
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceH/D exchange under mild conditions in arenes and unactivated alkanes with C6D6 and D2O using rigid, electron-rich iridium PCP pincer complexes†
Joel D.
Smith
 a,
George
Durrant
b,
Daniel H.
Ess
b,
Benjamin S.
Gelfand
a and
Warren E.
Piers
*a
a,
George
Durrant
b,
Daniel H.
Ess
b,
Benjamin S.
Gelfand
a and
Warren E.
Piers
*a
aDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: wpiers@ucalgary.ca
bDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, USA
First published on 16th June 2020
Abstract
The synthesis and characterization of an iridium polyhydride complex (Ir-H4) supported by an electron-rich PCP framework is described. This complex readily loses molecular hydrogen allowing for rapid room temperature hydrogen isotope exchange (HIE) at the hydridic positions and the α-C–H site of the ligand with deuterated solvents such as benzene-d6, toluene-d8 and THF-d8. The removal of 1–2 equivalents of molecular H2 forms unsaturated iridium carbene trihydride (Ir-H3) or monohydride (Ir-H) compounds that are able to create further unsaturation by reversibly transferring a hydride to the ligand carbene carbon. These species are highly active hydrogen isotope exchange (HIE) catalysts using C6D6 or D2O as deuterium sources for the deuteration of a variety of substrates. By modifying conditions to influence the Ir-Hn speciation, deuteration levels can range from near exhaustive to selective only for sterically accessible sites. Preparative level deuterations of select substrates were performed allowing for procurement of >95% deuterated compounds in excellent isolated yields; the catalyst can be regenerated by treatment of residues with H2 and is still active for further reactions.
Introduction
Metal-catalyzed hydrogen isotope exchange (HIE) is a key fundamental process for preparing isotopically labeled organic compounds necessary for studying reaction mechanisms and biological metabolic processes.1 Since the seminal work on homogeneous HIE by Garnett and Hodges2,3 as well as Shilov and co-workers,4,5 advances in analytical biology and chemistry have created an ever growing demand for deuterium6 and tritium7 labeled molecules.8 Consequently, an important body of research devoted to studying the homogeneously catalyzed exchange of hydrogen and deuterium atoms (H/D exchange) has developed over the last 25 years.9–12 Pioneering work by the Bergman group on pentamethylcyclopentadienyl supported iridium hydrides demonstrated low temperature C–H activation13–15 and it was subsequently shown that H/D exchange by cationic iridium complexes16–18 occurred under relatively mild conditions using C6D6 as a deuterium source. Most of these transformations involved selective deuterium incorporation into arene substrates10,11 with only a handful of catalysts capable of H/D exchange on unactivated alkane substrates under more forcing conditions.16,17,19–24Tridentate “pincer” ligands have played a prominent role in homogeneous catalysis.25,26 For example, polyhydridic pincer complexes of the late transition metals (particularly Ru and Ir) are known to be exemplary alkane dehydrogenation27 and CO2 reduction catalysts,28,29 but recent work has demonstrated their promise as competent catalysts for promoting homogenous H/D exchange. Leitner, Milstein and co-workers were the first to show that a Ru based PNP polyhydride, IChart 1, could use C6D6 or D2O for arene deuteration at 50 °C.30,31 Shortly after, Zhou and Hartwig revealed that the aliphatic iridium PCP polyhydride II could be used as a pre-catalyst to efficiently perform H/D exchange between olefins and C6D6 at room temperature without isomerization.32 Following these reports, PNP (III) and PCP (IV) complexes synthesized by Grubbs33 and Wendt,34 respectively, were also tested as HIE mediators. The iridium dihydride species III was able to selectively deuterate aromatic compounds and silanes, while the cyclohexyl-based PCP complex IV was found to successfully deuterate arenes and to a limited extent Csp3–H sites, albeit at elevated temperatures of 150 °C.
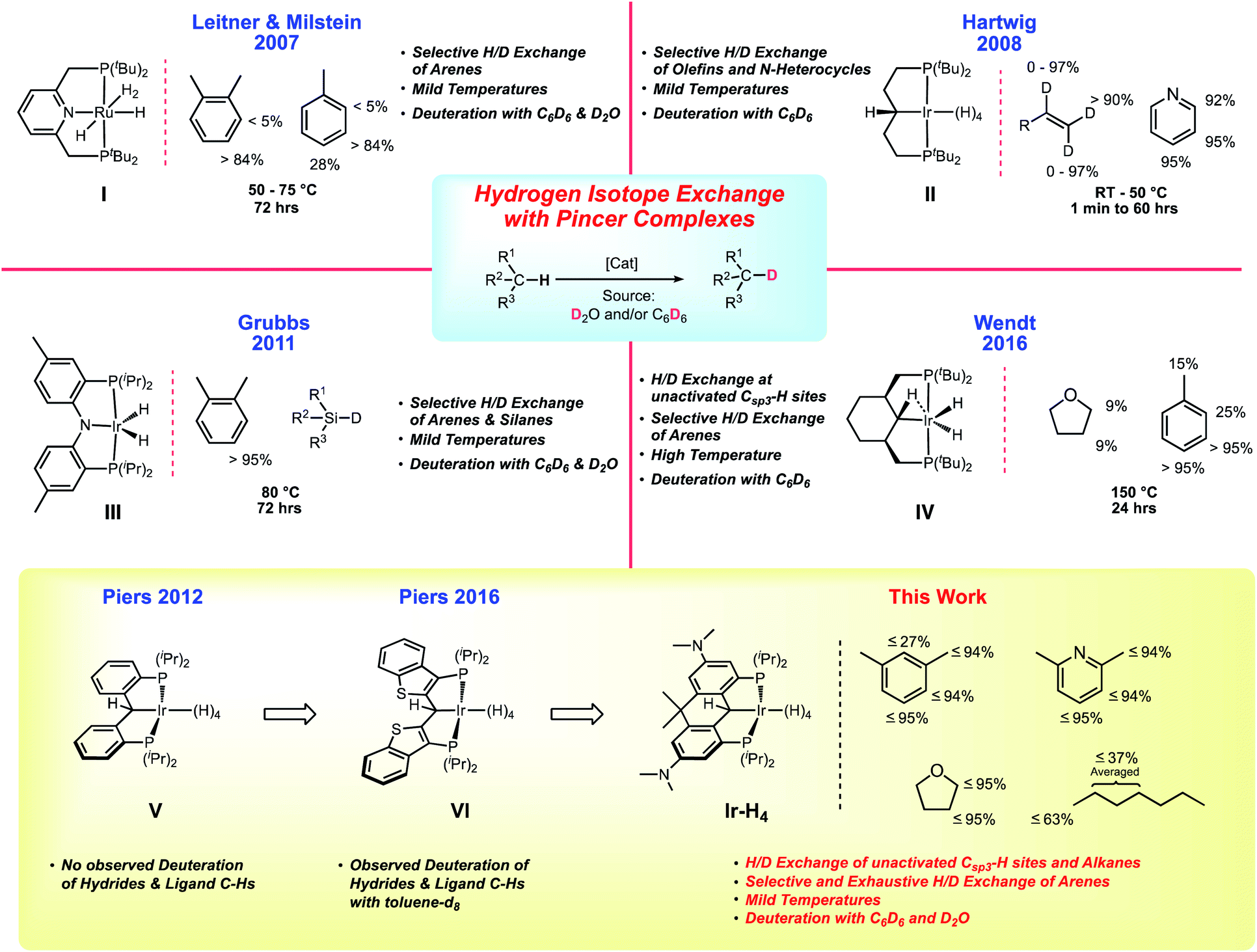 | ||
| Chart 1 Contextualization of this study. | ||
Design elements of the ligands II/IV and III have been incorporated into a family of PCP ligands V introduced by our group35 and also explored by others.36–39 An attractive feature of these ligands is their ability to assume PCsp3P or PCcarbeneP forms by the addition or removal of a hydrogen atom from the anchoring carbon of the pincer framework. This has precedence in the ligands II and IV but in the aryl-linked PCP systems V (and the related benzothiophene-based system VI40) this is potentially more controllable because of the lack of β-hydrogens in the PCP linker framework, and through manipulation of the electronic properties of the ligand via substitution on the aryl linkers and/or rigidification of the ligand via additional linking of the backbone aryl groups.41,42 With these two structural levers, a library of PCP ligands has been prepared and a substantial range of νCO values has been found for [(PCcarbeneP)IrCO]+ species.43 We have shown that the rate of addition of N2O to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ir bond40 can therefore be tuned substantially.44 Furthermore, the rigidification of the ligand tends to favor the PCcarbeneP form of the ligand vs. the PCsp3P form, while also providing greater protection against C–C bond cleavage reactions that destroy the integrity of the ligand in unlinked systems akin to V.45 The ability of the carbon atom in these PCP pincers to engage in ligand cooperativity has been exploited in hydrogen,35,46 silane47–49 and other small molecule40,50–53 activations in various systems. Furthermore, in polyhydridic systems, the dibenzylic hydrogen is actively involved in exchange with the metal hydrides,34,35 increasing the “hydrogen capacity” of these compounds relative to other (for example PNP) polyhydridic pincer complexes and potentially providing new pathways for catalysis.
Ir bond40 can therefore be tuned substantially.44 Furthermore, the rigidification of the ligand tends to favor the PCcarbeneP form of the ligand vs. the PCsp3P form, while also providing greater protection against C–C bond cleavage reactions that destroy the integrity of the ligand in unlinked systems akin to V.45 The ability of the carbon atom in these PCP pincers to engage in ligand cooperativity has been exploited in hydrogen,35,46 silane47–49 and other small molecule40,50–53 activations in various systems. Furthermore, in polyhydridic systems, the dibenzylic hydrogen is actively involved in exchange with the metal hydrides,34,35 increasing the “hydrogen capacity” of these compounds relative to other (for example PNP) polyhydridic pincer complexes and potentially providing new pathways for catalysis.
In both the parent, unsubstituted, unlinked system V,35 and that incorporating the benzothiophene-based ligand VI,46 the Ir polyhydride is only stable under an atmosphere of dihydrogen but it was observed that slow H/D exchange in the sterically exposed C–H bonds of the ligand aryl backbone occurred in C6D6. We hypothesized that a more electron rich system would accelerate this process and indeed, the linked dihydroanthracene PCP ligand framework fitted with electron donating dimethylamino groups para to the PCP carbon (bottom of Chart 1) supports a stable, isolable polyhydride species Ir-H4 (Chart 1). While stable as a solid, this complex readily loses molecular hydrogen in solution to give species that are highly active HIE catalysts at low loadings and under mild conditions. Both C6D6 and D2O can be used as deuterium sources and selectivity can be modulated to some extent by controlling the catalyst speciation.
