
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA breath of sunshine: oxygenic photosynthesis by functional molecular architectures
Thomas
Gobbato
 a,
Giulia Alice
Volpato
a,
Andrea
Sartorel
*a and
Marcella
Bonchio
*ab
a,
Giulia Alice
Volpato
a,
Andrea
Sartorel
*a and
Marcella
Bonchio
*ab
aDepartment of Chemical Sciences, University of Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: andrea.sartorel@unipd.it; marcella.bonchio@unipd.it
bITM-CNR Section of Padova, INSTM Unit of Padova, via Marzolo 1, 35131 Padova, Italy
First published on 4th October 2023
Abstract
The conversion of light into chemical energy is the game-changer enabling technology for the energetic transition to renewable and clean solar fuels. The photochemistry of interest includes the overall reductive/oxidative splitting of water into hydrogen and oxygen and alternatives based on the reductive conversion of carbon dioxide or nitrogen, as primary sources of energy-rich products. Devices capable of performing such transformations are based on the integration of three sequential core functions: light absorption, photo-induced charge separation, and the photo-activated breaking/making of molecular bonds via specific catalytic routes. The key to success does not rely simply on the individual components' performance, but on their optimized integration in terms of type, number, geometry, spacing, and linkers dictating the photosynthetic architecture. Natural photosynthesis has evolved along this concept, by integrating each functional component in one specialized “body” (from the Greek word “soma”) to enable the conversion of light quanta with high efficiency. Therefore, the natural “quantasome” represents the key paradigm to inspire man-made constructs for artificial photosynthesis. The case study presented in this perspective article deals with the design of artificial photosynthetic systems for water oxidation and oxygen production, engineered as molecular architectures then rendered on electrodic surfaces. Water oxidation to oxygen is indeed the pervasive oxidative reaction used by photosynthetic organisms, as the source of reducing equivalents (electrons and protons) to be delivered for the processing of high-energy products. Considering the vast and abundant supply of water (including seawater) as a renewable source on our planet, this is also a very appealing option for photosynthetic energy devices. We will showcase the progress in the last 15 years (2009–2023) in the strategies for integrating functional building blocks as molecular photosensitizers, multi-redox water oxidation catalysts and semiconductor materials, highlighting how additional components such as redox mediators, hydrophilic/hydrophobic pendants, and protective layers can impact on the overall photosynthetic performance. Emerging directions consider the modular tuning of the multi-component device, in order to target a diversity of photocatalytic oxidations, expanding the scope of the primary electron and proton sources while enhancing the added-value of the oxidation product beyond oxygen: the selective photooxidation of organics combines the green chemistry vision with renewable energy schemes and is expected to explode in coming years.
 Thomas Gobbato | Thomas Gobbato received his PhD in Chemistry at the University of Trieste in 2023 under the supervision of Professor Maurizio Prato and Professor Marcella Bonchio, working on bioinspired nanoarchitectures for artificial photosynthesis. His research interests are in supramolecular hybrids and organic–inorganic systems for photoelectrocatalysis. |
 Giulia Alice Volpato | Giulia Alice Volpato received her PhD in Science and Engineering of Materials and Nanostructures at the University of Padua in 2021, completing a thesis on photoactivation of small molecules for production of renewable solar fuels and valuable chemicals under the supervision of Professor Andrea Sartorel. Her research interests include nanomaterials for electrocatalytic HER, CO2RR and ORR applications. |
 Andrea Sartorel | Andrea Sartorel is Associate Professor of Organic Chemistry at the University of Padova, Department of Chemical Sciences. His research interests deal with photoelectrochemical processes exploiting molecular based systems for applications in small molecules activation and selective organic synthesis. |
 Marcella Bonchio | Marcella Bonchio is Full Professor of Organic Chemistry at the University of Padova, and Delegate Director of the CNR Institute of Membrane Technology section of Padova. Bonchio's team has established a highly interdisciplinary research program to address innovative, bio-inspired photosystems for water splitting and artificial photosynthesis (https://nanomolcat.wixsite.com/nanomolcat). |
Introduction
In the novel “The mysterious island” published between 1874 and 1875, the visionary French writer Jules Verne suggested that water, decomposed into its primitive elements, could be the fuel of the future, and provide an inexhaustible source of heat and light, of an intensity of which coal is not capable. In 1912, the Italian photochemist Giacomo Ciamician envisioned a quieter civilization based on the utilization of solar energy, contrasting the black and nervous civilization based on coal.1Water and solar energy are indeed the pillars of artificial photosynthesis, a long sought dream process for splitting water into hydrogen and oxygen by means of solar light, thus providing an inexhaustible resource of green and renewable fuels to satisfy the increasing global energy demand.2,3 The water splitting reaction (eqn (1) and Scheme 1) can be formally decomposed into two redox semi-reactions of proton reduction to hydrogen (eqn (1A)) and of water oxidation to oxygen (eqn (1B)), vide infra; accordingly, the ΔG0 for eqn (1) at 298 K can be reported to be 1.23 eV, being the normalized value per exchanged electron (4 in the case of eqn (1)).† It is useful to exploit this normalized value since light absorption events are mostly associated with single electron transfer, and therefore this value can be associated with the energy (or with the wavelength) of the electromagnetic radiation (i.e. 1.23 eV corresponds to a radiation wavelength of 1008 nm). This formalism also enables a direct comparison of the energetic requirement of photosynthetic processes: in the case of natural photosynthesis (eqn (2)), this value is 1.24 eV, very close to the one of water splitting.
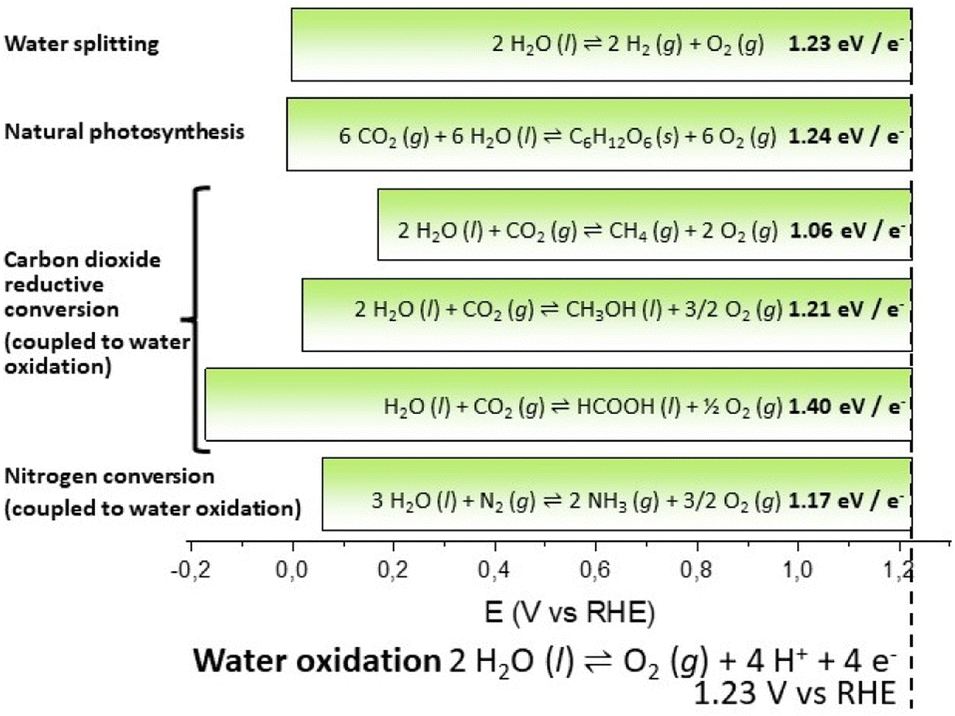 | ||
| Scheme 1 Schematic representation of the energetic requirements of selected photosynthetic reactions (see eqn (1)–(6) in the main text). | ||
Although water splitting for production of hydrogen is the most investigated process, the operating principle can be extended to CO2 or N2 reductive conversion (Scheme 1):4–6 some representative examples are reported in Scheme 1 and eqn (3)–(6), where water provides reducing equivalents to produce methane, methanol, formic acid (from CO2) or ammonia (from N2). Also for these reactions, the standard free energy at 298 K normalized per electron exchanged in the corresponding semireactions is reported (eqn (3)–(6)).
Water splitting
| 2H2O (l) ⇌ 2H2 (g) + O2 (g); 1.23 eV per e− | (1) |
| 4H+ + 4 e− ⇌ 2H2 (g) | (1A) |
| 2H2O (l) ⇌ O2 (g) + 4H+ + 4e− | (1B) |
Natural photosynthesis
| 6CO2 (g) + 6H2O (l) ⇌ C6H12O6 (s) + 6O2 (g); 1.24 eV per e− | (2) |
| 6CO2 (g) + 24H+ + 24e− ⇌ C6H12O6 (s) + 6H2O | (2A) |
| 12H2O (l) ⇌ 6O2 (g) + 24H+ + 24e− | (2B) |
Carbon dioxide reductive conversion (coupled to water oxidation)
| 2H2O (l) + CO2 (g) ⇌ CH4 (g) + 2O2 (g); 1.06 eV per e− | (3) |
| CO2 (g) + 8H+ + 8e− ⇌ CH4 (g) + 2H2O (l) | (3A) |
| 4H2O (l) ⇌ 2O2 (g) + 8H+ + 8e− | (3B) |
| 2H2O (l) + CO2 (g) ⇌ CH3OH (l) + 3/2O2 (g); 1.21 eV per e− | (4) |
| CO2 (g) + 6H+ + 6e− ⇌ CH3OH (l) + H2O (l) | (4A) |
| 3H2O (l) ⇌ 3/2O2 (g) + 6H+ + 6e− | (4B) |
| H2O (l) + CO2 (g) ⇌ HCOOH (l) + 1/2O2 (g); 1.40 eV per e− | (5) |
| CO2 (g) + 2H+ + 2e− ⇌ HCOOH (l) | (5A) |
| H2O (l) ⇌ 1/2O2 (g) + 2H+ + 2e− | (5B) |
Nitrogen conversion (coupled to water oxidation)
| 3H2O (l) + N2 (g) ⇌ 2NH3 (g) + 3/2O2 (g); 1.17 eV per e− | (6) |
| N2 (g) + 6H+ + 6e− ⇌ 2NH3 (g) | (6A) |
| 3H2O (l) ⇌ 3/2O2 (g) + 6H+ + 6e− | (6B) |
When reactions (1)–(6) are triggered by light, the radiative energy turns out to be stored in the product chemical bonds, yielding the so called “solar fuels” to feed fuel-cells and provide energy with an overall carbon zero-impact cycle. The development of efficient devices for the sustainable production of solar fuels represents the key challenge for a radical step forward in the field of renewable energy schemes. In particular, solar-energy conversion and storage has been recently demonstrated through promising technologies implementing artificial photosynthesis. These include:
(i) a consolidated photo-electrochemical approach (solar-to-hydrogen efficiency up to STH = 30%),7 implying the coupling of photovoltaic and electrochemical modules (PV–EC) or the fabrication of integrated photo-electrodes wired within photo-electrochemical cells (PEC);8–10
(ii) a recently assessed photocatalytic (PC) technology based on wireless and bias-free particulate materials suspended in photoreactors for the continuous extraction of solar fuels.11 This approach has been awarded with the European Innovation Council – Horizon Prize 2022, ‘Fuel from the Sun: Artificial Photosynthesis’ recognizing the promising potential of a 100 m2 prototype built with an array of panel photoreactors filled with particles based on aluminum-doped strontium titanate with co-loaded Rh, Cr and Co co-catalysts and enabling green hydrogen production, albeit with a limited STH efficiency of 0.76%.12 Very recently, a record STH value of 9.2% has been achieved using concentrated solar light conditions and particulate photocatalysts based on indium gallium nitride loaded with rhodium/chromium oxide and cobalt oxide co-catalysts, at an optimal temperature of 70 °C.13
In all these systems (PV–EC, PEC, PC), three main functionalities are pivotal to trigger the photosynthetic process: (i) visible light harvesting tuning the absorption properties; (ii) a cascade of charge separation events upon photo-induced electron transfer; (iii) orchestrated multi-redox catalytic routes to drive solar energy conversion into the chemical energy of new molecular bonds. Noteworthily, a bio-inspired compartmentalization of the photosynthetic complexity enables the breakdown of the overall process into separate modules that can be optimized separately.14,15 However, the key to success lies on the engineering of a-solo components that can be then operated within an integrated function, in order to maximize the photosynthetic efficiency.16
A source of inspiration is the natural photosynthetic system,17 consisting of an orchestrated functional architecture, where each step, from light absorption to electron/proton transport and multi-redox catalysis, is performed by a highly specialized unit incorporated in a structure-directing protein matrix. The great beauty of the natural process has thus inspired the invention of artificial replica based on novel photoactive molecules and materials, to be combined into an integrated photosynthetic assembly.
Herein we focus on the central role of photo-assisted water oxidation, evolving oxygen and liberating reduction equivalents (protons and electrons) to be used for the cascade production of solar fuels, including green H2 or CO2 or N2 reduction products (see eqn (1)–(6)). In this scheme, water activation and cleavage of what are among the strongest molecular bonds (Bond Dissociation Free Energy of the O–H bond in liquid H2O, BDFEOH = 115.8 kcal mol−1),18 provides the renewable and ubiquitous primary vector to drive solar energy conversion and storage, optimized along billions of years of aerobic life evolution. Indeed, oxygenic photosynthesis occurs in nature by the vital function of photosystem II (PSII), a unique membrane protein complex whose multi-site architecture and dynamics regulate the photosynthetic efficiency within the thylakoids of plants, cyanobacteria, and algae. In the same way, the artificial perspective relies on light-activated materials and molecular ensembles enabling photocatalytic water oxidation on tailored photoanodes for regenerative PEC technology, and/or as components of wireless photocatalytic systems (PC) mimicking the biological asset.
Since the seminal report in 2009 by Mallouk and co-workers,19 research on the oxygenic “artificial leaf” has exploded, with the last few years witnessing a crescendo of scientific achievements addressing the complexity of the photosynthetic design. In particular, new directions can be traced in the most recent literature regarding:
- The rapid and continuous progress in the field of water oxidation catalysis (WOC): T. J. Meyer remarked in 2008 that “catalysts for water oxidation are so rare that the discovery of a new family is cause for celebration”;20 since then, the number and type of molecular WOCs have been increasing exponentially21 and current state-of-the-art WOC based on a tda–ruthenium complex (tda = 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylate) reaches turnover frequency up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1,22 overarching by two orders of magnitude the tetra-manganate natural oxygen evolving centre (TOF = 100–400 s−1), while showing more than 1 million turnovers when probed under dark electrocatalytic conditions.23
000 s−1,22 overarching by two orders of magnitude the tetra-manganate natural oxygen evolving centre (TOF = 100–400 s−1), while showing more than 1 million turnovers when probed under dark electrocatalytic conditions.23
- The evolution of photosensitizer (PS) structures relative to their assembly behaviour and related photophysics.24 The first requirement for the PS choice is a broad light absorption in the visible region spanning the 380–740 nm range, which corresponds to the solar emission peak and accounts for 43% of the total solar radiation at the Earth surface. While photocatalytic systems performing in homogeneous solutions have been dominated by molecular PS operating through long-lived triplet excited state manifolds, as in the case of Ru(II) polypyridine complexes, the PS integration within heterogeneous/colloidal photosynthetic architectures opens novel routes engaging singlet excited states, as in the case of dye-sensitized semiconductors.25
- The use of redox-mediators (relays) to decouple light-induced electron transfer, charge separation and recombination events.26,27 Quinoid electron transfer relays have been recently exploited by Fukuzumi et al. enabling the combined water reduction/oxidation under homogeneous conditions mimicking the native electron/proton transport mechanism.28
- The use of non-covalent synthetic strategies to engineer the assembly of the photosynthetic components. This bio-inspired approach complements the synthetic design of covalent chromophore–catalyst conjugates to engineer photocatalytic dyads and/or the joint grafting of chromophores and catalysts on electrode surfaces.29 To this end, supramolecular strategies can include electrostatic assemblies favoured by multi-charge interactions,30,31 as in the case of tetravalent cations (such as ZrIV, vide infra) binding to carboxylate pendants,32,33 liposome membranes,34,35 polymeric coatings,36 and hierarchical photosynthetic architectures with controlled morphology, optimized for the light-quanta conversion.37–41
- Management of concomitant proton and electron transport so as to trigger favourable PCET mechanisms that are of fundamental importance to lower the energy requirement of photosynthetic reactivity.18,42,43
- Optimization of semiconductor (SC) technologies to boost charge transport and avoid energy losses. Innovation in the field is envisaged to control light harvesting, surface areas and porosity, electronic structures and the excited-state properties.44
Cutting-edge research in artificial photosynthesis builds on these innovative approaches as will be highlighted in the following sections addressing the state-of-the-art water splitting photoanodes from a molecular perspective and including relevant bio-hybrid case-studies based on photosystem II (PSII) as the natural paradigm.
Photosynthetic water oxidation at engineered electrode surfaces
The design of oxygen evolving photoanodes requires a careful design and assembly of some minimal components: a semiconductor, a photosensitizer and a water oxidation catalyst. Hence, the compliance of energetic requirements is essential, but the precise control of the interactions among the components and between interfaces is crucial to obtain a perfectly orchestrated chain of electron transfer events regulated by the photosynthetic assembly where we can pinpoint diverse roles and features as follows:The 4 electrons/4 protons mechanism of light-driven water oxidation by dye-sensitized/SC photoanodes generally involves the following steps:
(i) excitation of the photosensitizer (*PS, eqn (7)), (ii) *PS electron injection into the SC conduction band and generation of its oxidized state (PS+, eqn (8)), (iii) WOC sequential hole scavenging steps generating high valent WOC states (WOC+/WOC(n+1)+, eqn (9) and (9A)), (iv) water oxidation by the tetra-oxidized WOC4+, closing the catalytic cycle by regenerating the starting WOC state (eqn (10)):
| PS + hν → *PS | (7) |
| SC + *PS → SC(e−) + PS+ | (8) |
| PS+ + WOC → PS + WOC+ | (9) |
| PS+ + WOCn+ → PS + WOC(n+1)+ | (9A) |
| WOC4+ + 2H2O → WOC + O2 + 4H+ | (10) |
At variance with homogeneous photocatalysis using terminal sacrificial oxidants, PEC applications and SC technology suffer from parallel deactivation routes, deriving from electron–hole recombination pathways within the SC material and/or involving the oxidised dye (PS+ in eqn (11)), or the high valent WOC manifold (WOCn+ in eqn (12)),
| SC(e−) + PS+ → SC + PS | (11) |
| SC(e−) + WOCn+ → SC + WOC(n−1)+ | (12) |
Meanwhile, unproductive quenching of the PS excited state can also occur, via energy transfer (eqn (13)) or oxidative electron transfer (eqn (14)) involving the WOC sites:
| *PS + WOCn+ → PS + *WOCn+ → relax to the ground state | (13) |
| *PS + WOCn+ → PS+ + WOC(n−1)+ | (14) |
The main key performance indicators typically used to benchmark photoanodes for water oxidation include:48
(i) The onset potential of the oxygenic photocurrent, i.e. the minimum potential of the electrode at which a productive photocurrent response is observed (i.e. photoinduced electron injection is collected by the circuit); since the thermodynamic potential for the OER is 1.23 V vs. the reversible hydrogen electrode (RHE), efficient photoanodes should operate well below this potential value (underpotential regime), and ideally without any applied bias to improve the SC electron injection and counteract recombination pathways.
(ii) The photocurrent density plateau and saturation onset, i.e. the maximum photocurrent density value generally reached above an applied potential threshold (plateau or saturation photocurrent and onset). The photocurrent density should be determined after subtraction of any dark-current component registered at a given potential, so as to be correlated with the photo-catalytic reaction occurring at the photoelectrode.
(iii) The faradaic efficiency or yield (FE or FY) associated with water oxidation is a measure of the photocatalytic yield and selectivity, being the fraction of the observed photocurrent that is due to oxygen evolution. Non-unitary faradaic efficiencies are indicative of competitive processes occurring in concomitance with water oxidation to oxygen.
(iv) The light harvesting efficiency (LHE) represents the fraction of light that is absorbed by the photoanode, depending on the dye surface loading and on the resulting molar extinction coefficient (absorption cross section); it can be expressed as a function of the light wavelength namely LHE(λ) = 1–10−A(λ), with A = Γσ(λ) being the resulting absorbance related to Γ (the number of moles of sensitizer per cm2) and σ(λ) i.e. the absorption cross-section in cm2 per mole.
(v) The incident photon to current efficiency (IPCE) or external quantum efficiency (EQE) is the fraction of incident photons that are converted into photocurrent, defined as the mathematical product of light-harvesting efficiency, electron injection efficiency (φ), charge collection efficiency (ɳ) of the photoanode: IPCE = (LHE) × φ × ɳ.
(vi) The absorbed photon to current efficiency (APCE) or internal quantum efficiency (IQE) is defined as the fraction of absorbed photons that are converted into photocurrent, given by the ratio between IPCE and LHE.
(vii) The photocurrent stability under operation conditions (typically controlled potential photoelectrolysis at a given potential) can be given by the fraction of residual photocurrent after a certain amount of time, or by the time at which the initial photocurrent is halved.
Besides the photoanode performance metrics, parameters associated with the WOC performance are also considered, including the turnover frequency and number (TOF and TON). These parameters are generally estimated by correlating the oxygenic photocurrent density with the WOC loading, and/or with the resulting electroactive surface catalyst. However, the precise identification of the active WOC concentration is often elusive so that performance benchmarking via TOF and TON metrics might be misleading.49
In the next paragraphs, we will showcase the progress in the last 15 years (2009–2023) in the development of strategies for integrating the molecular photosensitizers, multi-redox water oxidation catalysts and semiconductor materials building blocks, highlighting how additional components such as redox mediators, hydrophilic/hydrophobic pendants, and protective layers can impact on the overall photosynthetic performance. We have chosen to group the literature reports in separate paragraphs, starting from the photosystem II machinery, and then collecting the artificial examples based on the photosensitizer nature, as its characteristics (absorption range, band levels, lifetime of the excited state, aggregation, proton-coupled electron transfer reactivity) often dictate the overall design of the device.
Finally, emerging directions in the field will be presented, exploiting the modular tuning of the multi-component device to target a diversity of photocatalytic oxidations, thus enhancing the added-value of the oxidation product beyond oxygen: the selective photooxidation of organic substrates incorporates the green chemistry vision with circular economy policies and is expected to explode in the near future.
