
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic studies of visible light-induced CO release from a 3-hydroxybenzo[g]quinolone†
Marina Popovaa,
Tomasz Borowski *b,
Josiah G. D. Elsberga,
C. Taylor Dedericha and
Lisa M. Berreau*a
*b,
Josiah G. D. Elsberga,
C. Taylor Dedericha and
Lisa M. Berreau*a
aDepartment of Chemistry and Biochemistry, Utah State University, 0300 Old Main Hill, Logan, UT 84322-0300, USA. E-mail: lisa.berreau@usu.edu
bJerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Science, Niezapominajek 8, Krakow 30-239, Poland
First published on 20th January 2022
Abstract
Organic compounds that can be triggered using light to release CO in biological environments are of significant current interest to probe the role of CO in biology and as potential therapeutics. We recently reported that a 3-hydroxybenzo[g]quinolone (5) can be used as a CO delivery molecule to produce anticancer and potent anti-inflammatory effects. Herein we report mechanistic studies of the visible light-induced CO release reaction of this compound. In wet CH3CN under aerobic conditions, 5 releases 0.90(2) equivalents of CO upon illumination with visible light (419 nm) to give a single depside product. Performing the same reaction under an 18O2 atmosphere results in quantitative incorporation of two labeled oxygen atoms in the depside product. Monitoring via 1H NMR and UV-vis during the illumination of 5 in CH3CN using 419 nm light revealed the substoichiometric formation of a diketone (6) in the reaction mixture. H2O2 formation was detected in the same reaction mixtures. DFT studies indicate that upon light absorption an efficient pathway exists for the formation of a triplet excited state species (5b) that can undergo reaction with 3O2 resulting in CO release. DFT investigations also provide insight into diketone (6) and H2O2 formation and subsequent reactivity. The presence of water and exposure to visible light play an important role in lowering activation barriers in the reaction between 6 and H2O2 to give CO. Overall, two reaction pathways have been identified for CO release from a 3-hydroxybenzo[g]quinolone.
Introduction
Organic carbon monoxide (CO)-releasing molecules are of current interest for use in evaluating the roles of CO in biological systems and as potential therapeutics.1,2 Of particular interest for biomedical applications are compounds that can be triggered so as to enable spatial and temporal control of CO release.3,4 Toward meeting this need, five general types of metal-free, visible-light induced CO releasing molecules have been reported (Scheme 1(a–e)).5–9 As a step toward advancing frameworks that are suitable for further applications (e.g., for subcellular targeting), it is important to have a detailed mechanistic understanding of the CO release reaction of each motif.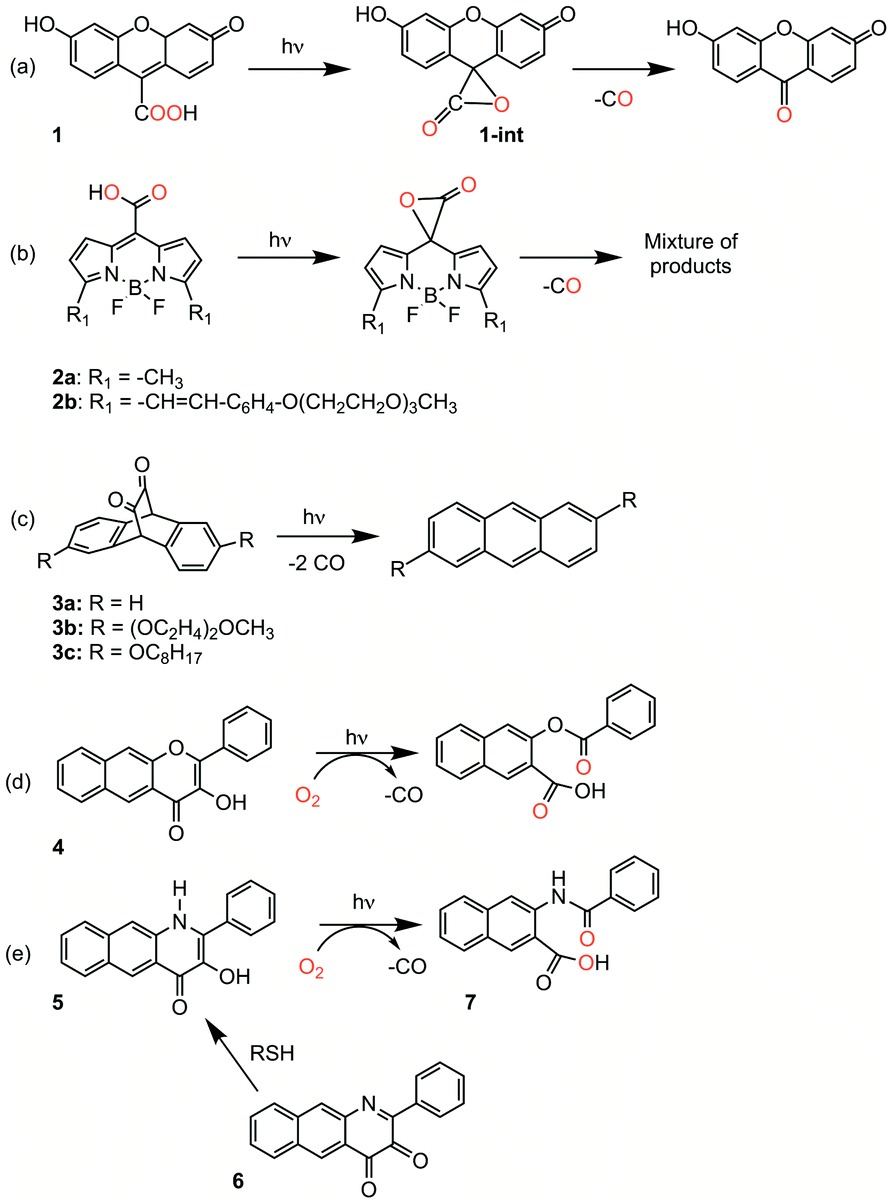 | ||
| Scheme 1 Metal-free CO releasing molecules. Red coloration indicates the results of 18O labelling studies. | ||
Compound 1 (Scheme 1(a)) exhibits visible light-induced CO release at pH = 5.7–7.4 in its monoanionic form to give a single organic product.5 The overall yield for CO release from 1 has not been reported. 18O-labeling studies provide evidence that the carboxyl group is the source of the CO oxygen atom (C18O). A CO release reaction pathway involving a putative α-lactone intermediate (Scheme 1(a), 1-int) has been proposed for 1. Notably, the CO release reaction of 1 is not affected by the presence of O2, suggesting that the photoinduced pathway either does not involve a triplet state, or if present, its lifetime must be very short.
