
Understanding cancer and the anticancer activities of naphthoquinones – a review
Kevin W. Wellington
CSIR Biosciences, PO Box 395, Pretoria, South Africa. E-mail: kwellington@csir.co.za; kwwellington@gmail.com
First published on 5th February 2015
Abstract
The non-communicable disease, cancer, is one of the major causes of death across the world and is forecast to increase by 75% to reach close to 25 million cases over the next two decades. Radiotherapy and surgical approaches have been unsuccessful in controlling the incidence of most cancers. The development of chemotherapeutic strategies involving novel small molecule antitumour agents has therefore been the focus area of cancer chemotherapy for several decades as another strategy to combat and control the incidence of cancer. Many natural products as well as several synthetic drugs have a naphthoquinone chromophore. The anticancer activities of naphthoquinones have been the focus of much research to discover novel anticancer agents. The naturally occurring 1,2-naphthoquinone-based compound, β-lapachone (ARQ 761), is currently being assessed for its anti-tumour activity against advanced solid tumours. This review describes the most recent applications of naphthoquinones and their derivatives in cancer drug discovery. The biology relevant to the design of novel naphthoquinone anticancer agents is also discussed. Furthermore, the discussion of the biology will contribute to understanding cancer as well as the applications of naphthoquinones as anticancer agents.
Introduction
Cancer, as a single entity, is the major cause of death globally with an estimated 8.2 million deaths being attributed to it in 2012.1 There was a 11% increase in cancer cases to reach an estimated 14.1 million cases in 2012.2 Lung (1.8 million cases, 13.0% of the total), breast (1.7 million, 11.9%) and large bowel (1.4 million, 9.7%) cancers were the most prevalent diagnosed cancers. Globally cancer cases are forecast to increase by 75% to reach close to 25 million cases over the next two decades. The most common causes of cancer death were due to lung (1.6 million, 19.4% of the total), liver (0.8 million, 9.1%), and stomach (0.7 million, 8.8%) cancers.In developing countries the growing and aging populations are inexplicably affected by the increasing numbers of cancer cases. Africa, Asia, Central and South America account for more than 60% of the world's total cases and about 70% of the world's cancer deaths. It is anticipated that 80% of the increase in the number of all cancer deaths will occur in less developed regions by 2025.3
The cancer burden is destructive to the economies of wealthy nations and is placing unmanageable pressure on health-care systems. Premature deaths (aged 30 to 69 years) across the world are currently estimated at 4.2 million and are anticipated to increase well beyond 5 million per annum by 2025 unless practical strategies are applied to address cancer.4
Cancer is a class of non-communicable diseases that is characterised by uncontrolled cell growth. To date, there are more than 100 different types of cancers of which each have been classified according to the cell type that was initially affected.
Uncontrolled growth of damaged cells results in the formation of lumps or masses of tissue called tumours. The latter can interfere with the nervous, digestive and circulatory systems.5 Tumours can also release hormones that alter body function. Tumours are considered benign when they are localised and have limited growth. Two things must happen for a tumour to be considered malignant. The first is that a cancer cell has to spread throughout the body and destroy healthy tissue by a process called invasion.5 Second is that it must divide, grow and form new blood vessels by angiogenesis.5 A tumour has metastasised when it has successfully spread to other areas of the body. This is a critical condition that is very hard to treat and is responsible for about 90% of human cancer deaths.6–8
Different treatments are available to patients who have been diagnosed with cancer. The conventional cancer treatments are surgery, radiation and chemotherapy.
Surgery is the oldest and the primary treatment modality. It is most effective in the treatment of localized primary tumours and associated regional lymphatics.9 Disadvantages are that healthy tissues or organs (lymph nodes) can be damaged, metastasised cancer and cancer cells or tumours that are not visible cannot be removed and that “latent” small tumours can be activated thus causing further proliferation.10,11
Radiation therapy (by X-rays and gamma rays) is an important treatment modality which shrinks tumours.12 A disadvantage is that healthy cells may be damaged.12 The most prevalent side effect is fatigue which may last through treatment and for many months afterwards. Other side effects include hair loss, skin irritation, possible hearing problems, nausea, vomiting, loss of appetite, and neurologic effects (permanent memory and speech problems).12
Chemotherapy is a systemic treatment which means that the anticancer drugs are circulated throughout the body by the blood circulatory system to reach cancer cells wherever they are. The goal of chemotherapy is to annihilate both cancerous colonies and metastasised cancer cells within a patient's body. The side effects of chemotherapy are anaemia, diarrhoea, nausea, vomiting, hair loss and weakening of the immune system. Resistance to chemotherapeutic drugs may also be developed by cancer cells.11,13,14 Most cancers cannot be cured with only chemotherapy.
Quinones are widely distributed in nature and occur in animals, plants and microorganisms. They often perform vital roles in the biochemistry of energy production by providing essential links in the respiratory chain of living cells.15 Quinones display various biological activities and have therefore been the subject of much research.16 Seminal studies on several synthetic and natural quinones conducted by the National Cancer Institute (NCI-USA), nearly four decades ago, showed that these compounds possess anticancer activity.17 Both natural quinones and their analogues are vital sources of cytotoxic compounds.18–21 The anthracycline antibiotics (daunorubicin, doxorubicin, idarubicin and mitoxantrone), bleomycins, dactinomycin and mitomycin-C are examples of quinones that have been used clinically for cancer chemotherapy.22–26
The most important and widely distributed chemical class in the quinone family is the 1,4-naphthoquinones. Their derivatives have exhibited a variety of biological responses which include antiallergic,27–29 antibacterial,30,31 antifungal,30–33 anti-inflammatory,27–33 antithrombotic,34,35 antiplatelet,27–29,36–39 antiviral,30,35,40,41 apoptosis,42–44 lipoxygenase,45,46 radical scavenging47 and anti-ringworm30 activities. Anticancer activity has also been reported for the 1,4-naphthoquinones.31,32,35–37 The biological activities and structural properties of these compounds have led to them being regarded as privileged structures in Medicinal Chemistry.48 This is because of biological activities particularly against pathogenic protozoa and cancer cells.49,50 The clinical importance of 1,4-naphthoquinones has stimulated enormous research interest in this class of compounds.51 A pertinent research area in cancer chemotherapy has been the development of novel antitumour agents.52,53
In this article we review the synthesis of naphthoquinone derivatives that have been synthesised over the last five years (2009–2013) to discover novel antitumour agents for cancer chemotherapy. We will also discuss other topics in this article that are of relevance to cancer drug discovery such as oxidative stress, reactive oxygen species (ROS), multidrug resistance, mechanisms of action of quinone anti-tumour agents, reductive activation of quinones, and quinone-based drugs in cancer therapy.
Oxidative stress and cancer
ROS are due to metabolic reactions in the mitochondria of eukaryotic cells. Specific subcellular processes require low concentrations of ROS for example disulfide bond formation, enzyme activation, signal transduction and gene expression. Low concentrations of ROS are also required when new proteins fold in the endoplasmic reticulum and for controlling caspase activity that is initiated in the apoptotic mechanism. ROS are the most abundantly produced compounds from oxidative metabolism with half-lives ranging from a few nanoseconds to hours depending on the stability of the molecule. ROS include the hydroxyl radical (˙OH), hydrogen peroxide (H2O2), superoxide radical anion (O2˙−), superoxide anion (O2−), singlet oxygen (1O2) and ozone (O3).54 After ROS has been used for the subcellular events they are eliminated from normal cells. Cancer cells, however, require high ROS concentrations to maintain their high proliferation rate.Internal sources of oxidative stress
These encompass peroxisomes and enzymes such as xanthine oxidase, the detoxifying enzymes from the P450 complex, and the nicotinamide adenine dinucleotide (NADPH) oxidase complexes which include the Nox family. The mitochondria, where the majority of these enzymes function, are the primary origin of oxidative stress.55External sources of oxidative stress
These encompass UV radiation and chemical compounds from smoking, alcohol, exercise and environmental pollutants. Reactive species have been classified into four groups based on the main atom involved: (i) reactive chloride species (RCS); (ii) reactive nitrogen species (RNS); (iii) ROS and; (iv) reactive sulfur species (RSS).55Damage by ROS in cells
ROS damage in cells depends on their intracellular concentration and on the equilibrium between the ROS and the endogenous antioxidant species. Oxidative stress is generated as a result of the loss of the pro-oxidant/anti-oxidant equilibrium. This oxidative stress alters and damages many intracellular molecules such as proteins, lipids, DNA and RNA.56ROS causes several types of DNA damage such as base modification, strand breakage and DNA-protein cross-linkage.57 8-Hydroxy-2′-deoxyguanosine (8-OH-dG) is one of the major oxidatively modified DNA base products in vivo which causes mutations in DNA resulting in enhanced aging and carcinogenesis (Fig. 1).57–60 The hydroxyl radical (˙OH), singlet oxygen (1O2)or photodynamic action is responsible for the formation of 8-OH-dG.57,61
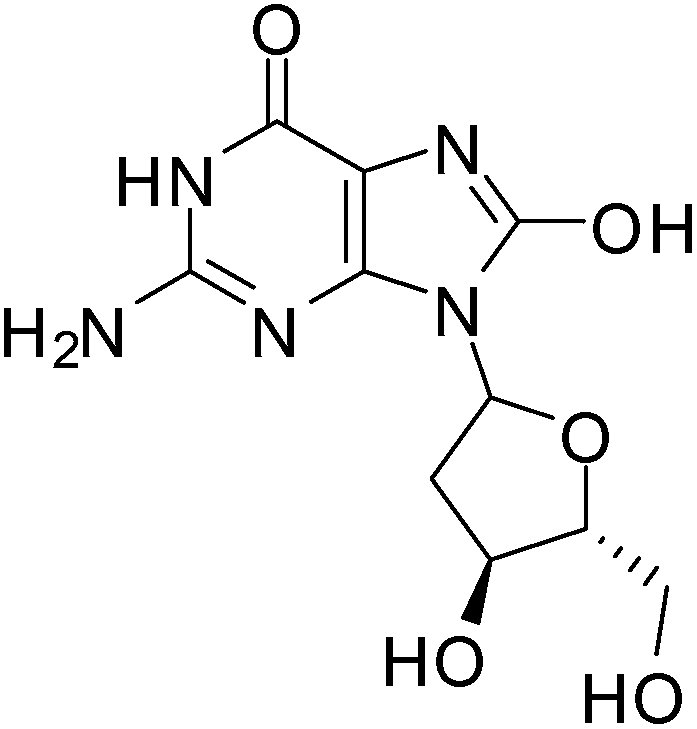 | ||
| Fig. 1 8-Hydroxy-2′-deoxyguanosine responsible for mutations in DNA. | ||
The cell membrane, which is rich in polyunsaturated lipids, is susceptible to oxidation by ROS. ROS induces lipid peroxidation reactions which increases the permeability of the cell membrane that could lead to cell death.62
The high concentration of ROS affects proteins the most and leads to a generation and accumulation of carbonyl (i.e., aldehydes and ketones) and thiol groups (–SH) within the proteins. The thiol groups may be converted into sulfur reactive radicals.63 Oxidation induced modification leads to an alteration in the protein structure resulting in changes or loss of protein function.
Tumour development and progression
Several aspects can be promoted by ROS which have been classified into the following biological processes: (i) angiogenesis [e.g., the release of vascular endothelial growth factor (VEGF) and angiopoietin]; (ii) cellular proliferation [e.g., ligand independent RTK activation and extracellular-regulated kinase 1/2 (ERK1/2) activation]; (iii) tissue invasion and metastasis [e.g., metalloproteinase (MMP) secretion into the extracellular matrix (ECM), Rho–Rac interaction, and Met overexpression], and; (iv) evasion of apoptosis or anoikis [e.g., Src, NF-κB and phosphatidylinositol-3 kinase (PI3K)/Akt activation].64–66Biochemical pathways affected by oxidative stress
Several biochemical pathways pertaining to cellular proliferation are affected. These include the epidermal growth factor receptor (EGFR) that encompasses vital signaling proteins such as Ras, Raf, PKC, nuclear factor erythroid 2-related factor 2 (Nrf2) and kelch-like protein 19 (Keap1). In addition, the mitogen activated protein kinases (MAPK) e.g. p38α, c-myc, ERK1/2, c-Jun N-terminal kinase (JNK), MEK and p53 are also involved.64–66 The master regulator of the antioxidant response is Nrf2 while p38α acts as a key sensor of oxidative stress. The redox sensing function of p38α is essential in the control of tumour development.67 Unlike other MAPKs, p38α suppresses tumourigenesis by either promoting apoptosis or blocking proliferation.Reactive oxygen species (ROS)
Formation in the cell
The production of ROS is a natural process of normal cells and these species are by-products of the oxidative phosphorylation process in the mitochondria. Electrons can escape from the mitochondria and react with molecular oxygen during electron transfer through the electron transport chain. Superoxide radical anions (O2−˙) anions are formed from up to 2% of the oxygen that is consumed during ATP synthesis in the mitochondria.68 ROS can also be produced from the detoxifying enzymes such as cytochrome P450s.A 1-electron reduction produces a superoxide radical anion (O2−˙). This radical has a short lifetime in the cell because it reacts quickly with antioxidants or is transformed to another ROS, such as hydrogen peroxide (H2O2).69 Amongst the common ROS species, peroxides are the least reactive and can therefore persist longer in the cell than the superoxide radical anion and the hydroxyl radical (˙OH).69 The uncharged hydroxide radical, despite having a short lifetime, can be very damaging since it can react readily with a variety of cellular macromolecules.69 Because ROS can cause damage to the cell, endogenous cellular systems for scavenging ROS have evolved.
Defence against ROS in cells
The redox balance can be maintained and the amount of ROS limited by endogenous cellular systems within the cell. The enzymatic and small molecule antioxidant defences involve superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase (CAT), ascorbic acid (vitamin C), α-tocopherol (vitamin E), β-carotene, glutathione (GSH), and vitamin A.70–74 In this way mammalian cells can defend themselves against ROS damage.ROS neutralizing enzymes such as catalase and superoxide dismutases (SOD) can detoxify radicals and maintain the cellular redox balance (Fig. 2).75
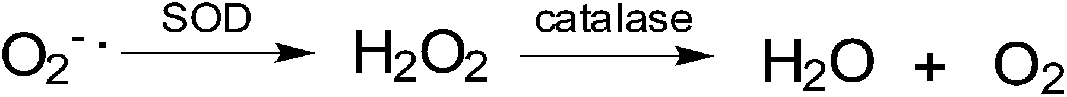 | ||
| Fig. 2 ROS neutralizing enzymes: (a) superoxide dismutase reduces superoxide radical anions to peroxide and; (b) catalase transforms peroxide into molecular oxygen and water. | ||
SOD catalyses the formation of hydrogen peroxide from superoxide while catalase transforms hydrogen peroxide into molecular oxygen and water.75 Hydrogen peroxide, even though it is not as reactive as superoxide, can damage the cell by oxidising DNA.76 NADPH provides the reducing power for many of the enzymes involved in ROS neutralization.
ROS scavengers, thioredoxin (a protein) and glutathione (a small molecule) neutralize ROS by the oxidation of dithiols (Fig. 3).75 Different enzymes, by reduction of the disulfide bond, recycle the thioredoxin and glutathione for re-use.
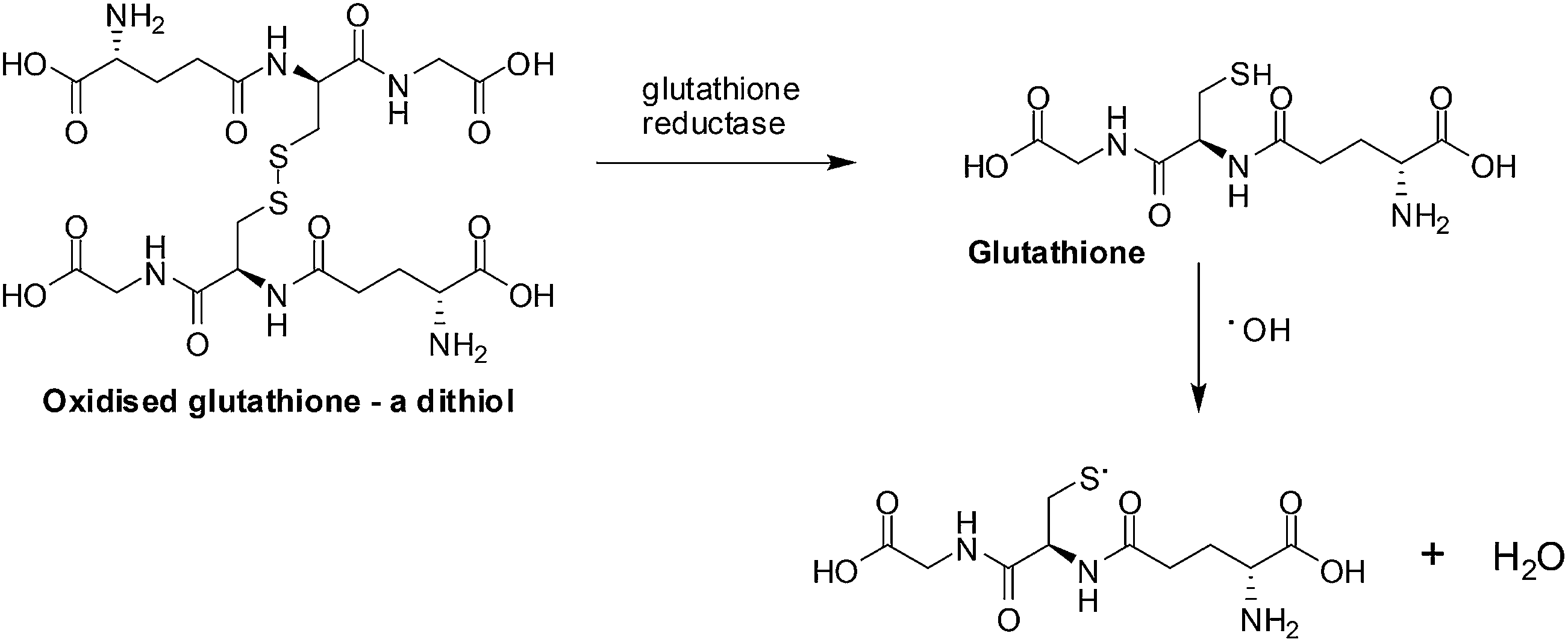 | ||
| Fig. 3 ROS scavengers. The neutralization of ROS is achieved by glutathione forming dithiols and the enzyme, glutathione reductase, reduces the oxidized glutathione. | ||
In more highly oxygenated locations where there is a greater probability of ROS damage Vitamin E, the enzymes SOD, CAT and GPX, as well as substrates (GSH) tend to be in a higher concentration.77
ROS in cancer cells
Cancer cells have a greater concentration of endogenous ROS compared to normal cells.77,78 Several theories have been proposed to explain this phenomenon. One theory proposes that because cancer cells are more metabolically active than normal cells, they require more ATP. As a result of the additional metabolic burden (stresses and respiration) on the electron transport chain, more superoxide radical anions (O2−˙) are formed.73ROS can damage mitochondrial DNA which leads to mutations in members of the oxidative phosphorylation process resulting in more ROS being produced. ROS is believed to increase cancer cell proliferation which leads to uncontrolled tumour growth. One mechanism that has been proposed is that ROS interferes with the MAPK signaling pathway thus disrupting normal metabolic regulation and allows uncontrolled metabolism and growth.69,79
A second mechanism by which ROS promotes cancer cell survival is through DNA damage (Fig. 4).
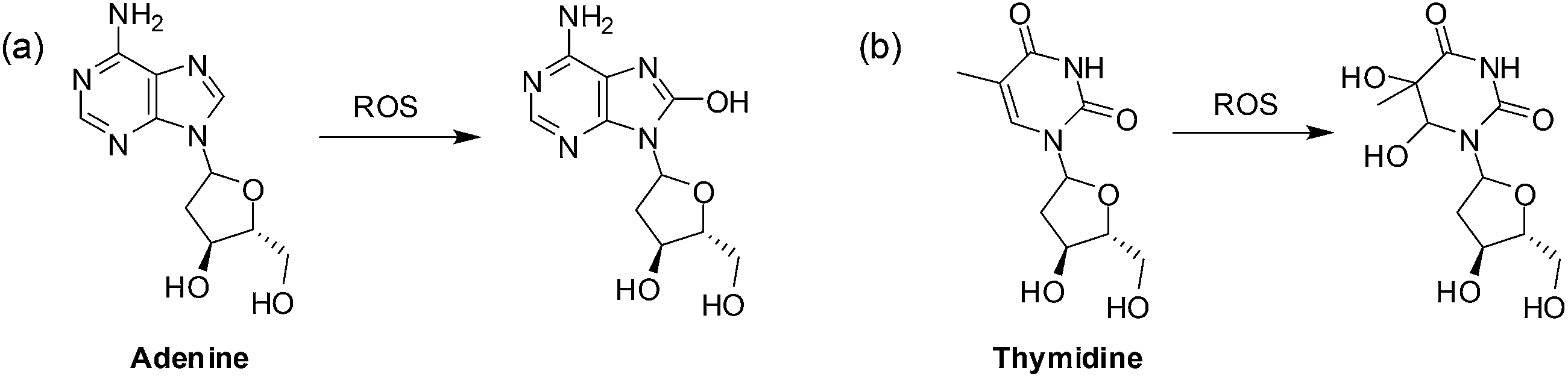 | ||
| Fig. 4 Oxidation of DNA bases by ROS: (a) purines can be monohydroxylated and; (b) pyrimidines can be dihydroxylated at the double bond. | ||
When the DNA is damaged beyond repair, mutations may occur during replication that may be advantageous to cancer cell growth. Faulty respiration proteins are coded for in the cycle of mitochondrial DNA damage which then allows for even greater ROS leakage.69 Proteins can also be oxidized by ROS thus damaging the mitochondrial membrane and the proteins involved in ATP synthesis thus promoting further ROS production.80
It is believed that the sustained imbalance in oxidative stress in cancer cells can be exploited for chemotherapeutic selectivity. By increasing the ROS in a cancer cell the already strained redox balance could be pushed to a critical level that could overpower the ROS buffering capacity of the cancer cell and cause cell death.69,75,80
Mechanisms of cell death involving ROS
Inducing cancer cell death by ROS is a direct method which is being employed as an anti-cancer strategy.69,75,80–82 There are three mechanisms by which ROS can affect a cell (Fig. 5).69,80,81,83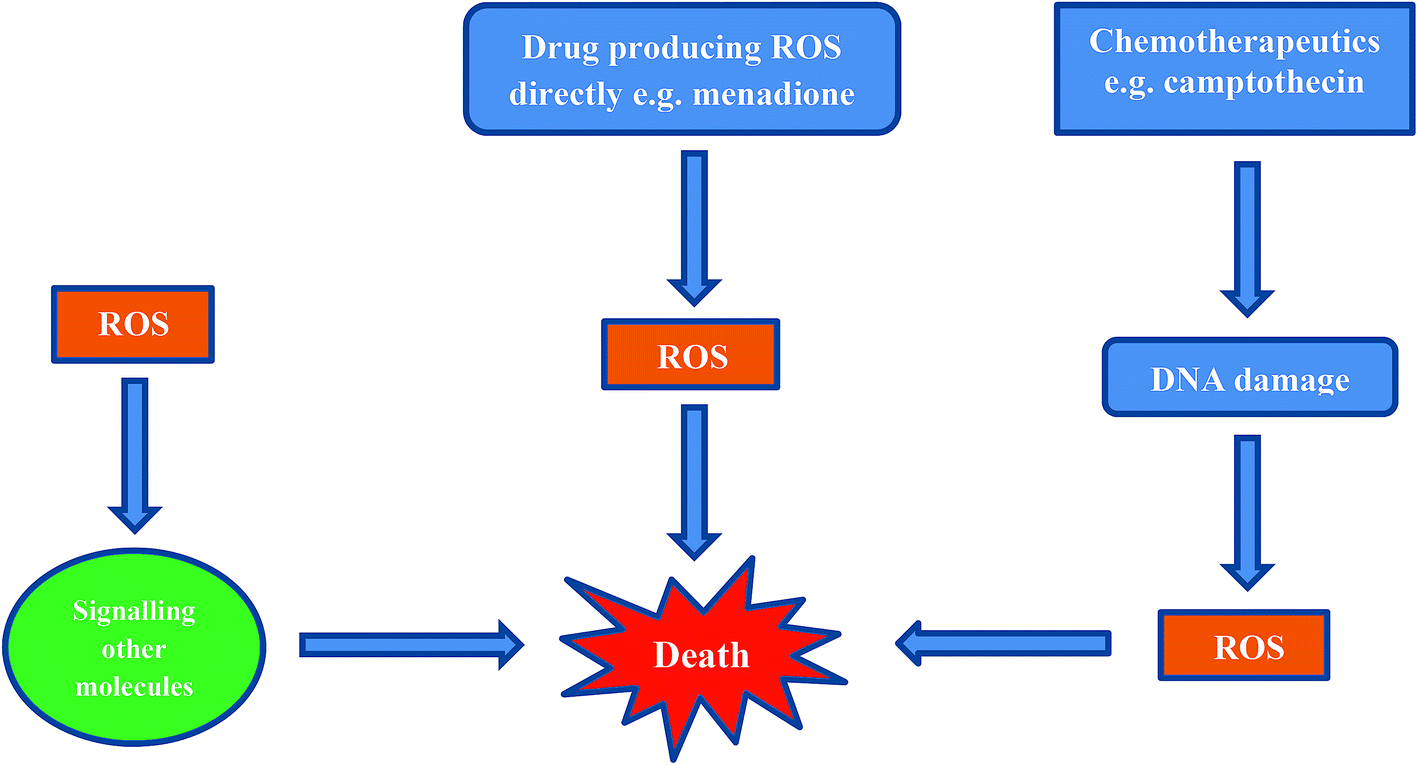 | ||
| Fig. 5 Death induced by cellular ROS by at least three mechanisms. | ||
The first mechanism entails cells exhibiting ROS damage as a by-product from cytotoxins in combination with another primary mechanism involving an anticancer agent (e.g. cisplatin, camptothecin). The ROS produced during cisplatin treatment as well as after DNA crosslinking by cisplatin, probably contribute to cell death, but these are not the primary cause for the induction of cell death.
The second mechanism by which cells die is from ROS exposure to compounds that induce a small amount of ROS. These ROS then cause downstream signaling for the actual molecules that are responsible for inducing cell death.
The third, and most direct mechanism, is when macromolecules in the cell are damaged by ROS as a result of reduction and oxidation of a compound e.g. menadione. When a sufficient amount of ROS is generated and intracellular stores of antioxidant molecules are exhausted, the cells cannot recover from ROS-induced damage.
Reductive activation of quinones
Enzymes and the oxygen environment modulate the biological activity of quinones and other molecules. Quinones undergo bio-reductive activation by two major enzymes such as the cytochrome P450's and the cytochrome P450 reductases. In addition, a hypoxic (oxygen deficient) environment also has an effect on some anticancer agents.Quinones can undergo bio-reductive activation by two pathways i.e. a 1 electron or a 2 electron reduction (Fig. 6). A 1 electron reduction affords a semiquinone while a 2 electron reduction affords the corresponding hydroquinone. The hydroquinone and quinone are in equilibrium. The semiquinone can be oxidized to the quinone under normal oxygen levels. In this process O2 is reduced resulting in the production of superoxide radical anions (O2−˙).