The native photosystem II (PSII) machinery and PSII wired bio-hybrid photoanodes
More than 3 billion years ago oxygenic photosynthesis triggered our aerobic life, originating at the PSII protein within thylakoid membranes of photosynthetic organisms such as bacteria, algae and higher plants.The crystal structure of PSII from Thermosynechococcus vulcanus was reported at 1.95 A resolution by Suga et al.;50 PSII exists in a dimeric form with 700 kDa molecular mass51,52, where each subunit contains the functional components enabling light absorption, energy transfer, and electron and proton separation, ultimately leading to oxygen evolution.53
The process occurs at the reaction centre RC, composed of D1 and D2 subunits, and initiates at a PD1/PD2 porphyrin dimer, characterized by weak electronic coupling (85–150 cm−1; for comparison in the anoxygenic purple bacterial reaction centre the coupling between PL and PM is 500–1000 cm−1). This weak coupling enables PD1 and PD2 to maintain properties typical of monomers: in particular, generation of the first excited state *P with 1.83 eV energy is possible through direct excitation of P or via energy transfer from the internal or external PSII antennas (CP47 and CP43 are the inner antennas in D1 and D2, respectively, while LHC1 and LHC2 are the outer antennas).54 *P is considered the primary electron donor in PSII: as represented in Fig. 1 for PD1, the excited state transfers one electron in a few ps to a neighbouring pheophytin PhD1, with the assistance of a chlorophyll ChlD1. The PD1˙+/PhD1˙− secondary radical pair represents thus the first charge separated state. The ground state of PD1 is restored upon electron transfer to PD1˙+ from a tyrosine residue; the process is proton-coupled with the assistance of a proximal histidine residue acting as a base, and allowing the generation of a neutral tyrosine radical; this step occurs within 50 to 250 ns, depending on the oxidation state of the oxygen evolving centre. Finally, the Tyr-O˙ acts as a 1e− oxidant towards the Mn4 oxygen evolving centre, Mn4-OEC (30–200 μs, depending on the oxidation state of the OEC); the stepwise hole accumulation at the Mn4-OEC is represented by the Joliot–Kok cycle, finally releasing O2 from the highest oxidized state S4 (the subscripts 0–4 indicate the oxidation level of oxidation of the Mn4-OEC, with the S1 being the dark stable state), and operating with a turnover frequency of the order of 102 s−1.55 From the acceptor side, the electron chain continues with a subsequent electron transfer from PhD1˙− to a quinone QA (within 400 ps), and from QA˙− to a quinone QB in 0.2 to 0.8 ms, depending on the redox state of QB.
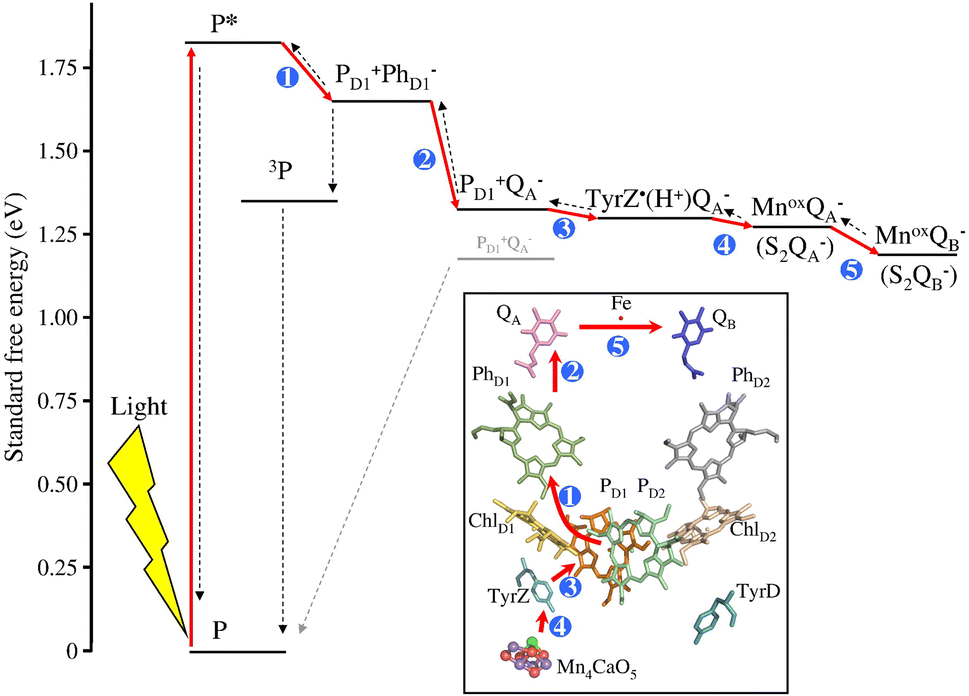 | ||
| Fig. 1 Energy scheme and schematic representation of the photoinduced charge separation in photosystem II (red arrows represent the electron transfer steps, the blue numbers represent the order in the sequence of electron transfer events). Reprinted from ref. 54 with permission from Elsevier, copyright 2012. | ||
Indeed, QB can be reduced twice through a proton coupled process and transforms into the two-electron reduced form QBH2, which is released from PSII (and replaced by a QB present in the membrane) and transfers the reducing equivalents to a membrane heme-cofactor Cyt b6f. The concomitant transfer of electrons and protons is assisted by “water channels” that connect the protein bulk surface with the protein interior.56
Clearly, the possibility of transferring reducing equivalents outside the PSII enzyme opens the possibility of engineering bio-hybrid interfaces with artificial systems, in particular by exploiting protein film photo-electrochemistry.57,58 Strategies for PSII wiring on photoelectrodes resulting in bio-hybrid PEC devices applied to water splitting have been recently reviewed;52 current state-of-the art performances of PSII based photoelectrodes reach photocurrent densities of ca. 1 mA cm−2.59
Reisner and co-workers reported several studies on PSII photoelectrodes, with PSII extracted from particular cyanobacteria and immobilised onto nanostructured indium tin oxide (ITO).48,59–62 Since photo-instability is a major issue for isolated PSII that cannot benefit from the continuous “self-healing” machinery of the in vivo system,48 a recent strategy is to wire the cyanobacterium Synechocystis sp. PCC 6803 live cells on ITO electrodes.63,64
Bio-hybrid PSII/ITO photoelectrodes are capable of oxidizing water using red-light low-energy photons (680 nm) with a low onset potential of 0.6 V vs. RHE at pH 6.5, reaching the photocurrent saturation below the OER thermodynamic limit, and therefore behaving as ideal photoanodes (Table 1).
| Entry and photoanode | Light intensity (mW cm−2) | E (V) vs. RHE | J (μA cm−2) and IPCE | F.E.(O2) (%) | TOF (s−1) | Ref. |
|---|---|---|---|---|---|---|
| a Coupled to a hydrogenase cathode. b Coupled to a formate dehydrogenase cathode. | ||||||
| 1, IO-mesoITO|PSII | 679 nm; 10 mW cm−2 | 0.88 | 20 (direct electron transfer, DET) no IPCE | 75 ± 4 | —(DET) | 59 |
| 930 ± 30 (mediated electron transfer, MET via DCBQ) (IPCE 17.0 ± 0.5% at 679 nm) | 12.9 ± 0.4 (MET via DCBQ) | |||||
| 2, IO-mesoITO|POs|PSII | 685 nm; 10 mW cm−2 | 0.88 | 381 ± 31 (DET) (IPCE 6.9 ± 0.9% at 685 nm) | 85 ± 9 | 4.0 ± 0.4 (DET) | 62 |
| 513 ± 29 (MET via DCBQ) (IPCE 9.3 ± 1.2% at 685 nm) | 6.7 ± 0.7 (MET via DCBQ) | |||||
| 3, IO-TiO2|dpp|POs–PSII | AM 1.5G, 100 mW cm−2, λ > 420 nm | 0.68a | 130–140 (DET) (IPCE 2.7% at 560 nm, Iph = 6 mW cm−2) | 88 ± 12 | — | 65 and 66 |
| 0.78b | 99 ± 4 (DET) | 70 (formate) | ||||
To address one major limitation due to the low PSII loading on mesoporous ITO (mesoITO) (ca. 20 pmol cm−2), limiting the photocurrent density to the sub-microampere range,48 ITO substrates with hierarchical porosity and the inverse opal (IO) morphology were used to allow higher PSII loading increasing the effective surface area and favouring a stable anchoring of the enzyme.59 The optimized IO-mesoITO|PSII photoanodes can reach up to 20 μA cm−2 photocurrent density in the absence of redox mediators, which highlights the challenge to improve the electrical communication between PSII and the electron-collecting ITO (entry 1, Table 1). This issue was later addressed through co-adsorption of PSII with an osmium-based redox active polymer (POs), allowing the collection of electrons from PSII, regardless of the relative orientation and proximity of the natural enzyme to the ITO surface (Fig. 2).62 Under irradiation with monochromatic red light, the improved IO-mesoITO|POs–PSII photoanodes showed an outstanding 50-fold increase of the saturation photocurrent (ca. 400 μA cm−2) and 15-fold increase of IPCE (4.4%), with a 90% faradaic efficiency for oxygen evolution and TOFmax of 4 s−1 per PSII center (entry 2, Table 1).
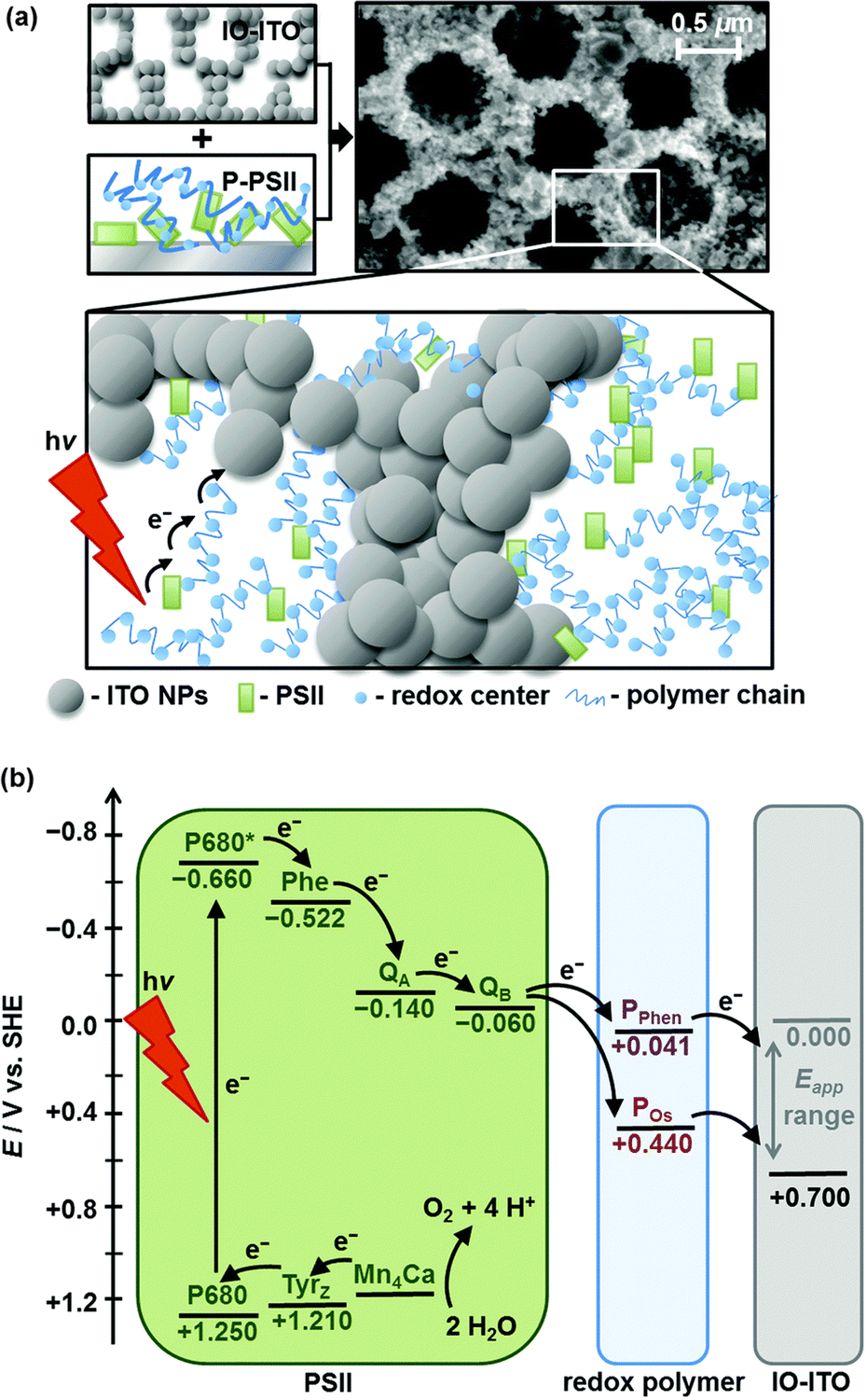 | ||
| Fig. 2 (a) PSII wired via a redox polymer to IO-ITO electrodes (SEM image of IO-ITO is also shown), and (b) energy diagram of electron transfer events among PSII, redox polymer and IO-ITO electrode. Reprinted from ref. 62 with permission from the Royal Society of Chemistry, copyright 2016. | ||
More recently, a further advancement was the integration of a diketopyrrolopyrrole dye (dpp) as a green light absorbing unit, integrated into inverse opal mesoporous titanium oxide (IO-TiO2) electrodes, functionalized with PSII and with the co-immobilized osmium polymer mediator (POs). In the IO-TiO2|dpp|POs–PSII photoanodes, an artificial Z-scheme takes place by simultaneous excitation of PSII and of the dpp dye; this latter is responsible for light induced electron injection into TiO2 from its excited state dpp* with formation of the oxidized dpp+, which is further restored to the neutral state by the electron chain photo-promoted by PSII, with the assistance of the POs redox mediator (Fig. 3). The photoelectrochemical process leading to water oxidation is associated with a photocurrent density of ca. 80 μA cm−2.66 The association of the photocurrent with the simultaneous excitation of the diverse chromophores was confirmed by the action spectrum response, which turns out to be consistent with the spectral overlap of PSII with dpp, peaking at 560 nm and characterized by an external quantum efficiency of 2.7%. Besides broadening the absorption spectrum of the electrode, a benefit of using the dpp dye is also an anticipation of the photoanodic current onset potential (−0.5 V vs. SHE, ca. 0.5 V more favourable than sole PSII photoelectrodes), which allows the bias-free application of IO-TiO2|dpp|POs–PSII electrodes.
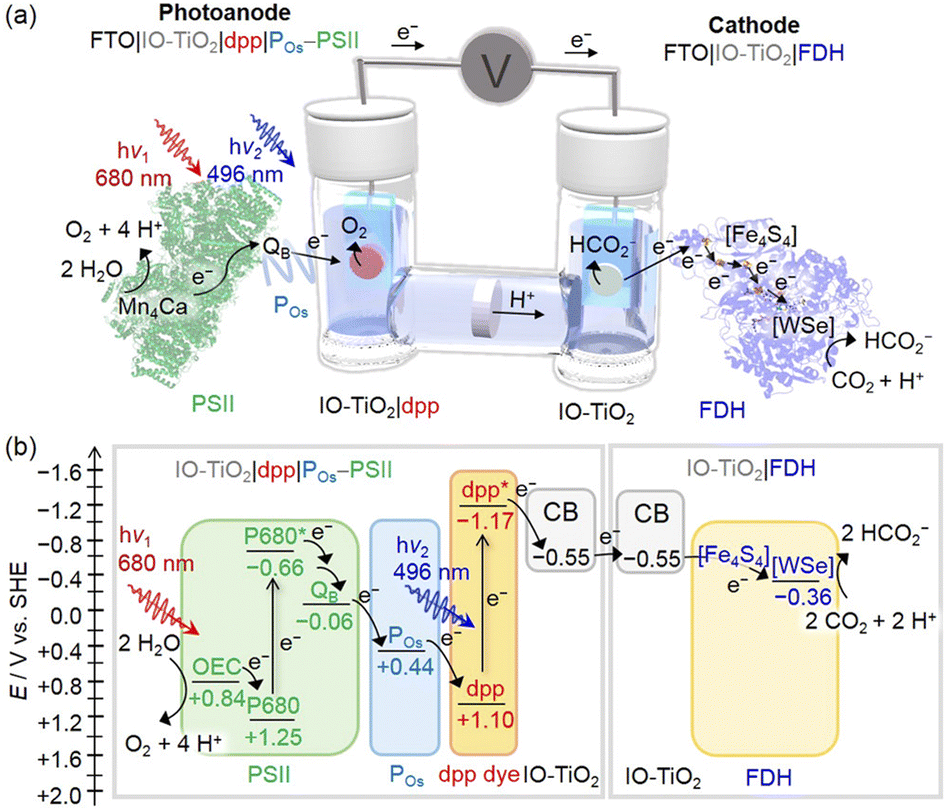 | ||
| Fig. 3 (a) Representation of the photoelectrochemical cell developed by Reisner and co-workers for bias-free water oxidation and carbon dioxide reduction to formate. The photoanode is an IO-TiO2|dpp|POs–PSII photoelectrode, while the cathode is an IO-TiO2|FDH. (b) Energy diagram. Reprinted from ref. 65 with permission from the American Chemical Society, copyright 2018. | ||
Indeed, the IO-TiO2|dpp|POs–PSII electrodes have been successfully employed in a bias-free photoelectrochemical cell combining [NiFeSe] hydrogenase66 or W-dependent formate dehydrogenase (FDH)65 photocathodes (Fig. 3 and entry 3, Table 1).
Ruthenium polypyridine photosensitizers for PEC technology applied to water oxidation
The established properties of Ru(II) polypyridine complexes as photosensitizers67 lead to an extensive investigation of this class of compounds in light driven water oxidation. The long lived triplet excited state of such species allowed their investigation for homogeneous photocatalysis, in water, in the presence of a sacrificial electron acceptor.25Moreover, ultrafast photoinduced electron injection is observed when these Ru-based sensitizers are anchored onto semiconductors, occurring in a timescale of pico-seconds (hundreds of fs to ps).‡68,69 This behaviour has been exploited to design a fully integrated photoanode for photoelectrochemical water oxidation in combination with suitable WOCs.