CO release from the BODIPY derivatives 2a and 2b (Scheme 1(b)) also occurs most readily from the monoanionic form, which is present at physiological pH.6 The overall yield of CO from 2b is reduced from 87% to 45% in the presence of oxygen, suggesting the involvement of a triplet excited state in the reaction pathway. Transient absorption spectral features of 2a and heavy atom-substituted analogs provide evidence that an excited state singlet is initially formed that undergoes rather inefficient intersystem crossing (ISC) to give a triplet state. From this triplet state, photoinduced electron transfer (PET) from the carboxylate to the BODIPY is proposed to give a triplet diradical species which undergoes ISC to give an α-lactone on the singlet energy surface. Release of CO from the α-lactone then occurs via non-photochemical fragmentation. Evidence for the proposed α-lactone intermediate in this system was put forth based on DFT studies involving a truncated analog.
The 1,2-diketone-type compounds 3a–3c (Scheme 1(c)) undergo fast double decarbonylation, but only under conditions wherein water is excluded, as the formation of carbonyl hydrates prevents CO release.7 The CO release reaction from 1,2-diketones such as 3a–3c occurs via Strating-Zwanenburg photodecarbonylation.10,11 This reaction occurs from both singlet and triplet manifolds via an initial step that involves either bond cleavage between a carbonyl and a methylene carbon, or homolytic cleavage between the carbonyl units, to give a diradical species. It is unclear whether CO release then occurs in a stepwise or concerted fashion, or whether 2 eq. CO or the CO dimer are initially released.11
Our recent research has focused on investigations of the 3-hydroxybenzo[g]flavone framework 4 (Scheme 1(d)) as a light-triggered CO-releasing molecule.4,8,12,13 This compound exhibits quantitative CO release upon illumination with visible light (λill = 419 nm). An 18O-labeling study showed quantitative incorporation of both atoms of 18O2 into the depside product. Klán and coworkers performed additional mechanistic investigations of the O2-dependent CO release reactivity of 4.14 They identified three visible-light induced reaction pathways leading to CO release: (1) reaction of a neutral triplet excited state tautomer of 4 with 3O2; (2) reaction of the ground state conjugate base of 4 with 1O2, which is produced via self-sensitization; and (3) inefficient photorearrangements of the triplet excited states of both 4 and its monoanion.
We recently reported light-induced CO delivery using the 3-hydroxybenzo[g]quinolone 5 (Scheme 1(e)).9 A key feature of 5 is its tight binding to bovine serum albumin protein (Ka = 2.9 × 106) which is ∼900 fold higher than that of 4 and enables the use of a protein:5 complex for CO delivery to cells. Additionally, an oxidized prodrug form of this molecule (6, Scheme 1(e)), is reduced by thiols, providing an approach toward activation in the reducing environment of some cancer cells. Use of the protein:5 complex produced cytotoxicity values among the lowest reported to date for CO delivery to cancer cells. Additionally, the protein:5 complex is the first CO delivery molecule to be reported to produce significant anti-inflammatory effects at nanomolar concentrations. Based on these results, and its non-toxic properties in cells, 5 is particularly attractive for further development as a light-triggered CO releasing molecule for biological and biomedical applications.
Herein we report combined experimental and computational studies of the mechanistic pathway of the visible light-induced CO release reaction of 5. Interestingly, these studies revealed two pathways for CO release, with one pathway involving initial oxidation of the quinolone framework to 6 prior to CO release.
Materials and methods
Compounds 5 and 6 have been synthesized according to literature procedures.9,15 All other reagents were purchased from common vendors and used as received unless otherwise noted.Physical methods
1H NMR spectra were collected using a Bruker Avance III HD Ascend-500 spectrometer. UV-vis spectra were recorded at ambient temperature using a Cary 50Bio or a Hewlett-Packard 8453A diode array spectrophotometer. Fluorescence emission spectra were recorded using a Shimadzu RF-530XPC spectrometer using 1.0 cm quartz cells. The excitation and emission slit widths were set at 3.0 nm. Mass spectral data were collected at the Mass Spectrometry Facility, University of California, Riverside. CO gas was detected and quantified using an Agilent 3000A micro gas chromatograph with molecular sieve and Plot U columns and a thermal conductivity detector. H2O2 formation in the conversion of 5 to 6 was detected using commercial H2O2 test strips.Computational methods
