
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilicon quantum dots: surface matter, what next?
Deski
Beri

Chemistry Department, Faculty of Mathematics and Natural Science, Universitas Negeri Padang, Jl. Hamka, Air Tawar, Padang, 25132, Indonesia. E-mail: deski.beri@fmipa.unp.ac.id
First published on 13th March 2023
Abstract
Silicon quantum dots (SiQDs) are of great interest because they are believed to be harmless to living organisms, mainly due to their low toxicity. They have great potential for various applications such as in optoelectronics, photonics, and photodynamic therapy. As the Si core is susceptible to oxidation, efforts are needed to protect the core or to passivate it. This passivation/functionalization is aimed at both the protection of the core and the enhancement of the light emission. The importance of surface modification of SiQDs concerning the effect of quantum confinement and ligand type on their photoluminescence quantum yield is discussed in this review. Different functionalization reactions are described along with the precursors used. Among these are hydrosilylation reactions between the silicon core and the carbon of alkyl ligands and chromophoric ligands. Alkyl ligands protect the silicon core and provide chemical and light stability to the silicon. Ligands containing chromophoric groups also play a role in increasing the absorption of light, which in turn can increase the emission of PL light. These chromophoric ligands can also be used for optoelectronic, photonic, energy conversion, and biological purposes to enhance photophysical properties through energy transfer mechanisms. The energy transfer between the SiQDs and the ligands allows the triplet–triplet annihilation to enhance the emission and the generation of singlet oxygen. Finally, a brief description is given of the opportunities that SiQDs can offer and the progress made in recent years in the potential applications of SiQDs. They can be used as materials for light-harvesting antennas, bioimaging, luminescent solar concentrators, LEDs, and photosensors by exploiting triplet–triplet annihilation and singlet oxygen.
 Deski Beri | Deski Beri studied chemistry at the Universitas Negeri Padang (UNP) and the Institut Teknologi Bandung (ITB) in Indonesia. He left Indonesia to continue his PhD at the Faculty of Chemistry and Biosciences at the Karlsruhe Institute of Technology (KIT) in Germany, where he graduated in 2020. His research focused on quantum dot synthesis for photonic applications, which was conducted at the Institute of Microstructure Technology at the Helmholtz Research Centre, Karlsruhe. Upon his return to Indonesia, he was promoted to Assistant Professor at the Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Negeri Padang. He has published several papers in international journals, especially in the field of quantum dot synthesis and its applications. |
A. Introduction
The functionalization of silicon quantum dots (SiQDs) significantly improves their resulting physical and chemical properties.1–5 The synthesis of SiQDs begins with the formation of hydrogen-terminated SiQDs (H-SiQDs), which is a metastable state before passivation with more inert species such as alkyl, amine, carboxyl, et cetera. It is well known that H-SiQDs are highly susceptible to oxidation that destroys their ability to emit light, as the band gap of silicon dioxide (SiO2) is much more extensive than SiQDs functionalized with, e.g., alkyl groups.6,7 When H-SiQDs undergo oxidation, the emission properties of SiQDs will slowly blue shift along with the formation of a SiO2 layer – as a QDs envelope – continues to expand inward until the SiQDs lose their emission intensity, and the emission ultimately vanishes.8 Finally, the SiQDs experience instability as the reaction with oxygen makes the core size smaller due to the formation of the SiO2 layer on the surface. It is eventually followed by a significant reduction of the core size, resulting in particles losing their ability to emit light – due to the formation of a trap state at the boundary of the Si–core and SiO2 layers. Experimental data shows that the light intensity generated fades over time until it disappears thoroughly after a few hours or days of storage.9The FT-IR data in Fig. 1a shows that the vibrational bands at ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) : 950–1250 cm−1 and 2250 cm−1 – the characteristic vibrational peaks for Si–O stretching – gradually increased when H-SiQDs were kept under room/air atmosphere. Simultaneously the enhancement in these vibrational peaks is followed by the fading of the SiHx vibration peaks at : 2100 cm−1. So it is reasonable to suggest that the formation of Si–OH, Si–O–Si, and Si
: 950–1250 cm−1 and 2250 cm−1 – the characteristic vibrational peaks for Si–O stretching – gradually increased when H-SiQDs were kept under room/air atmosphere. Simultaneously the enhancement in these vibrational peaks is followed by the fading of the SiHx vibration peaks at : 2100 cm−1. So it is reasonable to suggest that the formation of Si–OH, Si–O–Si, and Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O occurs to replace hydrogen in Si–Hx (at the surface of SiQDs). Fig. 1b depicts the emission quenching due to oxidation. Freshly hydrogen-terminated SiQDs perform high photoluminescence (PL) and gradually fade due to oxidation. The insert in Fig. 1b shows the normalized PL spectrum blue shift due to oxidation.10,11
O occurs to replace hydrogen in Si–Hx (at the surface of SiQDs). Fig. 1b depicts the emission quenching due to oxidation. Freshly hydrogen-terminated SiQDs perform high photoluminescence (PL) and gradually fade due to oxidation. The insert in Fig. 1b shows the normalized PL spectrum blue shift due to oxidation.10,11
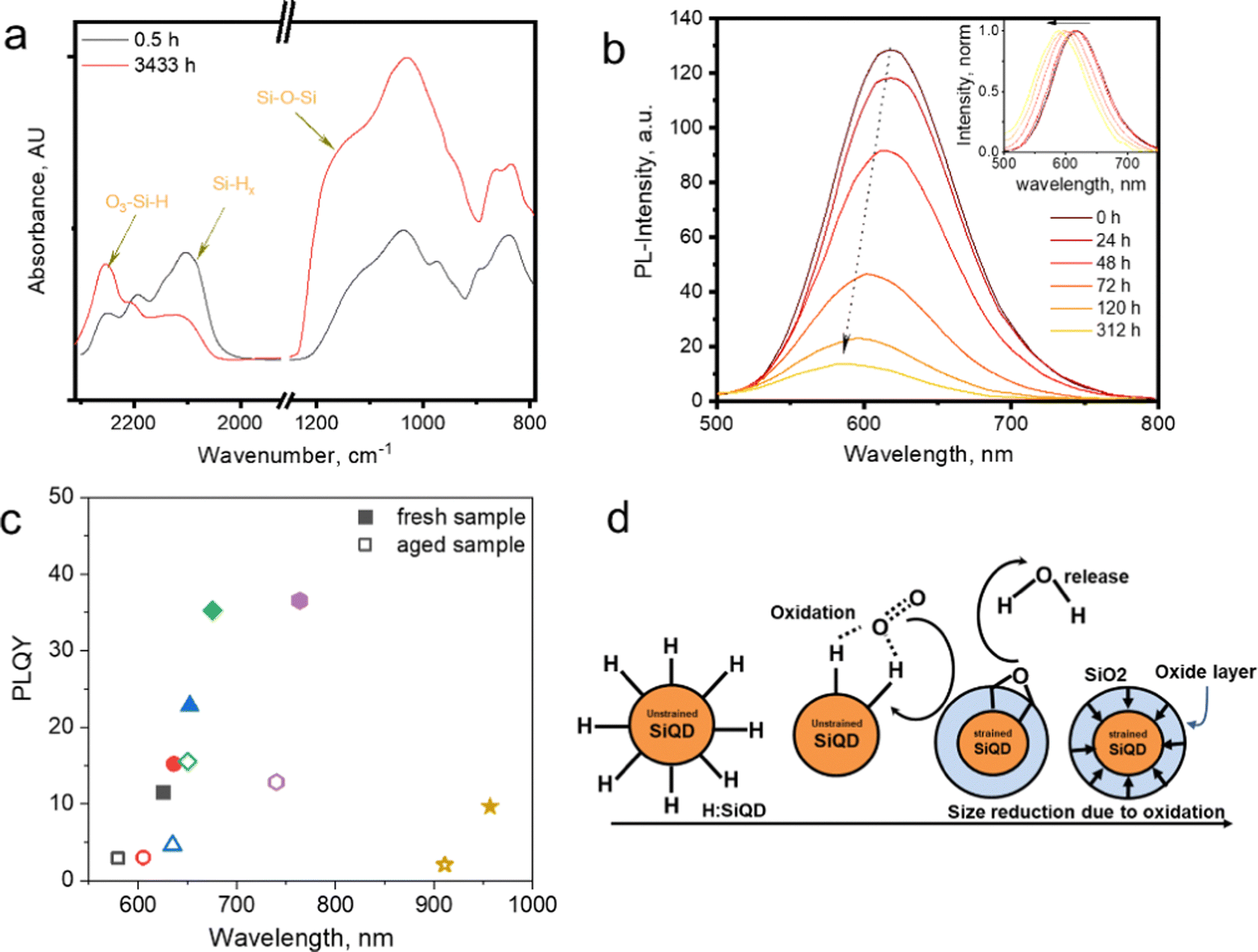 | ||
| Fig. 1 (a) FT-IR spectra of prolonged exposure of hydrogen functionalized SiQDs in the air. Adapted with permission from ref. 10. Copyright (2023) American Physical Society. (b) PL-Emission of long-time exposure H-SiQDs in the air, in the inset: PL-shift. Adapted with permission from ref. 12. Copyright (2014) John Wiley and Sons. (c) PLQY vs. PL-spectra maximum fresh and an aged sample of allyl phenyl sulphide-SiNCs. Adapted with permission from ref. 12. Copyright (2014) John Wiley and Sons, and (d) cartoon representation of SiO2 shell formation after oxidation, Adapted with permission from ref. 10. Copyright (2023) American Physical Society. | ||
In Fig. 1c, the Photoluminescence Quantum Yields (PLQY) of the six samples in the allylsulfide-SiQDs ensembles show emission quenching after prolonged storage in an ambient atmosphere.12 PLQY measurements for each ensemble were measured at maximum PL. After aging, each ensemble shows a similar trend, i.e., blue-shift and quenching. This tendency is well described in the cartoon representations in Fig. 1d. Hydrogen-terminated SiQDs react with oxygen and water vapor in the air to form Si–O bonds (e.g., SiO2).
Furthermore, long-term oxidation makes the core of SiQDs stiffen due to the formation of a SiO2 shell on the outer layer of the quantum dot. The formation reaction (oxidation) at the surface results in SiQDs shrinking and the emission blue shift. The construction of the SiO2 shell could explain the gradual fading of the PL, i.e., the formation of the trapping site.
The SiQDs produce two types of emission depending on the origin of the emission. The first type is called fast band (F-band) emission. It is a typical emission that spans around λ 250–600 nm. Some authors have reported resulting synthesis SiQDs that emit blue (λem ∼ 450 nm), green (λem ∼ 530 nm), and yellow/orange (λem ∼ 580 nm). The resulting emission does not correspond to quantum confinement theory but the ligand attached to the synthesized SiQD core. Among these emission characteristics is that the PL-decay (τPL) is so fast, on the order of nanoseconds instead of micro – to milliseconds as typical alkyl-SiQD.13,14
Fujimoto et al. (2022)15 elaborated on the difference between the F-band and S-band. The authors synthesized three types of SiQDs based on their emission.15 In short, the emission produced is in the RGB colors of red (λem 670 nm), green (λem 530 nm), and blue (λem 400 nm) for the manufacture of light-emitting displays (LEDs). Alkyl-functionalized SiQDs (R-SiQDs) perform the red emission (λem 670 nm)—the green emission (λem 530 nm) served by carbazole-functionalized SiQDs (Ca-SiQDs). At the same time, the blue emission (λem 400 nm) was performed by siloxane-functionalized SiQDs (Si-SiQDs). The conventional knowledge in quantum confinement dictates that the emission produced by carbazole-SiQDs and siloxane-SiQDs should not be green and blue since the particle sizes are ϕ 6.8 and ϕ 3.0 nm, respectively. However, due to the influence of the ligand, the emission produced is green (λem 530 nm) and blue (λem 430 nm). PL-decay measurements show that carbazole and siloxane functionalized SiQDs give a value of 5 ns independent of particle size as opposed to alkyl-SiQDs which are highly dependent on particle size. It should be underlined here that this F-band or fast decay hardly obeys the quantum confinement but is solely influenced by the nature of the ligand that binds to the SiQD core.15
In contrast, the second emission type is called the slow band (S-band). S-Band is typical emission that spans from orange/red to near-infrared electromagnetic radiation (590–1200 nm). The name slow is attributed to the PL-decay of the materials. Instead of the nanoseconds, the PL-emission of the materials is 103 magnitudes slower than the F-band. In short, materials belonging to the S-band have PL-decay in the order of micro – to the millisecond and are highly dependent on the quantum dot particle size. Generally, the larger the particle size, the slower the PL-decay. This S-band emission behavior fully complies with the rules of quantum confinement theory.
Fig. 2a shows the relationship between the PLQY measurement value of alkyl functionalized SiQDs (R-SiQDs) and the resulting particle size. PLQY measurements were performed on particle sizes from ϕ ∼ 1–10 nm.16–18 The alkyls used by the authors essentially have carbon chains length ranging from six (C6), eight (C8), ten (C10), twelve (C12), fourteen (C14), and sixteen (16). There is no separation for each alkyl based on chain length in the graph. The synthesis methods used in the charting are also diverse, such as plasma synthesis,7,18–26 Tilley method with silicon-rich precursors,27,28 disproportionation reactions of silicon-rich precursors (e.g., SiO, HSQ, and TES)4,6,11,12,15,17,29–70 followed by chemical etching, electrochemical synthesis, electrodeposition, chemical vapor deposition, and physical vapor deposition. Hydrosilylation reactions may vary, e.g., by thermal reactions in microwave reactors,53–55,68,69 organometallic catalysts, and radical initiator reactions.29,31,48,51 It is worth noting that there are no differentiated R-SiQDs based on alkyl chain length, precursors, and reaction methods to design Fig. 2.