In 2009, the first water splitting dye-sensitized PEC (DS-PEC) was assembled by T. E. Mallouk,70 with a Ru trisbipyridine photosensitizer-IrOx NPs catalyst covalent dyad anchored onto a nanostructured TiO2 photoanode (entry 1, Table 2). This ground-breaking result was the forerunner of further technology development including many diverse and increasingly sophisticated strategies to optimize both the overall stability and efficiency of the resulting DS-PEC.71
| Entry and photoanode | Light intensity (mW cm−2) | E (V) vs. RHE and pH | J (μA cm−2) | F.E.(O2) (%) | Ref. |
|---|---|---|---|---|---|
| a 2-IrOx = benzimidazole-phenol (BIP) and 2-carboxyethylphosphonic acid (CEPA) capped Ir NPs. b Catalyst with silatrane-terminated anchoring group. c RuP and Ru(bda) are vinyl terminated. d Electrode synthesized via co-loading. e Electrode prepared by layer-by-layer deposition. f Photosensitizer functionalized with R = H. g Photosensitizer functionalized with R = Me. | |||||
| 1, TiO2|Ru(bpy)-IrOx | 450 nm, 7.77 mW cm−2 | 0.55, pH 5.75 | 30–10 (IPCE 0.9% at 450 nm) | 20 | 70 |
| 2, TiO2|3P–Ru-2-IrOxa | White light AM 1.5G, >410 nm | 0.75, pH 5.8 | 100 (2.3% internal quantum yield) | >85 | 72–74 |
| TiO2|RuP|IrO2 | 0.70, pH 6.8 | Up to 225 (IPCE up to 6.75% when integrated from 410 to 700 nm) | 98 | ||
| 3, TiO2|RuP|Co3O4 | AM 1.5G, 100 mW cm−2, λ > 400 nm | 0.90, pH 6.8 | 135 (IPCE not reported) | — | 78 |
| 4, TiO2|-RuPNa2-Ru4POM | 450 nm (LED), 33 mW cm−2 | 0.55 (pH = 5.8) | 54.8 (IPCE 0.392% at 450 nm) | 86 | 84 |
| 0.63 (pH = 7.2) | 34.2 (IPCE 0.228% at 450 nm) | 80 | |||
| 5, SnO2/TiO2|(PAA/PS-Ru)5/(PAA/RuC)5 | White light 100 mW cm−2, λ > 400 nm | 0.85, pH = 7 | 18 (IPCE not reported) | 22 | 86 |
| 6, TiO2|RuP/Ru(bda)b | White light, 300 mW cm−2, λ > 400 nm | 0.60, pH = 6.8 | 1700 (IPCE 14% at 450 nm) | 83 | 88 |
| 7, nanoSnO2|TiO2 (3 nm)|-RuP-Ru(bda)c | White light, 100 mW cm−2, λ > 400 nm | 1.05, pH = 7 | 400 (IPCE 3.75% at 430 nm) | 22 | 89 |
| 8, SnO2 (5.5 μm)/TiO2 (4.3 nm)|RuP-R2+/Ru(bda)d | 100 mW cm−2 | 0.64, pH = 5.7 | 1500f, 1440g (IPCE not reported) | 88f, 97g | 90 |
| SnO2 (5.5 μm)/TiO2 (4.3 nm)|RuP32+-Zr(IV)–Ru(bda)e | 1450 (IPCE not reported) | 74 | |||
| 9, nanoSnO2|TiO2 (3 nm)|RuP22+ ALD SnO2-1 | White light, 100 mW cm−2, λ > 400 nm | 0.7, pH = 4.65 | 800 (IPCE 17.1% at 440 nm) | 80–90 | 91 |
| 10, SnO2|TiO2|-RuP-LO-C10-Ru(bda) | White light, 100 mW cm−2, λ > 400 nm | 0.82, pH = 7 | 1400 (IPCE 24.8% at 440 nm) | 83 | 92 |
| 11, TiO2|-(RuP2+)5-Ru(bda) | White light, 100 mW cm−2, λ > 400 nm | 0.75, pH = 5.8 | 1700 (IPCE 25% at 450 nm) | 90 | 93 |
| 12, nanoITO|-MV2+-S-Fe(II)–Ru(II) | White light, 100 mW cm−2, λ > 400 nm | 0.77, pH = 4.65 | 250 (IPCE 2.3% at 440 nm) | 67 | 94 |
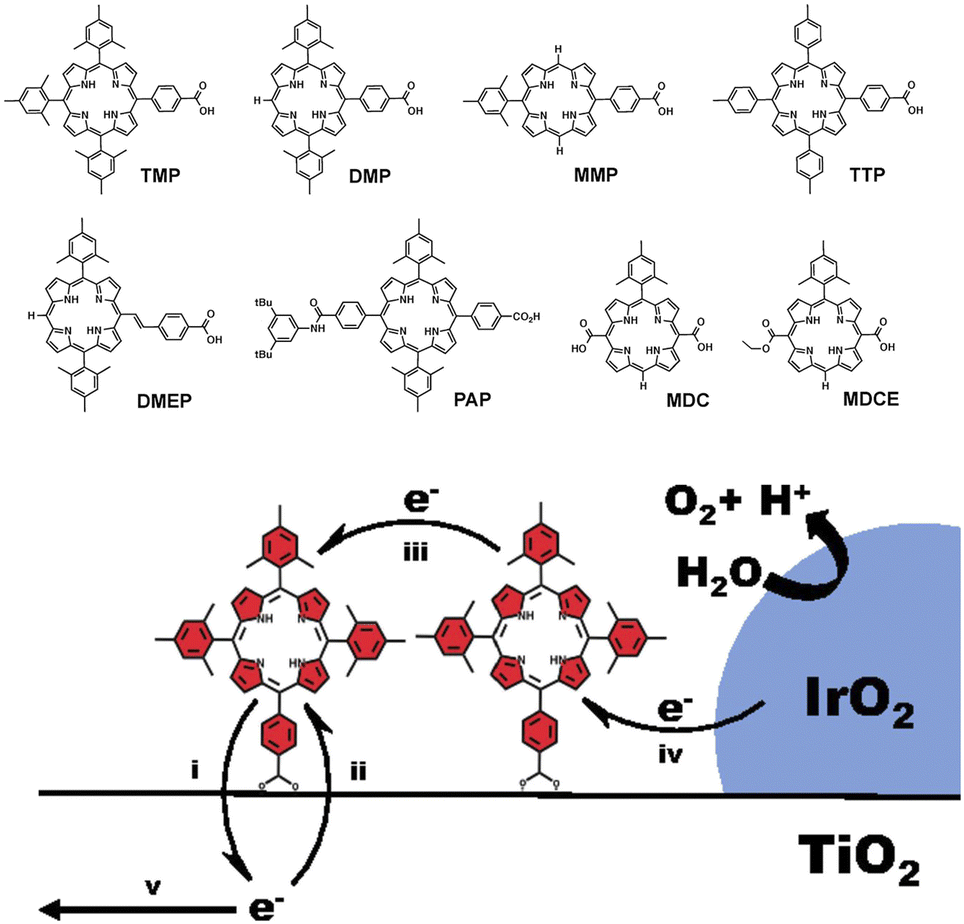
One major issue of the [Ru(bpy)3]-IrOx assembly was indeed the slow electron transfer from IrOx to the oxidized form of the Ru photosensitizer (hole scavenging), occurring in a ms timescale. The introduction of a benzimidazole-phenol based redox mediator grafted on the IrOx nanoparticles did not significantly alter the ET rate domain, although an increase of the internal quantum efficiency up to 2.3% was observed (Fig. 4: in this case both the ruthenium polypyridyl complex and the IrOx were independently anchored onto TiO2via covalent phosphonate binding, entry 2, Table 2), and the photoelectrode's resulting charge transport dynamics was indeed affected by the choice of the deposition solvent.72–74
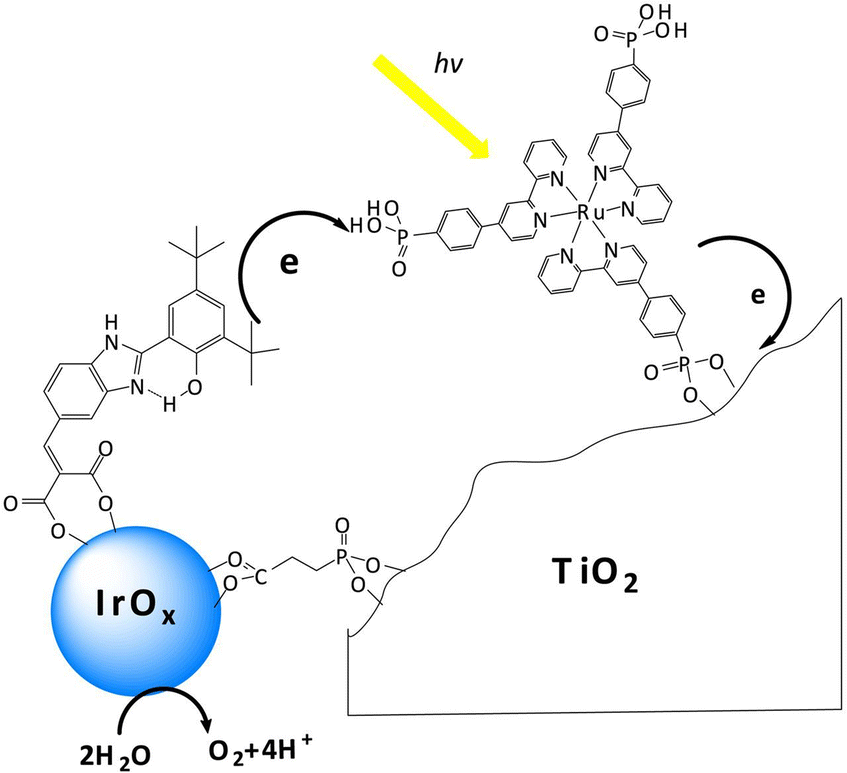 | ||
| Fig. 4 Schematic representation of the photosynthetic system proposed by Mallouk and co-workers, constituted by a Ru polypyridine photosensitizer, IrOx WOC and a benzimidazole-phenol (BIP) based redox mediator, embedded onto a TiO2 surface. Reprinted from ref. 72 with permission from the Proceedings of the National Academy of Sciences (PNAS), copyright 2012. | ||
Slow hole scavenging processes seem to be a common feature characterizing also other metal oxide nanoparticles, including very active Co3O4;75,76 in this case the transfer of one electron is accompanied by transfer of one proton (proton coupled electron transfer, PCET), and the nature of the aqueous medium and in particular the presence of bases can impact the overall PCET rate, through general base catalysis (Fig. 5).77 This pathway is likely occurring also in the photoanode developed by Na and coworkers, combining a Ru(II) trisbipyridine photosensitizer and Co3O4 nanoparticles anchored to a TiO2 semiconductor through a 3-amino-propyltriethoxysilane linker, providing photocurrent densities up to 135 μA cm−2 (entry 3, Table 2).78
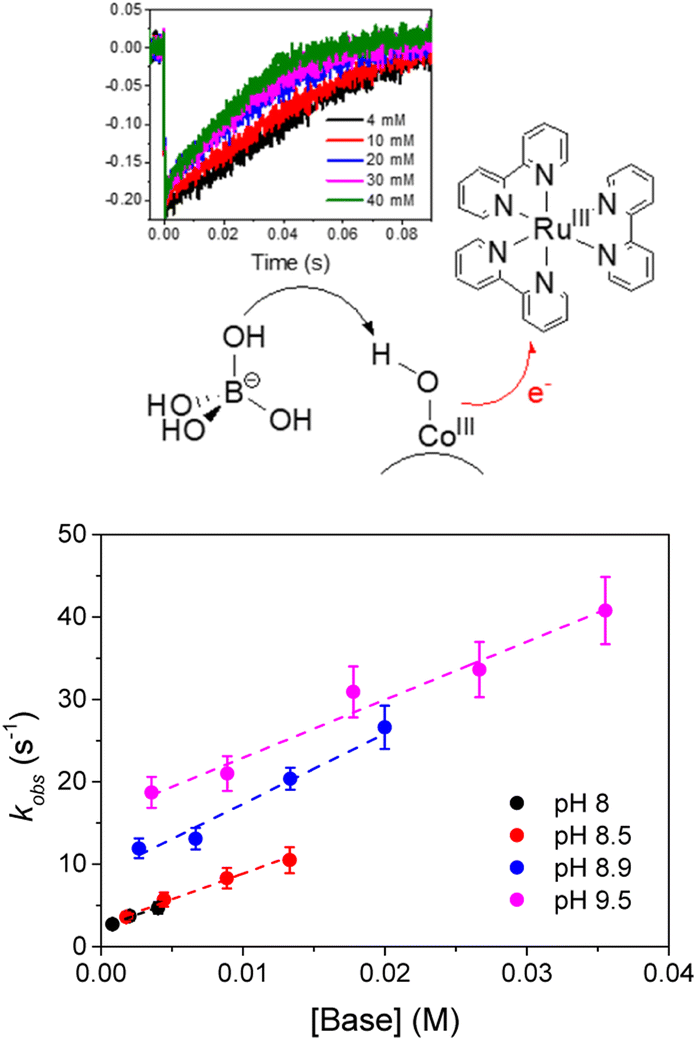 | ||
| Fig. 5 Representation of photoinduced proton coupled electron transfer occurring at the surface of Co3O4 nanoparticles WOC, where the electron is transferred to RuIII(bpy)33+ and the proton to borate; below is reported the dependence on the rate constant on the pH and on the concentration of the borate base, indicative of a general base catalysis. Reprinted from ref. 77 with permission from the Royal Society of Chemistry, copyright 2018. | ||
These results lead other groups to further investigate the dynamics of the hole scavenging process, in order to identify WOCs possibly capable of fast electron transfer.
In 2010, in collaboration with Franco Scandola and Sebastiano Campagna, we reported the application of a robust inorganic tetraruthenate Ru4POM WOC on a Ru(II) polypyridine DS-photoanode, providing evidence of fast hole-scavenging (Fig. 6).79 The Ru4POM WOC, {Ru4(μ-OH)2(μ-O)4(H2O)4(γ-SiW10O36)2}10−,79–84 features a tetraruthenate catalytic core embedded within two oxidatively stable polyoxotungstate ligands; since the four ruthenium atoms at the WOC core are connected through μ-oxo and μ-hydroxo bridges, Ru4POM is capable of undergoing consecutive one-electron oxidations through PCET within a narrow potential range, resulting in oxygen evolution at overpotentials between 200 and 300 mV, depending on the pH.85 Ru4POM readily adsorbs on TiO2 photoanodes sensitized with Ru(bpy)2(dpbpy) (dpbpy = [4,4′-(PO3H2)2bpy]) through electrostatic interaction with the positively charged dye, and exhibits very fast hole scavenging in the sub-ns timescale, as evidenced by laser flash photolysis studies.79
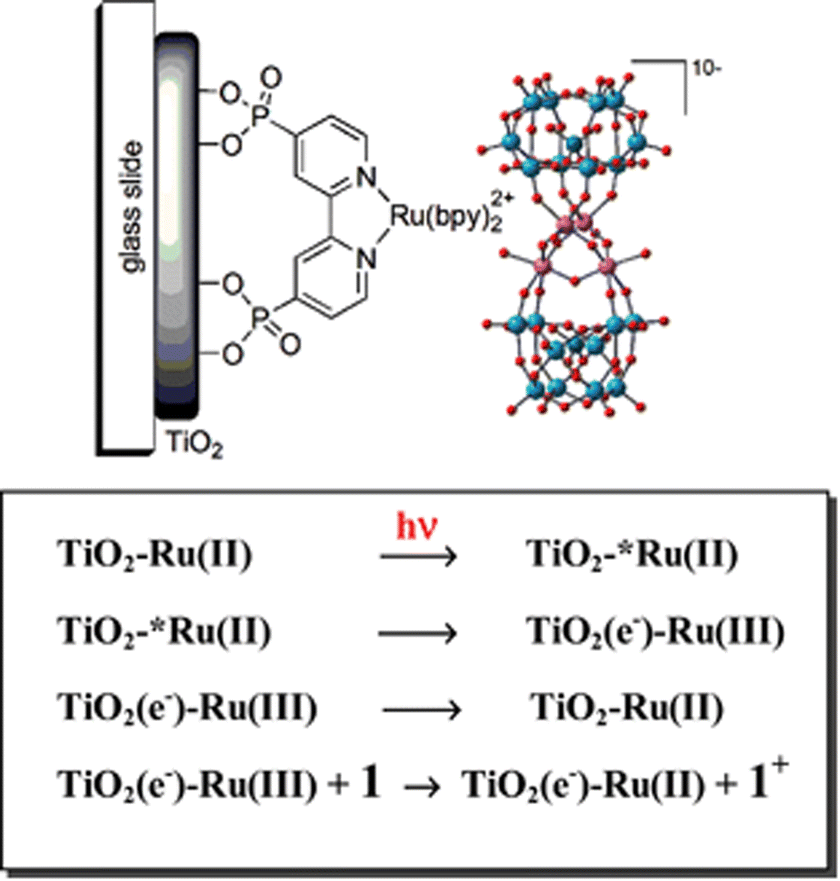 | ||
| Fig. 6 Representation of a Ru trisbipyridine sensitized TiO2 photoanode with a Ru4POM catalyst assembled by electrostatic interactions, and schematic view of the photoinduced electron transfer events (1 is the Ru4POM catalyst). Reprinted from ref. 79 with permission from the Royal Society of Chemistry, copyright 2010. | ||
A later study by Hill and co-workers84 aimed at the enhancement of the dye–catalyst coupling by functionalization of a Ru-polypyridyl dye with two crown ether moieties capable of binding Na+ or Mg2+ cations, and acting as a tweezer-like recognition group for Ru4POM; they reported an almost three-fold increase of photocurrent and APCE between the pristine and the crown ether-functionalized dye (entry 4, Table 2). The assembly of catalyst/sensitizer through complementary charge was also exploited by Meyer and Schanze, via a layer-by-layer deposition onto FTO|SnO2/TiO2 core shell structures of a cationic polystyrene-based Ru polypyridine chromophore and a [Ru(tpy)(2-pyridyl-N-methylbenzimidazole)(OH2)]2+ water oxidation catalyst codeposited with a poly(acrylic acid) polyanion; the resulting device provided limited photocurrent densities lower than 20 μA cm−2 (0.4 V vs. NHE applied bias, phosphate buffer pH 7) and a modest faradaic yield for oxygen evolution of 22% (entry 5, Table 2).86
Besides the fast electron transfer to the oxidised dye, another important feature required for the WOC is the high turnover frequency of oxygen evolution. Concerning this point, state-of-the-art catalysts are Ru(II) derivatives with the 2,2′-bipyridine-6,6′-dicarboxylate ligand (bda)87 or [2,2′:6′,2′′-terpyridine]-6,6′′-dicarboxylate (tda) ligands.22 These coordination complexes operate with turnover frequency up to 50000 s−1,22 operating through high valent Ru(IV) or Ru(V)-oxo intermediates.
In 2013, Sun and co-workers88 reported one of the currently top-performing molecular photoanodes by co-absorbing on TiO2 the ruthenium trisbipyridine dye through phosphonate linkers and [RuII(bda)] catalyst functionalized with a long insulating silatrane-terminated anchoring group (Fig. 7). Photocurrent densities as high as 1.7 mA cm−2 and 80% faradaic efficiency were obtained at pH 6.8 with a low applied bias (0.6 V vs. RHE), though under 3 sun irradiation, resulting in a 14% IPCE at the 450 nm absorption maximum (entry 6, Table 2).
 | ||
| Fig. 7 Photoanode reported by Sun and co-workers, based on the ruthenium trisbipyridine dye anchored to TiO2 through phosphonate linkers and a RuII(bda) WOC functionalized with a long insulating silatrane-terminated anchoring group. Reprinted from ref. 88 with permission from the American Chemical Society, copyright 2013. | ||
Meyer and co-workers tried then to improve the performance of this system through an engineered design.89 The RuP photosensitizer and WOC components were integrated in a covalent dyad: both photosensitizer and catalyst were functionalized with vinyl groups, and the dyad was then electro-assembled directly on the sensitized electrode, through a layer-by-layer method. Secondly, core–shell SnO2|TiO2 photoanodes were engineered by depositing a thin (3 nm) TiO2 overlayer on 8 μm thick mesoporous SnO2 film by the atomic layer deposition technique (ALD). A photocurrent density of 400 μA cm−2 was achieved at 1 V vs. RHE in pH 7 phosphate buffer, almost doubled with respect to SnO2|TiO2 (3 nm)|RuP and ten-fold higher with respect to the photocurrent registered for the same electro-assembly on TiO2 photoanodes, confirming the successful effect of the core–shell design in limiting back-electron transfer. However, a marked instability of the electro-assembly was evidenced by the low faradaic efficiency for oxygen evolution of 22% (entry 7, Table 2).
An alternative approach to synthetically demanding PS/WOC covalent binding was based on the use of Zr(IV) linkers, exploiting phosphonate binders both at the PS and WOC (Fig. 8).90 The PS/WOC layer-by-layer zirconate assembly was compared to the co-loaded approach (Fig. 8) on SnO2/TiO2 core–shell electrodes under 1 sun illumination in pH 5.7, 0.1 M acetate buffer solutions. Under the optimized conditions, the two approaches displayed similar performance, reaching photocurrent densities of 1.44 and 1.45 mA cm−2 for the co-loaded and LBL assembly, respectively, associated with faradaic efficiencies for O2 evolution of 97 and 74% for the co-loaded and LBL assembly, respectively. An advantage of the Zr(IV) based assembly appeared to be the stability under operating conditions, for up to 20 minutes (entry 8, Table 2).
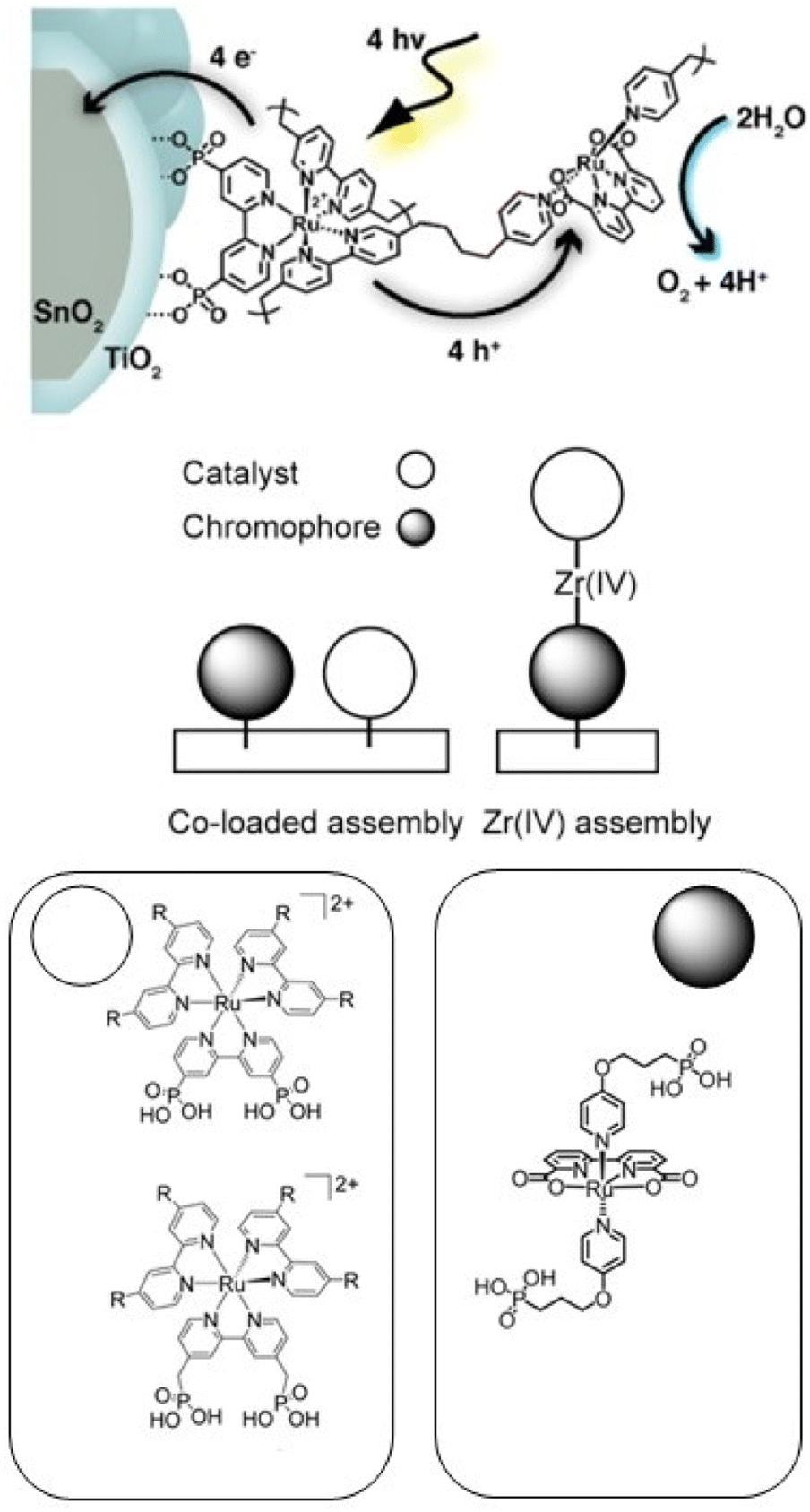 | ||
| Fig. 8 Top: Covalent approach to anchor the Ru trisbipyridine photosensitizer and Ru(bda) WOC. Reprinted from ref. 89 with permission from the American Chemical Society, copyright 2015. Bottom: Co-loaded and Zr(IV) assembly approaches. Adapted from ref. 90 with permission from the American Chemical Society, copyright 2016. | ||
The layer-by-layer strategy for cation mediated self-assembly of the photosensitizer and catalyst was further tuned in 2017, employing an atomic layer deposition for derivatizing phosphonate linkers in the gas phase, embedding Al(III), Ti(IV), Zr(IV) or Sn(IV) ions.91 The same core–shell SnO2|TiO2 (3 nm) photoanodes were used, and the best performance was achieved by the photoanode with the SnOx-bridged dyad, delivering ca. 800 μA cm−2 at pH 4.6 with an applied bias of 0.7 V vs. RHE (1 sun, λ > 400 nm), with 90% faradaic yield and 17% IPCE at 440 nm (entry 9, Table 2). The same approach was also extended to embed a nickel hydrogen evolving catalyst.
Nevertheless, Sun, Meyer and co-workers recently achieved record-breaking efficiencies upon improvement of the former co-loaded design.92,93 In the first case,92 a simpler synthetic strategy was exploited for the covalent anchoring of the Ru(bda) WOC: in particular, the MOx surface was pre-functionalized by anchoring through a phosphonate bridge a long pyridine-terminated alkyl linker (LO-C10) that was exploited to coordinate and anchor the WOC (Fig. 9, top). In pH 7 phosphate buffer, the resulting SnO2|TiO2|-RuP-LO-C10-Ru(bda) photoanodes achieved a photocurrent of 1.4 mA cm−2 under an applied bias of 0.8 V vs. RHE, with 83% faradaic efficiency and a 24.8% IPCE at 440 nm.