All computations were performed employing the ωB97XD16 exchange–correlation functional and the Def2-TZVP17 basis set. The only exception is T1TSOO, which was fully optimized with the def2-SVP basis set, but despite many various attempts we were not able to re-optimize it with the def2-TZVP basis set. Hence, the activation energy connected with this transition structure was computed with the def2-TZVP basis using geometries of T1INTC2P and T1TSOO optimized with the def2-SVP basis set. Lowest lying triplet and broken symmetry singlet spin states were described with the unrestricted formalism. Excited singlet and triplet states were described with the TD-DFT18 method using a closed-shell ground state singlet as a reference. Minimum energy crossing points (MECP) between various potential energy surfaces (PESs) were optimized with the use of the meta-program crossing obtained courtesy of Prof. J. Harvey.19 The polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM) was used in all calculations to model the solvent (acetonitrile). All electronic structure computations were done with the Gaussian 16 program.20Treatment of 6 with H2O2 (30%) in acetonitrile
H2O2 (30%, 5 μl) was added to a solution of 6 (1.5 × 10−4 M, 3 ml) in acetonitrile. The absorption spectral features of the solution were monitored in the absence or presence of illumination (419 nm). The same reaction under illumination conditions was also examined using 1H NMR.CO quantification from the reaction of 6 with H2O2
The diketone 6 (10−5 mol) was dissolved in acetonitrile (5 ml) in a 50 ml round bottom flask. Excess H2O2 (30%, 0.1 ml) was added and the flask was then sealed with a septum. The solution was stirred for 48 h with or without illumination at 419 nm. A 10 ml sample of the headspace gas was used to determine the CO generated using gas chromatography. Approximately 0.6 eq. (illuminated) and 0.3 eq. of CO (non-illuminated), respectively, were produced. As a control for CO quantification under these conditions, light induced CO release from 4 produces 0.99 eq.Results
Spectroscopic studies of the CO release reaction of 5
Illumination of a wet CH3CN solution of 5 under aerobic conditions results in the formation of 7 (Scheme 1(e)) and the release of 0.90(2) eq. of CO as determined using 1H NMR and gas chromatography. To gain further insight into this reaction, a solution of 5 in CD3CN was illuminated using 419 nm light at ∼35 °C and was monitored periodically using 1H NMR as shown in Fig. 1. After 15 min of illumination (Fig. 1(b)), in addition to starting material, two species are present, the CO release product 7 and the diketone 6 (Scheme 1(e)).15 The formation of the diketone 6 indicates that upon illumination at least some of the reactant (5) undergoes two-electron oxidation with the release of H2O2. The formation of H2O2 was confirmed by examining the reaction mixture using H2O2 test strips (Fig. S1†) which turned blue indicating formation of a peroxide species. We note that an analogous experiment involving illumination of the extended flavonol 4 did not produce the change in the peroxide strip color (Fig. S1†), indicating the unique formation of H2O2 in the illumination of 5. Continuing with the reaction of 5, after 30 min (Fig. 1(c)), the 1H NMR resonances associated with the starting material 5 are gone, and only the resonances for 6 and the carboxylic acid-containing CO release product 7 remain. After 2 h the integrated intensity for the signals for 6 diminished further while those of 7 increased suggesting a second reaction pathway wherein 6 may react with H2O2 to give 7. Notably, use of 18O2 in the reaction results in quantitative incorporation of two labelled oxygen atoms in 7 (Fig. S2†). This provides evidence that the reaction pathway involving 6 results in the incorporation of labelled oxygen atoms derived from O2. Illumination of 5 using 465 nm lamps produces similar results (Fig. S3†). A mixture of 6 and 7 is formed in the first few hours of illumination followed by the disappearance of the resonances associated with 6 at long illumination times (24 h) to give resonances only associated with 7.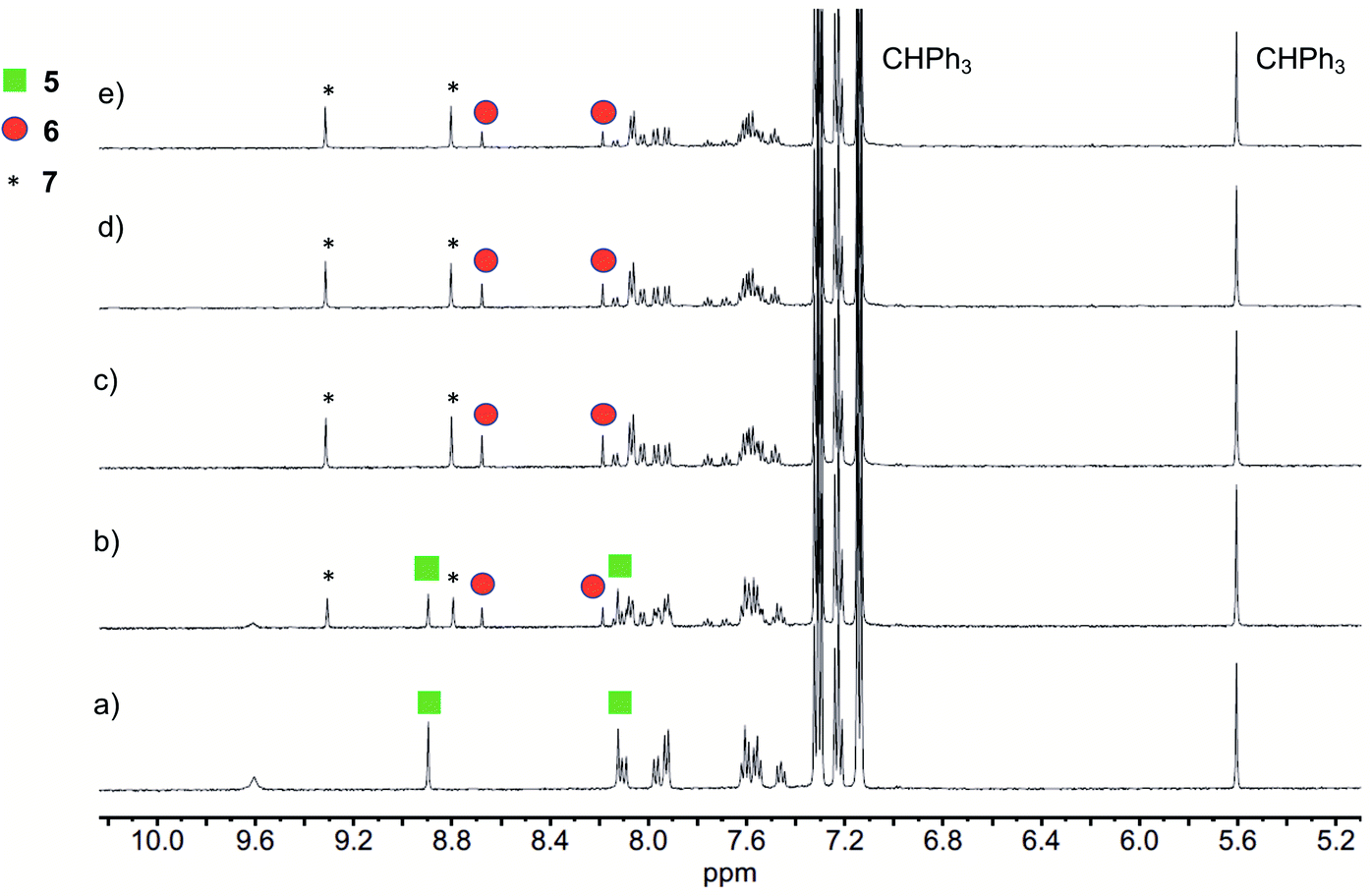 | ||
| Fig. 1 1H NMR features of a solution of 5 in wet CD3CN with illumination (419 nm) at ∼35 °C under O2: (a) Prior to illumination; (b) 15 min; (c) 30 min; (d) 1 h and (e) 2 h. The combined integrations of the indicated resonances of 5, 6 and 7 are consistent versus the internal standard CHPh3 over the course of illumination. | ||
The visible light-induced CO release reaction of 5 (at 419 nm) in wet acetonitrile was also monitored using UV-vis absorption spectroscopy. As shown in Fig. 2, loss of the absorption features of 5 over the first ∼12 min of illumination coincides with the appearance of a new broad absorption band at ∼480 nm, which is consistent with the formation of a substoichiometric amount of the diketone 6 based on the individual absorption spectral features of the compounds.9 Two isosbestic points at ∼380 and 465 nm suggest clean conversion of 5 to 6. After ∼20 min of growth of the band at ∼480 nm, this feature begins to decay.