Results and discussion
Synthesis and characterization of iridium PCP pincer polyhydride Ir-H4
Using methodology established previously,46,54 the iridium PCP pincer complex Ir-OH was prepared by heating the previously reported Ir-Cl44 to 120 °C in toluene with an excess of CsOH for 24 hours (Scheme 1, red arrows). The resulting brown solution was filtered through Celite® and a brown powder was isolated in 80% yield after removing all volatiles in vacuo. The Ir-OH complex displays a characteristic –OH triplet (3JHP = 3.8 Hz) at 3.62 ppm in the 1H NMR spectrum as well as a singlet in the 31P{1H} NMR at 51.9 ppm. The presence of an iridium-carbene moiety was indicated by a triplet signal at 196.0 ppm (2JCP = 2.2 Hz) in the 13C{1H} NMR spectrum.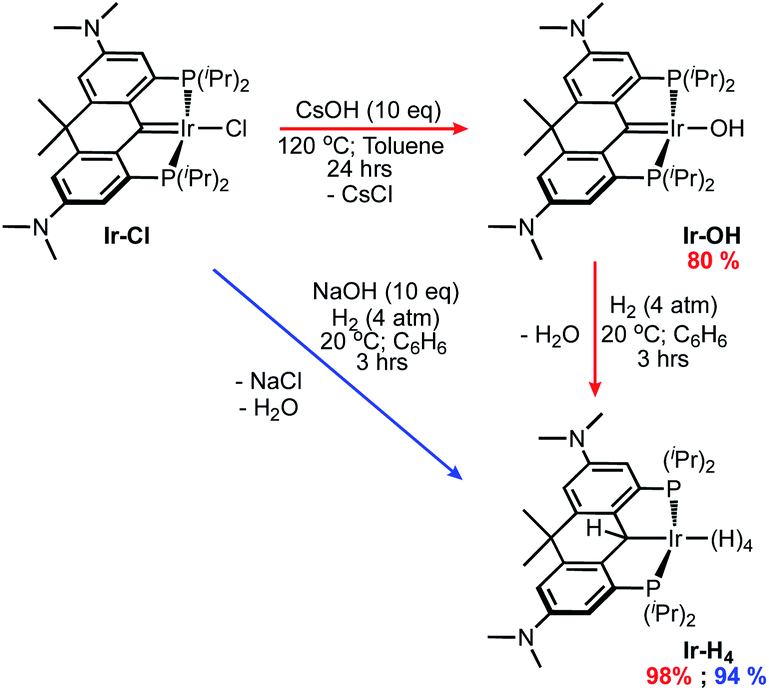 | ||
| Scheme 1 Synthetic routes to iridium PCP polyhydride Ir-H4. | ||
Dissolving Ir-OH in benzene and exposing it to 4 atm of H2 gas for 3 hours resulted in a color change from dark reddish-brown to yellow-orange. The 31P{1H} NMR spectrum for the solution exhibited a single peak at 56.5 ppm and upon solvent removal a brown residue was formed. This residue was then stirred in n-pentane under 1 atm of H2 for 20 minutes producing a beige powder, Ir-H4, that was isolated in 98% yield following the removal of all volatiles in vacuo. Alternatively, we found stirring the precursor Ir-Cl with NaOH (10 eq.) under H2 gas (4 atm) in benzene for 3 hours produced a similar red-orange solution (Scheme 1, blue arrow). Following filtration through a pad of Celite®, the solution was stirred for 20 minutes under H2 gas. All volatiles were removed in vacuo and a beige powder was again isolated in 94% yield. Compared to the parent ortho-phenylene V35 (Chart 1) and the benzothiophene-based ligand congener VI,46 which both could not be isolated due to decomposition via loss of H2 under vacuum, complex Ir-H4 is remarkably stable as a solid and can be stored indefinitely at room temperature under an inert atmosphere. Furthermore, it can be interrogated via NMR spectroscopy in cyclohexane-d12 or methylcyclohexane-d14 under argon, whereas the others (V and VI) must be kept under an atmosphere of H2 in order to avoid decomposition. The 1H NMR spectrum for Ir-H4 in cyclohexane-d12 displays a single triplet (average JHP = 10.2 Hz) resonance at −10.32 ppm and a singlet at 5.52 ppm, integration indicating the presence four rapidly exchanging hydridric nuclei and one α-C(H)–Ir proton, respectively. Moreover, the ligand resonances in the spectrum clearly point to Cs symmetry with 12 equivalent dimethyl amino protons represented by a singlet at 2.85 ppm and two signals at 1.23 ppm and 1.84 ppm for the inequivalent –CH3 groups attached to the carbon backbone linker. The 13C{1H} NMR also corroborates the formation of Ir-H4 showing the loss of the carbene triplet associated with the Ir-Cl and Ir-OH complexes and the presence of a signal at 21.0 ppm coupling to the α-C–H proton at 5.52 ppm in the 1H–13C HSQC spectrum.
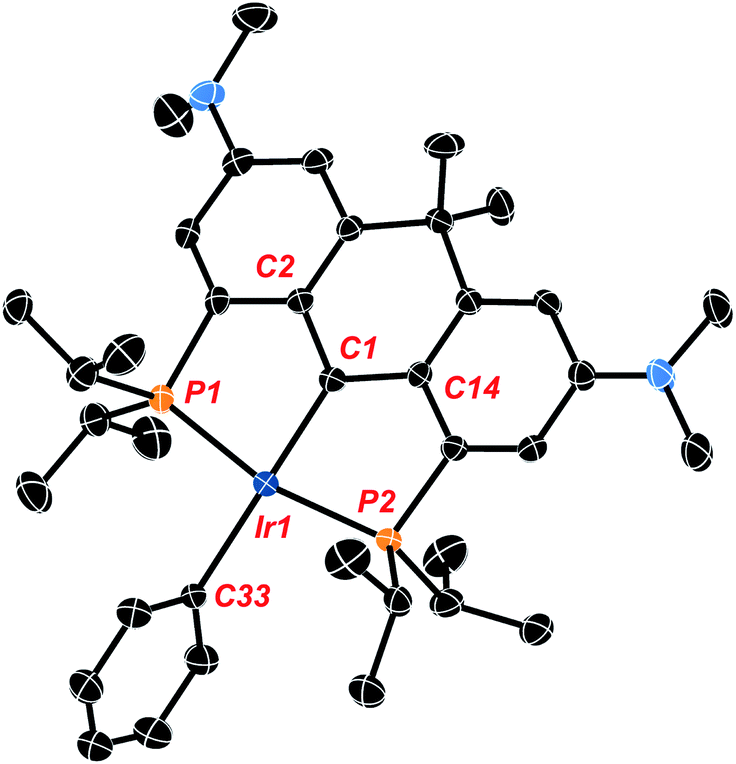
Crystals of the compound were obtained by slowly cooling a supersaturated solution of Ir-H4 in isopropanol from 50 °C to 20 °C and crystallographic analysis corroborates the solution data described above. The hydrogen atoms associated with the metal center were located from the Fourier map but low accuracy in Ir-H and H–H distances preclude conclusions concerning their arrangement as hydride or dihydrogen ligands. The data show the anchoring α-carbon (C1 in Fig. 1) is pyramidalized (the sum of the angles about C1 is 335.2(8)°), and the central 6 membered ring containing this α-carbon has also adopted a “boat”-like conformation. The sp3 hybridization of the α-carbon is also indicated by the central Ir1–C1 bond length of 2.184(5) Å which is similar to other sp3 hybridized Ir–C bonds we have observed with these NR2 substituted ligand frameworks44,45 and lengthened significantly from the values of ≈1.93 Å seen in PCcarbeneP complexes of this ligand.44
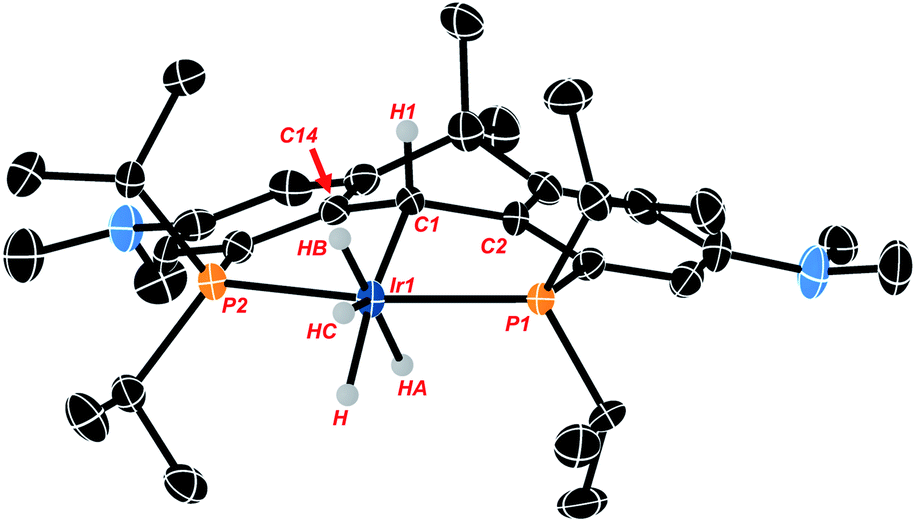 | ||
| Fig. 1 Thermal ellipsoid diagram of lr-H4 (ellipsoids drawn to 50% probability; all hydrogen atoms except H, H1, HA, HB and HC have been omitted for clarity). Selected metrical data for lr-H4: bond distances (Å); lr1–C1, 2.186(5); Ir1–P1, 2.2703(16); lr1–P2, 2.2786(16), C1–C2, 1.496(8); C1–C14 1.519(8). Bond angles (°); P1–lr1–P2, 163.45(5); lr1–C1–C2, 112.9(4); lr1–C1–C14, 112.0(4); C2–C1–C14, 110.2(5). | ||
Given the lack of crystallographic data concerning the iridium hydrides in Ir-H4, we turned to criteria developed by Halpern and co-workers55 to shed some light on what form dominates the speciation of Ir-H4 in solution. While it is typically difficult to unambiguously categorize the hydride bonding mode for these complexes, we were able to rule out the possibility that Ir-H4 is a “non-classical” Ir(I) bis-dihydrogen species (Scheme 2). The T1 (min) relaxation for the hydridic nuclei of Ir-H4 in methylcyclohexane-d14 (Fig. S1†) was found to be 186 ms at 600 MHz (10 °C ± 0.5 °C), approximately 155 ms when adjusting to a 500 MHz field strength. These values are comparable to those obtained for other PCP pincer polyhydride complexes reported by Heinekey,56 Wendt34 and us;35 a collection of comparative data is given in Table S1.† These data point to an equilibrating mixture of a “classical” Ir(V) tetrahydride and the “intermediate” Ir(III) dihydrogen–dihydride isomers shown in Scheme 2. The experimental T1 of 155 ms is somewhat lower than the calculated T1 value of 203 ms calculated from the parameters obtained from the M06 density functional theory (DFT) optimized structure of Ir-H4, suggesting a significant contribution from any or all of the dihydrogen–dihydride isomers, which also can be optimized using DFT and give rise to T1 values of ≈15 ms using the Halpern methodologies. Significantly, the non-classical bis-dihydrogen structure could not be optimized by DFT, suggesting that this formally Ir(I) isomer is not favored with this electron rich pincer ligand and does not contribute to the speciation of Ir-H4 in solution.
 | ||
| Scheme 2 (a) Outline of isomerism in Ir-H4 with benzene-solvated enthalpies (Gibbs energies) in kcal mol−1 (b) Ir-H and H–H distances (Å) found in M06 DFT structures. | ||
Another key metric in this family of compounds is the averaged JH–D coupling constant in the Ir-HD3 isotopologue. For Ir-H4, this species is trivially prepared by dissolving Ir-H4 in C6D6 and stirring for 15 minutes at room temperature (vide infra), removing the solvent and redissolving the sample in methylcyclohexane-d14. All four iridium hydrides and the benzylic position on the ligand are rapidly deuterated under these conditions, but by halting before full deuteration, the residual hydride resonance in methylcyclohexane-d14 at −10.25 ppm displays a resolved seven line pattern with an average observed JH–D of 3.1 ± 0.1 Hz when collected at 50 °C (Fig. S2†). This coupling correlates to an approximated average H–H atomic distance of 1.02–1.25 Å (Table S1†),56 and is consistent with equilibrating classical tetrahydride (H–H > 1.2 Å) and intermediate dihydrogen (H–H ≈ 1.0–1.2 Å) dihydride structures (Table S1†)57–59 as depicted in Scheme 2. Unlike the related system based on IV, attempts to freeze out the exchange between the isomers of the latter and/or the equilibrium between the two via variable temperature 1H{31P} NMR spectroscopy on Ir-H4 in methylcyclohexane-d14 showed no decoalescence of the hydride resonance to temperatures as low as −110 °C (Fig. S3†). This indicates a lower energetic barrier for hydride exchange than the estimated 8.7 kcal mol−1 at −70 °C reported by Wendt et al. in the system IV.34 Indeed, M06 DFT calculations indicate the potential energy surface for postulated tetrahydride and dihydrogen–dihydride complexes is relatively flat with an energy range of <2 kcal mol−1 between these four structures (Scheme 2). For the full covalent Ir-H bonds, the distances are ∼1.6 Å. In the dihydrogen–dihydride structures, the dihydrogen distances are ∼0.85 Å, which are only slightly stretched compared to free H2.59 The barriers are <12 kcal mol−1 for conversion between tetrahydride and dihydrogen–dihydride complexes. The transition-state distances for the forming H–H bonds are ∼1.1 Å. Overall, these DFT thermodynamic and kinetic estimates suggest that all intermediate and classical structures are accessible in solution and the spectroscopically observed complex lies within the dihydrogen–dihydride tetrahydride continuum. While the benzylic hydrogen on the Csp3 carbon of the ligand certainly exchanges with the four iridium hydrogens rapidly on the chemical timescale, the two peaks only undergo slight broadening at temperatures below which loss of H2 and conversion to other species ensues (vide infra). Line shape analysis to probe the barrier was not possible with the spectral data available in this system, unlike that of IV.34
Solution behavior of Ir-H4
While Ir-H4 is quite stable in the solid state, evidence exists for reversible loss of H2 in solution (Scheme 3). When dilute solutions in benzene were exposed to vacuum, the pale-yellow color darkened slowly to a maroon-tinged hue. Readmission of H2 gas to the vessel gave back the yellow solution; monitoring this transformation by NMR spectroscopy indicated that the maroon solution was essentially spectroscopically identical to the yellow solutions of Ir-H4. However, when heating solutions of Ir-H4 to 80 °C under a static vacuum and monitoring by NMR spectroscopy, other resonances became apparent. In the 1H NMR spectrum, a new resonance at 2.71 ppm in the NMe2 region and a faint peak at −10.30 ppm were observed in about a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. In the 31P{1H} NMR spectrum, resonance at 66.4 ppm grew in relative to that of Ir-H4 at 56.5 ppm (Scheme 3 and Fig. S4 and S5†). Cooling the sample resulted in loss of this resonance and a return to solutions that were >98% Ir-H4. We hypothesized that this maroon species was either the PCsp3P supported Ir(III) dihydride Ir-H2 or the PCcarbeneP ligated Ir(III) trihydride Ir-H3 shown in Scheme 3. These should be distinguishable by the 13C resonance of the PCP ligand, but because it is produced in such small amounts via this method, we sought ways to prepare or at least enrich the speciation of this compound in order to obtain more concentrated samples.
:3 ratio. In the 31P{1H} NMR spectrum, resonance at 66.4 ppm grew in relative to that of Ir-H4 at 56.5 ppm (Scheme 3 and Fig. S4 and S5†). Cooling the sample resulted in loss of this resonance and a return to solutions that were >98% Ir-H4. We hypothesized that this maroon species was either the PCsp3P supported Ir(III) dihydride Ir-H2 or the PCcarbeneP ligated Ir(III) trihydride Ir-H3 shown in Scheme 3. These should be distinguishable by the 13C resonance of the PCP ligand, but because it is produced in such small amounts via this method, we sought ways to prepare or at least enrich the speciation of this compound in order to obtain more concentrated samples.
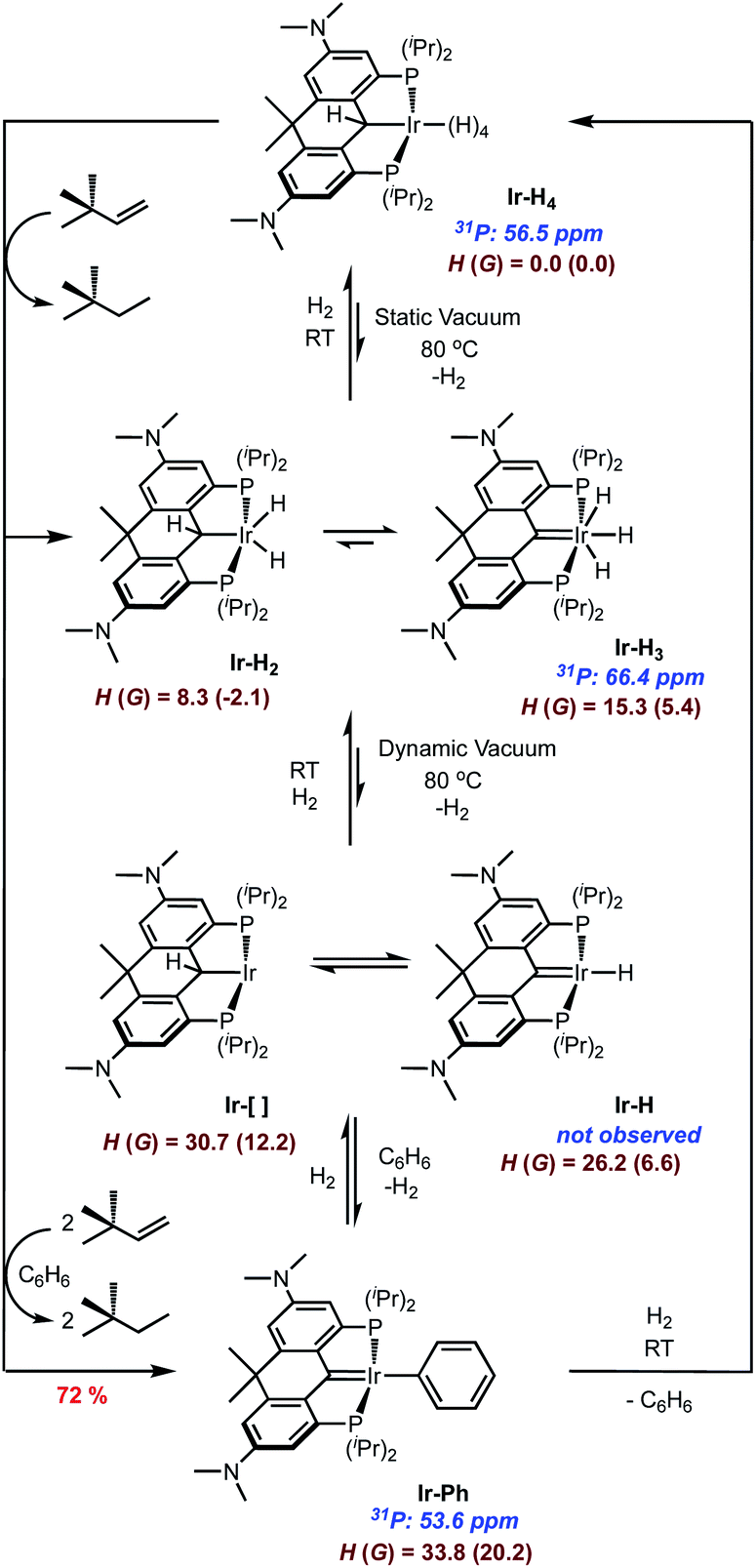 | ||
| Scheme 3 Solution chemistry of Ir-H4. Energy units in dark red are kcal mol−1. | ||
Heating Ir-H4 in benzene under dynamic vacuum did lead to a much darker colored solution, but the 31P{1H} NMR spectra indicated that a third species with a resonance at 53.6 ppm was dominant in these samples. Separate synthesis, NMR spectroscopy and X-ray crystallography confirmed this product to be the PCcarbeneP Ir(I) phenyl complex Ir-Ph (Scheme 3). In attempts to generate Ir-H2/Ir-H3 by treating Ir-H4 with the dehydrogenation agent tert-butyl ethylene, varying mixtures of the three components were obtained depending on the equivalency of olefin employed; when >3 equivalents were used, Ir-Ph was cleanly produced and isolated as a green solid in 72% yield. The 13C{1H} NMR spectrum of Ir-Ph in benzene-d6 shows a distinct carbene triplet at 221.1 ppm (JCP = 4.2 Hz) while the 1H NMR spectrum shows three separate aryl proton signals for the new phenyl group as well as characteristic ligand resonances. Crystallographic data (Fig. 2) show a central Ir1–C1 distance of 1.965(4) Å. This is shorter than that found in Ir-H4 and indicative of an iridium carbene bond similar to analogous structures.35,40,43–45 Accordingly, unlike the boat-like shape found in the Ir-H4 the central 6-membered ring of the ligand framework in Ir-Ph adopts a planar conformation, as a result of the sp2 hybridization found at the carbene center. The rigidity of the ligand employed here stabilizes this phenyl complex with respect to the intriguing C–C bond formation reaction observed by Wendt in a the more flexible IV.60
 | ||
| Fig. 2 Synthesis and thermal ellipsoid diagram of Ir-Ph (ellipsoids drawn to 50% probability; hydrogen atoms have been omitted for clarity). Selected metrical data for Ir-Ph: bond distances (Å): Ir1–C1, 1.965 (4); Ir–C33, 2.118(4); Ir1–P1, 2.2581(13); Ir1–P2, 2.2671(17), C1–C2, 1.467(6); C1–C14 1.469(6). Bond angles (°); P1–Ir1–P2, 166.46(4); C1–Ir1–C33, 179.53(19); P1–Ir1–C1, 83.46(13); P1–Ir1–C33, 96.38(13); Ir1–C1–C2, 122.6(3); Ir1–C1–C14, 122.9(4); C2–C1–C14, 114.5(4). | ||
Taken together, the above observations suggest that Ir-H4 rather easily loses one equivalent of H2, even thermally, to give Ir-H2/Ir-H3, but when exposed to vacuum and heat, a second equivalent of H2 is eliminated to form the (unobserved) compounds Ir-[]/Ir-H depicted in Scheme 3. Consistent with the relative ease for H2 loss, the DFT calculated energy of Ir-H3 is only endergonic by ∼5 kcal mol−1 relative to Ir-H4. Calculations suggest that formation of the dihydrogen–dihydride complex and dihydrogen dissociation is required before 1,2-hydrogen shift from the ligand to Ir to form Ir-H3, which only requires a barrier of ∼15 kcal mol−1. The Ir-H/Ir-[] structures are calculated to be 6.6 and 12.2 endergonic relative to Ir-H4. Overall, the thermodynamic and kinetic DFT calculations suggest that Ir-H3, Ir-H2, Ir-H, and Ir-[] structures are all viable. These compounds rapidly C–H activate benzene and, after elimination of another equivalent of H2, Ir-Ph is produced; this compound rapidly converts back to Ir-H4 upon treatment with H2, with loss of C6H6. The speciation of the Ir-H2/Ir-H3 manifold was solidified as being mainly Ir-H3 by mixing isolated Ir-H4 and Ir-Ph in a ≈2:3 ratio in benzene; this experiment resulted in an equilibrium mixture of these two compounds along with about 40% Ir-H3 which allowed us to acquire meaningful 1H and 13C NMR data on the latter. Both are consistent with C2v symmetry and the presence of a PCcarbeneP ligand. In the 1H NMR spectrum, no resonance in the region between 3 and 6 ppm consistent with the presence of an α-C(H)–Ir proton was observed, while a characteristic resonance at 245.0 ppm in the 13C{1H} NMR spectrum is diagnostic of a carbene carbon. This signal correlated with the triplet integrating to 3 hydrogens at −10.12 ppm in the 2D 1H–13C HMBC spectrum. At room temperature, the single resonance for the three hydrides in Ir-H3 indicate they are in rapid exchange, presumably via a 1,2-hydride shift from the iridium to the carbene carbon, i.e., viaIr-H2. The apparent thermodynamic preference for Ir-H3 over Ir-H2 in this system contrasts with observations reported by Wendt and co-workers,34,61 but these structures are very close in energy and in rapid exchange. DFT calculations using a variety of functionals (M06, wB97X-D, M11, and others) all indicate that the Ir-H2 structure is slightly lower in energy than Ir-H3, the methodology has limitations for distinguishing these types of isomers accurately.
