
Peptide based folding and function of single polymer chains†
Henrik
Kalmer
ab,
Federica
Sbordone
ab and
Hendrik
Frisch
 *ab
*ab
aCentre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
bSchool of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: H.Frisch@qut.edu.au
First published on 15th September 2022
Abstract
The folding of synthetic polymers into single chain nanoparticles draws inspiration from the folding of polypeptides into the functional macromolecular architectures of proteins. The building blocks of their natural inspiration, amino acids and peptides, are surprisingly underutilised in the design of synthetic folded architectures. Based on N-terminal cysteines, methylmethacrylate derived monomers were designed that are readily copolymerizable with PEGMA and MMA to provide water soluble polymer-peptide conjugates. Upon acidic deprotection of the peptide side chains, disulphide bridges crosslink the parent polymer into a folded architecture. Providing access to functional folded macromolecular architectures, the presented synthetic strategy allows for a facile incorporation of functional amino acid sequences. Embedding of the catalytic triad into pentapeptides containing N-terminal cysteines enabled a one step folding and activation of their catalytic activity as exemplified by hydrolysis of para-nitrophenylacetate.
Introduction
Over the last decade, intramolecularly crosslinked synthetic polymers have been established as a distinct class of functional synthetic macromolecular architectures.1–5 These so called Single Chain Nanoparticles (SCNPs) are investigated for numerous applications spanning sensing,6 drug delivery7 and catalysis.8–11 Their compacted structure has largely been inspired by the reversible folding of polypeptides into functional proteins that is observed in nature.12,13 The precise architecture, which is obtained upon folding of these sequence-defined biomacromolecules, is encoded into their primary sequence, i.e. their monomer sequence. Synthetic efforts have thus also investigated how sequence control can be used to create specifically folded domains within SCNPs and thereby enable control over their 3D-architecture.14,15To introduce the intramolecular crosslinking, a wide range of functional groups has been explored,16 utilizing for instance photochemistry,3,17–19 metal-complexation9,20 or non-covalent interactions.21,22 Surprisingly, the fundamental monomers of their natural counterparts, i.e. amino acids and peptides, have rarely been applied for the folding of synthetic polymer chains.
The enormous potential of peptide derived functional groups to control SCNPs folding has been highlighted by the Knight group. The use of di(phenylalanine) moieties as polymer sidechains enabled the folding of polymers into hydrogen bonded macromolecular architectures.23 Next to hydrogen-bonds, disulphide bonds are key interactions that stabilise secondary and tertiary structures of proteins. Berda24 and Thayumanavan25 implemented disulphide containing crosslinks into SCNPs by either incorporating disulphide containing crosslinkers or disulphide exchange. Elegant work by Lutz26 attached cysteine–arginine–cysteine (C–R–C) containing linkers to sequence defined polymers in order to obtain bicyclic, 8-shaped polymers.
In solid phase peptide synthesis (SPPS), disulphide formation is often observed upon deprotection of cysteine containing peptides and is usually addressed through cleavage conditions.27 We report herein a synthetic strategy that exploits disulphide formation upon deprotection of polymer tethered N-terminal cysteines to provide a modular platform to fold single polymer chains in one step (Fig. 1). Seizing the modularity of cysteine terminal monomers, a short catalytically active peptide sequence was introduced into the peptide-based monomer yielding SCNPs with functional folded architectures. The peptide-based sidechain is acting hereby as both the crosslinker and the catalytically active moiety.