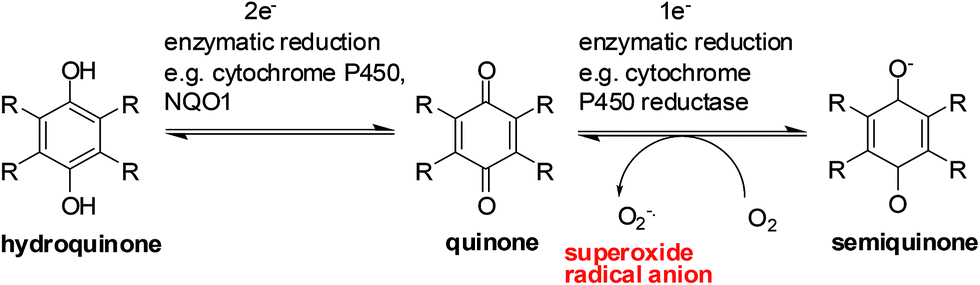 | ||
| Fig. 6 A general scheme for quinone reduction. | ||
Quinone activation by a 1-electron reduction
The cytochrome P450 reductases are the main enzymes responsible for the 1-electron reduction of anticancer drugs.84–86 The cofactors, FADH and FMNH, are utilised by P450 reductase to transfer electrons from NADP for a 1 electron quinone reduction.87 Since Cytochrome P450 reductase does not have a metal center only a 1 electron reduction of a molecule can be done.87Cytochrome P450 enzymes are found in nearly all tissues in the body and have many different functions. They accept various substrates that can be part of biosynthesis, degradation as well as activation of both xenobiotics and endogenous compounds.88 It is the Cytochrome P450s that are responsible for the activation of some investigational drugs to active molecules. The enzymatic heme core (a mononuclear Fe center) of cytochrome P450s uses NADPH to reduce molecules by 2 electrons resulting in the formation of the active species of some drugs.89,90
Quinone activation by a 2-electron reduction
The enzyme, NAD(P)H: quinone oxidoreductase 1 (NQO1, DT-diaphorase), reduces a quinone by 2 electrons to afford a hydroquinone (Fig. 7).91 The catalytic action is accomplished through the cofactors FAD, NAD(P)H, and the enzyme pocket since it does also not contain a metal center.91 The detoxification of many quinones may be due to NQO1 which results in the excretion of the less reactive hydroquinone (Fig. 8).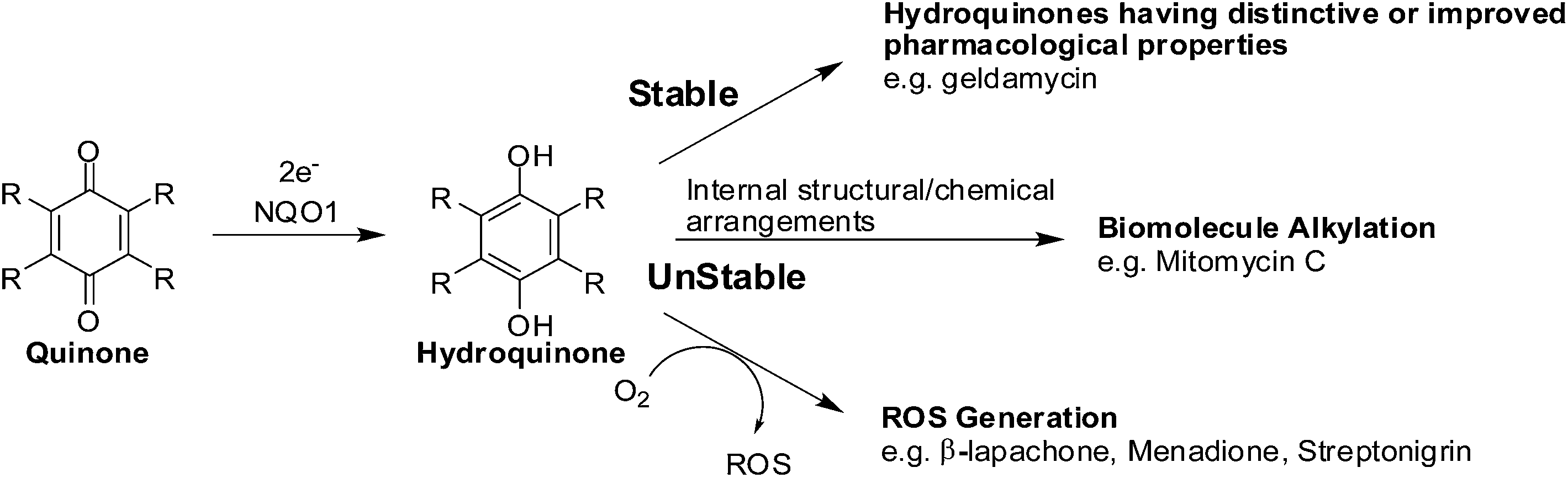 | ||
| Fig. 7 The bio-reductive activation of antitumour quinones by NQO1. | ||
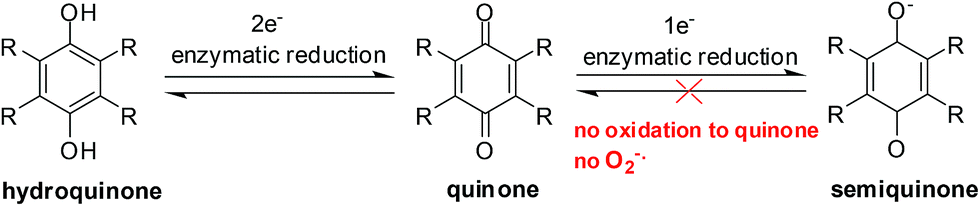 | ||
| Fig. 8 Semiquinones cannot re-oxidize to quinones without oxygen and can thus accumulate in cells. | ||
The 2-electron reduction process has made some compounds biologically active (Fig. 7).92 Since quinones exert their activities after reduction many are regarded as pro-drugs. For a particular target the corresponding hydroquinone may have greater pharmacological activity than the parent quinone. Semiquinones or hydroquinones may be formed after quinone reduction followed by subsequent generation of ROS and oxidative stress.
Activation in hypoxic environments
Hypoxia (also known as hypoxiation or anoxemia) is a condition in which the body or a region of the body is deprived of adequate oxygen supply. It is commonly defined at 0.1–2% oxygen in the local environment, compared to ∼20% oxygen in normoxic tissues.93 Hypoxia has been identified as a major contributor to cancer progression and to treatment failure. By decreasing the availability of oxygen patient treatment resistance increases and tumour progression is favoured.Hypoxia, a major feature of solid tumours, offers several treatment challenges. Hypoxia, in rapidly growing tumours, is a result of either a reduced number or incorrectly formed blood vessels.93 Availability of a drug is reduced when the blood vessels are reduced in number and in integrity. The resistance to drugs also increases due to slower cellular metabolism. Furthermore, solid tumours also become resistant to radiation treatments as oxygen is not present to repair DNA damage.93
In the absence of oxygen, semiquinones, created by a 1-electron reduction, cannot be oxidized to quinones and results in the accumulation of semiquinone and less hydroquinone (Fig. 4). Differences between normoxic and hypoxic cells offer a method of selective cancer treatment.93
Mechanisms of action of quinone anti-tumour agents
The mechanisms of action of these compounds have been the subject of intense investigation.18,94 In the structure of most of the anti-tumour agents it is the para-quinone moiety that participates in the cell redox cycle and acts as a precursor of ROS which leads to oxidative stress.18Under aerobic conditions in organs with a sufficient blood supply (normoxia) the highly redox active quinones undergo a one-electron reduction resulting in free-radical intermediates that undergo back oxidation in the presence of oxygen, releasing ROS.95,96 The ROS are generated via their intermediate semiquinone or hydroxy radicals. It is the hydroxyl radicals that are responsible for DNA strand breaks.97
Under anaerobic conditions (hypoxia), a two-electron reduction of the quinone to the hydroquinone, is followed by its inactivation through subsequent glucuronidation and/or sulfation. The in situ reduction of the quinone to the hydroquinone is an alternative pathway.96 This alternative pathway leads to conjugated intermediates which are powerful alkylating agents.97–101 This is believed to be the dominant mechanism under anaerobic conditions.102
When severe oxidative stress occurs within cells as a result of the formation of ROS, oxidized cellular macromolecules such as proteins, lipids and DNA can be formed and a number of signaling pathways can also be activated.103–106
Quinone-based drugs in cancer therapy
The goal of anticancer drug design is to develop drugs which are selectively toxic to tumour cells with minimal toxicity to normal cells. A requirement for a drug to be used in therapy is that it must replace the natural chelators that bind to natural cellular receptors.107 Natural regulative agents (agonists) may be imitated by the drug or the activity of natural ligands may be inhibited by blocking the receptor spot.Some drugs can be administered in an inactive form into the tissue and become activated upon interaction with the cytoplasmic reticulum e.g. doxorubicin (adriamycin) and mitomycin. These drugs only become active when they have been reduced in the cytoplasmic reticulum. Drug entry into the cell occurs by passive diffusion.108 Quninone-based drugs used in chemotherapy such as the anthracyclines, anthraquinones, mitomycin C and streptonigrin will be discussed in this section.
1. Anthracyclines
The anthracyclines (doxorubicin, daunorubicin and idarubicin) are quinone-containing antitumour agents used in treatment of cancers such as leukemias, lymphomas, breast, uterine, ovarian, and lung cancers (Fig. 9). Doxorubicin has been used clinically to treat solid tumours,109 daunorubicin is used to treat acute lymphoblastic and myeloblastic leukaemias,110 and idarubicin is a first line treatment for acute myeloid leukemia.111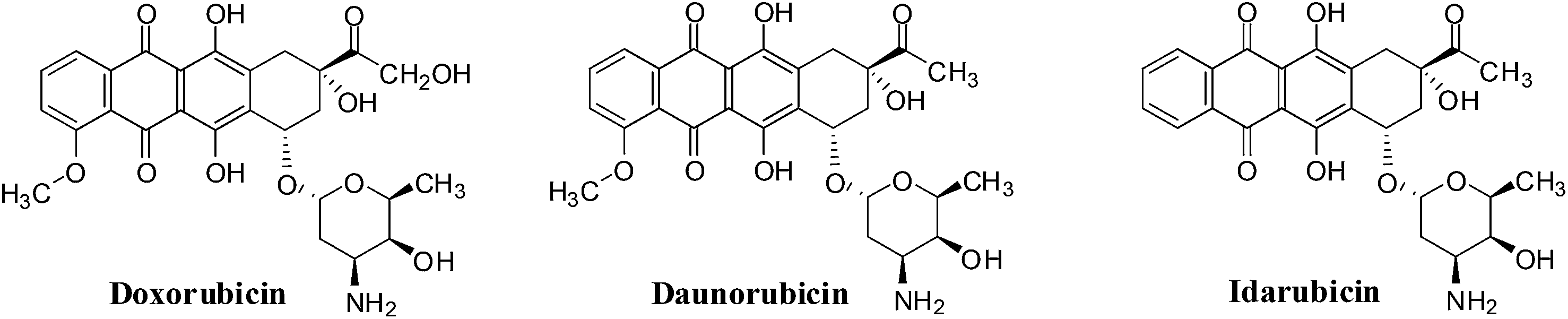 | ||
| Fig. 9 The anthracycline drugs. | ||
Another mechanism proposes that anthracyclines can also undergo redox cycling to generate ROS by a process that involves enzyme catalyzed one-electron reduction to the semiquinone radical which, on interaction with oxygen, generates the superoxide anion radical (O2−˙) and hydrogen peroxide (H2O2). Damage to tumour cells is due to the ability to undergo enzymatic reduction to the semiquinone radical.112,113
The action of doxorubicin (adriamycin) was shown to be more complicated.114 Formaldehyde was formed from the oxidation of the keto side chain by hydrogen peroxide which subsequently affected covalent bonding of the drug via its amino group with the 2-amino moiety of a DNA guanine. The anthracycline thus also functioned as an alkylating agent. It was shown that the synthetic anthracycline formaldehyde conjugates, precursors for formaldehyde generation, could circumvent multi-drug resistance and could potentially be used in the treatment of resistant cancers.114
A major disadvantage of all the anthracyclines is the cardiac damage that is produced which can result in serious and even life-threatening complications.115
2. Anthracenediones
They were developed in a program to find a cytotoxic agent with less cardiotoxicity than that of the anthracyclines.116 The anthracenediones differ in structure from the anthracyclines since they do not have a glycoside substituent.The synthetic anthracenedione, mitoxantrone, is used to treat mostly metastatic breast cancer, acute myeloid leukemia, and non-Hodgkin's lymphoma (Fig. 10).117–119 The survival rate of children suffering from first relapse of acute lymphoblastic leukaemia was also improved by mitoxantrone treatment.120
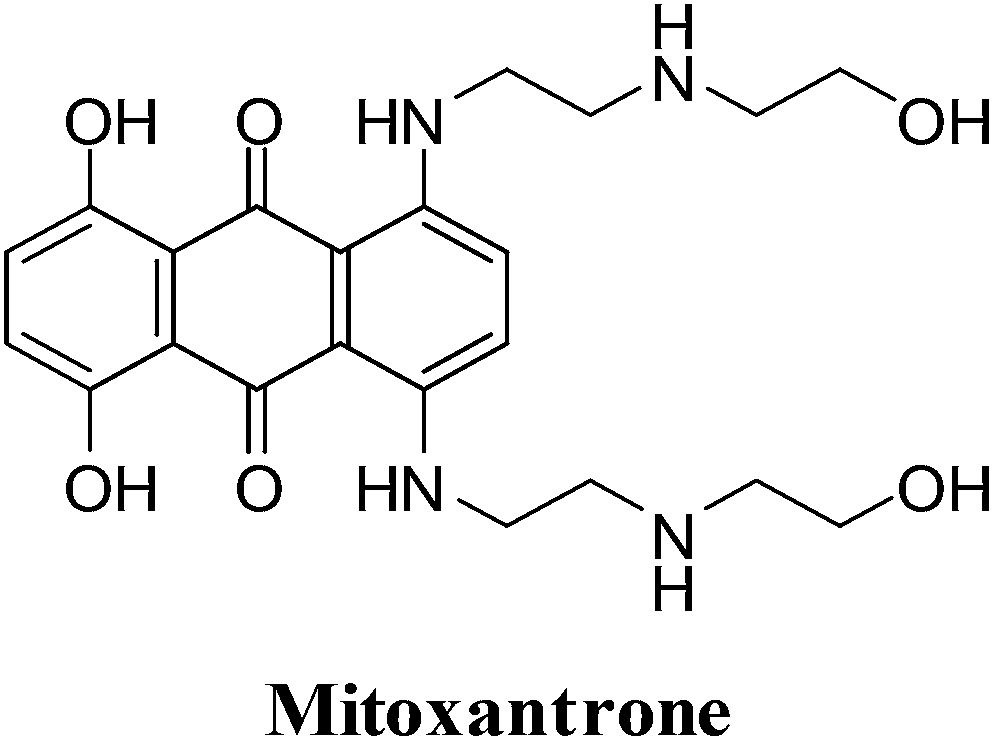 | ||
| Fig. 10 A potent topoisomerase II inhibitor and DNA intercalator. | ||
The antitumour activity spectrum of mitoxantrone is less than that of doxorubicin. It is also has less cardiac toxicity as a result of a decreased ability to participate in ROS generation. The main disadvantage of mitoxantrone is its cardiac toxicity.121 It has a similar activity to that of doxorubicin but produces significantly less cardiac toxicity in patients with previously treated breast cancer.122 Mitoxantrone has also been used clinically to treat solid tumours.109
3. Mitomycin C
The mitomycins from Streptomyces sp. are quinone-containing alkylating agents. Mitomycin C (MMC) is a quinone containing antibiotic that was isolated from Streptomyces caespitosus (Fig. 11). MMC is considered a prototype bio-reductive alkylating drug.125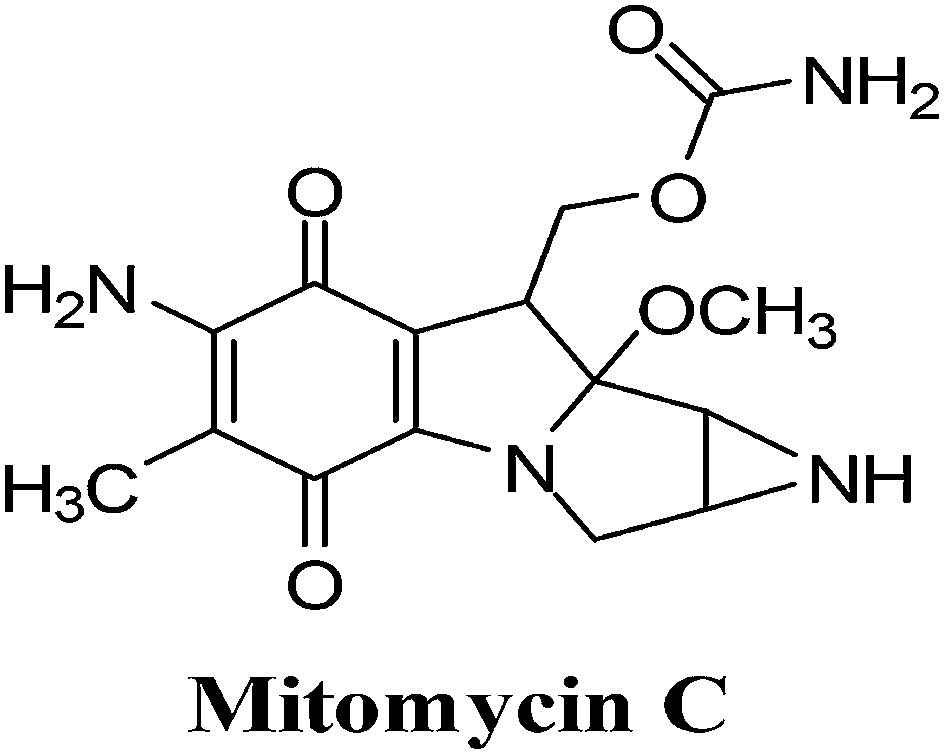 | ||
| Fig. 11 A bio-reductively activated anticancer drug. | ||
The term “bio-reductive alkylating agent” refers to drugs which generate electrophilic species upon reduction which then bind covalently to cellular macromolecules126 and was first used by Sartorelli and co-workers.127 Enzymatic reduction, by both one- and two-electron reductases, is required to achieve intracellular activation of bio-reductive alkylating agents. Bioactivation affords mono- and bifunctional alkylating species (bifunctional alkylating agents can cross-link DNA) and/or generates ROS.
Bio-reductive chemotherapy exploits the differences in oxygen content and cellular pH between normal tissue and tumour tissue and can therefore be utilised to selectively attack carcinomas.128 This treatment is most successful in certain cancers such as breast, colon, head and lung cancer that are rich in reductive activation proteins.
Hypoxic cells of solid tumours, though resistant to most chemotherapeutic agents, create an environment which favours activation of MMC through reductive processes. MMC exhibits a greater cytotoxicity to oxygen-deficient cells than to their oxygenated counterparts and are thus ideal for attacking hypoxic regions of solid tumours.
This FDA approved drug, which has been used clinically for more than 30 years, is used for the treatment of solid tumours including stomach, pancreas, breast and lung. Studies showed that there was an excellent correlation between tumour cell kill and formation of ROS and that the drug, covalently bound to DNA, remained redox active resulting in oxy radicals.130
4. Streptonigrin
This quinoline analogues of naphthoquinone shows a broad range of activity against breast, lung, lymphoma, melanoma cancer as well as head and neck cancer (Fig. 12).131–136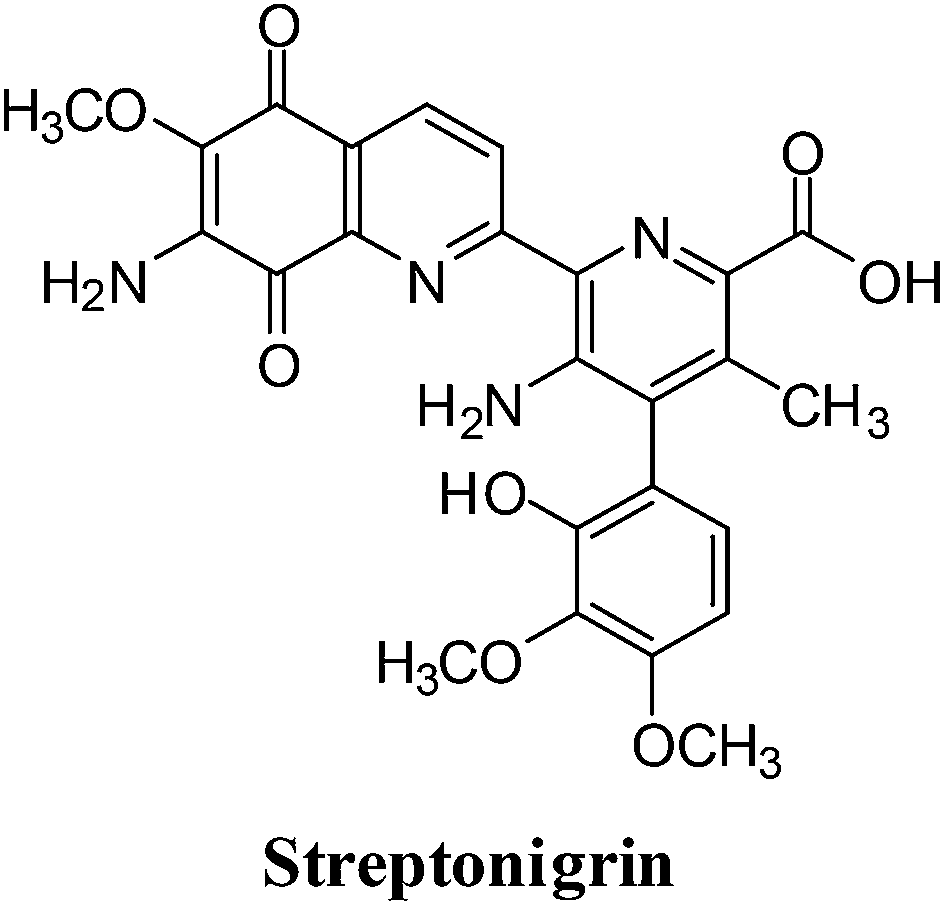 | ||
| Fig. 12 An aminoquinone containing anticancer agent. | ||
A side-effect of streptonigrin (SN), which has limited its use in cancer therapy, is prolonged bone marrow depression.137–142 Positive results were, however, reported for SN in the treatment of leukemias, lymphomas and melanomas.143–147 DT-diaphorase, an enzyme showing increased activity in certain tumours such as human non-small cell lung cancer, efficiently bio-activates SN.148,149 It was therefore proposed SN could be used against tumours with increased DT-diaphorase function.
For SN to produce its DNA-damaging effects, autoxidation of the quinone moiety to semiquinone in the presence of NADH is required which leads to the production of free radical species.150,153,156,157
Multidrug resistance (MDR) in cancer
Cancer multidrug resistance is defined as the cross-resistance or insensitivity of cancer cells to the cytostatic or cytotoxic actions of various anticancer drugs which are structurally or functionally unrelated and have different molecular targets.158 This phenomenon is prevalent in various types of cancers and is a major impediment to the effective chemotherapeutic treatment. The resistance of cancer cells to multiple cancer drugs is also the main reason for the failure of cancer treatments.159 Resistance entails cellular and non-cellular mechanisms which are utilised by cancer cells to overcome the cytotoxic actions of various drugs. These mechanisms form part of the strategy that confers resistance to cancer cells.Mechanisms of multidrug resistance in cancer
The numerous mechanisms that have been proposed to mediate cancer multidrug resistance can be categorized as cellular or non-cellular depending on the factors contributing to MDR development.160 Cellular mechanisms involve enzymes and transport systems while non-cellular mechanisms involve factors that are extracellular such as the cell growth environment or limited vascular accessibility.Cellular MDR mechanisms
There are two cellular MDR mechanisms which are utilised by the cell for the removal of a drug. The first has been classified as classical/transport based and the second, non-classical/non-transport based.The first mechanism involves the efflux of a drug from the cell by various energy dependant membrane transport proteins and occurs in the classical/transport based cellular mechanism of MDR. This prevents the drug from reaching its therapeutic concentration inside the cell.161
The superfamily of proteins that mediate this MDR via the ATP-dependent drug efflux pumps are the ATP-binding cassette (ABC) transporters.162 ABC transporters are proteins that occur throughout the cell membrane and can actively transport both endogenous compounds and xenobiotics. MDR is a result of overexpression of the ABC transporters.163
P-glycoprotein (Pgp), multidrug resistance-associated protein-1 (MRP1), its homologs MRP2-6 and the breast cancer resistance protein (BCRP) are all transport proteins of the ABC superfamily.164–166 These proteins are overexpressed in cancer cells and pump anticancer drugs out of the cell. Among the ABC transporters Pgp confers the strongest resistance to a wide variety of compounds and has been expressed in cancers of the gastrointestinal (GI) tract (small and large intestine, liver cancer, and pancreatic cancer), cancers of the genitourinary system (kidney, ovary, testicle), cancers of the hematopoietic system (myeloma, lymphoma, leukemia), as well as childhood cancers (neuroblastoma, fibrosarcoma).167
Many cellular processes require transport of metabolites or substrates across the cell membrane and utilise the ABC transporters. The ABC transporters are thus key elements to consider in the discovery and development of drugs targeting MDR in cancer cells.
The second mechanism, the non-classical/non-transport based one, employs enzymatic systems that restrict the required drug activity without changing its effective concentration within the cell. The enzyme, glutathione-S-transferase (GST), is an important enzyme responsible for xenobiotic metabolism. The excretion of organic molecules can be expedited through biotransformation by GST which catalyses their conjugation with polar molecules. Drugs are modified into end products with reduced activity and an enhanced rate of excretion. Elevated levels of GST have been reported in various resistant cancer cell-lines like MCF-7.168,169
Non-cellular MDR mechanisms
In vivo tumour growth leads to non-cellular drug resistance which is typically associated with solid tumours. For cells to be considered cancerous, growth beyond their natural boundaries is required. This growth is usually accompanied by a vasculature that is well developed. In certain solid tumours, where angiogenesis has been compromised, the vasculature is poor.170 Poor vasculature can lead to limited drug access to regions within these solid tumours and thus protect them from cytotoxicity thus conferring resistance. Lactic acid generation by hypoxic tumour cells generates an acidic environment in tumours which can confer a resistance mechanism against weak bases where cellular uptake across membranes is dependent on the pH gradient. The physiological properties of solid tumours can result in tumour regions that lack oxygen (hypoxia) and nutrients which can confer resistance to cancer cells particularly against drugs that act on actively dividing cells.171The three main mechanisms responsible for cancer MDR
The three mechanisms identified are as follows: the membrane mechanism determining the low drug level in resistant cells associated with the existence of specific membrane proteins (P-gp and MRP);172–174 the mechanism associated with change in the effectiveness of the glutathione cycle;168,169 the mechanism associated with the decrease in DNA topoisomerases expression.161,175Knowledge about mechanisms of cancer drug resistance may aid in the design and development of strategies to circumvent resistance. This knowledge may also contribute to the development of new drugs that are less prone to the known resistance mechanisms. Several reviews have been written on cancer MDR and these can be consulted for further information.161,176–182
Studies on synthetic naphthoquinones and derivatives of natural naphthoquinones
In this section we will discuss the anticancer activity of the synthesised synthetic naphthoquinones as well as synthesised derivatives of natural naphthoquinones. The reaction schemes are only shown for reports where the synthetic route to the target molecule is short.1. Aminonaphthoquinones
A subject of study for many years have been 1,4-naphthoquinones possessing an amino or a substituted amino group in the 2-position. This was inspired by various medical and biological applications such as antituberculars, antimalarials, antibacterials, antitumour agents, larvicides and molluscicides, herbicides, and fungicides.183–186 The nitrogen atom in this position enables geometric modification of the neutral molecules and of their reduction intermediates and modulation of the substituent's effects on the electronic properties of the quinone system.The commercial anti-neoplastic agents actinomycin187 and streptonigrin187 as well as the antibiotics, mitomycin188 and rifamycin,189 are also based on an aminoquinone. The amino group is in the 2-position in mitomycin C, streptonigrin,187 actinomycin189 (and the structurally related aurantins190) as well as in the ansa-antibiotics, rifamycin189 and geldanamycin.191 The biological activity exhibited by these drugs have inspired research into new routes to the synthesis of aminoquinones.