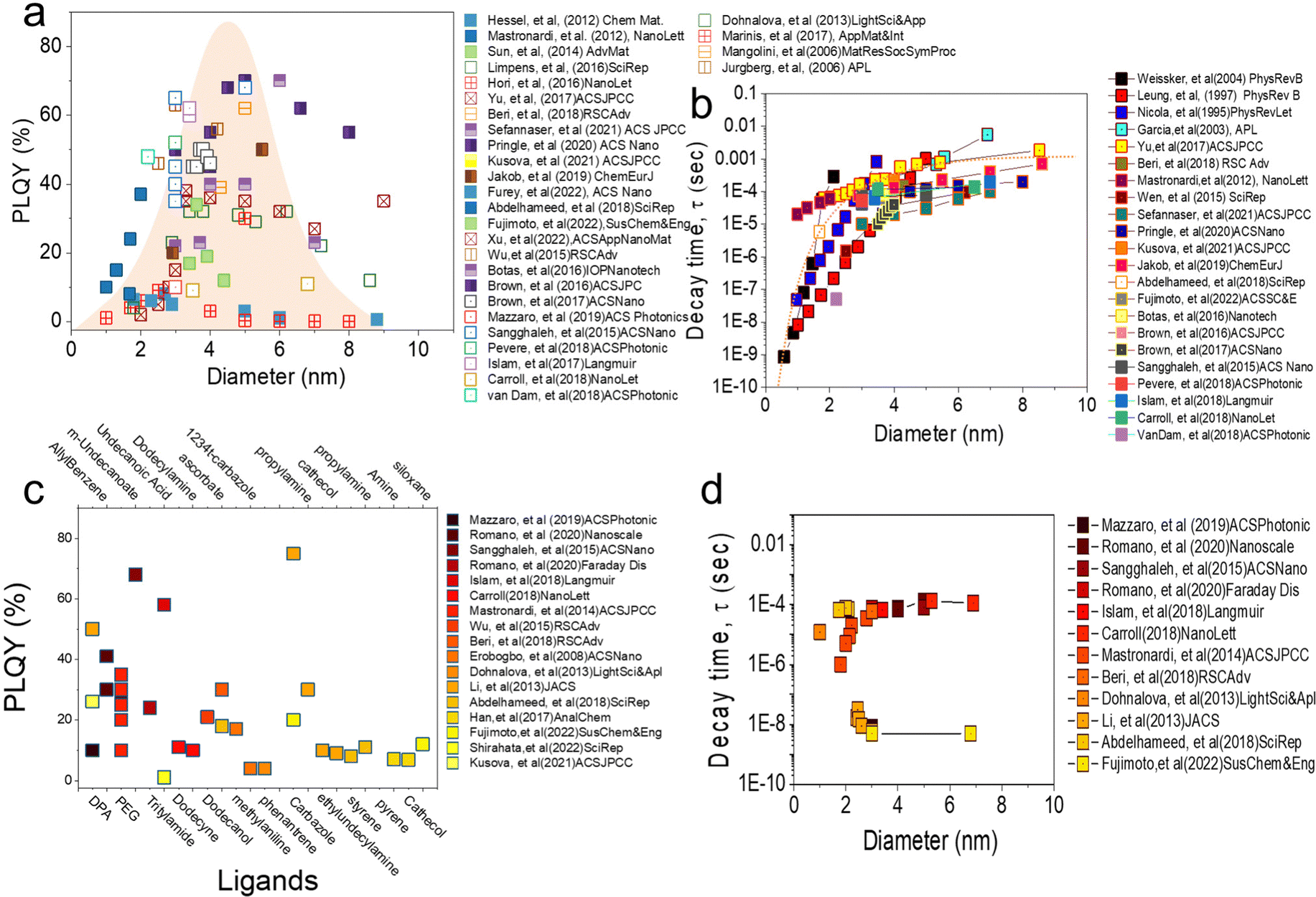 | ||
| Fig. 2 (a) The relationship between PLQY and diameter for alkyl-functionalised SiQDs reported by various authors in recent years. It can be seen that most papers report PLQY for particles between 1–10 nm with distribution patterns mostly concentrated between 2.5–6 nm. The imaginary distribution curve in the centre of the figure provides insight that most authors are working within this range. (b) The relationship between PL decay and the diameter of SiQDs reported by the authors in graph (a). It can be seen that the PL decay of alkyl-functionalised SiQDs is size dependence (10−9 to 10−6 s). (c) The relationship between PLQY and ligand for non-alkyl functionalized SiQDs reported by various authors in recent years. As in (a), the particle size is in the same range but the PLQY value can be very much different depending on the type of ligand connected to the SiQDs core. Finally (d) is a graph of the relationship between PL decay and diameter as supporting (c). The PL decay reported by these authors is in the nano second range regardless of particle size, however there are also reports that it depends on particle size. | ||
The relationship between PLQY and the diameter of alkyl functionalized SiQDs is shown in Fig. 2a. It can be seen that the PLQY tends to increase as the particle size increases from ϕ 1.0 nm to ϕ 3.0–6.0 nm. It then decreases when the particle size exceeds ϕ ∼ 6.0 nm. In other words, it resembles a bell curve. The maximum is distributed between ϕ 3.0–6.0 nm. This phenomenon has been reported before and is in line with the quantum confinement theory that has been the subject of many authors.65,71 As reported by many authors,18,21,44,72,73 the maximum height of PLQY so far is in the 70% range for alkyl-functionalized SiQDs. For example, recently Sefannaser et al. (2021)72 reported PLQY is ∼70% for dodecane (C12)-SiQDs colloids dispersion. It was worth noting that although the colloidal dispersion was encapsulated in the OSTE polymer, the PLQY of the SiQDs could be maintained.72 Previously, Pringle et al. (2020)18 also reported PLQY ∼70% for C12-SiQDs for ϕ ∼ 5 nm particle size.18 Previous reports have also shown the measured PLQY with a range of >40% for R-SiQDs having a particle size of ϕ ∼ 3–6 nm.7,19–21,24,26,38,44,73,74 This PLQY is very size dependent, as when the particle size is less than ϕ 2 nm, the measurement value tends to be small due to strong light absorption at the particle size ϕ ≤ 2 nm. As a result, the fraction of light emitted divided by the light absorbed is also tiny (hence, PLQY becomes small).70 On the other hand, when the particle size is larger than ϕ ∼ 6 nm, the fraction of light emitted is small, so the PLQY also gets smaller.63,70
Fig. 2b shows the relationship between PL-decay time and particle diameter reported by previous authors. Generally, there is a strong trend between the PL-decay behavior of R-SiQDs and their core size. For particles ϕ ∼ 1 to 2 nm in size, the PL-decay is in the order of nanoseconds (ns). The smaller the particle, the faster the PL-decays (i.e., only in a few ns). When the particle size is ϕ ∼ 3 to 4 nm, the PL-decay is tens to several hundred microseconds (μs). Then the curve becomes sloping for particles of ϕ ∼ 4 to 10 nm, even for particles ϕ ≥ 7 nm, data shows that the PL-decay rate is in milliseconds (ms). PL-Decay data from R-SiQDs is fully in agreement with the quantum confinement theory. For instance, Pringle et al. (2020) synthesized different diameter sizes of C12-SiQDs (i.e., ϕ ∼ 3 to ∼8 nm), and PL-decay measurements gave values from τPL ∼ 45 to 200 μs.18 Similarly, Sefannaser et al. (2021) also synthesized different sizes of C12-SiQDs (i.e., ϕ ∼ 2.8 to 7.0 nm), and the resulting PL-decay measurements were τPL ∼ 10 to 120 μs.72 Of the dozens of articles on R-SiQDs that the authors analyzed, their PL-decay is in the order of (μs to ms), and it is worth noting that almost all of them the quantum confinement theory.7,19–21,24,26,38,44,73–75
Fig. 2c shows the relationship between the PLQYs as a function of the type of ligand attached to the SiQDs. Contrary to our previous discussion, it turns out that the PLQY measurements deviate from quantum confinement theory for the ligands that are not derived from alkyl groups.37,76–78 Reports from various authors9,27,28,53–55,62,74,79–81 indicate that the attachment of more electronegative ligands causes a shift in the emission of SiQDs.82 For example, SiQDs attached to carbazole (Ca) shows a blue emission shift from λmax 780 nm for an average ϕ 3.0 nm diameter of R-SiQDs to λmax 530 nm.15,83 It is noteworthy that this green emission is also very characteristic, with a PLQY of ∼80%, as Li et al. (2013)84 previously reported.
Interestingly, by pairing different ligands, the emission can be shifted without changing the diameter of the QDs particles. Ideally, simply swapping the ligands could produce a full emission spectrum at the visible range. For example, to fabricate LEDs capable of emitting red, green, and blue light, Fujimoto et al. (2022)15 performed ligand tuning of ϕ ∼ 3 to 6 nm diameter SiQDs. As a result, red emission (λmax 780 nm) was produced by R-SiQDs, green emission (λmax 530 nm) by Ca-SiQDs, and blue emission (λmax 400 nm) by siloxane SiQDs. The PL-emission is quite stable with a relatively high PLQY and does not lose much of its PLQY when encapsulated in a polymer matrix. As it produces emissions in a wide range of wavelengths, the resulting PLQY values are pretty variable. Without considering the position of the PL-emission, the author's analysis shows that the resulting PLQY varies from 1% to close to 90%,44 depending on the ligand, PL-emission, and λ-excitation. It should be noted that the attachment of more electronegative ligands to SiQDs causes energy transfer and electron transfer to and from SiQDs. This transfer process can have positive and negative effects on the PLQY, depending on the direction in which the transfer occurs. There are still very open possibilities for further research into the photophysical properties of ligand tuning on SiQDs.37,44
Fig. 2d shows the relationship between the PL-emission decay time as a function of SiQD diameter. A more detailed analysis of the relationship between PLQY results as a function of diameter can be seen from the PL-emission decay time measurement. For the results reported in Fig. 2c, the PL-emission decay time might be in μs and can fulfill the quantum confinement rule as written by Romano et al. (2020),76 Mazzaro et al. (2019),82 and Beri et al. (2020).55 In their research, the authors report the PL-decay in the order of μs and rely heavily on the quantum confinement rule. It is worth noting that the ligands attached to SiQDs are alkyl ligands alternating with electronegative ligands (dyes). It might explain why the PL-decay is at μs.
In contrast, for SiQDs attached to electronegative ligands (i.e., carbazole and siloxane), one might study the research conducted by Fujimoto et al. (2022).15 The measurement results of PL-decay were in the order of ns (i.e., τPL 5 ns). The authors measured for two different particle sizes (ϕ 3.0 and 6.0 nm) and ligands (carbazole and siloxane) and concluded that the PL-decay of SiQDs binding to carbazole and siloxane ligands is on the order of ns and does not follow the quantum confinement rules.
To get a more comprehensive study on how the ligands play an essential role in the photophysical properties of SiQDs, Beri et al. (2018)53 conducted a systematic study of the effect of carbon length in ligands on the photophysical properties of SiQDs and found that the size of the alkyl chain length affects the PL-emission properties of SiQDs.53 The findings show a negative trend in the PLQY value against the alkyl chain length. In short, the longer the alkyl chain, the lower the PLQY value (Fig. 3a). The authors suspect that the surface coverage area significantly affects light emission. The conjecture is that SiQDs with less coverage tend to absorb more water vapor and oxygen from the environment, so they will be oxidized, resulting in these SiQDs emitting less intensity than SiQDs with more coverage. According to the authors,53 longer alkyl chains will find it challenging to find reaction points on the surface of the QDs due to the head's low flexibility and the tail's curling, which significantly reduces the effectiveness of the reaction. Thus, according to the authors, instead of bonding in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (one alkene head to one Si atom on the surface), the curled tail will reduce the chance of the alkene head on the long ligand reacting with the SiQDs core. As a result, this surface is not covered and will be oxidized. Lifetime measurements and PLQY measurements during storage support this claim.53 In addition to improving photoluminescent properties, ligand passivation has another function to maintain environmental and photostability. The second term is closely related to oxygen, water (hydrolytic), thermal, and photochemical reactions.53,85Fig. 3b depicts the long-term stability of difference alkyl-SiQDs in ambient.
:1 ratio (one alkene head to one Si atom on the surface), the curled tail will reduce the chance of the alkene head on the long ligand reacting with the SiQDs core. As a result, this surface is not covered and will be oxidized. Lifetime measurements and PLQY measurements during storage support this claim.53 In addition to improving photoluminescent properties, ligand passivation has another function to maintain environmental and photostability. The second term is closely related to oxygen, water (hydrolytic), thermal, and photochemical reactions.53,85Fig. 3b depicts the long-term stability of difference alkyl-SiQDs in ambient.
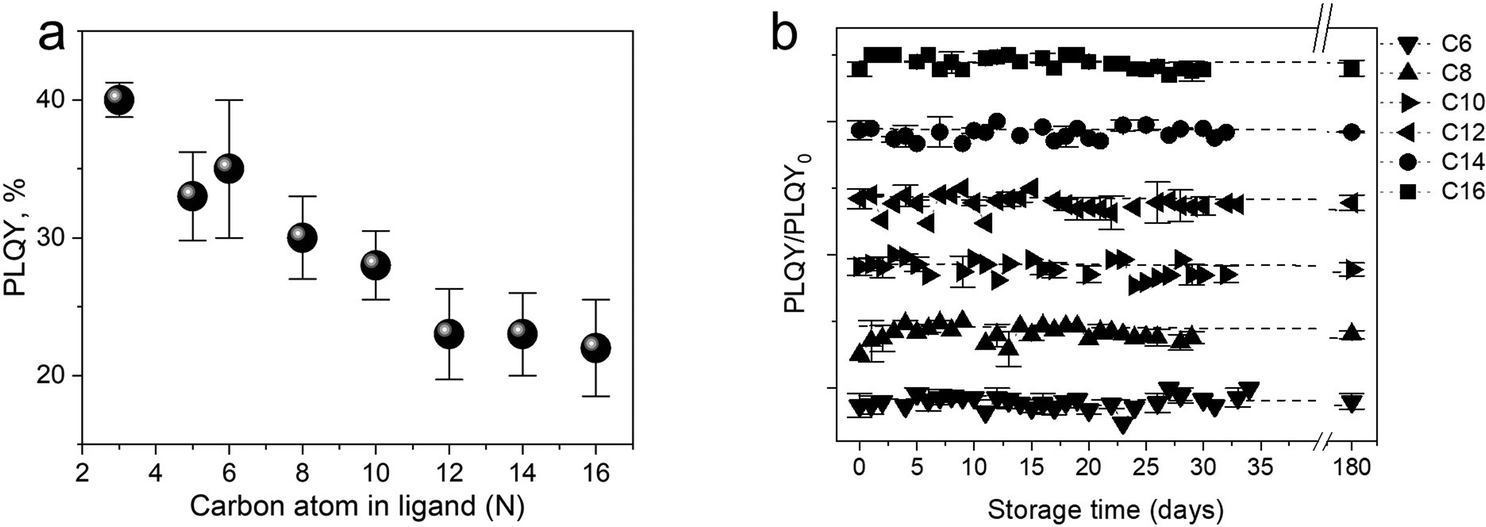 | ||
| Fig. 3 (a) PLQY trends as a function of alkyl chain length. (b) Photophysical stability of alkyl functionalized SiQDs as a function of storage. Adapted with permission from ref. 53. Copyright (2018) Royal Society of Chemistry. | ||
Research conducted by Sieval et al. (2000)86 demonstrated that monolayer alkyl functionalized SiQDs protect against oxidation for four months.86 X-ray photoelectron spectroscopy (XPS) measurements were performed to investigate the formation of SiO2 on the surface of alkyl-SiQDs. The results show that under ambient atmospheric storage conditions, proper alkyl-functionalized SiQDs showed almost no sign of SiO2 formation. Bhairamadgi et al. (2014)87 conducted a systematic study on hydrolytic ability (pH resistance) and thermal stability for monolayer functionalization of alkyl chain ligands with 6 (six) different sets of functional groups, namely -alkene, -alkynes, -chloro-silane, -thiol, -amine, and -phosphine (C–R, C–R, –SiCl3, –SO3, –NH3, and PO2−) on Si(111) and Si(100) surfaces.87 Hydrolytic stability was monitored using a static contact angle and XPS measurement. Silicon functionalized with an alkene and alkyne demonstrated good stability against acid (pH = 3.0) and base (pH = 11) for at least 30 days. In addition, thermal stability was conducted with XPS at elevated temperatures from room temperature (25 °C) up to 600 °C. The result shows that covalent bonding of alkene and alkyne functionalized Si demonstrated good stability from room temperature until Si–C bonding was degraded at 260 °C.87
Finally, photostability studies of SiQDs after being functionalized with alkyl(dodecyl) were performed by Wu et al. (2015).7 The results showed that PLQY SiQDs degraded by ∼20% (from the initial ∼60%) after four hours of illumination using UV light with a wavelength of 365 nm (power density similar to AM 1.5G of the Sun). It is suspected that the breaking of Si–H dangling bonds causes the degradation of PLQY due to illumination. The formation of dangling bonds creates electron traps, resulting in reduced emission. To prove this hypothesis about photostability, the author re-functionalized the degraded SiQDs, and the result was that the photo-degraded PLQY SiQDs could be fully recovered, which showed that his hypothesis was proven.7
B. Hydrogen terminated SiQD synthesis
There are many methods have been developed to produce hydrogen-terminated SiQDs. It is worth to mentions: silane gas plasma synthesis (SiH4),22,23,25,88,89 electrochemical etching,90–92 halosilane reduction reaction,93,94 oxide-rich silicon thermal synthesis,53–55 hydrogen silsesquioxane thermal synthesis,4,6,11,17,29–37,39–52,95 tetraethoxysilane (TES) thermal synthesis,96,97 pulsed laser ablation, and recently also the green synthesis of rice husk thermal pyrolysis.85 In general, the photoluminescence of the resulting material depends on the type of synthesis carried out. Because the photoluminescence properties of the synthesized SiQDs are strongly influenced by the particle size (that represents the quantum confinement effect) and the chemical characteristics of the species that cover its surface. As an illustration for the quantum confinement effect, SiQD particles with decyl functionalization (C10) with a particle size of ϕ ∼ 3.0 nm will provide PL-max at around λmax 600 nm, while SiQD particles with the same functionalization and size of ϕ ∼ 5.0 nm will provide PL-max at about λmax 800 nm. So it is very noticeable when we tune the particle size from ϕ ∼ 2.5 nm to ϕ ∼ 15 nm; PL-max is redshift gradually from λmax 590 nm to λmax 1100 nm.4,6,11,12,17,29–37,39–52,56–71,77,95,98–107Sychugov's group (2021)96 researched the synthesis of silicon quantum dots from three kinds of commonly used precursors, namely: silicon monoxide (SiO), silicon silsesquioxane (HSQ), and tetraethoxysilane (TES). In terms of cost, HSQ is ranked the highest because it is produced in limited quantities at a high price, so commercialization of this material in the future is quite limited. On the other hand, TES and SiO have competitive prices, so both precursors will have economic value in the future. The three precursors technically have almost the same synthesis treatment, namely heated in an Ar/H; 95%/5% atmosphere at a relatively high temperature of 600–1200 °C and then chemically etched with concentrated hydrogen fluoride (48–49% v/v) to deliberate H-SiQD from SiO2. The synthesis results show that among the three precursors, SiO shows the worst performance in terms of the solidity of the results. The particle size interval of SiQD spans from ϕ ∼ 3–6 nm, with a broad distribution, as seen in the TEM results. The PL-max is λmax 835 nm, with the PLQY value of the resulting SiQD also the lowest (∼15% in toluene) for the same treatment. The non-uniformity of the Si/SiOx material present in the material and the tendency to oxidize, as well as the instability of the material itself, makes the resulting SiQD tend to produce non-emissive materials.96
In comparison, HSQ gives more brilliant results because of the almost monodisperse particles (ϕ 4.0–5.0 nm) with a relatively narrow distribution. Its PLQY performance is also very good at 35% in toluene and ∼50% when embedded in the polymer (OSTE) with a PL-max of λmax 810 nm. In the meantime, the TES precursor gave the best performance, as the synthesis results showed that alkyl functionalized SiQDs derived from this TES precursor produced quantum dot particles that were nearly monodisperse, with a standard distribution spanned from ϕ 3.5–6.0 nm. The PL-max is λmax 810 nm with PLQY values of 40% in toluene and 55% when embedded in the polymer. The performance shown by TES is remarkable because, with a retail price of only ∼10% of the retail price of HSQ, it offered an awe-inspiring result, so in the future, TES is a precursor worth developing.96,97
C. Hydrosilylation reaction
Surface modification of nanocrystals or QDs has been a key factor in the development of nanotechnology. Surface modification has become possible by covalently attaching functionalized monolayers to silicon surfaces.86 The hydrosilylation reaction could be tailored to the creation of a functionalized monolayer. Hydrosilylation is an addition reaction of the unsaturated chemical group to hydrosilane. The reaction could be carried out by thermal induction or by the use of photochemistry. Thermally-induced hydrosilylation is carried out at elevated temperatures under a controlled atmosphere, while photochemical hydrosilylation is carried out at low temperatures using high-energy photons.108 Thermally-induced hydrosilylation requires relatively more energy and time than photochemical hydrosilylation. It also requires advanced chemistry and experienced scientists to carry out the experiments. To overcome these limitations, a different approach has been developed. The catalytic hydrosilylation using the metal complex catalyst and the radical-initiated hydrosilylation using radical initiators have been developed to reduce energy consumption.109 Another approach is by using microwave-assisted hydrosilylation. The microwave-assisted hydrosilylation reaction could be performed at a higher temperature in the bench-top reactor for less experienced chemists.2,53,107,110Fig. 4 depicts the proposed hydrosilylation reaction of SiQDs and alkene. | ||
| Fig. 4 Proposed mechanism of functionalization reaction of SiQDs under thermal and radical hydrosilylations. Successful hydrosilylation marked by substitution of hydrogen-terminated SiQDs demonstrated with alkyl-terminated SiQDs. Adapted with permission from ref. 111. Copyright (1993) American Chemical Society. | ||
The first work on thermally induced hydrosilylation was carried out by pyrolysis of diacyl peroxide in the presence of a hydride-terminated Si film. The introduction of alkene in the presence of radical diacyl peroxide results in a monolayer of alkyl-terminated silicon.111 The formation of alkyl radicals from the homolytic cleavage of diacyl peroxide initiated the proposed mechanism. The alkyl radical then abstracts H˙ from a surface Si–H group to form a silicon radical. Silyl radicals react rapidly with the olefins and perform a chain reaction on the propagation step. The termination step was performed when carbon-based radicals abstracted a hydrogen atom. The proposed mechanism of the hydrosilylation reaction of alkyl functionalized SiQDs by pyrolysis of diacyl peroxide is illustrated in Fig. 5.