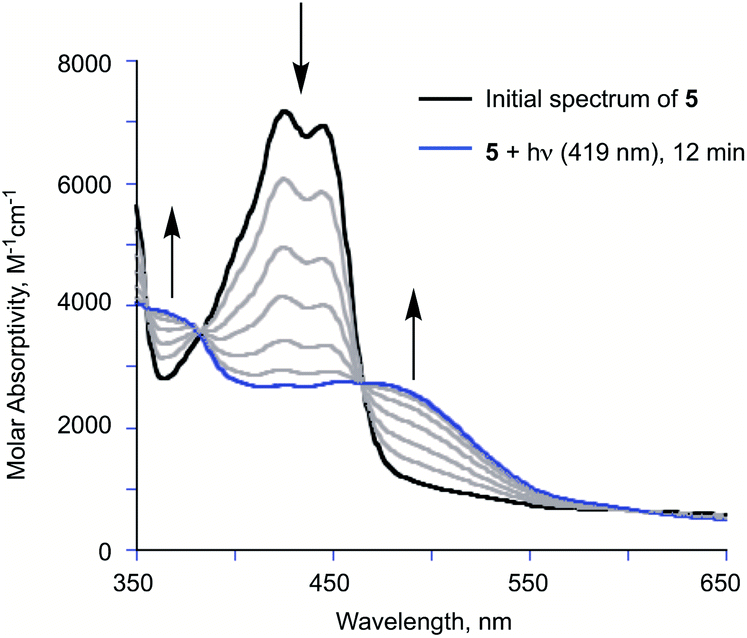 | ||
| Fig. 2 Changes in the absorption spectrum of 5 in acetonitrile upon illumination with 419 nm light. | ||
Reactivity of 6 with H2O2
To evaluate the reactivity of 6 with H2O2, independent experiments were performed. Treatment of the diketone 6 dissolved in acetonitrile with excess aqueous H2O2 (30%) under ambient conditions results in the disappearance of its absorption band at 480 nm over 8 h in absence of illumination and within 45 min with illumination at 419 nm (Fig. 3(a) and (b), respectively). Compound 7 is formed in the illuminated sample as determined by 1H NMR (Fig. S4†). CO gas is generated under both thermal and illuminated conditions (∼0.3 and 0.6 eq., respectively). The low amount of CO generated under these conditions may be due to hydration of the diketone or other water-dependent decomposition pathways as 6 undergoes spectroscopic changes in the presence of added D2O to produce unidentified species (Fig. S5†).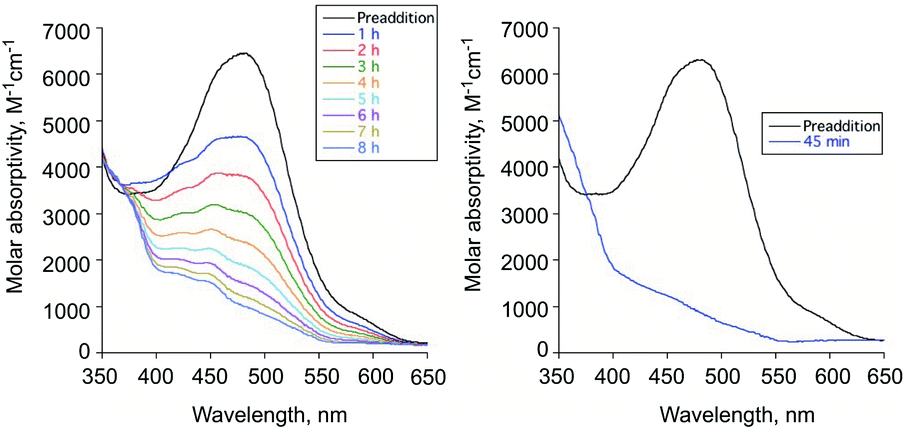 | ||
| Fig. 3 Absorption spectral features for 6 prior to and following addition of H2O2 (30%) under thermal (left) and illumination conditions (right). The decay of the absorption band for 6 occurs over 8 h under thermal conditions and in under an hour under illumination (419 nm). | ||
Based on these investigations, we hypothesized that the light-driven CO release reactivity of 5 to produce 7 could be proceeding via at least two light-driven pathways. The primary pathway could be similar to those of 414 and 3-hydroxyflavone.21,22 These previously reported pathways involve excited state proton-transfer tautomer triplet state reactivity with triplet oxygen. The reaction of 5 with O2 that leads to formation of diketone 6 and H2O2, which then appears to subsequently undergo reaction leading to CO release, has not been previously reported for a quinolone compound. To gain further insight into the visible-light induced CO release pathways involving 5, we turned to DFT calculations.
Computational studies; low-lying excited states of 5
TD-DFT results indicate that the first excited singlet state and two lowest lying triplet states of 5 originate from one electron excitations within four frontier molecular orbitals (HOMO-1, HOMO, LUMO and LUMO+1), whose contours are shown in Fig. 4. In the S1 and T1 states the HOMO → LUMO excitation is the leading one, whereas in the T2 state the two major excitations are HOMO-1 → LUMO and HOMO → LUMO+1.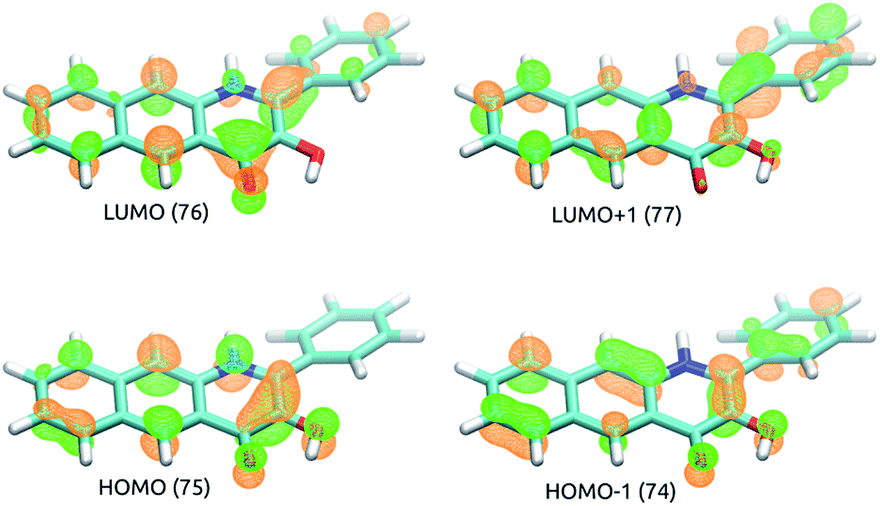 | ||
| Fig. 4 Contours of frontier molecular orbitals of 5. | ||
Tautomeric forms with a proton located on the C3- or C4-bound oxygen atoms, which are labelled with 5 and 5a, respectively, differ in relative stability depending on the nature of the excited state. For the S1 and T1 states the S15a and T15a forms are the most stable, whereas for the S0 and T2 states the S05 and T25 forms are the lower energy ones (Fig. 5). Adiabatic barriers computed for the proton transfer steps (5 → TSPT → 5a) are rather low, i.e. amount to 14.2, 5.0, 3.0 and 9.9 kcal mol−1 for S0, S1, T1 and T2 potential energy surfaces (PESs), respectively. For the S15 species one can envisage several potential channels for its decay. First, an internal conversion process, i.e. S1 → S0, may lead to de-excitation and dissipation of the excess energy. Second, an intersystem crossing may lead to the triplet manifold. Interestingly, a direct transition from S1 to T1 is not an easy process. For the tautomer 5 we could not locate a minimum energy crossing point (MECP) between S15 and T15, most likely due to the fact that S1 and T1 have PESs of very similar