A series of side chain homologated derivatives of 2-chloro-3-(n-alkylamino)-1,4-naphthoquinone {n-alkyl: pentyl; hexyl; heptyl; and octyl} were synthesised by Pal et al. (Scheme 1).192
 | ||
| Scheme 1 Synthesis of aminonaphthoquinones 1–8. | ||
Antiproliferative activities of 1 to 8 {n-alkyl: methyl; 1, ethyl; 2, propyl; 3 and butyl; 4} were studied in a panel of cancer cells consisting of colon (COLO205), brain (U87MG) and pancreas (MIAPaCa2) cell-lines using the XTT assay.
Compounds 1, 2 and 3 were active against MIAPaCa2 (1 = 2 < 3) and COLO205 (2 = 3 > 1) and inactive against U87MG. Compound 3 was the most active compound (IC50 = 1.3 ± 2 mM). It was established that homologation of 2-chloro-3-(n-alkylamino)-1,4-napthoquinone with saturated methyl groups afford tissue specific compounds such as 1 (for MIAPaCa2) and 3 (for COLO205) with ideal activity.
Bhasin et al. used 1,4-naphthoquinone, a 1,4-naphthoquinone derivative, Juglone, Menadione and Plumbagin as lead molecules in structure–activity relationship studies of a series of substituted 1,4-naphthoquinones.193 The synthetic routes are shown in Scheme 2 below.
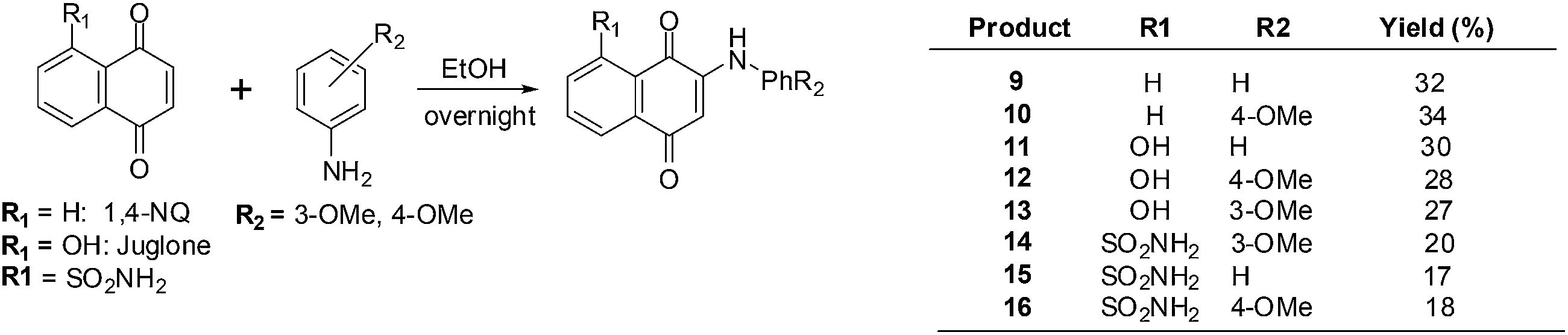 | ||
| Scheme 2 Synthesis of aminonaphthoquinones 9–16. | ||
The compounds were screened against a panel of cell-lines using the Promega CellTiter 96 Aqueous Non-Radioactive Cell Proliferation assay. Enhanced antiproliferative activity was observed in prostate (DU-145), breast (MDA-MB-231) and colon (HT-29) cancer cell-lines in contrast with juglone, the lead molecule. Compounds 9 and 10 only exhibited potent activity against prostate cancer, but compounds 11–13 exhibited potent activity against all three cancer cell-lines. The most potent compounds were 12–16 which had IC50 values in the 1–7 μM range in all three cancer cell-lines.
Wellington et al. reported on the synthesis of a series of aminonaphthoquinones obtained by laccase catalysis (Scheme 3).194
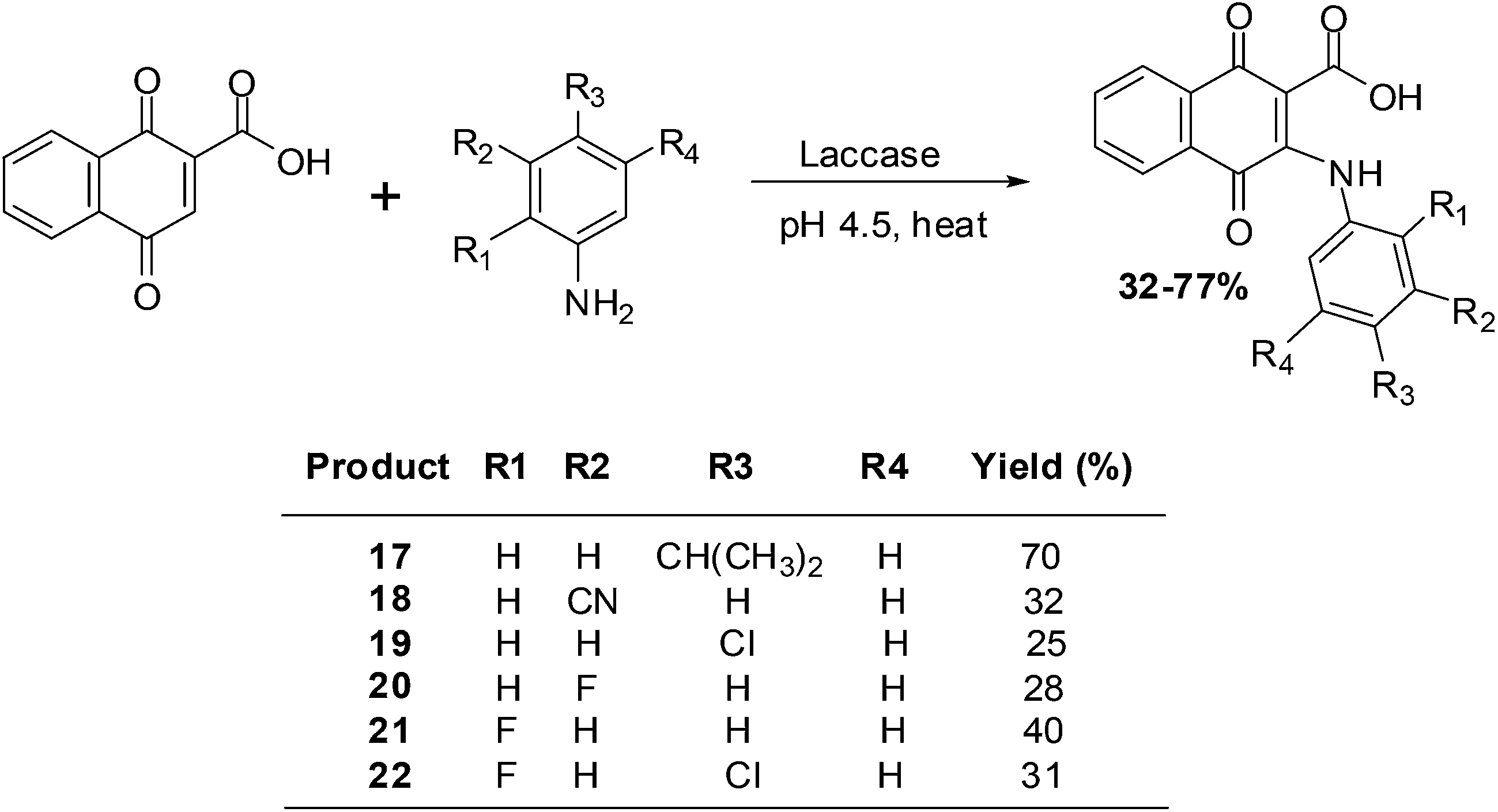 | ||
| Scheme 3 Laccase-catalysed synthesis of aminonaphthoquinones. | ||
The compounds were screened against renal (TK10), melanoma (UACC62), breast (MCF7) and cervical (HeLa) cancer cell-lines using the sulforhodamine assay to determine their growth inhibitory activity. From the screening results it was apparent that the aminonaphthoquinones exhibited potent cytostatic effects mainly against the melanoma cancer cell-line (GI50 = 3.98–7.54 μM).
Potent cytostatic effects against both the renal and the melanoma cancer cell-lines were exhibited by compound 17. Against the renal cell-line the activity (GI50 = 8.38 μM) was nearly as good as that of the anticancer agent, etoposide (GI50 = 7.19 μM). Potent cytostatic effects against both the melanoma and the breast cancer cell-lines were also exhibited by compounds 20 and 21 (GI50 = 5.28–9.84 μM).
Most compounds exhibited a total growth inhibition (TGI) which was better than that of etoposide against the melanoma cell-line. The potent cytostatic effects of compounds 18, 19 and 22 (TGI = 7.17–7.94 μM) against the melanoma cell-line was 7- to 8-fold better than that of etoposide (TGI = 52.71 μM).
The melanoma cancer cell-line was the most susceptible to the aminonaphthoquinones.
Benites et al. synthesised a series of phenylamino-1,4-naphthoquinones to investigate structure–activity relationships (SARs) and the anticancer activity of this scaffold (Scheme 4).195
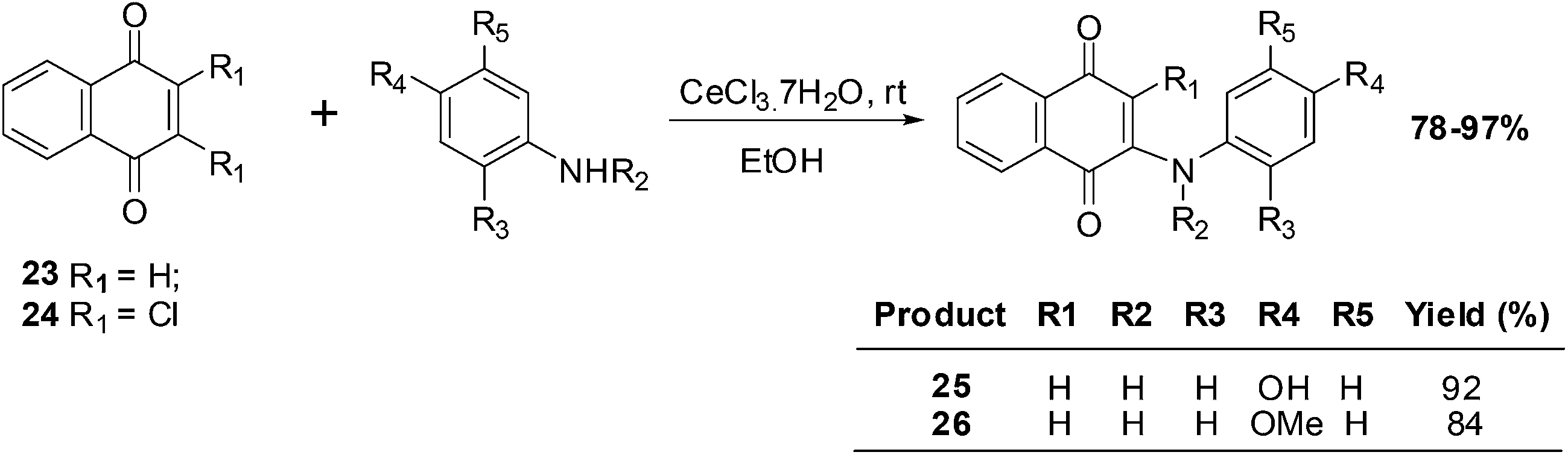 | ||
| Scheme 4 Synthesis of phenylaminonaphthoquinones. | ||
The cytotoxic effects of the aminoquinones were evaluated in vitro using the MTT reduction assay against breast (MCF7), prostate (DU145) and bladder (T24) cell-lines as well as healthy fibroblasts (BALB/3T3). The half maximal effective concentration (EC50) was determined for each of the compounds.
From a SAR analysis of the aminonaphthoquinone series it was shown that insertion of a methyl group at the nitrogen atom of the donor phenylamino group and/or insertion of a chlorine atom in the acceptor quinone nucleus induced significant changes in cytotoxic activity.
The aminoquinones, 25 and 26, exhibited a high safety index (5.73 and 6.29, respectively), low hydrophobicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of 1.16 and 1.43, respectively), and low redox potential (−775 and −856 mV, respectively). Since they had a high safety index they were further investigated in various assays: cell proliferation (clonogenic assay), caspase-3 activation (DEVDase activity), and the intracellular content of ATP. Aminoquinone 25 impaired the proliferative capacity of T24 cells without activating caspase-3 and also strongly influenced ATP levels.
P of 1.16 and 1.43, respectively), and low redox potential (−775 and −856 mV, respectively). Since they had a high safety index they were further investigated in various assays: cell proliferation (clonogenic assay), caspase-3 activation (DEVDase activity), and the intracellular content of ATP. Aminoquinone 25 impaired the proliferative capacity of T24 cells without activating caspase-3 and also strongly influenced ATP levels.
PI-083, an aminonaphthoquinone from the NCI chemical library (Fig. 13), was discovered in a search for inhibitors against the chymotrypsin-like (CT-L) activity of the proteasome. It is a non-peptidic proteasome inhibitor that has exhibited broader antitumour activity than bortezomib, the first 20S proteasome inhibitor approved by the US FDA (Fig. 13).
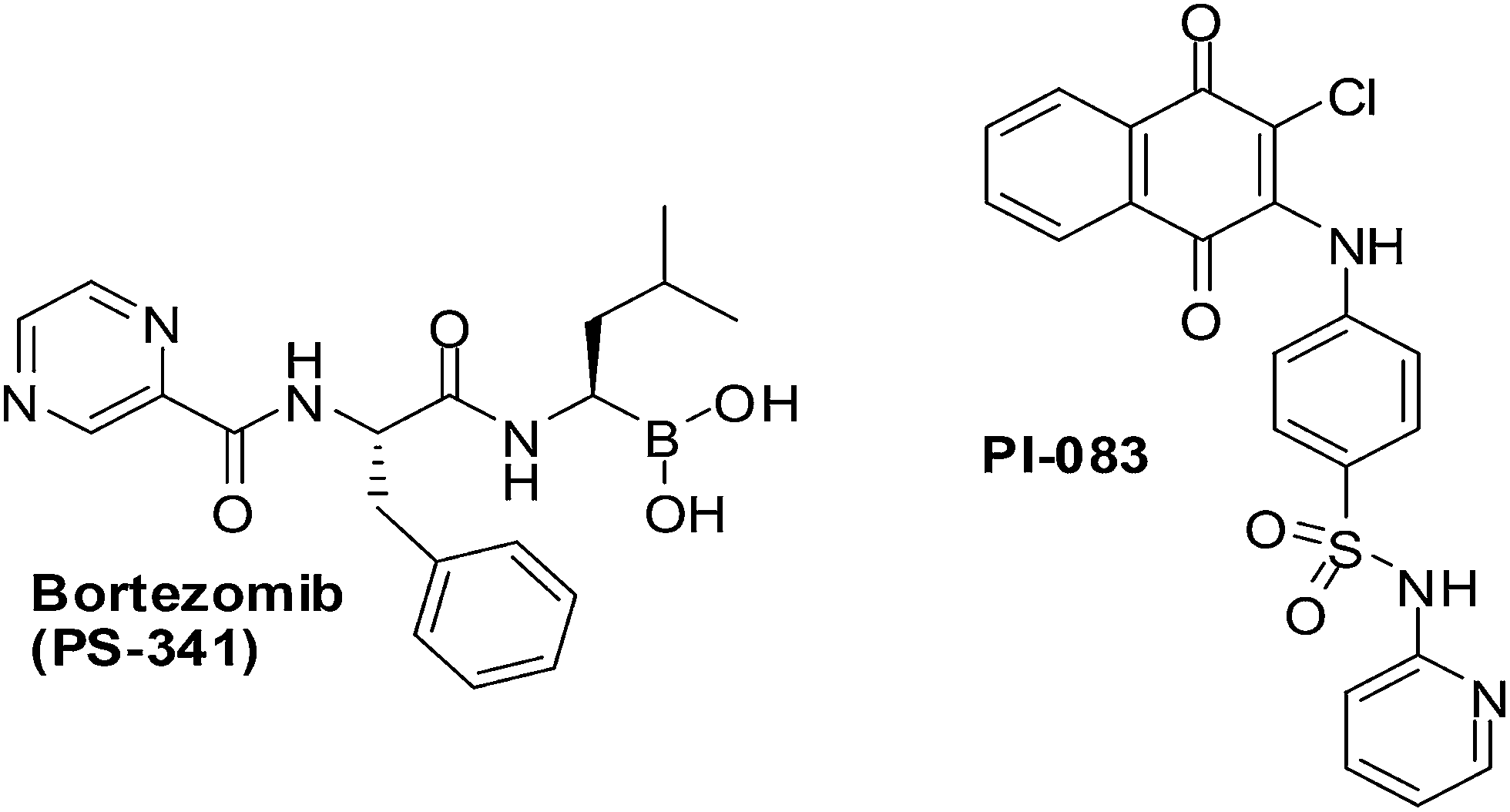 | ||
| Fig. 13 The chemical structures of PI-083 and Bortezomib. | ||
CT-L activity and cell proliferation is inhibited by PI-083 which also induces apoptosis selectively in cancer cells (ovarian T80-Hras, pancreatic C7-Kras and breast MCF-7) as compared to their normal/immortalized counterparts (T80, C7 and MCF-10A, respectively). PI-083 was found to induce its antitumour effects quicker than Bortezomib in a range of cancer cells such as Multiple Myeloma (MM), breast, pancreatic, ovarian, lung, prostate cancer cell-lines as well as fresh MM cells from patients.196,197
Xu et al. synthesised a series of naphthoquinone derivatives that were designed as analogues of the reputed proteasome inhibitor, PI-083 (Fig. 14).198
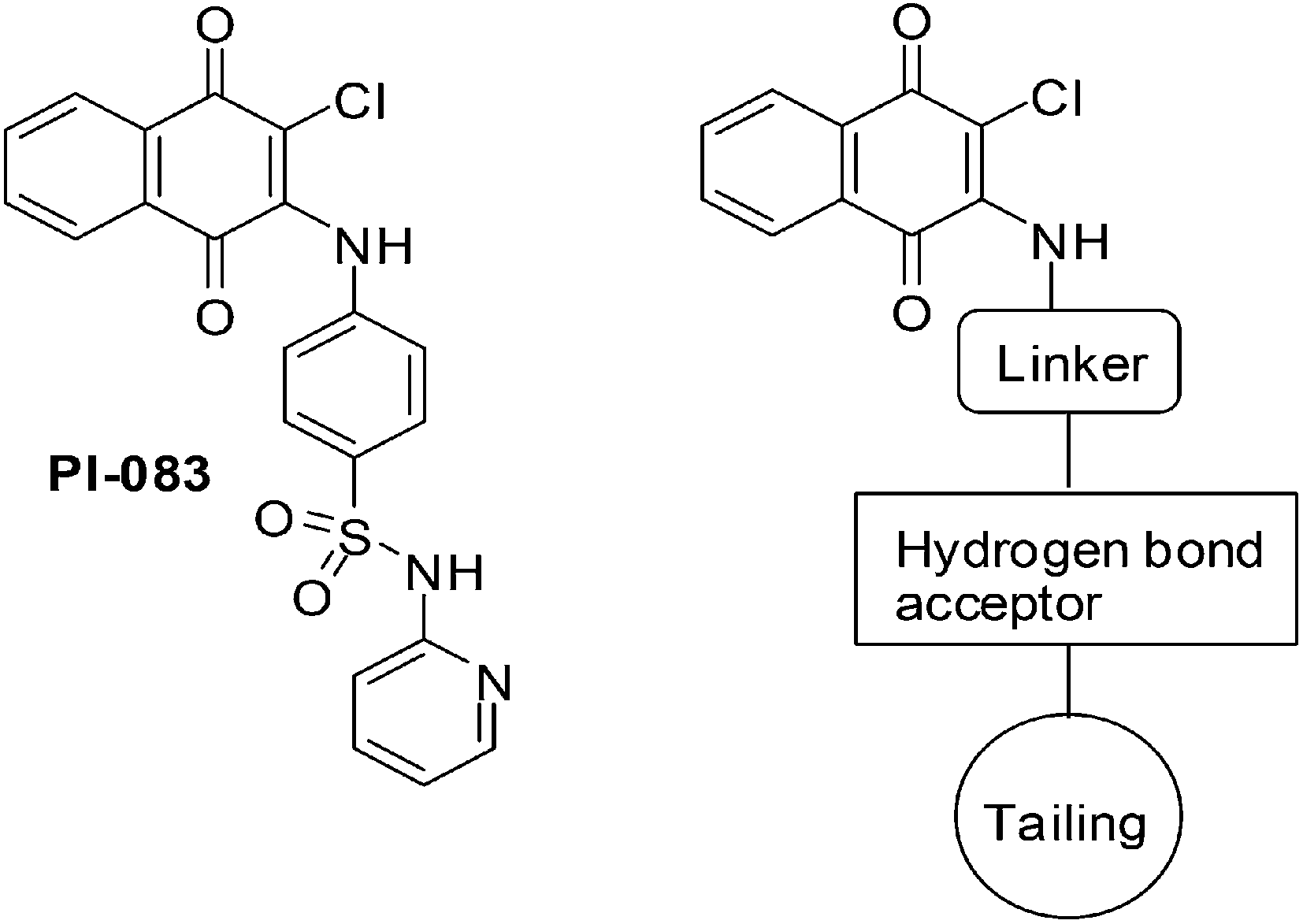 | ||
| Fig. 14 Structure of PI-083 and the general formula of the target compounds. | ||
The synthesis route for the aminonaphthoquinones is shown in Scheme 5 below.
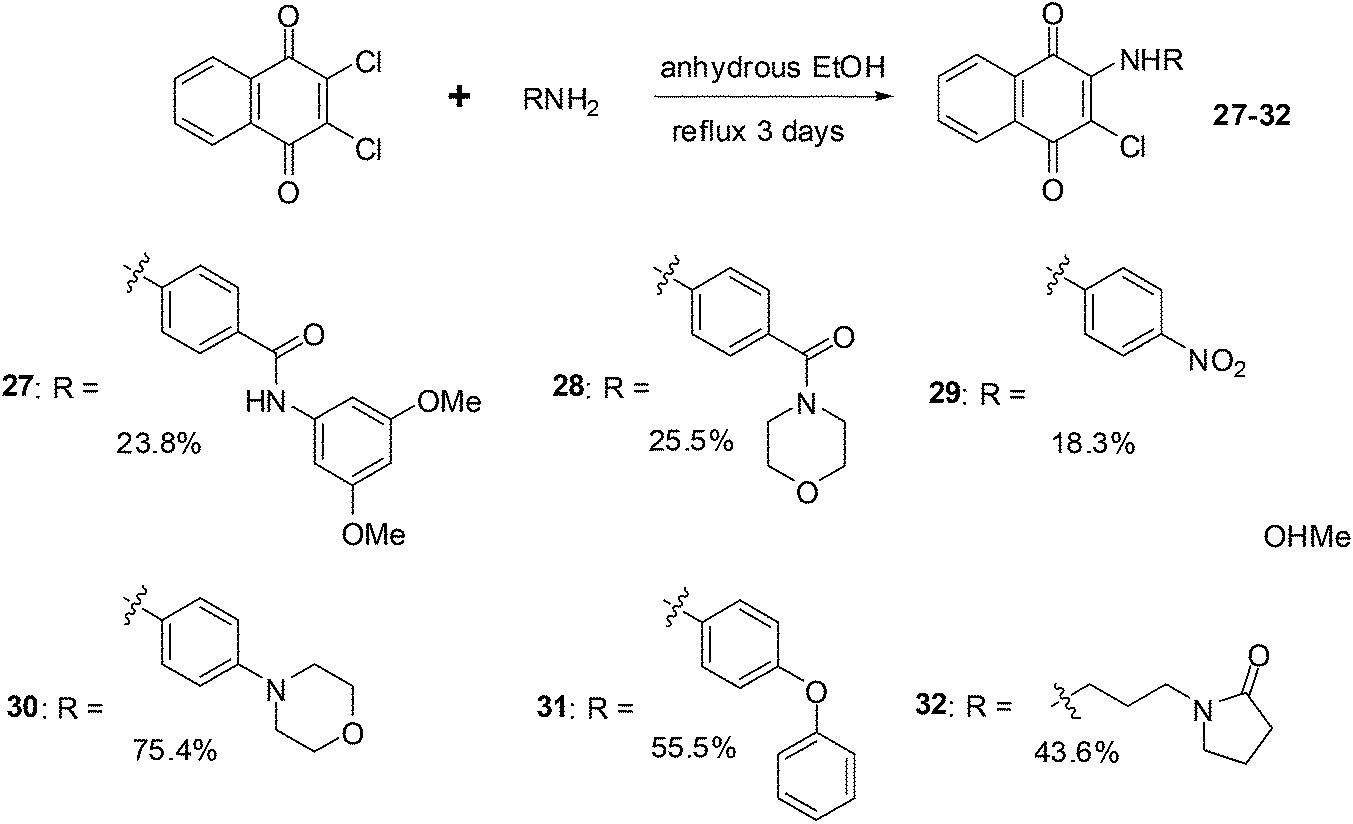 | ||
| Scheme 5 Synthesis of the aminonaphthoquinones – analogues of the proteasome inhibitor PI-083. | ||
The compounds were evaluated for antiproliferative activity against lung (A549), prostate (DU145), nasopharyngeal (KB), and vincristine-resistant nasopharyngeal (KBVIN) cancer cell-lines. Antiproliferative activities comparable to that of PI-083 were obtained for six compounds (27, 28, 29, 30, 31, and 32). Compound 31 exhibited potent activity (IC50 = 1.3–8.12 μM) against all four cancer cell-lines. Compound 29 was confirmed as a 20S proteasome inhibitor in both in vitro and in cell-based assays (Scheme 5).
2. 2-Arylnaphtho[2,3-d]oxazole-4,9-dione derivatives
It has been shown that naphthoquinones with fused five-membered rings reduce multidrug resistance and also potentially increases cytotoxicity.199 This prompted the synthesis of compounds 34–38 which were synthesised by refluxing 2-amino-3-bromo-1,4-naphthoquinone with appropriate benzoyl chloride analogues at elevated temperatures (Scheme 6).200 The reaction is thought to progress by first forming the 2-amido-3-bromo-1,4-naphthoquinone derivative 33 (Scheme 6).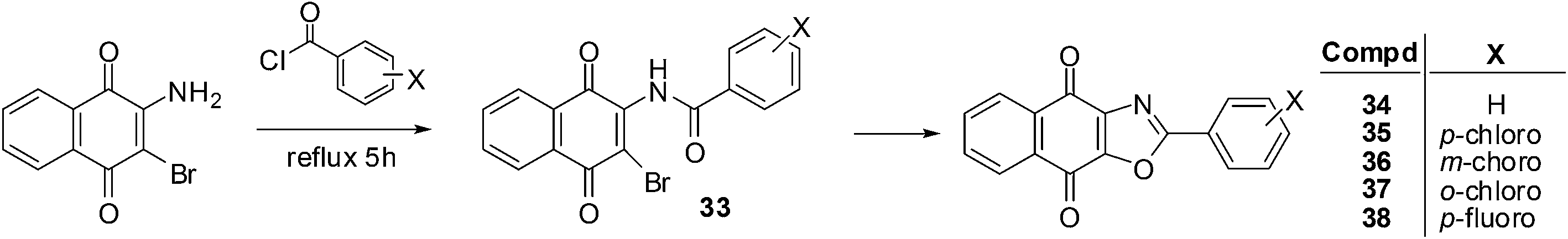 | ||
| Scheme 6 Synthesis of aryl-substituted oxazolonaphthoquinones. | ||
These compounds were evaluated in vitro for their cytotoxic effects by screening against androgen-dependent, LNCaP, and androgen-independent, PC3, human prostate cancer cell-lines using the MTT assay. The aryl-substituted oxazolonaphthoquinones displayed potent cytotoxicity against both LNCaP (IC50 = 0.01–0.40 μM) and PC3 (IC50 = 0.08–0.36 μM) prostate cancer cell-lines after 5 days of drug exposure. These compounds exhibited slightly stronger cytotoxicity against the LNCaP cell-line than on the PC3 cell-line.