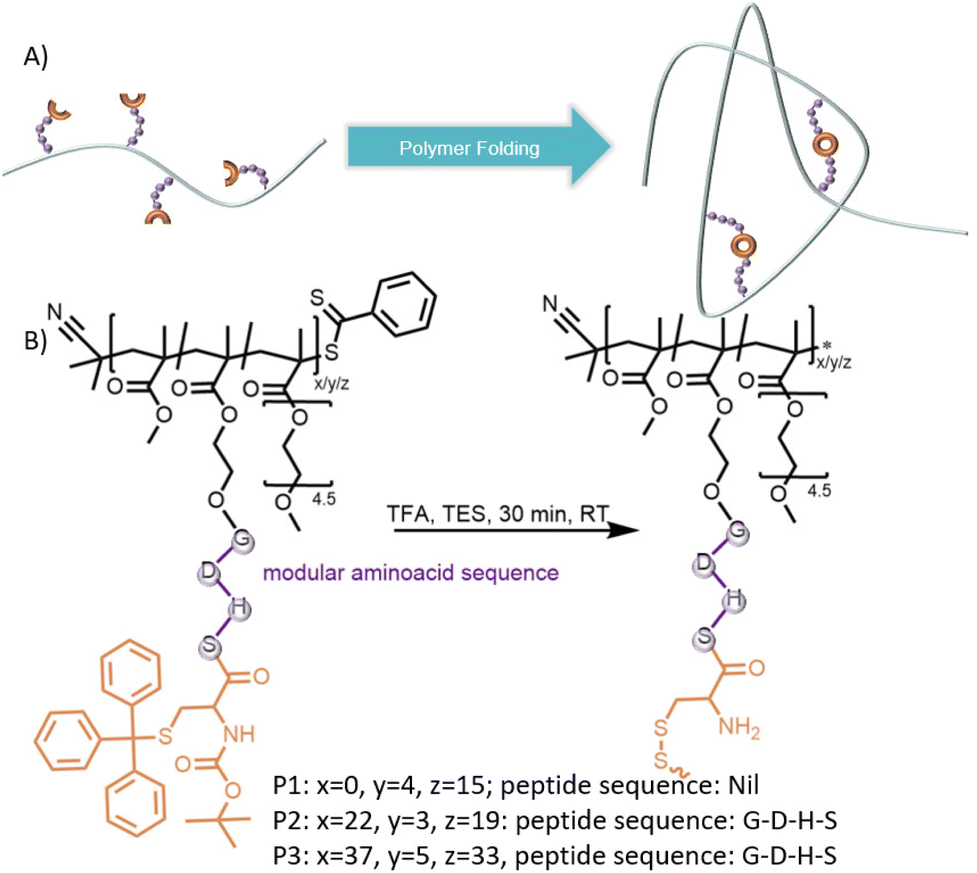 | ||
| Fig. 1 (A) Schematic representation folding of single polymer chains through cysteine-terminal peptide side chains. (B) Deprotection and concomitant folding of P1–P3 to SCNP1–SCNP3. | ||
Results and discussion
A cysteine-based monomer (M1) was synthesized by esterification of protected cysteine (N-(tert-butoxycarbonyl)-S-trityl-L-cysteine) with 2-Hydroxyethylmethacrylate (HEMA, ESI, Chapter 2.1†). Upon reversible addition–fragmentation chain-transfer (RAFT) polymerization with poly(ethylene glycol) methacrylate (PEGMA), a water-soluble polymer P1 (Mn = 7000 g mol−1, Đ = 1.2) was obtained, containing on average 4 cysteine units. To initiate deprotection of the acid labile protecting groups and concomitant disulphide formation, P1 was dissolved in a TFA/TES mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, V:V) at 25 mg mL−1 and stirred for 30 minutes at ambient temperature while exposed to air, which has been observed to induce disulphide formation of Cysteinemethylester (Fig. S18†). After precipitation, the folding of the single polymer chains was monitored via Size Exclusion Chromatography (SEC, Fig. 2A, top). The obtained SCNP1 showed a significantly reduced apparent molecular weight from Mp = 7900 (P1) to 7000 (SCNP1) g mol−1 as a result of the decrease in hydrodynamic volume and loss of protecting groups. A high molecular weight shoulder in the SEC trace of SCNP1 indicates intermolecular crosslinking as a minor side reaction. While the concentration of 25 mg mL−1 is high for SCNP folding and facilitates upscaling of the SCNP synthesis,28 intermolecular crosslinking could not be avoided by lowering the polymer concentration in the folding reaction by more than half to 10 mg mL−1 (Fig. S9†). On a molecular level, the successful deprotection of the thiol was monitored via1H nuclear magnetic resonance (NMR) spectroscopy by the significant decrease of aromatic resonances of the trityl-protecting group at δ = 7.5–7.0 ppm and tert-butyl resonances at δ = 1.4 ppm (Fig. 2C). The residual aromatic resonances indicate a deprotection efficiency of close to 90%. Longer reaction times lead to the full disappearance of aromatic resonances (Fig. S7 and S13†), however, increased intermolecular crosslinking was observed when extending the reaction time too long (Fig. S7 and S14†). Hence, the 30 min deprotection was found preferential to achieve more selective intra chain reactivity.