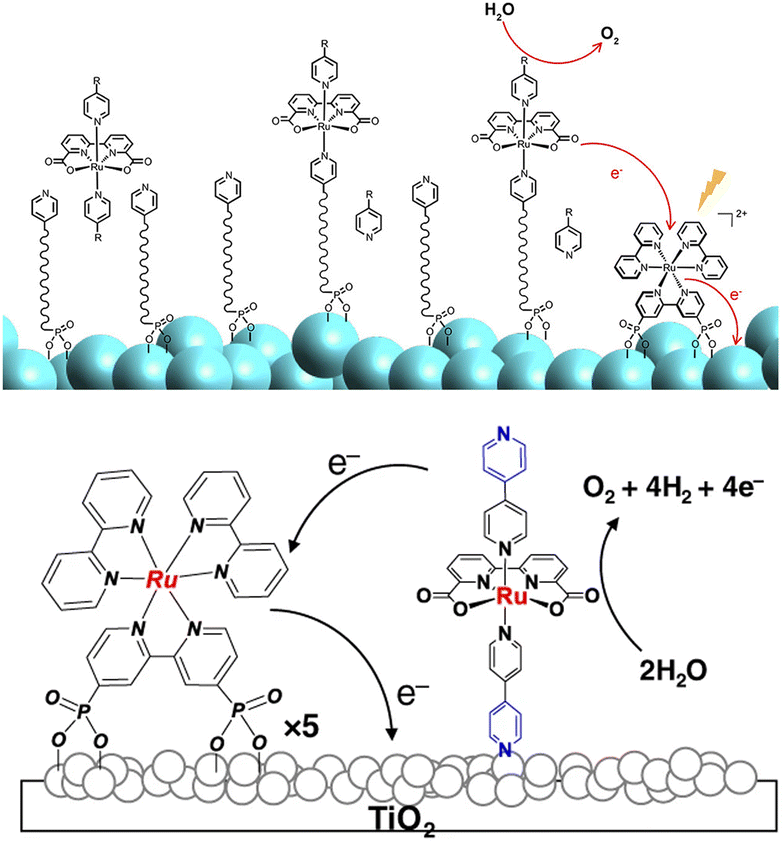 | ||
| Fig. 9 Top: Coordination of a Ru(bda) WOC at MOx surfaces exploiting a long pyridine-terminated alkyl linker; the Ru trisbipyridine photosensitizer is anchored through phosphonate linkers at the bpy. Bottom: Immobilization of a Ru(bda) WOC on TiO2 through a pyridine based anchoring group. Reprinted from ref. 92 with permission from the American Chemical Society, copyright 2018 and from ref. 93 with permission from the Nature Publishing Group, copyright 2020. | ||
The hydrophobic alkyl chain showed a protecting effect towards hydrolysis of the phosphonate bridges of both the Ru photosensitizer and Ru(bda) catalyst (entry 10, Table 2). Recently, superior performances and increased stability were found when employing an unprecedented pyridine anchor for the immobilization of Ru(bda) (Fig. 9 bottom):93 the pyridyl-derivatized Ru(bda) displayed an increased hydrolytic stability at near neutral pH; upon stepwise loading of RuP and Ru(bda) on TiO2, a 5:1 chromophore to catalyst ratio was obtained, and the resulting photoanodes achieved a photocurrent of 1.7 mA cm−2 under an applied bias of 0.55 V vs. RHE at pH 5.8, with a faradaic efficiency over 90% for 2 hours and a record-breaking IPCE of 25% at 440 nm (entry 11, Table 2).To conclude this section, it is worth mentioning the promising performances obtained by a truly biomimetic molecular photoanode, recently published by Meyer and co-workers:94 the photoanode is based on a molecular assembly which can autonomously achieve photoinduced charge separation, supported on mesoporous ITO, similarly to the hybrid PSII photoelectrodes. In close parallelism with the main components of PSII, the photoanode (Fig. 10) is structured as follows: the mesoITO collector plays a role as acceptor of the plastoquinone QA; a methyl viologen (MV2+) mediator, anchored to ITO through a phosphonate bridge, acts as the pheophytin primary electron acceptor; a [Ru(bpy)(dpbpy)2] sensitizer (S) acts as the primary donor P680, transferring the photoexcited electron to MV2+; an Fe(II)-terpyridine (Fe) acts as the tyrosine mediator, regenerating the sensitizer and slowly withdrawing electrons from a [RuII(bda)] WOC (Ru). Noteworthily, the assembly is easily generated on the ITO surface through a layer-by-layer method, exploiting the methodology of Zr(IV)-phosphonate bridges. The introduction of MV2+ significantly hinders back electron-transfer from ITO to the oxidized sensitizer or WOC: indeed, ITO|MV-S-Ru shows an 8-fold increase in photocurrent with respect to ITO|-S-Ru. A maximum photocurrent of ca. 250 μA cm−2 was obtained by the complete ITO|-MV-S-Fe-Ru photoanode at 0.77 V vs. RHE in pH 4.65 buffer (AM 1.5G light, λ > 400 nm), coupled with a 67% faradaic efficiency and a 2.3% IPCE at 440 nm (entry 12, Table 2).94
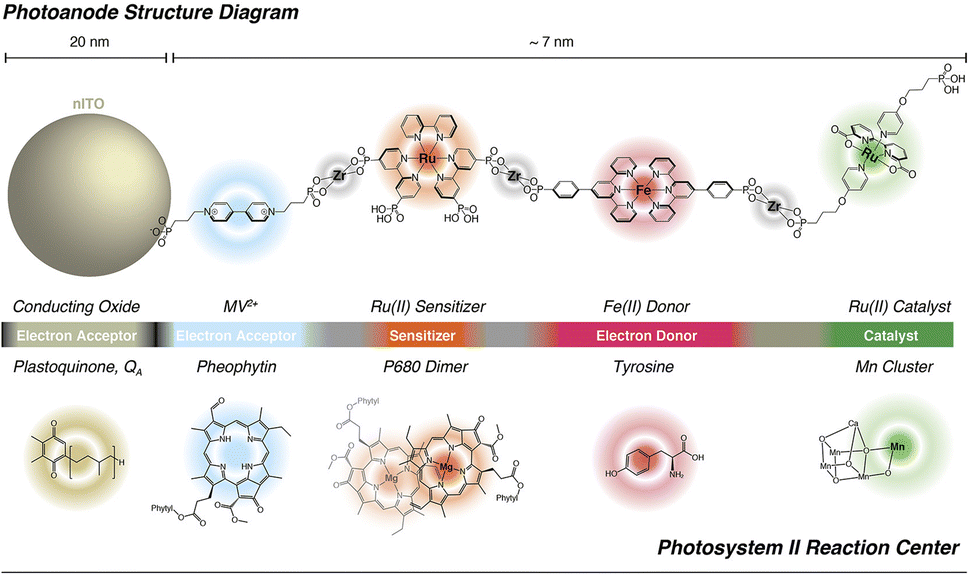 | ||
| Fig. 10 The bioinspired assembly proposed by Meyer and co-workers, exploiting MV and Fe(II) redox mediators combined with a Ru trisbipyridine photosensitizer and a Ru(bda) WOC, mimicking the natural architecture in PSII. Reprinted from ref. 94 with permission from the American Chemical Society, copyright 2019. | ||
The potential of totally organic photosensitizers
Despite the unique photophysical properties which make ruthenium trisbipyridine derivatives the most investigated family of sensitizers, this class of dyes is definitely not suitable for a sustainable DS-PEC technology, due to the high cost and narrow blue-centered absorption range: moreover, the photogenerated Ru(III)bpy derivatives are susceptible to attack by water and buffer anions, leading to irreversible degradation under photocatalytic conditions.47 However, SC-based photoanodes where electron injection occurs from the photosensitizer excited state in the ultrafast (fs-to-ps) regime can work with totally organic photosensitizers displaying short-lived singlet excited states. This behaviour shows a marked difference from homogeneous systems, where long lived excited states are typically required to promote diffusion-controlled electron transfer events.Indeed, the potential of dye chemistry for photosynthetic processes has been recently demonstrated in several case-studies. Moreover, the complex supramolecular systems that can originate from the dye self-assembly in solution and on electrodic surfaces open new possibilities in terms of the electrochemical and photochemical properties, since these can be dependent on the molecular aggregation state.24 These aspects will be highlighted for representative classes of organic photosensitizers that are receiving a great deal of attention for PEC technology.
Bioinspired photosensitizers: the class of porphyrinoid chromophores
Taking direct inspiration from biological photosystems and building on recent progress in the dye-sensitized solar cells (DSSC) research,95 porphyrinoid chromophores have been considered for PEC technology applied to artificial photosynthesis. Brudvig and co-workers reported in 2011 the first example of a DS-PEC displaying a fluorinated Zn-porphyrin with a high oxidation potential (ZnPor, 1.35 V vs. NHE for the first reversible one-electron oxidation) in combination with a molecular Cp*Ir precatalyst (Cp* = pentamethylcyclopentadienyl). Both photosensitizer and Ir-based WOC were functionalized with terminal carboxylates in order to be covalently anchored onto TiO2 (Fig. 11, top).96 In this first case-study, the photocurrent density reached up to 30 μA cm−2 albeit no oxygen measurement was performed (entry 1, Table 3).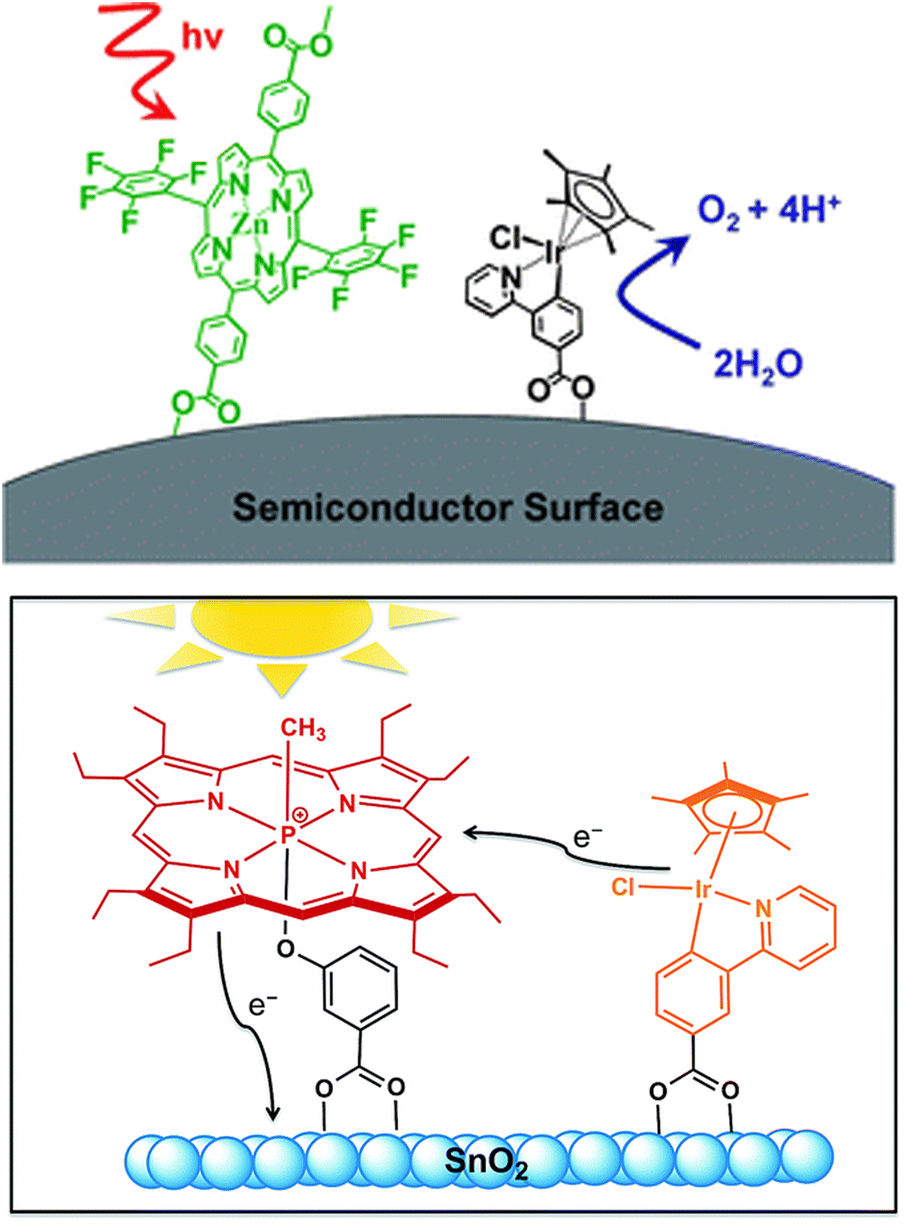 | ||
| Fig. 11 Pioneer examples of porphyrinoid derivatives coupled to Ir precatalysts. Reprinted from ref. 96 and 97 with permission from the Royal Society of Chemistry copyright 2011 and 2015. | ||
| Entry and photoanode | Light intensity (mW cm−2) | E (V) vs. RHE and pH | J (μA cm−2) | F.E. (O2) (%) | Ref. |
|---|---|---|---|---|---|
| (1) TiO2|ZnPor-Cp*Ir | White light, 200 mW cm−2, λ > 400 nm | 0.91, pH = 7 | 30 (IPCE not reported) | — | 96 |
| (2) TiO2|PPor/CpIr | White light, 200 mW cm−2, λ > 400 nm | 0.91, pH = 7 | 20–30 (IPCE not reported) | — | 97 |
| (3) TiO2–IrO2/Por | λ > 410 nm (power not reported) | 0.70, pH = 6.8 | ≈50 (IPCE 0.014–0.032% integrating the photon flux from 410 nm to 700 nm) | 100 ± 2 | 98 |
| (4) SnO2|P/Ir | White light, ≈100 mW cm−2, λ > 420 nm | 1.15, pH = 6 | 50 for the 2:1 material |
80 ± 10 | 101 |
| 60 for the 8:1 material (IPCE 0.9% at 520 nm) |
|||||
| (5) SnO2/TiO2/ZnTPPF20-CN/IrWOC | White light, 100 mW cm−2, λ > 450 nm | 1, pH = 1 | 100 (IPCE not reported) | >95 | 102 |
| (6) TiO2|ZnP–Ru | Halogen lamp 100 W, 35 mW cm−2, λ > 380 nm | 0.24, pH = 7.3 | 100 (IPCE 17 ± 1% at 424 nm) | 33 | 104 |
| (7) TiO2/SP-Ru(bda) | White light, 100 mW cm−2, λ > 420 nm | 0.63, pH = 7.3 | 60 (IPCE 6% at 400 nm) | 64 | 105 |
| (8) TiO2/BODIPY-Ru(bda) | White light, 200 mW cm−2, λ > 400 nm | 0.55, pH = 5.8 | 80 (IPCE 4% in the 400–700 nm range) | 77 | 106 |
In another pioneering work, van der Est and co-workers exploited highly oxidizing P(V) porphyrin sensitizers (oxidation potential in the range 1.62–1.65 V vs. NHE), bound to an SnO2 semiconductor through benzoate-like pendants in the axial coordination of the hexavalent P(V) center (Fig. 11, bottom) so as to avoid aggregation of the porphyrin dyes on the electrode surface.97 By combining time-resolved tetrahertz spectroscopy and electron paramagnetic resonance measurements, the authors demonstrated the occurrence of electron injection into SnO2 from the S1 and S2 excited states of the P-porphyrins in a 3–30 ps timescale and generated the oxidized radical cation, PPor˙+, followed by electron transfer to PPor˙+ from an iridium(III) phenylpyridine derivative co-deposited as a precatalyst onto the SnO2 surface. This was supported by the abatement of a photoinduced EPR signal at g = 2 attributed to the oxidized PPor˙+ when the Ir-species was co-anchored onto SnO2, while a new rhombic EPR spectrum arises (g-values g1 = 2.57, g2 = 2.09 and g3 = 1.83), attributable to an Ir(IV) species. For this system, photocurrent densities of the order of 20–30 μA cm−2 were registered, although oxygen measurement and quantification was not reported (entry 2, Table 3). Mallouk and co-workers considered free base porphyrins for sensitizing mesoporous TiO2 (Fig. 12).98 Typically, free base porphyrins are ca. 200 mV more oxidizing than the corresponding Zn(II) counterparts.99 This property enables access to a suitable oxidizing porphyrin radical cation from the free base systems even in the absence of electron-withdrawing groups: in particular in this study a potential in the range 1.02–1.29 V vs. Ag/AgCl was measured in a series of seven free-porphyrins chemisorbed to TiO2 through benzoic acid linkers.98 These dye sensitized photoelectrodes generated photocurrent densities of ca. 50 μA cm−2 with quantitative faradaic yield for oxygen evolution, when combined with a co-deposited IrOx catalyst (entry 3, Table 3).
 | ||
| Fig. 12 Free base porphyrins considered by Mallouk and co-workers for sensitization of TiO2, in combination with an Ir oxide WOC. Reprinted from ref. 98 with permission from the Proceedings of the National Academy of Sciences (PNAS), copyright 2015. | ||
Interestingly, porphyrin and Ru-polypyridine sensitizers supported on photoelectrodes have been compared in terms of electron injection, hole transport and charge recombination behaviour. Electron injection from a porphyrin-type dye is 3–10 times less efficient with respect to the performance of Ru-polypyridine analogs, as determined by the resulting photon-to-current efficiency (APCE). Concerning the hole transport, and considering the cross-surface electron diffusion coefficients, Dapp, Ru-polypyridine sensitizers outperform porphyrins by two-orders of magnitude (Dapp = 10−9–10−10 cm2 s−1 and 10−11 cm2 s−1 respectively for Ru-polypyridine and porphyrin-type dyes). Besides these negative aspects, porphyrin sensitized electrodes generally display a more favourable, slower back electron transfer recombination with respect to the Ru-polypyridine analogs.98 Progress in this field by Brudvig and co-workers includes a redesign of porphyrin sensitized photoelectrodes by addressing various aspects: (i) a –CF3 meso-substituted free-base tetraphenyl porphyrin to increase the sensitizer stability against photo-oxidation; (ii) the installation of the oxidatively robust 2-(2′-pyridyl)-2-propanoate (pyalc) ligand on the Cp*Ir WOC, further stabilizing the high-valent Ir states by electron-donating effects;100 (iii) the engineering of the covalent anchoring of both the porphyrin sensitizer and the Ir WOC, using a conjugated hydroxamic acid-terminated anchor to increase the electronic coupling of the dye with the SnO2 surface, while a long insulating silatrane anchor was used for the Cp*Ir(pyalc) WOC in order to favour hole accumulation, by decreasing the probability of unproductive back-electron transfer; (iv) the tuning of the dye/WOC ratio to maximize the rate of hole accumulation at the WOC. Improved photocurrents up to 50–60 μA cm−2 and faradaic yield up to 80% were achieved by the 2:1 and 8:1 SnO2|ZnPor|Cp*Ir(pyalk) photoanodes at 1.16 V vs. RHE; however, recombination and long-term photodegradation remain one major issue (entry 4, Table 3).101
In a recent work by Tessore and co-workers,102 perfluorinated ZnPor dyes bearing a conjugated electron-acceptor linker, terminating with a cyanoacrylic anchor (ZnPor-CN), were combined with the bridged μ-oxo dimer of Ir(pyalc) (Ir-blue catalyst):103 the two components were co-loaded on SnO2|TiO2 photoanodes, which were engineered both to reduce recombination and to favour the dye absorption. This system achieved a photocurrent density of 100 μA cm−2 and 95% faradaic yield in pH 1 electrolyte at 1.0 V vs. RHE: however, transient absorption spectroscopy evidenced an incomplete recovery of the ground state of the dye, due to limited electronic communication between the dye and the Ir-WOC (entry 5, Table 3).
In 2016 the groups of Sun and Imahori reported a TiO2 photoanode sensitized by a photocatalytic covalent dyad, formed by a pegylated zinc porphyrin (ZnP) connected to a Ru(bda) WOC through a π-conjugated linker (Fig. 13).104
 | ||
| Fig. 13 A Zn-porphyrin/Ru(bda) photosensitizer/WOC dyad covalently bound to TiO2. Reprinted from ref. 104 with permission from the Royal Society of Chemistry, copyright 2016. We thank Prof. Hiroshi Imahori for providing the figure in high resolution. | ||
Photocurrents higher than 100 μA cm−2 and outstanding IPCE of 17% were achieved at neutral pH with just 0.24 V vs. RHE applied bias at λ > 380 nm (which does not exclude direct excitation of the TiO2). However, a low faradaic efficiency for oxygen evolution of 33% was reported in contrast with the high IPCE record value, thus suggesting that a competitive oxidative degradation of the ancillary PEG moiety was occurring (entry 6, Table 3).
Interestingly, the same authors have reported a novel push–pull dye resulting from a sub-porphyrin (SubPor) dye, decorated with two triphenylamine electron-donating groups while a carboxyphenyl electron-acceptor terminal serves as an anchoring group to the semiconductor (Fig. 14, top). Indeed, the molecular LUMO turns out to be conveniently localized on the carboxyphenyl terminal unit, thus favouring electron injection into the semiconductor, while the HOMO is located on the triphenylamine moiety, far from the semiconductor surface, thus preventing back recombination (Fig. 14, top). When co-anchored on TiO2 with the Ru(bda) WOC, at neutral pH with an applied bias of 0.6 V vs. RHE, the resulting photoanodes showed photocurrents of 60 μA cm−2, 6% IPCE at 400 nm and 2% at 520 nm, though a 64% faradaic yield for oxygen evolution was obtained due to the concomitant degradation of the dye (entry 7, Table 3).105
 | ||
| Fig. 14 Photosynthetic systems combining a Ru(bda) WOC and a subporphyrin (top) or BODIPY (bottom) photosensitizer, anchored onto a TiO2 semiconductor. Reprinted from ref. 105 and 106 with permission from the Royal Society of Chemistry, copyright 2016 and 2017. | ||
A somehow related boron-dibenzopyrromethene dye (BODIPY) was later applied for the first time to DSPECs by Kubo and co-workers.106 The π-conjugated skeleton of BODIPYs can be considered as a porphyrinoid fragment, displaying similar photophysical properties, i.e. broad absorption in the visible reaching the near IR regions (400–500 nm and 600–750 nm), with molar extinction coefficients as high as 105 M−1 cm−1, and showing an excellent photostability. The Kubo group specifically aimed at the development of a water oxidizing DSPEC exploiting low-energy radiation, and therefore successfully synthesized a π-extended BODIPY absorbing up to 800 nm, with a strong absorption maximum at ca. 690 nm (ε = 1.09 × 105 M−1 cm−1). Upon co-anchoring with Ru(bda) on TiO2 with a 5:1 ratio (Fig. 14 bottom), the cell achieved anodic photocurrents of 80 μA cm−2 in neutral medium at 0.6 V vs. RHE, coupled with a 77% and 66% faradaic yield for oxygen and hydrogen, respectively, with an IPCE at 400 nm up to 4%, extending up to 700 nm (entry 8, Table 3).
Perylene bisimide, when aggregation matters
Perylene bisimide (PBI) dyes are characterized by strong and broad absorption in the visible region (400–700 nm) due to π–π* transitions, with easily tunable HOMO and LUMO levels, and are conveniently obtained via low-cost synthetic protocols, featuring high thermal and oxidative stability.107 In addition, PBIs show a vast supramolecular chemistry, arising from directional π-stacking interactions (H- and J-aggregates) that regulate the collective optical and photophysical properties of these multichromophoric supramolecular polymers.Another important feature of PBI dyes is that the energy of the singlet excited state S1 (optical band gap, estimated from the intersection of normalized absorption and emission spectra) matches the electrochemical band gap (estimated from the difference of the oxidation and reduction potentials). This means that an excited state in aggregates (consider a PBI*|PBI dimer for simplicity) can evolve almost isoergonically into a radical ion pair state PBI+|PBI−, through a symmetry-breaking charge separation event (SBCS).108–110 The SBCS typically occurs in a ps timescale, being competitive to other relaxation pathways, and more energetically favourable in high polar solvents. These characteristics make PBI-based materials excellent n-type organic semiconductors (OSC) with high electron affinity and mobility. For these reasons PBI-based OSCs have been exploited in the field of organic field-effect transistors and as electron acceptor layers in organic photovoltaics.