Analogue 37, having the ortho-chloro substituent, had the strongest cytotoxicity (IC50 = 0.01 ± 0.002 μM) followed by the meta-chloro-analogue 36 (IC50 = 0.03 ± 0.002 μM) against the LNCaP cell-line. For the PC3 cell-line, the most potent compounds were the meta-chloro-analogue 36 (IC50 = 0.08 ± 0.007 μM) and the para-chloro-analogue 35 (IC50 = 0.08 ± 0.01 μM). The meta-substituted product 36 had the best cytotoxicity against both cell-lines (IC50 = 0.03 μM for LNCaP and 0.08 μM for PC3). When the cytotoxicities of the two para-substituted analogues on the androgen-independent PC3 cell-line were compared it was found that the p-chloro-analogue 35 (IC50 = 0.08 ± 0.01 μM) showed better activity than the p-fluoro- analogue 38 (IC50 = 0.20 ± 0.012 μM). It appears that both the position and the electronegativity of the substituent on the aryloxazole group are important in modulating the cytotoxicity of this class of compounds.
3. 2-Aryl-1,4-naphthoquinone-1-oxime methyl ethers
The cytotoxicity of a variety of quinone monooxime derivatives was evaluated against a HeLa S3 cell-line in an initial screen to determine SARs.201 The active compounds were then further screened against a panel of eight cancer cell-lines comprised of ovarian cancer (HeLa), renal cancer (Caki-1and 786-O), lung cancer (A549), breast cancer (MCF-7) and mesothelioma cancer (H28, H2052, and MSTO-211H). The cytotoxic activity was evaluated against HeLa S3 cells by a methylene blue staining method and the IC50 values were determined using the Litchfield and Wilcoxon method.These naphthoquinone oximes effectively inhibited HeLa (IC50 = 0.10 ± 0.02 μM for 39 and 0.23 ± 0.04 μM for 40) and MCF-7 (IC50 = 0.20 ± 0.07 μM for 39 and 0.51 ± 0.09 μM for 40) cancer cell-lines (Fig. 15).
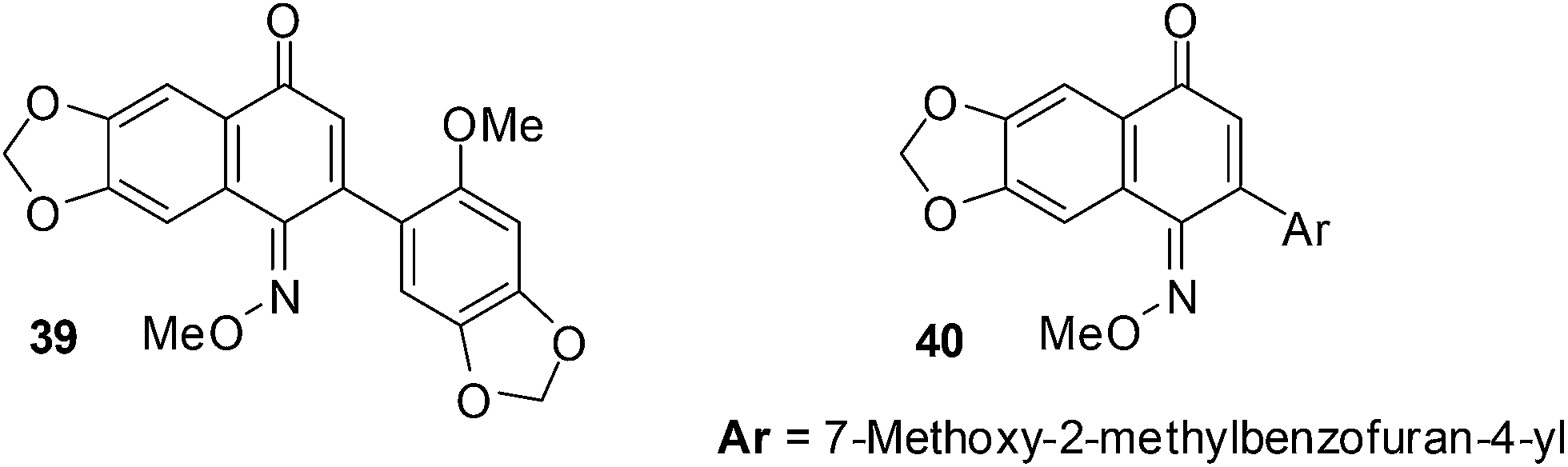 | ||
| Fig. 15 Compounds exhibiting activity against cancer. | ||
The 2-aryl-6,7-methylenedioxy-1,4-naphthoquinone-1-oxime methyl ethers showed cytotoxic activity against a wide range of cancer cell-lines of which the HeLa and MCF-7 cell-lines were most susceptible. The quinone monooxime core is therefore a promising scaffold for the discovery of bioactive small molecules against cancer.
4. Azanaphthoquinone pyrrolo-annelated derivatives
Shanab et al. reported on the synthesis of a series of azanaphthoquinone pyrrolo-annelated derivatives with basic side chains (Fig. 16).202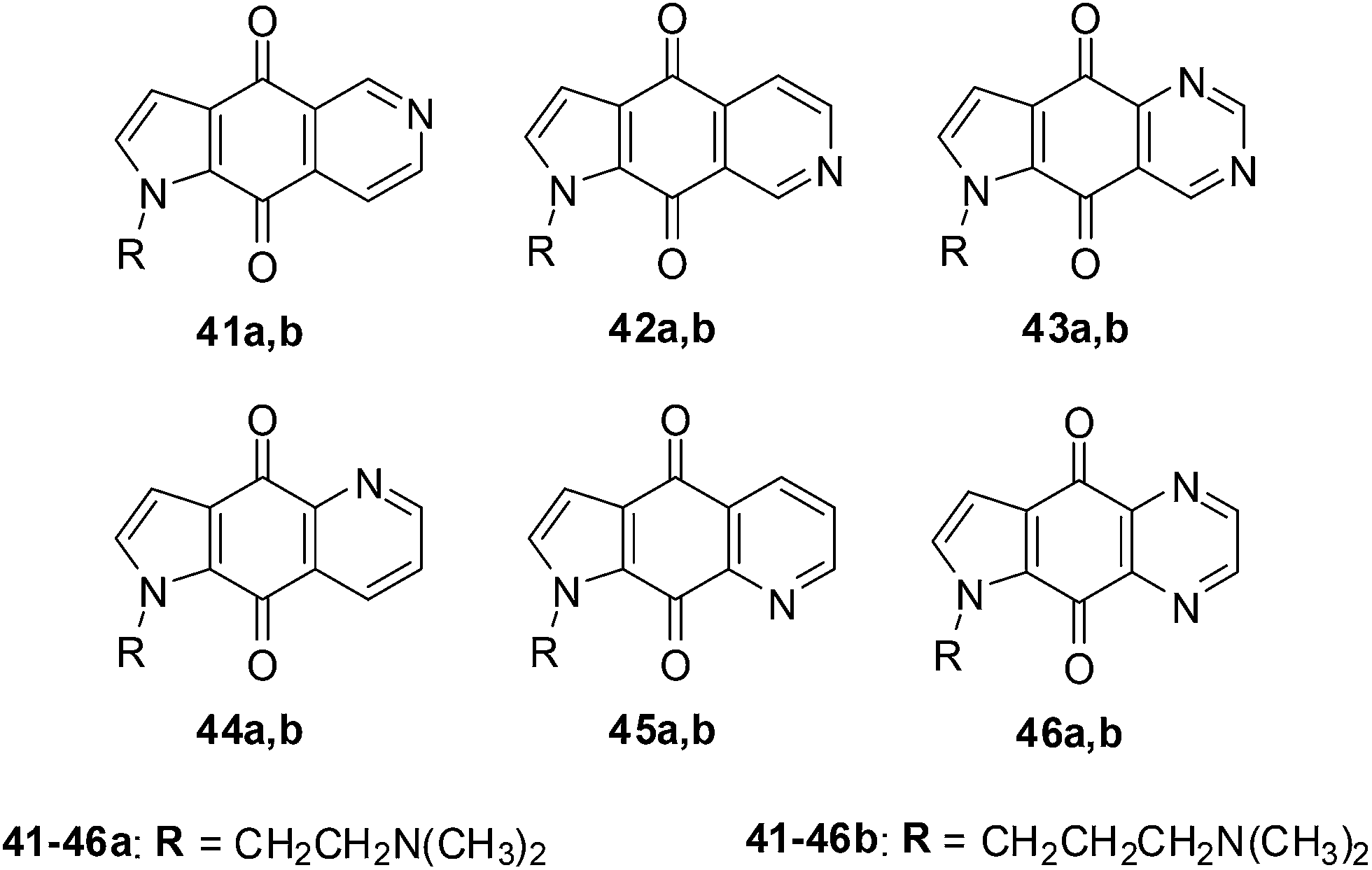 | ||
| Fig. 16 Compounds 41a,b–46a,b exhibiting significant antiproliferative activity. | ||
Amongst this series compound 42a exhibited significant antiproliferative activity which was due to the induction of caspase 3/7 activity and not due to intercalation. The rationale for the reported synthesis was to determine whether different annelation patterns of the pyridine ring to the quinone nucleus and substitution by equally electron-withdrawing heteroaromatic ring systems such as pyrazine or pyrimidine would enhance cytotoxicity.
The compounds were evaluated against five different cancer cell-lines such as cervical carcinoma (KB/HeLa), ovarian carcinoma (SKOV-3), CNS glioma (SF-268), non-small cell lung carcinoma (NCI-H460), and colon adenocarcinoma (RKOp27, RKOp27IND) using two independent XTT cytotoxicity assays.
The quinazolines 43 exhibited the highest inhibition across all the cell-lines with 43a, having the dimethylaminoethyl chain, displaying the highest inhibition (EC50 = 0.082 ± 0.006 μg mL−1 against NCI-H460 and 0.107 ± 0.020 μg mL−1 against SF-268). The quinazoline derivative 43b having a dimethylamino-propyl chain was slightly less active (EC50 = 0.177 ± 0.007 μg mL−1 against NCI-H460 and 0.198 ± 0.025 μg mL−1 against SF-268). The quinoxaline, 46a, was the next most active compound within the series and was nearly 10-fold less active. The isoquinolines 41a,b and 42a,b were at least two-fold less potent than 46a. Compound 45b, having a dimethylaminopropyl chain was inactive as well as the quinolines 44a,b and 45a,b.
From the results it was apparent that activity clearly falls into groups of positional isomers. The best cytotoxicity was exhibited by ring systems containing two nitrogens (quinazolines and quinoxalines), while ring systems containing one nitrogen (with the exception of 42) exhibited lower cytotoxicity.
The effect of the most active compound 43a, on cell cycle and intercalation was also investigated. Standard DNA intercalating UV experiments showed that 43a does not intercalate into DNA.
These nitrogen heterocyclic compounds have exhibited interesting anticancer activity that can be further investigated for potential application as anticancer agents.
5. Naphthoquinone amides and esters
The synthesis of fourteen new naphthoquinone aliphatic amides and seventeen naphthoquinone aliphatic esters were reported by Kongkathip et al.203 The synthesis was inspired by the anticancer activity reported for the long chain rhinacanthins (Fig. 17). The nine to ten steps synthesis from 1-hydroxy-2-naphthoic acid afforded a 9–25% overall yield for the amides, and a 16–21% overall yield for the esters.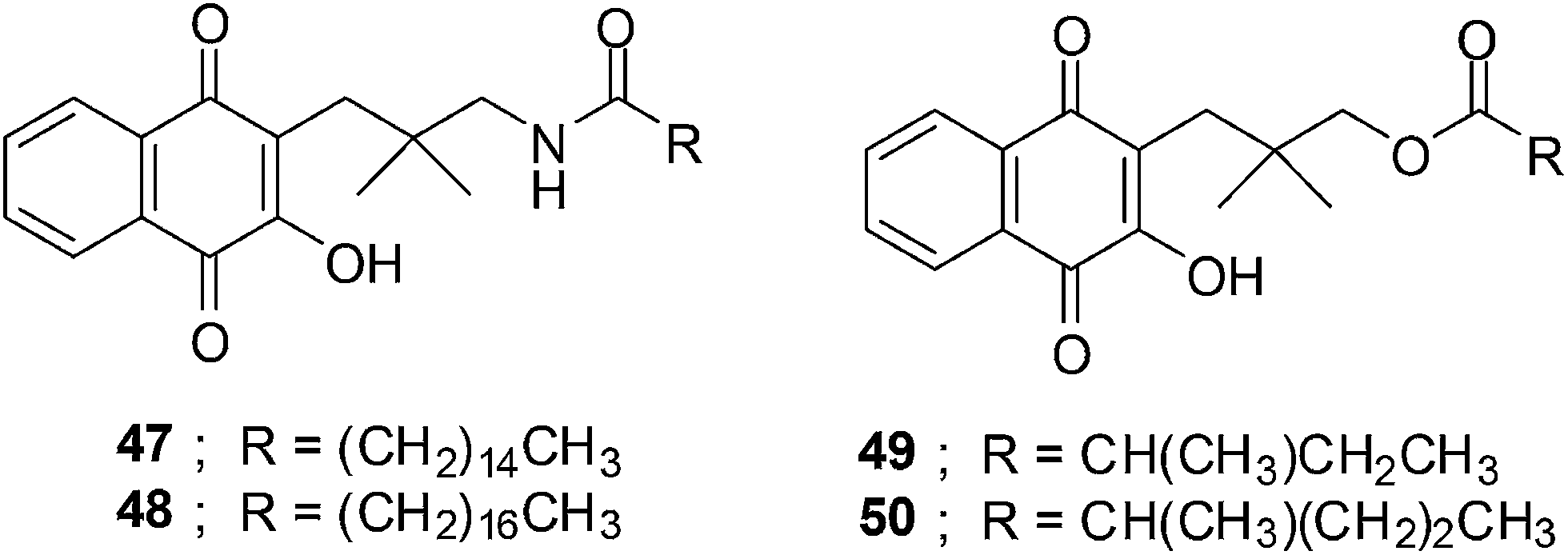 | ||
| Fig. 17 The structures of the naphthoquinone amides and aliphatic esters. | ||
For the amide synthesis the crucial step was the coupling reaction between amine and various aliphatic acids using the coupling agent, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM). DCC/DMAP or CDI was used as the coupling reagent between aliphatic acids and naphthoquinone alcohol for the ester synthesis.
The compounds were evaluated for their anticancer activity against oral cavity cancer (KB), small cell lung cancer (NCI-H187), using the resazurin microplate assay (REMA) and normal Vero cell-lines by a green fluorescent protein (GFP)-based assay.
The ester series having chain lengths between 3- to 7-carbon atoms (IC50 = 1.76–41.01 μM) inhibited KB cells stronger than the amide series (IC50 = 28.21–101.86 μM) having the same chain length. Compounds 49 and 50 exhibited potent activity against the KB cancer cell-line i.e. 1.76 and 4.65 μM, respectively.
The naphthoquinone aliphatic amides exhibited better anticancer activity than the esters when the chain length exceeds 7-carbon atoms. The amides 47 (14-carbon atoms) and 48 (16-carbon atoms) exhibited very strong activity against the KB cancer cell-line with IC50 values of 5.12 and 6.35 μM, respectively. The optimum chain length of amides is expected to be 16-carbon atoms. Much stronger anticancer activity was observed for naphthoquinone aliphatic esters having an α-methyl group on the ester moiety compared to the straight chains.
Compounds 47 and 48 have almost the same potency against Vero cells (IC50 = 5.60 and 5.76 μM, respectively) as against KB cells. The naphthoquinone esters 49 and 50 were more potent against Vero cells (IC50 = 0.87 and 0.12 μM respectively) than against the KB cancer cell-line.
The decatenation assay was used to determine whether the compounds can inhibit human topoisomerase II alpha (hTopoIIα). The enzyme, hTopoIIα, catalyzes the ATP-dependent decatenation of long-chain, catenated DNA molecules into free, relaxed and supercoiled forms. The assay results showed that a naphthoquinone amide 47 with a 16-carbon atom chain can completely inhibit hTopoIIα activity at 15 and 20 mM. Only moderate inhibition was obtained at 10 mM.
The same trend was observed in molecular docking studies as that in the cytotoxicity and decatenation assays.
Overall, the potent compounds are not selective for cancer cells over Vero cells. Further structural modification is required to make these compounds more selective for KB cancer cells.
Pradidphol et al. reported the synthesis of sixteen novel naphthoquinone aromatic amide analogues of rhinacanthin (naphthoquinone ester, Fig. 18).204 The synthesis entailed a new synthetic route which started from 1-hydroxy-2-naphthoic acid. The products were obtained in nine or ten steps in good to excellent yield (54–98%). The amide formation reaction was carried out by using the coupling reagent, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM). DMTMM is a useful condensing agent that affords carboxamides in high yield. Oxidation with Fremy's salt followed by hydroxylation with tert-butyl hydroperoxide and triton B was the key step for converting naphthol to 3-hydroxynaphthoquinone.
 | ||
| Fig. 18 Naphthoquinone amides, analogues of the rhinacanthin ester. | ||
The anticancer activity of the naphthoquinone amides were evaluated against human cancer cell-lines such as oral cavity cancer (KB), small cell lung cancer (NCI-H187), and breast cancer (MCF-7) using the resazurin microplate assay (REMA) and normal Vero cell-lines by a green fluorescent protein (GFP)-based assay. Benzamide 51 (IC50 = 8.83 ± 1.79 μM) showed potent inhibition against small cell lung cancer (NCI-H187) cell-line while naphthamides 52 (IC50 = 9.00 ± 2.17 μM) and 54 (IC50 = 8.62 ± 2.26 μM) were the most potent against the oral cavity cancer (KB) cell-line. The activity of compound 55 (IC50 = 41.23 ± 15.93 μM) was at least 4.5-fold less than that of 54. Most of the naphthoquinone amides showed weak toxicity against normal Vero cell-lines.
The decatenation assay was used to determine the inhibitory activity of novel naphthoquinone aromatic amides against human Topoisomerase IIα (hTopoIIα). The results showed that compound 53 and 54 inhibit hTopoIIα activity at a concentration of 20 mM while compounds 51, 52 and 55 exhibited hTopoIIα inhibitory activity at 50 mM. The same trend was observed in the docking experiment as the cytotoxicity and decatenation assay.
From this study it was concluded that the naphthoquinone amides 53 and 54 are promising target molecules for anticancer drug development.
6. Dimeric naphthoquinones
The anticancer activity of a novel series of twelve dimeric naphthoquinones against prostate cancer was reported by Ross et al.205–209The compounds were screened against androgen-independent (PC3, DU145) and androgen-responsive (LNCaP, 22RV1) prostate cancer cell-lines as well as prostate epithelial cells (PrECs). Cytotoxicity at 15 μM against all the prostate cancer cell-lines were observed for 56–60 (Fig. 19).209
 | ||
| Fig. 19 Dimeric naphthoquinones. | ||
A study in clonogenic assays showed that the ability of prostate cancer cells to form colonies was limited after treatment with the compounds. It was found that cellular ROS formation increased after exposure of prostate cancer to these compounds. ATP production decreased and apoptosis was promoted.209 Cytotoxicity was dependent on NADP(H):quinone oxidoreductase activity in DU-145 cells but not in PrECs, PC3 and 22RV1 cells.
The accumulation of ROS and mitochondrial dysfunction probably contributed to the cytotoxic effect of the dimeric naphthoquinones.
7. Dioncoquinones and dioncoquinone derivatives
A family of natural naphthoquinones that have exhibited promising anti-tumoural and anti-infective activities are the dioncoquinones. Amongst these the dioncoquinones A 61 and B 62 are highly active against Leishmania major and multiple myeloma cells with low toxicity toward normal blood cells (Fig. 20).210 Their activities against multiple myeloma cell-lines were comparable to melphalan. The latter is a documented DNA-alkylating agent effective against both multiple myeloma and B cell lymphoma.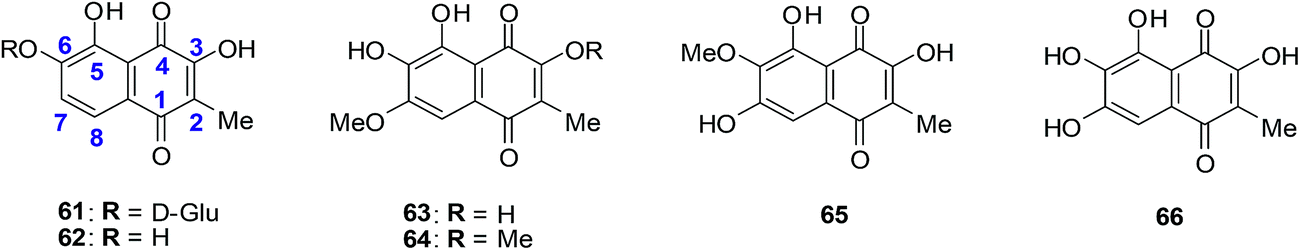 | ||
| Fig. 20 Higher-oxygenated naphthoquinones 61–65 isolated from T. peltatum cell cultures and compound 66 exhibiting anticancer activity. | ||
The first total synthesis of the highly oxygenated anticancer agent dioncoquinone B 62 and the isolation of its new, even higher-oxygenated analogues, dioncoquinones C 63, D 64 and E 65, from cell cultures of Triphyophyllum peltatum was reported (Fig. 20).210 Several derivatives of these compounds were synthesised, including dioncoquinone C 63.
The dioncoquinones B 62 and C 63 and related naphthoquinones were screened against a malignant human multiple myeloma cell-line (INA-6) and non-malignant peripheral blood mononuclear cells (PBMC). Apoptotic and viable cell fractions were determined by staining with annexin V-FITC and propidium iodide (PI). Dioncoquinones A 61 and B 62 showed promising results with B 62 exhibiting especially high (EC50 = 11 μM) and selective activity against multiple myeloma (MM) cells. Dioncoquinone C 63 (EC50 = 14 μM) showed a similar activity against the MM cells as that of dioncoquinone B 62 which was inactive against PBMCs. The 7-hydroxydioncoquinone B 66 (EC50 = 7 μM) was 10-times more selective for MM INA-6 than for PBMCs (EC50 = 70 μM) (Fig. 20).
From a SAR study it was determined that all three hydroxyl groups (at C-3, C-5, and C-6) are necessary for enhanced anticancer activities and reduced cytotoxicities. This can be seen from the similar activities of dioncoquinone B 62, dioncoquinone C 63, and 7-hydroxyl-dioncoquinone B 66. Dioncoquinone C 63 was the only new compound that strongly induced apoptosis in the MM cell-line (INA-6) without any noteworthy toxicity against PBMC. Dioncoquinones B 62 and C 63 are two encouraging elementary structures for developing anti-MM candidates.
8. Glycosylated derivatives of juglone and related 1,4-naphthoquinones
Antifungal, immunomodulatory, and antitumour properties have been reported for the glycosylated derivatives of the physiologically active natural compound juglone as well as related 1,4-naphthoquinones. There were, however, no reports on their antileukemic properties and their SARs have been insufficiently studied. In light of this, a series of fifty 1,4-naphthoquinone derivatives were therefore evaluated for their antileukemic effects and subjected to a structure–activity study (SAR).211The compounds were screened against human promyelocytic leukemia (HL-60) cells using the MTS method for determining cell viability. The compounds inhibited the HL-60 cells at a wide range of concentrations.
From the SAR study it was apparent that the juglone derivatives were among the most active compounds and that the hydroxyl group at position 5 of the quinone moiety was essential for the increase of cytotoxicity. The most active compounds were the glycosides of juglone 67–77 (IC50 = 0.6–4.8 μM). The structures of compounds with IC50 value less than 1 μM are shown in Fig. 21.
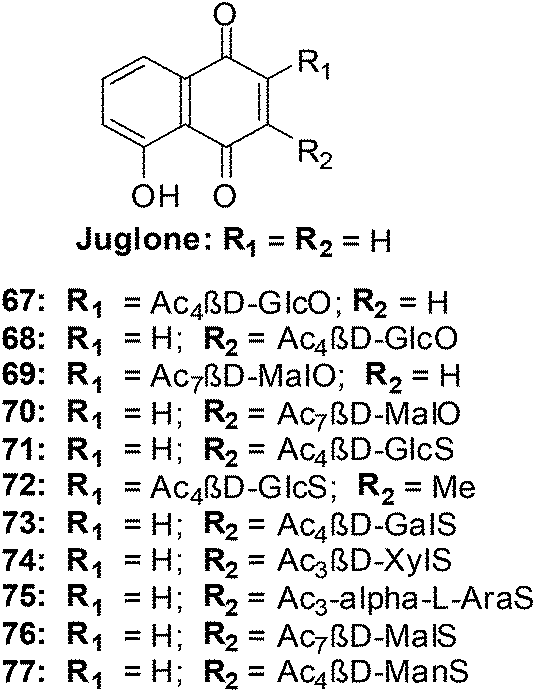 | ||
| Fig. 21 Juglone glycosides 67–77 highly active against leukemia (HL-60) cells. | ||
The products of intramolecular cyclisation of the S-glycosides of 1,4-naphthoquinones were also very active (IC50 = 1–5 μM). It was found that electronegative groups like –OH, –Cl, –NH2, –OMe, –OAc at the α-position to the sugar moiety dramatically reduced antileukemic activity of the compounds. The structures of compounds with an IC50 value less than or equal to 1.5 μM are shown in Fig. 22.
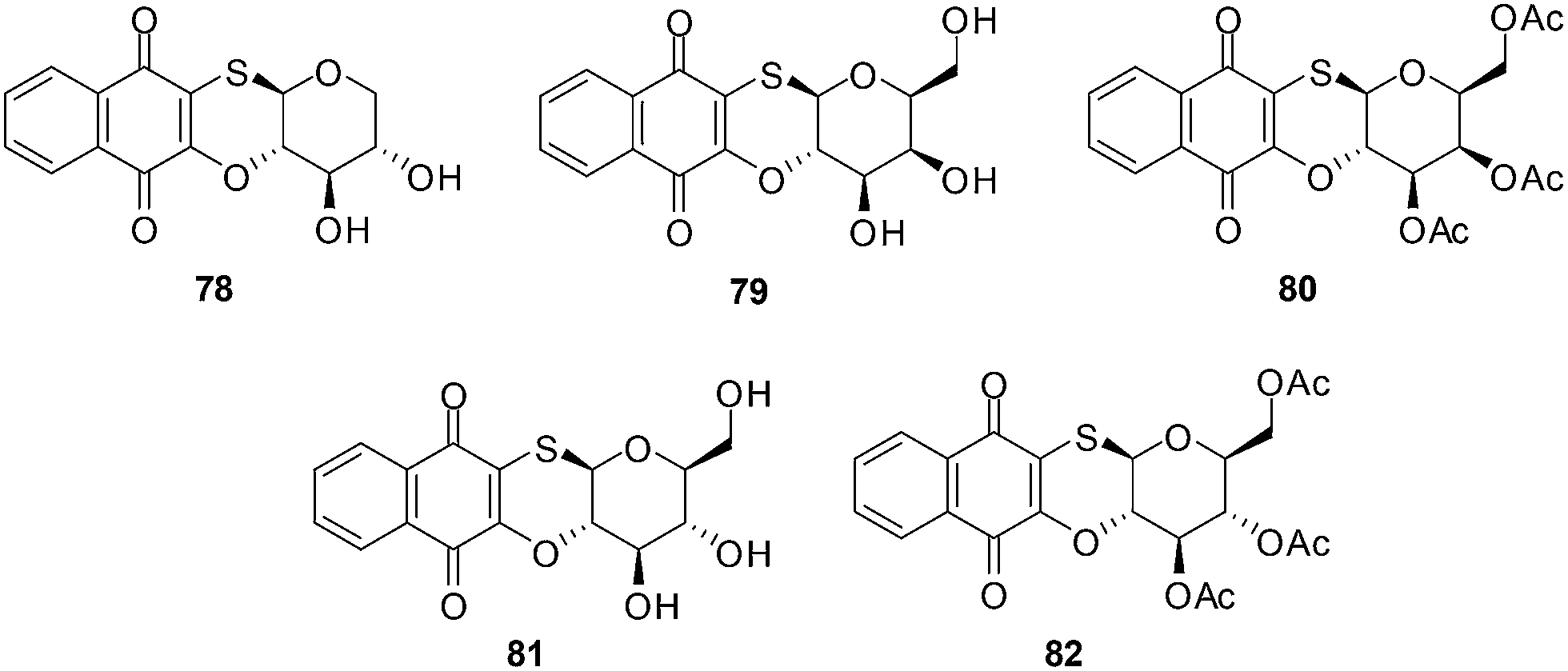 | ||
| Fig. 22 S-glycosides of 1,4-naphthoquinones highly active against leukemia (HL-60) cells. | ||
There is potential for the development of O- or S- glycosylated derivatives of juglone and related 1,4-naphthoquinones as new antileukemic agents.