:1, V:V) at 25 mg mL−1 and stirred for 30 minutes at ambient temperature while exposed to air, which has been observed to induce disulphide formation of Cysteinemethylester (Fig. S18†). After precipitation, the folding of the single polymer chains was monitored via Size Exclusion Chromatography (SEC, Fig. 2A, top). The obtained SCNP1 showed a significantly reduced apparent molecular weight from Mp = 7900 (P1) to 7000 (SCNP1) g mol−1 as a result of the decrease in hydrodynamic volume and loss of protecting groups. A high molecular weight shoulder in the SEC trace of SCNP1 indicates intermolecular crosslinking as a minor side reaction. While the concentration of 25 mg mL−1 is high for SCNP folding and facilitates upscaling of the SCNP synthesis,28 intermolecular crosslinking could not be avoided by lowering the polymer concentration in the folding reaction by more than half to 10 mg mL−1 (Fig. S9†). On a molecular level, the successful deprotection of the thiol was monitored via1H nuclear magnetic resonance (NMR) spectroscopy by the significant decrease of aromatic resonances of the trityl-protecting group at δ = 7.5–7.0 ppm and tert-butyl resonances at δ = 1.4 ppm (Fig. 2C). The residual aromatic resonances indicate a deprotection efficiency of close to 90%. Longer reaction times lead to the full disappearance of aromatic resonances (Fig. S7 and S13†), however, increased intermolecular crosslinking was observed when extending the reaction time too long (Fig. S7 and S14†). Hence, the 30 min deprotection was found preferential to achieve more selective intra chain reactivity.
 | ||
| Fig. 2 (A) Overlay of the SEC traces of P1 and SCNP1 (top) and SCNP1 before and after addition of DTT (bottom). (B) Schematic representation of the formation of SCNP1via disulphide bridges and unfolding of SCNP1 with DTT. (C) 1H-NMR of P1 and SCNP1 in acetonitrile-d3. | ||
To confirm that all cysteine moieties have formed intramolecular disulphide crosslinks upon deprotection, an excess of iron(III)chloride hexahydrate, which has been reported to efficiently induce disulphide formation from thiols in SCNPs,24 was added to the obtained SCNP1. However, no shift towards lower apparent molecular weights was observed via SEC (ESI, Chapter 2.6†). In contrast, a shoulder towards higher molecular weights appeared, indicating intermolecular crosslinking of the remaining cysteine moieties. DTT, a common disulphide bridge reducing agent in proteins, was added after the SCNP-formation to unfold the polymer chain (SEC, Fig. 2A, bottom). The unfolding lead to a shift in the SEC trace due to the larger hydrodynamic volume of the unfolded chain, to an apparent molecular weight of Mp = 7300 g mol−1, a behaviour that is in agreement with previously reported disulphide based SCNP unfolding.24,25
To investigate the modularity of the developed synthetic approach, a short tetrapeptide (G–D–H–S) was juxtaposed to the terminal cysteine. The D–H–S sequence, often labelled catalytic triad, is found in the active site of a wide range of enzymes able to catalyse for example ester hydrolysis.29,30 The propensity of D–H–S and derived peptide sequences to cleave esters as both individual peptides31 and peptide-polymer conjugates32,33 has shown great potential for the design of artificial enzyme mimetics.
