
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Clinical translation of microfluidic sensor devices: focus on calibration and analytical robustness†
Sally A. N.
Gowers‡
 a,
Michelle L.
Rogers‡
a,
Marsilea A.
Booth
a,
Chi L.
Leong
a,
Isabelle C.
Samper
a,
Tonghathai
Phairatana
ac,
Sharon L.
Jewell
a,
Clemens
Pahl
b,
Anthony J.
Strong
b and
Martyn G.
Boutelle
*a
a,
Michelle L.
Rogers‡
a,
Marsilea A.
Booth
a,
Chi L.
Leong
a,
Isabelle C.
Samper
a,
Tonghathai
Phairatana
ac,
Sharon L.
Jewell
a,
Clemens
Pahl
b,
Anthony J.
Strong
b and
Martyn G.
Boutelle
*a
aDepartment of Bioengineering, Imperial College London, UK. E-mail: m.boutelle@imperial.ac.uk
bDepartment of Basic and Clinical Neuroscience, Kings College London, UK
cInstitute of Biomedical Engineering, Faculty of Medicine, Prince of Songkla University, Hat Yai, Thailand
First published on 10th July 2019
Abstract
We present approaches to facilitate the use of microfluidics outside of the laboratory, in our case within a clinical setting and monitoring from human subjects, where the complexity of microfluidic devices requires high skill and expertise and would otherwise limit translation. Microfluidic devices show great potential for converting complex laboratory protocols into on-chip processes. We demonstrate a flexible microfluidic platform can be coupled to microfluidic biosensors and used in conjunction with clinical microdialysis. The versatility is demonstrated through a series of examples of increasing complexity including analytical processes relevant to a clinical environment such as automatic calibration, standard addition, and more general processes including system optimisation, reagent addition and homogenous enzyme reactions. The precision and control offered by this set-up enables the use of microfluidics by non-experts in clinical settings, increasing uptake and usage in real-world scenarios. We demonstrate how this type of system is helpful in guiding physicians in real-time clinical decision-making.
Introduction
The field of microfluidics has expanded rapidly in recent years from point-of-care devices to continuous measurement systems, providing many solutions to lab-based processes.1–6 Additionally, many of the components for wearable sensing devices are already in place.7–10 The increasing use of droplet-based microfluidics and the use of multi-phase flow devices has enabled optimisation and miniaturisation of many systems, increasing the throughput and reducing the cost.2,11 A commonly acknowledged problem with lab-on-a-chip devices is the issue of chip-in-a-lab,12,13 where highly optimised miniaturised devices require large external support systems. This issue of the microfluidic external environment presents a barrier to adoption in a clinical setting. In addition, to be practical in a clinical setting lab-on-a-chip devices must be stand-alone and portable, simple to use by non-experts, and reliable. Analogous to how the development of electronic circuitry from complex, hand-crafted components to standardised, off-the-shelf components has revolutionised the field of electronics, we propose the first step towards the translation of microfluidics into general use.In vivo clinical measurements can provide clinicians with vital information about a patient's state of health in real time. However, there are many analytical challenges associated with making measurements in unknown samples or tissue, making it difficult to translate important technological advances in online analysis from the laboratory to the clinical environment. For laboratory-based instruments to be regularly used by non-experts, all processes need to be tightly controlled to ensure data quality and reliability. Consequently, a simple and robust automated platform is required, with the flexibility to address each of these different challenges and to enable optimisation of a laboratory analysis system for use within a specific clinical application.
Chemical sensors and biosensors have the potential to enable real-time continuous concentration measurements for the monitoring, diagnosis and, ultimately, the control of complex systems.14,15 We have previously shown that, when combined with microfluidic devices and microdialysis sampling, chemical sensors can provide important insights into human disease and clinical treatments and with significantly improved temporal resolution compared with commercial offline analysers.16–19 However, a major problem that persists for all bioanalytical systems is that the sensor/biosensor element suffers a loss of sensitivity over time, leading to instability in measurements.20,21 Much research has concentrated in vain on the search for a perfectly stable sensor. Ultimately, this unreached target has inhibited the translation of chemical sensors from laboratory use to routine, real-time clinical measurements. Facilitating the translation of lab-on-a-chip devices to real-world applications has the potential to address this important and as yet, unmet need in bioanalysis.
In this paper we address the problems associated with taking microfluidic biosensor devices from the laboratory into clinical use. We describe an adaptable computer-controlled platform arranged in numerous configurations to create novel solutions to common analytical problems outside of the laboratory. Complex sets of tasks can be carried out and repeated automatically without the need for a skilled operative to be present and without interrupting the microfluidic biosensor-based measurement system. The system is ideal for accurately manipulating low-volume flow streams and therefore is particularly well suited for analysis systems with small, precious samples such as clinical microdialysis, or for addition of expensive reagents such as enzymes. We present a number of important example methods to address key analytical challenges such as automated calibration, standard addition, reagent addition and lab-on-a-chip reaction optimisation. The general applicability covers a wide range of measurement systems used routinely in laboratories and can greatly aid in translating lab-based systems into the clinic.
Experimental
2.1 Materials and reagents
Glucose oxidase (GOx) from Aspergillus niger (HPS300) and recombinant lactate oxidase (LOx) from Aerococcus viridans were purchased from Sekisui Diagnostics. Pyruvate oxidase (POx) from Aerococcus viridans was purchased from Asahi Kasei Enzymes. Glycerol-3-phosphate oxidase from Streptococcus thermophilus and glycerokinase from Cellulomonas sp. were purchased from Sigma. Polyurethane (Texin 985) was obtained from Bayer. All other reagents were obtained from Sigma-Aldrich.2.2 Microfluidic chip fabrication
Sensors were housed in one of two types of custom-made microfluidic chip, either a soft poly(dimethylsiloxane) (PDMS) chip or a hard 3D-printed chip. Both systems have been described in detail previously. Briefly, PDMS chips19,22 were fabricated by soft lithography using an SU-8 master made using photolithography. The PDMS silicone elastomer was thoroughly mixed with a curing agent (Sylgard 184, Dow Corning, USA) in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by weight and baked at 65 °C for 1 h, after which holes for the connection tubing and electrodes were punched using a blunt needle. The chips were placed channel-side down onto a semi-cured PDMS base and the two parts cured together at 65 °C overnight. PDMS chips were used for the data presented in sections 3.1.1, 3.1.2 and 3. The 3D-printed chip18,23 was fabricated using an ULTRA 3SP printer and ABS 3SP White resist. Electrode holders were printed using an Objet260 Connex 3D-printer, which can print hard and soft material on the same component. VeroWhitePlus (RGD835) and TangoBlack (FLX973) were used for the hard and soft materials, respectively. All parts were printed based on schematics generated in SolidWorks.18 3D-printed chips were used for the data collected in sections 3.1.3 and 3.3.
:1 ratio by weight and baked at 65 °C for 1 h, after which holes for the connection tubing and electrodes were punched using a blunt needle. The chips were placed channel-side down onto a semi-cured PDMS base and the two parts cured together at 65 °C overnight. PDMS chips were used for the data presented in sections 3.1.1, 3.1.2 and 3. The 3D-printed chip18,23 was fabricated using an ULTRA 3SP printer and ABS 3SP White resist. Electrode holders were printed using an Objet260 Connex 3D-printer, which can print hard and soft material on the same component. VeroWhitePlus (RGD835) and TangoBlack (FLX973) were used for the hard and soft materials, respectively. All parts were printed based on schematics generated in SolidWorks.18 3D-printed chips were used for the data collected in sections 3.1.3 and 3.3.