9. Lapachol derivatives
Lapachol 83, a naturally occurring 1,4-naphthoquinone, has its origins in several tree species from the Bignoniaceae family found in the Western Hemisphere including Brazil and has a history of cancer cell cytotoxicity.212,213 β-Lapachone 84, a secondary metabolite, has shown anticancer activity against various cancer molecular targets.214–216 It is in phase II clinical trials in the USA for the treatment of advanced solid tumours.217A series of synthetic 1,4-naphthoquinones related to the secondary metabolites lapachol, α- and β-lapachone were synthesised (Fig. 23).218 The secondary metabolites lapachol, α- and β-lapachone and the synthetic 1,4-naphthoquinones were screened against the oesophageal cancer cell-line (WHCO1) using the MTT assay. Most of the compounds exhibited potent activity (IC50 = 1.6–7.3 μM).
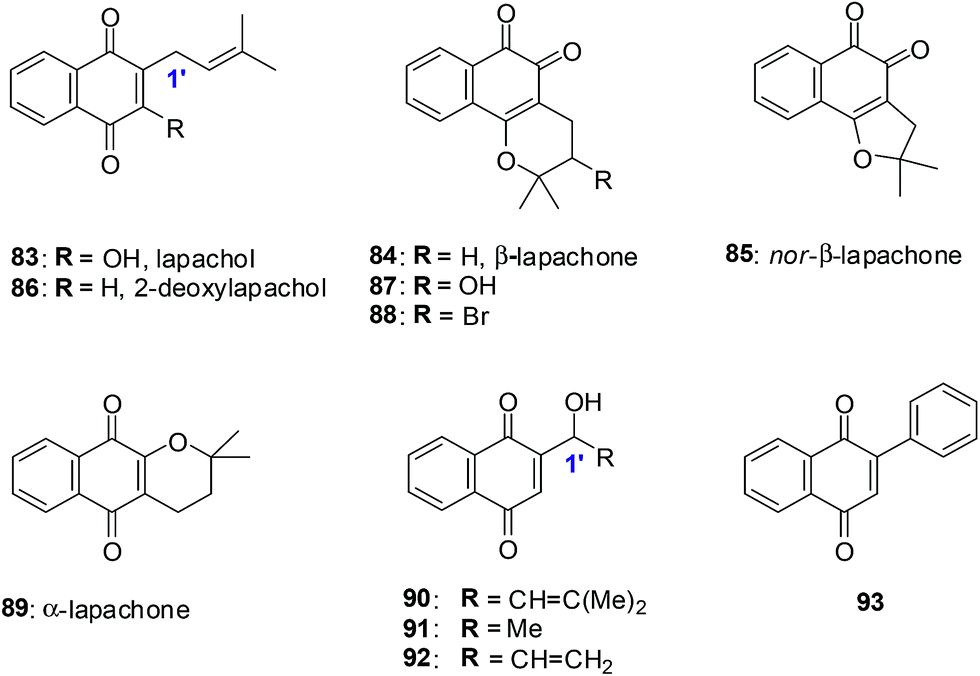 | ||
| Fig. 23 Para- and ortho-naphthoquinones evaluated against the oesophageal cancer cell-line. | ||
The synthetic compounds 94, 95 and 96 were screened against a normal fibroblast cell-line (NIH3T3). The two new synthetic halogenated compounds 95 and 96 (IC50 = 3.0 and 7.3 μM, respectively) and the previously reported compound 94 (IC50 = 3.9 μM), were found to be non-toxic (Fig. 24). Compounds 95 and 96 are also more cytotoxic than cisplatin the current drug of choice against oesophageal cancer.
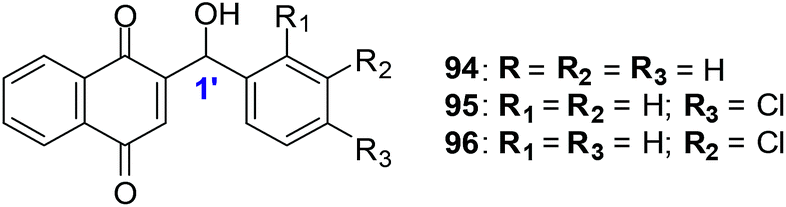 | ||
| Fig. 24 Synthetic 1,4-naphthoquinones related to lapachol, and α- and β-lapachone. | ||
Compound 94 was used to determine the mechanism of action of cell death in oesophageal cancer cells. Cell death entailed processes involving PARP cleavage (indicative of apoptosis) caused by 94 which was shown to be associated with elevated c-Jun levels.
This investigation identified compounds that are useful as a starting point for new and improved pharmacologically improved chemotherapeutic agents against oesophageal cancer.
The natural naphthoquinone, Lapachol 83, has been investigated mainly due to its antibacterial,219–221 antifungal,222 trypanocidal223 and anticancer activities (Fig. 25).224 The hydroxyl derivative, 5-hydroxylapachol 97, was isolated from the root heart wood of Tectona grandis. This compound, like lapachol 83, was also found to be cytotoxic to Artemia salina (brineshrimp) with an LC50 of 5 ppm.225 The cyclisation product of lapachol 83 is β-Lapachone 84 which has been under intense investigation for clinical use in cancer chemotherapy (Fig. 25).214,226,227
 | ||
| Fig. 25 The chemical structures of natural quinones. | ||
Bonifazi et al. reported on a series of 5-hydroxy-1,4-naphthoquinone analogues which were synthesised from juglone.228 The antiproliferative activity of these compounds was evaluated against a panel of six human cancer cell-lines such as ovarian (A2780), breast (HBL-100), cervical (HeLa), non-small cell lung (SW1573), breast (T-47D), and colon (WiDr) using the sulforhodamine B (SRB) assay.
The most potent compounds, 5-hydroxylapachol 97, β-Lapachone 84 and 98 (Fig. 25), had GI50 values of 0.42–8.1 μM, 0.69–2.3 μM and 0.80–2.2 μM, respectively. The comparison between lapachol 83 and the hydroxyl derivative 97 showed that the occurrence and location of a hydroxyl group attached to the aromatic ring does affect the capacity to inhibit the progression of cancer cell-lines. Thus, it is concluded that the presence of a phenolic hydroxyl group at C-5 plays an important role in increasing the antiproliferative activity.
10. β-Lapachone-based 1,2,3-triazoles
A variety of β-lapachone derivatives were synthesised by different synthetic routes and were obtained in moderate to high yields (Fig. 26).229 The compounds were screened against leukemia (HL-60), melanoma (MDA-MB435), colon (HCT-8) and central nervous system (SF295) cancer cell-lines using the MTT assay. Some of the compounds were highly active exhibiting IC50 values below 2 μM. The β-lapachone-based 1,2,3-triazoles had the best activity.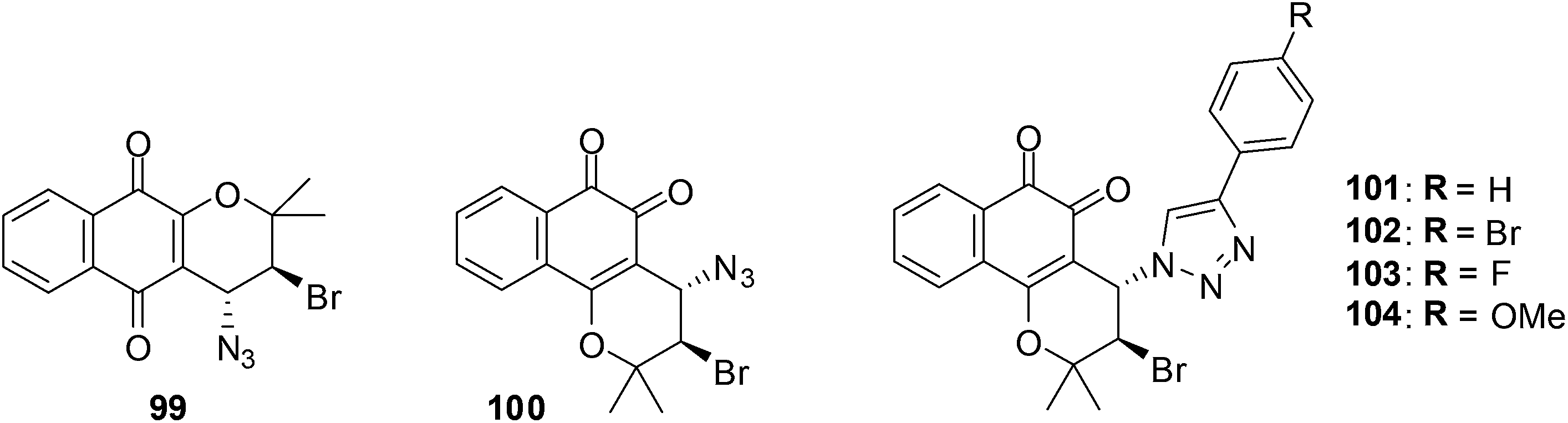 | ||
| Fig. 26 Lapachone derivatives. | ||
The most significant activity was observed toward HL-60 and the compounds 100, 101 and 103 were highly active with IC50 values of 1.43, 1.01 and 1.20 μM, respectively. These compounds were as active as the prototype, β-lapachone 84 (IC50 = 1.65 μM). Compounds 101–104 were highly active against MDA-MB435 with IC50 values below 2 μM. Only substance 101 was considered highly active against HCT-8 cell-lines with an IC50 value of 1.37 μM. The compounds 101–104 exhibited an activity between 1.55-3.01 μM against SF295.
The addition of 1-bromoheptane that deplete reduced glutathione (GSH) content and the use of N-acetylcysteine that protects cells against apoptotic cellular death gave an understanding of the ROS mechanism of anticancer action. An analysis for the formation of thiobarbituric acid reactive substances (TBARS) was also done. Furthermore the comet assay which utilises the bacterial enzymes, formamidopyrimidine DNA-glycosylase (FPG) and endonuclease III (ENDOIII), was used to detect for DNA damage after treatment. It was found that compounds 101 and 103 promoted cell death by apoptosis and genotoxicity and suggests that quinone-induced apoptosis is associated with ROS production.
The β-lapachone-based 1,2,3-triazoles are promising prototypes for application in cancer chemotherapy.
11. Nor-β-lapachone derivatives
Several 3-arylamino and 3-alkoxy-nor-β-lapachone derivatives were synthesised in moderate to high yields (Fig. 27).230 These compounds were evaluated for their anticancer activity against central nervous system (SF295), colon (HCT8), melanoma (MDA-MB435), and leukemia (HL60) cancer cell-lines using the MTT assay. Most of the arylamino-nor-β-lapachone derivatives exhibited potent activity against all cancer cell-lines. Several compounds were found to be highly active with IC50 values below 2 μM and were also more active than β-lapachone 84 an anti-prostate cancer prototype in phase II clinical studies.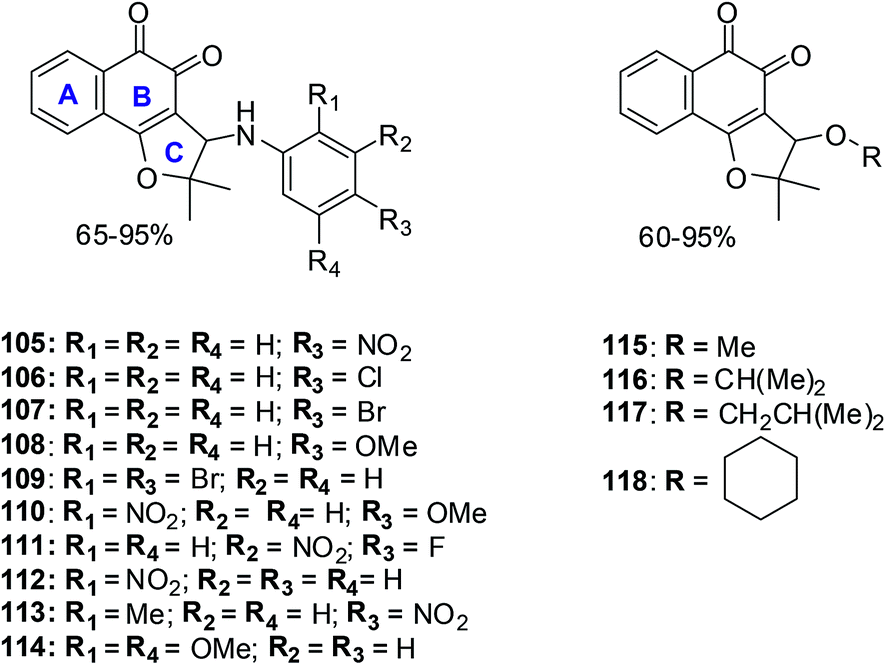 | ||
| Fig. 27 Compounds exhibiting potent anticancer activity. | ||
Compounds 105 and 114 were the two most active compounds against all cancer cells with IC50 values below 1 μM which was even better than that of the standard, doxorubicin (toward MDA-MB435).
MDA-MB435 was the most sensitive cell-line with compounds 105–109, 110, 111–118 exhibiting activity more potent than doxorubicin. Toward the HL60 cancer cell compounds 105, 106, 109–112, 114 and 118 were superior in activity when compared to β-lapachone 84 (IC50 = 1.65 μM) and nor-β-lapachone 85 (IC50 = 1.75 μM) with compounds 111 and 114 having similar activity (IC50 = 0.28 μM).
Toward the HCT8 and SF295 cancer cell-lines, two compounds 114 (IC50 = 0.15 μM and 0.39 μM, respectively) and 105 (IC50 = 0.76 μM and 0.82 μM, respectively) were more potent than nor-β-lapachone 85 (IC50 = 1.36 and 1.58 μM) and β-lapachone 84 (IC50 = 0.83 and 0.91 μM).
The arylamino para-nitro 105 and the 2,4-dimethoxy substituted naphthoquinones 114 exhibited the best activity while the ortho-nitro 112 and the 2,4-dimethoxy substituted compound 114 were more selective than doxorubicin and similar to the precursor lapachones. Compound 114 exhibited the best anticancer activity and selectivity index which was even better than doxorubicin, the positive control.
These compounds are promising new lead compounds for anticancer drug development.
12. Nor-β-lapachone arylamino derivatives
A variety of naphthoquinones such as nor-β-lapachone and its arylamino derivatives, iodinated and methylated naphthoquinones and nor-β-lapachone-based 1,2,3-triazoles were synthesised (Fig. 28).231 These compounds were evaluated for their anticancer activities against four human leukemia cell-lines (HL-60, K562, Molt-4 and Jurkat) using the MTT assay. It was found that nor-β-lapachones with arylamino substitutents exhibited potent activity. | ||
| Fig. 28 Compounds exhibiting anticancer activity. | ||
Naphthoquinones 119–122 (IC50 = 0.33–2.30 μM) were the most active. Compounds 123–125, the iodinated and methylated derivatives, were less active against the evaluated cancer cell-lines (IC50 = 4.05–3.44 μM) except for 125 for HL-60 (IC50 = 1.11 μM) and compound 124 for Jurkat (IC50 = 1.38 μM). Compounds 119–122 (for HL-60), 122 (for K562), 120 and 122 (for Molt-4) and 119, 120 and 122 (for Jurkat) were found to be more active than nor-β-lapachone, the naphthoquinoidal precursor.
The standard comet assay was used to determine how these compounds caused DNA damage. Compounds 119–122 were found to induce oxidative DNA damage by ROS generation and treatments with these compounds impair DNA repair activity resulting in the triggering of apoptosis. The mechanism of action of these compounds does not involve topoisomerase inhibition.
A study was conducted in Chinese hamster lung fibroblasts (V79 cells) to determine the capacity of the compounds to stimulate apoptosis and chromosomal aberrations as micronuclei. After drug treatment morphological apoptotic nuclei and micronuclei stimulation were detected signifying a link between DNA damage and apoptosis.
Since compounds 119–122 lack selectivity between cancer and non-cancer cells, their therapeutic potential may be limited as anticancer agents.
13. α- and β-lapachone derivatives
The synthesis of a series of compounds having α- and β-lapachone containing hydroxyl or methoxyl groups on the benzene ring was achieved through a selective acid catalysed cyclization of suitable lapachol analogues (Fig. 29).232 Compounds were evaluated for their anticancer activity against breast (HBL-100), cervical (HeLa), lung (SW1573), and colon (WiDr) cancer cell-lines using the SRB assay.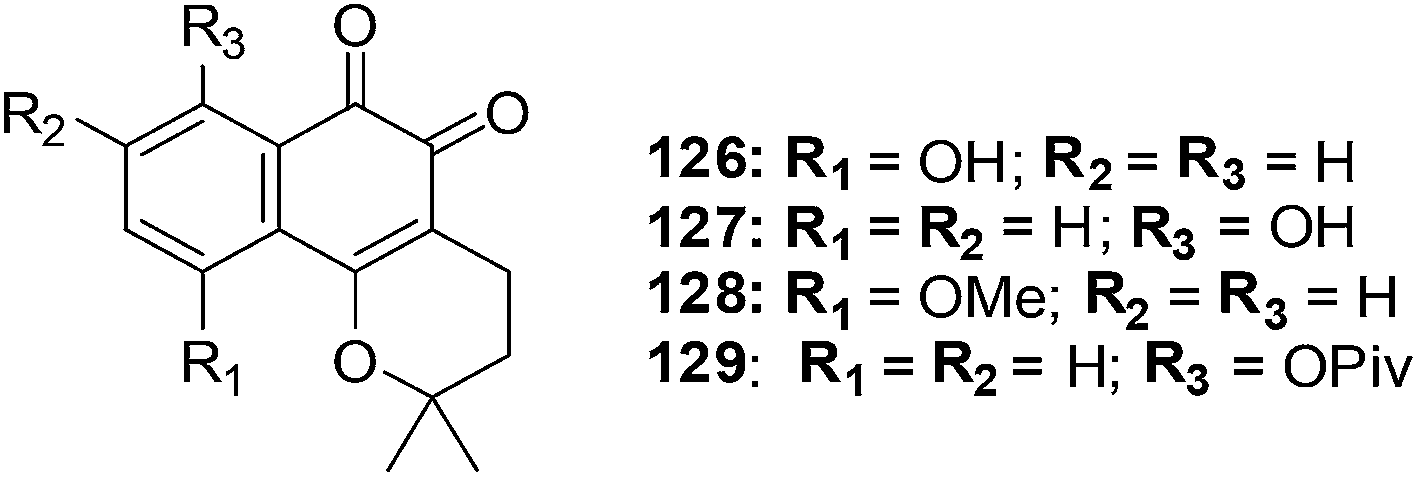 | ||
| Fig. 29 β-Lapachone analogues exhibiting potent anticancer activity. | ||
From the data it was apparent that there was a clear difference in activity between the three series of compounds. The activity order was as follows: β-lapachone analogues > α-lapachone analogues ≈ lapachol analogues. Compounds 126, 127, 128 and 129 exhibited activity less than 2.2 μM) against all the cell-lines. The sensitivity of the cell-lines was in the order HBL-100 > SW1573 > HeLa > WiDr. From this evaluation the β-lapachone analogue 127 was found to be the most active of the series (GI50 = 0.029–2.0 μM). This compound exhibited better activity than the parent drug, β-lapachone (GI50 = 0.38–2.0 μM).
From cell cycle studies, protein expression experiments, and ROS analysis it was determined that, likewise to β-lapachone, ROS formation and DNA damage are vital aspects in the cellular toxicity of 127. This may be the leading mechanism of action of this compound since human DNA topoisomerase IIα is not inhibited in the decatenation assay.
Compound 127 is promising for lead development in cancer chemotherapy.
14. Nor-β-lapachone-based 1,2,3-triazoles
da Silva Júnior et al. reported on the anticancer activity of five nor-β-lapachone-based 1,2,3-triazoles and a precursor azidonaphthoquinone against central nervous system (SF-295), colon (HCT-8), melanoma (MDAMB-435), leukaemia (HL-60), prostate (PC-3), murine melanoma (B-16) and a normal murine fibroblast L-929 using the MTT assay (Fig. 30).233 Most of the compounds are strongly or moderately active (IC50 = 0.43–9.48 μM) against all cancer cell-lines.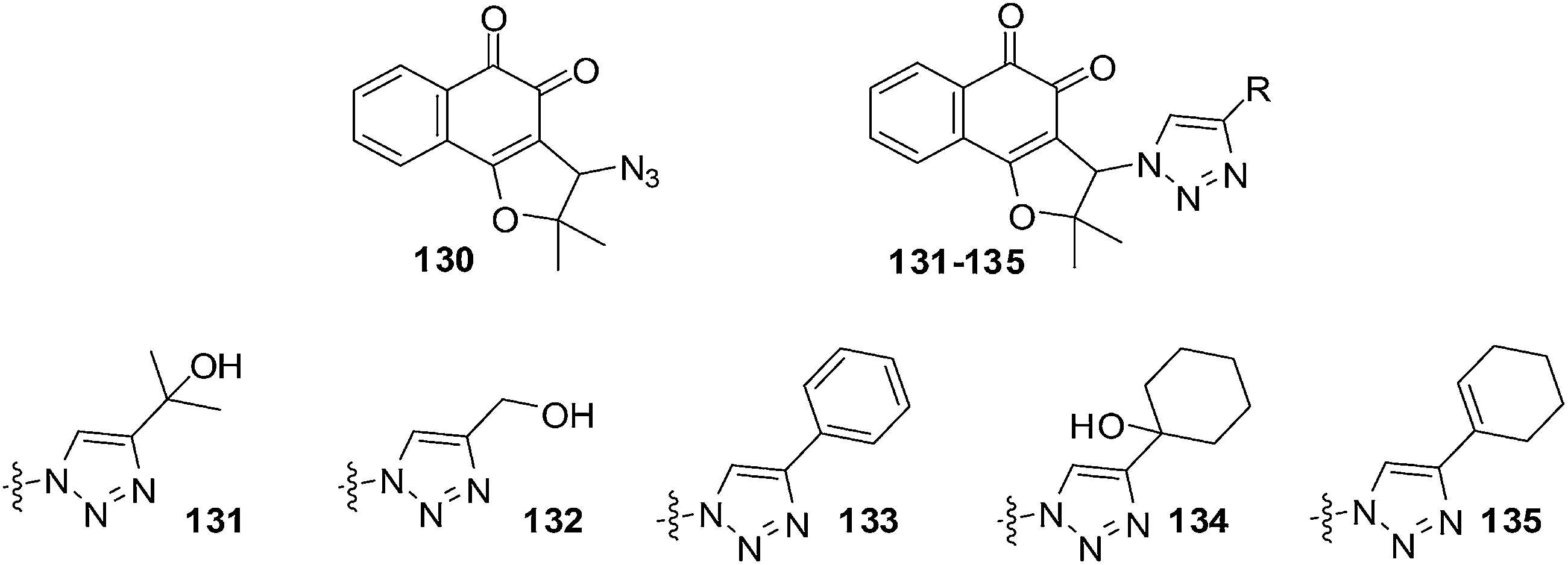 | ||
| Fig. 30 Compounds exhibiting anticancer activities. | ||
Compound 130 (IC50 = 1.19 μM) was selectively active against MDAMB-435. The triazolic naphthoquinone 134 exhibited the best activity against cancer cells (IC50 = 0.43–1.83 μM) with the strongest activity being against MDAMB-435. In addition, it also exhibited high selectivity (2–4 fold more selective) for cancer cells over normal cells (L-929, IC50 = 2.85 μM). The activity of compound 134 against melanoma cells was comparable to that of the initial quinones, nor-β-lapachone and β-lapachone, and better than that of doxorubicin. Compound 134 was also more active (IC50 = 1.17 μM) than nor-β-lapachone 85 (IC50 = 1.75 μM) and β-lapachone 84 (IC50 = 1.65 μM) towards HL-60.
An attempt was made to correlate physicochemical parameters (reduction potentials and calculated logP) with cytotoxic activity. There was no correlation found between reduction potentials and cytotoxicity. There was a correlation between cytotoxicity and the calculated logP: the higher the lipophilicity values (clogP) the better the cytotoxicity. Compounds 133–135 could be exploited for the preparation of analogues for the development of new anticancer drugs.
