, 36, 3475–3484 CrossRef CAS.
- Y. Vasilieva, D. Thomas, C. Scales and C. McCormick, Macromolecules, 2004, 37, 2728–2737 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, GPC and 1H NMR data of random copolymer synthesis. See DOI: 10.1039/c0py00300j |
| This journal is © The Royal Society of Chemistry 2011 |