shape. For the tautomer 5a the optimized crossing point (S1-T1MECP2) is quite high in energy, i.e. it is 16.4 kcal mol−1 higher than S15a (Fig. 5), which suggests this reaction channel is rather unlikely. On the other hand, MECPs between the S1 and T2 states have energies only slightly higher than the two minima on the S1 PES, i.e. by 3.9 and 0.3 kcal mol−1 for S1-T2MECP1 and S1-T2MECP2, respectively, and hence, they are the most likely gates to the triplet manifold. Importantly, the T2 state lies lower in energy than S1 and it features a low lying MECP with the T1 state, T2-T1MECP1, which lies only 1.7 kcal mol−1 higher than T25. Hence, it is suggested that the lowest lying triplet state is populated through a sequence of steps encompassing proton transfer on the S1 PES (S15 → S15a), intersystem crossing to the T2 state (S15a → T25a), another proton transfer step (T25a → T25) and finally an internal conversion from T2 to T1 (T25 → T15). The T15a form, easily formed via proton transfer T15 → T15a, is the lowest energy excited state species which is supposed to live long enough to have a chance to react with the triplet dioxygen, as described in the following subsections. Indeed, the optimized minimum energy crossing point between T1 and S0, i.e. T1-S0MECP2, lies 10.8 kcal mol−1 above the T15a minimum, which should lend this triplet species substantial lifetime.
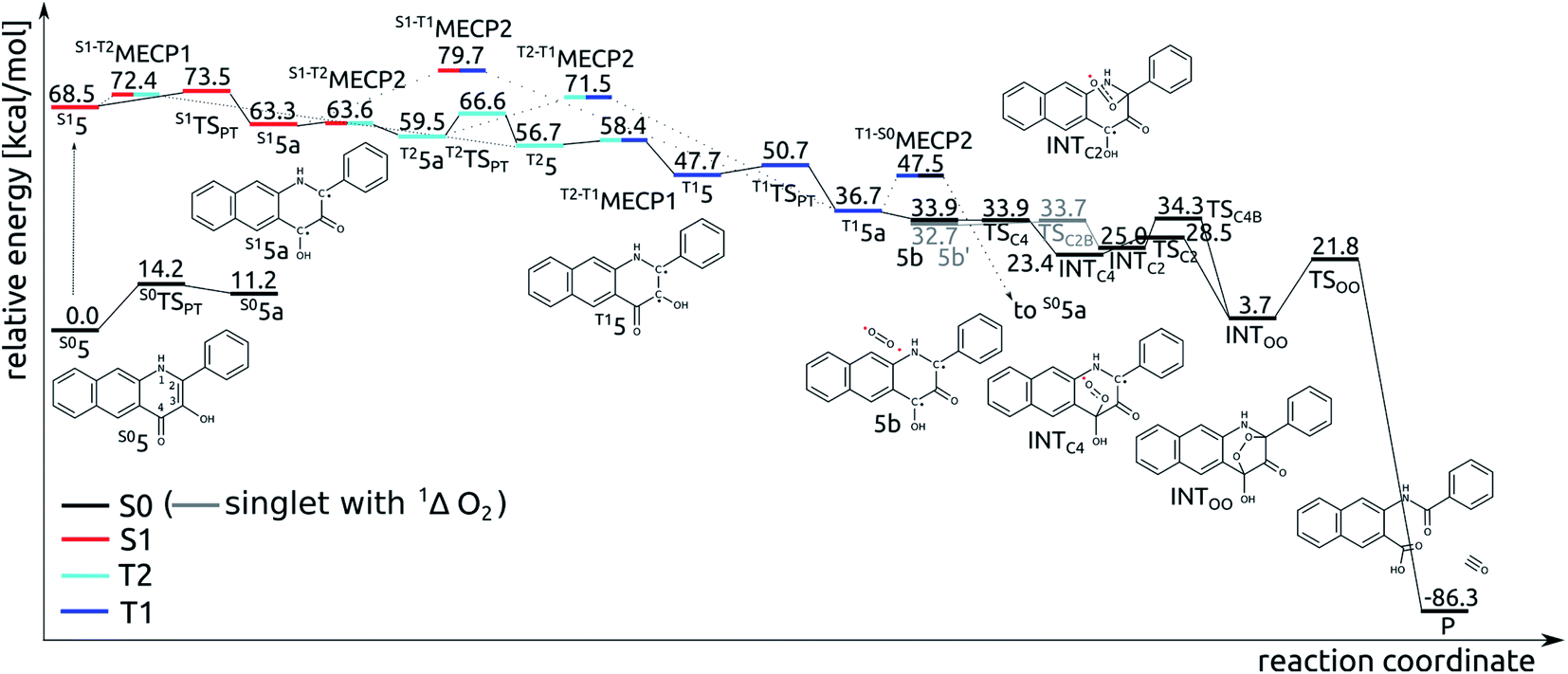 | ||
| Fig. 5 Relative energy of species in the calculated light-induced reaction pathway of 5 with O2. | ||
Addition of O2 to T15a
The T15a species forms an encounter complex with triplet dioxygen and two electronic states are almost degenerate for this complex. First, the unpaired electrons of the two triplet entities are coupled to a (broken symmetry) singlet spin state (5b; for spin density plots see Fig. 6). Second, the organic molecule returns to its ground closed-shell singlet state while dioxygen is excited to the 1Δg singlet state (5b′). The two states are computed to be only 1.2 kcal mol−1 apart and both are very reactive towards C–O bond formation. Irrespective of the electronic state of the encounter complex, O2 addition to the organic molecule is a stepwise process, whereby the two C–O bonds are formed in two successive elementary reactions. The computed barriers for binding of O2 at C2 (5b → INTC2) is only 1.0 kcal mol−1, whereas the barrier for attack of triplet O2 on C4 (5b → INTC4) vanishes completely. Spin density maps computed for resulting intermediates (INTC2 and INTC4; Fig. 6) indicate that the binding process engages one pair of electrons with anti-parallel spins, whereas the other two electrons remain localized on O2−˙ and the ring. The latter couple to a bonding pair when the second C–O bond forms yielding the endoperoxide intermediate INTOO. The decay of INTOO with the release of CO is a symmetry allowed reaction, which involves concerted and synchronous cleavage of two C–C and one O–O bonds.23 This reaction pathway is similar to those reported for 414 and 3-hydroxyflavone.21,22 | ||
| Fig. 6 Spin density maps of intermediates involved in C–O bond formation in the reaction of 5 with O2. | ||
Reaction of T15a with O2 leading to 6 and H2O2