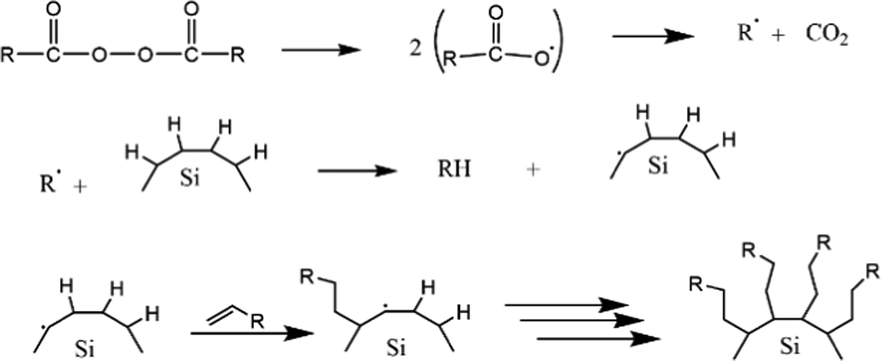 | ||
| Fig. 5 Proposed mechanism of thermal functionalization reaction of alkyl functionalized SiQDs. Adapted with permission from ref. 112. Copyright (2002) American Chemical Society. | ||
The hydrosilylation reaction could be tailored endothermically by thermal decomposition without a diacyl peroxide initiator at temperatures above 150 °C. At this temperature, the homolytic cleavage of Si–H proceeds independently according to the Si–H → Si˙ + H˙ reaction mechanism.112 The silicon radicals reacting with alkene yields correspond to the mechanism shown in Fig. 6. Based on the thermodynamic calculation, the bond formation of Si–H is thermodynamically stable with a bond energy of 80–90 kcal mol−1; therefore, this reaction will be prolonged at a temperature of 150 °C.113 However, the functionalization reaction at that temperature is much faster in the experiment. So, there must be another mechanism driving the reaction. A study by Coletti et al. (2006) proposed a new mechanism that could explain the high rate of reactions.113 The idea is that fluoride molecules left over from the etching step could act as promoters – in reaction, the F− works to acidify the silicon by forming intermediates. These intermediates help Si to capture alkene or alkyne molecules. The reaction mechanism is similar to nucleophilic substitution, and thermodynamically this reaction can occur at a significant rate at temperatures below 200 °C.113 The description of the hydrosilylation reaction is shown schematically in Fig. 6.
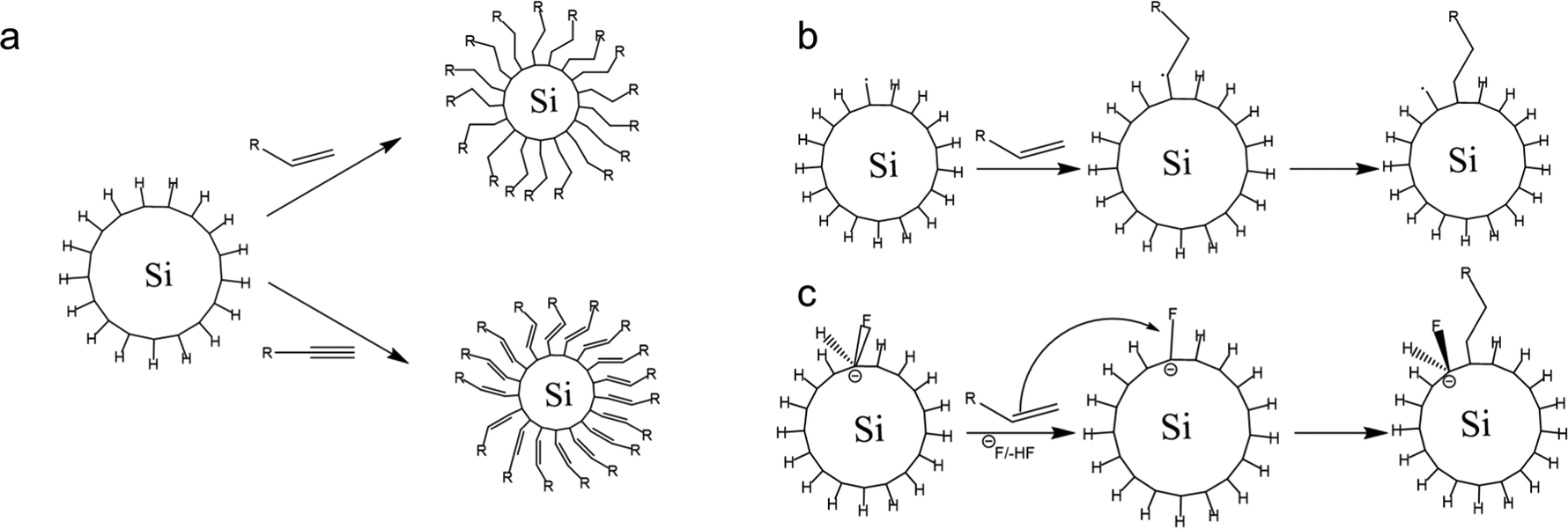 | ||
| Fig. 6 Schematic of (a). 1-Alkene and 1-alkyne hydrosilylation reaction of hydrogen-terminated SiQDs, (b). Radical-based hydrosilylation reaction, and (c). A fluoride-assisted hydrosilylation reaction mechanism. Adapted with permission from ref. 113. Copyright (2006) American Chemical Society. | ||
The other reaction is called photochemical hydrosilylation. High thermal exposure should be avoided in certain cases to maintain the expected results. Photochemical hydrosilylation was developed to minimize the use of thermal energy. High energy UV light has been used to induce homolytic cleavage of the Si–H bond to form a silyl radical and hydrogen radical. Silyl radicals will react with alkene or alkyne to form alkyl-functionalized silicon.112 The brief mechanism of photochemical hydrosilylation is shown schematically in Fig. 7.
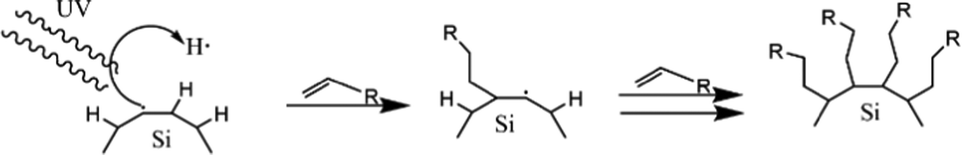 | ||
| Fig. 7 Proposed mechanism of photochemical hydrosilylation of alkyl functionalized SiQDs. Adapted with permission from ref. 112. Copyright (2002) American Chemical Society. | ||
In addition to thermal efficiency, photochemical hydrosilylation is also advantageous for forming monolayer surface functionalization.29,40,41 The drawbacks of the photochemical reaction are the limited number of reagents that can be used and that the reaction is only effective on SiQDs of ∼5 nm or less.28
Catalytic hydrosilylation could be performed using metal-complex catalysts such as platinum (Pt),114 rhodium (Rh),112 palladium (Pd),112 copper (Cu),115 boron (B),43 phosphor(P),37 and aluminum (Al).31,43,56,116 The main advantage of using metal-complex catalysts is that they significantly reduce the temperature reactions. However, the deposit metal complex on the surface of silicon is the problem so far. The catalyst deposit tends to reduce luminescent properties because it pronounces to absorb oxygen.112
Organolithium-mediated functionalization reactions are a type of chemical reaction that has been carried out for a long time. Since the pioneering work of Wilhelm Schlenk and Joanna Holtz in 1917, organolithium compounds have made a significant contribution to the development of chemical reactions. Nowadays, the widespread use of organo-alkali metal catalysts makes it possible to carry out functionalization reactions exclusively with organolithium, sodium, potassium, rubidium, and cesium. Recent findings show that organo-Na and organo-K have capabilities that are sometimes even better than organo-Li. The advantage is that Na and K are much more abundant and more evenly distributed on the surface of the earth than Li.1
An example of an organolithium-mediated functionalization reaction on SiQDs is an attachment of hexyl ligands on the surface of SiQDs cores. Because of its high volatility, hexene (C6) is very difficult to be functionalized to SiQDs by thermal functionalization. The use of an organolithium-mediated functionalization reaction is a very reasonable choice. Cheong et al. (2022) used hexyl-Li precursor in the functionalization reaction to obtain the results, as shown in Fig. 8.95 There is another method of functionalizing SiQDs called radical-initiated hydrosilylation. The reaction could be carried out at relatively low temperatures using materials called radical initiators. Moran & Carter (2009)117 conducted a functionalization reaction of hydride-terminated silicon using 2,2′-azobis(2-methylpropionitrile) (AIBN) as a radical precursor. The reactions were completed after 24 h at 60 °C and 30 min at 90 °C. The graft polymer layer formed on the surface of the silicon was investigated using XPS and contact angle measurements. The polymer layer was intended to guarantee surface passivation and coverage.117 Later, Yang et al. (2015)51 conducted a systematic study on the functionalization reaction of hydride-terminated SiQDs using radical AIBN initiators and benzoyl peroxide (BP).51 The significant results were that the hydrosilylation reaction could be performed at a slightly lower temperature of 60 °C for AIBN and 85 °C for BP. A radical initiator for different functional groups (i.e., alkene, alkyne, carboxylic acid, and ester) could be successfully performed, and the resulting functionalization was monolayer surface passivation.17,51,78 Meanwhile, Yu et al. (2015)107 used an undecanoic acid initiator to produce styrene-functionalized SiQDs.107 The main advantage of using undecanoic acid is that the reaction could be tailored at room temperature with promising results.107 PL-Emission of (undecanoic acid-promoted hydrosilylation) styrene-functionalized SiQDs was comparable to that of classical decyl-functionalized SiQDs.107Fig. 9 depicts the proposed mechanism of a radical-initiated hydrosilylation reaction.
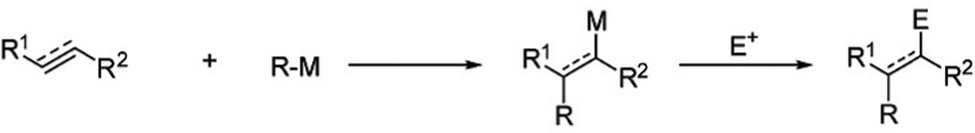 | ||
| Fig. 8 Proposed mechanism of organoalkali metals-mediated functionalization reaction to unsaturated substrates. Adapted with permission from ref. 1. Copyright (2020) Royal Society of Chemistry. | ||
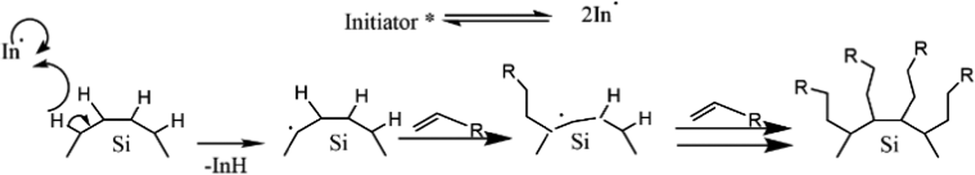 | ||
| Fig. 9 Proposed mechanism of radical initiated hydrosilylation of alkyl functionalized SiQDs. Adapted with permission from ref. 51. Copyright (2013) American Chemical Society. | ||
Beri et al. (2018) conducted alkyl functionalization using microwave-assisted hydrosilylation. In principle, the reaction is another type of thermally induced hydrosilylation by molecular heating stimulation.53 The reactant is irradiated with microwave radiation to stimulate vibration at the molecular level. Heat is generated by dipolar polarization and ionic conduction, where the first term is attributed to molecules having dipoles gradient (permanent or induced dipoles), and the second term is mainly attributed to charged particles (ions).118 When a microwave frequency is applied, the dipoles of the samples align in the applied field—the oscillation frequency of the microwave radiation forces the molecule to oscillate to compensate for the applied frequency. Molecular re-alignment across the applied field creates friction and dielectric loss, leading to elevated temperature (dielectric heating). The amount of heat generated by this method depends on the ability of the molecule to re-align itself against the frequency of the applied field. If the molecules can re-align faster than the applied field, no heating occurs.118 Similarly, ionic particles oscillate to a microwave frequency. Oscillation causes collisions with neighboring particles and excites a chaotic system to produce heat.119
Boukherroub et al. (2003) described the functionalization reaction of hydrogen-terminated porous silicon with 1-alkene under microwave irradiation to give monolayer alkyl functionalized porous silicon.120 The reaction rate was increased significantly, and the result was monolayer alkyl functionalized of porous silicon. By using a microwave reactor, there are opportunities to perform functionalization using a wide range of functional group materials. The functionalized product was robust, highly stable, and had higher surface coverage.120 The proposed mechanism of the microwave reactor depicts in Fig. 10.
 | ||
| Fig. 10 Proposed mechanism of microwave hydrosilylation reaction of alkyl functionalized SiQDs. Adapted with permission from ref. 120. Copyright (2003) American Chemical Society. | ||
There is a dispute regarding the role of microwave radiation in hydrosilylation. Some people believe that the increase in reaction rate is due to a microwave energy stimulating reaction process, whereas others believe that microwave radiation does not have any role in the reaction process. The second opinion argues that the reaction rate increases because it proceeds at a higher temperature. To study the effect of microwave radiation on a hydrosilylation reaction, Sun et al. (2013) conducted a kinetic study to understand the process by comparing the rate and extent of the reaction.69 Both reactions were tailored in the same condition except for hydrosilylation methods. One sample was functionalized using conventional thermal hydrosilylation, and the other was functionalized using a microwave reactor. However, the temperature and time were adjusted similarly at 170 °C for 60 minutes. The extent of the reaction was monitored by FT-infrared spectroscopy to see the number of Si–C formations as a function of time. The rate constant for both hydrosilylation methods was deduced from the quantitative measurement of Si–H replacement during the functionalization of decyl-SiQDs as a function of time relative to the original Si–H hydrogen-terminated SiQDs. In the result, both functionalization methods presented relatively similar rate constants: k = (15 ± 1) × 10−3 minutes−1 for conventional thermal functionalization, and k = (12.3 ± 0.6) × 10−3 minutes−1 for microwave reactor. Hence, microwave radiation does not influence a hydrosilylation reaction.69
The advantage of microwave hydrosilylation is the homogeneity of the internal heat, as the heating caused by molecular resonance towards microwave frequency intramolecularly, the heat efficiency is maximum, and heat loss is minimum. This also results in higher surface coverage of alkyl functionalization because every single bond is affected by vibrations and tends to be involved in the reaction. Because some reactions must be conducted for hours with conventional thermal hydrosilylation, the period could be compressed into minutes in a microwave reactor at higher temperatures. The reaction in a microwave reactor is, therefore, time-efficient.118,119 Reactions in the microwave reactor could proceed above the reactants' boiling point under so-called solvothermal conditions, so the reactants' volatility is not an issue. Likewise, the reaction is easy to handle because it is performed in the safety-proof bench-top reactor.
