
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA reconstructed porous copper surface promotes selectivity and efficiency toward C2 products by electrocatalytic CO2 reduction†
Jianyu
Han
a,
Chang
Long
ab,
Jing
Zhang
c,
Ke
Hou
a,
Yi
Yuan
ad,
Dawei
Wang
a,
Xiaofei
Zhang
ab,
Xueying
Qiu
ab,
Yanfei
Zhu
ae,
Yin
Zhang
a,
Zhongjie
Yang
ae,
Shuhao
Yan
ae and
Zhiyong
Tang
 *ae
*ae
aCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: zytang@nanoctr.cn
bSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150080, P. R. China
cInstitute of Applied Chemistry, Shanxi University, Taiyuan 030006, P. R. China
dSchool of Chemistry, Beihang University, Beijing 100083, P. R. China
eUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 19th May 2020
Abstract
Electrocatalytic synthesis of multicarbon (C2+) products from CO2 reduction suffers from poor selectivity and low energy efficiency. Herein, a facile oxidation–reduction cycling method is adopted to reconstruct the Cu electrode surface with the help of halide anions. The surface composed of entangled Cu nanowires with hierarchical pores is synthesized in the presence of I−, exhibiting a C2 faradaic efficiency (FE) of 80% at −1.09 V vs. RHE. A partial current density of 21 mA cm−2 is achieved with a C2 half-cell power conversion efficiency (PCE) of 39% on this electrode. Such high selective C2 production is found to mainly originate from CO intermediate enrichment inside hierarchical pores rather than the surface lattice effect of the Cu electrode.
Introduction
The electrochemical synthesis of value-added feedstocks by carbon dioxide (CO2) reduction is an appealing approach for permanent storage of renewable electricity and closing the carbon neutral cycle.1,2 Multicarbon (C2+) products including ethylene, ethanol and propanol are highly desirable owing to their diverse applications in industry.3–5 However, the multistep nature and the competing pathways make it a challenge to drive the reactions to the desired C2+ products.Copper (Cu) is currently known to be the most promising electrocatalyst that may improve the formation of C2+ products via CO2 reduction.6,7 One major approach attempting to endow Cu with the preference for C2+ products is to fabricate oxide-derived Cu (OD-Cu) surfaces. Thermal oxidation,8,9 anodic treatment,10,11 electrodeposition12,13 and hydrothermal14 synthesis have been reported to create surface oxide layers. Nevertheless, regardless of the methods employed, the enhanced C2 selectivity is typically attributed to two mechanisms, one where the residual subsurface Cu+ under the reduction conditions stabilizes the CO intermediates, thus lowering the overpotential and boosting C2 selectivity,9,15 and the other where the oxide-derived facets or undercoordinated surface Cu sites favor C2 formation.5,16
We note that the reconstructed surface morphology of OD-Cu is seldom considered, although it is essential in catalysis.17,18 The porous and inhomogeneous surfaces, which are generally generated upon rapid removal of lattice oxygen under a very negative reduction potential,12 would have large surface roughness and hence benefit C2+ formation.19 Previous studies have revealed that porous structures boost C2+ production from either CO or CO2 electroreduction through a confinement effect.20–22
In this work, halide anions are utilized to reconstruct the Cu surface via electrochemical redox treatment. A Cu film consisting of entangled Cu nanowires (Re-Cu–I) is formed when I− is involved, and this morphology is rarely reported by previous studies adopting similar methods.10,11,23–25 It exhibits better selectivity toward C2 generation compared with Cu nanoparticles obtained with Br− and Cl−. It is found that the hierarchical pores rather than the reconstruction-derived properties are responsible for this enhancement, which is distinct from the mechanism proposed in the literature.11,23,24 The porous structure from the disordered accretion of Re-Cu–I is capable of enriching CO to enhance the local *CO coverage at the surface, consequently boosting C2 species generation. A C2 faradaic efficiency (FE) of 80% is achieved with a partial current density of 21 mA cm−2 at −1.09 V vs. the reversible hydrogen electrode (RHE), corresponding to a C2 half-cell PCE of 39% on this electrode.