The C-terminus of otherwise fully protected peptide sequence (G–D–H–S–C) obtained via SPPS was esterified with HEMA, yielding monomer M2. After RAFT mediated co-polymerization with methyl methacrylate (MMA) and PEGMA, water-soluble polymer P2 was obtained (Mn = 11600 g mol−1, Đ = 1.2), containing on average 3 sidechain tethered peptide units. P2 was dissolved in a TFA/TES mixture (8:2, V:V) at 7.5 mg mL−1 and stirred for 30 minutes at ambient temperature while exposed to air to obtain SCNP2 after precipitation. The deprotection and folding was monitored via SEC showing a reduced apparent molecular weight from Mp = 12400 (P2) to 11200 (SCNP2) g mol−1 due to the polymer compaction (Fig. 3A and B, top). Intermolecular crosslinking, however, could also for SCNP2 not be avoided, hence the SEC traces show a slight high molecular weight shoulder. Similarly to SCNP1, SCNP2 reduction with DTT resulted in a shift towards larger apparent molecular weights (Mp = 12100 g mol−1, Fig. 3A and B, bottom). The deprotection was monitored via1H-NMR showing the loss of resonances of the trityl (δ = 7.4–7.2 ppm) and tert-butyl (δ = 1.4 and 1.1 ppm) protecting groups (Fig. 3C). To investigate the effect of the number of catalytically active peptide pendants per chain on the desired catalytic performance, a third polymer P3 was synthesized (Mp = 21200 g mol−1, ESI†) using the same procedure as described for P2. The deprotection lead to a shift to lower apparent molecular weights (Mp = 20000 g mol−1). Monitoring of monomer incorporation via 1H-NMR indicated a random copolymer of PEGMA and M2 with a slight gradient of MMA (Table S1†).
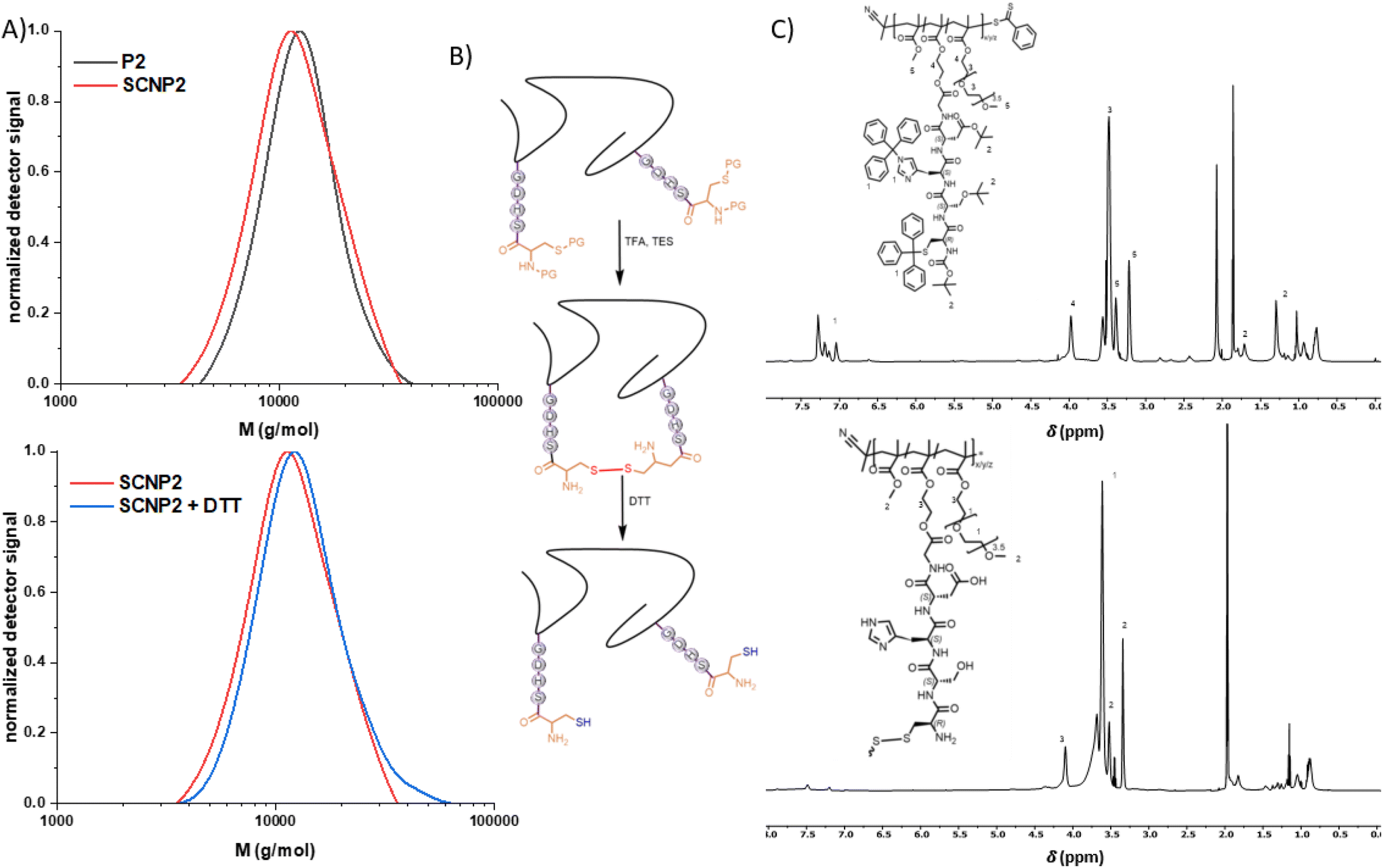 | ||