Last but not least, monolayer alkyl functionalized SiQDs can be achieved by strictly controlling the reaction conditions. However, the main drawback of using a microwave reactor so much depends on a relatively expensive instrument. Also, the reaction was conducted at a slightly higher temperature, ∼200–270 °C, and the grafted polymer layer is most likely to form outside the surface of SiQDs. Multilayer (corona) guarantees the ligand-to-surface ratio but also enhances the hydrodynamic ratio of individual SiQDs.
Nevertheless, the hydrosilylation reaction is conducted to complete the surface modification. The reaction could be performed using thermally induced hydrosilylation, metal-complex catalysts, and photochemical hydrosilylation. Thermally-induced hydrosilylation could be performed above 150 °C. However, the high reaction temperature tends to form multilayer passivation that negatively impacts photophysics. Therefore, some methods have been developed to lower the reaction temperature, including metal-complex catalysts or introducing initiators. Some metal ion complexes have been used as a catalyst and initiators, but the deposit of metal ions on the surface of SiQDs could create another problem. Hence, radical initiators, e.g., AIBN, AIBCN, BP, and undecanoic acid, have been developed as promising candidates for (room-temperature) thermal-initiated hydrosilylation.
D. Synthesis of dye-functionalized SiQDs
Chromophore-assisted photoluminescent SiQDs
One of the simple chromophores functionalized SiQDs is styrene terminated SiQDs, synthesized by Yu & Korgel (2015).107 The synthesis started from the annealing of silicon-rich oxide, HSQ (Fox-16 from Dow Corning), under an inert atmosphere and then followed by wet chemical etching using hydrogen fluoride and hydrochloric acid to form hydrogen-terminated SiQDs. Functionalization with styrene was performed at room temperature using undecanoic acids as a catalyst. The result was a monolayer styrene passivation with photophysical properties similar to the general alkyl passivation of SiQDs. PL-Spectra demonstrated a typical broadband emission with a maximum at λmax 615 nm under 295 nm λ-excitation. PLQY measurements gave a 12% yield for PL-emissions in the 490–1100 nm range. Compared to typical alkyl functionalized SiQDs (i.e., decyl-SiQDs), the absorption and the emission spectra have similar shapes, but the PL-maximum of styrene-SiQDs underwent a slight blue shift of ∼20 nm. Indeed, that subtle blue shift is because room temperature hydrosilylation is more efficient in forming a monolayer, whereas thermally induced hydrosilylation tends to embed SiQDs in the polystyrene layer, resulting in particle size enlargement. The PL lifetime measurements of styrene-SiQDs correspond to the PL lifetime of typical decyl or dodecyl-SiQDs. Therefore, the covalent attachment of styrene molecules to the surface of SiQDs does not change the photophysical properties of SiQDs.107The author compares his findings with their previous research group on SiQDs capped with polystyrene.49,107 The synthesis process was the same but was conducted thermally at 140 °C. For the same SiQDs core, the PLQY value is also the same, but the difference is that the PL-emission is redshifted by 20 nm. The authors proposed that due to the formation of multilayers on polystyrene encapsulated SiQDs, causing the dimensions of the QD's size are much more extensive. Redshift data on PL-emission and PL-decay fully support the conclusion. Herein, styrene-functionalized SiQDs have similar characteristics as typical monolayer alkene-functionalized SiQDs, while on the other hand, polystyrene-functionalized SiQDs have features relevant to typical multilayer capped SiQDs. Thermal Gravimetry Analysis (TGA) measurement data fully support the conclusion, where for octene functionalized SiQDs, the number of bound ligands is comparable to styrene functionalized SiQDs. Meanwhile, in terms of oxidation resistance, the authors showed that polystyrene functionalization is much more resistant than styrene functionalization. Since the formation of multilayers greatly helps to prevent oxidation of polystyrene functionalized SiQDs.49,107
Romano et al. (2020), Mazzaro et al. (2019), and Beri et al. (2020) show that organic dyes conjugated with SiQDs can enhance light absorption and PL-emission.50,55,105 The intense light absorption of the dye molecules can donate excitons to the SiQD core through energy transfer.28,83,121–123 The results show that the additional absorption from the ligand can enhance light absorption. PL-Emission of dye-SiQDs might be increased by excitons transfer. Locritani et al. (2014) demonstrated that SiQDs functionalized with pyrene groups can enhance the absorption of light-harvesting devices. The synthesis was carried out using the thermal decomposition of hydrogen silsesquioxane (HSQ) under an inert atmosphere and followed by wet chemical etching using hydrofluoric acid to produce H-SiQDs. The functionalization was carried out thermally at 170 °C utilizing a mixture of 1-(allylmethoxy) pyrene (py) and 1-dodecane (C12) given to two particle size classes of H-SiQDs (ϕ 2.6 and ϕ 5.0 nm). The resulting materials are ϕ 2.6 nm (C12)-py-SiQDs with a PL-spectra range of 600–900 nm and ϕ 5.0 nm (C12)-py-SiQDs with a PL-spectra range of 700–1200 nm. The absorption spectra show a significant increase at 300–400 nm due to the 3-fold light absorption of the pyrene group. PL-emission shows at λmax 400 nm and λmax 680 nm for a particle size of ϕ 2.6 nm and λmax 970 nm for ϕ 5.0 nm. Meanwhile, (C12)-SiQDs showed PL-emission at λmax 635 nm for a particle size of ϕ 2.6 nm and PL-emission at λmax 970 nm for ϕ 5.0 nm. Thus, the attachment of pyrene molecules is redshifted PL-emission of (C12)-SiQDs by ∼45 nm for a particle size of ϕ 2.6 nm but none for a particle size of ϕ 5.0 nm. The PLQY of ϕ 2.6 nm (C12)-SiQDs at λmax 635 nm is 11%, compared with the PLQY of ϕ 2.6 nm (C12)-py-SiQDs at λmax 680 nm is 8%. While the PLQY of ϕ 5.0 nm (C12)-SiQDs at λmax 970 nm is 45%, compared with the PLQY of ϕ 5.0 nm (C12)-py-SiQDs λmax 970 nm is 40%. Comparing the PLQY values of pyrene moieties attached and not attached to (C12)-SiQDs, the authors claim ∼95% energy transfer from the pyrene moieties to the ϕ 2.6 nm particle size SiQDs leading to a significant brightness increase of 300% in the visible range and an additional 78% in the near-infrared.103
Hence, the experiment demonstrated that pyrene (a chromophore) could enhance the light absorption and PL-emission of SiQDs. The light absorption enhancement was performed by additional strong absorption from the pyrene molecule at λmax 300–400 nm, whereas PL-emission enhancement was performed by energy transfer from the pyrene molecule to the core of SiQDs. Overall, a factor of 300% enhanced the brightness of conjugate materials due to the contributions of pyrene and (C12)-SiQDs.
Fermi et al. (2015) conducted a slightly similar experiment to enhance the absorption of SiQDs at a visible range of electromagnetic radiation.99 The chromophore of tetraphenylporphyrin Zn(II) (TPP-Zn) was used because it has a strong absorption at λmax 426 nm and λmax 560 nm. The SiQDs were synthesized using thermal decomposition of the hydrogen silsesquioxane (HSQ) and followed by wet chemical etching using hydrofluoric acid to yield two classes of H-SiQDs materials having particle sizes of ϕ 3 nm and ϕ 5 nm. The functionalization reactions were performed under thermal hydrosilylation by adding the given mixture of dodecene (C12) and tetraphenyl porphyrin Zn(II) (TPP-Zn) to the dispersion of H-SiQDs at a temperature of 170 °C. The resulting materials of ϕ 3 nm size (C12)-(TPP-Zn)-SiQDs exhibited two sorts of PL-emission maximum; at λmax 605 nm attributed to TPP-Zn emissions, and λmax 670 nm attributed to (C12)-SiQDs and some energy transfer from TPP-Zn. Whereas the resulting materials of ϕ 5 nm size (C12)-(TPP-Zn)-SiQDs also exhibited two sorts of PL-emission maximum at λmax 608 nm attributed to TPP-Zn emissions and PL-emission at λmax 905 nm attributed to (C12)-SiQDs and some energy transfer from TPP-Zn. PLQY of ϕ 3 nm size (C12)-(TPP-Zn)-SiQDs exhibited 7%, a reduction of 6% from the original 13% from (C12)-SiQDs. Whereas PLQY of ϕ 5 nm size (C12)-(TPP-Zn)-SiQDs exhibited 8%, which was significantly reduced from the original 42% of (C12)-SiQDs. Nanosecond lifetimes of dye molecules were quenched from τPL 2.3 ns to τPL 1.2 ns. Likewise, microsecond lifetimes of chromophore TPP-Zn attached to SiQDs were quenched from τPL 110 μs to τPL 70 μs for ϕ 3 nm particle size and from τPL 140 μs to τPL 100 μs for ϕ 5 nm particle size. Quenching PLQY and PL-decay demonstrated energy transfer from TPP-Zn to SiQDs. The calculation of efficient energy transfer leads to a 50% light synthesized process from TPP-Zn to SiQDs.99 In the experiment, the energy transfer efficiency of 50% was improved by covalently anchoring other chromophores such as perylene, phenanthrene, and other organic chromophores.
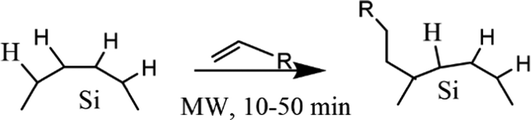
Beri et al. (2020)55 conducted a synthesis on dye-functionalized SiQDs. 3-Ethynyl perylene (p1) and ethylene-m-phenyl BODIPY (p2) were used in this experiment. The experiment was started by annealing SiO at a temperature of 900 °C to stimulate a disproportionation reaction. The optimized annealing conditions successfully produce hydrogen-terminated SiQDs (H-SiQDs) particles of ϕ ∼ 3.0 nm. Next, an etching process using 48% v/v HF was performed to eliminate the SiO2. The H-SiQDs were then functionalized in a microwave reactor using dyes as ligands. The resulting material showed that a low amount of dyes (2 mg) was used in the reaction to achieve 12-fold absorption enhancement at λmax 440 nm for the product of the reaction between 3-ethynyl perylene and H-SiQDs (i.e., C6-p1-SiQDs), and 3-fold enhancement at λmax 515 nm for the product of the reaction between ethylene-m-phenyl BODIPY and H-SiQDs (i.e., C6-p2-SiQDs). The efficient direct energy transfer (ηDET ≈ 99%) was measured from perylene and BODIPY chromophores to SiQDs. Notice PL enhancement of ∼270% and ∼140% was demonstrated for blue- and green-light excitation of the modified SiQDs (i.e., C6-p1-SiQDs and C6-p2-SiQDs). The absorption and PL-emission enhancement are summarised in Fig. 11.55
 | ||
| Fig. 11 (a) Absorbance of C6-SiQDs; C6-(p1)-SiQDs; C6-(p2)-SiQDs, (b) PL-emission of C6-SiQDs; C6-(p1)-SiQDs; C6-(p2)-SiQDs, under broadband excitation (λmax 435–550 nm). (c) Cartoon representation of attributed C6-SiQDs; C6-(p1)-SiQDs; C6-(p2)-SiQDs particles, and (d) PLQY values of C6-SiQDs; C6-(p1)-SiQDs; C6-(p2)-SiQDs measured at different excitation wavelengths. The PLQY values are the result of averaging for five independent synthetic batches. The error bars indicate the maximum and minimum values for five independent synthetic batches. Adapted with permission from ref. 55. Copyright (2020) Royal Society of Chemistry. | ||
Nevertheless, it is possible to enhance the light absorption and PL emission of SiQDs by energy transfer from chromophores (dyes) to SiQDs. The synthesis method could be started from H-SiQDs, followed by the functionalization of chromophores via hydrosilylations. The synthesis of H-SiQDs could be performed by thermal annealing of silicon-rich oxide followed by wet chemical etching or Tilley's method using precursors SiCl4 and TOAB reaction with reducing agent LiAlH4. This was followed by hydrosilylation using thermal hydrosilylation at 170 °C, radical-initiated hydrosilylation, and catalytic hydrosilylation. The resulting materials showed the redshift of PL spectra after functionalization with chromophores, except for one experiment carried out for styrene SiQDs. PLQY after chromophore attachments is generally down except for investigations conducted by Abdelhameed et al. (2018) that show up28 or remain constant.107 Hence, energy transfers from chromophore molecules to the core of SiQDs lead to PL enhancement.