Results and discussion
Catalyst preparation and characterization
The surface of the polycrystalline electropolished Cu (EP-Cu) electrode is reconstructed by electrochemical oxidative–reductive cycling in 0.1 M KHCO3 aqueous solution with the help of halide anions (Fig. 1a). Without addition of potassium halides, a small redox wave with the current density in microampere per square centimeter is discerned, suggesting that the electrode surface reaction is mild (black curve in Fig. S1†). Upon addition of KI, two orders of magnitude higher current density is observed at the oxidative potential (0.5 V to 0.8 V vs. RHE), which is ascribed to the formation of CuI (purple curve in Fig. S1†). The insoluble CuI formed passivates the Cu electrode surface and consequently prevents further dissolution of the Cu surface,23 causing a dramatically decreased current density at 0.9 V vs. RHE. The as-generated Cu is then reduced at potentials ranging from 0.3 V to −0.3 V vs. RHE to form metallic Cu. On the other hand, in the presence of KCl or KBr, the linearly increased current density is distinguished at oxidative potentials from 0.5 V to 0.9 V vs. RHE, indicating the continuous dissolution of Cu (green and orange curves in Fig. S1†). Subsequently, the as-dissolved Cu is transformed into Cu2O and redeposited onto the electrode surface under a reductive scan,10 finally reducing to metallic Cu at more negative potentials from −0.3 V to −0.6 V vs. RHE compared with the case in the presence of KI.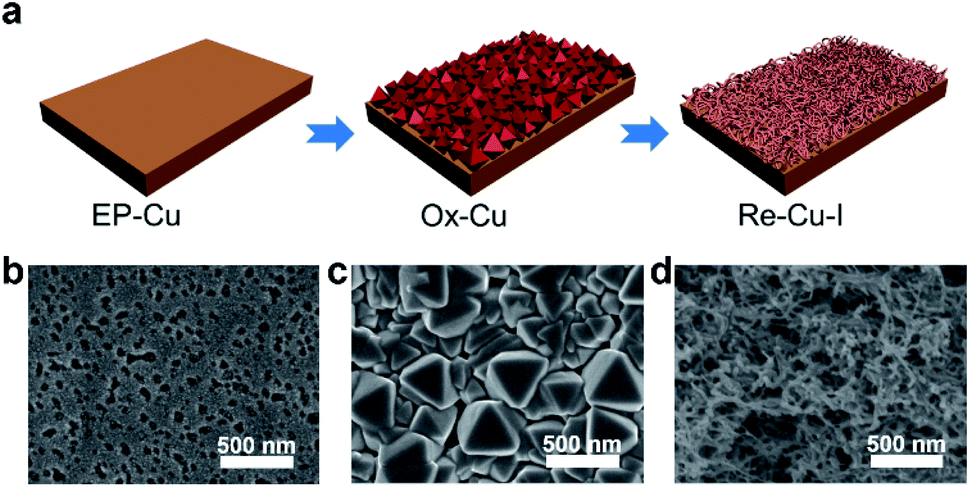 | ||
| Fig. 1 Schematic illustration and morphology characterization of the surface reconstruction process. (a) Scheme of the electrochemical redox reconstruction process. SEM images of EP-Cu (b), Ox-Cu (c) and Re-Cu–I (d). | ||
Grazing incidence X-ray diffraction (GIXRD), scanning electron microscopy (SEM) and the corresponding energy-dispersive X-ray spectroscopy (EDS) are conducted to monitor the phase transformation during the surface reconstruction process of the Cu electrode in the presence of KI. Initially, a rather flattened surface is observed for the Cu electrode after being electropolished (EP) in phosphoric acid (Fig. 1b and S2†). And the in-plane surface of EP-Cu is dominated by the Cu (200) facets (yellow curve in Fig. 2a). Under oxidative conditions, polyhedra are found to fully cover the electrode surface (Fig. 1c). The SEM images and the corresponding EDS elemental mapping indicate that these polyhedra are characteristic of Cu and I with a molar ratio of 2.7 to 1 (Fig. S3 and Table S1†). The XRD pattern of the oxidized Cu (Ox-Cu) suggests that the in-plane surface consists of CuI and metallic Cu (pink curve in Fig. 2a). After the reductive potential is applied, Cu nanowires with diameters of 23.4 ± 5.9 nm (Re-Cu–I) are produced and entangled all around the surface, giving rise to a hierarchical porous structure (Fig. 1d, S4 and S5†). Only Cu and O are detected by EDS elemental mapping with a molar percentage of 88.2% and 10.8% (Fig. S6 and Table S1†). After reduction, two phases are observed. The major Cu phase is dominated by Cu (220) along with Cu (200) facets, while Cu2O with (111) and (200) facets is the minor phase (purple curve in Fig. 2a).
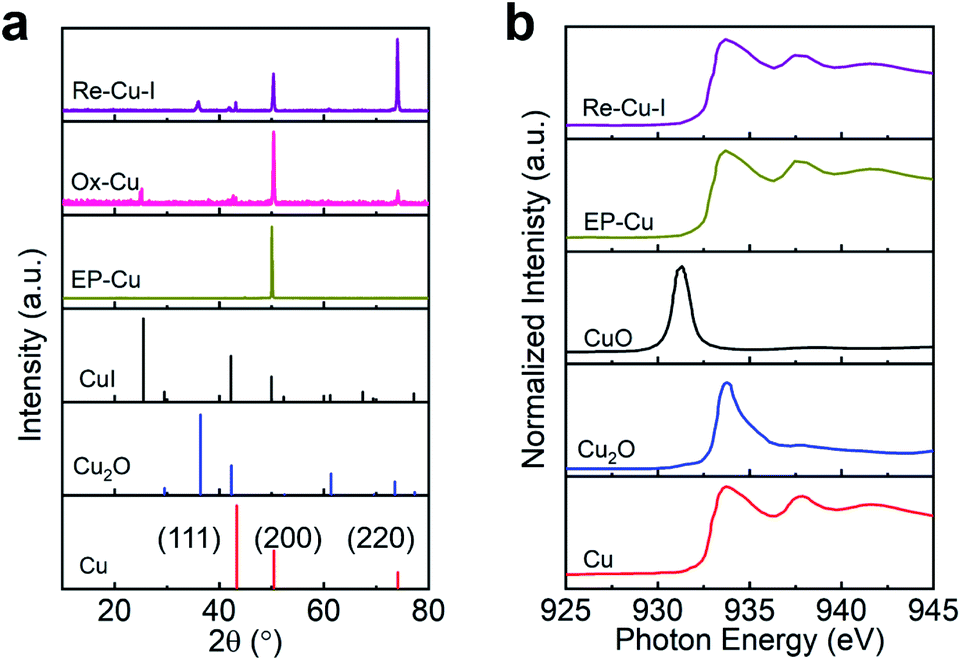 | ||
| Fig. 2 Structural characterization of EP-Cu, Ox-Cu and Re-Cu–I. (a) GIXRD patterns of Re-Cu–I (purple), Ox-Cu (pink), EP-Cu (yellow) and reference samples CuI (black, PDF#06-0246), Cu2O (blue, PDF#05-0667), and Cu (red, PDF#04-0836). (b) Cu L-edge XAS spectra of Re-Cu–I (purple), EP-Cu (yellow) and reference samples CuO (black), Cu2O (blue) and Cu (red). | ||
Obviously different from the case with KI as electrolyte, a rather dense structure consisting of quasi-cubic nanoparticles is obtained after electrochemical surface reconstruction of the Cu electrode in the presence of KBr or KCl (Fig. S7–S10†). And the molar percentage of O slightly increases with respect to that in Re-Cu–I (Table S1†). As displayed in Fig. S11,† the XRD patterns show that the major phase in Re-Cu–Br and Re-Cu–Cl is also metallic Cu accompanied by Cu2O as the minor phase. The difference is that Cu (200) is the dominating facet instead of Cu (220) in Re-Cu–I.
It is known that Cl− promotes the Cu dissolution by chelating with Cu+ to form [CuCl2]− complexes while the I− inhibits the Cu dissolution by generation of highly insoluble polyhedral CuI.26 Therefore, it can be concluded that the inhibitor nature of I− and the promoter role of Br− and Cl− toward Cu dissolution cause the distinct oxidized intermediates and ultimately induce quite different structures on the Cu electrode surface.
X-ray photoelectron spectroscopy (XPS) is performed to trace the oxidation state change of Cu during the reconstruction process. As shown in Fig. S12,† a strong peak at 932.4 eV in the Cu 2p3/2 region is found for EP-Cu, Ox-Cu and Re-Cu–I, which is ascribed to Cu0 or Cu+.27 The absence of satellite peaks excludes the existence of Cu2+ species, in agreement with XRD results.24 The strong I 2p peak at 619 eV is discerned in Ox-Cu, further confirming the formation of CuI in the oxidized state (Fig. S13†). The reduced (but not eliminated) signal of the I 2p peak in Re-Cu–I should come from I− adsorbed on the surface of the Cu electrode. Cu Auger spectra are also recorded to distinguish Cu0 from Cu+ in Re-Cu–I, Re-Cu–Br and Re-Cu–Cl. One main Auger peak at 568.1 eV corresponding to Cu0 and another small peak ascribed to Cu+ are observed for all three samples28 (Fig. S14†). It suggests that all three samples are composed of metallic Cu and Cu2O, which is in good agreement with the Cu 2p XPS analysis.
Cu L-edge X-ray absorption spectroscopy (XAS), which is more sensitive to the structural change, is employed to estimate the Cu oxidation state in the Cu electrode.29 Reference samples including Cu, Cu2O and CuO are also provided for quasi-quantitative analysis of the composition of the catalyst by linear combination fitting of the XAS data (Fig. 2b). The results demonstrate that EP-Cu is fully in the Cu0 state (Table S2†), and Re-Cu–I contains 75% Cu0 and 25% Cu+. The concentration of Cu0 determined in Re-Cu–Br and Re-Cu–Cl is 63% and 67%, respectively, where the remaining Cu species are also Cu+ species (Fig. S15 and Table S2†). It deserves to be noted that the measured ratio between Cu0 and Cu+ is in good agreement for XAS and EDS characterizations (Table S3†).
In short, the varied properties of I− with respect to Cl− and Br− lead to a different redox behavior of the Cu electrode, thus giving rise to a distinct surface structure. XAS analysis confirms that Re-Cu–I, Re-Cu–Br, and Re-Cu–Cl all consist of Cu0 and Cu+ with similar compositions. Thus, it is beneficial for correlating the electrocatalytic performance of these three catalysts with their surface structures.
Electrocatalytic CO2 reduction
To investigate the electrochemical performance of the surface reconstructed Cu electrode, CO2 reduction is conducted in 0.1 M KHCO3 aqueous solution utilizing an H-cell configuration. Prior to the electrochemical collection, all the Cu electrodes are firstly activated by electrolysis at −0.99 V vs. RHE for 15 min to reduce the Cu+. The three electrodes after the electrolysis are composed of metallic Cu as evidenced by the Cu LMM spectra, which rules out the possible influence of Cu oxidation state (Fig. S16†) Cyclic voltammetry (CV) curves of Re-Cu–I are firstly recorded in the above solution saturated with N2 or CO2. As shown in Fig. S17,† the current density under a CO2 atmosphere is 2 orders of magnitude higher than that under a N2 atmosphere, implying that Re-Cu–I is an excellent catalyst for CO2 reduction.Controlled potential electrolysis is then carried out at potentials ranging from −0.69 V to −1.19 V vs. RHE in CO2 saturated 0.1 M KHCO3 (pH 6.8). The evolved gaseous products are analyzed and quantified using an online gas chromatograph (GC) while the liquid products are determined by 1H NMR after the electrolysis. At an initial potential of −0.69 V, CO with a faradaic efficiency (FE) of 54.1% is detected as the major gaseous product and HCOOH with an FE of 14.3% is the liquid product accompanied by H2 as a byproduct (Fig. 3a). C2H4 begins to evolve at a potential of −0.79 V vs. RHE but the C1 species are still the main products from CO2 reduction. When more negative potentials are applied, the FE of C2H4 dramatically increases to 54.1% at −0.99 V vs. RHE and reaches the optimal value of 59.9% at a potential of −1.09 V vs. RHE. The FE of C2H4 is observed to decrease slightly to 53.6% under a more negative potential of −1.19 V due to the CO2 transportation limitation, which is also revealed by the current output (Fig. 3b). Noteworthily, at the potential of −1.09 V vs. RHE, the whole FE for C2 species (FEC2) reaches 80% with a partial current density of 21 mA cm−2 (Fig. 3b). Furthermore, the half-cell power conversion efficiency (PCE) of the entire C2 species reaches 39% (Fig. 3c), which is among the highest reported values under neutral conditions30–34 (Table S4†).
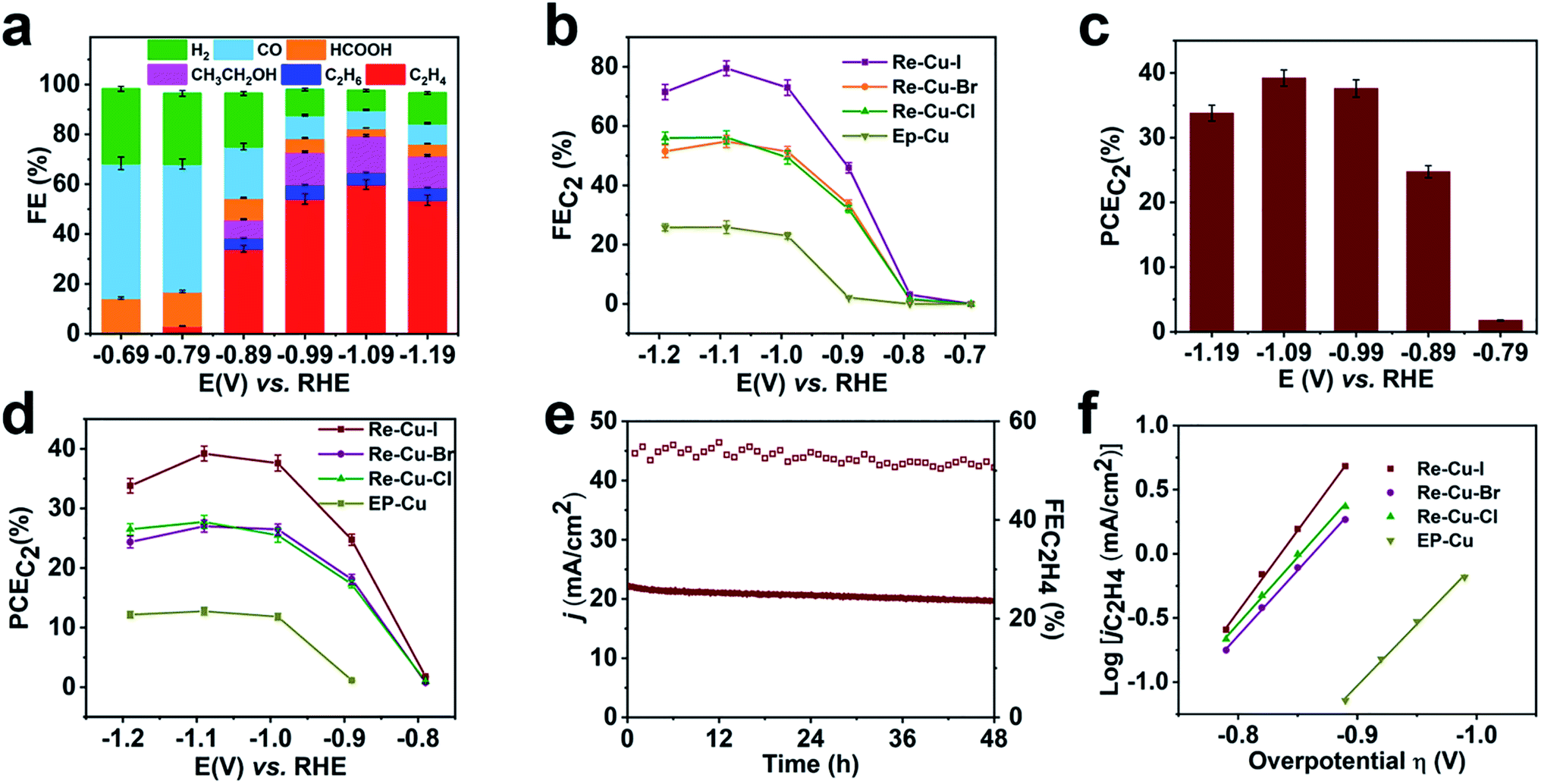 | ||
| Fig. 3 Electrochemical CO2 reduction performance of Re-Cu–I, Re-Cu–Br and Re-Cu–Cl. (a) FE of carbon products on Re-Cu–I at potentials ranging from −0.69 V to −1.19 V vs. RHE. (b) FE of C2 products on Re-Cu–I, Re-Cu–Br, Re-Cu–Cl and EP-Cu at potentials ranging from −0.69 V to −1.19 V vs. RHE. (c) PCE of C2 products on Re-Cu–I at potentials ranging from −0.79 V to −1.19 V vs. RHE. (d) PCE of C2 products on Re-Cu–I, Re-Cu–Br, Re-Cu–Cl and EP-Cu at potentials ranging from −0.79 V to −1.19 V vs. RHE. (e) Long term measurement of Re-Cu–I for electrocatalytic CO2 reduction at a potential of −0.99 V vs. RHE. (f) Tafel plot of C2H4 on Re-Cu–I, Re-Cu–Br, Re-Cu–Cl and EP-Cu for electrocatalytic CO2 reduction. | ||
As the contrast samples, Re-Cu–Br and Re-Cu–Cl show similar performance for electrochemical CO2 reduction but display poorer selectivity toward C2 species compared with Re-Cu–I at the same applied potentials (Fig. 3b, S18 and S19†). At a selected potential of −0.99 V, the FEs of C2 products for Re-Cu–Br and Re-Cu–Cl are 41.3% and 40.2%, respectively, with the corresponding PCEs of 26% and 25%, which are both lower than that of Re-Cu–I (Fig. 3d, S20 and S21†). As for EP-Cu, C1 species (CO and HCOOH) are preferentially produced (Fig. 3d, S22 and S23†). All the above results highlight that the surface reconstruction by electrochemical oxidation–reduction cycling in the presence of halide anions significantly promotes C2 product formation and Re-Cu–I exhibits the best performance.
Apart from activity and selectivity, stability is another key parameter to evaluate the catalyst performance. Therefore, extended electrolysis of Re-Cu–I is performed at −0.99 V vs. RHE (Fig. 3e). During continuous electrolysis for 48 hours, the overall current density gradually decreases from 22.3 mA cm−2 to 19.7 mA cm−2 but the FE of C2H4 remains above 50% during the whole period. To determine whether there is any structural change of the electrode surface, the catalysts after the reaction are characterized by GIXRD, Cu L-edge XAS and SEM coupled with EDS. The in-plane XRD pattern only exhibits Cu (200) and Cu (220) peaks, suggesting that the oxidized Cu+ is reduced during electrocatalytic CO2 reduction (Fig. S24†). Fitting of the Cu L-edge XAS spectrum reveals that the electrode after the reaction contains 96% Cu0 and 4% Cu+ (Fig. S25 and Table S2†). The residual Cu+ is likely attributed to rapid re-oxidation after removal of the reduction potential.35 Notably, a negligible morphology change is discerned for Re-Cu–I after the reaction, as evidenced by the SEM imaging (Fig. S26†). The trace amount of O detected by EDS is consistent with the XAS result.
The electrochemically active surface area (ECSA) of Re-Cu–I, Re-Cu–Br and Re-Cu–Cl is also determined by the double-layer capacitance method to exclude the roughness factor (RF) and facilitate direct comparison of their activity (Fig. S27†). The RF of Re-Cu–I, Re-Cu–Br and Re-Cu–Cl is calculated to be 262, 154 and 186, respectively (Fig. S28†). After being normalized by the ECSA, the total C2 partial current density (jC2) of Re-Cu–I is 79 μA cm−2 at −1.09 V vs. RHE, which is still 1.3-fold and 1.5-fold that of Re-Cu–Br and Re-Cu–Cl (Fig. S29†).
Why does Re-Cu–I possess the best electrocatalytic performance towards C2 products by CO2 reduction? The electrochemical CO2 reduction mechanism of all the reconstructed electrodes and EP-Cu is investigated by Tafel analysis. The Tafel plots are derived by plotting the overpotential (η) versus the logarithm of steady-state C2H4 partial current density [log(jC2H4)] (Fig. 3f). The Tafel slopes of Re-Cu–Br, Re-Cu–Cl and EP-Cu are determined to be 98 mV dec−1, 95 mV dec−1 and 114 mV dec−1 by linear fitting of the current density at low potentials from −0.79 V to −0.89 V vs. RHE. These values are close to 119 mV dec−1, implying that the electron transfer to *CO to form *OCCO is the rate-determining step (RDS). In sharp contrast, the Tafel slope of Re-Cu–I is calculated to be 76 mV dec−1 by the same method, demonstrating an improved 2*CO dimerization kinetics.36
It has been reported that the Cu (100) facet favors C2 species production through promoting the C–C coupling reaction while both the Cu(110) facet and the corresponding step preferentially generate CH4.37–39 In this respect, Re-Cu–Cl and Re-Cu–Br with a Cu nanocube structure contain higher percentages of Cu (100) facets compared with Re-Cu–I. However, either the faradaic selectivity or the Tafel analysis reveals that Re-Cu–I is more preferable for generating C2 products. Moreover, considering that all three electrodes have similar constituents (Cu0 and Cu+, Table S3†), the Cu oxidation state is excluded as the determining factor of the different electrocatalytic performances. The grain boundaries or some defect sites can also promote the C–C coupling reaction.40 But the active sites supported by grain boundaries have been reported to prefer to produce CH3COOH and CH3CH2OH. The product distribution is quite different from that in Re-Cu–I, implying that the grain boundaries contribute little to the excellent C2 selectivity in Re-Cu–I.8,16,41,42 The ECSA normalized total C2 partial current density (jC2) also demonstrates that Re-Cu–I exhibits better intrinsic activity toward electrocatalytic CO2 reduction. As a result, it is reasonable to correlate the electrochemical performances with the porous structure itself rather than the surface features of the Cu electrodes. Evidently, an entangled cobweb-like surface structure is achieved for Re-Cu–I after redox reconstruction due to the unique chemical properties of I−. Thus, we attribute the boosted selectivity toward C2 products of Re-Cu–I to its hierarchical porous structure, which entraps the key intermediate CO in the interior of the pores and consequently enhances the surface coverage of adsorbed *CO for the *CO–*CO dimerization reaction.
To verify the enrichment effect of the catalysts, the electrochemical CO reduction reaction with various flow rates is carried out in CO saturated 1 M KOH. At a CO flow rate of 6 mL min−1, C2 products including C2H4, CH3CH2OH and CH3COOH with a total FE of 19.5% are detected at a low onset potential of −0.26 V vs. RHE (Fig. 4a). The FE of C2H4 (FEC2H4) increases rapidly after a more negative potential is applied. The optimal value of FEC2H4 reaches 67.5% at a potential of −0.56 V vs. RHE. And at this potential, a total C2 selectivity (FEC2) of 85.1% is obtained with a partial current density (jC2) of 6.7 mA cm−2 (Fig. 4b and c). At more negative potentials, the surface CO coverage depletion induced by the CO transportation limitation causes a decrease of FEC2. It is clear that Re-Cu–I is also an excellent catalyst for C2 species generation in CO reduction.
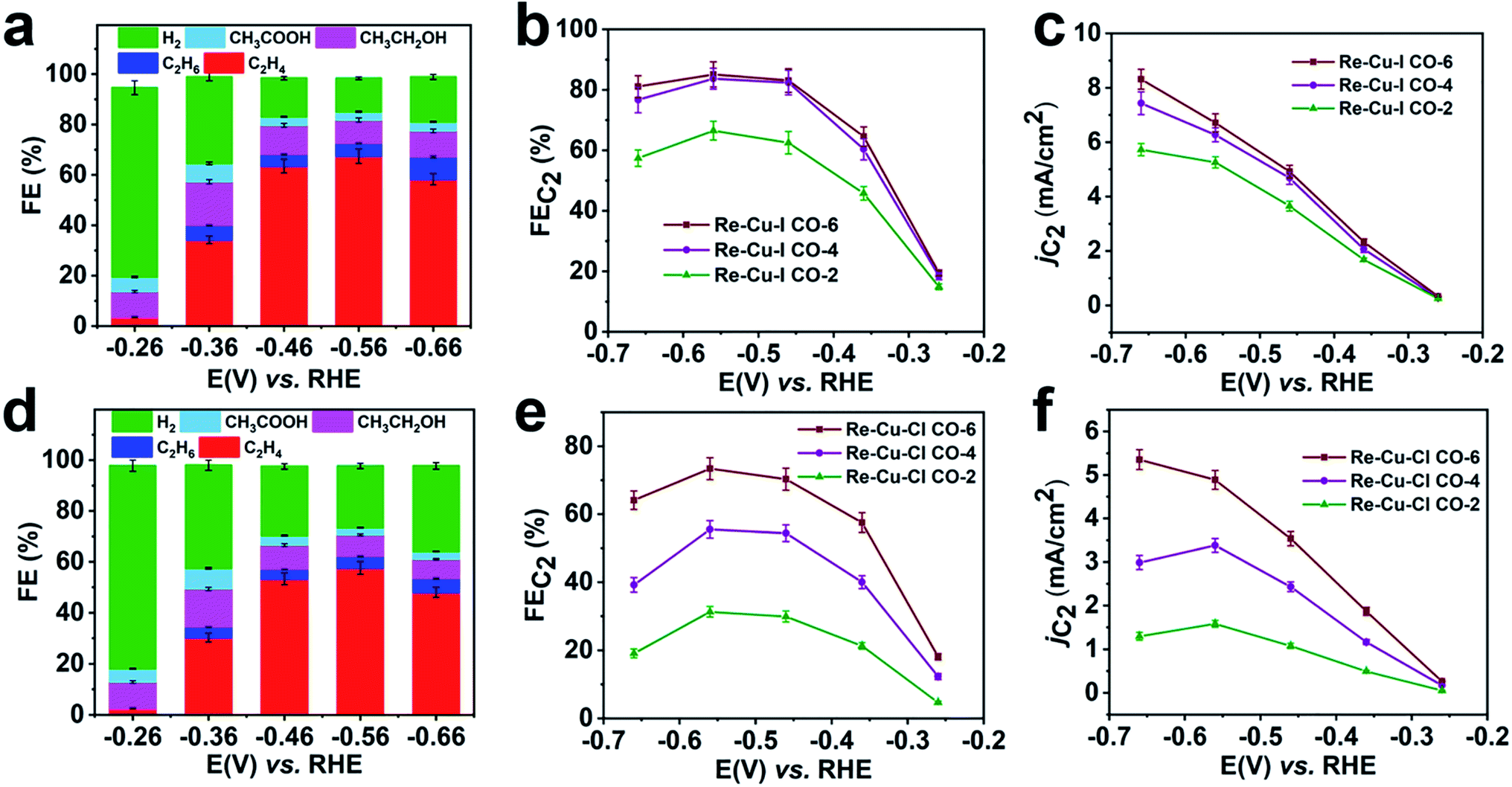 | ||
| Fig. 4 Electrochemical CO reduction performance of Re-Cu–I, Re-Cu–Br and Re-Cu–Cl. FE of carbon products from CO electroreduction on Re-Cu–I (a) and Re-Cu–Cl (d) with a CO flow rate of 6 mL min−1 at potentials ranging from −0.26 V to −0.66 V vs. RHE. FE of C2 products from CO electroreduction on Re-Cu–I (b) and Re-Cu–Cl (e) with CO flow rates of 6 mL min−1, 4 mL min−1, and 2 mL min−1 at potentials ranging from −0.26 V to −0.66 V vs. RHE. (c) Partial current density of C2 products from CO electroreduction on Re-Cu–I (c) and Re-Cu–Cl (f) with CO flow rates of 6 mL min−1, 4 mL min−1, and 2 mL min−1 at potentials ranging from −0.26 V to −0.66 V vs. RHE. | ||
When the CO flow rate is reduced, the CO reduction activity should be affected by the limited CO supply and the poor CO solubility in aqueous solution. Nevertheless, both FEC2 and jC2 only show a slight decrease when the CO flow rate is decreased to 4 mL min−1 (Fig. 4b and c, and S30a†). At a CO flow rate of 2 mL min−1, the FEC2 reduces by 22% and jC2 decreases by 20% at −0.56 V (Fig. 4c and S30b†). This demonstrates that the CO reduction performance of Re-Cu–I is independent of the CO flow rate to some extent. Under sufficient CO supply, Re-Cu–Cl as a typical contrast sample shows an FEC2 of 73.4% with jC2 of 4.9 mA cm−2 (Fig. 4e and f) at a potential of −0.56 V, and both values are lower that those of Re-Cu–I. Furthermore, the FEC2 diminishes by 26% and 56%, respectively, at CO flow rates of 4 mL min−1 and 2 mL min−1 and the jC2 decreases by 31% and 68% (Fig. 4e and S31†). Therefore, it is concluded that the electrocatalytic performance of Re-Cu–Cl is highly dependent on the CO supply.
It has been reported that the enhanced surface coverage of *CO will promote the C–C coupling reaction accompanied by suppression of H2 generation.43,44 In the electrocatalytic CO2 reduction, the activity and selectivity are highly dependent on the local CO concentration at the electrode surface.42 Under the CO-rich alkaline conditions, the electrocatalytic performance reflects the intrinsic nature of the catalyst for C2 species production apart from the effect of pH. Re-Cu–I displays higher C2 selectivity and partial current density, highlighting that the entangled cobweb-like surface structure is critical for the CO dimerization reaction. It is believed that the hierarchical porous structure contributes to enriching CO to maintain the local CO concentration at the electrode surface. As a result, it is reasonable that the C2 evolution activity is much less affected by the CO supply in Re-Cu–I (Fig. 4b and c). Analogously, during the electrochemical CO2 reduction, the CO is always in a deficient state since the concentration of the as-formed *CO on the surface is low. However, in the porous structure, the locally generated key intermediate CO has to take a long and complicated pathway to diffuse out as the final product of electrocatalytic CO2 reduction. Hence, the retention time of CO at the active surface is prolonged and the CO local concentration is enriched in the interior of the pores, which rationally enhance the surface coverage of *CO species. Since the C2 generation rate has a second-order dependence on the surface coverage of *CO,36 the reaction rate for the RDS (2*CO dimerization reaction) would be significantly improved because of the enhanced surface coverage of *CO species. In short, the raised surface coverage of *CO resulting from CO enrichment inside the hierarchical pores of Re-Cu–I boosts the dominating C2 production in electrocatalytic CO2 reduction.
Conclusions
In conclusion, we have developed a halide anion assisted electrochemical method to reconstruct the Cu electrode surface for promoting electrocatalytic CO2 reduction. The highly insoluble nature of CuI facilitates the formation of the entangled Cu nanowire structure on the Cu electrode surface via oxidation–reduction cycling. The resultant hierarchical pores in Re-Cu–I effectively enrich the key *CO intermediates for evolution of C2 products. A comprehensive comparison between the CO2 electroreduction performances of Re-Cu–I, Re-Cu–Br and Re-Cu–Cl demonstrates that the surface structure rather than the lattice facet is the determining factor for boosting the formation of C2 products. These results distinctly show that the interconnected porous structure on the electrode surface is crucial to improving C2+ generation by increasing the local CO availability. It is also anticipated that smartly tuning the pore size will achieve higher C2+ selectivity.Author contributions
J. H. and Z. T. conceived and designed this project. J. H. prepared the samples, participated in characterization, and conducted the electrochemical experiments. J. Z., K. H., D. W., Z. Y. and S. Y. joined the discussion of the data and gave useful suggestions. J. H., Y. Y., and Y. Z. participated in the characterization and helped to draft the manuscript. Z. T. supervised and guided the project. All authors commented on the data and the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge financial support from the National Key Basic Research Program of China (2016YFA0200700, Z. Y. T.), National Natural Science Foundation of China (21890381 and 21721002, Z. Y. T.), Frontier Science Key Project of Chinese Academy of Sciences (QYZDJ-SSW-SLH038, Z. Y. T.), and K. C.Wong Education Foundation (Z. Y. T.).Notes and references
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Norskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- A. D. Handoko, C. W. Ong, Y. Huang, Z. G. Lee, L. Lin, G. B. Panetti and B. S. Yeo, J. Phys. Chem. C, 2016, 120, 20058–20067 CrossRef CAS.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- K. J. Schouten, Z. Qin, E. Perez Gallent and M. T. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2018, 2, 86–93 CrossRef.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS PubMed.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- H. Yano, T. Tanaka, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2004, 565, 287–293 CrossRef CAS.
- S. Huemann, N. T. Minh Hai, P. Broekmann, K. Wandelt, H. Zajonz, H. Dosch and F. Renner, J. Phys. Chem. B, 2006, 110, 24955–24963 CrossRef CAS PubMed.
- F. A. Akgul, G. Akgul, N. Yildirim, H. E. Unalan and R. Turan, Mater. Chem. Phys., 2014, 147, 987–995 CrossRef CAS.
- L. Martin, H. Martinez, D. Poinot, B. Pecquenard and F. Le Cras, J. Phys. Chem. C, 2013, 117, 4421–4430 CrossRef CAS.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. Garcia de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed.
- T. T. H. Hoang, S. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- J. Kim, W. Choi, J. W. Park, C. Kim, M. Kim and H. Song, J. Am. Chem. Soc., 2019, 141, 6986–6994 CrossRef CAS PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Norskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- I. Takahashi, O. Koga, N. Hoshi and Y. Hori, J. Electroanal. Chem., 2002, 533, 135–143 CrossRef CAS.
- Y. Huang, Y. Chen, T. Cheng, L.-W. Wang and W. A. Goddard, ACS Energy Lett., 2018, 3, 2983–2988 CrossRef CAS.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS PubMed.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01202e |
| This journal is © The Royal Society of Chemistry 2020 |