The experimental results described above strongly suggest that in the photoreaction of 5 with O2 the diketone 6 and H2O2 are also formed. Insights into how this process might proceed were obtained via DFT calculations, with the results summarized in Fig. 7. First, the singlet encounter complex between T15a and 3O2 with O2 bound close to C4–OH (5c) forms. Its energy is only slightly higher (by 0.3 kcal mol−1) than for 5b. Second, formal hydrogen atom transfer from the phenolic oxygen to O2 leads to a significantly more stable diradical species INTHOO. The detailed mechanism of this step is most likely beyond the reach of DFT single determinant methods. Multiple attempts to locate a TS on the open-shell singlet PES failed, as the triplet converges to closed-shell S05a and 3O2, whereas the reaction might proceed on the open-shell triplet PES. Then, the peroxo radical migrates and forms H-bonding contacts with N1–H and the phenyl substituent. The INTHOO to INTOOH step is endoenergetic by 4.8 kcal mol−1. The transition state for the direct hydrogen atom transfer from N1 to OOH was optimized (S0TSNH), however its energy is very high (41.6 kcal mol−1 higher than for INTHOO), which renders this process very unlikely. However, a charge-transfer excitation within the singlet manifold leads to the closed-shell singlet species INTOOH*, which relaxes with no additional barriers directly to species 6a.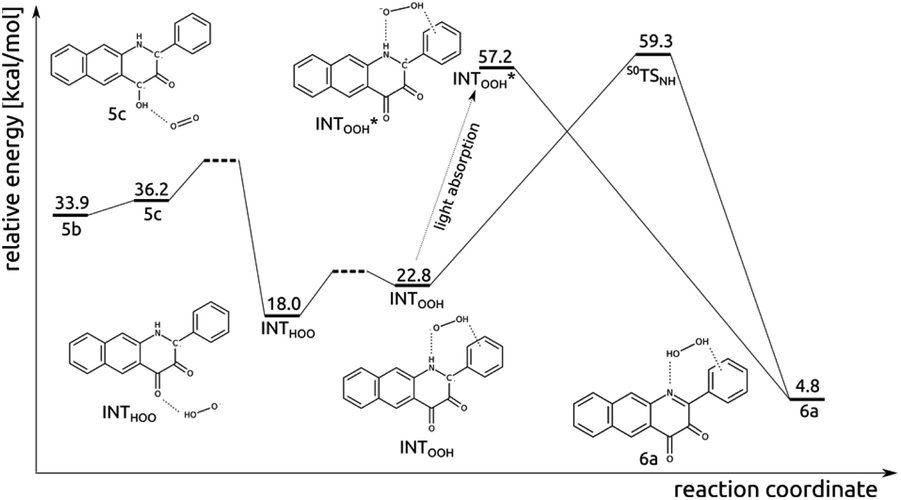 | ||
| Fig. 7 Relative energy of species in the light-induced reaction pathway of 5 with O2 to form 6a. | ||
Thermal reaction between diketone (6) and H2O2
Compound 6 and H2O2 form an encounter complex 6a that is stabilized by a hydrogen bond between H2O2 and the C4-bound oxygen. Two reaction channels lead from 6a to INTOO, one through INTC2P and the second though INTC4P (Fig. 8). To form either of them a proton needs to be transferred from the oxygen atom, which forms a bond with the respective carbon atom (C2 or C4), to N1 or O bound to C4. Simultaneous proton transfer and formation of the C–O bond proceed through a strained transition structure featuring a four-membered ring. In consequence, the computed barriers are very high, i.e. 37.3 and 38.8 kcal mol−1 for 6a → TSC2P → INTC2P and 6a → TSC4P → INTC4P, respectively. When a single water molecule acts as a catalyst and mediates the proton transfer, then the barriers drop significantly, to 28.6 and 26.1 for TSC2PWB and TSC4PW, respectively (Fig. 8). Similarly, the subsequent step whereby the second C–O bond is formed was modelled with a single water molecule as a catalyst. As it is hard to estimate how many water molecules might be available in the vicinity of the encounter complex 6a, the barriers computed here for the two channels leading from 6a to INTOO are to be considered only as qualitative, as they merely show that water can facilitate the process.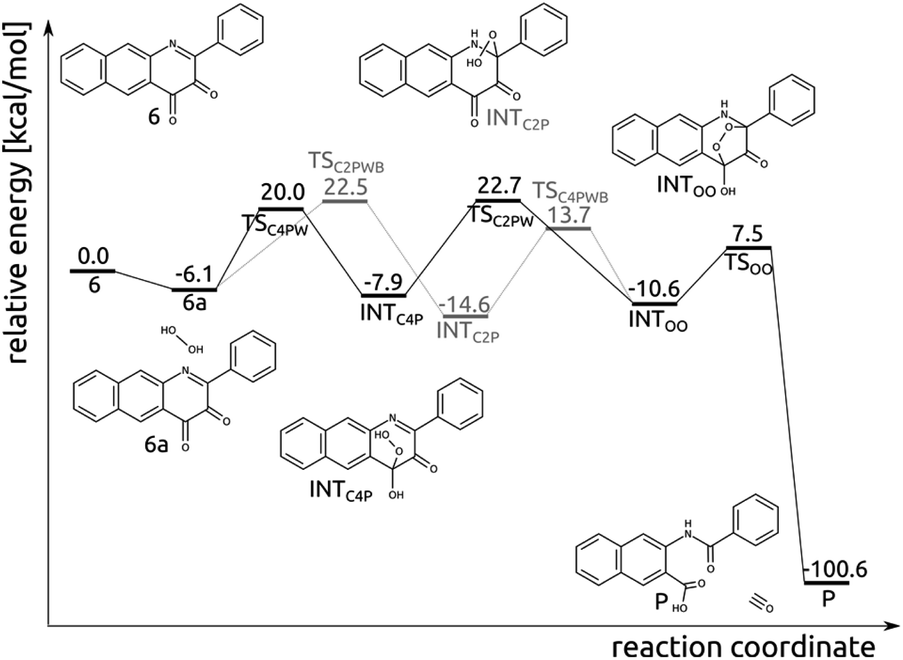 | ||
| Fig. 8 Relative energy of species in the thermal reaction pathway of 6 with H2O2. | ||
Photo-induced reaction between diketone (6) and H2O2
Excitation of the encounter complex 6a to the lowest lying triplet state (T16a), via singlet excitation and subsequent intersystem crossing, opens an efficient route for hydrogen transfer from H2O2 to N1 and subsequent formation of the C2–OOH bond (Fig. 9). In the triplet state N1 has significant radical character, which renders the H-atom transfer process to be fairly easy; T1TSNH is coupled with a barrier of 10.9 kcal mol−1. In the resulting intermediate INTOOH unpaired electrons are located on HOO and semiquinone radicals and triplet and open-shell singlet spin states are degenerate. Coupling of these radicals in the singlet state requires overcoming a tiny barrier and leads to the closed-shell intermediate INTC2P, encountered already in the thermal reaction between 6 and H2O2. Excitation of INTC2P to the lowest lying triplet state provides a feasible route for O–O bond cleavage and subsequent release of CO. In the triplet spin state, the aromatic ring can quite easily provide the OOH moiety with one electron, i.e. with barrier of 23.4 kcal mol−1 connected with T1TSOO, which aids O–OH bond cleavage. In the resulting intermediate INTOH the two unpaired electrons are again considerably separated, and triplet and singlet spin state are energetically degenerate. In the singlet state attack of the HO− group on C4 is very easy (S1TSC4O, barrier of 2.2 kcal mol−1) and leads directly to the depside product and CO.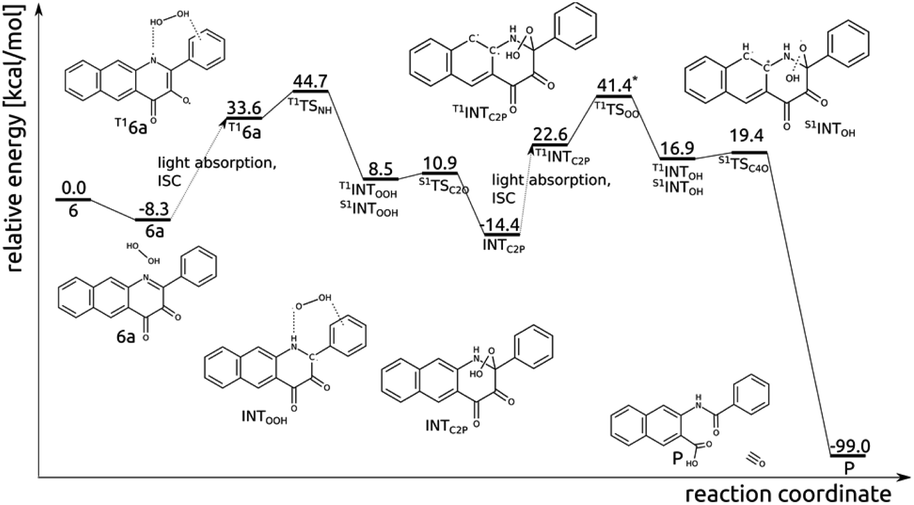 | ||