Energy transfer mechanism in dye-functionalized SiQDs
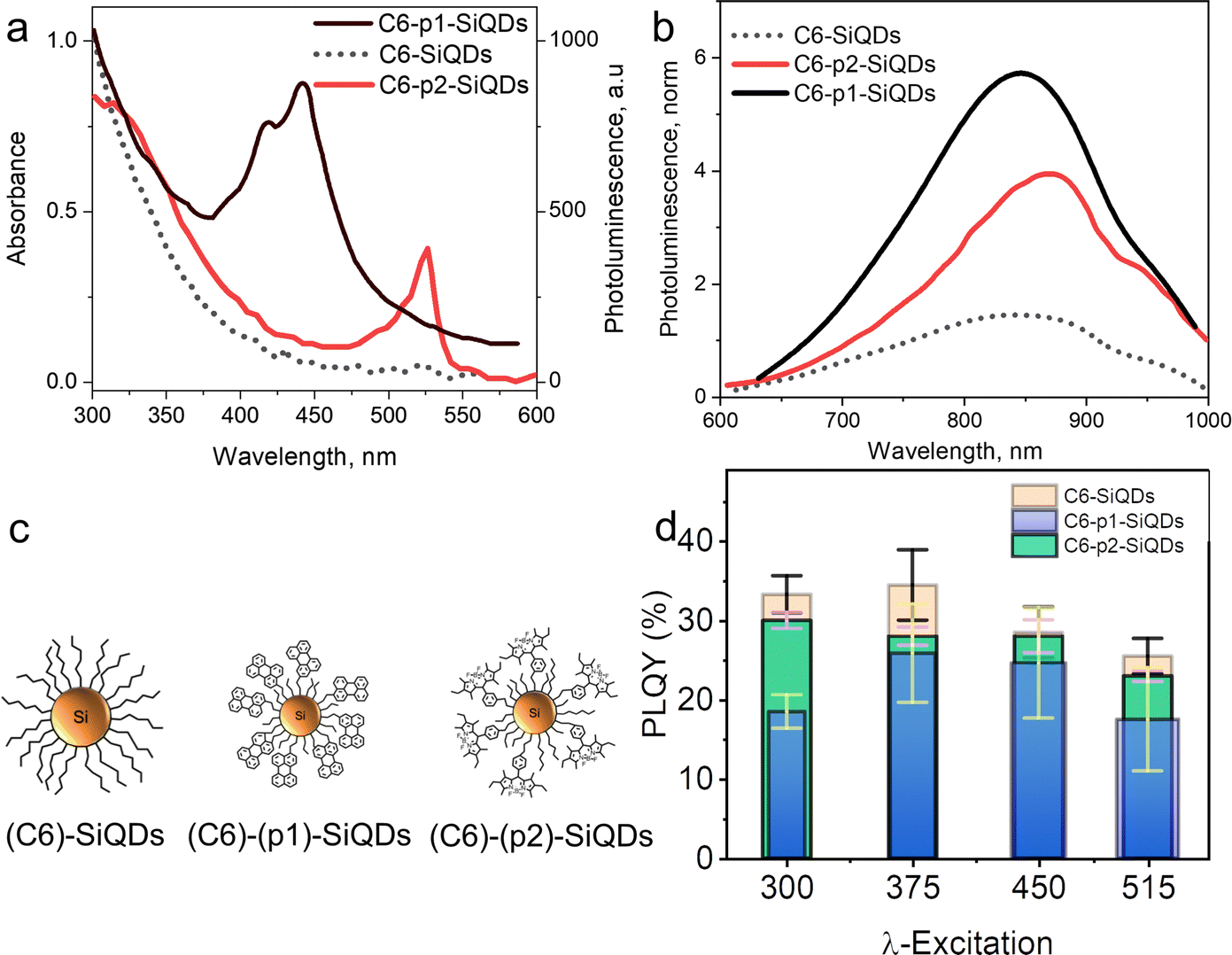
As demonstrated by Mongin et al. (2016), the possibility of exciton transfer in semiconductor QDs may open up new opportunities in physical and chemical sciences.124 Transient absorption spectroscopy was used to investigate the Dexter-like energy transfer from the QDs to the surface-anchored organic dyes. In addition, there is a possibility of triplet–triplet energy transfer from surface acceptor to dissolved organic dyestuff. There are indications that surface-anchored QDs or covalently anchored QDs could be used as organic sensitizers and that the sensitization process could be the result of a triplet–triplet energy transfer from the surface acceptor to free organic dyes (solutes). The sensitization process could lead to triplet–triplet annihilation upconversion (TTA-UC) and generate singlet oxygen.124,125 Singlet oxygen (1O2) was indicated by strong PL spectra at λmax 1270 nm.Xia et al. (2020)126 demonstrated that conjugated 9-ethyl anthracene (9EA)-functionalized SiQDs carry out the TTA-UC mechanism when dispersed in 9,10-diphenyl anthracene (DPA) The experiment was conducted under an inert nitrogen atmosphere using (9-EA)-SiQDs as a sensitizer and DPA as an emitter. The efficient Dexter-energy transfer was investigated by femtosecond transient absorption spectroscopy. Photoexciting sensitizer materials drove spin–triplet exciton transfer from silicon to anthracene through Dexter-like energy transfer with nearly 50% efficiency. With an excess of DPA molecules, these particles readily upconverted 488 nm photons to 420 nm photons with TTA-UC quantum yields exceeding ∼7%. The upconverting process occurred for (9EA)-SiQDs sensitizer only, whereas octadecene (C18)-SiQDs sensitizer with similar treatment did not show any upconverting phenomenon. The plotting of upconversion intensity as a function of excitation rate demonstrated that excitation rate threshold power fell from 0.95 W cm−2 for 488 nm excitation to 2 W cm−2 for 532 nm excitation. It was because the absorption of low-energy photons was significantly lower than that of high-energy photons. Another significant result was that the upconversion process depended on the particle size of SiQDs. SiQDs upconversion quantum yields increased from ∼0 to ∼7% as the particle size decreased from ϕ 3.6 to 3.1 nm. Authors described it as being due to reducing particle size, the bandgap energy laid above 9EA triplet state (∼1.8 eV), and the energy barrier was reduced for nanocrystals to molecule energy transfer and led to increasing upconversion quantum yields.126 One example of a TTA-UC mechanism illustrates in Fig. 12.
 | ||
| Fig. 12 (a) Triplet–triplet annihilation process of Perylene-functionalized SiQDs. (b). Triplet–triplet annihilation upconversion schematics by dye functionalized SiQDs immersed in the dye solvents. Adapted with permission from ref. 153. Copyright (2021) Royal Society of Chemistry. | ||
E. Applications of dye-functionalized SiQDs
As light-harvesting antenna
Research conducted by Locritani et al. (2014)103 attempted to attach chromophores (i.e., pyrene) to functionalized SiQDs. The pyrene group is tethered by (–C3H6–O–CH2–) to guarantee the long distance between SiQDs–core and pyrene groups. The authors investigated efficient energy transfers from chromophore to the SiQDs with two distinct particle sizes, ϕ 2.6 nm and ϕ 5.0 nm. The actual results of this experiment are efficient energy transfer from chromophore. For ϕ 2.6 nm particle size, it was ∼95%, leading to 300% brightness enhancement in the visible range, whereas for ϕ 5.0 nm particle size, it was ∼65%, leading to 78% brightness enhancement in near-infrared spectra.103Nevertheless, this experiment demonstrated that SiQDs could be used as a viable scaffold in light-harvesting antenna because chromophore enables the enhancement of molar absorption of SiQDs and leads to PL-emission or brightness enhancement.103,127 Besides pyrene, other chromophores could also be attached to SiQDs to get strong absorption at visible ranges. Ruthenium(II) bipyridine and Zinc(II) tetraphenylporphyrin were synthesized to manipulate SiQDs with strong absorption at visible ranges.76,77,128
In bioimaging
As light-harvesting materials, dye-functionalized SiQDs could be used as probe materials in photo imaging. Due to UV-excitation potentially damaging the biological tissues, other excitation methods must be developed in SiQDs probe-based bioimaging. One attractive method was a near-infrared photoexcitation via two-photon excitation (2P). Herein, 2P excitation was done by exciting dye-SiQDs using near-infrared radiation to produce emission at a PL-emission of SiQDs. Ravotto et al. (2017)105 conducted a systematic study on two-photon excitations of dye-SiQDs. SiQDs with particle size ϕ ∼ 5.0 nm were functionalized with 4,7-di(2-thienyl)-2,1,3-benzothiadiazole chromophores (TBT) and dodecyl chains exhibited PL-emission at near-infrared radiation (λmax 900 nm). The authors claimed that PL-emission could be obtained from 2P absorption by 960 nm excitations.105Furey et al. (2022)100 conducted two-photon excitation experiments in the near-infra-red (NIR) regions. The result in strong photoluminescence also at NIR gave a strong indication for potential applications in bioimaging. Two-photon excited photoluminescence spectra of dodecyl SiQDs with diameters ϕ 1.8 ± 0.2 nm and ϕ 2.3 ± 0.3 nm and dispersed in toluene. The cross-section is observed to be smaller for the smaller diameter SiQDs. SiQDs for bioimaging efficiency compared with other quantum dots and molecular fluorophores and found comparable.100 Later, Sarwat et al. (2022) conducted experiments to synthesize metal-functionalized SiQDs for bioimaging applications. Sc-SiQDs, Cu-SiQDs, and Zn-SiQDs were successfully synthesized, and their optimal PL and cytotoxicity were investigated. The Cu-SiQDs and Sc-SiQDs showed brighter PL and a lesser reduction in cell viability than the Zn-SiQDs. Notably, The Cu-SiQDs and Zn-SiQDs showed aggregation behavior. Hence, Sc-SiQDs appeared to be a better option for the future in vivo bioimaging of tear film.129 Although the PL and cytotoxicity of the materials seem promising, the hydrophobicity of the resulting compounds might be challenging.
Last but not least, Wei et al. (2022) conducted a synthesis of sulfhydryl-SiQD. The strong PL-emission might indicate that the material was suitable for photoimaging. The resulting emission was the characteristics of that long-lived photoluminescence of SiQDs and was completely different from the excitation wavelength.130 This invention might be useful as a probe in photo imaging.105 PL-Emission from 2P absorption demonstrated that dye-functionalized SiQDs could be used as a bioimaging probe.27,131,132
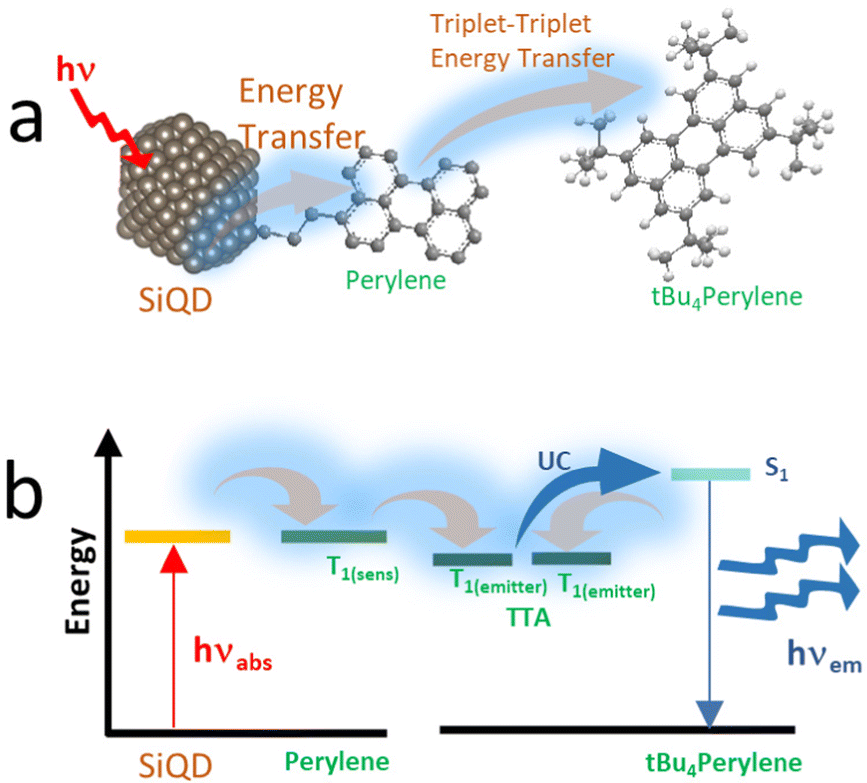
Fig. 13 shows PL images of zebrafish that have been treated with S-SiQD. The bright illumination of the zebrafish after treatment suggests that SiQDs have the potential for bioimaging. The authors report that these QDs have good photostability and biocompatibility in long-term measurements.130
 | ||
| Fig. 13 PL image of zebrafish after sulfhydryl-SiQD injections. Adapted with permission from ref. 130. Copyright (2022) Springer Nature. | ||
In luminescent solar concentrator
The SiQDs show similar behavior in luminescence properties compared with other direct semiconductors QDs. SiQDs demonstrate some advantages in toxicity and large Stokes shift compared to other semiconductors QDs. The significant Stokes shift makes it possible to design a semi-transparent solar luminescence concentrator (LSC) and aesthetically attractive.133 However, the main drawback is the relatively low absorption of SiQDs in the ultraviolet-A (UVA) (315–400 nm) region. Meinardi et al. (2017)26 demonstrated that an LCS device fabricated using alkyl functionalized SiQDs could exhibit power conversion efficiencies as high as 2.85% and 70% transmittance.26 Mazzaro et al. (2019) used 9,10-diphenylanthracene (DPA) functionalized SiQDs embedded in methyl methacrylate (MMA) polymer to enhance the absorption in the UVA range. The authors found that LSC's absorption was significantly enhanced due to dye attachment, and optical efficiency was enhanced as high as 4.25%, almost without sacrificing transparency.82 This remarkable result shows the possibility of dye-functionalized SiQDs as a lumiphore in LSC.One recent work was conducted by Ren et al. (2021)134 on tandem solar cells. Herein, a four-terminal tandem solar cell consisting of a luminescent solar concentrator (LSC) based on silicon quantum dots (SiQDs) in front of a perovskite solar cell (PSC). The LSC was covered with PMMA antireflection coating, which enhanced the transmission by up to 3% from the visible to the near-infrared range. The colloidal SiQDs in the LSC absorb the UV light and re-emit red PL, which propagates to the waveguide edges to generate electricity while allowing the rest of the incident sunlight to be absorbed in the back of the PSC. Notably, at 1.08 mg mL−1, although the tandem solar cell has about the same PCE as the bare PSC, the front SiQD-LSC absorbs 69% of the solar UV, making the back PSC more stable than the bare PSC.134 The arrangement of LSC-based SiQDs and Perovskite tandem solar cells illustrates in Fig. 14.
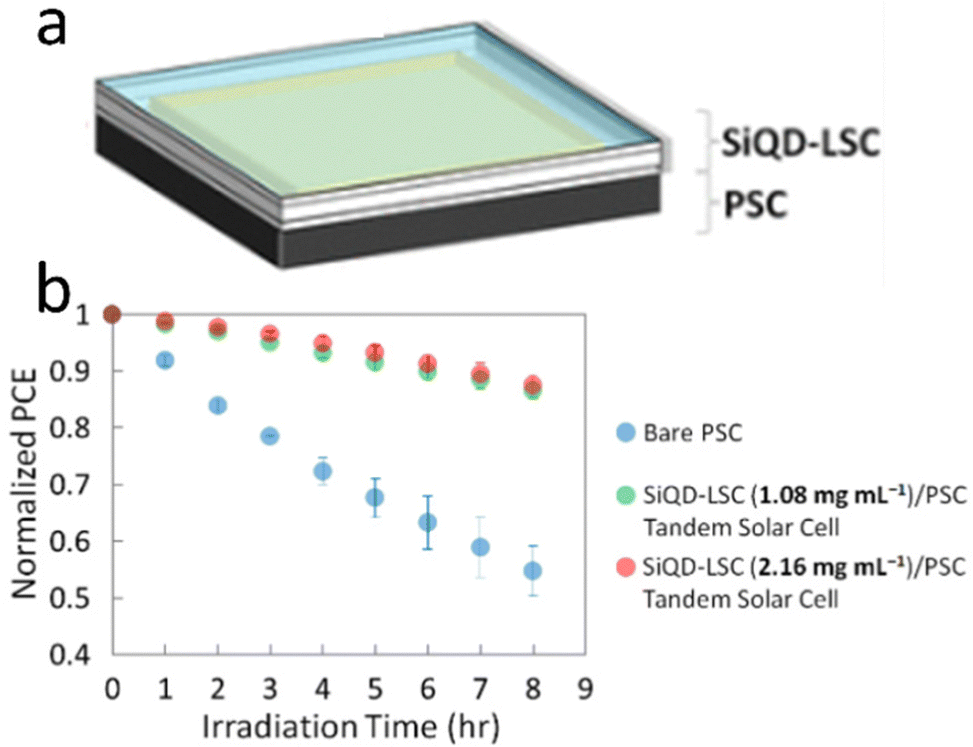 | ||
| Fig. 14 Tandem perovskite solar cell-LSC-based SiQD, (a) arrangement, and (b) PCE vs. Irradiation times. Reprinted with permission from ref. 134. Copyright (2021) American Chemical Society. | ||
In biosensors
Terbium-amine functionalized SiQDs was used as a biomarker to detect dipicolinic acid (DPA) as one constituent in Bacillus anthracis spores. By utilizing a biomarker made of terbium-amine-functionalized SiQDs, the detection of anthrax disease could be performed much more efficiently compared with polymerase chain reaction, which is a standard detection method today. The detection method compared the PL intensity of terbium-amine functionalized SiQDs in the interaction with DPA and PL intensity without interaction with DPA. The detection limit was about 24 nM, four orders of magnitude lower than the spores' infectious dosage (6 × 10−5 M required).135
Zhou et al. (2021)136 conducted a synthesis on acetic functionalized SiQDs to detect the imbalance of Zn2+/Cd2+˙ The imbalance reaction might lead to many severe diseases because antibiotics are deposited in animal products. Developing a functional material for detecting is challenging and attractive. The COOH-functionalized SiQDs were chelated with Eu(NO3)3 to form Eu3+-SiQDs and used as a PL probe. The emission wavelength of λmax 450 nm can be used sequentially to detect tetracycline (TCs) and Zn2+/Cd2+ by the PL resonance energy transfer (FRET) principle. The detection limit of TCs and Zn2+/Cd2+ are 0.2 × 10−6 m and 3 × 10−6 m, respectively, when the pH of the solution is 7.4. The result in Eu3+-SiQDs exhibited good stability (from 94.9% to 103.1%).136
Ma et al. (2021)137 synthesized water-dispersible silicon quantum dots (SiQDs) to quantify ascorbic acids in soda drinks. The synthesis was started with precursor 3-aminopropyltriethoxysilane (APTES) as a silicon source. MnO2 nanosheets (NS) are used as the quencher, while SiQDs is PL unit. The PL “switch-on” was attributed to ascorbic acid (AA) determination. MnO2 NS can effectively quench the PL of SiQDs because of the internal filtration effect. In the presence of AA, MnO2 is reduced to Mn2+, so the PL of SiQDs is partially recovered. The recovered PL intensity was related to the concentration of AA. Under the optimal experimental conditions, the linear response range of the assay to AA is 1–80 μM, and the detection limit is 0.48 μM. The method for determining AA has the advantages of simplicity, low cost, good selectivity, and sensitivity. The assay has been successfully applied to quantifying AA in beverage (mizone) samples, proving the assay's practicability.137 The illustrative model for SiQDs to detect ascorbic acids depicts in Fig. 15.