Noteworthily, a bis-phosphonate PBI sensitizer was employed by Finke in combination with cobalt oxide/phosphate catalyst Co-Pi for the fabrication of an oxygen evolving photoanode (Fig. 15).111 In particular, a thin film of bis(phosphonomethyl)PBI (PMPBI) was spin-coated onto an ITO substrate, and the Co-Pi catalyst was then photoelectrochemically grown on the PBI film. Thanks to the strong phosphonate bridges installing the PBI-thin film between the ITO substrate and Co-Pi and to the optimal film thickness (50 nm, i.e. lower than the exciton diffusion length), the ITO|PMPBI|Co-Pi photoanode exhibited photocurrents as high as 150 μA cm−2 and faradaic efficiency for oxygen evolution of ca. 80% at 1.6 V vs. RHE despite a light harvesting efficiency (LHE) as low as 12% (entry 1, Table 4). Attempts to increase the efficiency of PMPBI-based photoanodes112 aimed at substituting the flat ITO with a high surface area SnO2 substrate. SnO2 also has a lower energy conduction band with respect to other n-type semiconductors, thus making the electron injection from the excited state of the dye more energetically favourable. However, despite a nearly quantitative LHE, SnO2|PMPBI|Co-Pi photoanodes systematically showed lower photocurrent densities in the order of 20 μA cm−2 at 0.86 V vs. RHE in pH 7 phosphate buffer, and a low faradaic yield for oxygen evolution of ca. 30% (entry 2, Table 4). These results were ascribed to a so-called “anti-catalyst” effect of the cobalt oxide, due to major recombination issues and likely dependent on adventitious carbon impurities in the SnO2 material, originating from the organic precursor employed in the synthesis.113
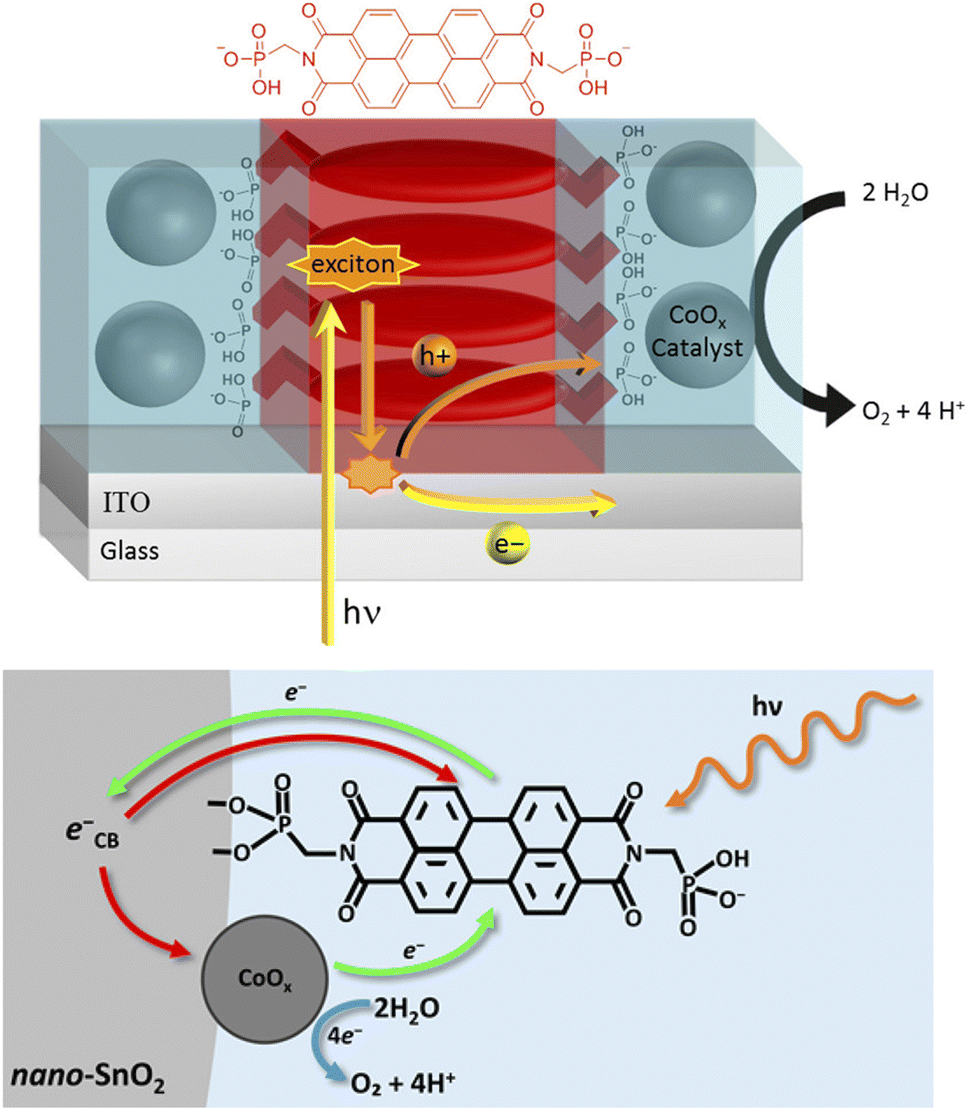 | ||
| Fig. 15 Perylene bisimide (PBI) photosensitizers combined with a cobalt oxide WOC onto ITO or mesoporous SnO2, developed by Finke and co-workers. Reprinted from ref. 111 and 112 with permission from the American Chemical Society, copyright 2017 and 2022. | ||
| Entry and photoanode | Light intensity (mW cm−2) | E (V) vs. RHE and pH | J (μA cm−2) | F.E.(O2) (%) | Ref. |
|---|---|---|---|---|---|
| 1, ITO|PMPDI|CoOx | 100 mW cm−2, λ = 315–700 nm | 1.62, pH = 7 | 150 (IPCE 0.12% at 480 nm) | 80 ± 15 | 111 |
| 2, SnO2|PMPDI|CoOx | 100 mW cm−2, λ = 400–700 nm | 0.86, pH = 7 | 20 (IPCE not reported) | 31 ± 7 | 112 |
| 3, WO3|PBI2+-IrO2 | AM 1.5G light, λ > 435 nm | 0.93, pH = 3 | 70 (IPCE 0.9% at 450 nm) | — | 114 |
| 4, nanoWO3|{[PBI]5Ru4POM}n | AM 1.5G, 100 mW cm−2, λ > 450 nm | 0.91, pH = 3 | 44 (IPCE 0.5% at 490 nm) | >97 | 117 |
| 5, IO-ITO|QS | AM 1.5G, 850 mW cm−2, λ > 450 nm | 1.52, pH = 7 | 290 ± 40 | >95 | 39 |
| IO-ITO|QS-TEGlock | 370 ± 30 (IPCE 1.2% at 500 nm) |
The first application of PBI-based non-covalent aggregates deposited onto conventional DS-photoanodes was reported by Prato, Caramori and Bignozzi114 using the dicationic [N,N′-bis(2-(trimethyl-ammonium)-ethylene)-3,4,9,10-perylenebisimide] (PBI2+) displaying a favourable electrostatic adhesion on the negatively charged metal oxide surface (Fig. 16). In fact, this approach was effective with mesoporous nanocrystalline films of WO3, SnO2 and TiO2, in combination with IrO2 nanoparticles as the water oxidation catalyst. According to transient absorption spectroscopy experiments electron injection was most effective on WO3, due to the higher driving force for injection and to the enhanced electronic coupling with the dye, determined by the low isoelectric point of WO3. The combined use of a PBI-based photogenerated oxidant with high oxidation potential (1.9 V vs. NHE) suitable for water oxidation and the co-deposition of IrO2 NPs resulted in a six-fold enhancement of the photocurrent density in 0.1 M NaClO4 pH 3 (AM 1.5G light, λ > 435 nm), reaching 70 μA cm−2 at 0.93 V vs. RHE (entry 3, Table 4).
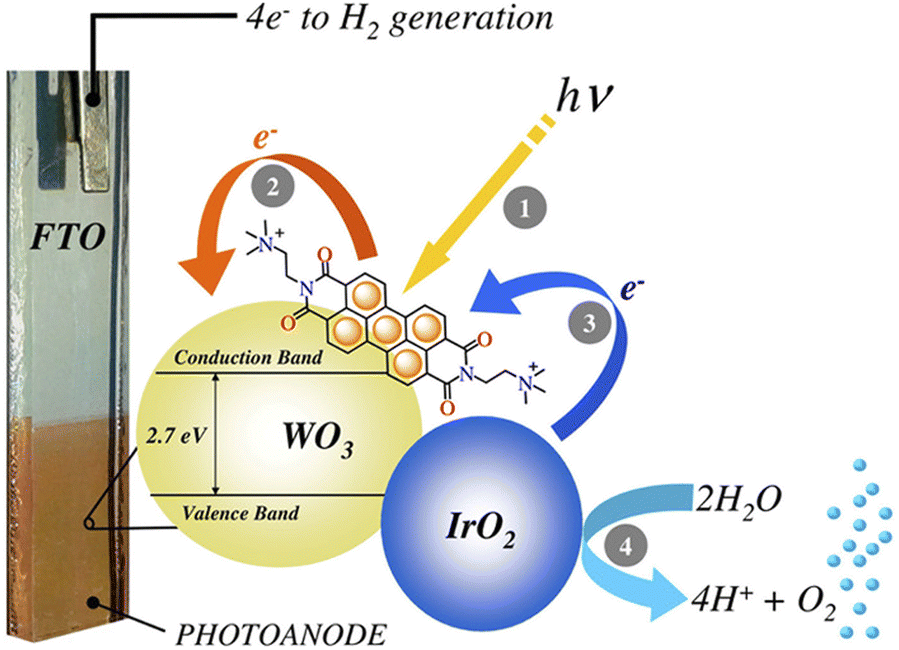 | ||
| Fig. 16 A cationic perylene bisimide derivative, PBI2+, absorbed on a nanostructured WO3 semiconductor and coupled to an IrOx nanoparticle WOC, developed by Prato, Caramori and Bignozzi. Reprinted from ref. 114 with permission from the American Chemical Society, copyright 2015. | ||
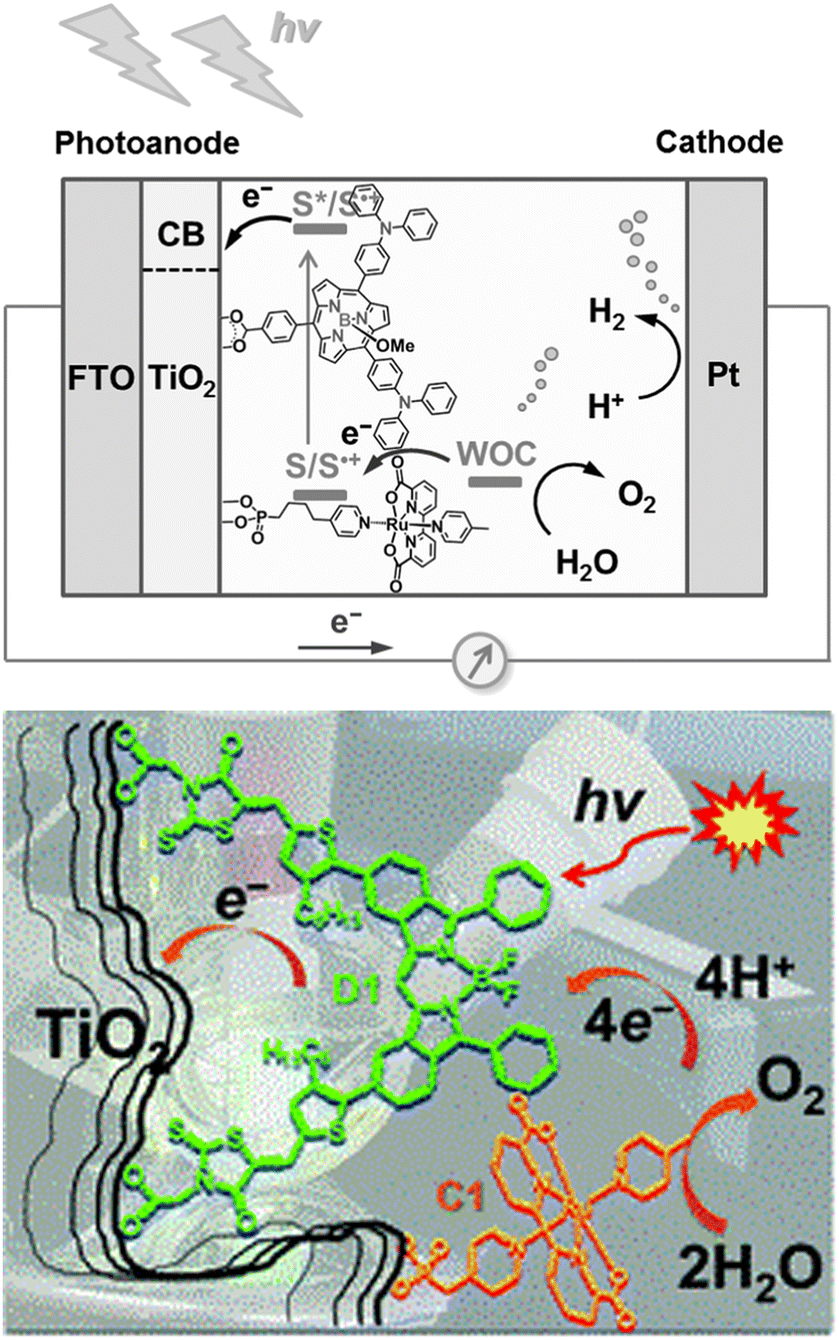
Building on the supramolecular chemistry of the bis-cationic PBI, in 2019, we designed a novel approach towards artificial photosystems resulting in the synthesis and characterization of the first PSII-inspired artificial “quantasome” evolving oxygen under visible light irradiation. The quantasome concept, firstly reported in 1932 and then reprised in 1964, describes the minimal photosynthetic unit responsible for ‘quantum’ solar energy conversion in natural photosystems (PSI and PSII), as an integrated “body” composed of light-harvesting antennas, reaction centers and catalytic cofactors.115,116 Following natural guidelines, the artificial quantasome is designed as a supramolecular photosynthetic material in which a dicationic perylene bisimide derivative (PBI2+) is shaped to a core–shell architecture embedding a tetraruthenium polyoxometalate (Ru4POM) as the oxygen evolving center. These two organic–inorganic molecular building blocks self-assemble in water, through complementary electrostatic interactions, with a definite charge-balanced stoichiometry of 5:1 (Fig. 17).
 | ||
| Fig. 17 Self-assembly in water of artificial quantasomes (QS) from a dicationic perylene bisimide PBI2+ and a decaanionic Ru4POM WOC. Reprinted from ref. 117 with permission from the Nature Publishing Group, copyright 2019. | ||
The supramolecular assembly consists of a POM-encapsulated structure in which the inorganic polyanion templates a corolla-like arrangement of five surrounding PBIs, yielding a [PBI2+]5Ru4POM unit that evolves oxygen with a multi-photon/electron/proton mechanism118 thus behaving as a natural quantasome (Fig. 17). Moreover, the core–shell organic–inorganic amphiphiles undergo hierarchical aggregation in water forming 2D lamellae nano-stacks with a striking resemblance to the appressed chloroplast membranes. When wired to a tungsten oxide substrate (nanoWO3|{[PBI2+]5Ru4POM}n), the photoanodic device reported a quantitative faradaic yield for oxygen production (>97%, λ > 455 nm), with photocurrents up to 44 μA cm−2 at 0.91 V vs. RHE at pH 3 thus anticipating the thermodynamic barrier of 1.23 V vs. RHE, incident photon to current efficiency of IPCE = 0.5% and APCE = 1.30% (at λ = 490 nm) that exceeds by 1 order of magnitude the nanoTiO2|[Ru(dpbpy)(L1)2]|Ru4POM photoanode using Ru(bpy) as sensitizer (entry 4, Table 4).117 A second-generation improved quantasome structure was reported in 2022, by installing cross-linked hydrophilic tetraethylene glycol chains (TEG) on the PBI terminals via click-chemistry (Fig. 18).39 This modification is inspired by the natural water channels in PSII119 while yielding an inter-locked assembly of the quantasome units with improved water access and reinforced proximity of the photocatalytic units, paired by the TEG cross-linkers (QS-TEGlock, Fig. 18). PSII pairing in the natural membrane, being reinforced by the overlap of peripheral antennas, is instrumental to its oxygenic efficiency.120 Along these lines, the second-generation QS-TEGlock performs as a supramolecular photosynthetic material with increased water solvation properties, controlled colloidal growth (particles with diameters of ca. 20 nm, estimated by dynamic light scattering) and up to 340% (at 1.12 V vs. RHE applied bias) enhancement of the oxygenic photocurrent compared to the parent QS, as probed on 3D-Inverse Opal Indium Tin Oxide (IO-ITO) electrodes under analogous conditions. The highest photocurrent densities are J(QS-TEGlock) = 370 ± 30 μA cm−2 and J(QS) = 290 ± 40 μA cm−2 at 1.52 V vs. RHE, respectively, associated with FEO2 > 95% (Fig. 18 and entry 5, Table 4).39 Indeed, the TEG-ylated quantasome displays the specific formation of TEG-templated hydration shells, probed by Raman microscopy, where water molecules undergo a structural “ordering” by effect of a H-bonding chain, as it results from the Raman peak (band area <3350 cm−1 compared to the disordered water region observed above 3350 cm−1).121 This behaviour confirm the importance of water transport and harvesting in the proximity of the OEC, and sets a key parallelism with the water channel function of the natural PSII.
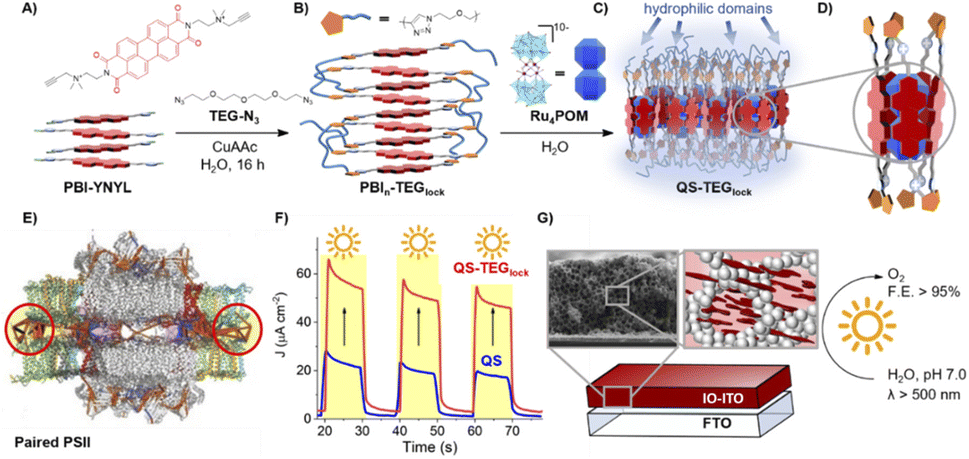 | ||
| Fig. 18 Second-generation quantasomes, exploiting an inter-locked multi-chromophoric antenna system based on PBI photosensitizers bearing hydrophilic tetraethylene glycol chains (TEG). Reprinted from ref. 39 with permission from the American Chemical Society, copyright 2022. | ||
Moving electrons and protons with dyes enabling PCET
The Z-scheme photosynthetic chain transports electron AND protons. In some cases, the electrons and the proton take different routes, in a multiple site proton coupled electron transfer (MS-PCET).18,122 This is the case of the oxidation of the tyrosine to tyrosine radical (Tyr-OH → Tyr-O˙) where the electron is transferred to the oxidized P680˙+, and the proton to a histidine residue. In other cases, electrons and protons convey to a single acceptor, as the quinone QB that is twice reduced to H2QB (see previous discussion in the “The Native Photosystem II (PSII) machinery and PSII wired bio-hybrid photoanodes” paragraph).In 2020, Sartorel, Galloni et al. reported the unprecedented application of KuQuinone (KuQ) dyes for dye-sensitized OER photoanodes.123 KuQuinones are biomimetic polyquinoid dyes, characterized by broad and intense absorption in the visible region (400–600 nm, ε up to 1.5 × 104 M−1 cm−1) owing to the fully conjugated pentacyclic core: furthermore, due to their quinoid nature, their redox behaviour follows a proton-coupled electron transfer mechanism, which is an unprecedented feature among dyes sensitized photoanodes (Fig. 19). Combining the electrochemical properties of the ground state and the E00 energy of the excited state, this latter turns out to be highly oxidizing with a redox potential for the 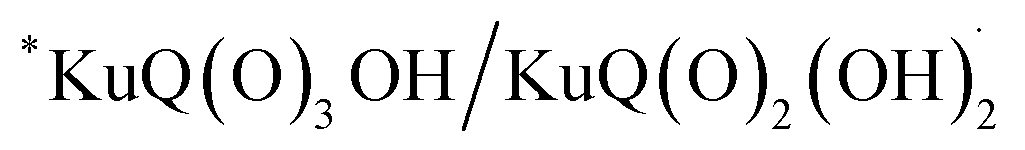 couple of 2.5 V vs. RHE, where
couple of 2.5 V vs. RHE, where 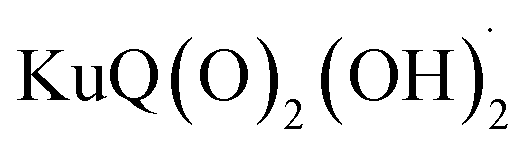 indicates the reduced form of the dye upon addition of one electron and one proton. This species was indeed characterized by transient absorption spectroscopy on mesoporous SnO2|KuQ electrodes in ascorbate aqueous solution, with the dye anchored on the semiconductor through a carboxylate bridge. This evidence speaks in favour of a reductive quenching mechanism, where the excited KuQ dye oxidizes ascorbate, and the generated, reduced
indicates the reduced form of the dye upon addition of one electron and one proton. This species was indeed characterized by transient absorption spectroscopy on mesoporous SnO2|KuQ electrodes in ascorbate aqueous solution, with the dye anchored on the semiconductor through a carboxylate bridge. This evidence speaks in favour of a reductive quenching mechanism, where the excited KuQ dye oxidizes ascorbate, and the generated, reduced 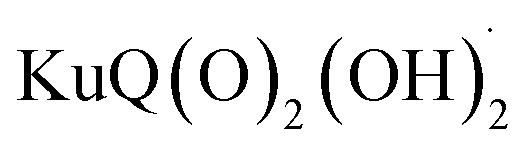 injects electrons into the SnO2 semiconductor, thus causing the photocurrent response. When integrated with a Ru4POM WOC embedded in a Nafion matrix (see Fig. 19), the resulting mesoSnO2|KuQ|Ru4POM photoanodes showed a remarkable light harvesting efficiency of 90% at 530 nm, and, under irradiation in pH 5.8 buffer, exhibited a low potential onset of 0.64 V vs. RHE and a photocurrent of 20 μA cm−2 at 1.14 V vs. RHE, coupled with a 70 ± 15% faradaic yield for O2. Conversely, no O2 was detected for mesoSnO2|KuQ, confirming the fundamental role of the Ru4POM catalyst in evolving oxygen. However, the mesoSnO2|KuQ|Ru4POM photoanodes showed a maximum IPCE of 0.09% at 490 nm, which was ascribed to the negative effect of the Nafion matrix, and a limited stability under operation (entry 1, Table 5).
injects electrons into the SnO2 semiconductor, thus causing the photocurrent response. When integrated with a Ru4POM WOC embedded in a Nafion matrix (see Fig. 19), the resulting mesoSnO2|KuQ|Ru4POM photoanodes showed a remarkable light harvesting efficiency of 90% at 530 nm, and, under irradiation in pH 5.8 buffer, exhibited a low potential onset of 0.64 V vs. RHE and a photocurrent of 20 μA cm−2 at 1.14 V vs. RHE, coupled with a 70 ± 15% faradaic yield for O2. Conversely, no O2 was detected for mesoSnO2|KuQ, confirming the fundamental role of the Ru4POM catalyst in evolving oxygen. However, the mesoSnO2|KuQ|Ru4POM photoanodes showed a maximum IPCE of 0.09% at 490 nm, which was ascribed to the negative effect of the Nafion matrix, and a limited stability under operation (entry 1, Table 5).
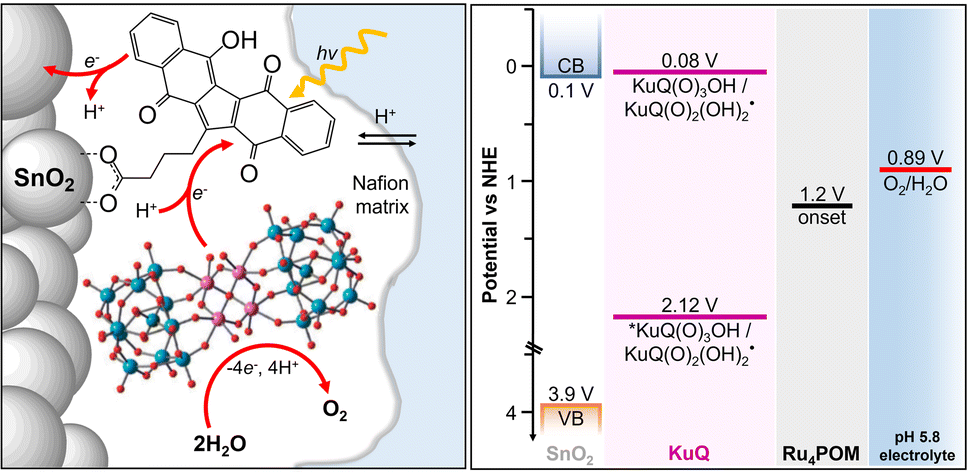 | ||
| Fig. 19 Anchoring of KuQ dye onto a mesoporous SnO2 surface, coupled with embedding of a Ru4POM WOC in a Nafion membrane. Energy levels of the photosynthetic assembly are shown on the right. Reprinted from ref. 123 with permission from the Royal Society of Chemistry, copyright 2020. | ||
| Entry and photoanode | Light intensity (mW cm−2) | E (V) vs. RHE and pH | J (μA cm−2) | F.E.(O2) (%) | Ref. |
|---|---|---|---|---|---|
| a Photoanode tested in a standard three electrode photoelectrochemical cell set-up. b Photoanode tested in a two-electrode set-up. c The two data points refer to different lengths of the spacer in the WOC Ru(bda)(py(CH2)xP(O3H)2)2 (bda is 2,2-bipyridine-6,6-dicarboxylate with x = 3 or 10). | |||||
| 1, mesoSnO2|KuQ|Ru4POM | White light, 100 mW cm−2, λ > 400 nm | 1.14, pH = 5.8 | 20 (IPCE 0.09% at 490 nm) | 70 ± 15 | 123 |
| 2, TiO2|L0 + Ru1 | White light, 100 mW cm−2, λ > 400 nm | 0.62, pH = 7a | 300 (IPCE 25% at 380 nm) | 73 | 124 and 125 |
| 0, pH = 7b | 70 (in the overall PEC configuration) | 55 (H2 FY yield) | |||
| 3, SnO2|TiO2|[P-A-π–D]–Ru(bda)2 | White light, 100 mW cm−2, λ > 400 nm | 0.85, pH = 7 | 1400 to 100 (decay in 60 s) (IPCE 17% at 420 nm with a hydroquinone donor) | 8 | 126 |
| 4, SnO2/TiO2|dye(Al2O3)–Ru(bda)-(PyP)2 | White light, 100 mW cm−2, λ > 400 nm | 0.68, pH = 4.8 | 500 (IPCE 33% at 400 nm) | 82 | 127 |
| 5, SnO2/TiO2/Org1-/1.1 nm Al2O3/-RuP2+-WOCc | White light, 100 mW cm−2, λ > 400 nm | 0.87, pH = 4.65 | 500–800c (IPCE 32% at 400 nm) | 100 | 128 |
| 6, TiO2|D1-CoF | Xenon arc lamp 300 W, 100 mW cm−2, λ > 400 nm | 1, pH = 7 | 100 (IPCE not reported) | 77 | 129 |
| 7, TiO2/[CoFe-JG] | White light, 100 mW cm−2, λ > 420 nm | 1.23, pH = 7 | 50 (IPCE 0.6% at 430 nm) | 83 | 130 |
| 8, SnO2/TiO2|-[T2-BTD]/Ru(bda) | White light, 100 mW cm−2, λ > 400 nm | 0.63, pH = 3.9 | 38 μA cm−2 (IPCE 20% at 360 nm with a hydroquinone donor) | 12 | 131 |
| 9, TiO2|QAP-C16|Zr4+|RuOEC | White light, 100 mW cm−2, λ > 400 nm | 0.71, pH = 7 | 253 μA cm−2 (IPCE 6% at 520 nm) | 27 | 132 |
| 10, TiO2/PH/CoO(OH)x | White light, λ > 420 nm | 1.12, pH = 7 | 400 μA cm−2 (IPCE 7% at 420 nm) | 34 | 133 |
Triarylamine-based push–pull sensitizers
In 2015 Sun and co-workers introduced the use of push–pull sensitizers and demonstrated the versatility of such organic dyes developing a p/n tandem DSPEC capable of unassisted water splitting under irradiation with visible light.124 The photoanode was composed of a simple triarylamine dye (electron-donor) adsorbed on TiO2 through an electron-accepting cyanoacrylate anchor and a co-loaded [Ru(pda)] WOC (pda = 2,6-pyridinedicarboxylate ligand) (Fig. 20). When irradiated with visible light in neutral buffer at 0.6 V vs. RHE, the photoanode delivered an initial photocurrent of 300 μA cm−2 (though a decay of 70% was observed over 1 h) with a 70% faradaic efficiency for oxygen evolution. The system was further associated with a NiO based molecular photocathode sensitized by a similar push–pull triarylamine dye and employing a co-loaded Co cobaloxime as a hydrogen evolving catalyst.125 The fully assembled cell, limited by the photocathode performance, delivered a photocurrent of 70 μA cm−2 with no applied bias, associated with a 55% faradaic efficiency for H2 production, corresponding to a 0.05% solar-to-hydrogen efficiency (entry 2, Table 5).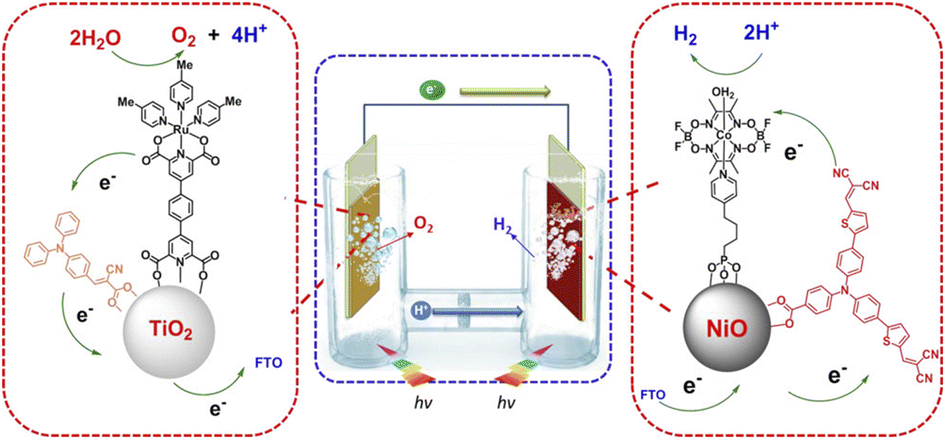 | ||
| Fig. 20 Example of an entire photoelectrochemical cell for water splitting, where the photoanode is designed from a triphenylamine photosensitizer and a Ru WOC. Reprinted from ref. 124 with permission from the American Chemical Society, copyright 2015. | ||
A similar triarylamine dye was exploited in combination with a Ru(bda) catalyst in a photosynthetic system proposed by Meyer et al. assembled onto FTO|SnO2/TiO2 (3 nm) electrodes with a core/shell SnO2/TiO2 (Fig. 21).126 Gaining inspiration from the DSSC field, the triarylamine dye was covalently bound to a dithiophene unit that integrated the phosphonate anchoring group for the semiconductor. Upon photoexcitation, the triarylamine donor transfers an electron to the dithiophene unit, which further injects the electron into TiO2. The core/shell SnO2/TiO2 junction is beneficial for retarding back electron transfer, which was observed to occur in 170 ns on TiO2, and in 1.02 μs on the core/shell oxide through transient absorption spectroscopy. This retarded recombination was instrumental for enhancing the photocurrent response of the photoelectrodes, when probed for the oxidation of hydroquinone. However, when investigating the water oxidation process through the combination of the Ru(bda) catalyst (co-anchored in a 1:5 ratio with respect to the dye), the photoelectrodes provided fast decreasing photocurrents associated with a very low faradaic yield for oxygen evolution of ca. 8%, due to the competitive oxidation of the dye under photoelectrolysis conditions (entry 3, Table 5).
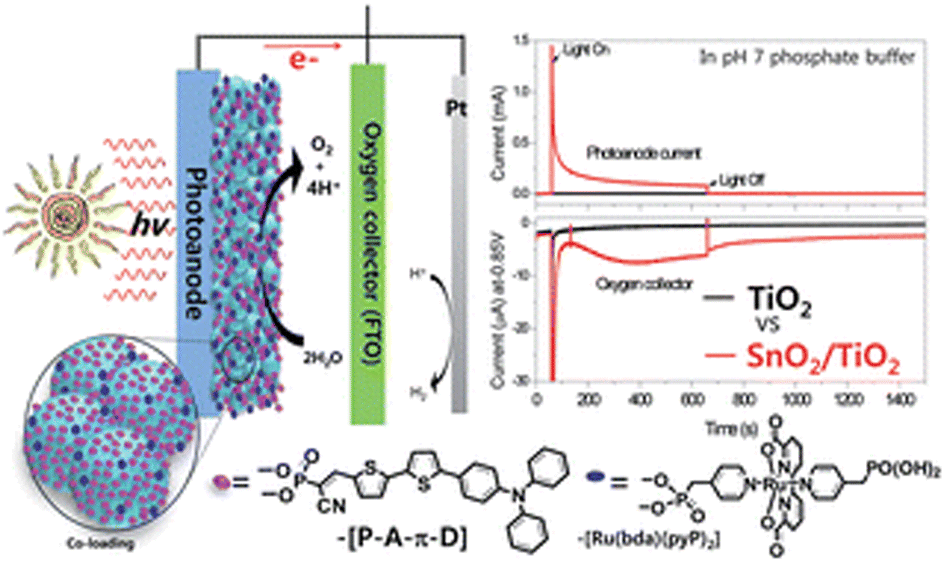 | ||
| Fig. 21 Photosynthetic electrode by co-adsorbing a triarylamine photosensitizer and a Ru(bda) WOC through phosphonate linkers. Reprinted from ref. 126 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Despite these discouraging results, the combination of a triarylamine dye and a Ru(bda) catalyst onto core shell SnO2/TiO2 was then successfully exploited for water oxidation by adding an Al2O3 layer as a further element of the electrode (Fig. 22). The role of the Al2O3 layer is twofold: protecting the triarylamine from hydrolysis and allowing sufficient loading of the Ru(bda) catalyst; the thickness of the Al2O3 layer is crucial for the oxygenic performance of the system. Under the optimized conditions (4 μm thickness of 4.5 nm SnO2/TiO2, 7–10 Å Al2O3), the photocurrent density was ca. 100 μA cm−2 over one hour, with 80% faradaic efficiency in oxygen evolution (entry 4, Table 5).127
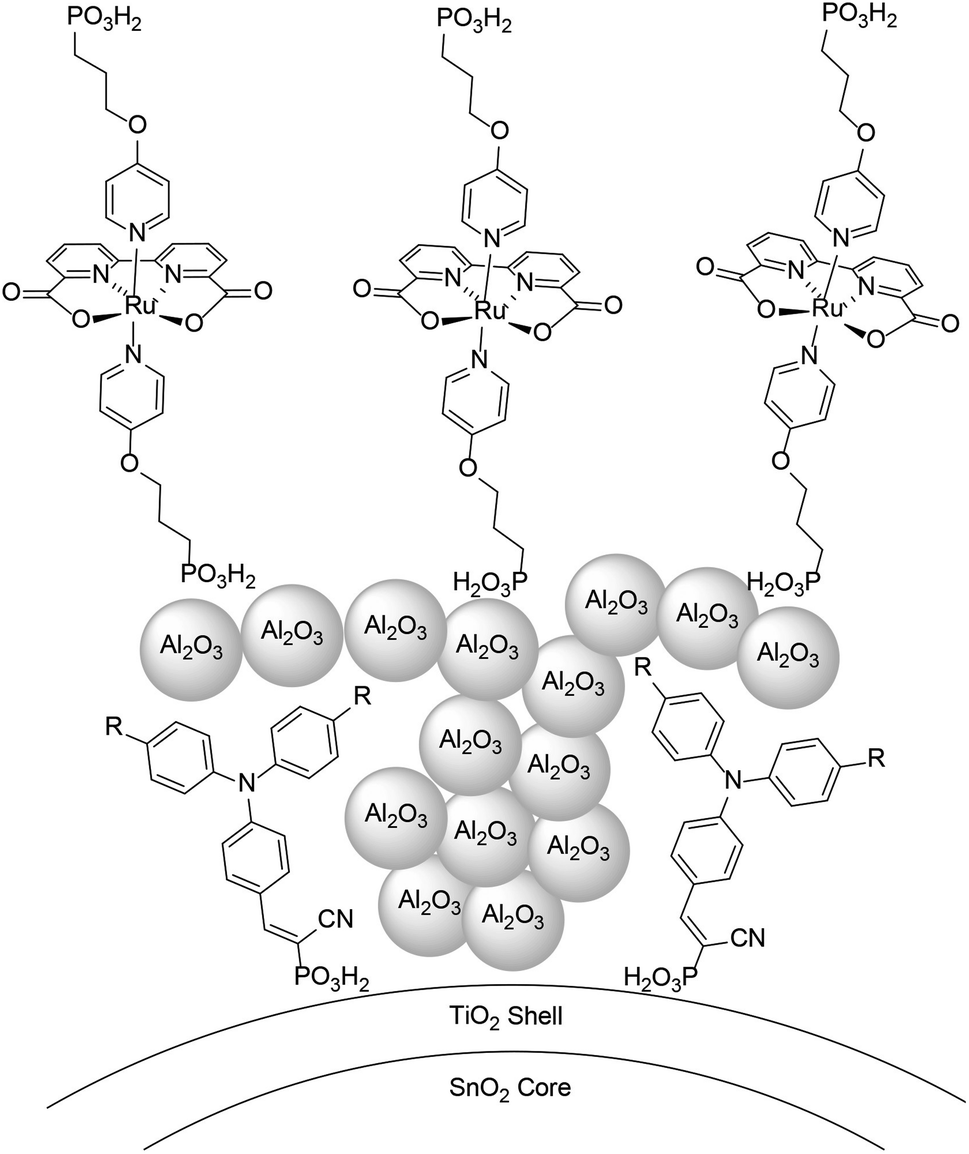 | ||
| Fig. 22 4.5 nm SnO2/TiO2 particles sensitized with a triphenylamine photosensitizer, embedding a 7–10 Å Al2O3 protecting layer, exploited to anchor a Ru(bda) WOC. Reprinted from ref. 127 with permission from the American Chemical Society, copyright 2017. | ||
Further development of photoanodes employing these molecular components envisaged the use of Ru trisbipyridine derivatives as electron transfer mediators between the oxidized triphenylamine dye and the Ru(bda) catalyst; the optimized assembly is shown in Fig. 23.128 In the postulated mechanism, corroborated by transient absorption spectroscopy measurements, photoinduced electron injection into the semiconductor is promoted by excitation of the triphenylamine dye (its absorption dominates with respect to the Ru polypyridine in the high energy portion of the visible spectrum, with a maximum around 400 nm), while there is no evidence of photoinduced electron injection from the Ru polypyridine chromophore (absorption maximum around 450 nm), prevented by a 1.1 nm thick Al2O3 insulating layer (by atomic layer deposition, ALD in Fig. 23). The role of the Ru trisbipyridine is instead to transfer an electron to the oxidized triphenylamine dye, with the generated Ru(III) trisbipyridine (E = 1.28 V vs. NHE) able to subsequently oxidize the Ru(bda) WOC, thus acting as an electron transfer mediator. In the optimized setup the photoelectrodes reach photocurrents of ∼500 μA cm−2 and quantitative O2 evolution at pH 4.65, with IPCE of 32% at 400 nm (entry 5, Table 5). The IPCE profile resembles the absorption of the TPA chromophore, supporting the postulated mechanism.
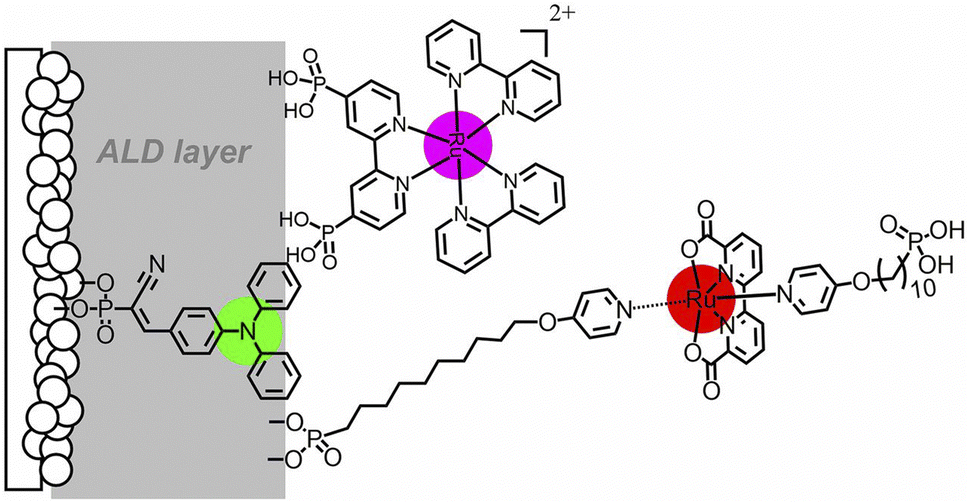 | ||
| Fig. 23 Photoanodes employing a triphenylamine photosensitizer (green ball) bound to the semiconductor, a protective ALD layer used to anchor a Ru(bda) WOC (red ball) and a Ru(II) trisbipyridine redox mediator (purple ball). Reprinted from ref. 128 with permission from the Proceedings of the National Academy of Sciences (PNAS), copyright 2018. | ||
Still exploiting triphenylamine push–pull dyes, Meyer, Sun et al. recently reported the first example of a molecular catalyst based, noble metal-free, dye-sensitized photoanode.129 In particular, the catalyst takes advantage of a bioinspired tetracobalt cubane, stabilized by pyridine and carboxylate ligands. These latter bear hydrophobic fluorinated aliphatic chains that help in stabilizing the organic dye on the TiO2 electrode surface (Fig. 24). Besides the performance of the optimized system, reaching photocurrent densities around 100 μA cm−2 and associated with a faradaic yield of 77% (pH 7 phosphate buffer, entry 6, Table 5), it is interesting to mention the mechanistic analysis, performed by transient spectroscopic measurements, which confirmed injection efficiencies for the dyes in the range 86–90%, and subsequent electron transfer from the cubane catalyst to the oxidized dye, as confirmed by a shortening of the lifetime of the oxidized dye from 169 to 60.4 μs in the presence of the cubane.
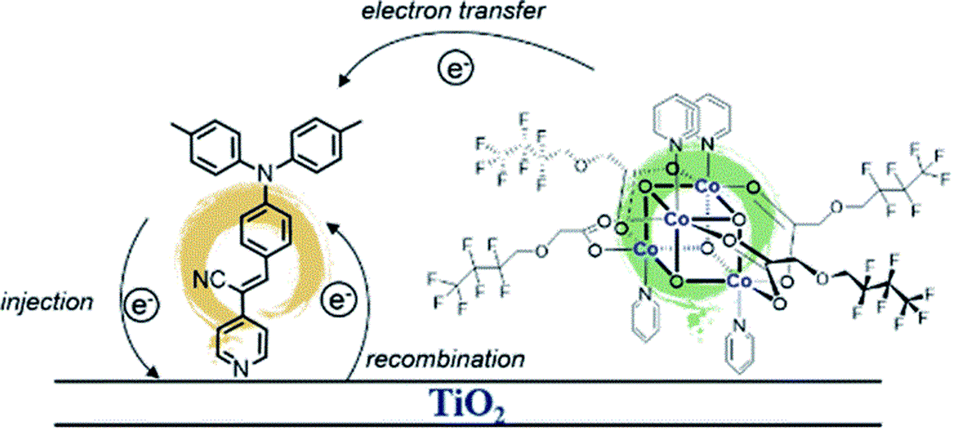 | ||
| Fig. 24 Representation of the photosynthetic assembly proposed by Meyer and co-workers based on a triphenylamine photosensitizer and a tetracobalt cubane WOC. Reprinted from ref. 129 with permission from the Royal Society of Chemistry, copyright 2022. | ||
Novel, recent classes of organic dyes employed in oxygenic photoanodes
Ghobadi et al. reported a dye-sensitized TiO2/[CoFe-JG] photoanode, where the molecular components constitute a triad composed of a Janus Green B dye (JG, i.e. a phenazine based dye with a quaternized N atom), a pentacyano ferrate group (Prussian blue analog) acting as an electron shuttle between the JG dye and a cobalt catalytic center (Fig. 25).130 The TiO2/[CoFe-JG] photoanode performs photoelectrochemical water oxidation with a stable photocurrent of 50 μA cm−2 along two hours of electrolysis at 1.23 V vs. RHE in aqueous phosphate buffer pH 7, associated with an 83% faradaic yield for oxygen evolution (entry 7, Table 5). Surprisingly, the IPCE of the system was found to be around 0.6% at 430 nm, while flattening off at 500 nm, thus not matching with the absorption spectrum of the [CoFe-JG] triad (reaching a maximum at around 650 nm). The authors attributed this mismatch to an improper alignment of the conduction band of TiO2 and the LUMO of the dye, which limits photoconversion efficiencies; nevertheless, fast injection into TiO2 was claimed based on the absence of ground state bleaching in femtosecond transient absorption experiments.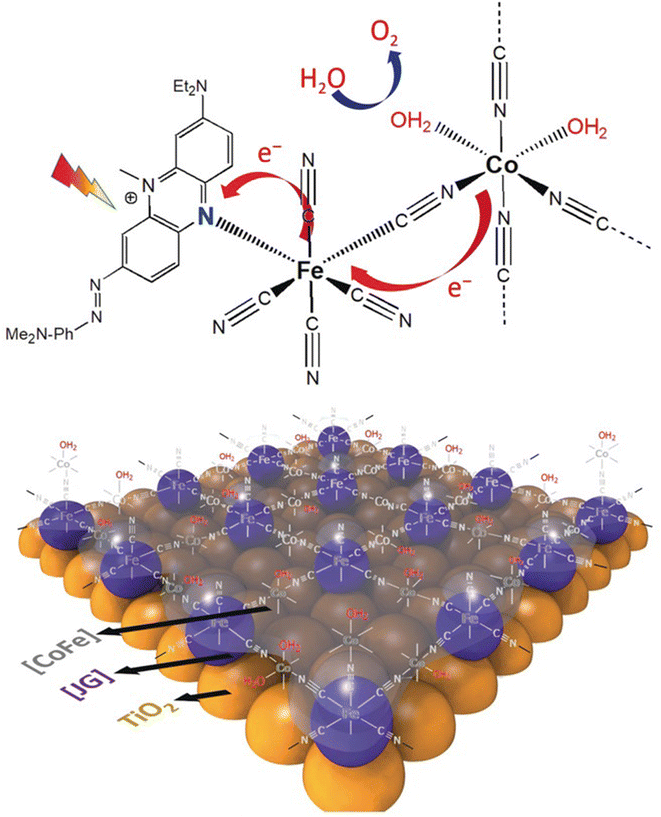 | ||
| Fig. 25 Structure of the photoanode reported by Karadas and co-workers, based on the incorporation of a Prussian blue structure for the sensitization of TiO2, acting as an electron shuttle between a Co based catalytic centre and a phenazine-based Janus green B dye with a quaternized nitrogen. Reprinted from ref. 130 with permission from Wiley, copyright 2020. | ||
A low faradaic yield of 12% was obtained employing the SnO2/TiO2 core shell structure with a 2,2′-(benzo[c][1,2,5]-thiadiazole-4,7-diyl)bis(thiophene-3-carboxylic acid) dye combined with a Ru(bda) catalyst derivative (giving 38 μA cm−2 in acetate buffer, pH 3.9, entry 8, Table 5).131
Hua and co-workers have recently reported the use of a quinacridone dye derivative in a bias-free photoelectrochemical cell for water splitting (Fig. 26).132 Quinacridone (pigment violet 19) is a commercially available dye used in paints; in their work, Hua et al. exploited a derivative of QNC embedding pyridine dicarboxylic acid pendants to anchor the dye to TiO2 and to NiO semiconductors in the anodic and cathodic compartments, respectively. Long alkyl chain substituents (C4–C16) at the nitrogen atom prevent flopping down of the dye to the hydrophilic TiO2 surface. In the photoanode, the pyridine dicarboxylic acid pendant was exploited also to anchor the dye to a Ru single site catalyst bearing the same linker, by exploiting Zr(IV) bridges. Concerning the photoanode, the QNC dye has sufficient oxidizing power (E in the range 1.59–1.77 V vs. RHE) to feed the water oxidation by the Ru catalyst (ca. 1.61 V vs. RHE) working at pH 7. Indeed, a photocurrent response was observed at an onset potential of −0.13 V vs. RHE, reaching a plateau photocurrent density of up to 250 μA cm−2, IPCE up to 6% at 520 nm, but a limited faradaic efficiency for oxygen evolution of ca. 30% (entry 9, Table 5). Nevertheless, the photoanode was combined with a photocathode based on the same dye (the photocathode was assembled by co-adsorbing a Co-cobaloxyme hydrogen evolving catalyst through phosphonate linkers) to achieve bias free hydrogen evolution with a photocurrent density of ca. 110 μA cm−2, a faradaic efficiency of 89% and a solar to hydrogen efficiency of 0.11% when irradiating both the photoanode and the photocathode.
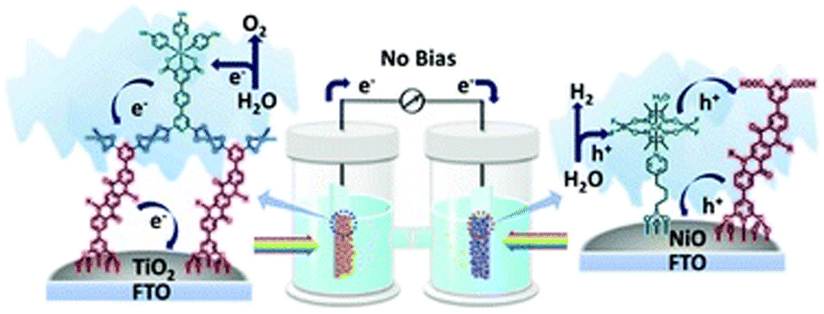 | ||
| Fig. 26 A bias free photoelectrochemical cell for water splitting into hydrogen and oxygen, based on a quinacridone dye both at the photoanode and at the photocathode. In particular, a Ru WOC is assembled at the photoanode exploiting carboxylic pendants through Zr(IV) bridges. Reprinted from ref. 132 with permission from the Royal Society of Chemistry, copyright 2021. | ||
Despite the many reports on photoanodes based on rare metal-free sensitizers, very few “fully rare metal-free” systems have been published to date. The first example was developed by Beranek and co-workers in 2012 and is based on a rather uncommon polyheptazine (PH, or graphitic carbon nitride, g-C3N4) sensitizer, coupled to photoelectrodeposited Co-Pi (Fig. 27):134,135 noteworthily, polyheptazine has an optical band gap of 430 nm, but, when directly grown on TiO2, the resulting TiO2|g-C3N4 composite shows broad light absorption up to 540 nm, suggesting a direct photoinduced electron transfer from the HOMO of g-C3N4 to the TiO2 conduction band (Fig. 27). Under irradiation with visible light (λ > 420 nm) in pH 7 phosphate buffer, the TiO2|g-C3N4|Co-Pi photoanodes showed a 2-fold increase in photocurrent (50 μA cm−2 at 1 V vs. RHE) with respect to TiO2|g-C3N4: O2 was only detected for the complete photoanode, though the faradaic efficiency was not reported; a maximum IPCE of 12% was registered at 350 nm, but ca. 1.7% IPCE was still obtained at 450 nm.135
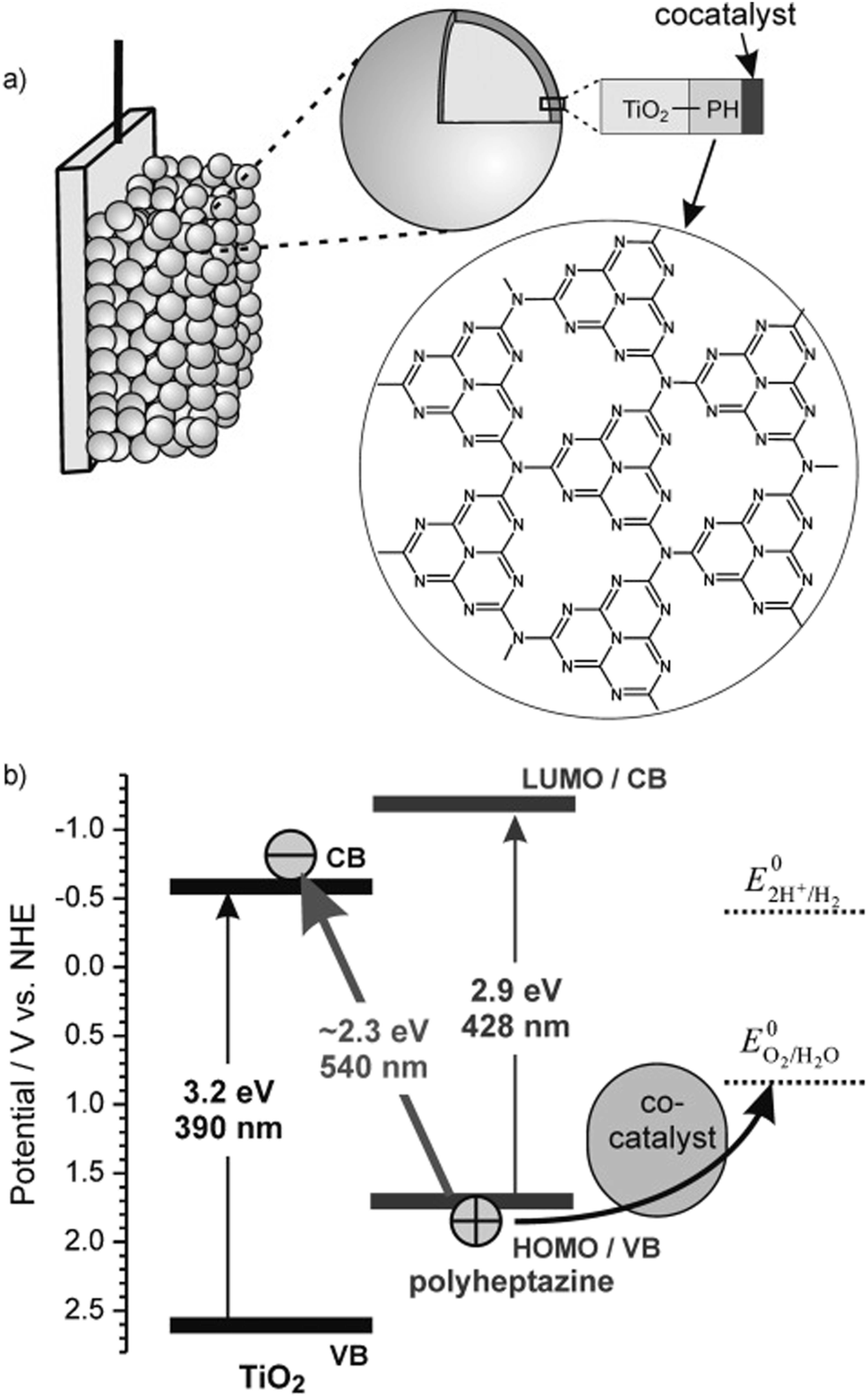 | ||
| Fig. 27 (a) Hybrid photoelectrodes based on nanocrystalline TiO2 sensitized with polyheptazine and loaded with a CoOx WOC, and (b) energy levels evidencing the direct optical charge-transfer excitation of an electron from the polyheptazine HOMO into the conduction band of TiO2. Reprinted from ref. 134 with permission from Springer, copyright 2013. | ||
The system was subsequently implemented by replacing the electrodeposited CoPi catalyst with ultrasmall (1–2 nm) CoO(OH)x nanoparticles via a two-step impregnation method.133 The ultrasmall nanoparticles provide the advantage of being highly transparent, thus allowing to reach high levels of loading without impacting on the g-C3N4 light absorption. The optimized TiO2|g-C3N4|CoO(OH)x photoanodes provided up to 400 μA cm−2 in borate buffer at 1.12 V vs. RHE, although with a limited faradaic efficiency of 34% (entry 10, Table 5).
Beyond the oxygen evolution reaction: selective organic photosynthesis at engineered photoanodes
The four-photon/four-electron manifold to accomplish water oxidation to dioxygen represents a huge kinetic bottleneck, for either natural or artificial photosystems. This is evident from the severe efficiency limitations shown by the current state-of-the-art photoanodes applied to water-oxidation as reported in the previous sections (Tables 1–5). In Fig. 28, we graphically represent the performance of these systems depending on the wavelength. We selected IPCE (circles; only maximum values are reported for the sake of simplicity, although some of the systems display activity in a broad range of the spectrum) and FY (empty squares) as the key performance parameters representing the photon-to-current and current-to-oxygen performance, respectively. From this plot, it is evident that: (i) best performing systems exploit high energy radiation (λ < 475 nm), as in the case of Ru-polypyridine chromophores; (ii) porphyrinoids and organic dyes overstepping this barrier (λ between 475 and 550 nm) are still associated with low IPCE values; (iii) the “green region” above 550 nm where the PSII operates is almost unexploited in artificial systems. | ||
| Fig. 28 Plot of IPCE (circles; only the maximum values are reported) and FE (empty squares) depending on the wavelength for the systems described in Tables 1–5. | ||
Despite the strong photocatalytic challenge, biological photosynthesis has evolved by using water as the primary, vast and abundant source of reducing equivalents (electron and protons) necessary to feed large scale (i.e. terrestrial and oceanic) photosynthetic machineries. In this process, O2 is generated as a side-product by the water oxidation semi-reaction, that, while being of paramount importance for a circular processing of our aerobic atmosphere, retains scarce commercial interest per se.7,120,121 Considering these economic and sustainability factors, man-made photosynthetic devices have been reconfigured to bypass water oxidation while engineering the photoanodic process with diverse aims: (i) to lower the energy-barrier of the oxidative transformation, (ii) to address relevant sustainability goals via advanced oxidation processes (AOP) and/or (iii) to direct the photocatalytic oxidation towards valuable synthetic targets.
As a simple and practical example, the low-energy bromide photo-oxidation can be used to efficiently store energy, since the Br−/Br2 or Br−/Br3− couples can be readily interconverted by electrochemical means.136,137 This principle is valid for halides in general, especially considering the high concentration of Cl− and Br− in the most abundant water source, i.e. seawater.102,138 In addition, a few examples of PECs performing H2 evolution in tandem with the oxidative degradation of organic pollutants have been reported.138
Increasing attention is being dedicated to selective photosynthesis by the photoelectrochemical oxidation of organic reagents to afford value-added chemicals using visible light. Moreover, the electro-oxidation of organic compounds (electrochemical reforming), by tailored electrocatalysts and/or redox mediators, has been proven also in tandem with the cathodic hydrogen evolving reaction (HER) or CO2-reduction reaction (CO2RR).139,140
To this end, the oxidation of alcohols is one of the most investigated strategies,141 that is often accomplished by integrating a photoanodic process with an aminoxyl radical catalyst,142 added in solution or chemically anchored to the photoelectrode. Primary and benzylic alcohols are generally oxidised between 0.7 and 1.5 V vs. NHE143,144 to afford the corresponding aldehyde as a two-electron oxidation product, eventually undergoing a sequential oxidation step to the corresponding carboxylic acid. The oxidation of hydroxymethylfurfural (HMF, easily derived from biomass) to 2,5-furandicarboxylic acid (FDCA) provides a valuable synthetic target for the polymer industry.138,139
The photoelectrochemical oxidation of HMF or of other benzyl alcohol derivatives was reported with BiVO4 (ref. 145 and 146) and with a graphitic carbon-coated TiO2 nanowire photoanode.147 In recent years the development of dye sensitized photoanodes has been considered for this application, and the subject has been recently reviewed.141 The subject was pioneered by T. J. Meyer and co-workers,148 who reported the dehydrogenation of benzyl alcohol (BnOH) to benzaldehyde, with concomitant cathodic production of H2, with a DSPEC based on a core–shell nanoITO|TiO2 photoanode co-derivatized with the classical [Ru(bpy)2(dpbpy)] sensitizer and a Ru(II)-polypyridyl catalyst, for which the RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O active intermediate responsible for BnOH oxidation to benzaldehyde is generated by a two-photon/two-electron oxidation. A 5-fold photocurrent increase, up to 250 μA, was indeed obtained upon addition of 0.1 M BnOH, and a 37% faradaic efficiency for benzaldehyde was registered over 3 h photoelectrolysis at 0.87 V vs. RHE in pH 4.6 aqueous buffer under monochromatic irradiation (3.7% APCE at 445 nm, 60 mW), with a concomitant 87% faradaic efficiency for H2 evolution at the cathode.148 Among the examples of noble-metal-free or organic photosensitizers employed in these systems are zinc porphyrins,149 thienopyrroledione,150 diketopyrroles,151 perylenes,152 and polyquinoids.153 The same concept was recently employed for the photoelectrochemical oxidation of glycerol, an archetypical biobased compound for which oxidation to glyceraldehyde or dihydroxyacetone is an extremely appealing target reaction.154,155
O active intermediate responsible for BnOH oxidation to benzaldehyde is generated by a two-photon/two-electron oxidation. A 5-fold photocurrent increase, up to 250 μA, was indeed obtained upon addition of 0.1 M BnOH, and a 37% faradaic efficiency for benzaldehyde was registered over 3 h photoelectrolysis at 0.87 V vs. RHE in pH 4.6 aqueous buffer under monochromatic irradiation (3.7% APCE at 445 nm, 60 mW), with a concomitant 87% faradaic efficiency for H2 evolution at the cathode.148 Among the examples of noble-metal-free or organic photosensitizers employed in these systems are zinc porphyrins,149 thienopyrroledione,150 diketopyrroles,151 perylenes,152 and polyquinoids.153 The same concept was recently employed for the photoelectrochemical oxidation of glycerol, an archetypical biobased compound for which oxidation to glyceraldehyde or dihydroxyacetone is an extremely appealing target reaction.154,155
Bruggeman et al. reported a mesoporous TiO2 electrode sensitized with a thienopyrroledione based organic dye (E = 1.8 V vs. NHE for the D+/D couple) and integrated with TEMPO catalyst for glycerol oxidation (Fig. 29).156 Interestingly, both the sensitizer and the catalyst are integrated in a 3 mm thick acetonitrile based gel at the surface of the TiO2 semiconductor, thus creating a biphasic system with the aqueous solution. When irradiated with one sun in a 0.1 M glycerol aqueous solution (saturated NaCl, NaHCO3 pH 8.3) the photocurrent density approached 200 μA cm−2, remaining stable for up to 48 hours, with an almost quantitative faradaic efficiency for the production of glyceraldehyde. The acetonitrile gel was pivotal in protecting and stabilizing the photoanode components from delamination under the alkaline conditions required for glycerol oxidation (pH 8.5; under a purely aqueous system the photocurrent density rapidly fell to tens of μA cm−2); crucial to the system is the diffusion and transport of the photogenerated TEMPO+ and related intermediates within the gel and crossing the solution interface.
 | ||
| Fig. 29 Photoelectrochemical cell for glycerol oxidation and simultaneous H2 evolution proposed by Reek and co-workers. At the photoanode, the yellow and azure colours represent the redox-gel layer and the aqueous solution, respectively, while oxidation of glycerol by oxoammonium TEMPO+ occurs at the interface (green area). Reprinted from ref. 156 with permission from Wiley, copyright 2022. | ||
Noteworthily, engineered photoanodes have been considered also for the oxidative activation of C–H bonds, the holy grail for selective photosynthesis.157–159 Activation of a C–H bond is regulated by the C–H bond dissociation free energy, which is associated with the homolytic breaking of the bond via a hydrogen atom transfer (HAT) mechanism. In nature, this mechanism occurs typically by a high valent Fe-oxo species in cytochrome P450;160–162 artificial systems for photogenerating bio-inspired metal-oxo species capable of C–H activation have been pioneered by Nam and Fukuzumi.163–165 Alternative strategies to promote photochemical HAT mechanisms include: (i) the use of photocatalysts enabling HAT by a radical-type excited state, with examples being aromatic ketones166,167 or the inorganic decatungstate (W10O324−) species;159,168 (ii) the use of a multiple site proton coupled electron transfer mechanism (MS-PCET),18,43 as the removal of the hydrogen atom is formally equivalent to the removal of an electron and a proton that can be promoted by a photogenerated oxidant (Ox+) and by a cooperative base function (B). For this case, J. Mayer18 and R. R. Knowles43 pioneered the identification of a formal effective Bond Dissociation Free Energy (BDFEeff) for the Ox+/B system regulated by the redox potential and by the pKa of the Ox+/Ox and BH+/B couples, respectively.
Photoelectrochemical C–H activation at photoanode surfaces has been proposed by direct irradiation of metal-oxide semiconductors such as WO3 (cyclohexane photooxidation cyclohexanol/cyclohexanone, i.e. KA oil),169 BiVO4 (benzylic/allylic photo-oxygenation of tetralin and cyclohexene to the corresponding carbonyl products)146 and Fe2O3 (arene C–H amination).170 In these cases, irradiation of the semiconductor promotes the photoexcitation of electrons from the valence to the conduction band, accompanied by the formation of holes at the valence band, mainly localized on the oxygen atoms of the semiconductor, which results in the photogeneration of metal-oxyl radicals thus enabling a photo-induced HAT mechanism.
More recently, a proof of concept for photoelectrochemical C–H activation was reported with dye-sensitized photoanodes, exploiting a quinacridone dye (QNC) sublimated under vacuum onto TiO2 or SnO2 semiconductors (Fig. 30). As demonstrated by cyclic voltammetry and the Pourbaix diagram, the key feature of QNC is its oxidative reactivity through a proton-coupled electron transfer, which implies the generation of a nitrogen-based radical. A BDFE of 80.5 ± 2.3 kcal mol−1 was estimated for the N–H bond in QNC by combining the redox potential of the QNC(N˙)/QNC(N–H) and H+/H2 couples and the ΔG0 of formation of H2.171
 | ||
| Fig. 30 Left: Schematic representation of quinacridone sensitized SnO2, capable of promoting C–H activation in organic substrates towards a Hydrogen Atom Transfer (HAT) mechanism. On the right, the photoelectrochemical response towards the organic substrate is shown, in terms of the Jsub/J0 parameter depending on the BDFE of the C–H bond. Reprinted from ref. 171 with permission from Wiley, copyright 2023. | ||
When embedded onto the surface of semiconductors, excitation of QNC promotes ultrafast electron injection in a ps timescale, likely accompanied by proton transfer from the dye to the metal-oxide surface. The photogenerated QNC(N˙) can react with C–H bonds, as demonstrated in the case of γ-terpinene where the photocurrent density is enhanced by a factor of 2.4 ± 0.7, while photoelectrolysis produces p-cymene through a radical chain mechanism involving dissolved oxygen. Interestingly, the photocurrent density associated with the C–H bond photooxidation (measured as Jsub/J0 photocurrent enhancement in the presence of the organic reagent) depends on the BDFE values of the allylic or benzylic C–H bond examined and of the N–H bond in the QNC photosensitizer. Specifically, a photoelectrochemical response was observed for substrates below the 80.5 kcal mol−1 BDFE threshold of QNC (xanthene, γ-terpinene, dihydroanthracene), while more challenging substrates (BDFE > 80.5 kcal mol−1 threshold) provided a null photocurrent enhancement (Jsub/J0ca. 1). This result combined with control experiments (negligible photocurrent enhancement with alkylated QNC derivatives or for electron rich substrates lacking C–H functionalities) enabled the authors to propose a hydrogen atom abstraction from the oxidized form of the dye QNC(N˙) as the primary step activating the C–H bond. This elegant strategy highlights the potential for the design of novel photoanodes with a tailored photo-induced HAT driving force.
Conclusions and perspectives
We have discussed herein a collection of case-studies that shed light on the key interplay of molecular and supramolecular chemistry with materials science to engineer functional photoelectrodic surfaces for oxygenic photosynthesis, which still remains a fundamental hurdle for solar fuel production via PEC technology. A survey of the major achievements reached in the last 15 years put under the spotlight the conceptual and experimental efforts in integrating components and functions to shape the light-phase machinery of the oxygenic engine. This is not trivial, since the single molecular components, such as the photosensitizer (PS), the water oxidation catalyst (WOC), or any redox-manifold relays, are optimized separately, according to their a-solo performance, and mostly under very diverse solution/environment homogeneous conditions that are not operative on the final photoelectrodic surface. This observation calls for a change of paradigm in the photosystem design, aiming at addressing the system complexity while targeting possible emerging properties related to the cooperative domains as a whole. In particular, orchestration of the multi-functional assembly requires the engineering of several interfaces by the introduction of mediators, hydrophilic/hydrophobic residues, and transport/protective layers, which can play a fundamental role in implementing the overall efficiency.Nowadays, the performances achieved with PEC devices based on molecular interfaces are still far from the ones targeted for real applications, both in terms of photocurrent density and stability: this suggests that there is much room for improvement.12 In this respect, the fundamental knowledge achieved in fabricating supramolecular polymers and materials controlled at the molecular level is expected to have a major impact on the development of novel and more efficient photosynthetic materials that can rival the best features of the biological machinery.
Finally, we expect that the possibility of designing and fine-tuning molecular components and their assembly will find space in photoelectrochemical devices targeting selective oxidation catalysis for new and light-powered processing of organic molecules.
Author contributions
All authors discussed the concepts and contributed to the final manuscript. T. G and G. A. V. contributed to writing parts of the original draft; A. S. and M. B. supervised the writing, and reviewed and edited the manuscript; A. S. and M. B. were responsible for funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Innovation Council (PLANKT-ON, Plankton-Like Protocells for Artificial Photosynthesis Targeting Carbon-Neutral Energy Vectors, Pathfinder-Open 2022, grant agreement no. 101099192), from Fondazione Cariparo (Project “SYNERGY”, Ricerca Scientifica di Eccellenza 2018), and from Ministero dell’Università e della Ricerca (PRIN 2022 PROMETEO 2022KPK8WM and NANOARC 20228YFRNL) is gratefully acknowledged. We thank Dr R. N. Dürr for the realisation of artwork.Notes and references
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS.
- International Energy Agency, IEA, https://www.iea.org/.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- B. M. Lindley, A. M. Appel, K. Krogh-Jespersen, J. M. Mayer and A. J. M. Miller, ACS Energy Lett., 2016, 1, 698–704 CrossRef CAS.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- C. Ros, T. Andreu and J. R. Morante, J. Mater. Chem. A, 2020, 8, 10625–10669 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- E. A. Reyes Cruz, D. Nishiori, B. L. Wadsworth, N. P. Nguyen, L. K. Hensleigh, D. Khusnutdinova, A. M. Beiler and G. F. Moore, Chem. Rev., 2022, 122, 16051–16109 CrossRef CAS PubMed.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed.
- K. T. Fountaine, H. J. Lewerenz and H. A. Atwater, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J. Xiong and C. E. Bauer, Annu. Rev. Plant Biol., 2002, 53, 503–521 CrossRef CAS PubMed.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- J. W. Youngblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef PubMed.
- T. J. Meyer, Nature, 2008, 27, 778–779 CrossRef PubMed.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem, 2019, 3, 331–341 CrossRef CAS.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060–1066 CrossRef CAS PubMed.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed.
- M. Natali, F. Nastasi, F. Puntoriero and A. Sartorel, Eur. J. Inorg. Chem., 2019, 2027–2039 CrossRef CAS.
- J. D. Megiatto, D. D. Méndez-Hernández, M. E. Tejeda-Ferrari, A. L. Teillout, M. J. Llansola-Portolés, G. Kodis, O. G. Poluektov, T. Rajh, V. Mujica, T. L. Groy, D. Gust, T. A. Moore and A. L. Moore, Nat. Chem., 2014, 6, 423–428 CrossRef CAS.
- N. S. Weliwatte, M. Grattieri and S. D. Minteer, Photochem. Photobiol. Sci., 2021, 20, 1333–1356 CrossRef CAS.
- Y. H. Hong, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS.
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70 RSC.
- F. Puntoriero, G. La Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725–4727 RSC.
- M. Natali, F. Puntoriero, C. Chiorboli, G. La Ganga, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, J. Phys. Chem. C, 2015, 119, 2371–2379 CrossRef CAS.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347–2350 CrossRef CAS.
- K. Hanson, D. A. Torelli, A. K. Vannucci, M. K. Brennaman, H. Luo, L. Alibabaei, W. Song, D. L. Ashford, M. R. Norris, C. R. K. Glasson, J. J. Concepcion and T. J. Meyer, Angew. Chem., Int. Ed., 2012, 51, 12782–12785 CrossRef CAS.
- A. Pannwitz, D. M. Klein, S. Rodríguez-Jiménez, C. Casadevall, H. Song, E. Reisner, L. Hammarström and S. Bonnet, Chem. Soc. Rev., 2021, 50, 4833–4855 RSC.
- H. Song, A. Amati, A. Pannwitz, S. Bonnet and L. Hammarström, J. Am. Chem. Soc., 2022, 144, 19353–19364 CrossRef CAS PubMed.
- B. L. Wadsworth, D. Khusnutdinova and G. F. Moore, J. Mater. Chem. A, 2018, 6, 21654–21665 RSC.
- B. K. Rugg, M. D. Krzyaniak, B. T. Phelan, M. A. Ratner, R. M. Young and M. R. Wasielewski, Nat. Chem., 2019, 11, 981–986 CrossRef CAS.
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 32, 1–32 CrossRef PubMed.
- T. Gobbato, F. Rigodanza, E. Benazzi, P. Costa, M. Garrido, A. Sartorel, M. Prato and M. Bonchio, J. Am. Chem. Soc., 2022, 144, 14021–14025 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- C. Li, A. Iscen, H. Sai, K. Sato, N. A. Sather, S. M. Chin, Z. Álvarez, L. C. Palmer, G. C. Schatz and S. I. Stupp, Nat. Mater., 2020, 19, 900–909 CrossRef CAS PubMed.
- G. A. Parada, Z. K. Goldsmith, S. Kolmar, B. P. Rimgard, B. Q. Mercado, L. Hammarström, S. Hammes-schiffer and J. M. Mayer, Science, 2019, 475, 471–475 CrossRef PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- A. Tacca, L. Meda, G. Marra, A. Savoini, S. Caramori, V. Cristino, C. A. Bignozzi, V. G. Pedro, P. P. Boix, S. Gimenez and J. Bisquert, ChemPhysChem, 2012, 13, 3025–3034 CrossRef CAS PubMed.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- Y.-H. Lai, M. Kato, D. Mersch and E. Reisner, Faraday Discuss., 2014, 176, 199–211 RSC.
- B. L. Wadsworth, A. M. Beiler, D. Khusnutdinova, E. A. Reyes Cruz and G. F. Moore, J. Am. Chem. Soc., 2019, 141, 15932–15941 CrossRef CAS.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem, 2020, 4, 6–21 CrossRef CAS.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 26–43 CrossRef CAS PubMed.
- P. Greife, M. Schonborn, M. Capone, R. Assuncao, D. Narzi, L. Guidoni and H. Dau, Nature, 2023, 617, 623–628 CrossRef CAS PubMed.
- N. Sakashita, H. C. Watanabe, T. Ikeda, K. Saito and H. Ishikita, Biochemistry, 2017, 56, 3049–3057 CrossRef CAS PubMed.
- J. Ye, A. Hu, G. Ren, M. Chen, S. Zhou and Z. He, iScience, 2021, 24, 102828 CrossRef CAS.
- N. Plumeré and M. M. Nowaczyk, Biophotoelectrochemistry: from bioelectrochemistry to biophotovoltaics, in Advances in Biochemical Engineering/Biotechnology, ed. L. Jeuken, 2016 Search PubMed.
- D. Mersch, C. Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332–8335 CrossRef CAS PubMed.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2013, 135, 10610–10613 CrossRef CAS PubMed.
- K. P. Sokol, D. Mersch, V. Hartmann, J. Z. Zhang, M. M. Nowaczyk, M. Rögner, A. Ruff, W. Schuhmann, N. Plumeré and E. Reisner, Energy Environ. Sci., 2016, 9, 3698–3709 RSC.
- J. Z. Zhang, P. Bombelli, K. P. Sokol, A. Fantuzzi, A. W. Rutherford, C. J. Howe and E. Reisner, J. Am. Chem. Soc., 2018, 140, 6–9 CrossRef CAS.
- T. K. Baikie, L. T. Wey, J. M. Lawrence, H. Medipally, E. Reisner, M. M. Nowaczyk, R. H. Friend, C. J. Howe, C. Schnedermann, A. Rao and J. Z. Zhang, Nature, 2023, 615, 836–840 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, A. R. Oliveira, J. Warnan, M. M. Nowaczyk, A. Ruff, I. A. C. Pereira and E. Reisner, J. Am. Chem. Soc., 2018, 140, 16418–16422 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, J. Warnan, N. Kornienko, M. M. Nowaczyk, A. Ruff, J. Z. Zhang and E. Reisner, Nat. Energy, 2018, 3, 944–951 CrossRef CAS.
- F. Puntoriero, S. Serroni, G. La Ganga, A. Santoro, M. Galletta, F. Nastasi, E. La Mazza, A. M. Cancelliere and S. Campagna, Eur. J. Inorg. Chem., 2018, 3887–3899 CrossRef CAS.
- T. A. Heimer, E. J. Heilweil, C. A. Bignozzi and G. J. Meyer, J. Phys. Chem. A, 2000, 104, 4256–4262 CrossRef CAS.
- F. Ronconi, M. P. Santoni, F. Nastasi, G. Bruno, R. Argazzi, S. Berardi, S. Caramori, C. A. Bignozzi and S. Campagna, Dalton Trans., 2016, 45, 14109–14123 RSC.
- J. W. Youngblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef PubMed.
- M. N. Collomb, D. V. Morales, C. N. Astudillo, B. Dautreppe and J. Fortage, Sustainable Energy Fuels, 2019, 4, 31–49 RSC.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 10974–10982 CrossRef CAS PubMed.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber, M. E. Strayer, N. M. Vargas-Barbosa and T. E. Mallouk, J. Phys. Chem. C, 2014, 118, 17046–17053 CrossRef CAS.
- I. Bazzan, A. Volpe, A. Dolbecq, M. Natali, A. Sartorel, P. Mialane and M. Bonchio, Catal. Today, 2017, 290, 39–50 CrossRef CAS.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS PubMed.
- G. A. Volpato, A. Bonetto, A. Marcomini, P. Mialane, M. Bonchio, M. Natali and A. Sartorel, Sustainable Energy Fuels, 2018, 2, 1951–1956 RSC.
- P. Wei, B. Hu, L. Zhou, T. Su and Y. Na, J. Energy Chem., 2016, 25, 345–348 CrossRef.
- M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC.
- A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- Y. V. V. Geletii, B. Botar, P. Kögerler, D. A. A. Hillesheim, D. G. G. Musaev and C. L. L. Hill, Angew. Chem., 2008, 120, 3960–3963 CrossRef.
- A. Sartorel, P. Miró, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. Di Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051–16053 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- J. Fielden, J. M. Sumliner, N. Han, Y. V. Geletii, X. Xiang, D. G. Musaev, T. Lian and C. L. Hill, Chem. Sci., 2015, 6, 5531–5543 RSC.
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC.
- G. Leem, B. D. Sherman, A. J. Burnett, Z. A. Morseth, K. R. Wee, J. M. Papanikolas, T. J. Meyer and K. S. Schanze, ACS Energy Lett., 2016, 1, 339–343 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- B. D. Sherman, D. L. Ashford, A. M. Lapides, M. V. Sheridan, K. R. Wee and T. J. Meyer, J. Phys. Chem. Lett., 2015, 6, 3213–3217 CrossRef CAS.
- M. V. Sheridan, B. D. Sherman, R. L. Coppo, D. Wang, S. L. Marquard, K. R. Wee, N. Y. Murakami Iha and T. J. Meyer, ACS Energy Lett., 2016, 1, 231–236 CrossRef CAS.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef CAS PubMed.
- D. Wang, S. L. Marquard, L. Troian-Gautier, M. V. Sheridan, B. D. Sherman, Y. Wang, M. S. Eberhart, B. H. Farnum, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 719–726 CrossRef CAS PubMed.
- Y. Zhu, D. Wang, Q. Huang, J. Du, L. Sun, F. Li and T. J. Meyer, Nat. Commun., 2020, 11, 4610 CrossRef CAS PubMed.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Gratzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389–2392 RSC.
- P. K. Poddutoori, J. M. Thomsen, R. L. Milot, S. W. Sheehan, C. F. A. Negre, V. K. R. Garapati, C. A. Schmuttenmaer, V. S. Batista, G. W. Brudvig and A. Van Der Est, J. Mater. Chem. A, 2015, 3, 3868–3879 RSC.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V. Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681–1686 CrossRef CAS PubMed.
- M. Natali, E. Deponti, D. Vilona, A. Sartorel, M. Bonchio and F. Scandola, Eur. J. Inorg. Chem., 2015, 2015, 3467–3477 CrossRef CAS.
- T. K. Michaelos, D. Y. Shopov, S. B. Sinha, L. S. Sharninghausen, K. J. Fisher, H. M. C. Lant, R. H. Crabtree and G. W. Brudvig, Acc. Chem. Res., 2017, 50, 952–959 CrossRef CAS PubMed.
- K. L. Materna, J. Jiang, K. P. Regan, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, ChemSusChem, 2017, 10, 4526–4534 CrossRef CAS PubMed.
- A. Orbelli Biroli, F. Tessore, G. Di Carlo, M. Pizzotti, E. Benazzi, F. Gentile, S. Berardi, C. A. Bignozzi, R. Argazzi, M. Natali, A. Sartorel and S. Caramori, ACS Appl. Mater. Interfaces, 2019, 11, 32895–32908 CrossRef CAS PubMed.
- J. M. Thomsen, S. W. Sheehan, S. M. Hashmi, J. Campos, U. Hintermair, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2014, 136, 13826–13834 CrossRef CAS PubMed.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC.
- M. Yamamoto, Y. Nishizawa, P. Chábera, F. Li, T. Pascher, V. Sundström, L. Sun and H. Imahori, Chem. Commun., 2016, 52, 13702–13705 RSC.
- O. Suryani, Y. Higashino, Y. Mulyana, M. Kaneko, T. Hoshi, K. Shigaki and Y. Kubo, Chem. Commun., 2017, 53, 6784–6787 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- E. Sebastian and M. Hariharan, ACS Energy Lett., 2022, 7, 696–711 CrossRef CAS.
- C. Lin, T. Kim, J. D. Schultz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2022, 14, 786–793 CrossRef CAS PubMed.
- Y. Hong, F. Schlosser, W. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2022, 144, 15539–15548 CrossRef CAS PubMed.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377 CrossRef CAS PubMed.
- J. T. Kirner and R. G. Finke, ACS Appl. Mater. Interfaces, 2017, 9, 27625–27637 CrossRef CAS PubMed.
- C. F. Jewell, A. Subramanian, C. Y. Nam and R. G. Finke, ACS Appl. Mater. Interfaces, 2022, 14, 25326–25336 CrossRef CAS PubMed.
- F. Ronconi, Z. Syrgiannis, A. Bonasera, M. Prato, R. Argazzi, S. Caramori, V. Cristino and C. A. Bignozzi, J. Am. Chem. Soc., 2015, 137, 4630–4633 CrossRef CAS PubMed.
- R. Emerson and W. Arnold, J. Gen. Physiol., 1932, 16, 191–205 CrossRef CAS PubMed.
- R. B. Park and J. Biggins, Science, 1964, 144, 1009–1011 CrossRef CAS PubMed.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS PubMed.
- S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS PubMed.
- M. D. Doyle, A. Bhowmick, D. C. Wych, L. Lassalle, P. S. Simon, J. Holton, N. K. Sauter, V. K. Yachandra, J. F. Kern, J. Yano and M. E. Wall, J. Am. Chem. Soc., 2023, 145, 14621–14635 CrossRef CAS PubMed.
- P. Albanese, S. Tamara, G. Saracco, R. A. Scheltema and C. Pagliano, Nat. Commun., 2020, 11, 1361 CrossRef CAS PubMed.
- B. A. Rogers, H. I. Okur, C. Yan, T. Yang, J. Heyda and P. S. Cremer, Nat. Chem., 2022, 14, 40–45 CrossRef CAS.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS.
- G. A. Volpato, M. Marasi, T. Gobbato, F. Valentini, F. Sabuzi, V. Gagliardi, A. Bonetto, A. Marcomini, S. Berardi, V. Conte, M. Bonchio, S. Caramori, P. Galloni and A. Sartorel, Chem. Commun., 2020, 56, 2248–2251 RSC.
- F. Li, K. Fan, B. Xu, Q. Daniel, L. Sun, L. Li and E. Gabrielsson, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- K. R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef CAS PubMed.
- D. Wang, M. S. Eberhart, M. V. Sheridan, K. Hu, B. D. Sherman, A. Nayak, Y. Wang, S. L. Marquard, C. J. Dares and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8523–8528 CrossRef CAS PubMed.
- Y. Zhu, D. Wang, W. Ni, G. G. Gurzadyan, L. Sun, T. J. Meyer and F. Li, J. Mater. Chem. A, 2022, 10, 9121–9128 RSC.
- T. G. U. Ghobadi, A. Ghobadi, M. Buyuktemiz, E. A. Yildiz, D. B. Yildiz, H. G. Yaglioglu, Y. Dede, E. Ozbay and F. Karadas, Angew. Chem., Int. Ed., 2020, 59, 4082–4090 CrossRef PubMed.
- Y. K. Eom, L. Nhon, G. Leem, B. D. Sherman, D. Wang, L. Troian-Gautier, S. Kim, J. Kim, T. J. Meyer, J. R. Reynolds and K. S. Schanze, ACS Energy Lett., 2018, 3, 2114–2119 CrossRef CAS.
- L. Shen, S. Zhang, H. Ding, F. Niu, Y. Chu, W. Wu, Y. Hu, K. Hu and J. Hua, Chem. Commun., 2021, 57, 5634–5637 RSC.
- L. Wang, D. Mitoraj, S. Turner, O. V. Khavryuchenko, T. Jacob, R. K. Hocking and R. Beranek, ACS Catal., 2017, 7, 4759–4767 CrossRef CAS.
- M. Bledowski, L. Wang, A. Ramakrishnan, A. Bétard, O. V. Khavryuchenko and R. Beranek, ChemPhysChem, 2012, 13, 3018–3024 CrossRef CAS PubMed.
- M. Bledowski, L. Wang, A. Ramakrishnan and R. Beranek, J. Mater. Res., 2013, 28, 411–417 CrossRef CAS.
- E. Benazzi, K. Rettenmaier, T. Berger, S. Caramori, S. Berardi, R. Argazzi, M. Prato and Z. Syrgiannis, J. Phys. Chem. C, 2020, 124, 1317–1329 CrossRef CAS.
- S. Berardi, V. Cristino, M. Canton, R. Boaretto, R. Argazzi, E. Benazzi, L. Ganzer, R. Borrego Varillas, G. Cerullo, Z. Syrgiannis, F. Rigodanza, M. Prato, C. A. Bignozzi and S. Caramori, J. Phys. Chem. C, 2017, 121, 17737–17745 CrossRef CAS.
- A. Y. Ru Ng, B. Boruah, K. F. Chin, J. M. Modak and H. Sen Soo, ChemNanoMat, 2020, 6, 185–203 CrossRef.
- L. M. Reid, T. Li, Y. Cao and C. P. Berlinguette, Sustainable Energy Fuels, 2018, 2, 1905–1927 RSC.
- A. Y. Ru Ng, B. Boruah, K. F. Chin, J. M. Modak and H. Sen Soo, ChemNanoMat, 2020, 6, 185–203 CrossRef.
- M. Natali, A. Sartorel and A. Ruggi, Front. Chem., 2022, 10, 907510 CrossRef CAS PubMed.
- H. A. Beejapur, Q. Zhang, K. Hu, L. Zhu, J. Wang and Z. Ye, ACS Catal., 2019, 9, 2777–2830 CrossRef CAS.
- Y. Kawamata, M. Yan, Z. Liu, D. H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 390 CrossRef.
- R. Zhang, M. Shao, Z. Li, F. Ning, M. Wei, D. G. Evans and X. Duan, Chem. – Eur. J., 2017, 23, 8142–8147 CrossRef CAS PubMed.
- W. Song, A. K. Vannucci, B. H. Farnum, A. M. Lapides, M. K. Brennaman, B. Kalanyan, L. Alibabaei, J. J. Concepcion, M. D. Losego, G. N. Parsons and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 9773–9779 CrossRef CAS PubMed.
- E. Nikoloudakis, P. B. Pati, G. Charalambidis, D. S. Budkina, S. Diring, A. Planchat, D. Jacquemin, E. Vauthey, A. G. Coutsolelos and F. Odobel, ACS Catal., 2021, 11, 12075–12086 CrossRef CAS.
- D. F. Bruggeman, T. M. A. Bakker, S. Mathew and J. N. H. Reek, Chem. – Eur. J., 2021, 27, 218–221 CrossRef CAS PubMed.
- D. Antón-García, E. Edwardes Moore, M. A. Bajada, A. Eisenschmidt, A. R. Oliveira, I. A. C. Pereira, J. Warnan and E. Reisner, Nat. Synth., 2022, 1, 77–86 CrossRef.
- J. L. Zhuang, Y. M. Shen, Y. Xue, M. Yan, H. Cheng, Z. Chen, X. J. Yu, X. B. Lian and S. Bin Zhu, ACS Appl. Energy Mater., 2020, 3, 9098–9106 CrossRef CAS.
- G. A. Volpato, E. Colusso, L. Paoloni, M. Forchetta, F. Sgarbossa, V. Cristino, M. Lunardon, S. Berardi, S. Caramori, S. Agnoli, F. Sabuzi, P. Umari, A. Martucci, P. Galloni and A. Sartorel, Photochem. Photobiol. Sci., 2021, 20, 1243–1255 CrossRef CAS.
- R. Ciriminna, A. Fidalgo, L. M. Ilharco and M. Pagliaro, ChemistryOpen, 2018, 7, 233–236 CrossRef CAS PubMed.
- D. Liu, J. C. Liu, W. Cai, J. Ma, H. Bin Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef PubMed.
- D. Bruggeman, A. Laoprte, R. J. Detz, S. Mathew and J. N. H. Reek, Angew. Chem., Int. Ed., 2022, 61, e202200175 CrossRef CAS PubMed.
- M. Galeotti, M. Salamone and M. Bietti, Chem. Soc. Rev., 2022, 51, 2171–2223 RSC.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS.
- T. H. Yosca, T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Science, 2013, 342, 825–829 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–938 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kishi, H. Kotani, Y.-M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed.
- Y. H. Hong, J. W. Han, J. Jung, T. Nakagawa, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2019, 141, 9155–9159 CrossRef CAS PubMed.
- S. Fukuzumi, Y. M. Lee and W. Nam, ChemSusChem, 2019, 12, 3931–3940 CrossRef CAS PubMed.
- Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- Y. Yang, G. A. Volpato, E. Rossin, N. Peruffo, F. Tumbarello, C. Nicoletti, R. Bonetto, L. Paoloni, P. Umari, E. Colusso, L. Dell'Amico, S. Berardi, E. Collini, S. Caramori, S. Agnoli and A. Sartorel, ChemSusChem, 2023, e202201980 CrossRef CAS PubMed.
Footnotes |
| † The ΔG0 at 298 K for eqn (1)–(6) are calculated from the difference between the ΔG0formation of the products and of the reactants. The following values taken from a physical chemistry handbook were used (values in kJ mol−1): H2O (l): −237.13; CO2 (g): −394.36; CH4 (g): −50.72; CH3OH (l): −166.27; HCOOH (l): −361.35; C6H12O6, β-D-glucose: −910; NH3 (g): −16.45. By definition, ΔG0formation of H2, O2, and N2 is 0. |
| ‡ Ru(II) trisbipyridine chromophores have also been found to be capable of injecting electrons into TiO2 semiconductors from their reduced form, in an “anti-biomimetic pathway”, where the excited state of the dye is first reductively quenched by electron acceptors in solution (i.e. ascorbate) (see ref. 69). |
| This journal is © The Royal Society of Chemistry 2023 |