15. Mansonone F derivatives
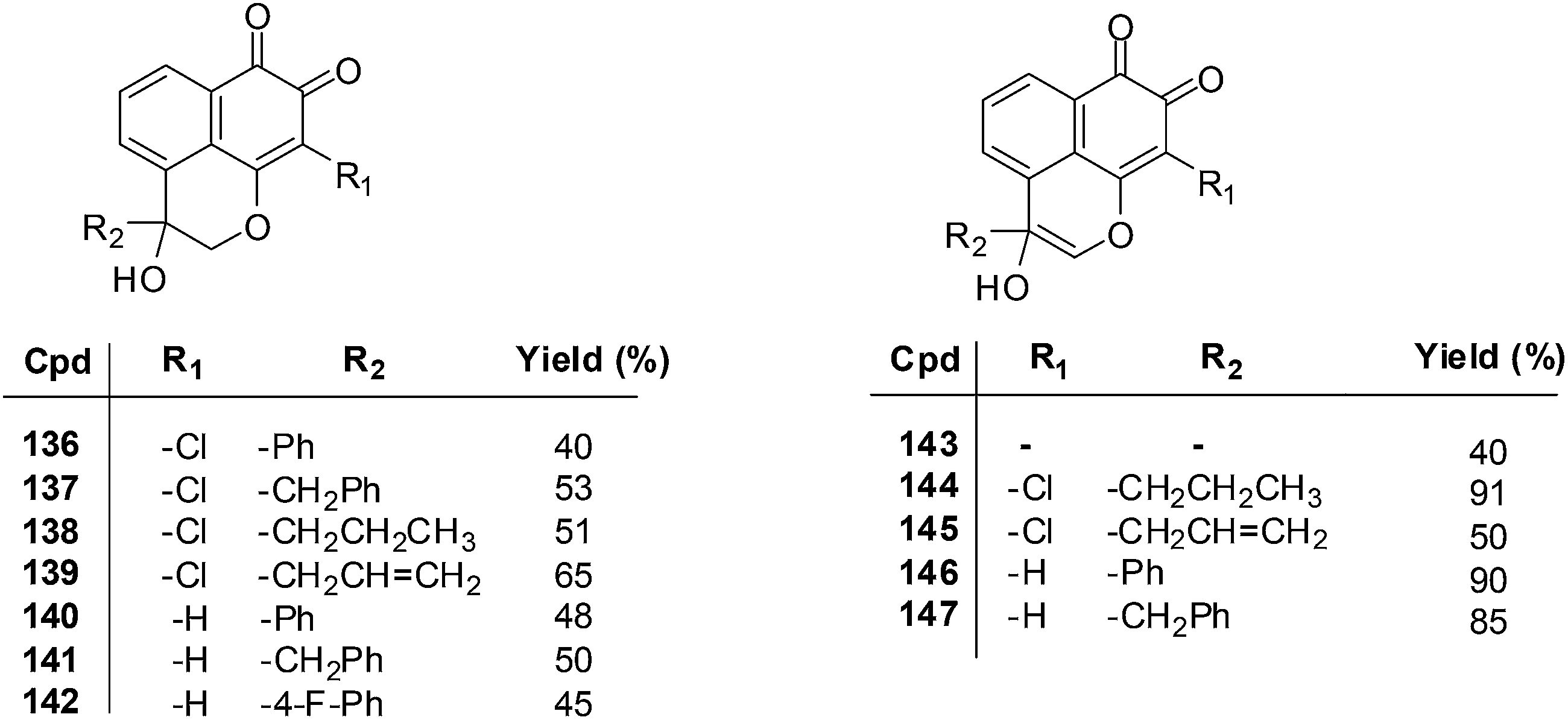
Naturally occurring Mansonone F (MF) is an o-quinone sesquiterpene that exists in plants such as Ulmus pumila and Mansonia altissima.234,235 It has antibacterial236,237 and anti-proliferative activities238,239 and also inhibits the activity of thioredoxin reductase, a potential target for anticancer drugs.240,241A series of MF derivatives were designed and synthesised by different synthetic routes and were obtained in moderate to high yields (Fig. 31).242 The compounds were screened against topoisomerase I and II and found to be potent inhibitors of topoisomerase II. The inhibitory activity exhibited by the best inhibitor against topoisomerase II was 20 times stronger than that of the positive control, Etoposide.
 | ||
| Fig. 31 Compounds exhibiting potent activity against both CNE-2 and Glc-82 cell-lines. | ||
The MTT assay was used to determine the growth inhibitory concentration (IC50) of the MF derivatives against the human lung adenocarcinoma cell-line (Glc-82) and the human nasopharyngeal carcinoma cell-line (CNE-2). Potent anticancer activity was exhibited by compounds 136–139 (IC50 = 2.75–7.93 μM), 140–142 (IC50 = 3.18–9.76 μM), and 146 and 147 (IC50 = 2.88–9.19 μM) against both CNE-2 and Glc-82 cell-lines. From a SAR study it was determined that the o-quinone group and the pyran ring are important for cytotoxic activity.
16. Naphthoquinone-based STAT3 inhibitors
The signal transducer and activator of transcription 3 (STAT3) is active in a large variety of cancers.243 It is activated by a number of cytokines and hormones.244–247 The STATs undergo homo- and/or hetero-dimerization after the phosphorylation of a specific tyrosine residue.248,249 STAT3 is activated upon phosphorylation at Tyr705 (ref. 249) and it signals as a homodimer or as a STAT1–STAT3 heterodimer.250Strategies to target STAT3 have been directed toward its aminoterminal, DNA-binding and SH2 domains. The SH2 domain is involved in both receptor binding and dimerization and has therefore received much attention.
The first small molecule inhibitor of the STAT3 SH2 domain was STA-21 148 (Fig. 32).251 The structural complexity of STA-21 has made the synthesis of analogues for structure–activity relationship (SAR) studies difficult. This led to the simplification of the structure and the design and synthesis of the initial compounds LLL3 149 and LLL12 150 which are substituted anthraquinones.252 LLL12 150 is an exceptionally effective compound that shows elevated levels of antiproliferative activity.
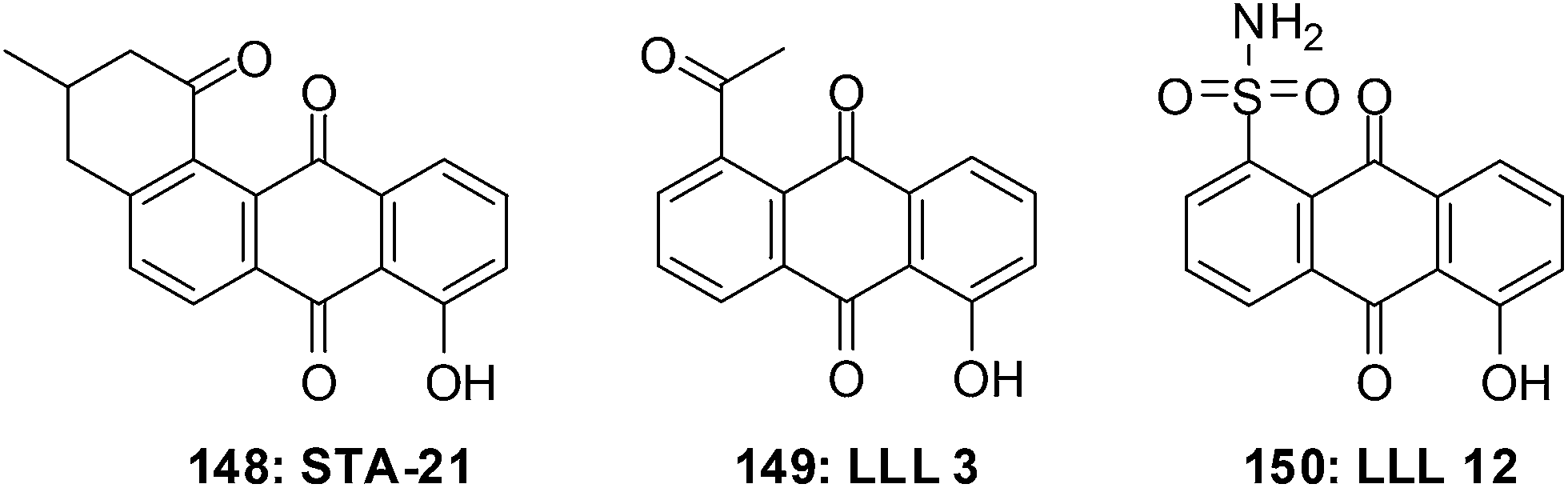 | ||
| Fig. 32 Small molecule inhibitors of the STAT3 SH2 domain. | ||
The synthesis and assessment of 1,4-naphthoquinone analogues targeting STAT3 was attempted to design more potent analogues with the 1,4-naphthoquinone scaffold (Fig. 33).253
 | ||
| Fig. 33 Compounds 151–154 were synthesised to bind to the STAT3 SH2 domain and inhibit STAT3. | ||
These analogues were evaluated against breast (MDA-MB-231), prostate (DU-145 and PC-3) and colon (HT-29) cancer cell-lines. The 1,4-naphthoquinone methyl ketone derivative 151 (DU145: IC50 = 6.4 ± 0.5 μM; HT29: IC50 = 4.2 ± 0.8 μM) and methyl ester derivative 152 (IC50 = 5.8 ± 1.4 μM, HT29) were found to be the most potent.
The STAT3 fluorescence polarization (FP) assay was used to evaluate selected 1,4-naphthoquinones for their abilities to bind to the STAT3 SH2 domain and inhibit STAT3. The compounds inhibited the FP signal and therefore STAT3 is one of their molecular targets.
The results from this study are promising for further development of these compounds for potential application in cancer chemotherapy.
17. Pentacyclic 1,4-naphthoquinones
Salustiano et al. conducted a study which entailed the comparison of the cytotoxic effects of pentacyclic 1,4-naphthoquinones, lapachol and α-lapachone on human leukemic cells.254 The pentacyclic 1,4-naphthoquinones of type 136 were designed as molecular hybrids of the cytotoxic naphthoquinones, lapachol and α-lapachone and natural pterocarpan255–257 (Fig. 34).
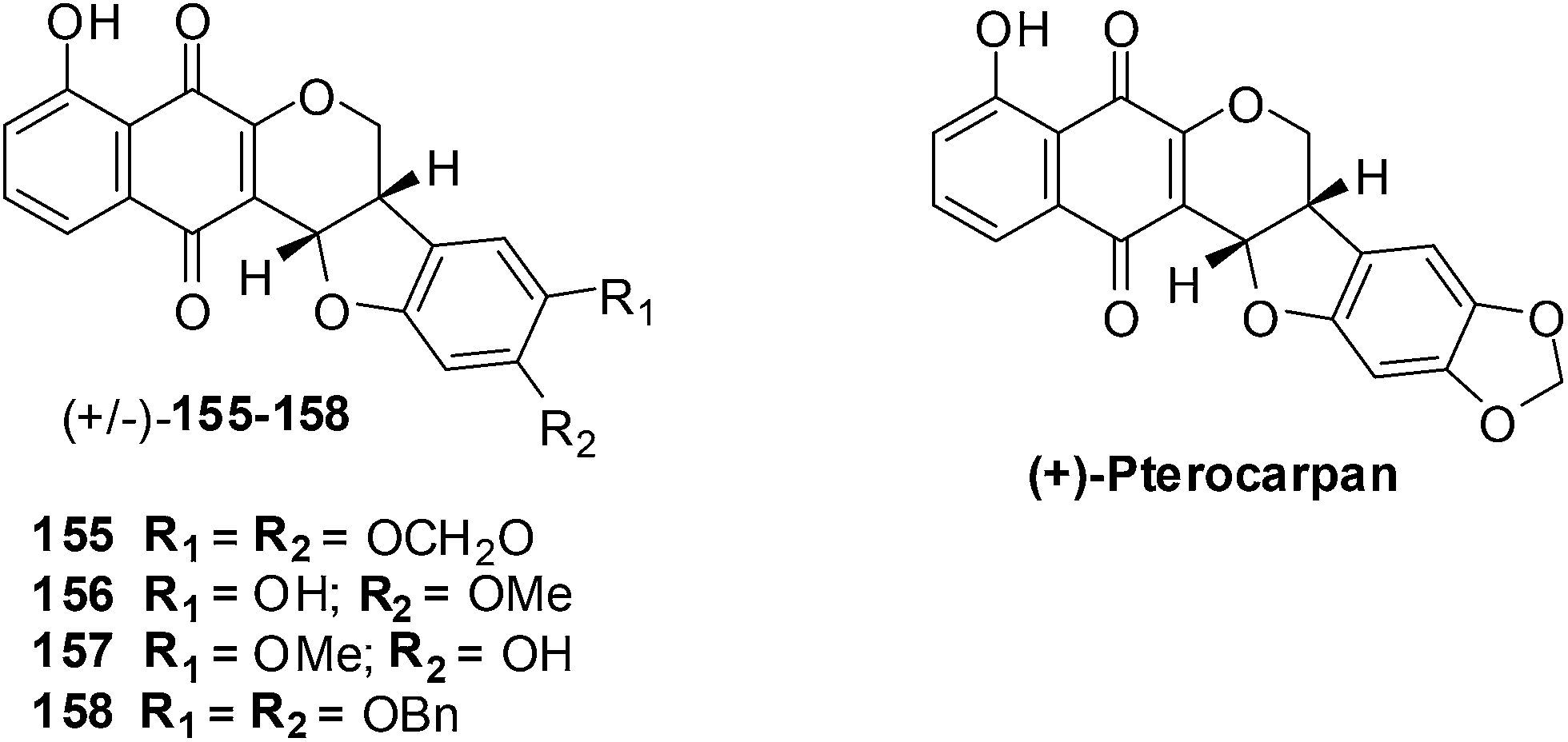 | ||
| Fig. 34 The pentacyclic naphthoquinones 155–158 and natural pterocarpan. | ||
The compounds were screened against two human leukemic cell-lines K562 (oxidative stress-resistant), Lucena-1 (MDR phenotype), Daudi, MCF-7 and PBMC using the MTT colorimetric assay. The pentacyclic 1,4-naphthoquinones 155–158 were found to be cytotoxic (IC50 = 2–7 μM) to the leukemic cell-lines including fresh leukemic cells obtained from patients, some with the MDR phenotype. Lapachol 83 and α-lapachone 89 were ineffective against these cell-lines while MMC inhibited cell proliferation at concentrations as low as 0.45 ± 0.04 μM. It was shown in previous work that in situ bioactivation activates these compounds and after rearrangement enones are formed that are powerful alkylating agents.
The little activity exhibited by lapachol 83 and β-lapachone 84 is because they cannot be activated by reduction. These compounds exhibited low toxicity against lymphocytes activated by phytohemagglutinin thus indicating that they are selective for the leukemic cancer cells.
Compounds 155–158 were active against oxidative stress-resistant K562 and Lucena-1 cell-lines which indicated that bioreductive activation of pentacyclic naphthoquinones type 1 could be responsible for the cytotoxic effect on these cells. By flow cytometry it was determined that the pentacyclic 1,4-naphthoquinones 155 and 157 induced cancer cell death by apoptosis.
18. Peptidomimetic compounds containing redox active quinones
Healthy cells are different from their cancer counterparts in that they do not exhibit a disturbed intracellular redox balance. Some cancers are naturally under oxidative stress (OS) such as prostate and breast cancer cells while others are hypoxic, i.e. their cells are more reducing than normal ones such as solid lung carcinoma. The ROS levels of these cells, when compared to healthy cells, are much closer to the critical redox threshold at which cell death is induced.258 Selective and effective redox drugs can be designed to exploit the biochemical differences between healthy and cancerous cells.259,260Shabaan et al. used the Passerini multicomponent reaction for the synthesis of di-, tri- and tetra-functional redox agents containing multiple chalcogen and quinone redox sites (Fig. 35).261
 | ||
| Fig. 35 The Passerini multicomponent reaction was used for the synthesis of di-, tri- and tetra-functional redox agents 159–168 containing multiple chalcogen and quinone redox sites. | ||
Compounds 162, 163, 165 and 167 were submitted for a one-dose screen at the National Institute of Health (NIH) which included 58 cell-lines. These cell-lines were grouped into breast cancer, renal cancer, colon cancer, prostate cancer, ovarian cancer, CNS cancer, non-small-cell lung cancer, leukemia, and melanoma cell-lines. All the evaluated compounds displayed a noteworthy cytotoxicity against the cancer cell-lines that were then selected for 5-dose testing. The compounds exhibited GI50 values that were in the low to sub-micromolar range.
From a COMPARE analyses it was determined that the compounds' pattern activities link with cisplatin which is used to treat numerous forms of cancers, including sarcomas, lymphomas, and germ cell tumours. The compounds' pattern activities also correlated with methyl MMC which has antitumour antibiotic activity against breast cancer, and with the anthracycline-based redox agents representing menogaril, deoxydoxorubicin, and MX2 HCl, which are used in the treatment of prostate cancer and leukemia.
The cytotoxic activity of the test compounds was further evaluated against human breast adenocarcinoma (MCF-7), human kidney carcinoma (A-498), and human epidermoid carcinoma (A-431). The MTT assay was used and after 24 h the IC50 values are estimated from the dose–response curves.
Compounds with four redox centers, 161 and 162, were more cytotoxic than their homologs with two and three redox centers. The most toxic compounds, 161, 162, 163, 165 and 167, possess a direct attachment of the selenium to the quinone. The exceptions were compounds 163 and 165 which have only two redox centers but were also among the most active compounds.
The compounds were also evaluated for their toxicity against primary human fibroblasts (HF) and human umbilical vein endothelial cells (HUVEC). Only compounds 159, 160, 165 and 166 were non-toxic, most were toxic. Amongst the non-toxic compounds 165 clearly showed a lower cytotoxicity against both primary cells than against the three cancer cell-lines.
The mediation of oxidative stress by the production of the superoxide radical (O2−˙) and hydrogen peroxide (H2O2) were also investigated. The DCF and DHE assays were used to determine ROS. It was found that the compounds were able to significantly increase the intracellular ROS levels in A-431 cells.
During cell cycle arrest there is an increase in ROS-induced DNA damage. A sub-lethal dose of a cytotoxic compound can prevent the progression of the cell cycle by causing a delay in G0/1. A significant delay of cell cycle progression was observed when MCF-7 cells were treated with 163, 164 and 168 at their respective IC50 concentrations for 24 h. In the G2/M phase there was a decrease in cells while in the G0/1 phase an increase in comparison to the negative control.
The ER stress-mediated apoptotic pathway or the mitochondrial-mediated apoptotic pathway or both pathways can be activated when a high level of ROS is present. These apoptotic pathways depend on the activation of caspases, particularly caspase-3 and -7, for the final execution of apoptosis. It was found that compound 168 (concentration = 2.8 μM) strongly activated caspase-3/7 activity in A-431 cells after 24 h of treatment.
Mitochondrial dysfunction and induction of apoptosis are linked to low levels of glutathione (GSH), thus decreasing chemoresistance. The intracellular levels of reduced GSH were estimated using the DTNB assay. Compounds 163, 164, 166 and 168, at a concentration of 5 μM, caused a decrease of the intracellular GSH level in breast cancer (MCF-7) cells by 46%, 59%, 62%, and 76%, respectively.
This study has afforded interesting results and thus opens the door to a range of follow-up studies in the areas of synthetic chemistry (organoselenium and organotellurium compounds), redox chemistry, biochemistry and, ultimately, drug development. Future studies will have to focus on improving the selectivity of these compounds for cancer cells.
19. Pyranonaphthoquinones
AKT,262 a serine/threonine kinase, is a key player in the signaling pathway that starts with phosphoinositide 3′-kinase (PI3Ka) activation. The latter generates phosphatidylinositol triphosphate (PIP3) which then binds and activates AKT. The phosphatase and tensin (PTEN) homologue, deleted on chromosome 10, dephosphorylates PIP3 and antagonizes AKT activation. Activated AKT has been found in a wide variety of human cancers and is often due to a mutational loss of PTEN activity. Its presence has been correlated with poor prognoses.263AKT has been the focus of intense research. It has been the target of small molecule inhibitors such as allosteric site binders, ATP-competitive active site binders, peptide or peptidomimetic pseudosubstrates, and analogues or mimics of PIP3.264
The known natural product antibiotic, lactoquinomycin 169, an amino-C-glycoside-pyranonaphthoquinone (PNQ) lactone (medermycin265) was found to inhibit AKT1 (IC50 = 149 nM) (Fig. 36). Kalafungin266 170, frenolicin B6 171, and related PNQ lactones, were found to have similar inhibitory activity (Fig. 36).
 | ||
| Fig. 36 Natural and semisynthetic PNQs 169–173 that inhibit AKT. | ||
A series of synthetic PNQs were synthesised for a SAR study (Fig. 37).267 Compounds were screened against the AKT and breast cancer (MDA468) cell-lines. The inhibition of AKT activity in cells was determined by the immunoblot assay of phosphorylation of downstream targets of AKT.
 | ||
| Fig. 37 Pyranonaphthoquinone lactones 174–186 that inhibit AKT and exhibit anticancer activity. | ||
It was found that PNQ lactones inhibit the proliferation of breast cancer containing activated AKT and show expected effects on cellular biomarkers. Compounds 174–186 exhibited potent activity and were highly active against breast cancer (IC50 = 0.23–1.60 μM). They also exhibited potent inhibitory activity against AKT (IC50 = 0.044–1.440 μM).
From the results of the screening assays it was apparent that there was no clear correlation between the level of enzyme inhibition and the cell antiproliferative activity in the series of analogues. Compounds 185 and 186, the least potent AKT inhibitors, had antiproliferative activities equal to or greater than some of the more active compounds.
A possible mechanism of action for AKT inhibition by the PNQs is the bioreductive alkylation mechanism. PNQ lactones, upon reduction in vivo, form a hydroquinone that can subsequently form a quinone methide which can then alkylate a nucleophilic site on a protein. Biochemical data supported the proposed bioreductive alkylation mechanism of action of a regulatory loop cysteine.
These pyranonaphthoquinone lactones are a highly selective new class of AKT kinase inhibitors.
20. Shikonin derivatives
The traditional oriental herb, Lithospermium erythrorhizon, is the source of the natural product, Shikonin, which is isolated from its roots (Fig. 38).268 Significant cytotoxic activity has been exhibited against various cancers both in vitro and in vivo by Shikonin and its derivatives.269,270 | ||
| Fig. 38 Shikonin 187 and its derivatives 188–190. | ||
For the mechanism of action the primary cause of cytotoxicity is the generation of ROS in which a semiquinone radical is believed to cause cell damage. Another plausible mechanism is bio-reductive alkylation which may also be the cause of their cytotoxic behavior.271 Shikonin and its derivatives could also strongly inhibit both topoisomerase I and II, and induce DNA cleavage and cell apoptosis.272–274 The acylated shikonin derivatives, acetylshikonin (188, Fig. 38) and isobutyrylshikonin (189, Fig. 38), have been reported to be more potent inhibitors of topoisomerases than shikonin.272
A novel series of shikonin analogues with various side chains have been synthesised in several synthesis steps (Fig. 39).275 These compounds were evaluated for their anticancer activity against cervical carcinoma cells (HeLa) and promyelocytic leukemia cells (HL60) using the SRB and MTT assays. Most of the compounds exhibited better inhibitory effects (IC50 = 0.02–0.35 μM) against HL60 than those of the positive controls (VP16: IC50 = 0.82 μM; shikonin 187: IC50 = 0.44 μM).
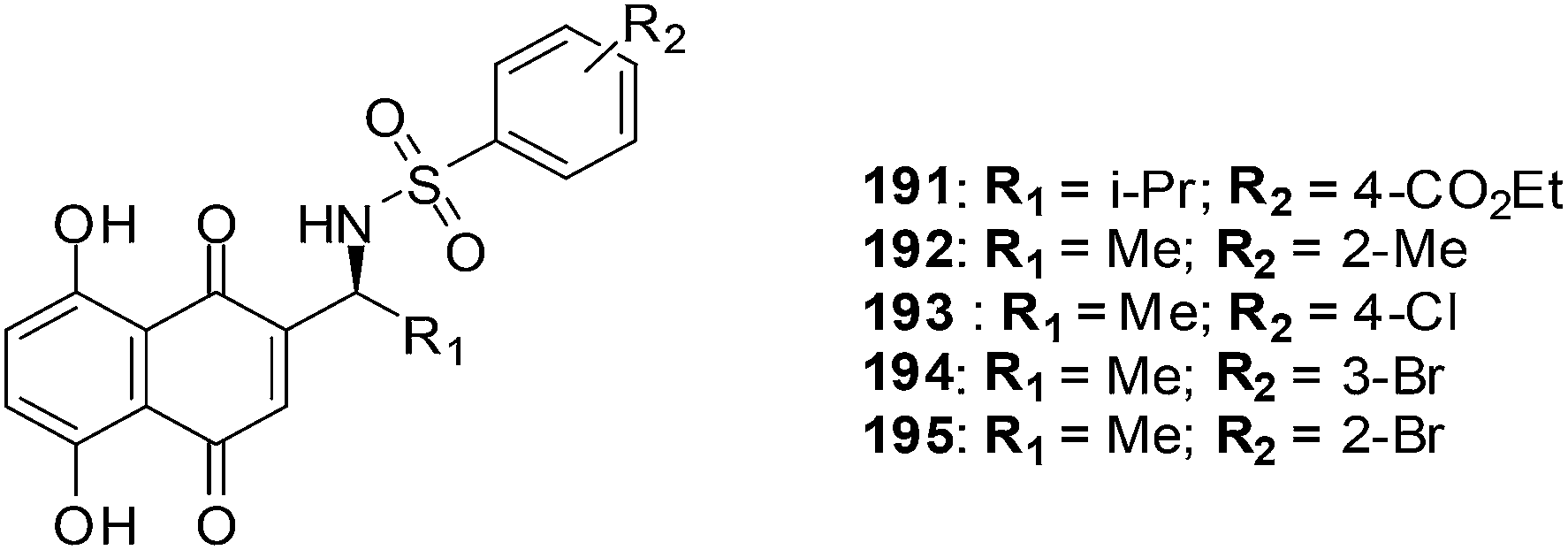 | ||
| Fig. 39 Shikonin analogues. | ||
Two compounds, 192 and 193, were selected for further evaluation for inhibitory activity against breast cancer (MDA-MB-231), colon cancer (HCT116), lung cancer (A549), rhabdomyosarcoma (RH30), oral epidermoid cancer (KB-3-1 and KB/VCR), and leukemia (K562). Both compounds 191 and 192 exhibited potent inhibitory activity against these cancer cell-lines with the average IC50 value of 8.6 μM and 7.3 μM, respectively. In addition, 191 and 193 also showed significant activity against KB/VCR (the vincristine-selected multidrug-resistant subline) with IC50 of 13.2 μM and 12.4 μM, respectively.
Hypoxia is a common characteristic of cancer cells and the hypoxia-inducible factor 1α (HIF-1α) is the most important transcript factor in response to intracellular oxygen pressure. It is enhanced to accumulate significantly in a hypoxic environment but under normoxia it is almost undetectable.
There is a correlation of HIF-1α with increased patient mortality in many different cancers such as brain, breast, cervix, colon, ovary and lung cancer. The overexpression of HIF-1α in human cancer cells increases tumour growth, angiogenesis and metastasis. HIF-1α is a validated therapeutic target.
Under hypoxic conditions compounds 192, 193, 194, 195 and shikonin 187 reduced the expression level of HIF-1α in breast cancer cells MDA-MB-231. These results demonstrate the potential of these compounds for further development as cancer chemotherapeutics.
21. Thiolated naphthoquinones
Thiolated derivatives of menadione, juglone and 1,4-naphthoquinone were synthesised for a structure–activity study of their anticancer properties (Scheme 7).193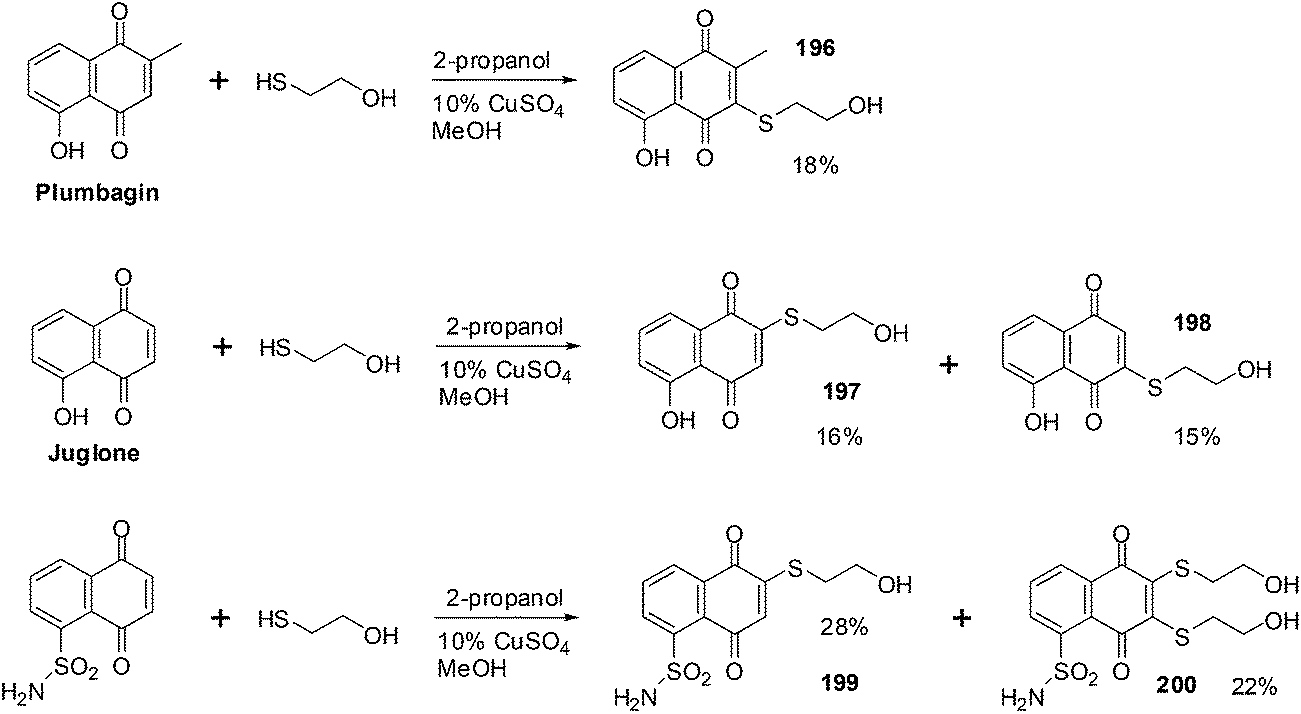 | ||
| Scheme 7 Synthesis of mono- and dithiolated naphthoquinone derivatives 196–200. | ||
These compounds were screened against prostate (DU-145), breast (MDA-MB-231) and colon (HT-29) cancer cell-lines using the Promega CellTiter 96 Aqueous Non-Radioactive Cell Proliferation assay. Compounds 196, 197, 198 and 199 (IC50 = 1.6–9.7 μM) exhibited potent activity against all three cancer cell-lines. Their antiproliferative activity was also better than that of the lead molecules. Inhibition of STAT3 dimerization was studied using the Fluorescent polarization assay and it was found that 196 and 197 were potent STAT3 dimerization inhibitors.
22. Promising candidates
The tumour-selective cytotoxic properties of the natural product, β-Lapachone 84 (ARQ 761), resulted in its advancement into clinical trials. It is currently being assessed for its anti-tumour activity against advanced solid tumours.276Conclusions
The synthesised synthetic naphthoquinones and derivatives of natural naphthoquinones have a broad range of structural diversity and anticancer activities. These compounds have also exhibited inhibitory activity against MDR cancer cell-lines. Some of the compounds were also shown to inhibit enzymes such as topoisomerase I and II, STAT3 as well as AKT kinases.The potent inhibitory activities coupled with low toxicities of some of these compounds have qualified them for further development as potential cancer chemotherapeutics. The results of these studies warrants continued investigation of the naphthoquinone class of compounds for the discovery of novel anticancer agents.
References
- WHO Global Health Observatory, http://www.who.int/gho/map_gallery/en/, accessed 28 October 2014.
- City Mayors, http://www.citymayors.com/statistics/largest-cities-population-125.html, accessed 28 October 2014.
- World Cancer Report 2014, IARC, http://apps.who.int/bookorders/anglais/detart1.jsp?codlan=1%26codcol=76%26codcch=31, accessed 28 October 2014.
- J. Ferlay, et al., GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC Cancer Base No. 11 [Internet], International Agency for Research on Cancer, Lyon, France, 2013, http://globocan.iarc.fr, accessed 28 October 2014 Search PubMed.
- P. S. Steeg, Nat. Med., 2006, 12, 895 CrossRef CAS PubMed.
- P. Mehlen and A. Puisieux, Nat. Rev. Cancer, 2006, 6, 449 CrossRef CAS PubMed.
- J. Monteiro and R. Fodde, Eur. J. Cancer, 2010, 46, 1198 CrossRef CAS PubMed.
- D. X. Nguyen, P. D. Bos and J. Massague, Nat. Rev. Cancer, 2009, 9, 274 CrossRef CAS PubMed.
- E. R. Pollock and D. L. Morton, et al., in Principles of Surgical Oncology, ed. D. W. Kufe, R. E. Pollock and R. R. Weichselbaum, B.C. Decker, Hamilton (ON), 2003, ch. 38 Search PubMed.
- J. C. Coffey, J. H. Wang, M. J. Smith, D. Bouchier-Hayes, T. G. Cotter and H. P. Redmond, Lancet Oncol., 2003, 4, 760 CrossRef CAS.
- M. Koehnlechner, Leben ohne Krebs. Knaur, Munich, 1987 Search PubMed.
- http://www.irsa.org/radation_therapy.html, accessed 28 October 2014.
- E. Barbounaki-Konstantakou, Chemotherapy, Beta Medical Arts, Athens, 1989 Search PubMed.
- A. Persidis, Nat. Biotechnol., 1999, 17, 94 CrossRef CAS PubMed.
- G. Powis, Free Radical Biol. Med., 1989, 6, 63 CrossRef CAS.
- (a) E. A. Hillard, F. C. de Abreu, D. C. M. Ferreira, G. Jaouen, M. O. F. Goulart and C. Amatore, Chem. Commun., 2008, 2612 RSC; (b) A. J. M. da Silva, C. D. Netto, W. Pacienza-Lima, E. C. Torres-Santos, B. Rossi-Bergmann, S. Maurel, A. Alexis Valentin and P. R. R. Costa, J. Braz. Chem. Soc., 2009, 20, 176 CrossRef CAS PubMed.
- J. S. Driscoll, Cancer Chemother. Rep., Part 2, 1974, 4, 3 CAS.
- (a) T. J. Monks and D. C. Jones, Curr. Drug Metab., 2002, 3, 425 CrossRef CAS; (b) J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Chem. Res. Toxicol., 2000, 13, 135 CrossRef CAS PubMed; (c) T. J. Monks, R. P.Hanslik, G. M. Cohen, D. Ross and D. G. Graham, Toxicol. Appl. Pharmacol., 1992, 112, 2 CrossRef CAS; (d) P. J. O'Brien, Chem.-Biol. Interact., 1991, 80, 1 CrossRef.
- C. Salas, R. A. Tapia, K. Ciudad, V. Armstrong, M. Orellana, U. Kemmerling, J. Ferreira, J. D. Maya and A. Morello, Bioorg. Med. Chem., 2008, 16, 668 CrossRef CAS PubMed.
- M. C. F. Linardi, M. M. de Oliveira and M. R. P. Sampaio, J. Med. Chem., 1975, 18, 1.159 CrossRef.
- M. P. M. Portela and A. O. M. Stoppani, Biochem. Pharmacol., 1996, 51, 275 CrossRef.
- D. A. Williams and T. L. Lemke, Foye's principles of medicinal chemistry, Lippincott Williams & Wilkins, Baltimore, 3rd edn, 2002, pp. 924–951 Search PubMed.
- U. Galm, M. H. Hager, S. G. V. Lanen, J. Ju, J. S. Thorson and B. Shen, Chem. Rev., 2005, 105, 739 CrossRef CAS PubMed.
- S. E. Wolkenberg and D. L. Boger, Chem. Rev., 2005, 102, 2477 CrossRef PubMed.
- (a) D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727 CrossRef CAS; (b) B. Pang, X. Qiao, L. Janssen, A. Velds, T. Groothuis, R. Kerkhoven, M. Nieuwland, H. Ovaa1, S. Rottenberg, O. van Tellingen, J. Janssen, P. Huijgens, W. Zwart and J. Neefjes, Nat. Commun., 2013, 4, 1908 CrossRef PubMed.
- G. Zhang, L. Fang, L. Zhu, Y. Zhong, P. G. Wang and D. Sun, J. Med. Chem., 2006, 49, 1792 CrossRef CAS PubMed.
- L.-J. Huang, F.-C. Chang, K.-H. Lee, J.-P. Wang, C.-M. Teng and S.-C. Kuo, Bioorg. Med. Chem., 1998, 6, 2261 CrossRef CAS.
- J.-C. Lien, L.-J. Huang, J.-P. Wang, C.-M. Teng, K.-H. Lee and S.-C. Kuo, Chem. Pharm. Bull., 1996, 44, 1181 CrossRef CAS.
- J.-C. Lien, L.-J. Huang, C.-M. Teng, J.-P. Wang and S.-C. Kuo, Chem. Pharm. Bull., 2002, 50, 672 CrossRef CAS.
- J. J. Inbaraj and C. F. Chignell, Chem. Res. Toxicol., 2004, 17, 55 CrossRef CAS PubMed.
- S.-T. Huang, H.-S. Kuo, C.-L. Hsiao and Y.-L. Lin, Bioorg. Med. Chem., 2002, 10, 1947 CrossRef CAS.
- V. K. Tandon, R. B. Chhor, R. V. Singh, S. Rai and D. B. Yadav, Bioorg. Med. Chem. Lett., 2004, 14, 1079 CrossRef CAS PubMed.
- K. Sasaki, H. Abe and F. Yoshizaki, Biol. Pharm. Bull., 2002, 25, 669 CAS.
- Y.-R. Jin, C.-K. Ryu, C.-K. Moon, M.-R. Cho and Y.-P. Yun, Pharmacology, 2004, 70, 195 CrossRef CAS PubMed.
- D.-Y. Yuk, C.-K. Ryu, J.-T. Hong, K-H. HongChung, W.-S. Kang, Y. Kim, H.-S. Yoo, M.-K. Lee, C.-K. Lee and Y.-P. Yun, Biochem. Pharmacol., 2000, 60, 1001 CrossRef CAS.
- A. J. M. da Silva, C. D. Buarque, F. V. Brito, L. Aurelian, L. F. Macedo, L. H. Malkas, R. J. Hickey, D. V. S. Lopes, F. Noel, Y. L. B. Murakami, N. M. V. Silva, P. A. Melo, R. R. B. Caruso, N. G. Castro and P. R. R. Costa, Bioorg. Med. Chem., 2002, 10, 2731 CrossRef CAS.
- A. G. Ravelo, A. Estevez-Braun, H. Chavez-Orellana, E. Perez-Sacau and D. Mesa-Siverio, Curr. Top. Med. Chem., 2004, 4, 241 CrossRef CAS.
- C.-Y. Ting, C.-T. Hsu, H.-T. Hsu, J.-S. Su, T.-Y. Chen, W.-Y. Tarn, Y.-H. Kuo, J. Whang-Peng, L. F. Liu and J. Hwang, Biochem. Pharmacol., 2003, 66, 1981 CrossRef CAS PubMed.
- Y.-H. Zhang, K.-H. Chung, C.-K. Ryu, M.-H. Ko, M.-K. Lee and Y.-P. Yun, Biol. Pharm. Bull., 2001, 24, 618 CAS.
- V. K. Tandon, R. V. Singh, S. Rai, R. B. Chhor and Z. K. Khan, Boll. Chim. Farm., 2002, 141, 304 CAS.
- T. V. Ilina, E. A. Semenova, T. R. Pronyaeva, A. G. Pokrovskii, I. V. Nechepurenko, E. E. Shults, O. I. Andreeva, S. N. Kochetkov and G. A. Tolstikov, Dokl. Biochem. Biophys., 2002, 382, 56 CrossRef CAS.
- H. J. Kim, S. K. Kang, J. Y. Mun, Y. J. Chun, K. H. Choi and M. Y. Kim, FEBS Lett., 2003, 555, 217 CrossRef CAS.
- H. J. Kim, J. Y. Mun, Y. J. Chun, K. H. Choi, S. W. Ham and M. Y. Kim, Arch. Pharmacal Res., 2003, 26, 405 CrossRef CAS.
- D. Gao, M. Hiromura, H. Yasui and H. Sakurai, Biol. Pharm. Bull., 2002, 25, 827 CAS.
- A. Richwien and G. Wurm, Pharmazie, 2004, 59, 163 CAS.
- G. Wurm and S. Schwandt, Pharmazie, 2003, 58, 531 CAS.
- G.-Y. Song, Y. Kim, Y.-J. You, H. Cho, S.-H. Kim, D.-E. Sok and B.-Z. Ahn, Arch. Pharm., 2000, 333, 87 CrossRef CAS.
- (a) L. Constantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65 CrossRef; (b) J. Li, K. K.-C. Liu and S. Sakya, Mini-Rev. Med. Chem., 2005, 5, 1133 CrossRef CAS; (c) R. W. De Simone, K. S. Currie, S. A. Mitchell, J. W. Darrow and D. A. Pippin, Comb. Chem. High Throughput Screening, 2004, 7, 473 CrossRef CAS.
- (a) A. V. Pinto and S. L. De Castro, Molecules, 2009, 14, 4570 CrossRef PubMed; (b) C. O. Salas, M. Faúndez, A. Morello, J. D. Maya and R. A. Tapia, Curr. Med. Chem., 2011, 18, 144 CrossRef CAS.
- C. Asche, Mini-Rev. Med. Chem., 2005, 5, 449 CrossRef CAS.
- (a) D. R. da Rocha, A. C. G. de Souza, J. A. L. C. Resende, W. C. Santos, E. A. dos Santos, C. Pessoa, M. O. de Moraes, L. V. Costa-Lotufo, R. C. Montenegro and V. F. Ferreira, Org. Biomol. Chem., 2011, 9, 4315 RSC; (b) V. R. Campos, E. A. dos Santos, V. F. Ferreira, R. C. Montenegro, M. C. B. V. De Souza, L. V. Costa-Lotufo, M. O. De Moraes, A. K. P. Regufe, A. K. Jordao, A. C. Pinto, J. A. L. C. Resende and A. C. Cunha, RSC Adv., 2012, 2, 11438 RSC; (c) S. J. Lucas, R. M. Lord, R. L. Wilson, R. M. Phillips, V. Sridharan and P. C. McGowan, Dalton Trans., 2012, 41, 13800 RSC; (d) E. Perez-Sacau, R. G. Dıaz-Penate, A. Estevez-Braun, A. G. Ravelo, J. M. Garcia-Castellano, L. Pardo and M. Campillo, J. Med. Chem., 2007, 50, 696 CrossRef CAS PubMed; (e) K. K. C. Liu, J. Li and S. Sakya, Mini-Rev. Med. Chem., 2004, 4, 1105 CrossRef CAS; (f) Y. Kumagai, Y. Shinkai, T. Miura and A. K. Cho, Annu. Rev. Pharmacol., 2012, 52, 221 CrossRef CAS PubMed; (g) M. Hallak, B. K. Thakur, T. Winn, O. Shpilberg, S. Bittner, Y. Granot, I. Levy and L. Nathan, Anticancer Res., 2013, 33, 183 CAS; (h) A. Ventura Pinto and S. Lisboa de Castro, Molecules, 2009, 14, 4570 CrossRef CAS PubMed.
- X. Mou, S. Kesari, P. Y. Wen and X. Huang, International Journal of Clinical and Experimental Medicines, 2011, 4, 17 CAS.
- D. S. Shewach and R. D. Kuchta, Chem. Rev., 2009, 109, 2859 CrossRef CAS PubMed.
- M. G. Simic, D. S. Bergtold and L. R. Karam, Mutat. Res., 1989, 214, 3 CrossRef CAS.
- J. Bannister, Free Radicals Biol. Med., 2007, 10, 250 Search PubMed.
- A. S. Veskoukis, A. M. Tsatsakis and D. Kouretas, Cell Stress Chaperones, 2012, 17, 11 CrossRef CAS PubMed.
- DNA and Free Radicals, ed. B. Halliwell and O. I. Aruoma, Ellis Horwood Ltd, Chichester, England, 1993 Search PubMed.
- H. Kasai and S. Nishimura, Nucleic Acids Res., 1984, 12, 2137 CrossRef CAS PubMed.
- S. Toyokuni, T. Mori and M. Dizdaroglu, Int. J. Cancer, 1994, 57, 123 CrossRef CAS.
- A. Matsui, T. Ikeda, K. Enomoto, K. Hosoda, H. Nakashima, K. Omae, M. Watanabe, T. Hibi and M. Kitajima, Cancer Lett., 2000, 151, 87 CrossRef CAS.
- H. Kasai, Z. Yamaizumi, M. Berger and J. Cadet, J. Am. Chem. Soc., 1992, 114, 9692 CrossRef CAS.
- B. Halliwell and S. Chirico, Am. J. Clin. Nutr., 1993, 57, 715S–724S CAS.
- R. L. Levine, Free Radicals Biol. Med., 2002, 32, 790 CrossRef CAS.
- A. Matsuzawa and H. Ichijo, Biochim. Biophys. Acta, 2008, 1780, 1325 CrossRef CAS PubMed.
- T. Nguyen, P. Nioi and C. B. Pickett, J. Biol. Chem., 2009, 284, 13291 CrossRef CAS PubMed.
- E. A. Wiemer, Nat. Med., 2011, 17, 1552 CrossRef CAS PubMed.
- Y. Luo, P. Zou, J. Zou, J. Wang, D. Zhou and L. Liu, Exp. Gerontol., 2011, 46, 860 CrossRef CAS PubMed.
- M. Y. Kim, T. Zhang and W. L. Kraus, Genes Dev., 2005, 19, 1951 CrossRef CAS PubMed.
- S. Smith, Trends Biochem. Sci., 2001, 26, 174 CrossRef CAS.
- J. M. Matés, I. Pérez-Gómez and C. Núnez de Castro, Clin. Biochem., 1999, 32, 595 CrossRef.
- J. M. Matés and F. Sánchez-Jiménez, Front. Biosci., 1999, 4, 339 CrossRef PubMed.
- B. Halliwell and J. M. C. Gutteridge, Free radicals in Biology and Medicine, Oxford University Press, New York, 1999 Search PubMed.
- B. Poljšak and R. Fink, Oxid. Med. Cell. Longevity, 2014, 671539 Search PubMed.
- M. Valko, D. Leibfritz, J. Moncola, M. T. D. Cronin, M. Mazura and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44 CrossRef CAS PubMed.
- P. Decker and S. Muller, Curr. Pharm. Biotechnol., 2002, 3, 275 CAS.
- J. Nakamura, E. R. Purvis and J. A. Swenberg, Nucleic Acids Res., 2003, 31, 1790 CrossRef CAS PubMed.
- M. T. Moslen, Free Radicals in Diagnostic Medicine, ed. D. Armstrong, Plenum Press, New York, 1994 Search PubMed.
- P. Jagtap and C. Szabo, Nat. Rev. Drug Discovery, 2005, 4, 421 CrossRef CAS PubMed.
- L. Liaudet, E. Szabo, L. Timashpolsky, L. Virag, A. Cziraki and C. Szabo, Br. J. Pharmacol., 2001, 133, 1424 CrossRef CAS PubMed.
- C. R. Calabrese, R. Almassy, S. Barton, M. A. Batey, A. H. Calvert, S. Canan-Koch, B. W. Durkacz, Z. Hostomsky, R. A. Kumpf, S. Kyle, J. Li, K. Maegley, D. R. Newell, E. Notarianni, I. J. Stratford, D. Skalitzky, H. D. Thomas, L.-Z. Wang, S. E. Webber, K. J. Williams and N. J. Curtin, J. Natl. Cancer Inst., 2004, 96, 56 CrossRef CAS PubMed.
- Y. Le Rhun, J. B. Kirkland and G. M. Shah, Biochem. Biophys. Res. Commun., 1998, 245, 1 CrossRef CAS.
- V. A. Kickhoefer, A. C. Siva, N. L. Kedersha, E. M. Inman, C. Ruland, M. Streuli and L. H. Rome, J. Cell Biol., 1999, 146, 917 CrossRef CAS.
- N. L. Kedersha, J. E. Heuser, D. C. Chugani and L. H. Rome, J. Cell Biol., 1991, 112, 225 CrossRef CAS.
- J. L. Vermillon and M. J. Coon, J. Biol. Chem., 1978, 253, 2694 Search PubMed.
- N. R. Bachur, S. L. Gordon, M. V. Gee and H. Kon, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 954 CrossRef CAS.
- M. I. Walton, C. R. Wolf and P. Workman, Biochem. Pharmacol., 1992, 44, 251 CrossRef CAS.
- A. Gutierrez, A. Grunau, M. Paine, A. W. Munro, C. R. Wolf, G. C. K. Roberts and N. S. Scrutton, Biochem. Soc. Trans., 2003, 31, 497 CrossRef CAS.
- P. B. Danielson, Curr. Drug Metab., 2002, 3, 561 CrossRef CAS.
- F. P. Guengerich and E. M. Isin, Acta Chim. Slov., 2008, 55, 7 CAS.
- K. M. Huttunen, N. Mahonen, H. Raunio and J. Rautio, Curr. Med. Chem., 2008, 15, 2346 CrossRef CAS.
- R. Li, M. A. Bianchet, P. Talalayt and L. M. Amzel, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8846 CrossRef CAS.
- D. Siegel, C. Yan and D. Ross, Biochem. Pharmacol., 2012, 15(83), 1033 CrossRef PubMed.
- J. M. Brown and A. J. Giaccia, Cancer Res., 1998, 58, 1408 CAS.
- F. C. de Abreu, P. A. M. Ferraz and M. O. F. Goulart, J. Braz. Chem. Soc., 2002, 13, 19 CrossRef CAS PubMed.
- U. M. Viswanathan, T. Burkholz and C. Z. Jacob, Zeitschrift für Physikalische Chemie, 2013, 227, 691 CrossRef CAS.
- F. A. Fitzpatrick, Int. Immunopharmacol., 2001, 1, 1651 CrossRef CAS.
- (a) R. B. Silverman, in The Organic Chemistry of Drug Design and Drug Action, Academic Press, New York, 1992 Search PubMed; (b) J. W. Lown, S. K. Sim, K. C. Majumdar and R. Y. Chang, Biochem. Biophys. Res. Commun., 1977, 76, 705 CrossRef CAS.
- A. J. Lin, L. A. Cosby, C. W. Shansky and A. C. Sartorelli, J. Med. Chem., 1972, 15, 1247 CrossRef CAS.
- A. J. Lin, R. S. Pardini, L. A. Cosby, B. J. Lillis, C. W. Shansky and A. C. Sartorelli, J. Med. Chem., 1973, 16, 1268 CrossRef CAS.
- H. W. Moore and R. Czerniak, Med. Res. Rev., 1981, 1, 249 CrossRef CAS.
- W. A. Denny, Lancet Oncol., 2000, 1, 25 CrossRef CAS.
- F. C. de Abreu, A. C. O. Lopes and M. O. F. Goulart, J. Electroanal. Chem., 2004, 562, 53 CrossRef CAS PubMed.
- F. H. Fry and C. Jacob, Curr. Pharm. Des., 2006, 12, 4479 CrossRef CAS.
- Y. Kumagai, Y. Tsurutani, M. Shinyashiki, S. Homma-Takeda, T. Nakai, Y. Yoshikawa and N. Shimojo, Environ. Toxicol. Pharmacol., 1997, 3, 245 CrossRef CAS.
- S. Rahimipour, L. Weiner, P. B. Shrestha-Dawadi, S. Bittner, Y. Koch and M. Fridkin, Lett. Pept. Sci., 1998, 5, 421 CAS.
- S. Rahimipour, L. Weiner, M. Fridkin, P. B. Shrestha-Dawadi and S. Bittner, Lett. Pept. Sci., 1996, 3, 263 CrossRef CAS.
- J. McKay and N. Hirano, Polish translation: D. Surdel Czym sa: chemioterapia iradioteriapia?, in Chemotherapy, radiotherapy, ed. E. Zubrzycka, Gdanskie Wydawnictwo Psychologiczne, Gdansk, 2002, pp. 21–30 Search PubMed.
- E. Espinosa, P. Zamora, J. Feliu and M. G. Baron, Cancer Treat. Rev., 2003, 29, 515 CrossRef CAS.
- (a) R. P. Verma, Adv. Anticancer Agents Med. Chem., 2006, 6, 489 CrossRef CAS; (b) P. Kovacic and R. Somanathan, Anti-Cancer Agents Med. Chem., 2011, 11, 658 CrossRef CAS; (c) E. Salustiano, C. Netto, R. Fernandes, A. da Silva, T. Bacelar, C. Castro, C. Buarque, R. Maia, V. Rumjanek and P. Costa, Invest. New Drugs, 2009, 28, 139 CrossRef PubMed.
- K. Lechner, K. Geissler, U. Jager, H. Greinix and P. Kalhs, Ann. Oncol., 1999, 10, s45–s51 CrossRef PubMed.
- A. Hollingshead and D. Faulds, Drugs, 1991, 42, 690 CrossRef PubMed.
- B. Kalyanaraman, K. M. Morehouse and R. P. Mason, Arch. Biochem. Biophys., 1991, 286, 164 CrossRef CAS.
- J. Schreiber, C. Mottley, B. K. Sinha, B. Kalyanaraman and R. P. Mason, J. Am. Chem. Soc., 1987, 109, 348 CrossRef CAS.
- D. Taatjes, D. J. Fenick, G. Gaudiano and T. H. Koch, Curr. Pharm. Des., 1998, 4, 203 CAS.
- D. D. Von Hoff, M. W. Layard, P. Basa, H. L. Davis Jr, A. L. Von Hoff, M. Rozencweig and F. M. Muggia, Ann. Intern. Med., 1979, 91, 710 CrossRef CAS PubMed.
- K. C. Murdock, R. E. Wallace and F. E. Durr, et al., J. Med. Chem., 1979, 22, 1024 CrossRef CAS.
- Z. A. Arlin, R. Silver, P. Cassileth, S. Armentrout, R. Gams, A. Daghestani, M. Coleman, I. Schoch and G. Dukart, Cancer Treat. Rep., 1985, 69, 61 CAS.
- P. A. Paciucci, T. Ohnuma, J. Cuttner, R. T. Silver and J. F. Holland, Cancer Res., 1983, 43, 3919 CAS.
- J. P. Dutcher, P. H. Wiernik and J. J. Strauman, Proc. Am. Soc. Clin. Oncol., 1985, 4, 170 Search PubMed.
- C. Parker, R. Waters, C. Leighton, J. Hancock, R. Sutton, A. V. Moorman, P. Ancliff, M. Morgan, A. Masurekar, N. Goulden, N. Green, T. Révész, P. Darbyshire, S. Love and V. Saha, Lancet, 2010, 376(9757), 2009 CrossRef CAS.
- W. Darko, A. L. Smith, E. L. King and S. J. Grethlein, Mitoxantrone-induced cardiotoxicity, J. Oncol. Pharm. Pract., 2001, 7, 47 CrossRef.
- E. H. Rubin, MD and W. N. Hait, MD, et al. Intercalating Drugs that Target Topoisomerases, in Holland-Frei Cancer Medicine, ed. D. W. Kufe, R. E. Pollock and R. R. Weichselbaum, 6th edn, BC Decker, Hamilton (ON), 2003 Search PubMed.
- M. O. Crepsi, S. E. Ivanier, J. Genovese and A. Baldi, Biochem. Biophys. Res. Commun., 1986, 136, 521 CrossRef.
- J. Mazerski, S. Martelli and E. Borowski, Acta Biochim. Pol., 1998, 45, 1 CAS.
- A. C. Sartorelli, W. F. Hodnick, M. F. Belcourt, M. Tomasz, B. Haffty, J. J. Fischer and S. Rockwell, Oncol. Res., 1994, 6, 501 CAS.
- A. C. Sartorelli, Cancer Res., 1988, 48, 775 CAS.
- A. J. Lin, L. A. Cosby, C. W. Shansky and A. C. Sartorelli, J. Med. Chem., 1972, 15, 1247 CrossRef CAS.
- H. A. Seow, P. G. Penketh, R. P. Baumann and A. C. Sartorelli, Methods Enzymol., 2004, 382, 221 CAS.
- Y. Wang, J. P. Gray, V. Mishin, D. E. Heck, D. L. Laskin and J. D. Laskin, Mol. Cancer Ther., 2010, 9, 1852 CrossRef CAS PubMed.
- B. K. Sinha, Chem.–Biol. Interact., 1989, 69, 293 CrossRef CAS.
- J. J. Oleson, L. A. Calderella, K. J. Mjos, A. R. Reith, R. S. Thie and I. Toplin, Antibiot. Chemother., 1961, 11, 158 CAS.
- M. N. Teller, S. F. Waghul and G. W. Woolley, Antibiot. Chemother., 1961, 11, 165 CAS.
- H. C. Reilly and K. Sigiura, Antibiot. Chemother., 1961, 11, 174 CAS.
- National Cancer Institute, Streptonigrin (NSC-45383), National Institute of Health, Clinical brochure, March 1968, pp. 1–20.
- H. D. Beall, R. T. Mulcahy, D. Siegel, R. D. Traver, N. W. Gibson and D. Ross, Cancer Res., 1994, 54, 3196 CAS.
- H. D. Beall, A. M. Murphy, D. Siegel, R. H. Hargreaves, J. Butler and D. Ross, Mol. Pharmacol., 1995, 48, 499 CAS.
- C. A. Hackethal, R. B. Golhey, C. T. C. Tan, D. A. Karnofsky and J. H. Burchenal, Antibiot. Chemother., 1961, 11, 178 CAS.
- E. W. Humphrey and N. Blank, Cancer Chemother. Rep., 1961, 12, 99 CAS.
- M. N. Harris, T. J. Medrek, F. M. Golomb, S. L. Gumport, A. H. Postel and J. C. Wright, Cancer, 1965, 18, 49 CrossRef CAS.
- T. J. McBride, J. J. Oleson and D. Woolf, Cancer Res., 1966, 26, 727 CAS.
- S. L. Rivers, R. M. Whittington and T. J. Medrek, Cancer, 1966, 19, 1377 CrossRef CAS.
- H. L. Davis Jr, D. D. Von Hoff, J. E. Henney and M. Rozencweig, Cancer Chemother. Pharmacol., 1978, 1, 83 CrossRef CAS.
- D. T. Kaung, R. M. Whittington, H. H. Spencer and M. E. Patno, Cancer, 1969, 23, 597 CrossRef CAS.
- D. T. Kaung, R. M. Whittington, H. H. Spencer and M. E. Patno, Cancer, 1969, 23, 1280 CrossRef CAS.
- P. Banzet, C. Jacquillat, J. Civatte, A. Puissant, J. Maral, C. Chastang, L. Israel, S. Belaich, J.-C. Jourdain, M. Weil and G. Auclerc, Cancer, 1978, 41, 1240 CrossRef CAS.
- R. J. Forcier, O. R. McIntyre, N. I. Nissen, T. F. Pajak, O. Glidewell and J. F. Holland, Med. Pediatr. Oncol., 1978, 4, 351 CrossRef CAS.
- N. I. Nissen, J. Ersboll, H. S. Hansen, S. Walbom-Jorgensen, J. Pedersen-Bjergaard, M. M. Hansen and J. Rygard, Cancer, 1983, 521 Search PubMed.
- D. Ross, H. Beall, R. D. Traver, D. Siegel, R. M. Phillips and N. W. Gibson, Oncol. Res., 1994, 6, 493 CAS.
- S. Y. Sharp, L. R. Kelland, M. R. Valenti, L. A. Brunton, S. Hobbs and P. Workman, Mol. Pharmacol., 2000, 58, 1146 CAS.
- R. Cone, S. K. Hasan, J. W. Lown and A. R. Morgan, Can. J. Biochem., 1976, 54, 219 CrossRef CAS.
- M. Levine and M. Borthwick, Virology, 1963, 21, 568 CrossRef CAS.
- H. L. White and J. R. White, Mol. Pharmacol., 1968, 4, 549 CAS.
- J. W. Lown and S. K. Sim, Can. J. Biochem., 1976, 54, 446 CrossRef CAS.
- H. D. Beall, Y. Liu, D. Siegel, E. M. Bolton, N. W. Gibson and D. Ross, Biochem. Pharmacol., 1996, 51, 645 CrossRef CAS.
- W. B. Kremer and J. Laszlo, Cancer Chemother. Rep., 1967, 51, 19 CAS.
- Y. Shiloh, E. Tabor and Y. Becker, Carcinogenesis, 1983, 4, 1317 CrossRef CAS PubMed.
- Y. Sugiura, J. Kuwahara and T. Suzuki, Biochim. Biophys. Acta, 1984, 782, 254–261 CrossRef CAS.
- M. M. Gottesman, Cancer Res., 1993, 53, 747 CAS.
- M. M. Gottesman and I. Pastan, Annu. Rev. Biochem., 1993, 62, 385 CrossRef CAS PubMed.
- D. Fan, P. J. Beltran and C. A. O'Brien, in Reversal of multidrug resistance, in Reversal of Multidrug Resistance in Cancer, ed. J. A.Kellen, CRC Press, Boca Raton, FL, 1994, pp. 93–24 Search PubMed.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615 CrossRef CAS PubMed.
- G. D. Leonard, T. Fojo and S. E. Bates, Oncologist, 2003, 8, 411 CrossRef CAS PubMed.
- C. H. Choi, Cancer Cell Int., 2005, 5, 30 CrossRef PubMed.
- J. R. Riordan, K. Deuchars, N. Kartner, N. Alon, J. Trent and V. Ling, Nature, 1985, 316, 817 CrossRef CAS.
- L. A. Doyle, W. Yang, L. V. Abruzzo, T. Krogmann, Y. Gao, A. K. Rishi and D. D. Ross, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15665 CrossRef CAS.
- P. Borst, R. Evers, M. Kool and J. Wijnholds, J. Natl. Cancer Inst., 2000, 92, 1295 CrossRef CAS PubMed.
- L. J. Goldstein, H. Galski, A. Fojo, M. Willingham, S. L. Lai, A. Gazdar, R. Pirker, A. Green, W. Crist, G. M. Brodeur, M. Lieber, J. Cossman, M. M. Gottesman and I. Pastan, J. Natl. Cancer Inst., 1989, 81, 116 CrossRef CAS.
- G. Batist, A. Tulpule, B. K. Sinha, A. G. Katki, C. E. Myers and K. H. Cowan, J. Biol. Chem., 1986, 33, 15544 Search PubMed.
- X. Y. Hao, M. Widersten, M. Ridderstrom, U. Hellman and B. Mannervik, Biochem. J., 1994, 297, 59 CAS.
- R. K. Jain, Cancer Res., 1987, 47, 3039 CAS.
- J. Demant, M. Sehested and P. B. Jensen, Biochim. Biophys. Acta, 1990, 1055, 117 CrossRef.
- C. F. Higgins, Res. Microbiol., 2001, 152, 205 CrossRef CAS.
- M. Dean, The human ATP-binding cassette (ABC) transporter superfamily, NCBI Monographs, 2002.
- A. Sparreboom, R. Danesi, Y. Ando, J. Chan and W. D. Figg, Drug Resist. Updates, 2003, 6, 71 CrossRef CAS.
- S. Naito, H. Koga, A. Yokomizo, N. Sakamoto, S. Kotoh, M. Nakashima, A. Kiue and M. Kuwano, World J. Surg., 2000, 24, 1183 CrossRef CAS.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714 CrossRef CAS PubMed.
- J. I. Fletcher, M. Haber, M. J. Henderson and M. D. Norris, Nat. Rev. Cancer, 2010, 10, 147 CrossRef CAS PubMed.
- M. F. Ullah, Asian Pacific Journal of Cancer Prevention: APJCP, 2008, 9, 1 Search PubMed.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219 CrossRef PubMed.
- M. Dean, T. Fojo and S. Bates, Nat. Rev. Cancer, 2005, 5, 275 CrossRef CAS PubMed.
- R. Krishna and L. D. Mayer, Eur. J. Pharm. Sci., 2000, 11, 265 CrossRef CAS.
- R. F. Silver and H. L. Holmes, Can. J. Chem., 1968, 46, 1859 CrossRef CAS.
- I. Oeriu, Biokhimiia, 1963, 28, 380 CAS.
- B. Prescott, J. Med. Chem., 1969, 12, 181 CrossRef CAS.
- E. M. Hodnett, C. Wongwiechintana, W. J. Dunn III and P. Marrs, J. Med. Chem., 1983, 265, 570 CrossRef.
- N. G. Clark, Pestic. Sci., 1984, 15, 235 CrossRef CAS.
- K. V. Rao, K. Biemann and R. B. Woodward, J. Am. Chem. Soc., 1963, 85, 2532 CrossRef CAS.
- (a) T. Hata, Y. Sano, R. Sugawara, A. Matsumae, K. Kanamori, T. Shima and T. J. Hoshi, J. Antibiot., Ser. A, 1956, 9, 141 CAS; (b) S. Wakaki, H. Marumo, T. Tomioka, G. Shimizu, E. Kato, H. Kamada, S. Kudo and Y. Fujimoto, Antibiot. Chemother., 1958, 8, 228 CAS; (c) S. Wakaki, Cancer Chemother. Rep., 1961, 13, 79 CAS; (d) J. S. Webb, D. B. Cosulich, J. H. Mowat, J. B. Patrick, R. W. Broschard, W. E. Meyer, R. P. Williams, C. F. Wolf, W. Fulmor, C. Pidacks and J. E. Lancaster, J. Am. Chem. Soc., 1962, 84, 3185 CrossRef CAS.
- P. Sensi, Res. Prog. Org.-Biol. Med. Chem., 1964, 1, 337 CAS.
- G. P. Georgiev, O. P. Samarina, M. I. Lerman, M. N. Smirnov and A. N. Severtzov, Nature, 1963, 200, 1291 CrossRef CAS.
- K. Sasaki, K. L. Rinehart, G. Slomp, M. F. Grostic and E. C. Olson, J. Am. Chem. Soc., 1970, 92, 7591 CrossRef CAS.
- S. Pal, M. Jadhav, T. Weyhermuller, Y. Patil, M. Nethaji, U. Kasabe, L. Kathawate, V. Badireenath Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2013, 1049, 355 CrossRef CAS PubMed.
- D. Bhasin, S. N. Chettiar, J. P. Etter, M. Mok and P.-K. Li, Bioorg. Med. Chem., 2013, 21, 4662 CrossRef CAS PubMed.
- K. W. Wellington and N. I. Kolesnikova, Bioorg. Med. Chem., 2012, 220, 4472 CrossRef PubMed.
- J. Benites, J. A. Valderrama, K. Bettega, R. C. Pedrosa, P. B. Calderon and J. Verrax, Eur. J. Med. Chem., 2010, 45, 6052 CrossRef CAS PubMed.
- A. Kazi, H. Lawrence, W. C. Guida, M. L. Mclaughlin, G. M. Springett, N. Berndt, R. M. Yip and S. M. Sebti, Cell Cycle, 2009, 12, 1940 CrossRef.
- H. R. Lawrence, A. Kazi, Y. T. Luo, R. Kendig, Y. Y. Ge, S. Jain, K. Daniel, D. Santiago, W. C. Guida and S. M. Sebti, Bioorg. Med. Chem., 2010, 18, 5576 CrossRef CAS PubMed.
- K. Xu, Z. Xiao, Y. B. Tang, L. Huang, C.-H. Chen, E. Ohkoshi and K.-H. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 2772 CrossRef CAS PubMed.
- M. Dzieduszycka, M. M. Bontemps-Gacz, B. Stefanska, S. Martell, A. Piwkowska, M. Arciemiuk and E. Borowski, Bioorg. Med. Chem., 2006, 14, 2880 CrossRef CAS PubMed.
- Y. Brandy, I. Ononiwu, D. Adedeji, V. Williams, C. Mouamba, Y. Kanaan, R. L. Copeland Jr, D. A. Wright, R. J. Butcher, S. R. Denmeade and O. Bakare, Invest. New Drugs, 2012, 30, 1709 CrossRef CAS PubMed.
- T. Ishikawa, T. Saito, A. Kurosawa, T. Watanabe, S. Maruyama, Y.-I. Ichikawa, R. Yamada, H. Okuzawa, H. Sato and K. Ueno, Chem. Pharm. Bull., 2011, 59, 472 CrossRef CAS.
- K. Shanab, E. Schirmer, H. Knafl, E. Wulz, W. Holzer, H. Spreitzera, P. Schmidt, B. Aicher, G. Müller and E. Günther, Bioorg. Med. Chem. Lett., 2010, 20, 3950 CrossRef CAS PubMed.
- B. Kongkathip, S. Akkarasamiyo, K. Hasitapan, P. Sittikul, N. Boonyalai and N. Kongkathip, J. Med. Chem., 2013, 60, 271 CrossRef CAS PubMed.
- N. Pradidphol, N. Kongkathip, P. Sittikul, N. Boonyalai and B. Kongkathip, Eur. J. Med. Chem., 2012, 49, 253 CrossRef CAS PubMed.
- K. W. Stagliano, A. Emadi, Z. Lu, H. C. Malinakova, B. Twenter, M. Yu, L. E. Holland, A. M. Rom, J. S. Harwood, R. Amin, A. A. Johnson and Y. Pommier, Bioorg. Med. Chem., 2006, 14, 5651 CrossRef CAS PubMed.
- K. W. Stagliano, Z. Lu, A. Emadi, J. S. Harwood and C. A. Harwood, J. Org. Chem., 2004, 69, 5128 CrossRef CAS PubMed.
- A. Emadi, J. S. Harwood, S. Kohanim and K. W. Stagliano, Org. Lett., 2002, 4, 521 CrossRef CAS PubMed.
- A. Emadi, Regiocontrolled Synthesis and HIV Inhibitory Properties of Binapthoquinone and Trimeric Naphthoqinone Derivates of Conocurvone, Illinois institute of Technology, Chicago, IL, 2004 Search PubMed.
- A. E. Ross, A. Emad, L. Marchionni, P. J. Hurley, B. W. Simons, E. M. Schaeffer and M. Vuica-Ross, BJU Int., 2010, 108, 447 CrossRef PubMed.
- G. Bringmann, G. Zhanga, A. Hagera, M. Moosa, A. Irmera, R. Bargoub and M. Chatterjee, Eur. J. Med. Chem., 2011, 46, 5778 CrossRef CAS PubMed.
- S. N. Fedorov, L. K. Shubina, A. S. Kuzmich and S. G. Polonik, Open Glycosci., 2011, 4, 1 CrossRef CAS.
- H. Hussain, K. Krohn, V. U. Ahmad, G. A. Miana and I. V. Green, ARKIVOC, 2007, 2, 145 CrossRef.
- E. R. de Almeida, Open Nat. Prod. J., 2009, 2, 42 CrossRef.
- C. J. Li, Y. Z. Li, A. V. Pinto and A. B. Pardee, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13369 CrossRef CAS.
- J. I. Lee, D. Y. Choi, H. S. Chung, H. G. Seo, H. J. Woo, B. T. Choi and Y. H. Choi, Exp. Oncol., 2006, 28, 30 CAS.
- Y. Z. Li, X. G. Sun, J. T. Lamont, A. B. Pardee and C. J. Li, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2674 CrossRef CAS PubMed.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012 CrossRef CAS PubMed.
- S. N. Sunassee, C. G. L. Veale, N. Shunmoogam-Gounden, O. Osoniyi, D. T. Hendricks, M. R. Caira, J.-A. de la Mare, A. L. Edkins, A. V. Pinto, E. N. da Silva Jr and M. T. Davies-Coleman, Eur. J. Med. Chem., 2013, 62, 98 CrossRef CAS PubMed.
- K. O. Eyong, G. N. Folefoc, V. Kuete, V. P. Beng, K. Krohn, H. Hussain, A. E. Nkengfack, M. Saeftel, S. R. Sarite and A. Hoerauf, Phytochemistry, 2006, 67, 605 CrossRef CAS PubMed.
- V. Kuete, K. O. Eyong, G. N. Folefoc, V. P. Beng, H. Hussain, K. Krohn and A. E. Nkengfack, Pharmazie, 2007, 62, 552 CAS.
- J. Breger, B. B. Fuchs, G. Aperis, T. I. Moy, F. M. Ausubel and E. Mylonakis, PLoS Pathog., 2007, 3, 168 CAS.
- A. Portillo, R. Vila, B. Freixa, T. Adzet and S. Cañigueral, J. Ethnopharmacol., 2001, 76, 93 CrossRef CAS.
- A. V. Pinto and S. L. de Castro, Molecules, 2009, 14, 4570 CrossRef PubMed.
- M. D. C. F. Linardi, M. M. De Oliveira and M. R. P. Sampaio, J. Med. Chem., 1975, 18, 1159 CrossRef CAS.
- R. M. Khan and S. M. Mlungwana, Phytochemistry, 1999, 50, 439 CrossRef CAS.
- J. J. Pink, S. M. Planchon, C. Tagliarino, M. E. Varnes, D. Siegel and D. A. Boothman, J. Biol. Chem., 2000, 275, 5416 CrossRef CAS PubMed.
- J. J. Pink, S. Wuerzberger-Davis, C. Tagliarino, S. M. Planchon, X. Yang, C. J. Froelich and D. A. Boothman, Exp. Cell Res., 2000, 255, 144 CrossRef CAS PubMed.
- E. L. Bonifazi, C. Ríos-Luci, L. G. León, G. Burton, J. M. Padrón and R. I. Misico, Bioorg. Med. Chem., 2010, 18, 2621 CrossRef CAS PubMed.
- E. N. da Silva Jr, B. C. Cavalcanti, T. T. Guimarães, M. do Carmo, F. R. Pinto, I. O. Cabral, C. Pessoa, L. V. Costa-Lotufo, M. O. de Moraes, C. K. Z. de Andrade, M. R. dos Santos, C. A. de Simone, M. O. F. Goulart and A. V. Pinto, Eur. J. Med. Chem., 2011, 46, 399 CrossRef PubMed.
- E. N. da Silva Jr, C. F. de Deus, B. C. Cavalcanti, C. Pessoa, L. V. Costa-Lotufo, R. C. Montenegro, M. O. de Moraes, M. do Carmo, F. R. Pinto, C. A. de Simone, V. F. Ferreira, M. O. F. Goulart, C. K. Z. Andrade and A. V. Pinto, J. Med. Chem., 2010, 53, 504 CrossRef PubMed.
- B. C. Cavalcanti, I. O. Cabral, F. A. R. Rodrigues, F. W. A. Barros, D. D. Rocha, H. I. F. Magalhães, D. J. Moura, J. Saffi, J. A. P. Henriques, T. S. C. Carvalho, M. O. Moraes, C. Pessoa, I. M. M. de Melo and E. N. da Silva Jr, J. Braz. Chem. Soc., 2013, 24, 145 CrossRef CAS PubMed.
- C. Ríos-Luci, E. L. Bonifazi, L. G. León, J. C. Montero, G. Burton, A. Pandiella, R. I. Misico and J. M. Padrón, Eur. J. Med. Chem., 2012, 53, 264 CrossRef PubMed.
- E. N. da Silva Jr, M. A. B. F. de Moura, A. V. Pinto, M. do Carmo, F. R. Pinto, M. C. B. V. de Souza, A. J. Araújo, C. Pessoa, L. V. Costa-Lotufo, R. C. Montenegro, M. O. de Moraes, V. F. Ferreira and M. O. F. Goulart, J. Braz. Chem. Soc., 2009, 4, 635 CrossRef PubMed.
- N. Tanaka, M. Yasue and H. Imamura, Tetrahedron Lett., 1966, 24, 2767 CrossRef.
- C. Galeffi, E. M. Delle Monache, C. G. Casinovi and G. B. Marini-Bettolo, Tetrahedron Lett., 1969, 40, 3583 CrossRef.
- Y.-G. Suh, S. N. Kim, D.-Y. Shin, S.-S. Hyun, D.-S. Lee, K.-H. Min, S. M. Han, F. Li, E.-C. Choi and S.-H. Choi, Bioorg. Med. Chem. Lett., 2006, 16, 142 CrossRef CAS PubMed.
- D.-Y. Shin, S. N. Kim, J.-H. Chae, S.-S. Hyun, S.-Y. Seo, Y.-S. Lee, K.-O. Lee, S.-H. Kim, Y.-S. Lee, J. M. Jeong, N.-S. Choi and Y.-G. Suh, Bioorg. Med. Chem. Lett., 2004, 14, 4519 CrossRef CAS PubMed.
- S.-L. Huang, Y. Luo, Z.-S. Huang, X.-Y. Wang, X.-Z. Bu, P.-Q. Liu, L. Ma, B.-F. Xie, Z.-C. Liu, Y.-M. Li, A. S. C. Chan and L.-Q. Gu, Synth. Commun., 2006, 36, 2667 CrossRef CAS.
- D. Wang, M. Y. Xia, Z. Cui, S. Tashiro, S. Onodera and T. Ikejima, Biol. Pharm. Bull., 2004, 27, 1025 CAS.
- Z. Liu, S. L. Huang, M. M. Li, Z. S. Huang, K. S. Lee and L. Q. Gu, Chem.-Biol. Interact., 2009, 177, 48 CrossRef CAS PubMed.
- H.-Y. Han, Y.-B. Kim, J.-S. Kim, H.-J. Park and M.-J. Noh, WO 01/51004, 2001, p. 28.
- W.-B. Wu, J.-B. Ou, Z.-H. Huang, S.-B. Chen, T.-M. Ou, J.-H. Tan, D. Li, L.-L. Shen, S.-L. Huang, L.-Q. Gu and Z.-S. Huang, Eur. J. Med. Chem., 2011, 46, 3339 CrossRef CAS PubMed.
- H. Yu and R. Jove, Nat. Rev. Cancer, 2004, 4, 97 CrossRef CAS PubMed.
- P. Sansone and J. Bromberg, J. Clin. Oncol., 2012, 30, 1005 CrossRef CAS PubMed.
- B. B. Aggarwal, A. B. Kunnumakkara, K. B. Harikumar, S. R. Gupta, S. T. Tharakan, C. Koca, S. Dey and B. Sung, Ann. N. Y. Acad. Sci., 2009, 1171, 59 CrossRef CAS PubMed.
- M. Hedvat, D. Huszar, A. Herrmann, J. M. Gozgit, A. Schroeder, A. Sheehy, R. Buettner, D. Proia, C. M. Kowolik, H. Xin, B. Armstrong, G. Bebernitz, S. Weng, L. Wang, M. Ye, K. McEachern, H. Chen, D. Morosini, K. Bell, M. Alimzhanov, S. Ioannidis, P. McCoon, Z. A. Cao, H. Yu, R. Jove and M. Zinda, Cancer Cell, 2009, 16, 487 CrossRef CAS PubMed.
- S. Dewilde, A. Vercelli, R. Chiarle and V. Poli, Front. Biosci., 2008, 13, 6501 CrossRef CAS PubMed.
- M. Ernst and B. J. Jenkins, Trends Genet., 2004, 20, 23 CrossRef CAS PubMed.
- P. C. Heinrich, I. Behrmann, G. Müller-Newen, F. Schaper and L. Graeve, Biochem. J., 1998, 334, 297 CAS.
- D. E. Levy and J. E. Darnell, Nat. Rev. Mol. Cell Biol., 2002, 3, 651 CrossRef CAS PubMed.
- H. Song, R. Wang, S. Wang and J. Lin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4700 CrossRef CAS PubMed.
- D. Bhasin, K. Cisek, T. Pandharkar, N. Regan, C. Li, B. Pandit, J. Lin and P.-K. Li, Bioorg. Med. Chem. Lett., 2008, 18, 391 CrossRef CAS PubMed.
- D. Bhasin, J. P. Etter, S. N. Chettiar, M. Mok and P.-K. Li, Bioorg. Med. Chem. Lett., 2013, 23, 6864 CrossRef CAS PubMed.
- E. J. S. Salustiano, C. D. Netto, R. F. Fernandes, A. J. M. da Silva, T. S. Bacelar, C. P. Castro, C. D. Buarque, R. C. Maia, V. M. Rumjanek and P. R. R. Costa, Invest. New Drugs, 2010, 28, 139 CrossRef CAS PubMed.
- S. K. Chaudhuri, L. Huang, F. Fullas, D. M. Brown, M. C. Wani and M. E. Wall, J. Nat. Prod., 1995, 58, 1966 CrossRef CAS.
- A. J. M. da Silva, C. D. Netto and P. R. R. Costa, J. Braz. Chem. Soc., 2004, 15, 979 CrossRef CAS PubMed.
- C. D. Netto, E. S. Santos, C. P. Castro, A. J. da Silva, V. M. Rumjanek and P. R. Costa, Eur. J. Med. Chem., 2008 and 2009, 44, 920 Search PubMed.
- H. Pelicano, D. Carney and P. Huang, Drug Resist. Updates, 2004, 7, 97 CrossRef CAS PubMed.
- A. T. Y. Lau, Y. Wang and J. Chiu, J. Cell. Biochem., 2008, 104, 657 CrossRef CAS PubMed.
- J. Wang and J. Yi, Cancer Biol. Ther., 2008, 7, 1875 CrossRef CAS.
- S. Shaaban, R. Diestel, B. Hinkelmann, Y. Muthukumar, R. P. Verma, F. Sasse and C. Jacob, Eur. J. Med. Chem., 2012, 58, 192 CrossRef CAS PubMed.
- E. Fayard, L. A. Tintignac, A. Baudry and B. A. Hemmings, J. Cell Sci., 2005, 118, 5675 CrossRef CAS PubMed.
- (a) I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489 CrossRef CAS PubMed; (b) A. Bellacosa, C. C. Kumar, A. DiCristofano and J. R. Testa, Adv. Cancer Res., 2005, 94, 29 CrossRef CAS.
- J. LoPiccolo, C. A. Granville, J. J. Gills and P. A. Dennis, Anti-Cancer Drugs, 2007, 18, 861 CAS.
- (a) S. Takano, K. Hasuda, A. Ito, Y. Koide, F. Ishii, I. Haneda, S. Chihara and Y. Koyama, J. Antibiot., 1976, 29, 765 CrossRef CAS; (b) N. Tanaka, T. Okabe, F. Isono, M. Kashiwagi, K. Nomoto, M. Takahashi, A. Shimazu and T. Nishimura, J. Antibiot., 1985, 38, 1327 CrossRef CAS; (c) T. Okabe, K. Nomoto, H. Funabashi, S. Okuda, H. Suzuki and N. Tanaka, J. Antibiot., 1985, 38, 1333 CrossRef CAS.
- (a) M. E. Bergy, J. Antibiot., 1968, 21, 454 CrossRef CAS; (b) H. Hoeksema and W. C. Krueger, J. Antibiot., 1976, 29, 704 CrossRef CAS.
- E. J. Salaski, G. Krishnamurthy, W.-D. Ding, K. Yu, S. S. Insaf, C. Eid, J. Shim, J. I. Levin, K. Tabei, L. Toral-Barza, W.-G. Zhang, L. A. McDonald, E. Honores, C. Hanna, A. Yamashita, B. Johnson, Z. Li, L. Laakso, D. Powell and T. S. Mansour, J. Med. Chem., 2009, 52, 2181 CrossRef CAS PubMed.
- S. Tanaka, M. Tajima, M. Tsukada and M. Tabata, J. Nat. Prod., 1986, 49, 466 CrossRef CAS.
- X. Chen, L. Yang, J. J. Oppenheim and O. M. Z. Howard, Phytother. Res., 2002, 16, 199 CrossRef CAS PubMed.
- V. P. Papageorgiou, A. N. Assimopoulou, E. A. Couladouros, D. Hepworth and K. C. Nicolaou, Angew. Chem., Int. Ed., 1999, 38, 270 CrossRef.
- B.-Z. Ahn, K.-U. Baik, G.-R. Kweon, K. Lim and B.-D. Hwang, J. Med. Chem., 1995, 38, 1044 CrossRef CAS.
- Z. F. Plyta, T. Li, V. P. Papageorgiou, A. S. Mellidis, A. N. Assimopoulou, E. N. Pitsinos and E. A. Couladouros, Bioorg. Med. Chem. Lett., 1998, 8, 3385 CrossRef CAS.
- N. Fujii, Y. Yamashita, Y. Arima, M. Nagashima and H. Nakano, Antimicrob. Agents Chemother., 1992, 36, 2589 CrossRef CAS.
- F. Yang, Y. Chen, W. Duan, C. Zhang, H. Zhu and J. Ding, Int. J. Cancer, 2006, 119, 1184 CrossRef CAS PubMed.
- W. Wang, M. Dai, C. Zhu, J. Zhang, L. Lin, J. Ding and W. Duan, Bioorg. Med. Chem. Lett., 2009, 19, 735 CrossRef CAS PubMed.
- Clinical Trial of ARQ 761 in Advanced Solid Tumors, https://clinicaltrials.gov/ct2/show/NCT01502800, accessed 22 January 2015.
| This journal is © The Royal Society of Chemistry 2015 |