| Fig. 3 (A) Overlay of the SEC traces of P2 and SCNP2 (top) and SCNP2 before and after addition of DTT (bottom). (B) Schematic representation of the formation of SCNP2via disulphide bridges and unfolding of SCNP2 with DTT. (C) 1H-NMR of P2 and SCNP2 in acetonitrile-d3. | ||
Finally, the propensity of SCNP1–SCNP3 towards the hydrolysis of esters was investigated. para-Nitrophenyl acetate (p-NPA) was dissolved in a phosphate buffered aqueous solution (pH = 7). The hydrolysis of p-NPA leads to the formation of para-nitrophenol, which absorbs visible light at λ = 405 nm. The absorption was recorded for 40 minutes via UV/VIS spectroscopy. Addition of SCNP2 lead to significantly faster linear increase in absorbance compared to the auto hydrolysis of p-NPA under the same conditions (Fig. 4). Since the concentration of the p-nitrophenol is proportional to the absorption at 405 nm, the steeper slope indicates a faster reaction rate due to the catalytic activity of SCNP2. In the presence of SCNP2 the hydrolysis is faster compared to the uncatalysed reaction. To demonstrate that the observed ester hydrolysis activity is a function of the amino acid sequence of SCNP2 and not the terminal cysteines linker, the same experiment was carried out using SCNP1, showing no impact on the hydrolysis rate. The origin of the catalytic activity of SCNP2 can therefore be directly assigned to the peptide sequence in its sidechains. Remarkably, the hydrolysis rate of SCNP2 was significantly faster compared to non-polymer tethered GDHSC peptide, which only showed very little catalytic activity at the same overall concentration of peptide. When the degradation rate of p-NPA in presence of the unprotected peptide sequence was investigated at different concentrations of GDHSC, the expected concentration dependence was, however, observed (Fig. S19†).
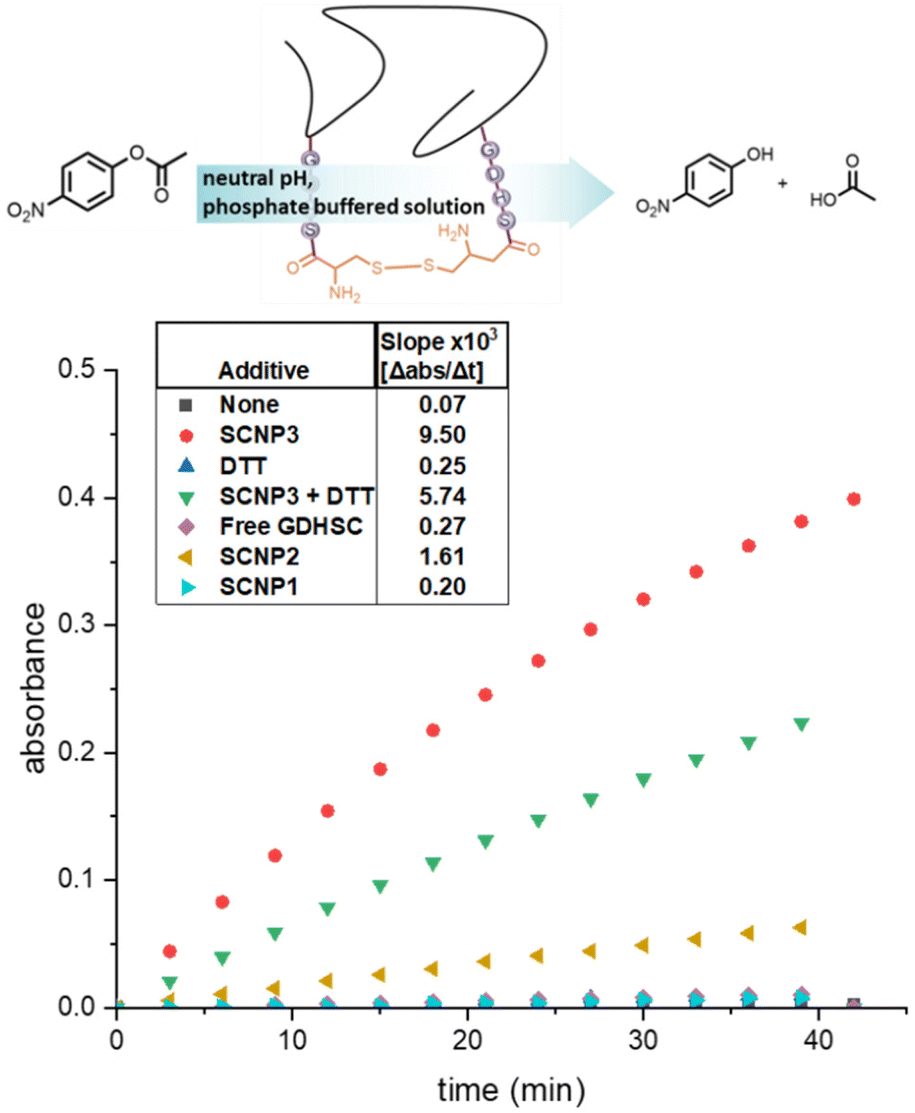 | ||
| Fig. 4 Schematic degradation of para-nitrophenylacetate to para-nitrophenol and acetic acid, catalysed by SCNP2 (top). Change of absorption at 405 nm over time (bottom) due to the hydrolysis of p-NPA for SCNP1–SCNP3, SCNP3 + DTT, and free GDHSC. | ||
Comparing SCNP2 to SCNP3 of a higher molecular weight and greater number of peptide moieties per chain, a considerably higher hydrolysis activity was observed for SCNP3 indicating a significant effect of polymer confinement on the hydrolysis activity of GDHSC. The addition of DTT to SCNP3, resulting in SCNP unfolding, lead to a decrease of the catalytic activity by close to 50%. Adding DTT to a reference experiment, in contrast, did not affect the degradation rate. Hence, the folded SCNP architecture appears to impact catalytic activity.