HIE with iridium polyhydrides Ir-Hn
As briefly alluded to above, dissolution of Ir-H4 into C6D6 (and indeed toluene-d8 or THF-d8) leads to the rapid deuteration of the α-C(H)–Ir and Ir-H positions at 20 °C to form Ir-D4 (Scheme 4a) and so it had to be characterized in deuterated cyclohexane or methylcyclohexane solvents which do not surrender deuterium at room temperature. Heating Ir-H4 in C6D6 to 65 °C for 24 hours lead to trace deuteration of the ligand NMe2 and iso-propyl methyl groups (Fig. S6†). Given the coordinative saturation of Ir-H4, we hypothesized that hydrogen loss to form Ir-H3 was required to initiate the HIE process. Indeed, mixing Ir-H4 and Ir-Ph as described above to generate solutions enriched in Ir-H3 leads to much higher deuteration activity. Heating solutions of Ir-H4/Ir-H3/Ir-Ph (3:2:2) in C6D6 to 65 °C for 24 hours gave the three species essentially exhaustively deuterated in the positions shown in Scheme 4b (Fig. S7†). Separate experiments showed that heating pure Ir-Ph under these conditions gave only 70% conversion to Ir-Phd5 after 24 hours and no deuteration of the ligand NMe2 or iso-propyl groups was observed (Fi. S8†), suggesting that Ir-Ph is not an effective HIE promoter in this system.
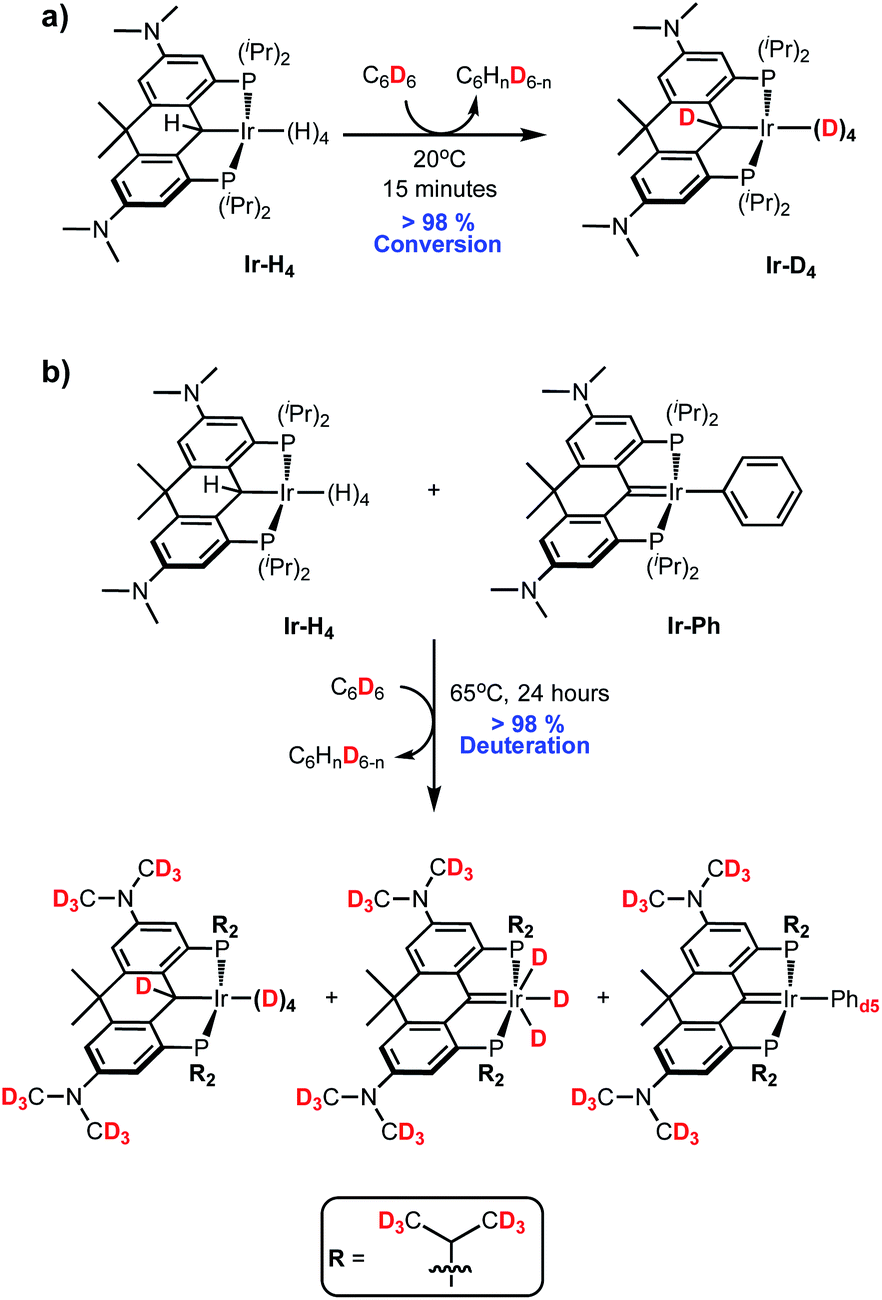 | ||
| Scheme 4 HIE in Ir-H4. | ||
While the iPr groups are likely deuterated via an intramolecular C–H activation, deuteration of the more remote NMe2 groups suggest that intermolecular catalytic deuteration of other substrates under mild conditions might be possible. We chose meta-xylene as a test substrate to evaluate the various iridium species as HIE catalysts due to the sterically varied aryl C–H bonds present, as well as the benzylic sp3 hybridized C–H bonds. The results of these experiments are summarized in Fig. 3. Solutions containing meta-xylene and 1 mol% of either Ir-H4 (argon, 1 atm), Ir-H4 (static vacuum), Ir-Ph (argon, 1 atm) or Ir-Ph activated with D2 gas (≈1 equivalent) were heated to 50 °C in benzene-d6 and monitored by 1H NMR spectroscopy. Each panel in Fig. 3 indicates the rate of loss of substrate protons as time progresses for a different C–H position in the substrate, as a function of the catalyst precursor employed (colored shapes). Cyclooctane was employed as an internal standard. As can be seen in panel a, none of the catalyst formulations deuterate the sterically encumbered 2 position of meta-xylene at 50 °C; the other positions, however, are deuterated to varying degrees. In general, Ir-H4 under argon and Ir-Ph behave similarly for deuteration of the C–H bonds in the benzylic, and aryl 4/5 positions, with moderate activity observed only for the sterically accessible aryl C–H bond in the 5 position. The catalyst systems which would be expected to generate Ir-[]/Ir-H, (Scheme 3) or Ir-H2/Ir-H3, namely Ir-Ph plus D2 (blue triangles) or Ir-H4 under vacuum (green inverted triangles) fared much better, with the former able to almost completely deuterate meta-xylene at all three positions under these mild conditions. Notably, when Ir-H4 is simply exposed to vacuum (green triangles), there is an acceleration in the rate of deuteration consistent with the slow formation of Ir-H3 through loss of H2 to the headspace, whereas when the active species is generated through activation of Ir-Ph with D2, a more typical first order time evolution is observed, further implicating Ir-H3 as the active catalyst or catalyst precursor.
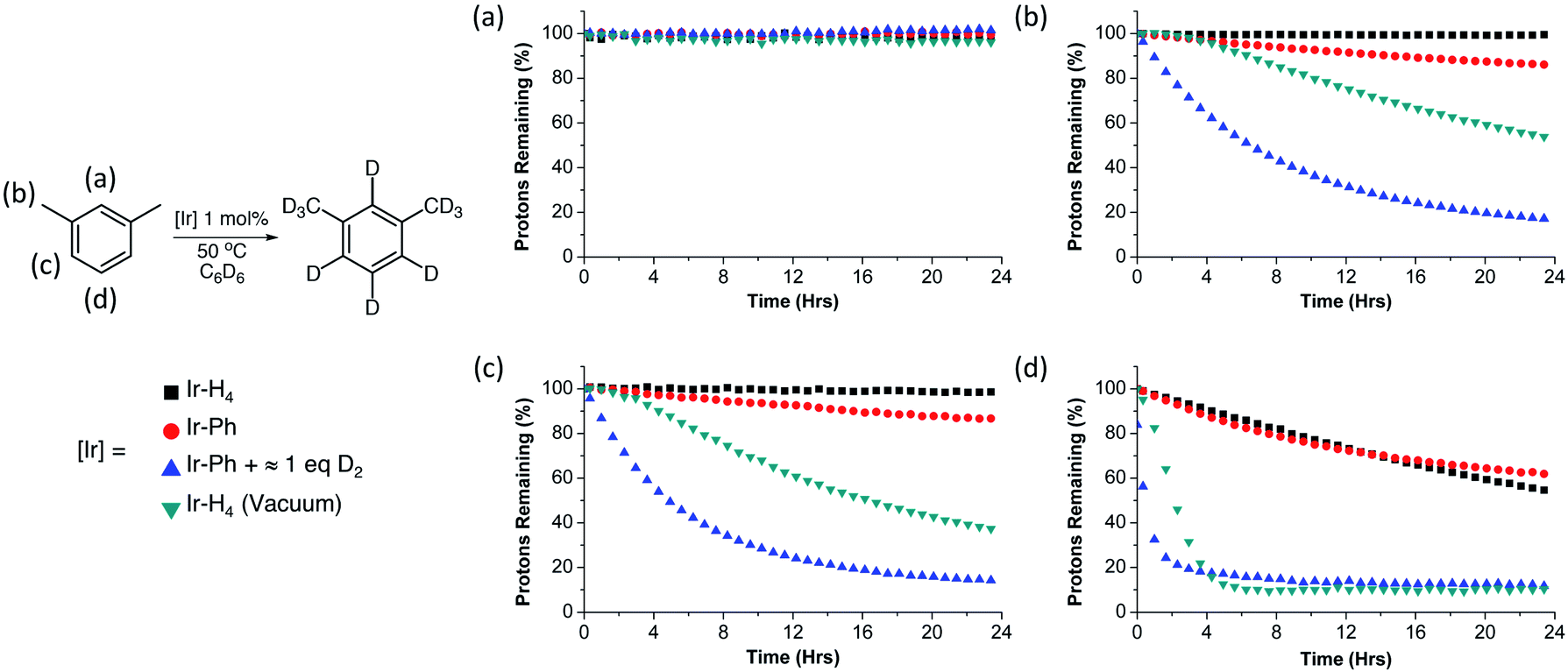 | ||
| Fig. 3 Reaction profiles of the H/D exchange at the 2 (graph a), benzyl (graph b), 4 and 6 (graph c) and 5 (graph d) position of m-xylene (0.38 mmol) in C6D6 (0.5 mL) using 1 mol% of either Ir-H4 (black square), Ir-H4 placed under static vacuum (green inverted triangle), Ir-Ph (red circle) or Ir-Ph activated with ≈1 equivalent of D2 gas (blue triangle). Conversion was measured via1H NMR using cyclooctane as an internal standard. | ||
Since it was clear from the above experiments that Ir-Ph activated with D2 gave the most active HIE catalyst, further optimization of conditions for this reaction were undertaken with this as a starting point (Table 1). Entries 1 and 2 reaffirm that a catalyst is required, and that Ir-Ph itself is a poor catalyst with low activity. Entries 3–6 show that there is an optimal amount of added D2 required to activate the catalyst of 1–3 equivalents. With excess D2 (entry 7), activity drops off markedly, presumably because the iridium speciation is now essentially 100% Ir-D4, which (as shown in Fig. 3) is not active under these conditions. Raising the temperature 15 degrees to 65 °C (entries 8–12) leads to greater HIE activity; comparison of entry 4 with entry 10 underscores this observation. Even the difficult 2-position sees some deuteration at this stage. As entries 11 and 12 show, near exhaustive deuteration of the benzylic, 4/6 and 5 positions can be achieved at 65 °C, with 1 mol% of Ir-Ph activated with 1.4 equivalents of D2; entry 12 uses double the amount of C6D6 in order to drive the deuteration in these positions to near completion.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Temp. (°C) | D2 (mol%) | Time (h) | Position % deuterated (TOF, h−1) | |||
| Benzyl | 2 | 4/6 | 5 | ||||
| a Optimization performed with meta-xylene (0.38 mmol) and Ir-Ph (1 mol%) in C6D6 (0.5 mL). D2 gas was injected into 0.015 M solutions of Ir-Ph in C6D6 and stirred for 10 minutes prior to its addition to m-xylene in C6D6 (0.25 mL). Conversion was measured via1H NMR using cyclohexane as an internal standard. b Volume of C6D6 increased to 1.0 mL. | |||||||
| 1 | 50 | No cat. | 24 | 0 (0) | 0 | 0 (0) | 0 |
| 2 | 50 | 0 | 3.5 | 3 (1) | 0 | 2 (1) | 11 (3) |
| 3 | 50 | 1.0 | 3.5 | 31 (53) | 0 | 40 (23) | 77 (22) |
| 4 | 50 | 1.4 | 3.5 | 32 (55) | 0 | 41 (23) | 81 (23) |
| 5 | 50 | 1.8 | 3.5 | 26 (45) | 0 | 34 (19) | 82 (23) |
| 6 | 50 | 3.0 | 3.5 | 23 (39) | 0 | 33 (19) | 87 (25) |
| 7 | 50 | 9.0 | 3.5 | 0 (0) | 0 | 0 (0) | 8 (2) |
| 8 | 65 | 9.0 | 3.5 | 0 | 0 | 0 | 20 (6) |
| 9 | 65 | 9.0 | 24 | 0 | 0 | 6 (<1) | 70 (3) |
| 10 | 65 | 1.4 | 3.5 | 69 (118) | 4 (1) | 77 (44) | 88 (25) |
| 11 | 65 | 1.4 | 24 | 89 (22) | 13 (<1) | 91 (8) | 89 (4) |
| 12b | 65 | 1.4 | 24 | 94 (24) | 27 (1) | 95 (8) | 95 (4) |
In comparison to other PCP (and PNP) pincer catalysts31–34 for HIE reactions (Chart 1), the iridium systems described here appear to be more active under milder conditions. Furthermore, through manipulation of the speciation of the active Ir-Hn compounds, the selectivity of deuteration can also be to some extent controlled. Finally, given the stability of these systems to water, D2O can also be used as a deuterium source rather than C6D6. The results of HIE with a number of substrates, under three sets of conditions are summarized in Fig. 4.
 | ||
| Fig. 4 Distribution of deuterium throughout various substrates following reactions using C6D6 or D2O as a deuterium source. Yields marked black and blue were obtained from reactions with substrates (0.38 mmol) using Ir-Ph (1 mol%) activated by 1.4 mol% (black) or 9.0 mol% (blue) of D2 gas in C6D6 (1.0 mL) and heating to 65 °C for 24 hours. Yields marked in red were obtained from reactions with substrates (0.38 mmol) using Ir-H4 (1 mol%) in C6H12 (0.750 mL) and heating to 80 °C in the presence of D2O for 24 hours. Conversion was measured via1H NMR using cyclohexane as an internal standard. For representative spectra for m-xylene, see Fig. S22–S24 in the ESI.†aConversion after 5 days at 65 °C. bReaction was not performed due to poor solubility. cThese positions are deuterated via uncatalyzed exchange in D2O. | ||
Using the optimized conditions of entry 11 in Table 1 developed for meta-xylene, HIE for several substrates were examined (Fig. 4, black data). Deuterium incorporation was found to be quite high for several arene based substrates with unhindered Csp2–H positions in fluorobenzene, naphthalene, aniline and 2,6-lutidine being deuterated almost quantitatively. In the case of naphthalene even the less reactive bay positions were fully deuterated in contrast to III33 and IV,34 which required longer reaction times and higher temperatures to achieve even partial deuteration of these positions in naphthalene. More interestingly, typically less active Csp3–H positions were significantly deuterated in substrates such as THF, triethylamine, 2,6-lutidine and meta-xylene under very mild conditions with no sign of catalyst decomposition. This level of deuteration for the Csp3–H sites, along with precedents from previous systems,17,24,62 prompted further investigation into the deuteration of aliphatic hydrocarbons. Despite no detectable H/D exchange occurring between benzene-d6 and cyclooctane at 50 °C the increased temperature of 65 °C resulted in mild deuteration of 9% after 24 hours which increased to 36% after 5 days. For n-heptane, deuteration of the terminal methyl groups was found to be 58% while internal methylene positions were deuterated to 37% after 5 days. The 13C{1H} NMR revealed that the majority of deuteration performed on the interior methylene groups occurred the 2 and 6 positions. This is intriguing considering reactivity of cyclopentadienyl Ru complex reported by Nikonov was limited to the terminal methyl C–H sites.24 Reduced activity was also observed with phenol while catalysis was completely halted in the presence of ethyl benzoate and pyridine. The 31P{1H} NMR spectra of these latter reactions showed that complexes Ir-Ph-d5, Ir-D3 and Ir-D4 were no longer present in solution.
While exhaustive deuteration is desirable in some situations, mild, selective deuteration has some interest for drug discovery and evaluation.6,8 The data in Table 1 indicates that, under conditions where Ir-D4 dominates, deuteration is emphasized at the more sterically accessible sites (see entry 9). We therefore examined HIE with the substrates in Fig. 4 under these conditions (blue data). Using these conditions, good selectivity is possible with compounds containing directing groups and less accessible Csp2–H sites. While fluorobenzene still showed almost quantitative deuterium incorporation, naphthalene, 2,6-lutidine and N,N-dimethylbenzamide showed higher levels of deuteration at accessible sites relative to less accessible ones. Catalytic HIE of aniline had a slightly greater preference for –NH and ortho-sites. The increased propensity of the iridium catalyst to bind at the more strongly donating –NH site likely resulted in this mild enhancement. While selectivity was observed in phenol, results were almost identical to the exhaustive conditions with the –OH and ortho-positions being deuterated to 42% and 37%, respectively. More surprisingly, deuteration for ethyl benzoate and pyridine became possible. Ethyl benzoate was significantly deuterated at both the meta- and para-positions while pyridine showed low level deuteration at all sites. For both pyridine and ethyl benzoate, the greater amount of Ir-D4 may stave off immediate catalyst decomposition by providing increased levels of molecular D2 present in solution which help prevent these substrates from strongly binding to the iridium center. Under these conditions deuteration at Csp3–H sites was essentially shut down in all cases.
The use of C6D6 as a deuterium source is convenient but in an effort to carry out the reaction with a more economical source of D, we took advantage of the high tolerance of the iridium complexes of this PCP framework towards water and explored the use of D2O as a deuterating agent for the HIE reaction. Table S2† gives the results of a short optimization study, again using meta-xylene as a substrate, and the red data in Fig. 4 shows the results HIE using D2O with Ir-H4 under static vacuum as the catalyst precursor. To solubilize the iridium species, reactions were performed in a biphasic mixture of D2O and cyclohexane.31,33 For meta-xylene, H/D exchange occurred in trace amounts at the benzyl position, 20% at the 4 and 6 positions and quantitatively at the 5 position. Deuteration of the other substrates in Fig. 4 using D2O gave broadly similar results to those obtained under the “selective” conditions giving rise to the blue data, with a few notable differences. Fluorobenzene was again exhaustively deuterated while naphthalene and 2,6-lutidine had deuterium unevenly distributed throughout the arene systems. Aniline was deuterated almost quantitively in all positions while deuteration in phenol was greatly enhanced to 95%, although limited to the ortho and –OH sites. Deuterium levels in pyridine were found to be slightly higher than in the previous attempts but again catalysis was hampered by catalyst degradation (detected via31P{1H} NMR). Aliphatic hydrocarbons were found to be mostly unreactive though trace H/D exchange had occurred at the terminal methyl positions of n-heptane. Unlike the results produced using Ir-Ph and 9 mol% D2 with benzene-d6, H/D exchange was found to be possible in both triethylamine and THF, albeit in low levels.
On a preparative scale, these biphasic conditions can be used with neat substrates as summarized in Fig. 5. Both benzene and fluorobenzene (8.3 mmol each) were deuterated at all positions to 90% and 94%, respectively, with D2O (305 mmol) at 80 °C using 1 mol% of Ir-H4. Following extraction of the organic layers and distillation of the liquids, Ir-H4 was recovered in excess of 90% and deuterated benzene and fluorobenzene were obtained. The experiments were repeated with the recovered Ir-H4 and deuterium with minimal loss of catalyst performance, as shown in Fig. 5.
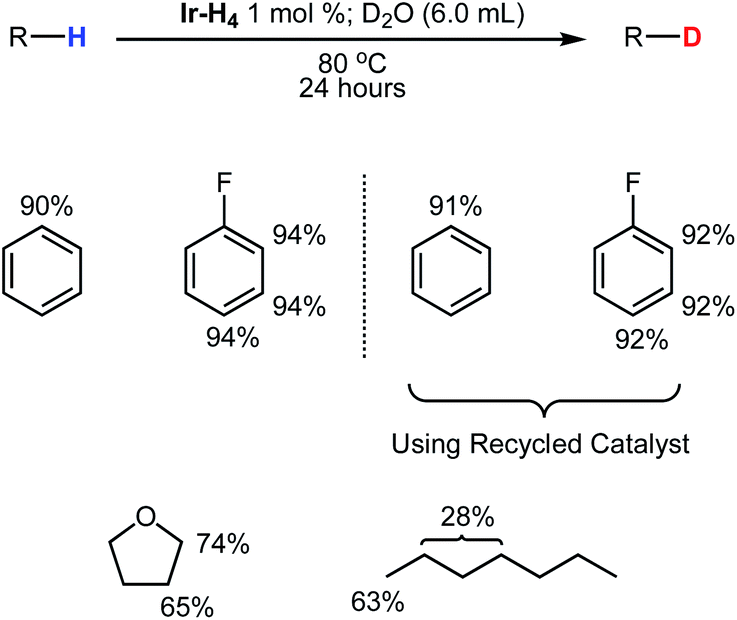 | ||
| Fig. 5 Distribution of deuterium throughout neat immiscible liquids following reactions using D2O. Reactions were heated under static vacuum a 80 °C except for THF (100 °C with NaOH (6 M) dissolved in D2O) and (120 °C). | ||
Using a slightly modified methodology, both THF and n-heptane were partially deuterated and isolated in good yields. Ir-H4 was not soluble in THF/D2O mixtures, so a 6 M solution of NaOH in D2O was employed to render the medium biphasic.63 Heating Ir-H4 (1 mol%) dissolved in neat THF (8.3 mmol) under static vacuum to 100 °C in the presence of a D2O with NaOH (6 M) solution resulted in both the 2 and 3 positions were deuterated to 74% and 65%, respectively. Lower temperatures produced significantly lower yields as did performing the reaction under 1 atmosphere of argon. Deuterated THF was isolated via distillation and the catalyst was again recovered in 90% yield. NMR analysis of the recovered iridium material in THF revealed the mixture was composed of Ir-H4, Ir-H3 and Ir-OD (76%, 3% and 21%, respectively). All material was easily converted back to Ir-H4 by placing the solution under 4 atmospheres of H2 gas. In order to partially deuterate n-heptane slightly higher temperature (120 °C) was required. After 24 hours the material was isolated and NMR analysis revealed n-heptane had been substantially deuterated to 63% at the terminal methyl positions and 28% among all methylene positions.
Conclusions
Since our initial report in 2012 (ref. 35) on iridium complexes of the PCcarbeneP framework, buttressed by robust aryl groups linking the arms of the pincer to the central carbene, the ligand design has evolved40,43,44 into the rigid, planar and highly electron-donating ligand employed here to prepare and isolate Ir-H4. Three features of this advanced ligand design make it a highly effective HIE catalyst system (Scheme 5). The electron richness of the ligand allows for the stabilization of the Ir-H4 form of the catalyst such that it is isolable, storable, and able to fully regenerate if the system is exposed to excess dihydrogen. However, loss of one equivalent of H2 is facile, allowing for access to the Ir-H2/Ir-H3 manifold, which is moderately active as an HIE catalyst and able to effect selective deuteration of sterically accessible positions on some substrates. We propose that further removal of H2, formally speaking, allows access to an even more active Ir-[]/Ir-H catalyst; this species, while not observed experimentally, is implied by the stoichiometry of the reactions that lead to its generation. This species is highly effective at activating a wide variety C–H and C–D bonds via oxidative addition, including bonds normally reticent towards activation. While highly reactive, this species is robust under HIE conditions by virtue of the electron richness of the ligand.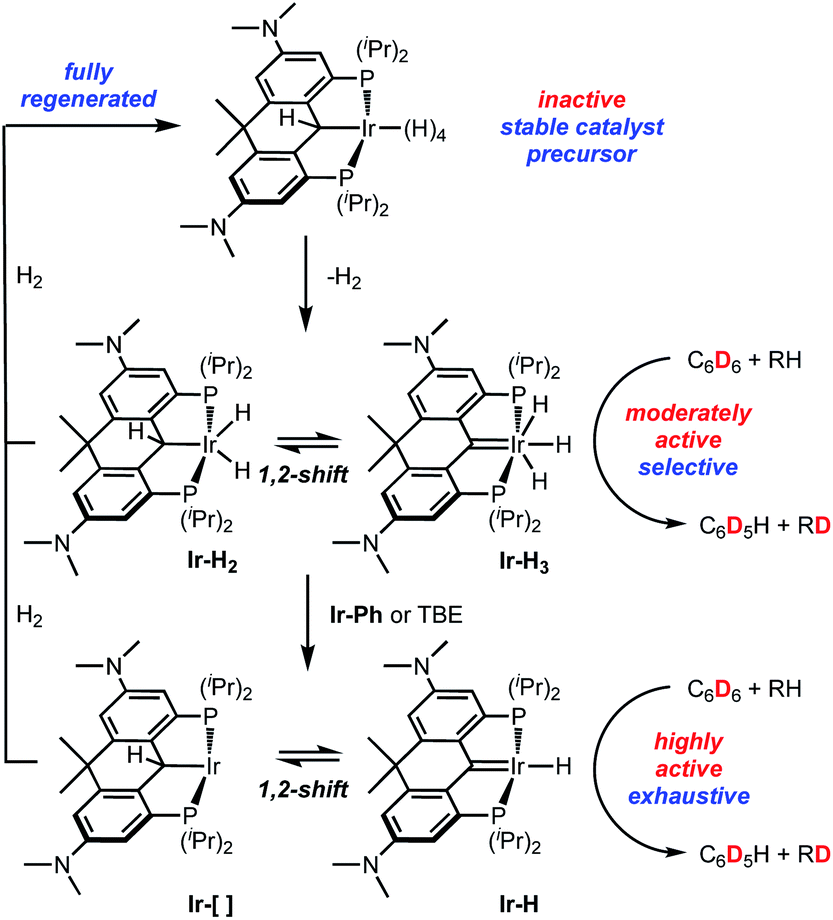 | ||
| Scheme 5 Summary of ligand design features and their implications for HIE activity in Ir-H4. | ||
A second useful feature in this context, is the ability of these compounds to cooperatively move hydrogen atoms from metal to pincer carbene via a low barrier 1,2-shift. This increases their hydrogen capacity in each HIE active manifold while also providing a mechanism for opening coordination space for bond-activating interaction with C6D6/D2O and substrate as is necessary for HIE. This also provides a low barrier path for the H/D scrambling that must occur in order to drive HIE reactions to completion. Finally, the third positive attribute is the rigidity of the ligand framework, as imposed by the aryl groups in the backbone and more crucially the linking Me2C group. This design element imparts thermal stability by avoiding undesirable catalyst deactivation pathways available to the unlinked variants.45 Furthermore, this rigidification also makes the 1,2-shift alluded to above more facile by discouraging pyramidalization of the pincer ligand anchoring carbon atom. This has the effect of “flattening the curve” on the energy surface joining the PCsp3P (as in Ir-H2 and Ir-[]) and PCcarbeneP (e.g.Ir-H3 and Ir-H) forms of the active species by making the C–H bond in the PCsp3P form weaker and rendering the overall HIE process more efficient. Thus, through advanced ligand design, we have developed a robust, recyclable catalyst for the selective or near exhaustive deuteration of a number of substrates, including on a preparative scale, under mild conditions and using C6D6 or D2O as a deuterium source.
Experimental section
General considerations
All compounds were stored, handled and manipulated, unless otherwise stated, using standard techniques in a VAC glovebox or on a double manifold high vacuum line under an atmosphere of argon purified via passage through an OxisorBW scrubber (Model 641-02 Matheson Gas Products). An M-Braun solvent purification system was used to dry n-pentane which was subsequently stored over sodium/benzophenone ketal in a 500 mL thick-walled glass pressure flask. Benzene, benzene-d6, toluene, toluene-d8, THF-d8 and cyclohexane were dried over sodium/benzophenone ketal and stored under vacuum in a 100 mL thick-walled glass pressure flask. All dried solvents were degassed and vacuum distilled prior to use. Triethylamine, aniline, pyridine and 2,6-lutidine were dried over CaH2 and distilled prior to use. Fluorobenzene and ethyl benzoate were dried over 3 Å molecular sieves and filtered off. Methylcyclohexane-d14, cyclohexane-d12, cyclooctane, and n-heptane were purchased from Sigma-Aldrich and degassed via freeze–pump–thaw prior to use. Phenol, N,N-dimethylbenzamide and naphthalene were placed under 1 atmosphere of argon prior to use. H2 (99.5%) and D2 (99.98%) were purchased from Praxair and used without further purification. Iridium(III) chloride hydrate was purchased from Pressure Chemicals Inc. and used as received. [Ir(COE)2Cl]2 and Ir-Cl were prepared as previously reported.44,641H and 13C{1H} NMR chemical shifts were referenced to residual solvent protons and naturally abundant 13C resonances for all deuterated solvents.65 Chemical shift assignments are based on 1H, 31P{1H}, 13C{1H}, 1H–1H–COSY, 1H–13C–HSQC and 1H–13C HMBC NMR experiments performed on Bruker Ultrashield 400, Ascend-500, or Avance-600 MHz spectrometers using TopSpin version 3.2 software. X-ray crystallographic analyses were performed on a Nonius system equipped with a Bruker Apex-II CCD using samples coated in Paratone 8277 oil (Exxon) and mounted on a glass fibre. Elemental analyses were performed by staff of the Instrumentation Facility in the Department of Chemistry, University of Calgary.Synthesis of Ir-OH
To a 100 mL thick walled round bottomed pressure flask charged with CsOH·H2O (0.866 g, 5.16 mmol) and a Teflon stir bar, Ir-Cl (0.388 g, 0.516 mmol) dissolved in toluene (25 mL) was added. The flask was sealed and heated to 120 °C. After stirring for 24 hours the green-brown solution had turned dark brown-red. This solution was cooled to room temperature and filtered through a pad of Celite® into a 50 mL round bottom flask. All volatiles were removed from the filtrate in vacuo and n-pentane (20 mL) was vacuum transferred into the flask at −78 °C. The flask was warmed to room temperature and the solution was stirred for 2 hours. All volatiles were removed in vacuo. A brown powder (0.301 g, 0.410 mmol) was isolated in 80% yield. Elemental analysis calcd (%): C, 52.37; H, 7.00; N, 3.82. Found (average of 4 trials): C, 53.41; H, 7.25; N, 3.59 (the carbon value is high due to small amounts of unremovable toluene). APCI-MS (M*+) for C32H51IrN2OP2: calcd: 734.3100. Found: 734.3070. 1H NMR (500 MHz, C6D6) δ 6.70 (dvt, 4JHH = 2.3 Hz, JHP = 3.7 Hz, 2H, ArH), 6.34 (d, 4JHH = 2.3 Hz, 2H, ArH), 3.64 (t, 2JHP = 3.8 Hz, 1H, Ir-OH), 2.95 (m, 4H, PCH(CH3)2), 2.36 (s, 12H, N(CH3)2), 1.65 (s, 6H (18H with PCH(CH3)2), –C(CH3)2–), 1.63 (dvt, 3JHH = 7.5 Hz, JHP = 7.3 Hz, 12H (18H with –C(CH3)2–), PCH(CH3)2), 1.40 (dvt, 3JHH = 7.1 Hz, JHP = 7.1 Hz, 12H, PCH(CH3)2). 13C{1H} NMR (126 MHz, C6D6) δ 197.0 (t, 2JCP = 2.2 Hz, IrC), 158.7 (vt, JCP = 18.8 Hz, ArC), 149.9 (vt, JCP = 3.9 Hz, ArC), 142.5 (vt, JCP = 20.0 Hz, ArC) 130.3 (vt, JCP = 7.8 Hz, ArC), 116.3 (s, ArCH), 114.4 (s, ArCH), 42.7 (s, –C(CH3)2–), 40.2 (s, N(CH3)2), 28.2 (s, –C(CH3)2–), 25.3 (vt, JCP = 12.8 Hz, PCH(CH3)2), 20.0 (vt, JCP = 2.7 Hz, PCH(CH3)2), 19.7 (s, PCH(CH3)2). 31P{1H} NMR (203 MHz, C6D6) δ 51.9 (s).
Synthesis of Ir-H4
Method 1: to a 100 mL thick walled glass pressure vessel equipped with a Teflon stirbar, Ir-OH (0.196 g, 0.267 mmol) dissolved in benzene (15 mL) was loaded. The mixture was degassed via the freeze–pump–thaw method and placed under 4 atm of H2 gas. After stirring for 3 hours all volatiles were removed in vacuo resulting in a dark red-brown residue. To this n-pentane (25 mL) were added via vacuum transfer at −78 °C. The solution was warmed to room temperature and stirred for 20 minutes under 1 atm of H2. The n-pentane was removed under high vacuum and a beige powder (0.188 g, 0.260 mmol) was isolated in 98% yield. Elemental analysis calcd (%): C, 53.24; H, 7.68; N, 3.88. Found: C, 53.48; H, 8.11; N, 3.73. Method 2: to a 100 mL thick walled glass pressure vessel charged with NaOH (0.266 g, 6.65 mmol) and a Teflon stirbar, Ir-Cl (0.500 g, 0.665 mmol) dissolved in benzene (35 mL) was loaded. The mixture was degassed via the freeze–pump–thaw method and then placed under 4 atm of H2 gas. After stirring for 3 hours the now orange-yellow solution was degassed and filtered through a pad of Celite® in a glovebox. The filtrate was placed in a 50 mL round bottom flask and all volatiles were removed in vacuo. To the dark-red residue n-pentane (25 mL) was vacuum transferred at −78 °C. The mixture was warmed to room temperature, placed under 1 atmosphere of H2 gas and stirred for 20 minutes. All volatiles were removed in vacuo and a beige powder (0.454 g, 0.627 mmol) was isolated in 94% yield. X-ray quality crystals were obtained by slowly cooling a supersaturated solution of Ir-H4 in isopropanol from 50 °C to 20 °C. Elemental analysis calcd (%): C, 53.24; H, 7.68; N, 3.88. Found: C, 53.43; H, 7.88; N, 3.80. 1H NMR (500 MHz, C6D12) δ 6.84 (d, 4JHH = 1.9 Hz, 2H, ArH), 6.48 (dvt, 4JHH = 1.9 Hz, JHP = 4.0 Hz, 2H, ArH), 5.52 (s, 1H, Ir–CH), 2.85 (s, 12H, N(CH3)2), 2.10 (m, 2H, PCH(CH3)2), 1.91 (m, 2H, PCH(CH3)2), 1.84 (s, 3H, –C(CH3)2–), 1.23 (s, 3H, –C(CH3)2), 1.16 (dvt, 3JHH = 7.9 Hz, JHP = 7.4 Hz, 6H, PCH(CH3)2), 1.07 (m, 12H, PCH(CH3)2), 0.78 (dvt, 3JHH = 8.0 Hz, JHP = 7.3 Hz, 6H, PCH(CH3)2), −10.32 (t, 2JHP = 10.7 Hz, 4H). 13C{1H} NMR (126 MHz, C6D12) δ 151.6 (vt, JCP = 15.4 Hz, ArC) 149.2 (vt, JCP = 4.3 Hz, ArC), 145.2 (vt, JCP = 7.4 Hz, ArC) 139.6 (vt, JCP = 24.4 Hz, ArC), 112.1 (s, ArCH), 111.6 (s, ArCH), 41.9 (s, N(CH3)2), 40.8 (s, –C(CH3)2–), 30.3 (s, –C(CH3)2–), 30.1 (vt, JCP = 14.8 Hz, PCH(CH3)2), 25.0 (s, –C(CH3)2–), 23.1 (vt, JCP = 16.7 Hz, PCH(CH3)2), 21.0 (s, Ir–CH), 20.2 (s, PCH(CH3)2), 20.2 (vt, JCP = 2.2 Hz, PCH(CH3)2), 19.4 (vt, JCP = 3.7 Hz, PCH(CH3)2), 18.0 (s, PCH(CH3)2). 31P{1H} NMR (203 MHz, C6D12) δ 56.5 (s).Synthesis of Ir-Ph
To a 25 mL round bottom flask equipped with a Teflon stirbar, Ir-H4 (0.180 g, 0.249 mmol) dissolved in benzene (10 mL) was added. The solution was stirred at room temperature in a glovebox and tert-butylethylene (0.105 g, 1.069 mmol) was added. After stirring for 10 minutes all volatiles were removed in vacuo. The dark green-brown residue was then sonicated in n-pentane (5 mL). The solid was then filtered through a fritted Büchner funnel and washed with n-pentane (3 × 2 mL). A dark green powder (0.143 g, 0.180 mmol) was isolated in 73% yield. X-ray quality crystals were obtained from a saturated solution of Ir-Ph in n-pentane that was sealed at room temperature in a 20 mL scintillation vial for 1 week. Elemental analysis calcd (%): C, 57.48; H, 6.98; N, 3.53. Found: C, 57.59; H, 6.99; N, 3.52. 1H NMR (500 MHz, C6D6) δ 7.54 (t, 3JHH = 7.4 Hz, 2H, Ph-H), 7.14 (d, 3JHH = 7.3 Hz, 2H, Ph-H), 6.97 (t, 3JHH = 7.3 Hz, 1H, Ph-H), 6.74 (m, 2H, Ar-H), 6.25 (d, 4JHH = 2.2 Hz, 2H, Ar-H), 3.16 (m, 2H, P(CH(CH3)2), 2.25 (s, 12H, N(CH3)2), 1.63 (s, 6H, –C(CH3)2), 1.40 (dvt, 3JHH = 7.0 Hz, 3JHP = 7.0 Hz 12H, PCH(CH3)2), 1.35 (dvt, 3JHH = 7.9 Hz, 3JHP = 7.3 Hz 12H, PCH(CH3)2). 13C{1H} NMR (126 MHz, C6D6) δ 221.1 (t, 2JCP = 4.0 Hz, CIr), 171.8 (vt, JCP = 8.8 Hz, PhC-Ir), 157.7 (vt, JCP = 19.2 Hz, ArC), 151.4 (vt, JCP = 3.9 Hz, ArC), 147.3 (vt, JCP = 17.9 Hz, ArC), 142.2 (vt, JCP = 7.3 Hz, ArC), 140.2 (s, PhCH), 126.6 (s, PhCH), 120.8 (s, PhCH), 116.6 (s, ArCH), 113.7 (s, ArCH), 43.5 (s, –C(CH3)2–), 40.2 (s, N(CH3)2), 27.4 (s, –C(CH3)2–), 24.3 (vt, JCP = 13.3 Hz, PCH(CH3)2), 20.1 (vt, JCP = 2.4 Hz, PCH(CH3)2), 19.3 (br s, PCH(CH3)2). 31P{1H} NMR (203 MHz, C6D6) δ 53.6 (s).
Generation of Ir-H3
To a J.-Young tube charged with Ir-Ph (0.010 g, 0.025 mmol) dissolved in C6H6 (0.25 mL), Ir-H4 (0.013 g, 0.037 mmol) in C6H6 (0.35 mL) was added dropwise over 5 minutes. The solution changed from dark green to a dark reddish brown color. Ir-H3 is generated in situ and is unisolable. 1H NMR (500 MHz, C6H6 spiked with C6D12) δ 6.78 (m, Ar-H), 6.63 (d, 4JHH = 2.5 Hz Ar-H), 2.58 (s, 12H, N(CH3)2), 2.25 (m, 4H, PCH(CH3)2), 1.63 (s, 6H, –C(CH3)2–), 1.46 (dvt, JHP = 7.5 Hz, PCH(CH3)2), 1.14 (m, PCH(CH3)2). 13C{1H} NMR (126 MHz, C6H6 spiked with C6D12) δ 245.0 (br s, CIr); 152.4 (vt, JCP = 19.8 Hz, ArC), 152.1 (vt, JCP = 19.0 Hz, ArC), 150.4 (vt, JCP = 3.3 Hz, ArC), 140.1 (vt, JCP = 6.9 Hz, ArC), 115.4 (s, ArCH), 112.3 (s, ArCH), 41.3 (s, –C(CH3)2–), 40.8 (s, N(CH3)2), 32.1 (s, –C(CH3)2–), 27.3 (vt, JCP = 15.6 Hz, PCH(CH3)2), 21.9 (s, PCH(CH3)2), 19.8 (s, PCH(CH3)2). 31P{1H} NMR (203 MHz, C6D6) δ 66.4 (s).
Preparation of stock solutions
Solution A: complex Ir-Ph (0.060 g, 0.076 mmol, 0.015 M) was dissolved with benzene-d6 in a volumetric flask (5 mL). Solution B: cyclohexane was added to a volumetric flask (5 mL) containing complex Ir-H4 (0.068 g, 0.094 mmol, 0.019 M) and cyclohexane-d12 (0.550 g, 5.07 mmol, 1.15 M). Solution C: cyclohexane was added to a volumetric flask (1 mL) containing Solution B (0.450 mL) and substrate (0.85 mmol).General procedure for deuteration of substrates in C6D6
Stock Solution A (0.80 mL) was added to a glass reaction tube (length: 8.5 cm length, inner diameter: 1.5 cm) equipped with a Teflon stirbar. The flask was sealed with a rubber stopper and 1.4 mol% (0.405 mL, 0.017 mmol at 20 °C) or 9 mol% (2.6 mL, 0.11 mmol) D2 gas was injected into the flask. After stirring for 10 minutes at 20 °C, the solution (0.250 mL, 0.0038 mmol of [Ir]) was injected into a J.-Young tube containing substrate (0.38 mmol), cyclohexane (0.046 mmol) and benzene-d6 (0.75 mL). The solution was heated to 65 °C for 24 hours and conversion was measured using cyclohexane as an internal standard. Cyclohexane was monitored without the use of an internal standard; no detectable traces of deuterium incorporation were visible using 1H, 2H or 13C{1H} NMR.General procedure for deuteration of substrates in cyclohexane with D2O
Stock Solution C (0.450 mL) was added to a pressure tube (10 mL) charged with cyclohexane (0.300 mL), argon sparged D2O (0.4 mL, 0.022 mmol) and a Teflon stirbar. Another sample was prepared by placing stock Solution C (0.450 mL) and cyclohexane (0.300 mL) in an NMR tube; this sample was used as a starting material reference. The flask was sealed under argon (1 atm) with a Teflon valve and heated to the appropriate temperature. After 24 hours the organic phase was carefully transferred to an NMR tube under argon without exposure to air. Conversion was measured via1H NMR using the cyclohexane signal as the internal standard. The reference samples containing water miscible/soluble compounds (THF, triethylamine, 2, 6-lutidine, pyridine, aniline and phenol) were extracted with D2O prior to running NMR. The 1H NMR spectra of the organic phases for these samples were compared to the organic phases of the reaction mixtures to measure conversion. Aliquots of the D2O fractions (0.2 mL) for both the reference and reaction mixtures were also taken and added to separate NMR samples containing D2O (0.4 mL) and DMSO (10 μL). The 1H NMR spectra for these samples indicated comparable levels of deuteration were present in the D2O phase.General procedure for deuteration of neat benzene, fluorobenzene and n-heptane with D2O
Substrate (8.31 mmol) was loaded into a pressure tube (10 mL) charged with Ir-H4 (0.060 g, 0.083 mmol) and a Teflon stirbar. Argon sparged D2O (6.0 mL, 330 mmol) was carefully added to the reaction flask under a steady flow of argon. The vessel was sealed with a Teflon valve and heated to 80 °C for 24 hours. The flask containing n-heptane was freeze–pump–thawed three-fold and heated to 120 °C under static vacuum for 24 hours. Organic phases were extracted under a flow of argon via syringe and liquids were distilled under reduced pressure from the iridium catalyst. Benzene was recovered in 81% yield (0.561 g, 6.72 mmol) with 90% deuterium incorporation. Fluorobenzene was recovered in 82% yield (0.725 g, 6.81 mmol) with 94% deuterium incorporation at all positions. n-Heptane was recovered in 88% yield (0.791 g, 7.40 mmol) with 63% deuterium at the terminal methyl positions and 28% deuterium incorporation throughout the methylene position.Deuteration of THF with D2O
THF (0.600 g, 8.32 mmol) was added to a pressure tube (10 mL) charged with Ir-H4 (0.060 g, 0.083 mmol) and a Teflon stirbar. Argon sparged D2O (6 mL) containing NaOH (1.45 g, 36.3 mmol, 6 M) was added to the flask under a flow of argon. The mixture was freeze–pump–thawed three-fold and sealed under static vacuum. After heating at 100 °C for 24 hours the organic phase was extracted under a flow of argon via syringe and distilled under reduced pressure from the iridium catalyst. Deuterated THF was recovered in 84% yield (0.547 g, 7.03 mmol) with deuterium incorporated in 74% and 65% at the 2 and 3 positions, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by NSERC of Canada (Discovery Grant) and the Canada Research Chair secretariat (Tier I CRC 2013–2020) to W. E. P. Author J. D. S thanks NSERC of Canada for PGSD Scholarship support. D. H. E acknowledges the United States National Science Foundation Chemical Structure, Dynamics, and Mechanisms B (CSDM-B) Program for support under award CHE 1952420. We thank Brigham Young University and the Office of Research Computing, especially the Fulton Supercomputing Lab. G. D. thanks the BYU Department of Chemistry and Biochemistry for undergraduate research awards.Notes and references
- J. Yang, Deuterium: Discovery and Applications in Organic Chemistry, Elsevier, Amsterdam, 2016 Search PubMed.
- J. L. Garnett and R. J. Hodges, J. Am. Chem. Soc., 1967, 89, 4546–4547 CrossRef CAS.
- J. L. Garnett and R. S. Kenyon, J. Chem. Soc. D, 1971, 1227–1228 RSC.
- N. F. Goldshleger, M. B. Tyabin, A. E. Shilov and A. A. Shteinman, Zh. Fiz. Khim., 1969, 43, 1222–1223 Search PubMed.
- A. E. Shilov and A. A. Shteinman, Coord. Chem. Rev., 1977, 24, 97–143 CrossRef CAS.
- T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed.
- R. Pony Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed. Engl., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406 RSC.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed. Engl., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed. Engl., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- R. H. Crabtree, H. Felkin and G. E. Morris, J. Organomet. Chem., 1977, 141, 205–215 CrossRef CAS.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1982, 104, 352–354 CrossRef CAS.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3929–3939 CrossRef CAS.
- B. A. Arndtsen and R. G. Bergman, Science, 1995, 270, 1970–1973 CrossRef CAS.
- J. T. Golden, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 5837–5838 CrossRef CAS PubMed.
- S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS PubMed.
- G. N. Nilsson and W. J. Kerr, J. Labelled Compd. Radiopharm., 2010, 53, 662–667 CrossRef CAS.
- S. R. Klei, T. D. Tilley and R. G. Bergman, Organometallics, 2002, 21, 4905–4911 CrossRef CAS.
- M. B. Skaddan, C. M. Yung and R. G. Bergman, Org. Lett., 2004, 6, 11–13 CrossRef CAS PubMed.
- C. M. Yung, M. B. Skaddan and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 13033–13043 CrossRef CAS PubMed.
- L. L. Santos, K. Mereiter, M. Paneque, C. Slugovc and E. Carmona, New J. Chem., 2003, 27, 107–113 RSC.
- J. L. Rhinehart, K. A. Manbeck, S. K. Buzak, G. M. Lippa, W. W. Brennessel, K. I. Goldberg and W. D. Jones, Organometallics, 2012, 31, 1943–1952 CrossRef CAS.
- S. H. Lee, S. I. Gorelsky and G. I. Nikonov, Organometallics, 2013, 32, 6599–6604 CrossRef CAS.
- Topics in Organometallic Chemistry: Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Heidelberg, 2013 Search PubMed.
- D. Morales-Morales, Pincer Compounds: Chemistry and Applications, Elsevier, Amsterdam, 2018 Search PubMed.
- A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed.
- N. A. Eberhardt and H. Guan, Pincer Compounds: Chemistry and Applications, Elsevier, Amsterdam, 2018 Search PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- M. H. G. Prechtl, M. Hoelscher, Y. Ben-David, N. Theyssen, D. Milstein and W. Leitner, Eur. J. Inorg. Chem., 2008, 3493–3500 CrossRef CAS.
- M. H. G. Prechtl, M. Hölscher, Y. Ben-David, N. Theyssen, R. Loschen, D. Milstein and W. Leitner, Angew. Chem., Int. Ed. Engl., 2007, 119, 2319–2322 CrossRef.
- J. Zhou and J. F. Hartwig, Angew. Chem., Int. Ed. Engl., 2008, 47, 5783–5787 CrossRef CAS PubMed.
- V. M. Iluc, A. Fedorov and R. H. Grubbs, Organometallics, 2011, 31, 39–41 CrossRef.
- A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Organometallics, 2016, 35, 2600–2608 CrossRef CAS.
- R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949–2952 CrossRef CAS.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059–6064 CrossRef CAS.
- S. Sung and R. D. Young, Dalton Trans., 2017, 46, 15407–15414 RSC.
- P. Cui and V. M. Iluc, Chem. Sci., 2015, 6, 7343–7354 RSC.
- S. Sung, Q. Wang, T. Krämer and R. D. Young, Chem. Sci., 2018, 9, 8234–8241 RSC.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef CAS PubMed.
- C. Yoo and Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 9502–9506 CrossRef CAS PubMed.
- D. Sahoo, C. Yoo and Y. Lee, J. Am. Chem. Soc., 2018, 140, 2179–2185 CrossRef CAS PubMed.
- J. D. Smith, J. R. Logan, L. E. Doyle, R. J. Burford, S. Sugawara, C. Ohnita, Y. Yamamoto, W. E. Piers, D. M. Spasyuk and J. Borau-Garcia, Dalton Trans., 2016, 45, 12669–12679 RSC.
- J. D. Smith, E. Chih, W. E. Piers and D. M. Spasyuk, Polyhedron, 2018, 155, 281–290 CrossRef CAS.
- J. D. Smith, J. Borau-Garcia, W. E. Piers and D. Spasyuk, Can. J. Chem., 2016, 94, 293–296 CrossRef CAS.
- L. E. Doyle, W. E. Piers, J. Borau-Garcia, M. J. Sgro and D. M. Spasyuk, Chem. Sci., 2016, 7, 921–931 RSC.
- E. A. LaPierre, W. E. Piers, D. M. Spasyuk and D. W. Bi, Chem. Commun., 2016, 52, 1361–1364 RSC.
- E. A. LaPierre, W. E. Piers and C. Gendy, Dalton Trans., 2018, 47, 16789–16797 RSC.
- C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048–9051 RSC.
- L. E. Doyle, W. E. Piers and D. W. Bi, Dalton Trans., 2017, 46, 4346–4354 RSC.
- E. A. LaPierre, W. E. Piers and C. Gendy, Organometallics, 2018, 37, 3394–3398 CrossRef CAS.
- S. Sung, T. Joachim, T. Krämer and R. D. Young, Organometallics, 2017, 36, 3117–3124 CrossRef CAS.
- H. Tinnermann, S. Sung, B. A. Cala, H. J. Gill and R. D. Young, Organometallics, 2020, 39, 797–803 CrossRef CAS.
- R. J. Burford, W. E. Piers, D. H. Ess and M. Parvez, J. Am. Chem. Soc., 2014, 136, 3256–3263 CrossRef CAS PubMed.
- P. J. Desrosiers, L. Cai, Z. Lin, R. Richards and J. Halpern, J. Am. Chem. Soc., 1991, 113, 4173–4184 CrossRef CAS.
- T. J. Hebden, K. I. Goldberg, D. M. Heinekey, X. Zhang, T. J. Emge, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 2010, 49, 1733–1742 CrossRef CAS PubMed.
- F. Maseras, A. Lledós, M. Costas and J. M. Poblet, Organometallics, 1996, 15, 2947–2953 CrossRef CAS.
- S. Gründemann, H.-H. Limbach, G. Buntkowsky, S. Sabo-Etienne and B. Chaudret, J. Phys. Chem. A, 1999, 103, 4752–4754 CrossRef.
- D. Devarajan and D. H. Ess, Inorg. Chem., 2012, 51, 6367–6375 CrossRef CAS PubMed.
- A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2015, 6, 2060–2067 RSC.
- A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem.–Eur. J., 2016, 22, 4078–4086 CrossRef CAS PubMed.
- R. Corberan, M. Sanau and E. Peris, J. Am. Chem. Soc., 2006, 128, 3974–3979 CrossRef CAS PubMed.
- Y. Xie, F. Yu, Y. Wang, X. He, S. Zhou and H. Cui, Fluid Phase Equilib., 2019, 493, 137–143 CrossRef CAS.
- J. L. Herde, J. C. Lambert, C. V. Senoff and M. A. Cushing, in Inorganic Syntheses, ed. G. W. Parshall, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, pp. 18–20 Search PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization data, experimental and computational details. CCDC 2002787 for Ir-H4 and 2002788 for Ir-Ph. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02694h |
| This journal is © The Royal Society of Chemistry 2020 |