| Fig. 9 Relative energy of species in the visible light-induced reaction pathway of 6 with H2O2. *The relative energy with respect to T1INTC2P was computed for geometries optimized with the def2-SVP basis set. | ||
Discussion and conclusions
The structurally similar 3-hydroxyflavone (3-HflH) and 3-hydroxyquinolone (3-HqH) are substrates for enzyme-catalyzed dioxygenase-type CO release reactions in bacteria and fungi.24,25 Compounds of this type have also been shown to undergo O2-dependent, UV-light-induced CO release. For example, 3-HflH undergoes incorporation of both atoms of O2 and expulsion of CO in the presence of a photosensitizer (e.g., Rose Bengal).26 In this reaction, 1O2 reacts with the ground state form of 3-HflH in a [3 + 2] cycloaddition to form a cyclic peroxide from which cleavage of two carbon–carbon bonds results CO release. In the absence of a photosensitizer, UV-light illumination has been proposed to result in the formation of an excited state triplet tautomeric form of 3-HflH, which then directly reacts with 3O2.21We are investigating the visible light induced CO release properties of structural analogs of 3HflH and 3HqH with extended conjugation (Scheme 1(d and e)).4,8,9,12,13 Notably, the 3-hydroxybenzo[g]quinolone 5 (Scheme 1(e)) binds tightly to bovine serum albumin which enables protein delivery of this CO releasing molecule to cancer cells. Visible light-induced CO release from the 5:albumin complex produces anti-cancer and potent anti-inflammatory effects.9
In this contribution we report spectroscopic and computational studies of the visible light-induced CO release reaction of 5 under aerobic conditions. Notably, in addition to the observed formation of the CO release depside product 7, the diketone 6 is identifiable in the reaction mixture. Over the course of illumination, 6 disappears from the reaction mixture. Independent studies show that 6 undergoes reaction with aqueous H2O2 to give 7 and CO. DFT studies of the direct reaction of 5 with O2 provide evidence for a light-driven CO release pathway (Path A, Scheme 2) that is akin to that previously described for 3-HFlH derivatives.21,22 Formation of an excited triplet tautomer enables reactivity with 3O2 resulting in C–C and C–O bond cleavage yielding CO and depside.
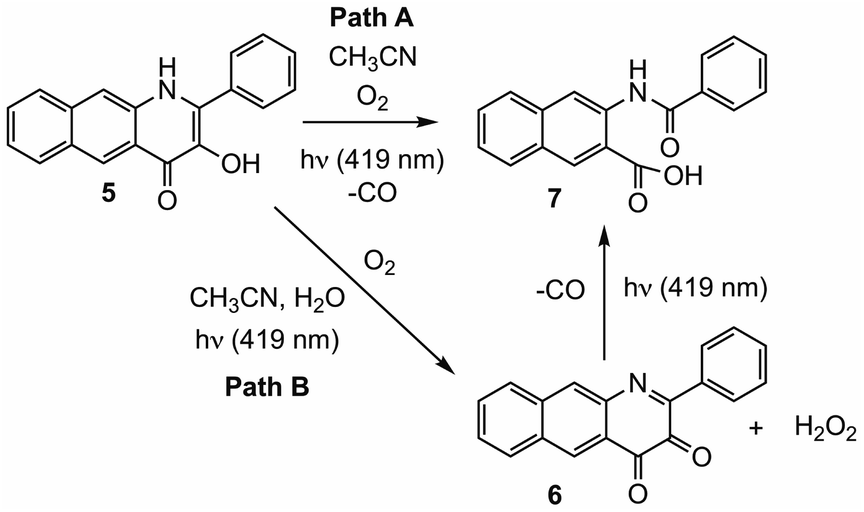 | ||
| Scheme 2 Reaction pathways leading to CO release from 5 under visible light illumination. | ||
The formation of 6 and H2O2 in the light-induced reaction of 5 is supported by DFT studies which provide initial insight into how these species can be generated (Fig. 7; Path B Scheme 2). Subsequent thermal reactivity between the diketone 6 and H2O2 appears to be feasible only in the presence of water, which lowers the activation barriers for C–O bond formation. The reaction between the diketone 6 and H2O2 to release CO is facilitated by excitation to the lowest triplet state. These combined results suggest that CO release via reaction of the diketone 6 with H2O2 likely proceeds via a light-driven process. The evidence provided herein suggests that this CO release reaction is less efficient, possibly due to decomposition pathways for the diketone involving water.
Quinolones are of current interest as anticancer compounds.27,28 The H2O2 induced CO release reactivity of 6 under illumination conditions is of interest in relation to the role that H2O2 is suggested to have in promoting tumor proliferation.29 Diketone molecules such as 6 may offer an approach toward reducing H2O2 levels in combination with CO delivery to produce anti-cancer effects. Our ongoing work is focused on advancing light-driven CO delivery using extended quinolones and their oxidized diketone forms, which are families of heterocyclic compounds that have received minimal attention to date.9,15
Data availability
Optimized structures were deposited in the ioChem-BD repository. See DOI: 10.19061/iochem-bd-4-32.Author contributions
MP, TB and LMB conceptualized the project; MP, TB, JGDE, and CTD performed the experimental investigations; MP, TB and LMB wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH (R15GM124596 to L. M. B.) and the NSF (CHE-1429195 for Brüker Avance III HD Ascend-500 spectrometer). This research was supported in part by PL-Grid Infrastructure. Computations were performed in the AGH Cyfronet Supercomputer Centre. T. B. acknowledges partial financial support of the project by the statutory research fund of ICSC PAS.References
- T. Slanina and P. Šebej, Photochem. Photobiol. Sci., 2018, 17, 692–710 CrossRef CAS PubMed.
- N. Abeyrathna, K. Washington, C. Bashur and Y. Liao, Org. Biomol. Chem., 2017, 15, 8692–8699 RSC.
- M. Kourti, W. G. Jiang and J. Cai, Oxid. Med. Cell. Longevity, 2017, 9326454 Search PubMed.
- L. S. Lazarus, A. D. Benninghoff and L. M. Berreau, Acc. Chem. Res., 2020, 53, 2273–2285 CrossRef CAS PubMed.
- L. A. Antony, T. Slanina, P. Šebej, T. Šolomek and P. Klán, Org. Lett., 2013, 15, 4552–4555 CrossRef CAS PubMed.
- E. Palao, T. Slanina, L. Muchová, T. Šolomek, L. Vítek and P. Klán, J. Am. Chem. Soc., 2016, 138, 126–133 CrossRef CAS PubMed.
- P. Peng, C. Wang, Z. Shi, V. K. Johns, L. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Org. Biomol. Chem., 2013, 11, 6671–6674 RSC.
- S. N. Anderson, J. M. Richards, H. J. Esquer, A. D. Benninghoff, A. M. Arif and L. M. Berreau, ChemistryOpen, 2015, 4, 590–594 CrossRef CAS PubMed.
- M. Popova, L. S. Lazarus, S. Ayad, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2018, 140, 9721–9729 CrossRef CAS PubMed.
- J. Strating, B. Zwanenburg, A. Wagenaar and A. C. Udding, Tetrahedron Lett., 1969, 10, 125–128 CrossRef.
- R. Mondal, A. N. Okhrimenko, B. K. Shah and D. C. Neckers, J. Phys. Chem. B, 2008, 112, 11–15 CrossRef CAS PubMed.
- L. S. Lazarus, H. J. Esquer, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2017, 139, 9435–9438 CrossRef PubMed.
- M. Popova, T. Soboleva, A. M. Arif and L. M. Berreau, RSC Adv., 2017, 7, 21997–22007 RSC.
- M. Russo, P. Štacko, D. Nachtigallová and P. Klán, J. Org. Chem., 2020, 85, 3527–3537 CrossRef CAS PubMed.
- M. D. Bilokin, V. V. Shvadchak, D. A. Yushchenko, A. S. Klymchenko, G. Duportail, Y. Mely and V. G. Pivovarenko, Tetrahedron Lett., 2009, 50, 4714–4719 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95–99 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. FoxGaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. L. Studer, W. E. Brewer, M. L. Martinez and P.-T. Chou, J. Am. Chem. Soc., 1989, 111, 7643–7644 CrossRef CAS.
- Z. Szakács, M. Bojtár, L. Drahos, D. Hessz, M. Kállay, T. Vidóczy, I. Bitter and M. Kubinyi, Photochem. Photobiol. Sci., 2016, 15, 219–227 CrossRef PubMed.
- A. M. Miłaczewska, E. Kot, J. A. Amaya, T. M. Makris, M. Zając, J. Korecki, A. Chumakov, B. Trzewik, S. Kędracka-Krok, W. Minor, M. Chruszcz and T. Borowski, Chem.–Eur. J., 2018, 24, 5225–5237 CrossRef PubMed.
- S. Fetzner, Appl. Environ. Microbiol., 2012, 78, 2505–2514 CrossRef CAS PubMed.
- U. Frerichs-Deeken, K. Ranguelova, R. Kappl, J. Hüttermann and S. Fetzner, Biochemistry, 2004, 43, 14485–14499 CrossRef CAS PubMed.
- T. Matsuura, H. Matsushima and R. Nakashima, Tetrahedron, 1970, 26, 435–443 CrossRef CAS.
- J. Rehulka, K. Vychodilova, P. Kejci, S. Gurska, P. Hradil, M. Hajduch, P. Dzubak and J. Hlavac, Eur. J. Med. Chem., 2020, 192, 112176 CrossRef CAS PubMed.
- K. Burglová, G. Rylová, A. Markos, H. Prichystalova, M. Soural, M. Petracek, M. Medvedikova, G. Tejral, B. Sopko, P. Hradil, P. Dzubak, M. Hajduch and J. Hlavac, J. Med. Chem., 2018, 21, 3027–3036 CrossRef PubMed.
- C. Hegedus, K. Kovács, Z. Polgár, Z. Regdon, E. Szabó, A. Robaszkiewicz, H. J. Forman, A. Martner and L. Virág, Redox Biol., 2018, 16, 59–74 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, mass spectrometry and UV-vis data. See DOI: 10.1039/d1ra07527f |
| This journal is © The Royal Society of Chemistry 2022 |