Noting that catalysis of DHS based enzymes occurs from the precise 3D arrangement of amino acids, which are usually not adjacent in the primary structure, these results raise questions for future investigations about how discrete SCNP architectures increase catalytic activity. Underlying effects could include changes in the conformation of individual peptide strands, closer proximities between peptides strands that enable catalysis involving multiple peptides as well as changes in local polarity within the polymer coil.
Conclusions
We herein reported a modular synthetic strategy to fold single polymer chains upon deprotection of pendent N-terminal cysteines (P1, P2, P3). One step deprotection and folding of single polymer chains via disulphide bridges was achieved at concentrations of up to 25 mg mL−1 (SCNP1). Demonstrating the modularity of the synthetic strategy, a short peptide sequence of G–D–H–S was juxtaposed to the N-terminal cysteine (P2). Deprotection of the amino acid sequence and folding of the polymer chain into SCNP2 was performed in one step, yielding a catalytically active D–H–S triad. The catalytic activity of SCNP2 was demonstrated through the hydrolysis of p-NPA, accelerating hydrolysis significantly compared to non-polymer tethered peptide of the same concentration. Increasing the number of pendent peptides per polymer chain (SCNP3) further increased the hydrolysis rate, whereas the addition of DTT induced unfolding and a substantial decrease in hydrolysis rate. The pendent peptide moieties thus induce folding and enable function of single polymer chains.In addition to the design of enzyme mimetic systems, the reported strategy may provide access to synthetic polymers with defined intramolecular architectures through the incorporation structurally encoded peptide sequences such as α-helices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. F. acknowledges the Australian Research Council for a Discovery Early Career Researcher Award (DECRA) as well as continued support from QUT through the Centre for Materials Science and School of Chemistry and Physics. H. F. acknowledges Prof. Christopher Barner-Kowollik (QUT) for sponsoring and mentorship of his research activities in the context of his ARC Laureate Fellowship. H. K. acknowledges a scholarship from the School of Chemistry and Physics. The authors are grateful to Dr Bryan Tuten (QUT), Dr Aaron Micallef (QUT) and Aidan Izuagbe (QUT) for insightful discussions on disulphide formation within SCNPs.References
- H. Frisch, B. T. Tuten and C. Barner-Kowollik, Isr. J. Chem., 2020, 60, 86–99 CrossRef CAS
.
- M. A. M. Alqarni, C. Waldron, G. Yilmaz and C. R. Becer, Macromol. Rapid Commun., 2021, 42, 2100035 CrossRef CAS PubMed
- O. Galant, H. B. Donmez, C. Barner-Kowollik and C. E. Diesendruck, Angew. Chem., Int. Ed., 2021, 60, 2042–2046 CrossRef CAS PubMed
- R. Chen, S. J. Benware, S. D. Cawthern, J. P. Cole, J. J. Lessard, I. M. Crawford-Eng, R. Saxena and E. B. Berda, Eur. Polym. J., 2019, 112, 206–213 CrossRef CAS
-
J. Rubio-Cervilla, E. onzález and J. A. Pomposo, in Single-Chain Polym. Nanoparticles, 2017, pp. 341–388 Search PubMed
- L. Deng, L. Albertazzi and A. R. A. Palmans, Biomacromolecules, 2022, 23, 326–338 CrossRef CAS PubMed
- A. P. P. Kröger and J. M. J. Paulusse, J. Controlled Release, 2018, 286, 326–347 CrossRef PubMed
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS PubMed
- N. D. Knöfel, H. Rothfuss, J. Willenbacher, C. Barner-Kowollik and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 4950–4954 CrossRef PubMed
- Z. Hu and H. Pu, Eur. Polym. J., 2021, 143, 110194 CrossRef CAS
- Y. Liu, P. Turunen, B. F. M. de Waal, K. G. Blank, A. E. Rowan, A. R. A. Palmans and E. W. Meijer, Mol. Syst. Des. Eng., 2018, 3, 609–618 RSC
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed
- M. H. Barbee, Z. M. Wright, B. P. Allen, H. F. Taylor, E. F. Patteson and A. S. Knight, Macromolecules, 2021, 54, 3585–3612 CrossRef CAS
- J. Zhang, G. Gody, M. Hartlieb, S. Catrouillet, J. Moffat and S. Perrier, Macromolecules, 2016, 49, 8933–8942 CrossRef CAS
- J. Zhang, J. Tanaka, P. Gurnani, P. Wilson, M. Hartlieb and S. Perrier, Polym. Chem., 2017, 8, 4079–4087 RSC
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS
- H. Frisch, D. Kodura, F. R. Bloesser, L. Michalek and C. Barner-Kowollik, Macromol. Rapid Commun., 2019, 1900414 Search PubMed
- D. Kodura, H. A. Houck, F. R. Bloesser, A. S. Goldmann, F. E. Du Prez, H. Frisch and C. Barner-Kowollik, Chem. Sci., 2021, 12, 1302–1310 RSC
- P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249–253 CrossRef CAS PubMed
- A. Levy, R. Feinstein and C. E. Diesendruck, J. Am. Chem. Soc., 2019, 141, 7256–7260 CrossRef CAS
- P. J. M. Stals, M. A. J. Gillissen, T. F. E. Paffen, T. F. A. de Greef, P. Lindner, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2014, 47, 2947–2954 CrossRef CAS
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085–5089 CrossRef CAS PubMed
- J. L. Warren, P. A. Dykeman-Bermingham and A. S. Knight, J. Am. Chem. Soc., 2021, 143, 13228–13234 CrossRef CAS
- B. Tuten, D. Chao, C. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC
- C. Song, L. Li, L. Dai and S. Thayumanavan, Polym. Chem., 2015, 6, 4828–4834 RSC
- O. Shishkan, M. Zamfir, M. A. Gauthier, H. G. Börner and J. F. Lutz, Chem. Commun., 2014, 50, 1570–1572 RSC
- P. R. Singh, M. Rajopadhye, S. L. Clark and N. E. Williams, Tetrahedron Lett., 1996, 37, 4117–4120 CrossRef CAS
- A. M. Hanlon, R. Chen, K. J. Rodriguez, C. Willis, J. G. Dickinson, M. Cashman and E. B. Berda, Macromolecules, 2017, 50, 2996–3003 CrossRef CAS
- E. Di Cera, IUBMB Life, 2009, 61, 510–515 CrossRef CAS PubMed
- L. Polgár, Cell. Mol. Life Sci., 2005, 62, 2161–2172 CrossRef
- Y. Li, Y. Zhao, S. Hatfield, R. Wan, Q. Zhu, X. Li, M. McMills, Y. Ma, J. Li, K. L. Brown, C. He, F. Liu and X. Chen, Bioorg. Med. Chem., 2000, 8, 2675–2680 CrossRef CAS PubMed
- M. D. Nothling, A. Ganesan, K. Condic-Jurkic, E. Pressly, A. Davalos, M. R. Gotrik, Z. Xiao, E. Khoshdel, C. J. Hawker, M. L. O'Mara, M. L. Coote and L. A. Connal, Chem, 2017, 2, 732–745 CAS
- C. Douat-Casassus, T. Darbre and J.-L. Reymond, J. Am. Chem. Soc., 2004, 126, 7817–7826 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00717g |
| This journal is © The Royal Society of Chemistry 2022 |