 | ||
| Fig. 15 Illustration of APTES functionalized SiQDs to detect ascorbic acids. Reproduced with permission from ref. 137. Copyright (2021) Elsevier. | ||
Another example of using SiQDs in bioimaging was done by Liu et al. (2022).138 The authors proposed a dual-signal fluorometric and colorimetric system based on silicon quantum dots (SiQDs) and 4-nitrophenol (4-NP) for pH and urease sensing. The SiQDs with PL-emission of λmax 460 nm were prepared in an aqueous phase. The absorption band of 4-NP at λmax 400 nm increased, and a color reaction from colorless to yellow occurred as the pH increased. The absorption of 4-NP overlapped quite well with the PL excitation spectrum of SiQDs, which can effectively quench the PL of SiQDs. The fluorometric and colorimetric pH-sensing systems exhibited a linear response to pH ranging from 6.0 to 7.8 with an interval of 0.2 pH. Urease could specifically hydrolyze urea to generate carbon dioxide and ammonia, causing a noticeable increase in the pH value. Thus, urease could also be detected quantitatively by the above dual-signal pH sensing system. The fluorometric and colorimetric methods' linear ranges for urease detection were 2–40 U L−1. The limits of detection were 1.67 and 1.07 U L−1, respectively.138
In light emitting diode
Yamada et al. (2020)139 aim at enhancing the electroluminescence of silicon quantum dot light emitting diode (SiQD-LED). The authors show that the LED emission can be adjusted from red to yellow by changing the particle size of SiQDs placed on the sandwich layer of the LED device. The SiQD particle sizes placed on the device are ϕ 2.7 nm, ϕ 2.2 nm, ϕ 1.9 nm, ϕ 1.7 nm, and ϕ 1.3 nm, giving a maximum PL-emission of λmax 755 nm, λmax 722 nm, λmax 670 nm, λmax 635 nm, and λmax 590 nm, respectively. At the same time, the peak external quantum efficiencies (EQE) for each particle size are 1.15%, 1.18%, 0.89%, and 0.12%, respectively.139 A schematic representation of the LED arrangement is depicted in Fig. 16.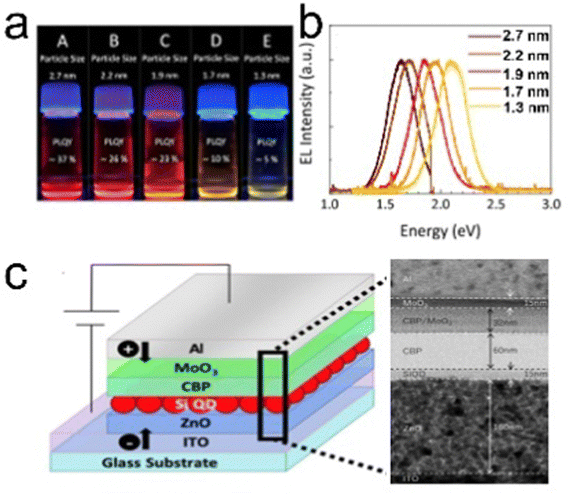 | ||
| Fig. 16 (a) Toluene dispersion of decane: SiQDs synthesized from disproportionation reaction of TES under 365 nm UV light irradiation, (b) PL-emission of toluene dispersion of different size SiQDs, and (c) potential arrangements of SiQD-LED and TEM image of sandwich layers. Adapted with permission from ref. 139. Copyright (2020) American Chemical Society. | ||
The synthesis of SiQDs begins with the disproportionation reaction of (HSiO1.5)n derived from triethoxysilane (TES) at high temperatures in an inert atmosphere. Separation of SiQDs from the SiO2 matrix was carried out by chemical etching using concentrated HF to obtain H-SiQDs. Functionalization with 1-decene was carried out through thermal hydrosilylation at 180 °C to obtain polydisperse decane-SiQD. Ensembles of various sample sizes were prepared through sedimentation using centrifuges. The results of sedimentation separation obtained ensemble samples with dimensions in order ϕ 2.7 nm, ϕ 2.2 nm, ϕ 1.9 nm, ϕ 1.7 nm, and ϕ 1.3 nm. PLQY measurements of the samples gave values of 37% for ϕ 2.7 nm, 26% for ϕ 2.2 nm, 23% for ϕ 1.9 nm, 10% for ϕ 1.7 nm, and 5% for ϕ 1.3 nm, respectively. The fabrication of the SiQDs layer on the equipment is done by spin coating with a thickness of ∼15 nm, which applies equally to each quantum dot size. Interestingly, the authors claimed that the wavelength of the LED produced could be adjusted by varying the size of the SiQD particles contained in the sandwich layer of the device.
In contrast, Cheong et al. (2022)95 recently demonstrated that the electroluminescence of colloidal SiQD particles with narrow emission in the range of ∼100–23 nm at visible light wavelengths (yellow, orange, red to infrared) could be tuned without changing the dimensions of the SiQD particles.95 Emission narrowing was achieved solely by adjusting the Fabry–Pérot cavities, and the voltage applied. The results show that the visual and spectral stability is remarkable at different voltages. The authors also showed that a wavelength interval in the visible (yellow to near-infrared) region could be achieved for the same SiQDs by adjusting the thickness of the SiO2 fill in the device array. This emission interval can then also be achieved by adjusting the Ag thickness. This arrangement in the LED configuration is a novelty that may be developed in the future. The SiQDs used in this study is hexyl-SiQDs synthesized by high annealing temperature of hydrogen silsesquioxane (HSQ) in an inert atmosphere to trigger the formation of SiQDs embedded in a silica matrix. Chemical etching using concentrated HF was required to separate the H-SiQDs from their SiO2 matrix to obtain SiQDs with a size of ϕ 2.78 ± 0.37 nm. The functionalization reaction was carried out using a technique called organolithium-mediated functionalization reaction. As a result, hexyl-SiQD was obtained, which gave significant emission with PLQY of 26% and λmax 726 nm. The thickness of the SiO2 and Ag layers in the device can be adjusted to optimize the emission according to the voltage used.95
Photothermal and nanotherapy (theranostics)
There was a report of carboxy-terminated silicon quantum dots (SiQDs) synthesized by Özbilgin et al. (2022)140 that exhibited high water solubility, luminescent light emission with high PLQYs, long-term stability of PL properties for cell monitoring, lower toxicity to cells and high photothermal response due to the high molecular coverage of the surface monolayer. Water-soluble SiQDs were prepared by thermal hydroxylation of 10-undecenoic acid as ligand, which was ensured by thermal disproportionation of TES hydrolyzed at pH 3 and subsequent etching with hydrogen fluoride. Functionalized SiQDs with 10-undecanoic acid (UA-SiQDs) were stable over time in hydrophilic solvents, including ethanol and water (pH 7). Their interaction with living cells was assessed by cell uptake, short-term toxicity, and long-term cytotoxicity for the first time. The results showed that UA-SiQDs are potential candidates for therapeutic products because they have good optical properties, allowing imaging over 18 days and a photothermal response with a photothermal conversion efficiency of 25.1% and direct detection of cell death by laser irradiation. UA-SiQDs have low cytotoxicity with full viability up to 400 μg mL−1 in the short term and 50% cell viability after 14 days of incubation at 50 μg mL−1.140 The illustrative photodynamic nanotherapy of 10-undecanoic acid functionalized SiQDs depicts in Fig. 17.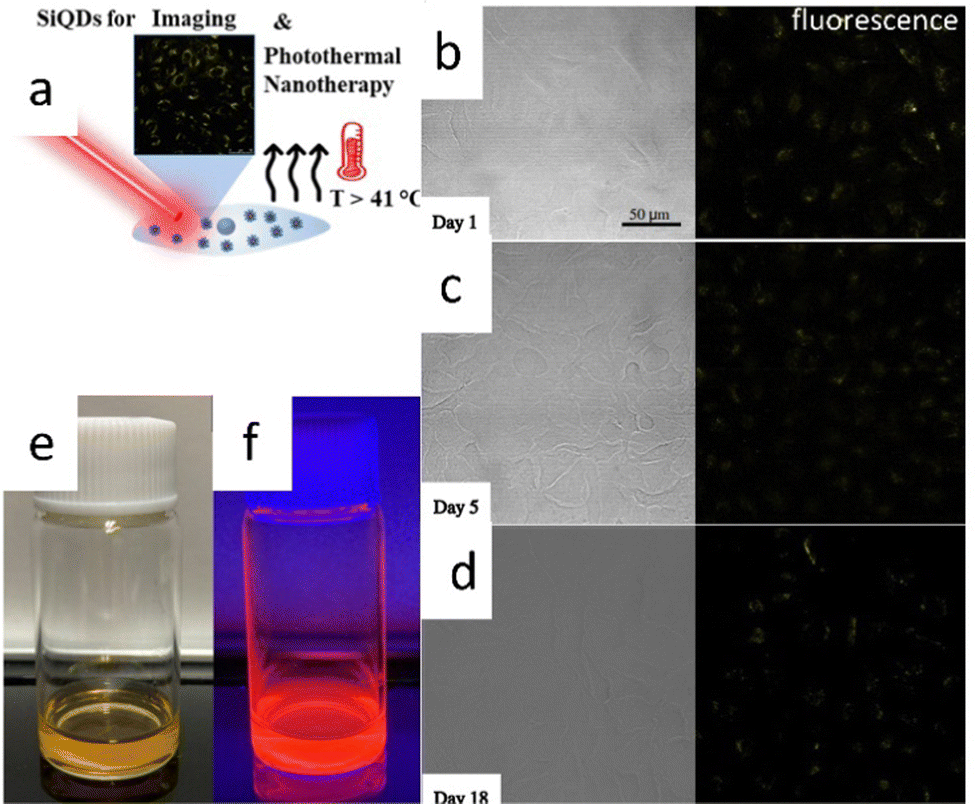 | ||
| Fig. 17 Photothermal nano therapy of 10-undecanoic acid functionalized SiQD. (a) Cartoon representation of Photothermal nanotherapy of SiQDs photoimaging. (b)–(d) fresh and aged SiQDs fluorescences. (e) and (f) Colloidal SiQDs under visible and UV light. Adapted with permission from ref. 140. Copyright (2022) American Chemical Society. | ||
Fig. 17a shows a cartoon illustration of photothermal nanotherapy. The UA-SiQDs absorb intense light so that the energy gained by the particles is partly converted into photons and heat. As a result of the irradiation, the temperature inside the cell rises above 41 °C, killing the cell internally. Fig. 17b–d show the PL of UA-SiQDs exposed to light. While Fig. 17e and f show the display of UA-SiQDs in room light and their emission when exposed to the laser.
As photosensitizer.
In triplet–triplet annihilation upconversion (TTA-UC)
Xia et al. (2021)141 demonstrated that hexadecane (C18) +9-ethyl anthracene (9EA) or C18 + 9EA-SiQDs are capable of performing triplet–triplet annihilation upconversion (TTA-UC) with upconversion quantum yields (QYuc) QYuc ∼ 7.5%. This photon upconversion could be sustained for a long time, even reaching 4 days with DPA ligand and methyl oleate solvent. The authors functionalized SiQDs with C18 and 9EA, which acts as a photon absorber (sensitizer), and then dispersed them in DPA emitters, resulting in photon upconversion from λmax 488 nm and λmax 532 nm to λmax 432 nm, with QYuc reaching a maximum of 7.5% when excited with a 1.0 W cm−1 CW laser at λmax 488 nm. The upconversion decay (τuc) has a different range depending on the solvent used. In toluene, mesitylene, and octadecene solvents, τuc is of the order of minutes, while in methyl oleate and oleic acid solvents, the τuc is of the order of days. The core size of the SiQDs was ϕ ∼ 3.0 nm, then became ϕ ∼ 9.0 nm after functionalization with C18 and 9EA. It is then aggregated into micelles so that the particle size becomes ϕ370 ± 70 nm after polymerization. The triplet–triplet annihilation process occurs when photons are absorbed by the photon absorber (i.e., SiQDs), then transferred to the 9EA triplet state and resonances with the DPA triplet state to undergo triplet annihilation to a higher energy state to be emitted via photon upconversion. Thus, the absorbed energy of λmax 488 nm and λmax 532 nm is upconverted to λmax 432 nm.141Previously, Xia et al. (2016)142 also described the process of spin–triplet exciton transfer between silicon and acceptor molecules for photon upconversion. According to the authors, photoexcitation of chemically functionalized silicon with anthracene ligand will lead to spin–triplet exciton transfer from silicon to anthracene through a single Dexter energy transfer step 15 ns with yields of nearly 50%. When coupled with the 9,10-diphenylantracene emitter, photon upconversion from λmax 488–640 nm to λmax 425 nm is possible with QYuc ∼ 7%. QYuc measurements were performed in a nitrogen environment. The authors also showed the relationship between QYuc and the particle size ϕ of SiQDs, where the smaller the particle size, the higher QYuc for the same λ-excitation.126
In singlet oxygen generation (1O2)
Singlet oxygen (1O2) is one of the key ingredients for photodynamic therapy (PDT) to treat oncological diseases. A suitable photosensitizer could be applied to form 1O2 if the following requirements are fulfilled: (1) having low toxicity in the absence of irradiation, (2) working specifically to the target cancer cells, (3) having relatively highly efficient energy transfer, (4) short retention time in the body, (5) having stability against coagulation/aggregation, and (6) exhibiting excellent photostability.143 Porous silicon has long been known to have the ability to generate singlet oxygen. Some reports from old articles mention singlet oxygen generation from non-passivated SiQDs.144–147 Due to the instability of non-passivated SiQDs, it is not very likely to implement the materials for long-term diagnostics. Therefore, it is challenging to generate singlet oxygen from completed functionalized SiQDs. Owing to the excellent behavior of SiQDs as a photosensitizer, it has the potential to be developed for nano-theranostics. Nano-theranostics is a term introduced for the comprehensive effort that integrates diagnostics and therapy in a single platform using nanomaterials. Dye-functionalized SiQDs is one of the potential nano-theranostics agents which could be applied in this platform. Applying dye-functionalized SiQDs as PS in vivo, the image of cancer and another disease could be resolved (photo imaging); simultaneously, 1O2 is released to cure and heal.148–150Osminkina et al. (2011)151 demonstrated that SiQDs with particle sizes ϕ ∼ 2 to 5 nm can act as photosensitizers for singlet oxygen generation. The luminescence detected at λmax 1270 nm is attributed to singlet oxygen emission and is dedicated to the presence of 1O2. The synthesis of SiQDs was done by grinding silicon wafers followed by electrochemical etching to obtain particles with sizes between ϕ 2–10 nm. PL measurements in the vacuum showed the presence of 1270 nm emission.151
Beri et al. (2020)54 have synthesized perylene-functionalized SiQDs to study singlet oxygen generation. The synthesis was done by annealing SiO at a relatively high temperature (i.e., 900–1100 °C), then H-SiQDs were deliberated by chemical etching using concentrated HF. The hydrosilylation reaction was carried out in a microwave reactor at 250 °C by mixing H-SiQDs, n-hexene, and 3-perylene to produce C6-p1-SiQDs. The resulting ϕ 3.0–8.0 nm diameter C6-p1-SiQDs were ready for direct singlet-oxygen quantum yield (QYSOG) measurements. Two excitation lasers were used, i.e., 317.5 nm and 405 nm, to hit the core of the SiQDs. The data show that although the PLQY of hexyl (C6) SiQDs is relatively high in the near-infrared, direct measurements of singlet oxygen emission at λmax 1270 nm show null emission. However, when the SiQDs surface was decorated with a mixture of hexyl (C6) and perylene, the λmax 1270 nm emission was detected. The singlet oxygen quantum yield for C6-p1-SiQDs relative to phenalenone is QYSOG ∼ 27%. The process of singlet oxygen generation is shown schematically in Fig. 18. Briefly, light is absorbed by the SiQDs core, a light absorber, and photons are transferred to the triplet state of perylene via the Dexter energy transfer mechanism. Photons in this triplet state then exchange energy with oxygen in the air to form singlet oxygen, which then decays to emit energy at λmax 1270 nm.54,152
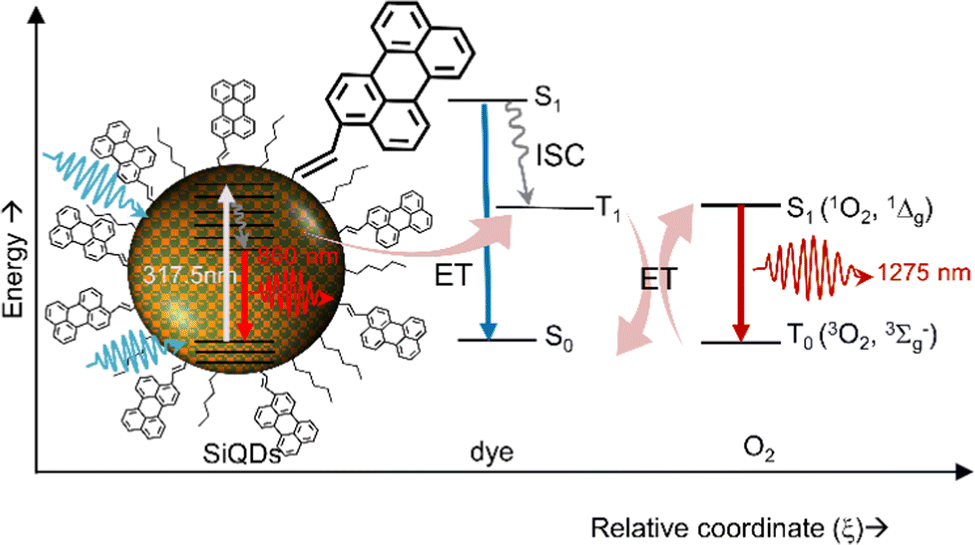 | ||
| Fig. 18 Singlet oxygen generation production via energy transfer from dye-functionalized SiQDs to Oxygen. Reproduced with permission from ref. 54. Copyright (2020) Frontier in Chemistry. | ||
F. Conclusions
Reaction with oxygen causes the photophysics of H-SiQDs to lose their uniqueness. Reaction with oxygen causes the SiO2 layer to envelop the SiQDs surface, causing the SiQDs core to shrink and lose its emission properties due to forming an electron-trapping layer of SiO2. Functionalization reactions or hydrosilylation with organic ligand groups is one solution to maintain the emission of SiQDs so that they can emit light in the long term. The hydrosilylation reaction can be performed thermally using conventional heat (reflux) or a microwave reactor. The advantage of this method is that it is relatively simple, but the disadvantage is the possibility of forming multilayers on the surface of the SiQDs, which reduces the emission. Another disadvantage is that it is almost impossible to do with highly volatile solvents using conventional thermal reaction methods. In addition to thermal reactions, hydrosilylation reactions can also use metal or organometallic catalysts, but the disadvantage is that the residual metal can interfere with the PL of the resulting SiQDs. Also, photochemical and radical initiation reactions can be carried out at low temperatures so that the ligand layer formed is a monolayer. The hydrosilylation reaction can also be carried out using dyes. Dyes can aid in the absorption of photons, which in turn can increase PL-emission. Because they have a large cross-section, dyes can act as an antenna that emits photons via the Dexter energy transfer mechanism from SiQDs to dyes. This energy transfer mechanism reaches the triplet state of the dyes, ready to be transferred to the nearest dye triplet state to perform triplet–triplet annihilation upconversion (TTA-UC) or to the closest oxygen triplet state to generate singlet oxygen (1O2). Due to their great potential, dye-functionalized SiQDs can be used as light-harvesting antennas, bioimaging, luminescence solar concentrators, biosensors, light emitting diodes, photothermal monotherapy (theranostics) and as sensitizers in TTA-UC and singlet oxygen generation (1O2).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to thank his PhD supervisors, Prof. Dr Bryce Richards and Dr Andrey Turshatov, who helped him to complete his studies at the Faculty of Chemistry and Biosciences, Karlsruhe Institute of Technology (KIT), especially at the Institute of Microstructure Technology. Of course, there are many bitter and sweet experiences that we went through during the author's education. The author would like to thank Prof. Ganefri, PhD (Rector of UNP), Prof. Yohandri, PhD as Head of the Institute for Research and Public Service (LPPM) and staff, and Dr Kasmita as Head of the UNP Internal Quality Assurance Agency (BPMI) and staff. The author would also like to thank all friends and colleagues at KIT and UNP who have played a role in the author's career. Finally, the author would also like to thank all the staff who have contributed to this success.Notes and references
- G. Marsico, P. Scafato, S. Belviso and S. Superchi, RSC Adv., 2020, 10, 32581–32601 RSC.
- J. Wang, Y. Liu, F. Peng, C. Chen, Y. He, H. Ma, L. Cao and S. Sun, Small, 2012, 8, 2430–2435 CrossRef CAS PubMed.
- S. Morozova, M. Alikina, A. Vinogradov and M. Pagliaro, Front. Chem., 2020, 8, 2–8 CrossRef PubMed.
- C. J. T. Robidillo and J. G. C. Veinot, ACS Appl. Mater. Interfaces, 2020, 12, 52251–52270 CrossRef CAS PubMed.
- F. Hua, M. T. Swihart and E. Ruckenstein, Langmuir, 2005, 21, 6054–6062 CrossRef CAS PubMed.
- H. Yu, A. N. Thiessen, M. A. Hossain, M. J. Kloberg, B. Rieger and J. G. C. Veinot, Chem. Mater., 2020, 32, 4536–4543 CrossRef CAS.
- J. J. Wu and U. R. Kortshagen, RSC Adv., 2015, 5, 103822–103828 RSC.
- S. Chinnathambi, S. Chen, S. Ganesan and N. Hanagata, Adv. Healthcare Mater., 2014, 3, 10–29 CrossRef CAS PubMed.
- K. Dohnalová, A. N. Poddubny, A. A. Prokofiev, W. D. A. M. de Boer, C. P. Umesh, J. M. J. Paulusse, H. Zuilhof and T. Gregorkiewicz, Light: Sci. Appl., 2013, 2, e47 CrossRef.
- B. P. Falcão, J. P. Leitão, M. R. Soares, L. Ricardo, H. Águas, R. Martins and R. N. Pereira, Phys. Rev. Appl., 2019, 11, 024054 CrossRef.
- T. K. Purkait, M. Iqbal, M. A. Islam, M. H. Mobarok, C. M. Gonzalez, L. Hadidi and J. G. C. Veinot, J. Am. Chem. Soc., 2016, 138, 7114–7120 CrossRef CAS PubMed.
- J. Rinck, D. Schray, C. Kübel, A. K. Powell and G. A. Ozin, Small, 2015, 11, 335–340 CrossRef CAS PubMed.
- L. Canham, Faraday Discuss., 2020, 222, 10–81 RSC.
- L. Ondič, K. Kůsová, M. Ziegler, L. Fekete, V. Gärtnerová, V. Cháb, V. Holý, O. Cibulka, K. Herynková, M. Gallart, P. Gilliot, B. Hönerlage and I. Pelant, Nanoscale, 2014, 6, 3837–3845 RSC.
- K. Fujimoto, T. Hayakawa, Y. Xu, N. Jingu and K.-I. Saitow, ACS Sustainable Chem. Eng., 2022, 10, 14451–14463 CrossRef CAS.
- X. Liu, Y. Zhang, T. Yu, X. Qiao, R. Gresback, X. Pi and D. Yang, Part. Part. Syst. Charact., 2016, 33, 44–52 CrossRef CAS.
- A. Marinins, Z. Yang, H. Chen, J. Linnros, J. G. C. Veinot, S. Popov and I. Sychugov, ACS Photonics, 2016, 3, 1575–1580 CrossRef CAS.
- T. A. Pringle, K. I. Hunter, A. Brumberg, K. J. Anderson, J. A. Fagan, S. A. Thomas, R. J. Petersen, M. Sefannaser, Y. Han, S. L. Brown, D. S. Kilin, R. D. Schaller, U. R. Kortshagen, P. R. Boudjouk and E. K. Hobbie, ACS Nano, 2020, 14, 3858–3867 CrossRef CAS PubMed.
- R. J. Anthony, D. J. Rowe, M. Stein, J. Yang and U. Kortshagen, Adv. Funct. Mater., 2011, 21, 4042–4046 CrossRef CAS.
- S. L. Brown, J. B. Miller, R. J. Anthony, U. R. Kortshagen, A. Kryjevski and E. K. Hobbie, ACS Nano, 2017, 11, 1597–1603 CrossRef CAS PubMed.
- S. L. Brown, D. J. Vogel, J. B. Miller, T. M. Inerbaev, R. J. Anthony, U. R. Kortshagen, D. S. Kilin and E. K. Hobbie, J. Phys. Chem. C, 2016, 120, 18909–18916 CrossRef CAS.
- N. J. Kramer, R. J. Anthony, M. Mamunuru, E. S. Aydil and U. R. Kortshagen, J. Phys. D: Appl. Phys., 2014, 47, 075202 CrossRef CAS.
- S. J. Lanham, J. Polito, Z. Xiong, U. R. Kortshagen and M. J. Kushner, J. Appl. Phys., 2022, 132, 073301 CrossRef CAS.
- L. Mangolini, D. Jurbergs, E. Rogojina and U. Kortshagen, J. Lumin., 2006, 121, 327–334 CrossRef CAS.
- L. Mangolini, E. Thimsen and U. Kortshagen, Nano Lett., 2005, 5, 655–659 CrossRef CAS PubMed.
- F. Meinardi, S. Ehrenberg, L. Dhamo, F. Carulli, M. Mauri, F. Bruni, R. Simonutti, U. Kortshagen and S. Brovelli, Nat. Photonics, 2017, 11, 177–185 CrossRef CAS.
- M. Abdelhameed, S. Aly, J. T. Lant, X. Zhang and P. Charpentier, Sci. Rep., 2018, 8, 17068 CrossRef PubMed.
- M. Abdelhameed, D. R. Martir, S. Chen, W. Z. Xu, O. O. Oyeneye, S. Chakrabarti, E. Zysman-Colman and P. A. Charpentier, Sci. Rep., 2018, 8, 3050 CrossRef PubMed.
- R. J. Clark, M. Aghajamali, C. M. Gonzalez, L. Hadidi, M. A. Islam, M. Javadi, M. H. Mobarok, T. K. Purkait, C. J. T. Robidillo, R. Sinelnikov, A. N. Thiessen, J. Washington, H. Yu and J. G. C. Veinot, Chem. Mater., 2017, 29, 80–89 CrossRef CAS.
- M. Dasog, G. B. De los Reyes, L. V. Titova, F. A. Hegmann and J. G. C. Veinot, ACS Nano, 2014, 8, 9636–9648 CrossRef CAS PubMed.
- M. Dasog, J. Kehrle, B. Rieger and J. G. C. Veinot, Angew. Chem., Int. Ed., 2016, 55, 2322–2339 CrossRef CAS PubMed.
- M. Dasog and J. G. C. Veinot, Phys. Status Solidi B, 2014, 251, 2216–2220 CrossRef CAS.
- M. Dasog, Z. Yang, S. Regli, T. M. Atkins, A. Faramus, M. P. Singh, E. Muthuswamy, S. M. Kauzlarich, R. D. Tilley and J. G. C. Veinot, ACS Nano, 2013, 7, 2676–2685 CrossRef CAS PubMed.
- G. B. De los Reyes, M. Dasog, M. Na, L. V. Titova, J. G. C. Veinot and F. A. Hegmann, Phys. Chem. Chem. Phys., 2015, 17, 30125–30133 RSC.
- C. M. Gonzalez and J. G. C. Veinot, J. Mater. Chem. C, 2016, 4, 4836–4846 RSC.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, J. Phys. Chem. C, 2007, 111, 6956–6961 CrossRef CAS.
- M. A. Islam, M. H. Mobarok, R. Sinelnikov, T. K. Purkait and J. G. C. Veinot, Langmuir, 2017, 33, 8766–8773 CrossRef CAS PubMed.
- M. Jakob, M. Javadi, J. G. C. Veinot, A. Meldrum, A. Kartouzian and U. Heiz, Chem. – Eur. J., 2019, 25, 3061–3067 CrossRef CAS PubMed.
- J. Kehrle, S. Kaiser, T. K. Purkait, M. Winnacker, T. Helbich, S. Vagin, J. G. C. Veinot and B. Rieger, Nanoscale, 2017, 9, 8489–8495 RSC.
- J. A. Kelly, A. M. Shukaliak, M. D. Fleischauer and J. G. C. Veinot, J. Am. Chem. Soc., 2011, 133, 9564–9571 CrossRef CAS PubMed.
- J. A. Kelly and J. G. C. Veinot, ACS Nano, 2010, 4, 4645–4656 CrossRef CAS PubMed.
- A. Marinins, R. Zandi Shafagh, W. van der Wijngaart, T. Haraldsson, J. Linnros, J. G. C. Veinot, S. Popov and I. Sychugov, ACS Appl. Mater. Interfaces, 2017, 9, 30267–30272 CrossRef CAS PubMed.
- T. K. Purkait, M. Iqbal, M. H. Wahl, K. Gottschling, C. M. Gonzalez, M. A. Islam and J. G. C. Veinot, J. Am. Chem. Soc., 2014, 136, 17914–17917 CrossRef CAS PubMed.
- F. Sangghaleh, I. Sychugov, Z. Yang, J. G. C. Veinot and J. Linnros, ACS Nano, 2015, 9, 7097–7104 CrossRef CAS PubMed.
- R. Sinelnikov, M. Dasog, J. Beamish, A. Meldrum and J. G. C. Veinot, ACS Photonics, 2017, 4, 1920–1929 CrossRef CAS.
- I. Sychugov, A. Fucikova, F. Pevere, Z. Yang, J. G. C. Veinot and J. Linnros, ACS Photonics, 2014, 1, 998–1005 CrossRef CAS.
- A. N. Thiessen, L. Zhang, A. O. Oliynyk, H. Yu, K. M. O’Connor, A. Meldrum and J. G. C. Veinot, Chem. Mater., 2020, 32, 6838–6846 CrossRef CAS.
- J. G. Veinot, Chem. Commun., 2006, 4160–4168, 10.1039/b607476f.
- Z. Yang, M. Dasog, A. R. Dobbie, R. Lockwood, Y. Zhi, A. Meldrum and J. G. C. Veinot, Adv. Funct. Mater., 2014, 24, 1345–1353 CrossRef CAS.
- Z. Yang, G. B. De los Reyes, L. V. Titova, I. Sychugov, M. Dasog, J. Linnros, F. A. Hegmann and J. G. C. Veinot, ACS Photonics, 2015, 2, 595–605 CrossRef CAS.
- Z. Yang, C. M. Gonzalez, T. K. Purkait, M. Iqbal, A. Meldrum and J. G. C. Veinot, Langmuir, 2015, 31, 10540–10548 CrossRef CAS PubMed.
- Z. Yang, M. Iqbal, A. R. Dobbie and J. G. Veinot, J. Am. Chem. Soc., 2013, 135, 17595–17601 CrossRef CAS PubMed.
- D. Beri, D. Busko, A. Mazilkin, I. A. Howard, B. S. Richards and A. Turshatov, RSC Adv., 2018, 8, 9979–9984 RSC.
- D. Beri, M. Jakoby, D. Busko, B. S. Richards and A. Turshatov, Front. Chem., 2020, 8, 567 CrossRef CAS PubMed.
- D. Beri, M. Jakoby, I. A. Howard, D. Busko, B. S. Richards and A. Turshatov, Dalton Trans., 2020, 49, 2290–2299 RSC.
- D. Chen, W. Sun, C. Qian, L. M. Reyes, A. P. Y. Wong, Y. Dong, J. Jia, K. K. Chen and G. A. Ozin, Adv. Funct. Mater., 2016, 26, 5102–5110 CrossRef CAS.
- K. K. Chen, M. L. Mastronardi, C. Kübel and G. A. Ozin, Part. Part. Syst. Charact., 2015, 32, 301–306 CrossRef CAS.
- D. O. Faulkner, J. J. McDowell, A. J. Price, D. D. Perovic, N. P. Kherani and G. A. Ozin, Laser Photonics Rev., 2012, 6, 802–806 CrossRef CAS.
- E. J. Henderson, A. J. Shuhendler, P. Prasad, V. Baumann, F. Maier-Flaig, D. O. Faulkner, U. Lemmer, X. Y. Wu and G. A. Ozin, Small, 2011, 7, 2507–2516 CAS.
- F. Maier-Flaig, E. J. Henderson, S. Valouch, S. Klinkhammer, C. Kübel, G. A. Ozin and U. Lemmer, Chem. Phys., 2012, 405, 175–180 CrossRef CAS.
- F. Maier-Flaig, J. Rinck, M. Stephan, T. Bocksrocker, M. Bruns, C. Kübel, A. K. Powell, G. A. Ozin and U. Lemmer, Nano Lett., 2013, 13, 475–480 CrossRef CAS PubMed.
- M. L. Mastronardi, K. K. Chen, K. Liao, G. Casillas and G. A. Ozin, J. Phys. Chem. C, 2014, 119, 826–834 CrossRef.
- M. L. Mastronardi, E. J. Henderson, D. P. Puzzo, Y. Chang, Z. B. Wang, M. G. Helander, J. Jeong, N. P. Kherani, Z. Lu and G. A. Ozin, Small, 2012, 8, 3647–3654 CrossRef CAS PubMed.
- M. L. Mastronardi, E. J. Henderson, D. P. Puzzo and G. A. Ozin, Adv. Mater., 2012, 24, 5890–5898 CrossRef CAS PubMed.
- M. L. Mastronardi, F. Hennrich, E. J. Henderson, F. Maier-Flaig, C. Blum, J. Reichenbach, U. Lemmer, C. Kubel, D. Wang, M. M. Kappes and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 11928–11931 CrossRef CAS PubMed.
- C. Qian, W. Sun, L. Wang, C. Chen, K. Liao, W. Wang, J. Jia, B. D. Hatton, G. Casillas, M. Kurylowicz, C. M. Yip, M. L. Mastronardi and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 15849–15852 CrossRef CAS PubMed.
- W. Sun, C. Qian, K. K. Chen and G. A. Ozin, ChemNanoMat, 2016, 2, 847–855 CrossRef CAS.
- W. Sun, C. Qian, X. S. Cui, L. Wang, M. Wei, G. Casillas, A. S. Helmy and G. A. Ozin, Nanoscale, 2016, 8, 3678–3684 RSC.
- W. Sun, C. Qian, M. L. Mastronardi, M. Wei and G. A. Ozin, Chem. Commun., 2013, 49, 11361–11363 RSC.
- W. Sun, C. Qian, L. Wang, M. Wei, M. L. Mastronardi, G. Casillas, J. Breu and G. A. Ozin, Adv. Mater., 2015, 27, 746–749 CrossRef CAS PubMed.
- M. L. Mastronardi, F. Maier-Flaig, D. Faulkner, E. J. Henderson, C. Kubel, U. Lemmer and G. A. Ozin, Nano Lett., 2012, 12, 337–342 CrossRef CAS PubMed.
- M. Sefannaser, S. A. Thomas, K. J. Anderson, R. J. Petersen, S. L. Brown, P. R. Boudjouk, T. A. Pringle and E. K. Hobbie, J. Phys. Chem. C, 2021, 125, 5824–5831 CrossRef CAS.
- F. Pevere, F. Sangghaleh, B. Bruhn, I. Sychugov and J. Linnros, ACS Photonics, 2018, 5, 2990–2996 CrossRef CAS.
- K. Kůsová, T. Popelář, I. Pelant, G. Morselli, S. Angeloni and P. Ceroni, J. Phys. Chem. C, 2021, 125, 2055–2063 CrossRef.
- R. J. Petersen, S. A. Thomas, K. J. Anderson, T. A. Pringle, S. May and E. K. Hobbie, J. Phys. Chem. C, 2022, 126, 12935–12943 CrossRef CAS.
- F. Romano, S. Angeloni, G. Morselli, R. Mazzaro, V. Morandi, J. R. Shell, X. Cao, B. W. Pogue and P. Ceroni, Nanoscale, 2020, 12, 7921–7926 RSC.
- F. Romano, Y. Yu, B. A. Korgel, G. Bergamini and P. Ceroni, Top. Curr. Chem., 2016, 374, 53 CrossRef PubMed.
- G. M. Carroll, R. Limpens and N. R. Neale, Nano Lett., 2018, 18, 3118–3124 CrossRef CAS PubMed.
- K. Dohnalová, T. Gregorkiewicz and K. Kůsová, J. Phys.: Condens. Matter, 2014, 26, 173201 CrossRef PubMed.
- K. Dohnalová, P. Hapala, K. Kůsová and I. Infante, Chem. Mater., 2020, 32, 6326–6337 CrossRef.
- K. Kůsová, Phys. Status Solidi A, 2018, 215, 1700718 CrossRef.
- R. Mazzaro, A. Gradone, S. Angeloni, G. Morselli, P. G. Cozzi, F. Romano, A. Vomiero and P. Ceroni, ACS Photonics, 2019, 6, 2303–2311 CrossRef CAS.
- B. Ghosh, M. Takeguchi, J. Nakamura, Y. Nemoto, T. Hamaoka, S. Chandra and N. Shirahata, Sci. Rep., 2016, 6, 36951 CrossRef CAS PubMed.
- Q. Li, Y. He, J. Chang, L. Wang, H. Chen, Y.-W. Tan, H. Wang and Z. Shao, J. Am. Chem. Soc., 2013, 135, 14924–14927 CrossRef CAS PubMed.
- S. Terada, H. Ueda, T. Ono and K.-I. Saitow, ACS Sustainable Chem. Eng., 2022, 10, 1765–1776 CrossRef CAS.
- A. B. Sieval, R. Linke, H. Zuilhof and E. J. R. Sudhölter, Adv. Mater., 2000, 12, 1457–1460 CrossRef CAS.
- N. S. Bhairamadgi, S. P. Pujari, F. G. Trovela, A. Debrassi, A. A. Khamis, J. M. Alonso, A. A. Al Zahrani, T. Wennekes, H. A. Al-Turaif, C. van Rijn, Y. A. Alhamed and H. Zuilhof, Langmuir, 2014, 30, 5829–5839 CrossRef CAS PubMed.
- M. Dworschak, N. Kohlmann, F. Matějka, P. Galář, L. Kienle, J. Schäfer and J. Benedikt, Plasma Processes Polym., 2023, 20(2), 2200129 CrossRef CAS.
- P. Wollny, J. Menser, L. Engelmann, J. Sellmann, C. Schulz, H. Wiggers, A. Kempf and I. Wlokas, Chem. Eng. J., 2023, 453, 139695 CrossRef CAS.
- N. S. Seroka, R. T. Taziwa and L. Khotseng, Appl. Sci., 2022, 12, 2310 CrossRef CAS.
- F. K. Başak and E. Kayahan, Opt. Mater., 2022, 124, 111990 CrossRef.
- Y. Park, J. Yoo, M.-H. Kang, W. Kwon and J. Joo, J. Mater. Chem. B, 2019, 7, 6271–6292 RSC.
- P. Singh, S. Srivastava and S. K. Singh, ACS Biomater. Sci. Eng., 2019, 5, 4882–4898 CrossRef CAS PubMed.
- A. D. Beck, S. Haufe, J. Tillmann and S. R. Waldvogel, ChemElectroChem, 2022, 9, e202101374 CAS.
- I. T. Cheong, J. Mock, M. Kallergi, E. Groß, A. Meldrum, B. Rieger, M. Becherer and J. G. C. Veinot, Adv. Opt. Mater., 2022, 11, 2201834 CrossRef.
- J. Zhou, J. Huang, H. Chen, A. Samanta, J. Linnros, Z. Yang and I. Sychugov, J. Phys. Chem. Lett., 2021, 12, 8909–8916 CrossRef CAS PubMed.
- S. Terada, Y. Xin and K.-I. Saitow, Chem. Mater., 2020, 32, 8382–8392 CrossRef CAS.
- D. S. English, L. E. Pell, Z. Yu, P. F. Barbara and B. A. Korgel, Nano Lett., 2002, 2, 681–685 CrossRef CAS.
- A. Fermi, M. Locritani, G. Di Carlo, M. Pizzotti, S. Caramori, Y. Yu, B. A. Korgel, G. Bergamini and P. Ceroni, Faraday Discuss., 2015, 185, 481–495 RSC.
- B. J. Furey, B. J. Stacy, T. Shah, R. M. Barba-Barba, R. Carriles, A. Bernal, B. S. Mendoza, B. A. Korgel and M. C. Downer, ACS Nano, 2022, 16, 6023–6033 CrossRef CAS PubMed.
- C. M. Hessel, D. Reid, M. G. Panthani, M. R. Rasch, B. W. Goodfellow, J. W. Wei, H. Fujii, V. Akhavan and B. A. Korgel, Chem. Mater., 2012, 24, 393–401 CrossRef CAS.
- J. D. Holmes, K. J. Ziegler, R. C. Doty, L. E. Pell, K. P. Johnston and B. A. Korgel, J. Am. Chem. Soc., 2001, 123, 3743–3748 CrossRef CAS PubMed.
- M. Locritani, Y. Yu, G. Bergamini, M. Baroncini, J. K. Molloy, B. A. Korgel and P. Ceroni, J. Phys. Chem. Lett., 2014, 5, 3325–3329 CrossRef CAS PubMed.
- R. Mazzaro, M. Locritani, J. K. Molloy, M. Montalti, Y. Yu, B. A. Korgel, G. Bergamini, V. Morandi and P. Ceroni, Chem. Mater., 2015, 27, 4390–4397 CrossRef CAS.
- L. Ravotto, Q. Chen, Y. Ma, S. A. Vinogradov, M. Locritani, G. Bergamini, F. Negri, Y. Yu, B. A. Korgel and P. Ceroni, Chem, 2017, 2, 550–560 CAS.
- Y. Yu, G. Fan, A. Fermi, R. Mazzaro, V. Morandi, P. Ceroni, D.-M. Smilgies and B. A. Korgel, J. Phys. Chem. C, 2017, 121, 23240–23248 CrossRef CAS.
- Y. Yu and B. A. Korgel, Langmuir, 2015, 31, 6532–6537 CrossRef CAS PubMed.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS.
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed.
- B. F. P. McVey and R. D. Tilley, Acc. Chem. Res., 2014, 47, 3045–3051 CrossRef CAS PubMed.
- M. R. Linford and C. E. D. Chidsey, J. Am. Chem. Soc., 1993, 115, 12631–12632 CrossRef CAS.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271–1308 CrossRef CAS PubMed.
- C. Coletti, A. Marrone, G. Giorgi, A. Sgamellotti, G. Cerofolini and N. Re, Langmuir, 2006, 22, 9949–9956 CrossRef CAS PubMed.
- H. Zai, Y. Zhao, S. Chen, L. Ge, C. Chen, Q. Chen and Y. Li, Nano Res., 2018, 11, 2544–2552 CrossRef CAS.
- M. L. Kantam, S. Laha, J. Yadav, P. R. Likhar, B. Sreedhar and B. M. Choudary, Adv. Synth. Catal., 2007, 349, 1797–1802 CrossRef CAS.
- R. J. Hofmann, M. Vlatković and F. Wiesbrock, Polymers, 2017, 9, 534 CrossRef PubMed.
- I. W. Moran and K. R. Carter, Langmuir, 2009, 25, 9232–9239 CrossRef CAS PubMed.
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
- M. Baghbanzadeh, L. Carbone, P. D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 11312–11359 CrossRef CAS PubMed.
- R. Boukherroub, A. Petit, A. Loupy, J.-N. Chazalviel and F. Ozanam, J. Phys. Chem. B, 2003, 107, 13459–13462 CrossRef CAS.
- E. Binetti, M. Striccoli, T. Sibillano, C. Giannini, R. Brescia, A. Falqui, R. Comparelli, M. Corricelli, R. Tommasi, A. Agostiano and M. L. Curri, Sci. Technol. Adv. Mater., 2015, 16, 055007 CrossRef PubMed.
- L. Qi and J. Rongchao, Nanotechnol. Rev., 2017, 6, 601–612 Search PubMed.
- X.-B. Shen, B. Song, B. Fang, X. Yuan, Y.-Y. Li, S.-Y. Wang, S.-J. Ji and Y. He, Nano Res., 2019, 12, 315–322 CrossRef CAS.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed.
- C. Mongin, P. Moroz, M. Zamkov and F. N. Castellano, Nat. Chem., 2018, 10, 225–230 CrossRef CAS PubMed.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Nat. Chem., 2020, 12, 137–144 CrossRef CAS PubMed.
- D. Roy, C. Fouzder, A. Mukhuty, S. Pal, M. K. Mondal, R. Kundu and P. Chowdhury, Bioconjugate Chem., 2019, 30, 1575–1583 CrossRef CAS PubMed.
- D. Soujon, K. Becker, A. L. Rogach, J. Feldmann, H. Weller, D. V. Talapin and J. M. Lupton, J. Phys. Chem. C, 2007, 111, 11511–11515 CrossRef CAS.
- S. Sarwat, F. J. Stapleton, M. D. P. Willcox, P. B. O’Mara, R. D. Tilley, J. J. Gooding and M. Roy, Nanomaterials, 2022, 12, 1965 CrossRef CAS PubMed.
- N. Wei, Y.-C. Sun, X.-F. Guo and H. Wang, Microchim. Acta, 2022, 189, 329 CrossRef CAS PubMed.
- G. Morselli, F. Romano and P. Ceroni, Faraday Discuss., 2020, 222, 108–121 RSC.
- Y. Nakahara, K. Machiya, T. Sato, N. T. Nwe, T. Furuike, H. Tamura and K. Kimura, Chem. Lett., 2013, 42, 498–500 CrossRef CAS.
- R. Mazzaro and A. Vomiero, Adv. Energy Mater., 2018, 8, 1801903 CrossRef.
- S. Ren, C. Shou, S. Jin, G. Chen, S. Han, Z. Chen, X. Chen, S. Yang, Y. Guo and C.-C. Tu, ACS Photonics, 2021, 8, 2392–2399 CrossRef CAS.
- L. Xu, Y.-N. Zhang, X.-H. Ji and Z.-K. He, Chem. Pap., 2019, 73, 1753–1759 CrossRef CAS.
- J. Zhou, R. Zhao, S. Liu, L. Feng, W. Li, F. He, S. Gai and P. Yang, Small Methods, 2021, 5, 2100812 CrossRef CAS PubMed.
- F. Ma, J. Luo, X. Li, S. Liu, M. Yang and X. Chen, Spectrochim. Acta, Part A, 2021, 249, 119343 CrossRef CAS PubMed.
- J. Liu, J. Zhang, Y. Zhang, Y. Wang, M. Wang, Z. Li, G. Wang and X. Su, Talanta, 2022, 237, 122956 CrossRef CAS PubMed.
- H. Yamada, N. Saitoh, B. Ghosh, Y. Masuda, N. Yoshizawa and N. Shirahata, J. Phys. Chem. C, 2020, 124, 23333–23342 CrossRef CAS.
- İ. N. G. Özbilgin, T. Yamazaki, J. Watanabe, H.-T. Sun, N. Hanagata and N. Shirahata, Langmuir, 2022, 38, 5188–5196 CrossRef PubMed.
- P. Xia, J. Schwan, T. W. Dugger, L. Mangolini and M. L. Tang, Adv. Opt. Mater., 2021, 9, 2100453 CrossRef CAS.
- H. Xia, J. Hu, J. Tang, K. Xu, X. Hou and P. Wu, Sci. Rep., 2016, 6, 36794 CrossRef CAS PubMed.
- R. Bakalova, H. Ohba, Z. Zhelev, T. Nagase, R. Jose, M. Ishikawa and Y. Baba, Nano Lett., 2004, 4, 1567–1573 CrossRef CAS.
- D. Kovalev, E. Gross, N. Künzner, F. Koch, V. Y. Timoshenko and M. Fujii, Phys. Rev. Lett., 2002, 89, 137401 CrossRef CAS PubMed.
- M. Fujii, S. Minobe, M. Usui, S. Hayashi, E. Gross, J. Diener and D. Kovalev, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 085311 CrossRef.
- D. Kovalev and M. Fujii, Adv. Mater., 2005, 17, 2531–2544 CrossRef CAS.
- D. Kovalev, E. Gross, J. Diener, V. Y. Timoshenko and M. Fujii, Appl. Phys. Lett., 2004, 85, 3590–3592 CrossRef CAS.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- L. Wang, D. Xu, J. Gao, X. Chen, Y. Duo and H. Zhang, Sci. China: Mater., 2020, 63, 1631–1650 CrossRef CAS PubMed.
- X. Y. Wong, A. Sena-Torralba, R. Álvarez-Diduk, K. Muthoosamy and A. Merkoçi, ACS Nano, 2020, 14, 2585–2627 CrossRef CAS PubMed.
- L. A. Osminkina, M. B. Gongalsky, A. V. Motuzuk, V. Y. Timoshenko and A. A. Kudryavtsev, Appl. Phys. B, 2011, 105, 665–668 CrossRef CAS.
- A. Ronchi, P. Brazzo, M. Sassi, L. Beverina, J. Pedrini, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2019, 21, 12353–12359 RSC.
- T. Huang, T. T. Koh, J. Schwan, T. T. Tran, P. Xia, K. Wang, L. Mangolini, M. L. Tang and S. T. Roberts, Chem. Sci., 2021, 12, 6737–6746 RSC.
| This journal is © The Royal Society of Chemistry 2023 |