2.3 Electrode construction
Electrode construction has been described in detail previously.24,25 Briefly, a 50 μm Teflon insulated platinum/iridium wire (Pt90/Ir10, PTFE coated, coating thickness 0.0125 mm, Advent Research Materials, USA) and a 50 μm insulated silver wire (polyester insulation, coating thickness 0.0075 mm, Goodfellow, UK) were threaded through a 27G hypodermic needle. Epoxy resin (Robnor resins, CY1301 & HY1300) was used to fill the internal volume of the needle to secure the wires in place. Once the epoxy had cured, the sharp end of the needle was ground away to a flat surface to expose disc electrodes. Alumina slurries (1.0, 0.30, and 0.05 μm) were used sequentially to polish the electrode surface. The platinum disc acted as the working electrode and the silver disc was chloridised using a potassium dichromate referencing solution (BASi Inc., US) to create a Ag|AgCl reference electrode by dipping the needle tip into the reference solution for 3 s and then into diluted 37% hydrochloric acid (1 in 10 dilution) for 20 s to remove the oxide layer from the working and counter electrodes. The needle shaft acted as a counter electrode.2.4 Biosensor fabrication
2.5 Potassium sensor
Fabrication of miniaturised potassium sensors has been described in detail elsewhere.16 Briefly, a polymer membrane consisting of potassium tetrakis(4-chlorophenyl)borate, bis(2-ethyl-hexyl) separate, poly(vinyl chloride) and the potassium ionophore valinomycin was cast at one end of a polymer electrode body. The electrode body consists of perfluoroalkoxyalkane tubing containing a Ag|AgCl reference electrode and an internal filling solution of physiological saline. The potassium sensors were stored in this physiological saline until use.2.6 Sensors used for reagent addition
For the examples presented in Fig. 4, where enzymes are added into the flow stream, electrodes are coated with a protective film (see ESI,† Fig. SI2, for more detail on enzyme fouling of electrodes). Electrodes were either coated with poly(mPD) as described in section 2.4.1 above or with poly(phenol), using the same process of electropolymerisation but applying +0.9 V vs. Ag|AgCl reference electrode for 20 minutes in 50 mM phenol solution in PBS.2.7 Clinical microdialysis
For clinical monitoring of traumatic brain injury (TBI) patients, written assent for study participation was obtained from legally authorised representatives, as patients were comatose at the time of inclusion. Written consent was obtained from patients themselves following this when possible. All human research procedures were approved by the King's College Hospital NHS Foundation Trust Ethical review board and were conducted in accordance with the Declaration of Helsinki.For TBI patient monitoring, a sterile clinical microdialysis catheter (CMA 70, 10 cm flexible shaft, 10 mm membrane length, 20 kDa cut-off, mDialysis Stockholm, Sweden) was inserted obliquely to the surface of the brain and to the full membrane length to ensure that the catheter membrane was located within the cortex. The microdialysis probe was secured using double suturing to the skin beside the point of exteriorisation. For online microfluidic analysis, the microdialysis catheter was perfused with sterile physiological fluid, CNS perfusion solution, at 2 μl min−1 using a CMA 107 microinjection syringe pump (mDialysis, Stockholm, Sweden). Perfusion of the microdialysis catheter was started immediately, allowing initial baseline dialysate levels to stabilise quickly, typically 10 minutes. The outlet tubing of the probe was adapted to connect to our online microfluidic analysis system.
Results and discussion
We present clinical results from both PDMS and 3D-printed chips. Our first clinical microfluidic chips were fabricated using soft lithography with PDMS. Whilst allowing small channels to be fabricated with relative ease and low cost, the chip was elastic and deformed easily. In a controlled environment, this is not a factor most users consider and PDMS still remains a popular material choice. Outside of a laboratory, however, the lack of robustness quickly becomes a problem. Sensors need to be housed reproducibly and the channel in which they are sensing cannot change shape during recording, as this will change the flow and sensor diffusion profiles and hence create artefacts in the data. We have also found that PDMS chips tend to tear and leak when used extensively in areas where they are frequently handled and moved. Therefore, we developed and now routinely use 3D-printed chips, which do not easily deform and are more robust for a hardwearing environment.The system described herein is based on microfluidic components that include actively controlled low-volume microfluidic pumps and valves, precision unions and tubing and a controlling computer interface. These are used to build modular microfluidic instruments (here, using components manufactured by LabSmith, Inc.) that are simply controlled via C++ drivers using a tablet or computer-based user interface. Coordinated control of several devices simultaneously to carry out a specific set of commands is achieved using automated, programmable sequences. Photographs of the instrumentation and the two types of chips used are shown in Fig. 1a. We now describe a series of methods used to address key issues regarding the translation of a lab based microfluidic device, fabricated from routine commercial parts, into clinical use.
 | ||
| Fig. 1 Simple modular microfluidic module arrangements for continuous and droplet-based basic calibration systems. a: Photographs of the LabSmith® devices connected to either a PDMS sensor chip or a 3D-printed sensor chip. b: Schematic of basic calibration layout consisting of a bidirectional syringe pump connected to a three-port two-position valve and reservoir. When the valve is switched towards the reservoir the pump can be programmed to pull solution quickly from the reservoir to refill the syringe. The valve can then be switched towards the analysis chip and programmed to slowly dispense solution at a precisely controlled flow rate. A larger external pump (shown in green) can be connected into the system to deliver an oil phase and create droplets at the interconnect (shown in blue). c: Continuous calibration at constant 2.0 μl min−1 total flow rate. Standard 1 is 0.0 mM glucose and standard 2 is 2.0 mM glucose. Each step change is 0.5 mM. The final glucose concentration and flow rates of each pump are shown for each step below the calibration data. d: Droplet calibration. Total aqueous flow rate 2.0 μl min−1. Oil flow rate 4.0 μl min−1. Standard 1 is 0.0 mM glucose and standard 2 is 2.0 mM glucose. Each step change is 0.5 mM. The final glucose concentration and flow rates of each pump are shown for each step below the calibration data. | ||
3.1 Automated sensor calibration
The simplest element of our platform is the setup for calibration (layout schematic shown in Fig. 1b) which consists of routine microfluidic pumps and valves. The ability to programme flow rates of two standards that are joined at a tee interconnect allows a set of calibration standards to be passed to the sensors. Upper and lower limits of calibration can be defined by the two reservoir solutions. The resulting output concentration is defined by the respective flow rates and hence the mixing ratio of the two flow modules. Automatically varying this ratio delivers a precise multi-step calibration stream to the analysis system without common errors such as pipetting errors. Critically, for sensors that have an output that is flow-rate sensitive, the total flow rate can remain constant. The user can pre-define the number of steps and their duration in a purpose-written script, which can be set to repeat at regular time intervals (an example calibration script is given in the ESI† (section 4.1)). Fig. 1c shows the current response for an automatic glucose sensor calibration and the corresponding calibration curve. This simple setup can easily be extended with additional flow modules for calibration over a wider concentration range if required. Moreover, careful selection of standards allows simultaneous calibration of multiple sensors with different size step changes if necessary.
Multi-phase flow, where discrete droplets of an aqueous sample are created in an immiscible fluid, is often employed in microfluidic applications to improve temporal resolution by mitigating the effects of Taylor dispersion.22 This is highly relevant clinically where connection tubing from patients are often long. Multi-phase flow systems are hard to set up as they require close balancing of pressure at the droplet generation point and this is a particular problem when using an additional injection stream to calibrate. However, a simple adaptation of our autocalibration system, changing the interconnecting tee junction for a cross junction, allows calibrations to easily and reliably be carried out in multi-phase flow. Fig. 1d shows example data for a glucose sensor calibrated using the same stepwise functions as for Fig. 1c but within a multi-phase droplet flow stream.
This simple implementation of automated and reliable calibration and the ability to pre-program the precise delivery of calibration standards at set intervals means that no intervention is required from any user once the system is in place clinically, thereby removing a previously complex barrier to patient data.
For long clinical monitoring periods, the basic calibration setup can be adapted to include a further programmable valve for introduction of a clinical microdialysate stream, Fig. 2a. This allows the patient sample to be diverted to a collection vial during calibrations ensuring no interruption to the dialysate flow, an essential requirement as a change in flow rate will change the recovery of the microdialysis probe and thus produce measurement artefacts. This collected sample can also be used periodically for additional analysis using traditional offline microdialysis measurements if required. During sample analysis, the calibration path can be blocked by closing the valves. This simple variation in layout allows the user to switch between calibration and analysis streams and this action can be automated to occur at regular time intervals. Critically, this pre-programming ensures reliable data are recorded even when a skilled operative is not present, such as overnight. We have built on this to develop a system that tracks the performance of individual biosensors; importantly, calibration information can be fed into a feedback loop creating an alert when the sensitivity becomes unacceptably low and the biosensors require replacing.
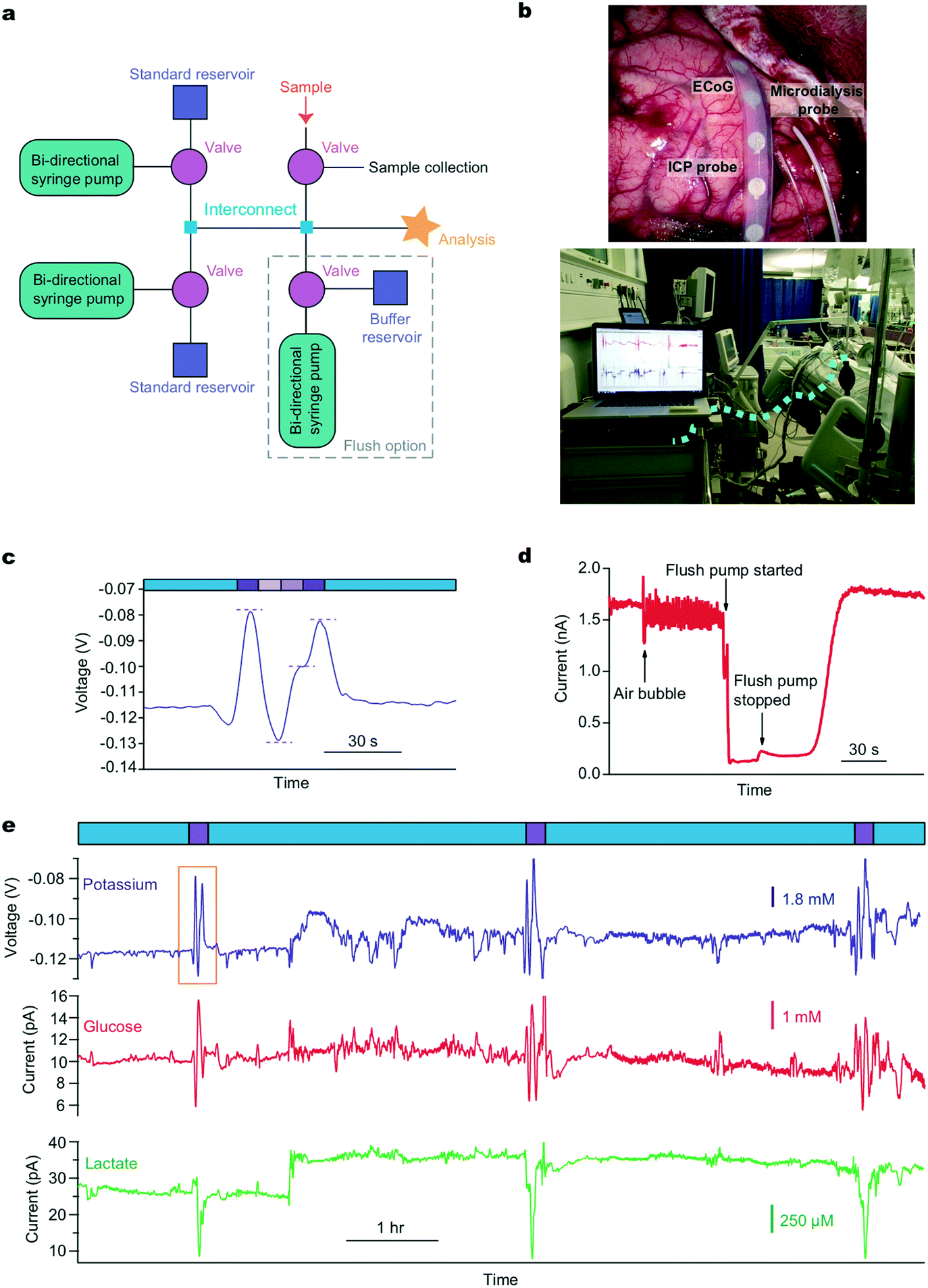 | ||
| Fig. 2 Clinical system to monitor TBI patients in ITU. a: Schematic of microfluidic platform layout. b: Top photograph shows an electrocorticography strip electrode (ECoG), the microdialysis probe and the intracranial pressure (ICP) probe which are inserted into human brain tissue during a craniotomy. Bottom photograph depicts use in King's College Hospital ITU. The dotted blue line shows where a length of fine-bore tubing connects the implanted microdialysis probe to the analysis system. c: An example potassium calibration carried out during TBI monitoring in the hospital. Dialysate measurements are indicated by blue bars and multi-point calibration by purple bars. Concentrations are 10.0, 2.70, 6.35 and 10.0 mM (levels indicated by dotted lines). This is an expanded view of the calibration highlighted by a yellow box in (e). d: Removal of air bubble in chip using the larger capacity flow module. An air bubble caused a sudden decrease in the signal-to-noise ratio of a sample perfused at 2 μl min−1. PBS was flushed through the system at 10 μl min−1 on top of the glucose flow for 20 seconds, removing the air bubble and the signal returned to the same level. e: Clinical automatic calibration example in TBI patient monitoring. Potassium (purple), glucose (red) and lactate (green) sensors were continually analysing dialysate (blue boxes) in between programmed calibration cycles (purple boxes). Known standards were introduced every 3 h (glucose: 0.0, 2.0, 1.0 mM, lactate: 1.0, 0.0, 0.5 mM, potassium: 10.0, 2.70, 6.35 mM). The changes in potassium and lactate seen at around 3 h are not artefactual but possibly represent a pathophysiological event that is not picked up by other monitoring modalities. | ||
We use this system clinically to monitor traumatic brain injury (TBI) patients in the intensive therapy unit (ITU) in King's College Hospital. A schematic of the system layout is shown in Fig. 2a and a photograph of the probes implanted during surgery and the system placed on a trolley beside the bedside in intensive care are shown in Fig. 2b. In this case, the system is set to automatically calibrate every 3 h. This time window can be adjusted for each application to ensure high quality data analysis while minimising the loss of online recorded data during calibrations. In this case, 10 minutes of data are lost to calibration every 180 minutes. Fig. 2c and e shows example extracts of raw clinical data collected from monitoring a TBI patient using microdialysis. The data in Fig. 2e shows continuous human dialysate measurements with three calibrations carried out automatically throughout this monitoring period (expanded view of potassium calibration shown in Fig. 2c). In this case we dataset collected over 39 hours of online microdialysis measurement, with a total of 14 successful automated calibrations.
Noise artefacts are a major problem in analytical measurements, particularly in a clinical environment. These artefacts can be caused by anything from patient movement, general administration of medication, external machinery such as bedside X-ray machines, to internal system factors. For microfluidic devices in a clinical environment a major potential issue is noise caused by air bubbles trapped within the microfluidic system. Whilst post processing of the data using various filters can reduce/eliminate much of the noise,28 it cannot help when the problem is internal to the system. To combat these problems we have devised an easy to use de-bubbling system that is automated using a pre-programmed script. We added a larger capacity flow module into the system, Fig. 2a. When filled with physiological buffer, it can be used to flush the system using a higher flow rate (10 μL min−1). This, in effect, resets the system, as seen in Fig. 2d where an air bubble was dislodged from the system and the low-noise glucose biosensor signal returned quickly. Trapped air bubbles can interfere with the signal and cause loss of important clinical data if not removed. As the system can be accessed and controlled remotely, from outside the lab or hospital, the user can also check the status of the system remotely and can use the ‘flush mode’ to reset the system if noise is becoming an issue. This ability to fix system problems remotely is vital for a clinical monitoring system where it is not possible for skilled operatives to be present 24 h a day for typically up to 5 days. In a recent monitoring case, the sensors were connected and ran continuously for 37 hours. The system autocalibrated successfully throughout the 12 hour overnight periods, as well as during the day, ensuring reliable clinical data were recorded. We are also developing a corresponding tablet app that will have the option of a simple button trigger so that a non-skilled operative can run the flush to address any critical issues if the need arises.
We have previously shown that events known as spreading depolarisations can be detected within the human brain using microdialysis with analysis by microfluidic sensors. Spreading depolarisations are an independent indicator of secondary brain injury and are a predicator of poor patient outcome29 and as such there is an increasing clinical awareness of the need to monitor spreading depolarisations. However, having a bedside analysis system to monitor the brain tissue after a TBI is currently not routine. What we now aim to address are the limiting factors for moving research-based technology into the real-world to aid physicians in real-time clinical decision making. Using a system such as that described herein, is the first step towards adoption of new technology outside of laboratories.
As an example, we have drawn from our experience of monitoring kidneys, both human30 and porcine,31Fig. 3a. The ultimate goal of this project is to create a kidney monitoring system to assess the transplant viability of individual kidneys, thereby addressing the vast difference between supply and need of human kidney transplants.
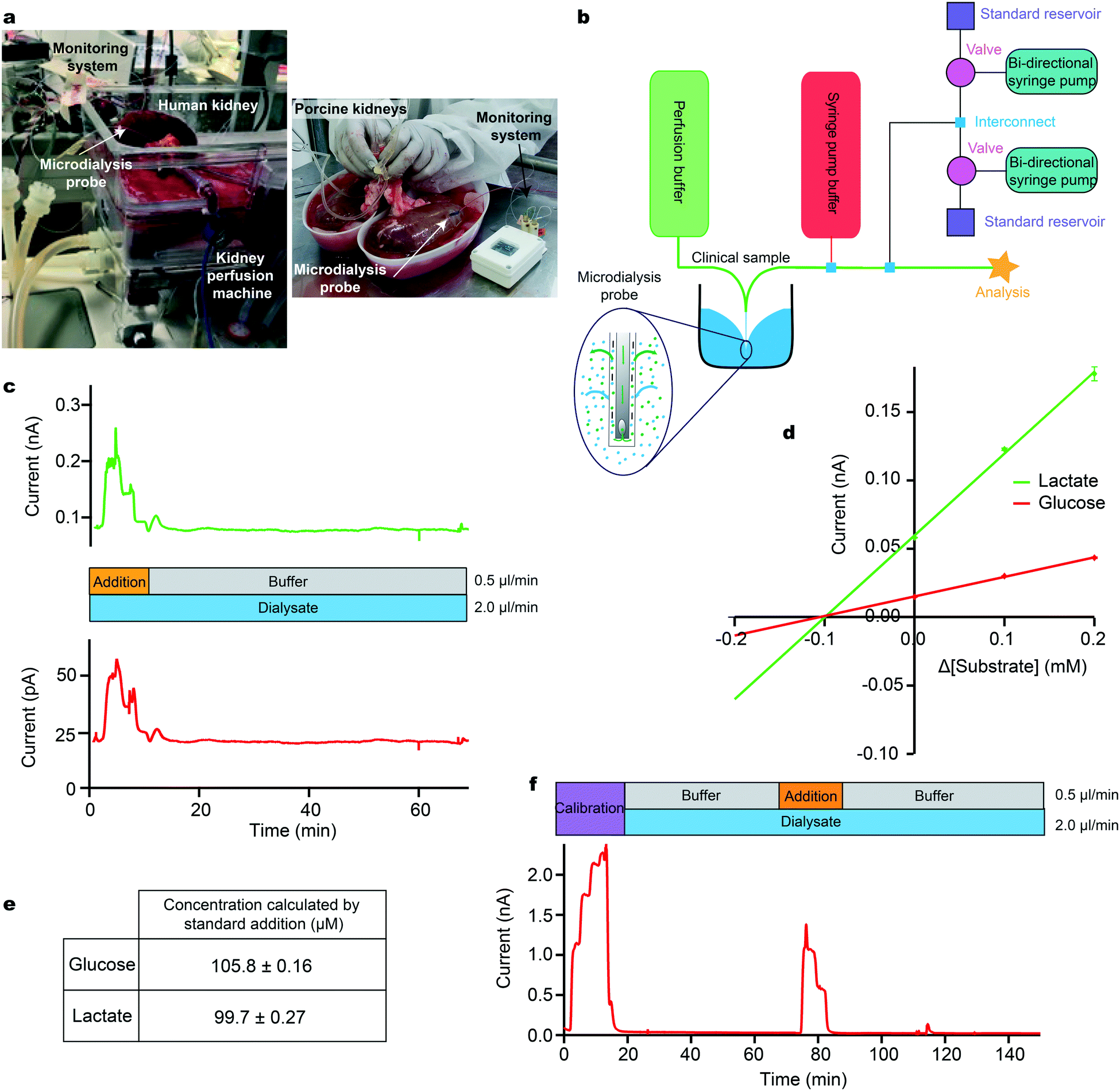 | ||
| Fig. 3 Standard addition to calibrate sensors during human and porcine kidney monitoring. a: A photograph of human kidney microdialysis monitoring during reperfusion and of online monitoring of porcine kidneys at the abattoir b: Schematic of the instrumentation layout used in human and porcine studies. Larger external pumps for continuous buffer delivery and the addition of the clinical sample were used in conjunction with the calibration setup previously described. Inset: Schematic showing exchange of analytes across microdialysis membrane. c: Initially, standard addition was run by adding a 3-point calibration (0.2, 0.1 and 0.0 mM) of glucose (red) and lactate (green) to the clinical sample stream. Analyte concentrations were chosen so that when they are diluted by the sample stream the final concentrations covered the appropriate physiological range. The dialysate was then measured. d: Current versus change in concentration for standard addition of glucose (red) and lactate (green). Extrapolation to where the lines cross the x-axis gives the sample concentration in the final volume. e: Concentrations for glucose and lactate calculated using standard addition, corresponding to a recovery of approx. 25%. f: Dialysate glucose levels in an ex vivo porcine kidney during cold preservation. A 5-point glucose calibration was carried out (steps: 0.00, 1.25, 2.50, 3.75 and 5.00 mM) at 2.5 μl min−1, after which the dialysate was analysed. Midway through, a 3-point standard addition equivalent to 0.00, 0.625 and 1.25 mM glucose in the final solution was carried out. | ||
Our instrumentation can be configured to carry out a multi-point standard addition by automatically adding known analyte concentrations to a biological sample stream (Fig. 3b). In this example, a microdialysis sampling probe was placed in a stirred beaker containing 400 μM glucose and 400 μM lactate in T1 physiological solution, simulating the tissue. The MD probe was perfused with T1 at a flow rate of 2 μl min−1. The outflowing dialysate was analysed using microfluidic biosensors and a standard addition was run by adding a 3-point calibration of glucose (red) and lactate (green) at 0.5 μl min−1 on top of the dialysate stream during analysis (Fig. 3c). Factoring in dilution, the final concentration of standards added were 0.2, 0.1 and 0.0 mM for both analytes. An additional T1 buffer flow stream was introduced at a flow rate of 0.5 μl min−1 during the dialysate measurement to maintain a constant final flow rate over the sensor and maintain the temporal integrity of the data (an example script is given in the ESI† (section 4.2)). The measured current during the standard addition is plotted versus the change in concentration from the unknown sample. The calibration plots are extrapolated to y = 0 to give an estimate of the concentration in the sample (Fig. 3d). Given the concentration of glucose and lactate in the beaker, the measured dialysate concentration indicates a probe recovery of approximately 25%, Fig. 3e. This recovery is due to the relatively fast perfusion rate and the small membrane size and is consistent with previously measured recovery for the same probe type and perfusion rate (data not shown).
As a clinical example, we have used this methodology to measure glucose levels in a sample of dialysate collected from a porcine kidney during cold preservation, Fig. 3f. Briefly, a microdialysis probe (4 mm membrane length) was inserted into the cortex of an ex vivo porcine kidney during cold storage at 4 °C. Dialysate was collected for 2 h in fine-bore collection tubing and frozen for analysis at a later time using the setup in Fig. 2a.30 In cases such as this, where sample measurements indicate that the analyte levels are close to the detection limit of the sensors, this methodology allows the user to ascertain whether the measurement is accurate or whether the sensor has failed or is being affected by the sample medium. Here, we were able to conclude that kidney dialysate glucose levels were unexpectedly low, below the detection limit of the sensors. The ability to troubleshoot the performance of the system without any interruption to the recording of clinical data is invaluable and will aid translation by increasing the confidence of the clinicians in the new technology.
3.2 Reagent addition
The ability to create homogeneous biosensing systems in which precise volumes of enzymes are dosed into a sample stream to react with the substrate in solution offers many advantages. For biosensing applications, an obvious application would be to protect the signal integrity, for example if ascorbate interference was a problem then ascorbate oxidase could be added to the sample stream or if there was in issue of cross-talk between biosensors, catalase could be added to remove this effect. Examples are shown in the ESI† (Fig. SI2). The methodology of precisely dosing in enzymes has many potential uses for complex clinical samples and microfluidics is ideally placed as only small amounts of expensive enzymes are required, minimising analysis costs. Alternatively, other substances can be added to the sample stream, such as a buffer stream to condition and regenerate the electrode surface during electrochemical detection schemes.17Reagent addition creates the unique opportunity to optimise the biorecognition and detection elements of the biosensing system separately. This can be particularly useful, for example, where detection mechanisms require multiple reagents or where complex reaction mechanisms involve multi-enzyme cascades.
Here, the instrumentation is configured to optimise the detection of pyruvate, Fig. 4a, an analyte of interest in monitoring human subjects who have a TBI. The reaction (ESI† eqn (S3)) requires the enzyme, pyruvate oxidase (POx), however, this enzyme needs an additional cofactor, thiamine pyrophosphate (TPP), to work effectively and its catalytic activity has been shown to be enhanced in the presence of Mg2+.32 Optimal concentrations of these cofactors vary greatly in the literature,33–35 however, our modular platform allows easy optimisation of each reaction component independently; a task that would otherwise be highly complex and time-consuming.
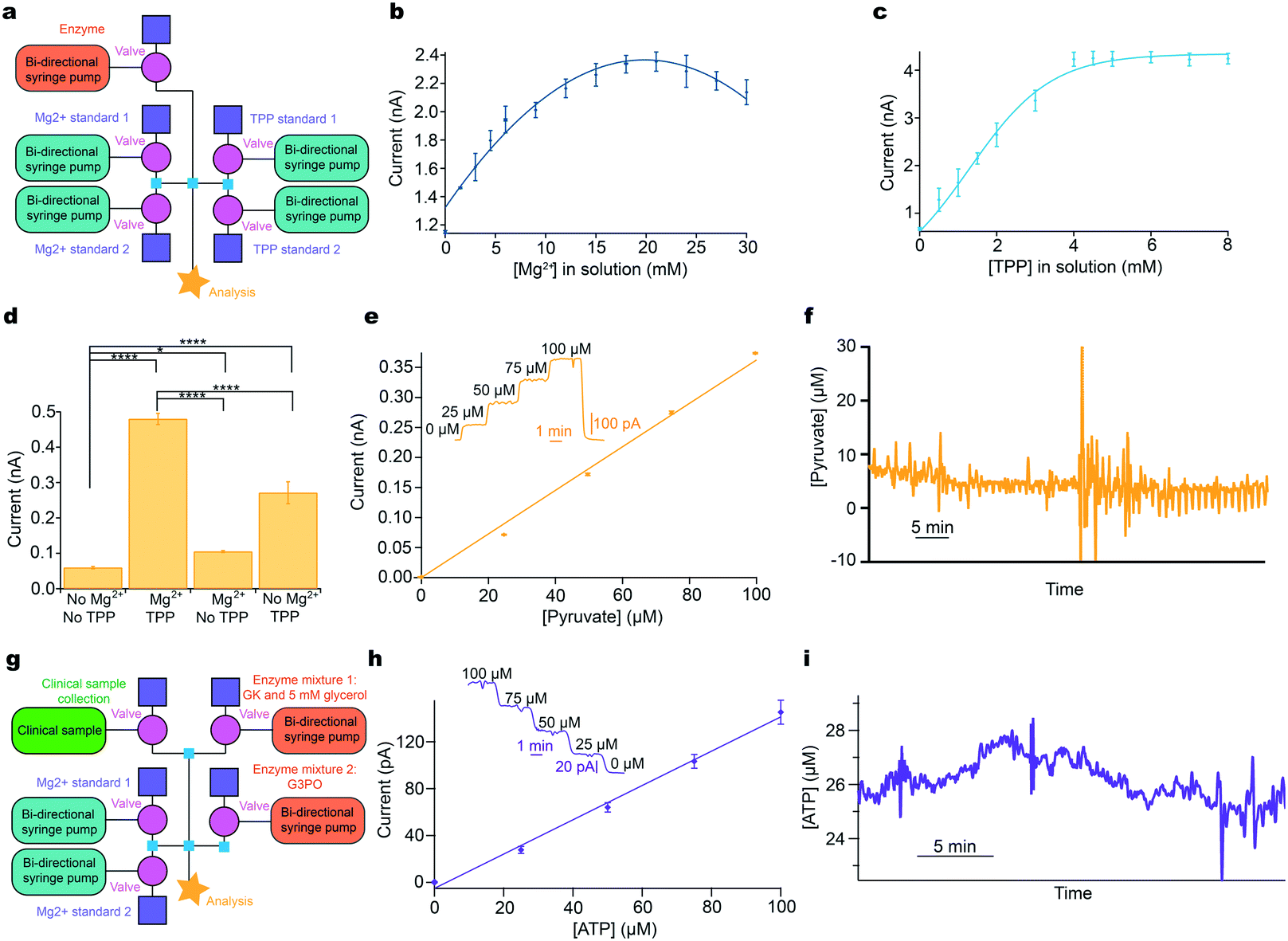 | ||
| Fig. 4 Addressing system problems: optimisation and reaction cascades. a: Schematic of modular platform to individually optimise cofactor concentration for pyruvate detection, comprising two sets of basic layouts with an additional module for enzyme addition (pink). b: The concentration of Mg2+ was varied and the current recorded for 0.5 mM pyruvate and 0.9 mM TPP. c: The concentration of TPP was varied and the current recorded for 0.5 mM pyruvate and 16.8 mM Mg2+. d: Significant improvement of current response following addition of optimised cofactors for 0.5 mM pyruvate. Tested using Mann–Whitney U test. Significance: * p < 0.05 and **** p < 0.0001. e: 5-Point pyruvate calibration at 2 μL min−1 in physiological range dosing in POx (30 mg ml−1, 0.5 μl min−1) and optimised cofactor concentrations. Markers represent mean ± standard deviation and are fitted with a straight line. Inset shows raw data for the 5-point pyruvate calibration. f: Raw data excerpt for dialysate brain pyruvate levels in a TBI patient at King's College Hospital. g: Schematic of the modular platform to sequentially dose in two enzyme mixtures, illustrated for ATP detection. Adaptation from the basic calibration layout is shown in pink (enzyme additions) and green (clinical sample). h: 5-Point ATP calibration in physiological range at 2 μL min−1, consecutively dosing in glycerol (5 mM) and glycerokinase (GK, 6.25 mg ml−1, 0.5 μl min−1), and glycerol-3-phosphate oxidase (G3PO, 6.55 mg ml−1, 0.5 μl min−1). Markers represent mean ± standard deviation and are fitted with a straight line. Inset: Raw data for the 5-point ATP concentration. i: ATP levels in kidney dialysate, collected in storage tubing before analysis.30 | ||
The concentration of each cofactor was individually varied using a computer-controlled script, while all other reagent concentrations were kept constant. The resulting currents were recorded for a fixed pyruvate concentration, Fig. 4b and c. The optimal concentration of Mg2+ and TPP was found to be 16.8 mM and 4.8 mM, respectively (taking account of dilution). Fig. 4d shows the significant improvement in sensitivity obtained by this optimisation process. Having optimised the detection parameters, this in-flow sensing methodology was used to detect pyruvate in the physiological range with good sensitivity, Fig. 4e. This methodology has been successfully used to monitor dialysate brain pyruvate levels in a TBI patient and an excerpt of raw data are shown in Fig. 4f. The patient was monitored for a total of 60.5 hours, with online recording for 39.5 hours. The system correctly functioned with four different sensors, one of which was sensing the pyruvate by addition of POx into the flow stream, for the total monitoring period. Automated calibrations ran successfully every 3 hours. The discrepancy between total and online monitoring hours is the system offline time for calibrations, predominantly automatic but also includes manual calibrations, changing of sensors to improve sensitivity, patient disconnection for a CT scan and accidental disconnections from the microdialysis probe overnight when only the nurses were present. External issues such as accidental disconnections are being addressed by further miniaturisation through wireless technology. Once in place, the analytical device can be placed on the patient rather than upon a large bedside monitoring trolley, which requires long connection tubing. Throughout monitoring, the dialysate basal level of pyruvate, in this patient, remained between 5–10 μM. This is the first time pyruvate has been continuously monitored in the brain of an acutely injured TBI patient.
Furthermore, this methodology is ideal for more complex systems such as multi-enzyme cascades, as it allows the reaction to be carried out in sequence, adding enzymes consecutively. Here, we briefly demonstrate its use for ATP detection, which involves a two-enzyme cascade and requires glycerol at the first stage at concentrations high enough to saturate the glycerokinase (ESI† eqn (S4) and (S5)).36 This was achieved by dosing glycerokinase and glycerol into the sample, followed by glycerol-3-phosphate oxidase, Fig. 4g. An optimised 5-point ATP calibration in the physiological range is shown in Fig. 4h, together with the corresponding calibration curve. To demonstrate the potential, the instrumentation has been used in a proof-of-concept experiment to measure ATP concentration in dialysate collected from a porcine kidney during haemoperfusion, Fig. 4i. ATP levels were found to be low and fairly stable throughout.
The ability to accurately dose in low volumes of enzymes and reagents to create complex analysis systems is an advantage clinically as relevant analytes often have complex detection schemes. One analyte rarely provides the clinician with a clear assessment of patient health and a system that can cope with the complexities of multi-analyte monitoring is more likely to be adopted into clinical environments.
3.3 System optimisation and clinical simulation
Each new clinical application imposes specific analytical challenges on the analysis system to obtain good quality data. Moreover, as biological processes operate on a range of timescales, temporal resolution is an important consideration. Before testing the system with real high-value samples, it is necessary to rigorously verify that the analysis system is capable of resolving dynamic changes of any analyte in the necessary concentration range and on the required timescale.To test and evaluate sensor performance in conditions mimicking the real clinical monitoring application, the modular platform can be configured to simulate expected dynamic chemical changes. As an example, in Fig. 5 we simulate changes occurring during maxillofacial reconstructive free flap surgery. Free flap surgery involves the transplantation of healthy tissue, together with its blood vessels, from one site of the body to another; further details are given in the supplementary information. We have previously measured glucose and lactate levels during free flap surgery using custom valve-based flow injection analysis, known as rapid sampling microdialysis, and have shown that online microdialysis can provide useful clinical information regarding the health state of the tissue.37 Whilst we saw excellent results, the technology was large and cumbersome, and the delay in transferring the sample from the tissue to our sampling machine meant that the analyte levels were typically displayed 20 minutes behind realtime. During surgery the decisions are time-critical and hence surgeons could not implement the technology for routine patient monitoring.
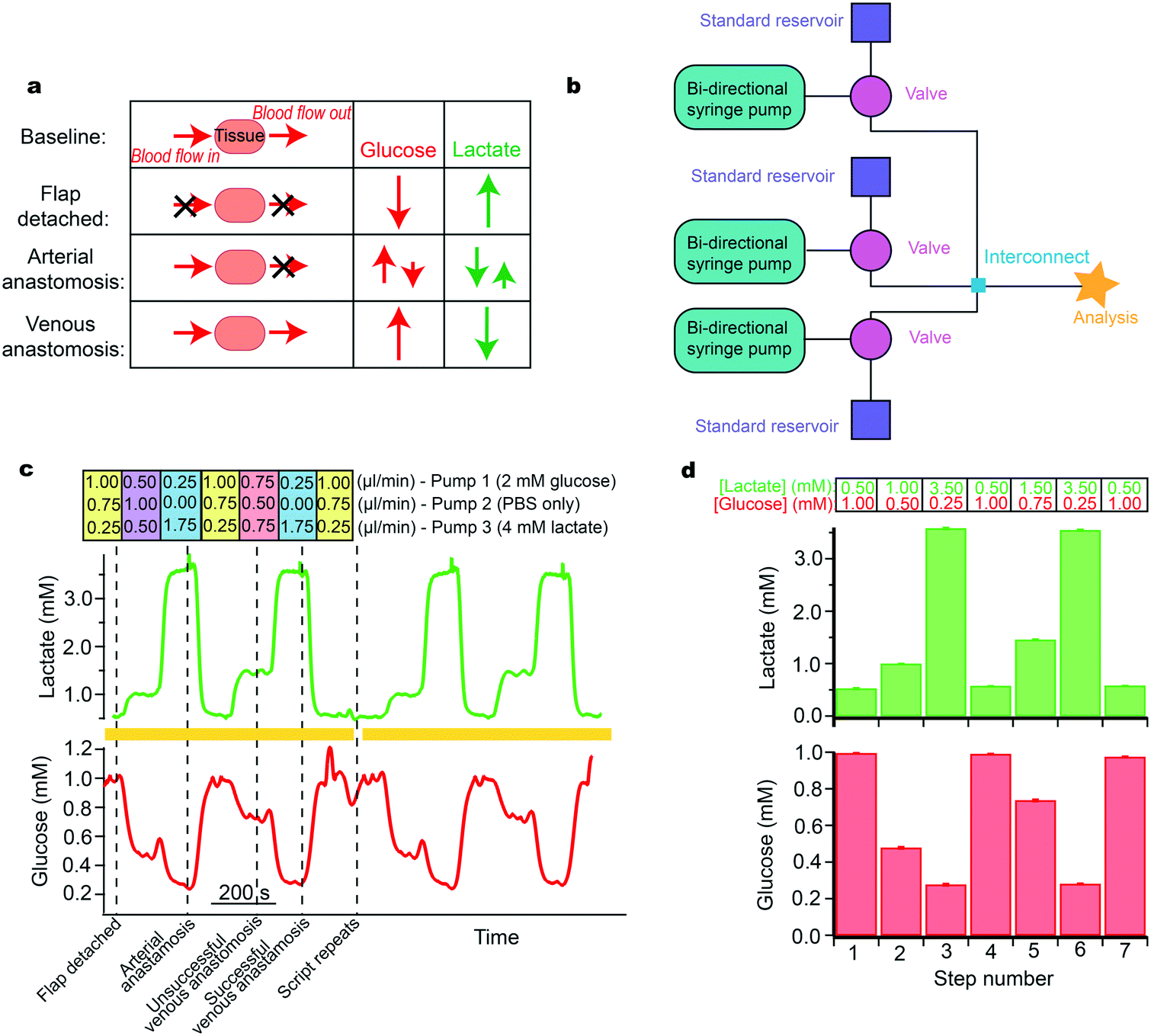 | ||
| Fig. 5 Simulation of free flap surgery for system optimisation. a: Schematic of the various stages of free flap surgery and the relative magnitude and direction of corresponding typical changes in glucose and lactate. b: Schematic of the layout used. c: Lactate (green) and glucose (red) response during a computer-controlled automatic simulation of typical metabolite changes during free flap surgery. Dotted lines indicate surgical events that these levels typically correspond to. The simulation script was set to automatically repeat; yellow bars indicate two complete script cycles given as an example. The respective flow rates for each flow module are given above the trace. Pump 1 contained 2 mM glucose and 0 mM lactate, pump 2 contained 0 mM glucose and 0 mM lactate, and pump 3 contained 0 mM glucose and 4 mM lactate. d: Histograms of glucose and lactate concentrations detected for each step of simulation over 4 repeats. Histograms show mean ± standard deviation. Input concentrations are indicated above the histograms for each step for lactate in green and for glucose in red. | ||
We have since developed a new microfluidic monitoring system that is smaller and more robust and has the potential to be wireless. This continuous, wireless monitoring system would be more suitable for this application and therefore has greater potential for translation. The system provides real-time data, which would allow surgeons to respond to physiological changes quickly and effectively. However, for this application, it is necessary to verify that the new sensing system can detect analyte levels over a wide concentration range for both glucose and lactate. This is because glucose levels change from high in healthy tissue to very low when the tissue becomes ischaemic, while lactate levels increase from low in healthy tissue to relatively high in ischaemic tissue. To verify the monitoring system before trialling it in a real clinical situation we can use our microfluidic platform to simulate expected analyte changes (based on the previously obtained clinical results).
The changes at each stage of the surgery are summarised in Fig. 5a.37 The simulation required analytes to change by different magnitudes and in different directions. The layout of instrumentation is shown in Fig. 5b. Levels of glucose were changed in different steps between 0.2 and 1.0 mM and lactate between 0.5 and 3.5 mM (Fig. 5c). For each pump, the flow rates used to generate these concentrations are indicated above the steps (Fig. 5c). The computer-controlled script was set to automatically change between each stage and to repeat when the end of the script was reached, allowing for sensor reproducibility to be tested. Validation of the detected concentrations against input concentrations is shown in the histogram in Fig. 5d. The relatively small error bars demonstrate the high reproducibility of this system over several script repeats. This is essential for delivering a controlled sensor environment and provides an ideal tool for optimisation of system components. This has proven useful within our group during development and optimisation of a new 3D-printed microfluidic chip as it provided reproducibility, which is essential in evaluating design iterations, particularly over long time periods.23
The computer-controlled simulation allowed us to verify that the biosensors met the analytical requirements of the monitoring application before testing them in the high-pressure clinical situation. This exercise not only assesses the new flow cell and sensors but highlights an important ability to fully understand device performance at physiological levels and during physiologically relevant changes. We are in the process of implementing the new technology within the area of free flap monitoring during surgery.
Conclusions
We have addressed an important previously unmet need in the translation of bioanalysis into clinical environments – robust microfluidic processes and devices. Whilst there are many excellent lab-based monitoring devices, there are very few reports of these devices being used in real-world clinical situations. The main hindrance being the ability to simplify the complex analytical device for non-experts to use and control and for device uptake to be successful. We have addressed the first barriers, including a robust microfluidic chip with sensors and the ability to automate calibration outside of the laboratory.23 We have successfully shown that the use of computer-controlled automated instrumentation can recreate important analytical laboratory methodologies that ensure data quality whilst facilitating the use of biomolecular sensors by non-experts. The modular nature of these commercial devices means that the instrumentation can be adapted for different analytical problems and clinical applications, beyond the examples given here, with relative ease. We have shown that more complicated analysis methods, such as those that rely on accurate reagent additions, are easily implemented, even in complex environments. Programs can be scripted for specific sets of tasks allowing accurate, precise manipulation of multiple flow streams simultaneously, thereby allowing non-experts to simply reset or troubleshoot the system. The various clinical examples demonstrate that we can meaningfully overcome analytical challenges and as such can aid the translation of early stage analytical devices from the laboratory to the clinic.Author contributions
SG, MLR, MAB, CLL, ICS and TP worked on developing the technology. SG and MLR planned and carried out the experiments and wrote and revised the manuscript. MAB planned and carried out the ascorbate removal experiments and revised the manuscript. CLL and ICS were involved in collection of clinical TBI patient data and revised the manuscript. TP revised the manuscript. SLJ, CP and AJS facilitated gathering of patient data and revised the manuscript. MGB planned experiments and wrote and revised the manuscript. Microfluidic platform, automatic calibration setup and pyruvate detection are described in patent application number: US20180136247A1, published 2018, Imperial Innovations (inventors: M. G. Boutelle, M. L. Rogers, C. L. Leong and S. A. N. Gowers).Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was funded by the Wellcome Trust DOH (HICF-0510-080). In addition, we would like to thank NIH for support via 1R01NS102725-01A1 (MLR) and 1R21NS109875-01 (SANG), the Freemasons Foundation of New Zealand through the Royal Society of New Zealand-Rutherford Foundation (MAB, RFT-ICT 1501-FF), EPSRC (SG, CLL, ICS), Imperial BRC (MLR), the Royal Thai Government (TP) and MRC (SLJ, MR/R00112X/1). We would like to thank Dr. Karim Hamaoui, Dr. Natalie Vallant and Prof. Vassilios Papalois for facilitating the kidney preservation experiments.References
- C. Chen, B. T. Mehl, A. S. Munshi, A. D. Townsend, M. Spence and R. S. Martin, Anal. Methods, 2017, 8, 6005–6012 RSC.
- L. Shang, Y. Cheng and Y. Zhao, Chem. Rev., 2017, 117, 7964–8040 CrossRef CAS.
- M. M. Gong and D. Sinton, Chem. Rev., 2017, 117, 8447–8480 CrossRef CAS PubMed.
- K. N. Han, C. A. Li and G. H. Seong, Annu. Rev. Anal. Chem., 2013, 6, 119–141 CrossRef CAS PubMed.
- P. N. Nge, C. I. Rogers and A. T. Woolley, Chem. Rev., 2013, 113, 2550–2583 CrossRef CAS PubMed.
- S. Nayak, N. R. Blumenfeld, T. Laksanasopin and S. K. Sia, Anal. Chem., 2017, 89, 102–123 CrossRef CAS PubMed.
- J. R. Windmiller and J. Wang, Electroanalysis, 2013, 25, 29–46 CrossRef CAS.
- G. Matzeu, L. Florea and D. Diamond, Sens. Actuators, B, 2015, 211, 403–418 CrossRef CAS.
- Y. Liu, M. Pharr and G. A. Salvatore, ACS Nano, 2017, 11, 9614–9635 CrossRef CAS.
- J. Kim, A. S. Campbell and J. Wang, Talanta, 2018, 177, 163–170 CrossRef CAS.
- S.-Y. Teh, R. Lin, L.-H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- M. I. Mohammed, S. Haswell and I. Gibson, Procedia Technol., 2015, 20, 54–59 CrossRef.
- P. Neužil, C. D. M. Campos, C. C. Wong, J. B. W. Soon, J. Reboud and A. Manz, Lab Chip, 2014, 14, 2168–2176 RSC.
- M. A. Booth, S. A. N. Gowers, C. L. Leong, M. L. Rogers, I. C. Samper, A. P. Wickham and M. G. Boutelle, Anal. Chem., 2018, 90, 2–18 CrossRef CAS PubMed.
- M. C. Frost and M. E. Meyerhoff, Annu. Rev. Anal. Chem., 2015, 8, 171–192 CrossRef CAS.
- M. L. Rogers, C. L. Leong, S. A. Gowers, I. C. Samper, S. L. Jewell, A. Khan, L. McCarthy, C. Pahl, C. M. Tolias, D. C. Walsh, A. J. Strong and M. G. Boutelle, J. Cereb. Blood Flow Metab., 2016, 37, 1883–1895 CrossRef PubMed.
- T. Phairatana, C. L. Leong, S. A. N. Gowers, B. A. Patel and M. G. Boutelle, Analyst, 2016, 141, 6270–6277 RSC.
- S. A. N. Gowers, V. F. Curto, C. A. Seneci, C. Wang, S. Anastasova, P. Vadgama, G. Z. Yang and M. G. Boutelle, Anal. Chem., 2015, 87, 7763–7770 CrossRef CAS PubMed.
- M. L. Rogers, C. L. Leong, S. A. Gowers, I. C. Samper, S. L. Jewell, A. Khan, L. McCarthy, C. Pahl, C. M. Tolias, D. C. Walsh, A. J. Strong and M. G. Boutelle, J. Cereb. Blood Flow Metab., 2017, 37, 1883–1895 CrossRef CAS PubMed.
- D. W. Kimmel, G. LeBlanc, M. E. Meschievitz and D. E. Cliffel, Chem. Sens. Biosens., 2011, 84, 685–707 CrossRef PubMed.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473–4491 RSC.
- M. Rogers, C. Leong, X. Niu, A. de Mello, K. H. Parker and M. G. Boutelle, Phys. Chem. Chem. Phys., 2011, 13, 5298–5303 RSC.
- I. C. Samper, S. A. N. Gowers, M. L. Rogers, D.-S. R. K. Murray, S. L. Jewell, C. Pahl, A. J. Strong and M. G. Boutelle, Lab Chip, 2019, 2038–2048 RSC.
- B. A. Patel, M. Rogers, T. Wieder, D. O'Hare and M. G. Boutelle, Biosens. Bioelectron., 2011, 26, 2890–2896 CrossRef CAS PubMed.
- M. L. Rogers, D. Feuerstein, C. L. Leong, M. Takagaki, X. Niu, R. Graf and M. G. Boutelle, ACS Chem. Neurosci., 2013, 4, 799–807 CrossRef CAS PubMed.
- A. Viggiano, S. Marinesco, F. Pain, A. Meiller and H. Gurden, J. Neurosci. Methods, 2012, 206, 1–6 CrossRef PubMed.
- N. Vasylieva, B. Barnych, A. Meiller, C. Maucler, L. Pollegioni, J. Lin, D. Barbier and S. Marinesco, Biosens. Bioelectron., 2011, 26, 3993–4000 CrossRef CAS PubMed.
- D. Feuerstein, K. H. Parker and M. G. Boutelle, Anal. Chem., 2009, 4987–4994 CrossRef CAS PubMed.
- J. A. Hartings, M. R. Bullock, D. O. Okonkwo, L. S. Murray, G. D. Murray, M. Fabricius, A. I. R. Maas, J. Woitzik, O. Sakowitz, B. Mathern, B. Roozenbeek, H. Lingsma, J. P. Dreier, A. M. Puccio, L. A. Shutter, C. Pahl and A. J. Strong, Lancet Neurol., 2011, 10, 1058–1064 CrossRef PubMed.
- S. A. N. Gowers, K. Hamaoui, P. Cunnea, S. Anastasova, V. F. Curto, P. Vadgama, G.-Z. Yang, V. Papalois, E. M. Drakakis, C. Fotopoulou, S. G. Weber and M. G. Boutelle, Analyst, 2018, 143, 715–724 RSC.
- K. Hamaoui, S. Gowers, S. Damji, M. Rogers, C. L. Leong, G. Hanna, A. Darzi, M. Boutelle and V. Papalois, J. Surg. Res., 2015, 200, 332–345 CrossRef PubMed.
- D. A. Jones, Development of a pyruvate assay for neurochemical studies, 1996 Search PubMed.
- K. Ikebukuro, R. Nishida, H. Yamamoto, Y. Arikawa, H. Nakamura, M. Suzuki, I. Kubo, T. Takeuchi and I. Karube, J. Biotechnol., 1996, 48, 67–72 CrossRef CAS.
- M. A. Rahman, D.-S. Park, S.-C. Chang, C. J. McNeil and Y.-B. Shim, Biosens. Bioelectron., 2006, 21, 1116–1124 CrossRef CAS PubMed.
- M. Mascini and F. Mazzei, Anal. Chim. Acta, 1987, 192, 9 CrossRef CAS.
- E. Llaudet, S. Hatz, M. Droniou and N. Dale, Anal. Chem., 2005, 77, 3267–3273 CrossRef CAS PubMed.
- M. L. Rogers, P. A. Brennan, C. L. Leong, S. A. N. Gowers, T. Aldridge, T. K. Mellor and M. G. Boutelle, Anal. Bioanal. Chem., 2013, 405, 3881–3888 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00400a |
| ‡ Contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |