
Precise and non-invasive circulating tumor cell isolation based on optical force using homologous erythrocyte binding†
Xuejia
Hu‡
ac,
Daoming
Zhu‡
a,
Ming
Chen‡
b,
Keke
Chen
a,
Hailiang
Liu
a,
Wei
Liu
*a and
Yi
Yang
 *ac
*ac
aSchool of Physics & Technology, Key Laboratory of Artificial Micro/Nano Structure of Ministry of Education, Wuhan University, Wuhan 430072, China. E-mail: yangyiys@whu.edu.cn; wliu@whu.edu.cn
bDepartment of Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan University, Wuhan, 430072, China
cShenzhen Research Institute, Wuhan University, Shenzhen 518000, China
First published on 25th June 2019
Abstract
Precise isolation of circulating tumor cells (CTCs) is proved to be significant for early cancer diagnosis and downstream analysis. Most of the existing strategies yield low purity or cause unexpected damage to cells because of foreign material introduction. To avoid foreign material caused damage and achieve high efficiency simultaneously, this work presents an innovative strategy using tumor cell targeting molecules to bind homologous red blood cells (RBCs) with tumor cells, which results in obvious optical constant differences (both size and mean refractive index) between CC-RBCs (RBC conjugated CTCs) and other blood cells. Then the modified CTCs can be precisely separated under laser illumination in an optofluidic system. Experiments show that CTCs are efficiently modified with erythrocytes and finally isolated from blood at high purity (more than 92%) and a high recovery rate (over 90%). In the whole process, CTCs are proved to keep membrane and function integrity. The combination of homologous RBC binding and an optofluidic system will provide a convenient tool for cancer early diagnosis and treatment monitoring, which exhibits good performance in CTC non-invasive and precise isolation, thus showing great potential.
Introduction
Clinical capture and analysis of rare CTCs is a key premise of clinical monitoring of tumor metastasis and chemotherapeutic response, because the seeds of metastatic CTCs in peripheral blood are believed to be the major reason for dissemination from the primary tumor to distant organ sites.1–3 Hence, occult tumor cells (about 1–10 cells per mL of blood) that contain important information can be used as an indicator of patient disease progression.4,5 Extensive technologies have been developed to isolate and enumerate CTCs, such as hydrodynamic approaches,6 immunoaffinity-based methods,7–10 and dielectrophoretic methods.11 Most of the passive or active strategies are based on the cell's intrinsic characteristics (such as size, density and dielectric constant), resulting in low purity due to the obvious overlap in these properties between CTCs and leukocytes.12 The presence of leukocytes will decrease the purity and also cause unexpected interference in CTC counting and molecular analysis.13,14 A specific anti-epithelial cell adhesion molecule (EpCAM) based chip can selectively adhere CTCs with many advantages, but the passive adhesion has low efficiency and precision.15 Existing commercial devices use immune-affinity magnetic beads to target tumor cells and an external magnetic field to enrich cells, enabling high precision and considerable throughput to be achieved. However, this strategy is limited by the inevitable problem of introduction of foreign magnetic beads, which may affect cell viability and further cellular analysis.16,17 These problems restrict the applications in research and clinic. So, it is necessary to find a solution to both low purity and foreign material introduction of traditional methods.Optical force is an ideal tool for micro-substance identification and separation,18–20 and has great advantages for precise, non-contact and non-invasive cell manipulation.21,22 In particular, it can be very useful in biological applications, such as leukocyte subpopulation label-free and precise separation,23 microfluidic sorting of mammalian cells,24 and optical chromatography.25 But for CTC isolation, being limited by similar optical constants between tumor cells and leukocytes, it is still a challenge for optical force to precisely distinguish them from normal blood cells.
Here, we developed a new strategy to achieve precise and non-invasive CTC isolation by combining targeted molecular binding technology with optical force separation in a microfluidic system. With the help of FA, multiple homologous RBCs bind with tumor cells; the RBCs with a much larger refractive index (RI)26,27 contribute to a significant optical constant increase (including size and mean RI) of the newly formed CC-RBCs. Hence, optical force supported by an infrared laser can accurately discriminate them from other cells. Compared with magnetic beads, RBCs are obtained from the same donor, innovatively avoiding foreign material introduction.28 Besides, RBCs are an ideal bio-friendly material for clinical application for they are easy to obtain and to be cleared from tumor cells.29 This new strategy is proved to be precise and non-invasive for CTC isolation with a considerable throughput, showing broad application prospects in clinic and research.
Results and discussion
Schematic of the CC-RBC construction and optofluidic system
Fig. 1 shows the schematic of precise and non-invasive circulating tumor cell sorting in the microfluidic system based on optical force using homologous erythrocyte binding. Obviously, the CC-RBCs experience an obvious volume increase; meanwhile, the RBCs with a higher RI will make a significant contribution to the mean RI increase of the CC-RBCs. To better reveal these changes, the size and RI distribution of different cells are depicted in ESI† Fig. S1. Generally, tumor cells have a larger size than white blood cells (WBCs) and RBCs,30 while WBCs have a wide size and RI distribution due to different subpopulations.31 The mean RI of nucleated cells mainly depends on the nucleus and cytoplasm; for leukocytes, the RI of the nucleus is about 1.43 ± 0.05 refractive index units (RIU) and that of the cytoplasm is 1.348 ± 0.004 RIU.32 Meanwhile RBCs and tumor cells have a much larger mean RI (about 1.399 ± 0.006 RIU) than WBCs. Both the cell's RI and volume are key factors that determine the optical force; thus this RBC caused optical constant increase will allow optical force to clearly distinguish tumor cells from other components.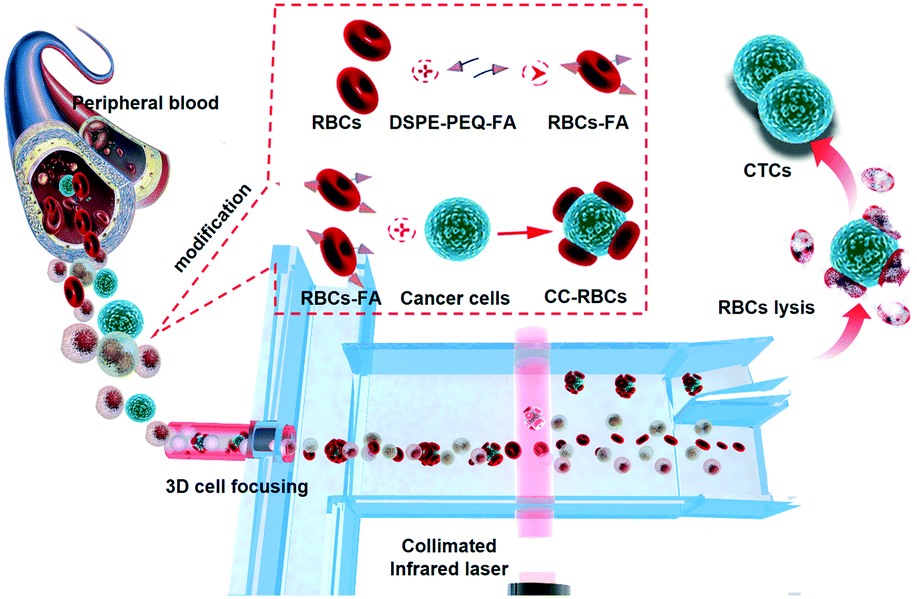 | ||
| Fig. 1 The schematic of the CTC modification process and the optofluidic system. Based on the obviously amplified optical characteristics, CC-RBCs are precisely isolated from blood cells in the optofluidic system. Homologous RBCs are artificially attached to CTCs, and DSPE-PEG-FA was used to form a stable connection. | ||
The optical separation process and the optofluidic system are depicted in Fig. 1. Optofluidics is flexible for biological applications and especially suitable for cell manipulation and detection.33–35 As shown in Fig. 1, a micro capillary is used in the microfluidic channel for facile and efficient hydrodynamic 3D cell focusing.36,37 The inner diameter of the capillary that is sealed in the center of the channel (120 μm deep and 150 μm wide) is 50 μm, and two sheath flows (PBS buffer) are injected into the channel through two sides. The micro glass capillary is inserted into the channel and kept in the horizontal center of the channel under a microscope. A prism is used to facilitate alignment in the vertical direction, while the capillary is controlled at the center of the channel both horizontally and vertically; UV epoxy is used for capillary sealing and fixing. The ratio of the sheath flow rate and sample flow rate is set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, so that the sheath flow will wrap the core flow and focus the cells in a tiny bundle. Rhodamine 6G and Rhodamine B are used to dye the core flow and sheath flow, respectively. A bright field image of the focusing part and 3D liquid spatial profiles are shown in ESI† Fig. S2, which are captured by laser scanning confocal microscopy (Nikon, A1R); the fluorescence is excited by an argon ion laser (wavelength 488 nm) and a DPSS laser (wavelength 561 nm), respectively. As a result, the cells are focused in the channel center in both lateral and vertical sections. The laser is collimated by a PDMS lens near the channel wall. In the collimated laser beam (50 μm wide), the cell's trajectories are mainly affected by optical radiation force compared to optical gradient force.21,38,39 As CC-RBCs flow through the laser beam region, they would be easily separated from the blood because of the obviously enhanced optical constant, while the RBCs and leukocytes travel little distance, and finally flow through the lower outlet. By obviously improving the physical properties of CTCs, the optofluidic system can provide much higher isolation precision than traditional methods, meaning fewer CTC omissions in the sorting process and lower sample volume demand for clinical use, although CTCs are rare in real blood samples. The driving optical force is produced by a NIR laser (Changchun New Industries Optoelectronics Technology, China) to prevent damage to cells. Many studies focused on the effect of near-infrared laser exposure on living cells.6,40–42 These studies indicate that the NIR laser induced a negligible effect on cell viability, proliferation or DNA expression, and the power intensity level observed to cause detectable damage in these studies is more than two orders of magnitude higher than the power intensity used in this device.
:2, so that the sheath flow will wrap the core flow and focus the cells in a tiny bundle. Rhodamine 6G and Rhodamine B are used to dye the core flow and sheath flow, respectively. A bright field image of the focusing part and 3D liquid spatial profiles are shown in ESI† Fig. S2, which are captured by laser scanning confocal microscopy (Nikon, A1R); the fluorescence is excited by an argon ion laser (wavelength 488 nm) and a DPSS laser (wavelength 561 nm), respectively. As a result, the cells are focused in the channel center in both lateral and vertical sections. The laser is collimated by a PDMS lens near the channel wall. In the collimated laser beam (50 μm wide), the cell's trajectories are mainly affected by optical radiation force compared to optical gradient force.21,38,39 As CC-RBCs flow through the laser beam region, they would be easily separated from the blood because of the obviously enhanced optical constant, while the RBCs and leukocytes travel little distance, and finally flow through the lower outlet. By obviously improving the physical properties of CTCs, the optofluidic system can provide much higher isolation precision than traditional methods, meaning fewer CTC omissions in the sorting process and lower sample volume demand for clinical use, although CTCs are rare in real blood samples. The driving optical force is produced by a NIR laser (Changchun New Industries Optoelectronics Technology, China) to prevent damage to cells. Many studies focused on the effect of near-infrared laser exposure on living cells.6,40–42 These studies indicate that the NIR laser induced a negligible effect on cell viability, proliferation or DNA expression, and the power intensity level observed to cause detectable damage in these studies is more than two orders of magnitude higher than the power intensity used in this device.
The entire RBC binding process is also revealed in Fig. 1. The ligand molecule, folate receptor (FR), is overexpressed on the membrane of many tumor cells, which allows folic acid (FA) to be a specific targeting molecule for CTCs.43,44 As a highly selective tumor cell targeting agent, FA has many advantages compared to most antibodies including being widely suitable for targeting many cancers, highly stable, and easy to operate and synthesize, and has been widely used in many therapeutic applications.45,46 Hence, FA is used as a targeting agent to bind RBCs and tumor cells by non-invasive and tight chemical connection. Here we use 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethyleneglycol)-2000] (ammonium salt) (DSPE-PEG-FA) to form the FA-RBCs. DSPE can be inserted into the RBC membrane's lipid bilayer, and due to the hydrophobic interaction between RBCs and DSPE, we are able to obtain DSPE-bound RBCs by physically mixing RBCs with DSPE-PEG-FA.47 Then the FA-RBCs are cultivated with CTCs to form the conjunction between FA and the receptor; finally RBC-FA-CTCs are established.
The success of this strategy depends on the highly efficient RBC attachment. To verify the DSPE binding performance, DSPE-PEG-Cy5 is used to label the RBCs. After mixing and combining RBCs with DSPE-PEG-Cy5, the mixture is washed and re-suspended in PBS buffer to remove free DSPE and fluorescent dye. Fig. 2a shows the fluorescence image captured using a confocal laser scanning microscope (CLSM) of labeled RBCs under the microscope, under illumination of the laser; almost all the cells emit red fluorescence, indicating the strong and highly efficient incorporation between the plasma membrane and DSPE molecule. Then, in order to reveal the connection between FA and tumor cells, MCF-7 cells are mixed with RBCs labeled with DSPE-PEG-FA (Fig. 2b) and DSPE-PEG-Cy5 (Fig. 2c), respectively. After incubation with modified RBCs for one hour, MCF-7 cells cultured with FA-RBCs in Fig. 2b are covered by many engineered RBCs, while the MCF-7 cells cultured with RBCs-Cy5 in Fig. 2c are isolated from RBCs, indicating the stable connection between RBCs, DSPE-PEG-FA and tumor cells. Fig. 2d shows the CLSM images of MCF-7 and RBCs labeled with Hoechst (tumor cell nucleus) and Cy5 (RBC membrane) dye, respectively. With this harmless chemical conjunction, numerous RBCs are tightly bound on the membranes of CTCs. The average number of attached RBCs on every CTC is counted under the microscope, and statistical results indicate that almost all the tumor cells are covered by more than five RBCs and 98% of the tumor cells are covered by ten to twenty RBCs, as shown in Fig. 2b. In addition, RBC membrane proteins are examined using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).48 SDS could break the connection between cell proteins and other molecules, and different cell proteins will travel different distances in the polyacrylamide gel; as a result, the proteins of RBCs are separated and revealed using this method. The protein analysis results are shown in Fig. 2e, with the modified and unmodified RBCs remaining the same in membrane proteins, showing that the modification process will keep the cell membrane integrity and is friendly to RBCs.
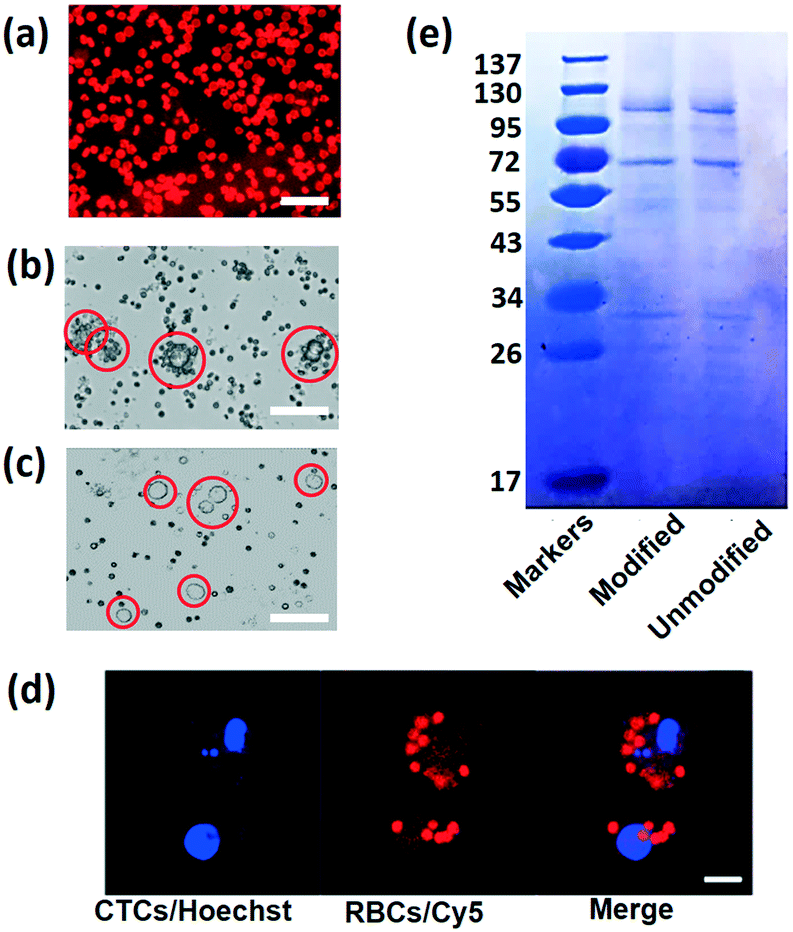 | ||
| Fig. 2 (a) To reveal the binding performance of the RBC membrane and DSPE, a CLSM photo of RBCs modified with DSPE-PEG-Cy5 is captured; RBCs that bind with DSPE will emit red fluorescence. Scale bar: 20 μm. (b) Bright field image of MCF-7 cultured with RBCs modified with DSPE-PEG-FA. The scale bar is 30 μm. (c) Bright field image of MCF-7 cultured with RBCs modified with DSPE-PEG-Cy5. The scale bar is 30 μm. (d) Two-fluorescence image of single HCT116 cells after incubation with FA-RBCs; the cell nucleus is labelled with Hoechst (blue) and the RBC membrane is labeled with Cy5 (red). The scale bar is 20 μm. (e) SDS-PAGE protein analysis of RBCs before and after modification. | ||
CTC separation in the optofluidic system
The size and mean RI magnification of CC-RBCs provide an ideal basis for optical force separation. We conducted different blood cell optical mobility tests under illumination of a collimated infrared laser.23,38,39 To compare the mobility, the laser power is set to 2 W and the sample flow rate is set to 100 μL h−1. Under these conditions, the optical mobility of RBCs and leukocytes is measured and shown in ESI† Fig. S3. Although leukocytes are larger than RBCs in size, RBCs are much more sensitive to light irradiation force due to their higher RI, and experience a larger displacement distance.Subsequent to the verification of different blood cell mobilities, we conduct CTC isolation from artificial CTC peripheral blood samples. Before MCF-7 cells are added to 2 mL blood samples, redundant erythrocytes are removed with RBC lysis buffer to reduce the sample volume and increase the handling efficiency, and the rest of the sample is re-suspended in PBS buffer, then a small amount of FA-RBCs is spiked to the treated sample to target MCF-7 cells. As erythrocytes account for more than 99% of blood,49 the sample is reduced by about ten times. As a result, CC-RBCs are formed after the mixing and incubating procedure, and the sample concentration is maintained at 2 × 106 cells per milliliter. The CC-RBC moving process is captured using a camera and arranged in time sequences, as demonstrated in Fig. 3a. When the time is 100 ms, the CC-RBCs flow through the center of the laser beam and experience the highest optical force; meanwhile the scattering light is too strong to exceed the CCD camera detection threshold. For better observation of the cell sorting process under the microscope, both the flow rate and laser intensity are appropriately decreased. The samples are delivered into the channel at a flow rate of 30 μL h−1, and the PBS sheath flow is set to 60 μL h−1 to confine the cells in the channel center. Under these conditions, the cells are focused in the channel center, with the cell stream width decreasing to less than 30 μm. The laser intensity is set to 0.8 mW μm−2. When tumor cells move through the laser beam region, they are quickly pushed to the upper location and finally travel more than 70 μm. Fig. 3b shows the image of the separation region, and the trajectory during the separation process is revealed by the red dotted line, while other blood cells' trajectories including leukocytes and erythrocytes are marked by the yellow dotted line. Due to their much smaller optical constants, these blood cells remain in the lower laminar flow. Hence, tumor cells exit the device from the upper outlet, while leukocytes and RBCs flow through the lower outlet, as shown in Fig. 3c. The dynamic sorting process can be found in ESI† Movie 1.
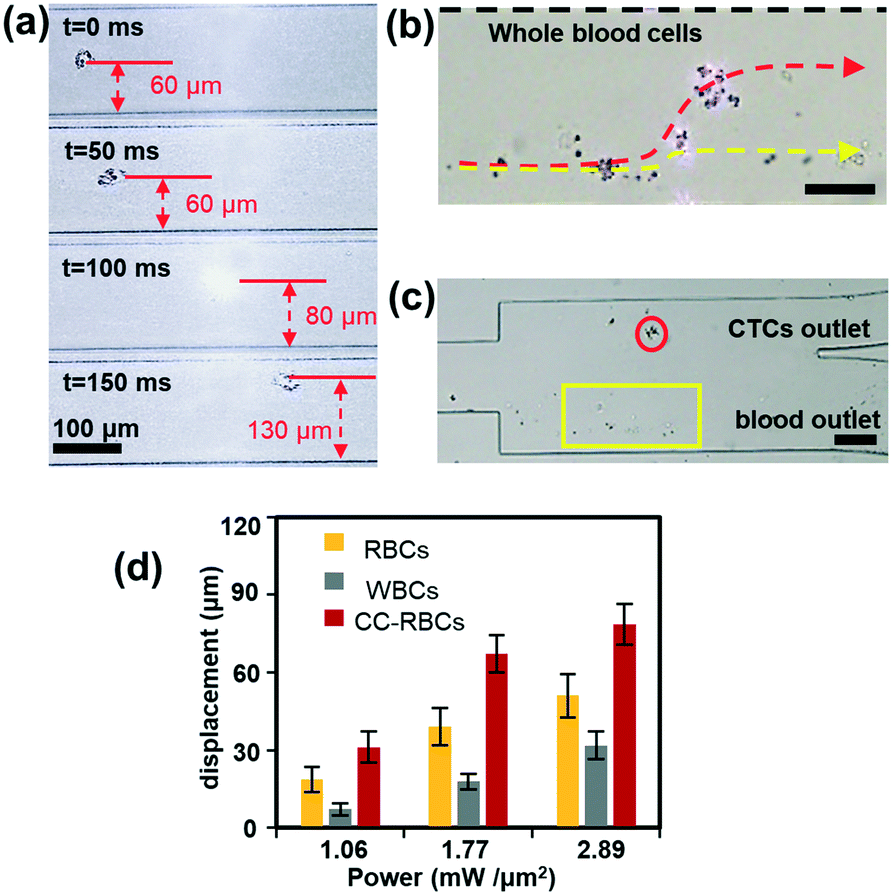 | ||
| Fig. 3 The optical sorting performance. (a) Optical mobility of CC-RBCs when the sample flow rate and infrared laser intensity are artificially reduced to 30 μL h−1 and 0.8 mW μm−2. (b) The separation process of modified MCF-7 from other blood components; because of their obviously magnified size and RI, CC-RBCs (trajectory marked by the red dotted line) are precisely pushed to the upper flow while leukocytes and RBCs (trajectory marked by the yellow dotted line) remain in the lower flow. Scale bar: 50 μm. (c) Microscopy image of the chip's outlet. The red circle marks the modified CTCs and the yellow box marks other blood cells. Scale bar: 100 μm. (d) Cell displacement when they flow through the laser sorting region. The sample flow rate is set to 100 μL h−1. The error bars represent mean ± standard deviation (n = 3). | ||
To demonstrate the motion of different cells, we investigated the influence of laser intensity on cell displacement in the Y direction. In the experiment, the sample flow rate is set to about 100 μL h−1, and the cell concentration is also 2 × 106 cells per milliliter. Statistical results are shown in Fig. 3d; with the increase of laser intensity, all the cells are pushed to a further distance, but the gap between CC-RBCs and other blood cells is obviously enlarged. To precisely segregate target cells, the laser intensity should match the flow rate carefully. When the intensity increases to 2.9 mW μm−2, the distance between leukocytes and RBCs reaches about 50 μm, so only CC-RBCs are sent to the upper flow. As the sample volume and cell number are reduced by several fold (about ten times smaller than before), about 2 mL whole blood is processed per hour under these conditions. In the clinic, tumor cell detection and analysis need to be conducted using a lower amount of blood sample, so that this system can meet clinic demands with much higher precision and considerable throughput. What's more, by optimizing device operating parameters and assembling parallel devices, the throughput can be enhanced many times to meet many real clinical demands. This statistical result reveals that the size and RI amplification using homogenous RBCs will effectively change the CC-RBCs' optical constant and assist the optical force distinguishing and sorting. It is important for a cell isolation tool to enrich target cells with high purity; the presence of unwanted cells will affect the counting and analysis process and may lead to incorrect results. Here, following the separation process, the CC-RBCs collected from the device are treated with RBC lysis buffer and re-suspended in PBS, for the upcoming detection and counting procedure. Next, the collected sample is stained with fluorescein isothiocyanate (FITC) conjugated anti-CK antibodies and Cy5 conjugated anti-CD45 antibodies for identification of MCF-7 and leukocytes. Besides, Hoechst is used to identify the cell nucleus. The collected sample is imaged and counted under a fluorescence microscope. Cells that are CK and DAPI positive and CD 45 negative are counted as CTCs, while cells that are CD 45 and DAPI positive and CK negative are counted as leukocytes, as shown in Fig. 4a. Isolated tumor cells have high purity and recovery rate when the sample flow rate is properly matched with the laser intensity; the recovery rate is defined as the collected CTCs divided by the total number of spiked cells. Fig. 4b and c respectively show that the purity and recovery rate of MCF-7 depend on the flow rate when the laser intensity is set to 2.9 mW μm−2. Under a certain laser intensity, the results reveal that before the sample flow rate reaches 200 μL h−1, this method gave high purity (92%) and recovery rate (90%), but when the flow rate continues to increase, the recovery rate is observed to significantly decrease. The measured values are reported as an average of three experiments, which is indicated by error bars in the graph. It reveals that the CTC size and mean RI amplification by RBCs greatly increase the cell's optical sensitivity, leading to obviously increased separation precision compared to conventional methods.
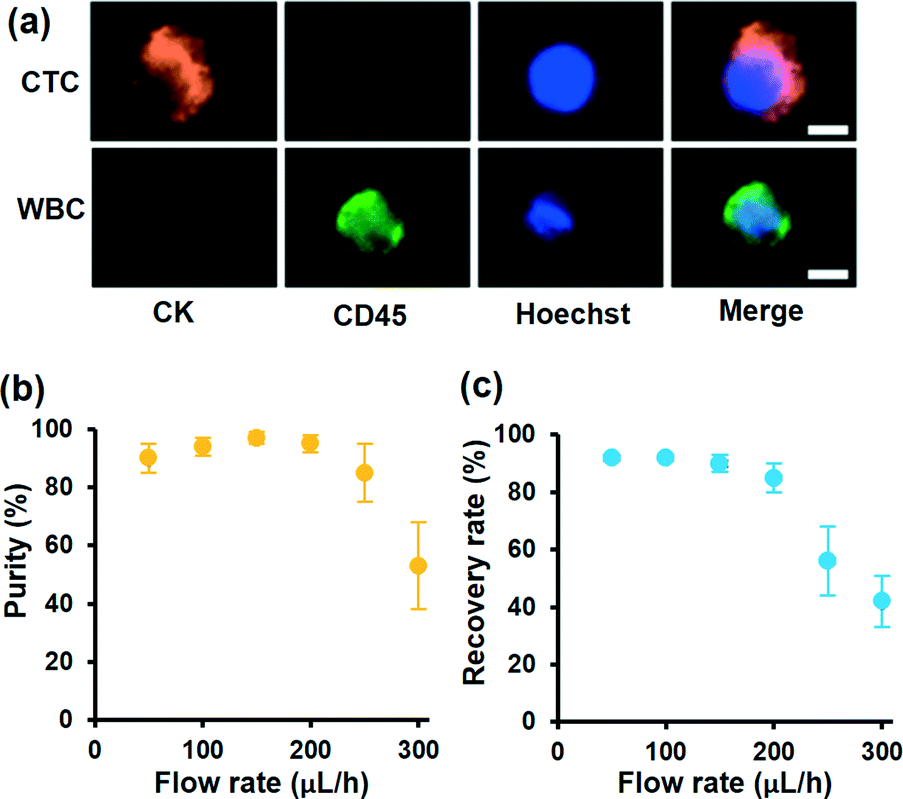 | ||
| Fig. 4 (a) Fluorescence images of isolated cells on this optofluidic system by three-color immunofluorescent staining. The scale bar is 10 μm. (b and c) The performance of the optofluidic chip for RBC modified CTC separation. The purity (b) and recovery rate (c) of MCF-7 collected from the target outlet varied with different flow rates when the laser intensity is set to 2.9 mW μm−2. The error bars represent mean ± standard deviation (n = 3). | ||
During the separation process, it is important for the hydrophobic interaction between RBCs and DSPE to remain stable. In order to test this binding, the RBCs modified with DSPE-PEG-Cy5 are injected into the optofluidic system three times in succession, to flow through the chip under the same conditions as those for the previous optical sorting procedure (laser intensity is 2.9 mW μm−2 and sample flow rate is 100 μL h−1). After each test, RBCs are collected to observe the fluorescence intensity, and then re-injected into the chip. As shown in ESI† Fig. S4, after flowing through the chip three times, the fluorescence intensity on the RBC's membrane is similar to the first test, indicating that DPSE with Cy5 is still tightly bound to RBCs.
Cell viability and proliferation test
Collected CTCs contain important information for understanding the molecular mechanism in clinic and research. Thus, it is important for the isolated CTCs to maintain viability and integrity. Here the demonstrated strategy will minimize cell damage and immunoreaction. Compared to magnetic beads, RBCs from the same donor have the advantages of being easily accessible and bio-friendly. The isolated CC-RBCs are shown in Fig. 5a, showing a negligible RBC loss on the membrane of tumor cells. RBCs can also be easily cleared with RBC lysis buffer; as shown in Fig. 5b, all the RBCs are cleared. Released cells are then tested using FDA/PI (fluorescein diacetate/propidium iodide) fluorescence, and cells showing green fluorescence are considered alive, while cells showing red PI fluorescence are considered dead. The statistical result is shown in Fig. 5c; in the four tests, more than 96% of the cells remain alive when different numbers of cells are spiked before processing. The proliferation ability of cells unprocessed using this strategy and those isolated in the experiment is examined; the unprocessed cells are tested as control groups. In Fig. 5d and e, the isolated MCF-7 cells show a similar proliferation tendency to that of initially untreated cells. These results show that this strategy has a negligible influence on cells in both viability and proliferation, verifying the biocompatibility of our strategy. This non-invasive advantage will meet the requirements of many biological applications which need isolated cells with function integrity.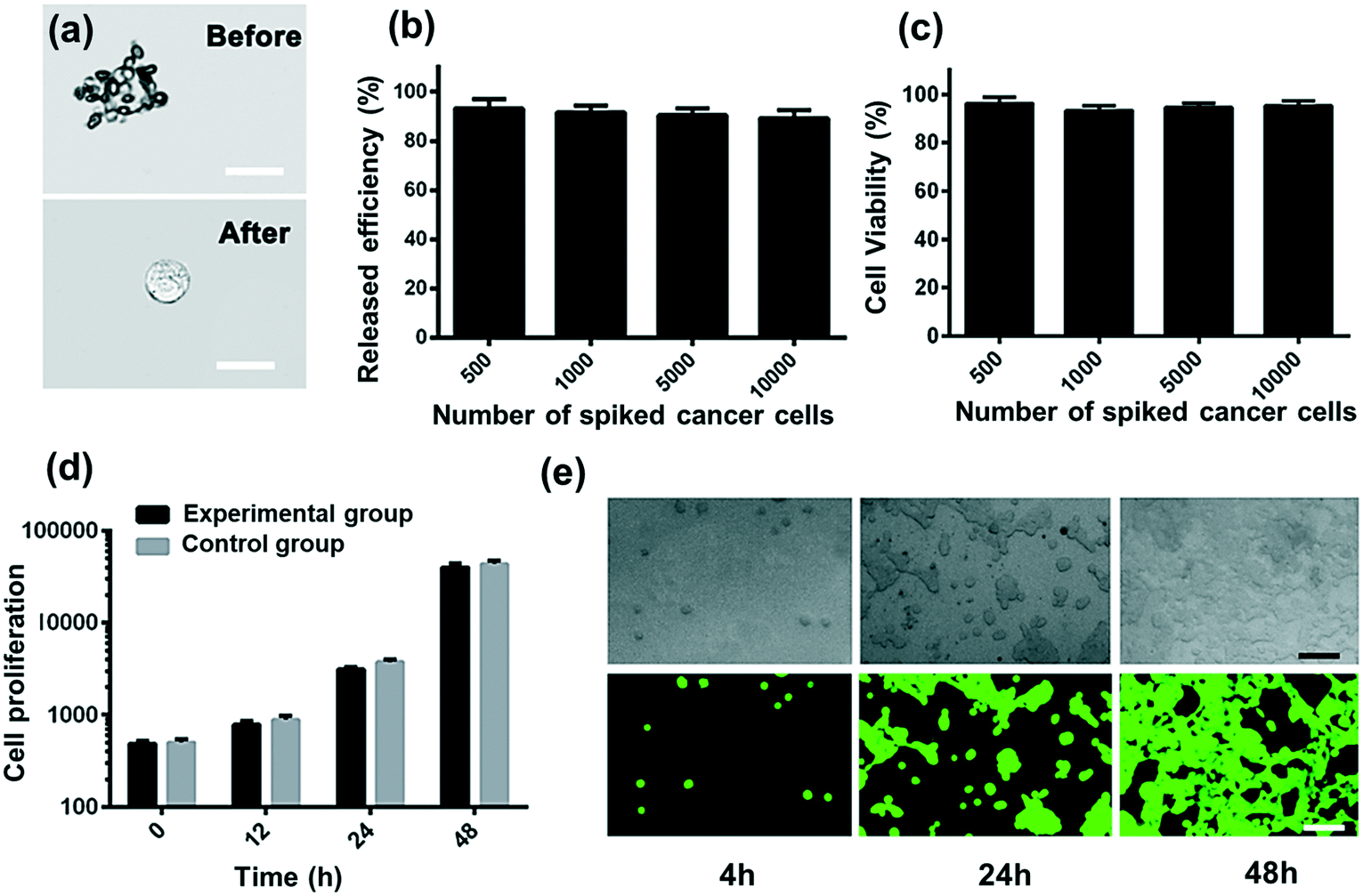 | ||
| Fig. 5 The cell viability and proliferation test. (a) Images of tumor cells before and after RBC lysis. Scale bar: 20 μm. (b) The release efficiency of different quantities of spiked cancer cells with red blood cell lysis buffer. (c) The viability of cells captured and released by the optofluidic device; it shows that cells maintain more than 90% viability in different tests. (d) Cell proliferation test of control and experimental groups at different times (n = 3). (e) Representative images of MCF-7 cells re-cultured for 2 days. Scale bar: 100 μm. | ||
Experimental
Materials
Paraformaldehyde (PFA, 36% in water), Triton X-100, bovine serum albumin (BSA), fluorescein diacetate (FDA), propidium iodide (PI), normal goat serum, lymphocyte separation medium (LSM) and bisbenzimide H 33258 (Hoechst 33258) were purchased from Sigma-Aldrich (USA). Sodium dodecyl sulfate (SDS)-polyacrylamide gel and SDS buffer were purchased from Beyotime (China) and Invitrogen (USA). Dulbecco's modified Eagle's medium (DMEM, Hyclone, high glucose) and PBS (Hyclone) were obtained from Thermo-Fisher Scientific. Fetal bovine serum (FBS) and 0.25% Trypsin-EDTA (Gibco, 1X) were obtained from Invitrogen (USA). Phycoerythrin (PE)-labeled anti-cytokeratin (CAM5.2) and fluorescein isothiocyanate (FITC)-labeled anti-human CD45 (Ms IgG1, clone H130) were purchased from BD Biosciences. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethyleneglycol)-2000] (ammonium salt) (DSPE-PEG-FA) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[Cy5 (poly ethylene glycol)-2000] (ammonium salt) (DSPE-PEG-Cy5) were obtained from Shang Hai Ponsure Biotech, Inc (China). All aqueous solutions were prepared using deionized (DI) water, which was purified on an experimental water purification system (Direct-Q3, Millipore, USA). All other solvents used in the present work were obtained from Aladdin-Reagent (China) and Sinopharm Chemical Reagent (China).Cell line and blood sample processing
The human breast cancer cell line MCF-7 and blood samples were provided by Zhongnan Hospital of Wuhan University. The blood samples were preserved after collection (under an approved institutional review board (IRB) protocol). The samples were delivered to our laboratory within 1 h after the draw and processed according to a DGC protocol. Firstly, 1 mL blood sample was 1:1 diluted using PBS. Next, diluted blood was carefully added onto the surface of LSM in a 15 mL centrifuge tube to form an obvious liquid–liquid interface. The tube was then centrifuged horizontally at 400g at RT for 30 min to separate the liquid into four layers. 50 μL of RBCs were extracted from the bottom layer and subsequently suspended in 1 mL of PBS for the capture procedure. Cancer cells were cultivated in Dulbecco's modified Eagle medium (DMEM) at 37 °C under a humidified 5% CO2 atmosphere, and the culture medium was supplemented with 10% bovine serum (FBS) and 1% penicillin/streptomycin. All the experiments were performed in compliance with the relevant laws and institutional guidelines of China and approved by the ethics committee at Wuhan University. Informed consent was obtained from human participants of this study.
Preparation and characterization of FA-RBCs
Firstly, about 50 μl of RBCs were extracted from healthy human blood, then the collected RBCs were mixed and cultured with 50 μL (1 mg ml−1) DSPE-PEG-FA for 30 min, so that the RBCs inserted with DSPE-PEG-FA are formed. Then, the samples were centrifuged at 1000g for 10 min and washed using PBS buffer three times. To ensure successful DSPE-PEG-FA insertion, DSPE-PEG-Cy5 was used instead of DSPE-PEG-FA. The fluorescence images of RBCs with DSPE-PEG-Cy5 were captured by employing a fluorescence microscope (IX81, Olympus, Japan). In order to check the protein change in the RBC membrane during the modification process, SDS-PAGE was employed. RBCs before modification and FA-RBCs were loaded into SDS buffer and measured using the BCA assay kit. Then the samples were heated at 95 °C for 5 min, and 20 μg of these two samples were added into wells in a 10% SDS-polyacrylamide gel. The samples were run at 120 V for 2 h; after that, the gel was obtained and stained using Commassie blue, then washed using DI water, and finally imaged. In addition, Hoechst labeled CTCs were incubated with Cy5 labeled engineered RBCs at 37 °C for 1 hour and subsequently observed by employing a confocal laser scanning microscope (CLSM; IX81, Olympus, Japan).Fabrication of the optofluidic isolating system
The optofluidic system consists of two main components, the micro channel and optical drive sorting system. The micro channel is made of PDMS, which is fabricated using the standard soft lithography technique; the channel height is 100 μm, and the sample inlet is half the width of the sheath flow inlet, while the two outlets have the same size. The sample and PBS flow streams are transported into the channel by glass syringes which are driven by syringe pumps (LSP01-2A, Longer Pump, China). For the optical sorting system, 1064 nm infrared laser light is produced by a solid state laser (Changchun New Industries Optoelectronics Tech Co, China); the laser is coupled to the channel with a multi-mode fiber, and the laser beam is collimated by a PDMS micro lens before the fiber.Cell viability and proliferation test
Cell viability was evaluated using both propidium iodide (PI) and fluorescein diacetate (FDA). PI is a red fluorescent nucleic acid dye that cannot enter cells that have intact plasma membranes, whereas FDA is a green fluorescent nucleic acid dye that can pass through intact cell membranes and accumulate inside the cells. PI and FDA solutions were both diluted to 5 μg mL−1 for the viability test of the cells. To test the cell viability, we spiked 500, 1000, 5000 and 10000 of the breast cancer cell line (MCF-7) into PBS and captured them by the optofluidic isolating system; live and dead cells were stained with PI and FDA, respectively, and subsequently counted under a fluorescence microscope (IX81, Olympus, Japan). Finally, the released MCF-7 cells were further re-cultured in the cell incubator and stained with FDA to observe their proliferation. The percentage of cell viability was defined as the ratio of viable cells against the total number of spiked cells. The release efficiency was defined as the ratio of a single tumor cell against the total number of spiked cells.
Fixation and staining of captured cells
After being purified through the microfluidic device, captured cancer cells were released and collected after RBC lysis buffer stimulation. Subsequently, 4% PFA was added into the tube to fix cells for 10 min. Further the cells were immersed in 0.2% Triton X-100 for 10 min to induce cellular permeability and allow for intracellular staining. Then, blocking was carried out for 30 min using 5% normal goat serum, 3% bovine serum album and 0.1% Tween-20 in PBS as a blocker, which decreased nonspecific bonding during immunofluorescence staining. Finally, cells were immersed in 10 μL FITC-labeled anti-CD45 stock solution (a marker for leukocytes) and 10 μL PE-labeled anti-CK stock solution (a protein marker for epithelial cells) at 4 °C in the dark for about 10 h and then stained with Hoechst 33258 for another 10 min before observation. All the bright field and fluorescence images were taken under an optical microscope (IX81, Olympus, Japan) and analyzed using Image-Pro Plus 6.0 software.Conclusions
In conclusion, we developed a new strategy using an optofluidic system to achieve precise and non-invasive CTC isolation by optical force using homologous erythrocyte binding. RBCs modified with FA can effectively bind to tumor cells, and form the CC-RBCs with an obviously larger volume and mean RI, so that the cluster becomes highly sensitive to optical force. Compared to immunomagnetic beads, engineered homologous FA-RBCs exhibit good performance in CTC rapid and non-invasive modification/releasing; the experiments based on MCF-7 spiked in donor blood samples indicate that more than 98% of the tumor cells are covered by ten to twenty erythrocytes, and 100% are released after processing. When the flow rate and laser intensity are properly matched, about 2 mL whole blood can be processed with over 90% recovery rate and 92% purity. Viability and proliferation tests indicate that this method is very friendly to cells, which is important for downstream applications. This strategy achieved convenient, non-invasive and quite precise CTC isolation, which is hard to realize using traditional methods. With these characteristics, we expect that this technology can be applied in clinic and research for early cancer diagnosis, personalized anticancer therapy and CTC based molecular profiling.Author contributions
Xuejia Hu, Daoming Zhu, Wei Liu and Yi Yang designed and performed experiments. Ming Chen helped with the blood sample preparation. Xuejia Hu, Daoming Zhu and Hailiang Liu analyzed the experimental results. Keke Chen helped with the device fabrication. All the authors took part in regular discussions and were involved in the completion of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 11774274, 81572860, and 81770179), National Key R&D Program of China (2018YFC1003200), Open Financial Grant from Qingdao National Laboratory for Marine Science and Technology (No. QNLM2016ORP0410), and Foundation Research Fund of Shenzhen Science and Technology Program (No. JCYJ20170818112939064).Notes and references
- V. Plaks, C. D. Koopman and Z. Werb, Science, 2013, 341, 1186–1188 CrossRef CAS PubMed.
- B. Mostert, S. Sleijfer, J. A. Foekens and J. W. Gratama, Cancer Treat. Rev., 2009, 35, 463–474 CrossRef CAS PubMed.
- R. Li, F. F. Chen, H. Q. Liu, Z. X. Wang, Z. T. Zhang, Y. Wang, H. Cui, W. Liu, X. Z. Zhao, Z. J. Sun and S. S. Guo, ACS Appl. Mater. Interfaces., 2018, 10, 16327–16334 CrossRef CAS PubMed.
- P. Paterlini-Brechot and N. L. Benali, Cancer Lett., 2007, 253, 180–204 CrossRef CAS PubMed.
- Q. Huang, F. B. Wang, C. H. Yuan, Z. Gelatin, L. Rao, B. Cai, B. Chen, S. Jiang, Z. Li, J. Chen, W. Liu, F. Guo, Z. Ao, S. Chen and X. Z. Zhao, Theranostics, 2018, 8, 1624–1635 CrossRef CAS PubMed.
- M. Hosokawa, T. Hayata, Y. Fukuda, A. Arakaki, T. Yoshino, T. Tanaka and T. Matsunaga, Anal. Chem., 2010, 82, 6629–6635 CrossRef CAS PubMed.
- L. Rao, Q. F. Meng, Q. Huang, Z. Wang, G. T. Yu, A. Li, W. Ma, N. Zhang, S. S. Guo, X. Z. Zhong, K. Liu, Y. Yuan and W. Liu, Adv. Funct. Mater., 2018, 28, 1803531 CrossRef.
- Y. Song, Y. Shi, M. Huang, W. Wang, Y. Wang, J. Cheng, Z. Lei, Z. Zhu and C. Y. Yang, Angew. Chem., Int. Ed., 2019, 58, 2236–2240 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- Q. Huang, B. Cai, B. Chen, L. Rao, Z. He, R. He, F. Guo, L. Zhao, K. K. Kondamareddy, W. Liu, S. S. Guo and X. Z. Zhao, Adv. Healthcare Mater., 2016, 5, 1554–1559 CrossRef CAS PubMed.
- F. F. Becker, X. B. Wang, Y. Huang, R. Pethig, J. Vykoukal and P. R. Gascoyne, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 860–864 CrossRef CAS PubMed.
- J. M. Park, J. Y. Lee, J. G. Lee, H. Jeong, J. M. Oh, Y. J. Kin, D. Park, M. S. Kim, H. J. Lee, J. H. Oh, S. S. Lee, W. Y. Lee and N. Huh, Anal. Chem., 2012, 84, 7400–7407 CrossRef CAS PubMed.
- D. A. Smirnov, D. R. Zweitzig, B. W. Foulk, M. C. Miller, G. V. Doyle, K. J. Pienta, N. J. Meropol, L. M. Weiner, S. J. Cohen, J. G. Moreno, M. C. Connelly, L. W. M. M. Terstappen and S. M. O'Hara, Cancer Res., 2005, 65, 4993–4997 CrossRef CAS PubMed.
- A. M. Sieuwerts, J. Kraan, J. B. Vries, P. Spoel, B. Mostert, J. W. M. Martens, J. W. Gratama, S. Sleijifer and J. A. Foekens, Breast Cancer Res. Treat., 2009, 118, 455–468 CrossRef CAS PubMed.
- E. S. Lianidou, A. Markou and A. Strati, Cancer Metastasis Rev., 2012, 31, 663–671 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- N. N. Lu, M. Xie, J. Wang, S. W. Lv, J. S. Yi, W. G. Dong and W. H. Huang, ACS Appl. Mater. Interfaces., 2015, 7, 8817–8826 CrossRef CAS PubMed.
- A. Ashkin, Phys. Rev. Lett., 1970, 24, 156–159 CrossRef CAS.
- M. P. MacDonald, G. C. Spalding and K. Dholakia, Nature, 2003, 426, 421–424 CrossRef CAS PubMed.
- Y. Z. Shi, S. Xiong, Y. Zhang, L. K. Chin, Y.-Y. Chen, J. B. Zhang, T. H. Zhang, W. Ser, A. Larson, L. S. Hoi, J. H. Wu, T. N. Chen, Z. C. Yang, Y. L. Hao, B. Liedberg, P. H. Yap, D. P. Tsai, C.-W. Qiu and A. Q. Liu, Nat. Commun., 2018, 9, 815 CrossRef CAS PubMed.
- D. G. Grier, Nature, 2003, 424, 810–816 CrossRef CAS PubMed.
- Y. Z. Shi, S. Xiong, L. K. Chin, Y. Yang, J. B. Zhang, W. Ser, J. H. Wu, T. N. Chen, Z. C. Yang, Y. L. Hao, B. Liedberg, P. H. Yap, Y. Zhang and A. Q. Liu, Lab Chip, 2017, 17, 2443–2450 RSC.
- X. J. Hu, H. L. Liu, Y. X. Jin, L. Liang, D. M. Zhu, X. Q. Zhu, S. S. Guo, F. L. Zhou and Y. Yang, Lab Chip, 2018, 18, 3405–3412 RSC.
- M. M. Wang, E. Tu, D. E. Raymond, J. M. Yang, H. Zhang, N. Hagen, B. Dees, E. M. Mercer, A. H. Forster, I. Kariv, P. J. Marchand and W. F. Butler, Nat. Biotechnol., 2005, 23, 83–87 CrossRef CAS PubMed.
- S. J. Hart, A. Terray, T. A. Leski, J. Arnold and R. Stroud, Anal. Chem., 2006, 78, 3221–3225 CrossRef CAS PubMed.
- Y. Park, M. Diez-Silva, G. Popescu, G. Lykotrafitis, W. Choi, M. S. Feld and S. Suresh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13730–13735 CrossRef CAS PubMed.
- P. Y. Liu, L. K. Chin, H. F. Chen, C.-M. Hsieh, C.-H. Lee, K.-B. Sung, T. C. Ayi, P. H. Yap, B. Liedberg, K. Wang, T. Bourouina and Y. Leprince-Wang, Lab Chip, 2016, 16, 634–644 RSC.
- L. Rao, L. L. Bu, J. H. Xu, B. Cai, G. T. Yu, X. Yu, Z. He, Q. Huang, A. Li, S. S. Guo, W. F. Zhang, W. Liu, Z. J. Sun, H. Wang, T. H. Wang and X. Z. Zhao, Small, 2015, 11, 6225–6236 CrossRef CAS PubMed.
- D. M. Zhu, L. Wu, M. Suo, S. Gao, W. Xie, M. H. Zan, A. Liu, B. Chen, W. T. Wu, L. W. Ji, L. B. Chen, H. M. Huang, S. S. Guo, W. F. Zhang, X. Z. Zhao, Z. J. Sun and W. Liu, Nanoscale, 2018, 10, 6014–6023 RSC.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd Jr, A. J. Gilman, J. B. Lord, D. W. Simeon Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- G. I. Ruban, S. M. Kosmacheva, N. V. Goncharova, D. V. Bockstaele and V. A. Loiko, J. Biomed. Opt., 2007, 12, 044017 CrossRef PubMed.
- V. P. Maltsev, A. G. Hoekstra and M. A. Yurkin, Exp. Tech., 2011, 4, 63–93 Search PubMed.
- L. Liang, Y. X. Jin, X. Q. Zhu, F. L. Zhou and Y. Yang, Lab Chip, 2018, 18, 1422–1429 RSC.
- H. L. Liu, Y. Shi, L. Liang, L. Li, S. S. Guo, L. Yin and Y. Yang, Lab Chip, 2017, 17, 1280–1286 RSC.
- Y. Yang, A. Q. Liu, L. K. Chin, X. M. Zhang, D. P. Tsai, C. L. Lin, C. Lu, G. P. Wang and N. I. Zheludev, Nat. Commun., 2012, 3, 651 CrossRef CAS PubMed.
- A. S. Utada, E. Lorenceau, D. R. Link, P. D. Kaplan, H. A. Stone and D. A. Weitz, Science, 2005, 308, 537–541 CrossRef CAS PubMed.
- A. Bayram, M. Serhatlioglu, B. Ortac, S. Demic, C. Elbuken, M. Sen and M. E. Solmaz, Sens. Actuators, A, 2018, 269, 382–387 CrossRef CAS.
- W. Wu, X. Zhu, Y. Zuo, L. Liang, S. Zhang, X. Zhang and Y. Yang, ACS Photonics, 2016, 3, 2497–2504 CrossRef CAS.
- S. Xiong, A. Q. Liu, L. K. Chin and Y. Yang, Lab Chip, 2011, 11, 1864–1869 RSC.
- G. Leitz, E. Fällman, S. Tuck and O. Axner, Biophys. J., 2002, 82, 2224–2231 CrossRef CAS PubMed.
- S. K. Mohanty, A. Rapp, S. Monajembashi, P. L. Gupta and K. O. Greulich, Radiat. Res., 2002, 157, 378–385 CrossRef CAS PubMed.
- Y. Liu, G. J. Sonek, M. W. Berns and B. J. Tromberg, Biophys. J., 1996, 71, 2158–2167 CrossRef CAS PubMed.
- L. E. Kelemen, Int. J. Cancer, 2006, 119, 243–250 CrossRef CAS PubMed.
- W. Pan, H. Yang, T. Zhang, Y. Li, N. Li and B. Tang, Anal. Chem., 2013, 85, 6930–6935 CrossRef CAS PubMed.
- W. Pan, H. Yang, T. Zhang, Y. Li, N. Li and B. Tang, Anal. Chem., 2013, 85, 6930–6935 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 197–206 CrossRef CAS PubMed.
- X. Sun, C. Wang, M. Gao, A. Hu and Z. Liu, Adv. Funct. Mater., 2015, 25, 2386–2394 CrossRef CAS.
- L. Rao, L. L. Bu, Q. F. Meng, B. Cai, W. W. Deng, A. Li, K. Li, S. S. Guo, W. F. Zhang, W. Liu, Z. J. Sun and X. Z. Zhao, Adv. Funct. Mater., 2017, 27, 1604774 CrossRef.
- P. Sethu, A. Sin and M. Toner, Lab Chip, 2005, 6, 83–89 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00361d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |