
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmuno-capture of extracellular vesicles for individual multi-modal characterization using AFM, SEM and Raman spectroscopy†
Pepijn
Beekman‡
 ab,
Agustin
Enciso-Martinez‡
c,
Hoon Suk
Rho
bd,
Sidharam Pundlik
Pujari
a,
Aufried
Lenferink
c,
Han
Zuilhof
ae,
Leon W. M. M.
Terstappen
c,
Cees
Otto‡
*c and
Séverine
Le Gac‡
*b
ab,
Agustin
Enciso-Martinez‡
c,
Hoon Suk
Rho
bd,
Sidharam Pundlik
Pujari
a,
Aufried
Lenferink
c,
Han
Zuilhof
ae,
Leon W. M. M.
Terstappen
c,
Cees
Otto‡
*c and
Séverine
Le Gac‡
*b
aLaboratory of Organic Chemistry, Wageningen University, The Netherlands
bApplied Microfluidics for BioEngineering Research, MESA+ Institute for Nanotechnology and TechMed Center, University of Twente, The Netherlands. E-mail: s.legac@utwente.nl
cMedical Cell BioPhysics, TechMed Center, University of Twente, The Netherlands. E-mail: c.otto@utwente.nl
dDepartment of Instructive Biomaterials Engineering, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht University, The Netherlands
eSchool of Pharmaceutical Sciences and Technology, Tianjin University, 92 Weijin Road, Tianjin, China
First published on 11th July 2019
Abstract
Tumor-derived extracellular vesicles (tdEVs) are promising blood biomarkers for cancer disease management. However, blood is a highly complex fluid that contains multiple objects in the same size range as tdEVs (30 nm–1 μm), which obscures an unimpeded analysis of tdEVs. Here, we report a multi-modal analysis platform for the specific capture of tdEVs on antibody-functionalized stainless steel substrates, followed by their analysis using SEM, Raman spectroscopy and AFM, at the single EV level in terms of size and size distribution, and chemical fingerprint. After covalent attachment of anti-EpCAM (epithelial cell adhesion molecule) antibodies on stainless steel substrates, EV samples derived from a prostate cancer cell line (LnCAP) were flushed into a microfluidic device assembled with this stainless steel substrate for capture. To track the captured objects between the different analytical instruments and subsequent correlative analysis, navigation markers were fabricated onto the substrate from a cyanoacrylate glue. Specific capture of tdEVs on the antibody-functionalized surface was demonstrated using SEM, AFM and Raman imaging, with excellent correlation between the data acquired by the individual techniques. The particle distribution was visualized with SEM. Furthermore, a characteristic lipid–protein band at 2850–2950 cm−1 was observed with Raman spectroscopy, and with AFM the size distribution and surface density of the captured EVs was assessed. Finally, correlation of SEM and Raman images enabled discrimination of tdEVs from cyanoacrylate glue particles, highlighting the capability of this multi-modal analysis platform for distinguishing tdEVs from contamination. The trans-instrumental compatibility of the stainless steel substrate and the possibility to spatially correlate the images of the different modalities with the help of the navigation markers open new avenues to a wide spectrum of combinations of different analytical and imaging techniques for the study of more complex EV samples.
Introduction
Liquid biopsies have been proposed as an alternative to conventional approaches (e.g., magnetic resonance imaging or solid biopsies) for the disease management of cancer patients. In this non-invasive approach, a blood sample (7.5 ml) is analyzed for the presence and amount of circulating tumor cells (CTCs), tumor-derived EVs (tdEVs), cell free DNA (cf-DNA), miRNA and/or tumor-associated proteins or peptides.1,2 CTCs are well suited to characterize a tumor and to evaluate the heterogeneity for subsequent selection of the optimal treatment.3 The concentration of CTCs is however extremely low (∼1 CTC ml−1), especially when compared with that of blood cells (∼109 ml−1).4 In contrast, tdEVs are much more abundant with concentrations up to 1010 tdEVs ml−1.5 Importantly, the presence and amount of tdEVs in blood has been proven to strongly correlate with the survival of patients with metastatic prostate cancer.6 EVs are membrane-bound biological carriers of biomolecules, which are shed by all cell types. They are found in all body fluids,7 and exhibit a size ranging from 30 nm to 1 μm.8,9 EVs are of great interest because of their implication in intercellular communication and pathogenesis;7,10,11 they show great promises not only for disease diagnosis but also for drug delivery.4,11–13 Altogether, EV analysis offers a promising approach for non-invasive cancer patient management as a result of the wealth of biological information they carry, some of which being potential biomarkers.14However, blood is a highly complex fluid15 that contains lipoproteins, cell debris and protein aggregates, as well as EVs of non-cancerous origin, which are all in the same size and density range as tdEVs, and the same applies for less complex samples originating from cell culture media. In all these cases, tdEVs need to be selectively isolated and/or distinguished from EVs of non-cancerous origin and other small objects.16,17 Several methods have been proposed for EV isolation, among which ultracentrifugation and size-exclusion chromatography are the most popular.8,16,18–20 However, these isolation approaches yield highly heterogeneous samples containing tdEVs, other EVs, cell debris and molecular aggregates. Therefore, alternative approaches have been introduced that rely on the immuno-capture of targeted EVs, using either generic membrane markers (e.g., CD9, CD63 and CD81) to retrieve all exosomes/EVs from a sample,21,22 or specific membrane markers (e.g., EpCAM,6 EGFR,12 HER223) to selectively isolate tdEVs. Microfluidic technology has proven to be instrumental for the immunocapture of EVs by controlling the surface dynamics (e.g., controlling flow rate when washing non-specifically bound species), and drastically reducing the distances over which EVs have to migrate before coming in contact with the functionalized surface. In addition, microfluidics facilitates controlled and sequential handling of (very small amounts of) samples.8,24
A second main challenge is the high heterogeneity found in any purified EV sample, in terms of size and from a molecular perspective. Therefore, EVs must be thoroughly characterized for their possible and reliable recognition in heterogeneous samples. For that purpose, it is important to study individual EVs and not populations to avoid that ensemble averaging obscures differences. EVs have been analyzed using a great variety of techniques25–28 such as flow cytometry,29–31 confocal and non-confocal (fluorescence) microscopy,22,32 scanning electron microscopy (SEM),33 atomic force microscopy (AFM),34 Raman spectroscopy,35 surface plasmon resonance (SPR),36,37 mass spectrometry (MS)38 and micro nuclear magnetic resonance (μNMR).39 However, not all techniques allow the collection of information at the single EV level. Furthermore, to get comprehensive information on heterogeneous samples, different techniques yielding complementary information must be combined. In that context, Raman spectroscopy, SEM and AFM are of great interest. Raman spectroscopy provides chemical information on a sample of interest in a label-free manner.40,41 SEM enables characterization of the size and morphology of intact EVs.42,43 Correlating this size and morphology information with Raman fingerprints confirms the cellular origin of individual EVs, and, in previous work, we have demonstrated that using this combination cancer cells could be distinguished from non-cancer cells.44,45 Finally, AFM yields more detailed information on the size and morphology of EVs, and possibly, on their mechanical properties.30,46
In this paper, we report the specific isolation of tdEVs obtained from a human prostate cancer cell line (LNCaP) on functionalized stainless-steel substrates followed by their in situ multi-modal characterization with SEM, Raman and AFM imaging (Fig. 1). Stainless steel substrates were selected for their suitability for all considered modalities: this material gives little background in Raman (see ESI 1†);44,45 it is conductive; and mirror-polished stainless steel substrates have a low surface roughness level of ca. 7 nm, which is well-suited for the analysis of EVs by AFM. Here, and as depicted in Fig. 2, stainless steel substrates were first functionalized with a monolayer of carboxydecyl phosphonic acid (CDPA),47–49 onto which antibodies targeting tdEVs were covalently anchored through carbodiimide-based bioconjugation chemistry.50 The resulting monolayers were characterized with X-ray photoelectron spectroscopy (XPS) and infrared reflection–absorption spectroscopy (IRRAS), to optimize their formation with respect to the initial CDPA concentration. Next, LNCaP-derived EVs were injected in a microfluidic channel assembled onto the functionalized stainless steel substrate, for their capture, which was confirmed using individual imaging techniques. For their multi-modal analysis, and to easily track individual EVs in the different instruments, navigation markers were fabricated on the functionalized substrates next to a region of interest (ROI). Finally, the captured EVs were successively analyzed by Raman imaging, SEM, and AFM, and data acquired by the different techniques correlated. The trans-instrumental compatibility of the stainless steel substrate and the tracking possibility offered by the navigation markers give the opportunity to apply a wide spectrum of combinations of different analytical and imaging techniques.
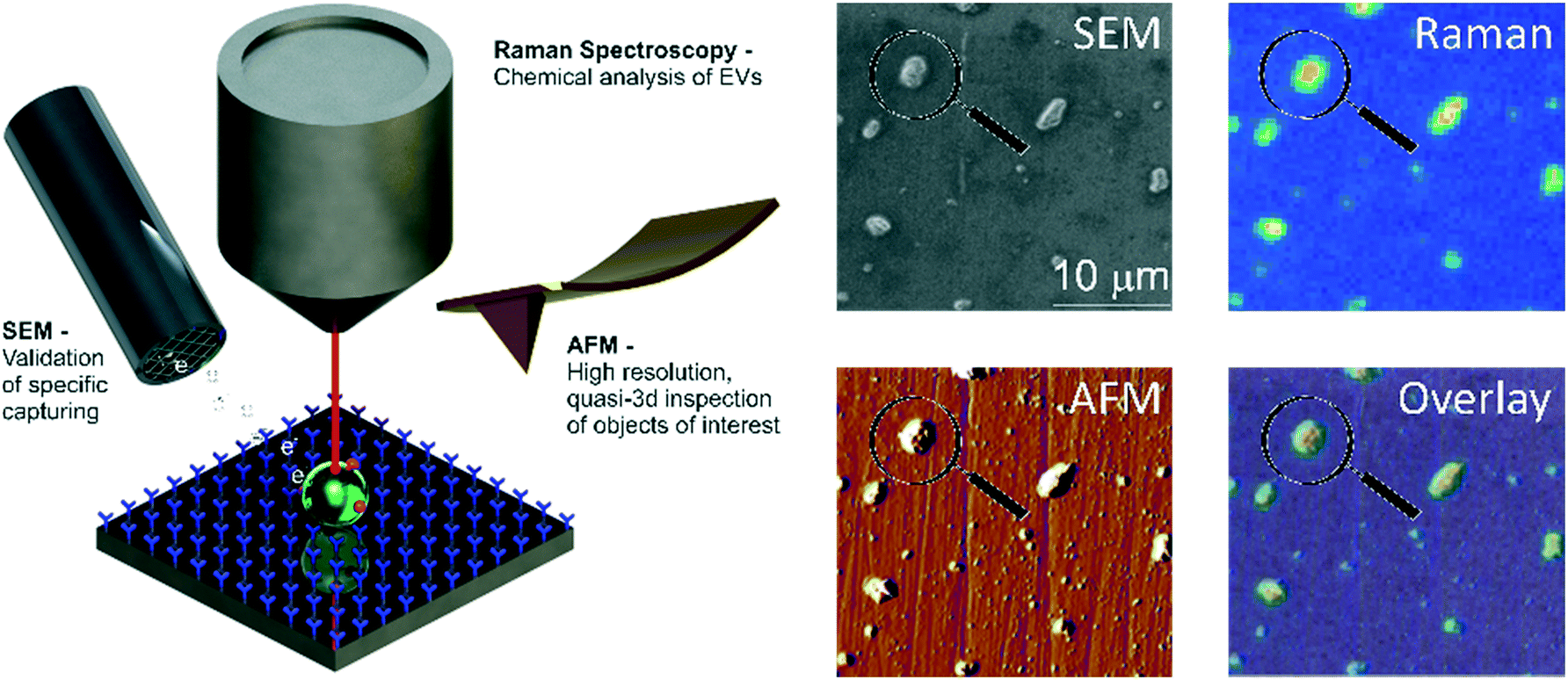 | ||
| Fig. 1 Multi-modal analysis of tdEVs on antibody-functionalized stainless steel substrates. After their selective capture on a stainless steel substrate functionalized with antibodies of interest (here anti-EpCAM antibodies targeting tdEVs), EVs are successively imaged using Raman spectroscopy, SEM and AFM, and information collected from these different imaging modalities correlated to get a comprehensive picture on the captured objects. | ||
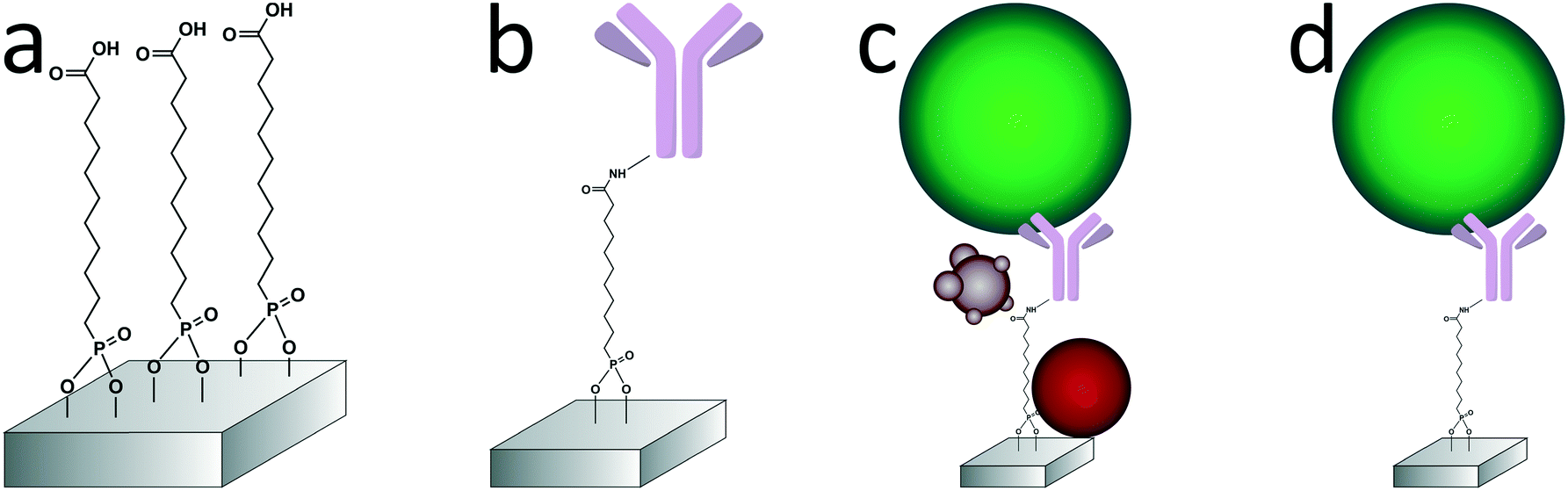 | ||
| Fig. 2 Different steps of surface modification and capture of the tdEVs on stainless steel substrates. a) An oxygen plasma-treated stainless steel substrate is functionalized with a carboxydecyl phosphonic acid (CDPA) monolayer. b) Anti-EpCAM antibodies are conjugated to the CDPA monolayer using NHS/carbodiimide chemistry. c) tdEVs (in green here) are specifically immuno-captured on the antibody-functionalized surface. d) The substrate is washed to remove non-specifically bound materials (in red and purple here), before retained EVs are fixed and dehydrated. | ||
Experimental
Materials
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), acetone (VLSI grade), paraformaldehyde (PFA), phosphate-buffered saline (PBS), and 2-(N-morpholino)ethanesulfonic acid were purchased from Merck (Zwijndrecht, The Netherlands). Ethanol (VLSI grade) and dichloromethane were purchased from VWR (Amsterdam, The Netherlands). Carboxydecyl phosphonic acid (CDPA) was purchased from Sikémia (Montpellier, France). Sylgard 184 poly(dimethylsiloxane) (PDMS) was purchased from Farnell (Utrecht, The Netherlands). SS316L Stainless steel foils (0.9 mm thickness, one side mirror polished) were purchased from Goodfellow Inc. (Bad Nauheim, Germany). Anti-EpCAM antibodies were produced at the University of Twente, The Netherlands (Medical Cell BioPhysics Laboratory) from VU1D9 hybridoma cells.PDMS handling devices
Three different PDMS devices (a 6 mm diameter reservoir, a xurography channel and a navigation marker device) were used for different steps of the sample preparation, as depicted in Fig. 3. For all devices, PDMS was prepared and cured according to the same procedure. PDMS precursor and cross linker (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) were first thoroughly mixed, and subsequently degassed by centrifugation at 1000 × g for 1 min. The resulting mixture was poured on different molds for the different devices, and degassed again in a desiccator for 15 min. Curing was performed at 80 °C overnight.
:1 weight ratio) were first thoroughly mixed, and subsequently degassed by centrifugation at 1000 × g for 1 min. The resulting mixture was poured on different molds for the different devices, and degassed again in a desiccator for 15 min. Curing was performed at 80 °C overnight.
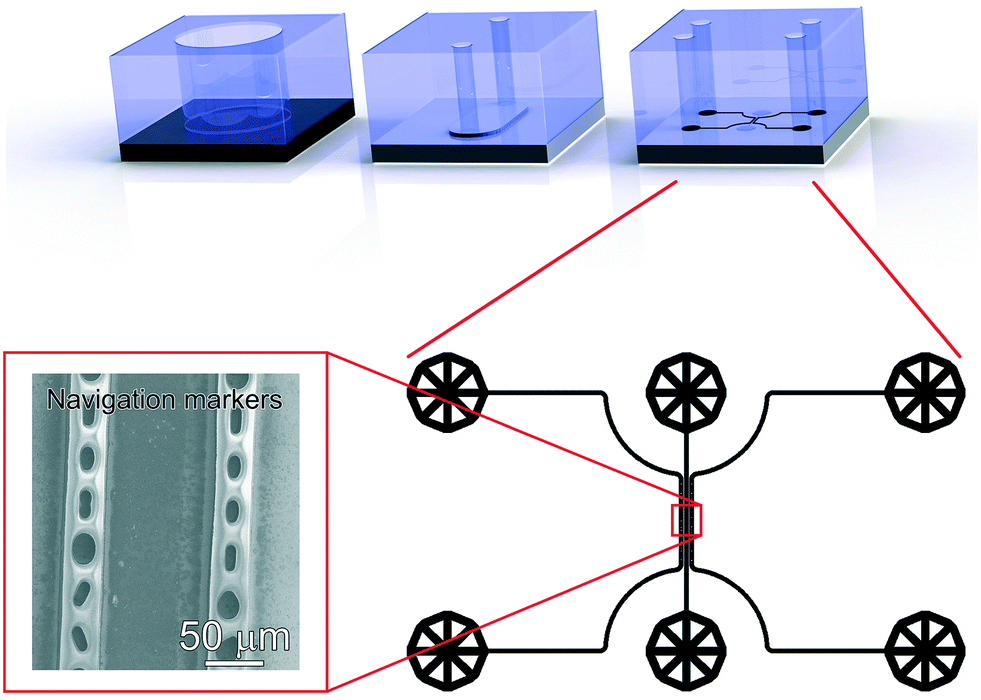 | ||
| Fig. 3 Various PDMS devices used in this work. Top, and from left to right: 6 mm diameter reservoir used for the immobilization of antibodies under static conditions on a CDPA monolayer; xurography microchannel (6 × 3 × 0.2 mm3) used for the capture of EVs and subsequent washing under mild flow conditions (400 μl min−1); microfluidic device used for the fabrication of navigation markers next to a 50 μm × 1 mm sample region, subsequently considered for analysis using SEM, Raman and AFM. Bottom, right: Design of the microfluidic device used to fabricate the navigation markers, consisting of three microfluidic channels, two microchannels comprising pillars with various geometries, flanking one sample microchannel, all channels being 30 μm height × 50 μm width × 1 mm length, and (Left) actual SEM image showing the navigation markers fabricated from cyanoacrylate glue injected in the side-channels and used to retrace the captured objects in the different imaging instruments. | ||
The design of this microfluidic device was drawn in CleWin (WieWeb, Hengelo, The Netherlands), and the mold fabricated in the Nanolab cleanroom of the MESA+ Institute for Nanotechnology. Briefly, a <100> Si wafer was spin-coated with AZ-40XT resist (Microchemicals, Ulm, Germany) at 3000 RPM for 1 min to yield a 30 μm thick layer. The photoresist was exposed, baked and developed according to the manufacturer's specifications. After PDMS casting on the finished mold, fluidic accesses were punched using a 1 mm diameter Harris Uni-Core biopsy punch. The PDMS device was placed on top of a functionalized stainless steel substrate after EV capture and dehydration. No specific care was required for alignment of the device, since the width of the sample region is much smaller than that of the xurography channel. Cyanoacrylate superglue (Tesa SE, Norderstedt, Germany) was injected in the side channels and cured for 30 min to create the navigation markers. After PDMS delamination, the designed micro-features were transferred to the stainless steel surface with high fidelity (Fig. 3, bottom left).
CDPA monolayer formation and characterization on stainless steel substrates
Antibody conjugation onto the CDPA monolayer
Anti-EpCAM antibodies were conjugated onto the CDPA monolayer using EDC/NHS chemistry at room temperature (Fig. 2b). In the PDMS reservoir, a solution of 40 mM NHS, 130 mM EDC and 50 mM 2-(N-morpholino)ethanesulfonic acid in Milli-Q (pH 5) was pipetted and left to react with the CDPA monolayer for 30 min. The substrate was subsequently rinsed with 50 μl of a 5 mM acetic acid solution in Milli-Q to stop the reaction, followed by 100 μl PBS. Next, the antibody solution at 20 μg ml−1 in PBS was pipetted in the reservoir and incubated with the surface for 1 h, followed by extensive washing with PBS to remove unreacted chemicals. Finally, unreacted NHS ester groups were blocked by a 0.1 M ethanolamine solution in Milli-Q for 30 min. The reservoirs were filled with PBS buffer until their use within 24 h.Vesicle capture and dehydration
EVs were isolated from culture medium (see ESI 3† for the isolation protocol) of the human prostate cancer cell line LNCaP, which is known to express epithelial cell adhesion molecules (EpCAM). To avoid any EV contamination from the serum added to the culture medium, the LNCaP cells were cultured in serum-free medium for 48 h before their isolation. Nanoparticle tracking analysis (NTA)51 of those isolated EVs revealed a concentration of 1.06 × 109 EVs ml−1 (see ESI 4†). 25 μl of this EV-containing suspension was introduced into the xurography channel (channel volume: 3.6 μl) using a capillary pipette tip acting as an inlet reservoir, and left under static incubation for 1 h at room temperature. Channels were subsequently washed with 200 μl of PBS at a flow-rate of 400 μl min−1 using a syringe-pump connected to the outlet reservoir and operated in withdrawal mode (Fig. 2d). Captured EVs were next fixed in the channel with a 1% PFA solution in PBS. After fixation of the EVs, the PDMS device was removed, the sample rinsed in Milli-Q, dehydrated by immersion in a solution of 70% ethanol in Milli-Q (5 min) followed by immersion in pure ethanol (5 min), and finally dried overnight under ambient conditions. Various negative control experiments were conducted in this study, as summarized in Table ESI 1:† (i) no activation of the CDPA layer with EDC/NHS; (ii) no immobilization of anti-EpCAM antibodies; and (iii) no incubation with any EV sample.Multi-modal analysis of the captured EVs on anti-EpCAM-conjugated stainless steel substrates
Results and discussion
Monolayer formation and characterization
To optimize the monolayer formation, different concentrations of carboxydecyl phosphonic acid (CDPA) were tested and the resulting CDPA monolayers analyzed using IRRAS to evaluate the surface coverage, molecular ordering, and the configurations of the carboxyl groups. Spectra recorded for stainless steel substrates functionalized with CDPA (1 mM solution), as well as for CDPA powder, are presented in Fig. 4a. Bands assigned to the anti-symmetric CH2 stretch were found around 2914 cm−1 for all tested CDPA concentrations (see ESI 5†). These anti-symmetric CH2 stretch bands are typically found between 2914 and 2930 cm−1, and their exact values reflect the packing density of the monolayer. Low values, as observed here, suggest densely packed monolayers displaying a short-range inter-chain monolayer ordering.47,48,53–56 In all samples, the carboxyl band was detected at ∼1720 cm−1, which is attributed to acyclically dimerized carboxyl groups,57i.e., hydrogen-bonding dimerization with nearest neighbors. Higher absorption frequencies for this band (towards 1740 cm−1) indicate non-hydrogen-bonded species, and therefore a less dense monolayer. Lower frequencies (∼1700 cm−1) would suggest cyclic dimerization as a result of multilayer formation. In the 1700–1740 cm−1 region, peak broadening was observed in the substrates functionalized with 0.1 and 10 mM solutions (see ESI 5†), indicating a lesser degree of ordering than for the substrates prepared with a 1 mM solution. The baseline across the relative samples revealed that the signal-to-noise ratio for the substrate prepared with a 1 mM solution was also significantly improved compared to the other samples. Therefore, on the basis of these results, further experiments were conducted using a 1 mM CDPA solution for the 1st step functionalization of the stainless steel substrates.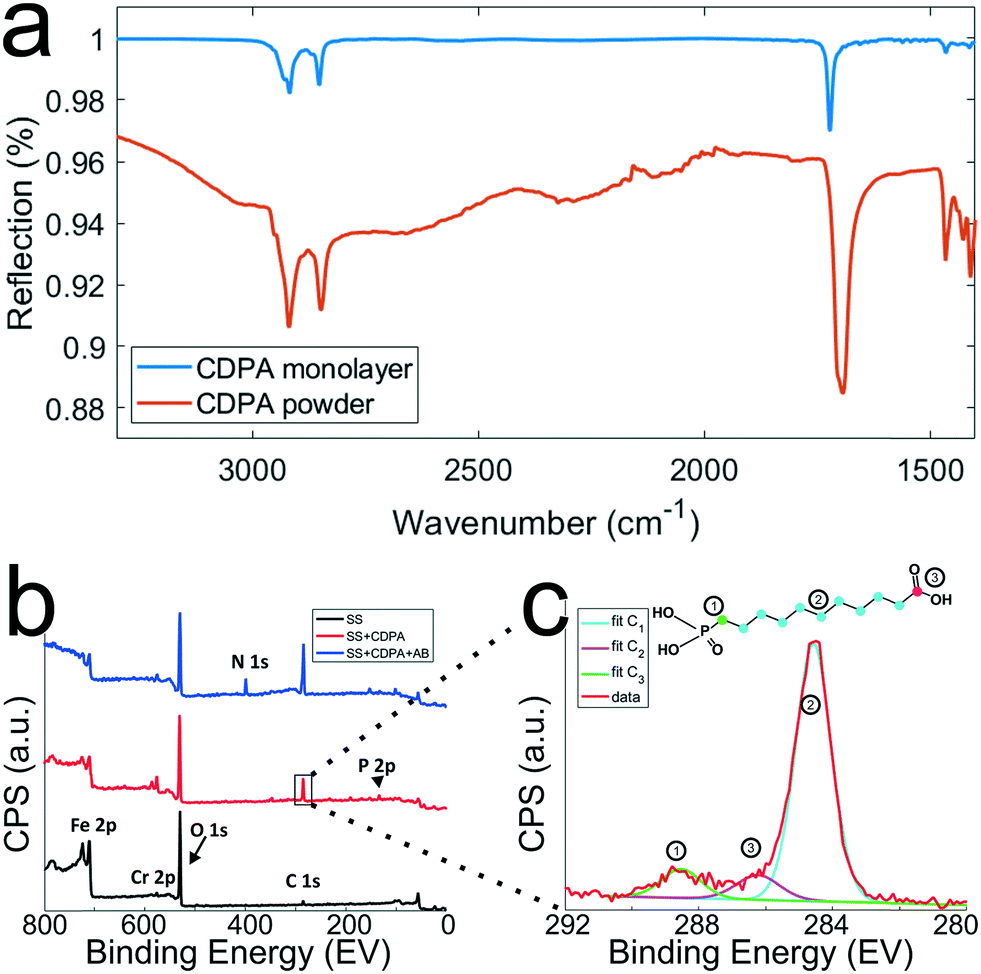 | ||
| Fig. 4 Chemical characterization of the stainless steel surfaces after the different surface modification steps. a) IRRAS spectra (which are background corrected) of CDPA monolayers prepared on SS316L stainless steel substrates using a 1 mM CDPA solution (blue trace) and CDPA powder (red trace). b) XPS spectra recorded after the various surface modification steps, as depicted in Fig. 2. Wide range scans acquired on a O2 plasma-treated stainless steel substrate (SS, black trace), a stainless steel substrate functionalized with a CDPA monolayer (SS + CDPA, red trace); and after antibody conjugation on the CDPA monolayer (SS + CDPA + AB, blue trace). c) C 1s narrow scan showing fitted peaks corresponding to the different carbon species found in a CDPA molecule (inset). | ||
After each surface modification step (oxygen plasma treatment, CDPA functionalization, antibody modification), the substrates were also analyzed using XPS (Fig. 4b). Integration of the peak surface areas provides quantitative information about the proportion of elements found on the substrate. After O2 plasma (black line, Fig. 4b), relatively little carbon (C 1s signal at ∼285 eV) was found on the stainless steel substrates, and this corresponds to adventitiously adsorbed carbon. A significant oxygen peak (O 1s peak at 532 eV) was detected as a result of the plasma treatment. Finally, various metals were present, such as Fe 2p (710 eV) and Cr 2p (575 eV). After formation of the CDPA monolayer, the signal corresponding to carbon became more intense, and a peak appeared at 134 eV, corresponding to P 2p. Integration of these two peaks reveals a C:P ratio of 11.2:1, which is in excellent agreement with the theoretically expected 11:1 ratio according to the molecular formula of CDPA (Fig. 4c). Carbon atoms experiencing different electronic environments are characterized by different binding energies, and CDPA molecules comprise carbon atoms in three distinct environments, as depicted in Fig. 4c. The C atom in the carbonyl group is observed at ∼289 eV; the phosphorous-bound carbon at ∼286.2 eV; and the alkyl chain carbon atoms at 285 eV. Integrating these different C 1s signals yields a ratio of ∼8.8:1.1:1, which is again in good agreement with the molecular structure of CDPA (9:1:1).
After formation of the monolayer (red line, Fig. 4b), signals originating from the metal elements decreased by a factor of ∼1.5, indicating successful coverage of the surface by the CDPA monolayer. Using these XPS data, the monolayer thickness can be derived, together with the tilt angle of the CDPA molecules on the surface. The thickness (t) of the CDPA layer was calculated using  ,58,59 with λ being the attenuation length estimated for Fe 2p (1.4 nm), θ = 80°, and Fe0 and FeCDPA the signal intensities (counts per s) for Fe 2p before and after grafting of the CDPA monolayer, respectively. A CDPA monolayer thickness of 1.2 ± 0.1 nm was found. Considering a molecular length of 1.30 nm for CDPA as determined by Chem3D (PerkinElmer Informatics, Inc.), this monolayer thickness corresponds to a tilt angle of 20 ± 10°, which supports the IRRAS data that suggested the formation of a densely packed and ordered monolayer.
,58,59 with λ being the attenuation length estimated for Fe 2p (1.4 nm), θ = 80°, and Fe0 and FeCDPA the signal intensities (counts per s) for Fe 2p before and after grafting of the CDPA monolayer, respectively. A CDPA monolayer thickness of 1.2 ± 0.1 nm was found. Considering a molecular length of 1.30 nm for CDPA as determined by Chem3D (PerkinElmer Informatics, Inc.), this monolayer thickness corresponds to a tilt angle of 20 ± 10°, which supports the IRRAS data that suggested the formation of a densely packed and ordered monolayer.
Finally, after antibody conjugation (Fig. 4b, blue line), an N 1s signal appeared at 400 eV. The metal signal was further attenuated, which can be accounted for by the formation of a thicker layer on the substrates due to the size of the antibody molecules, which is in the same order of magnitude as the probing depth of the technique, i.e., ∼10 nm.
Capture of LNCaP-derived EVs on antibody-conjugated stainless steel substrates and uncorrelated analysis
The antibody-conjugated surfaces were incubated with an EpCAM-positive tdEV sample prepared from LNCaP culture medium, and subsequently imaged with SEM to demonstrate their ability to immuno-capture tdEVs. As shown in Fig. 5a, tdEVs were successfully and specifically captured on the antibody-conjugated stainless steel surfaces, onto which quasi-spherical objects in the 100 nm–1 μm size range were identified. In contrast, in negative controls, for which one step of surface functionalization was omitted or which were not exposed to tdEV sample (see Table ESI 1, and Fig. ESI 6†), nothing was captured in the surface (Fig. 5a). Collectively, this experiment demonstrates the ability of our antibody-functionalized stainless steel substrates to successfully capture tdEVs.
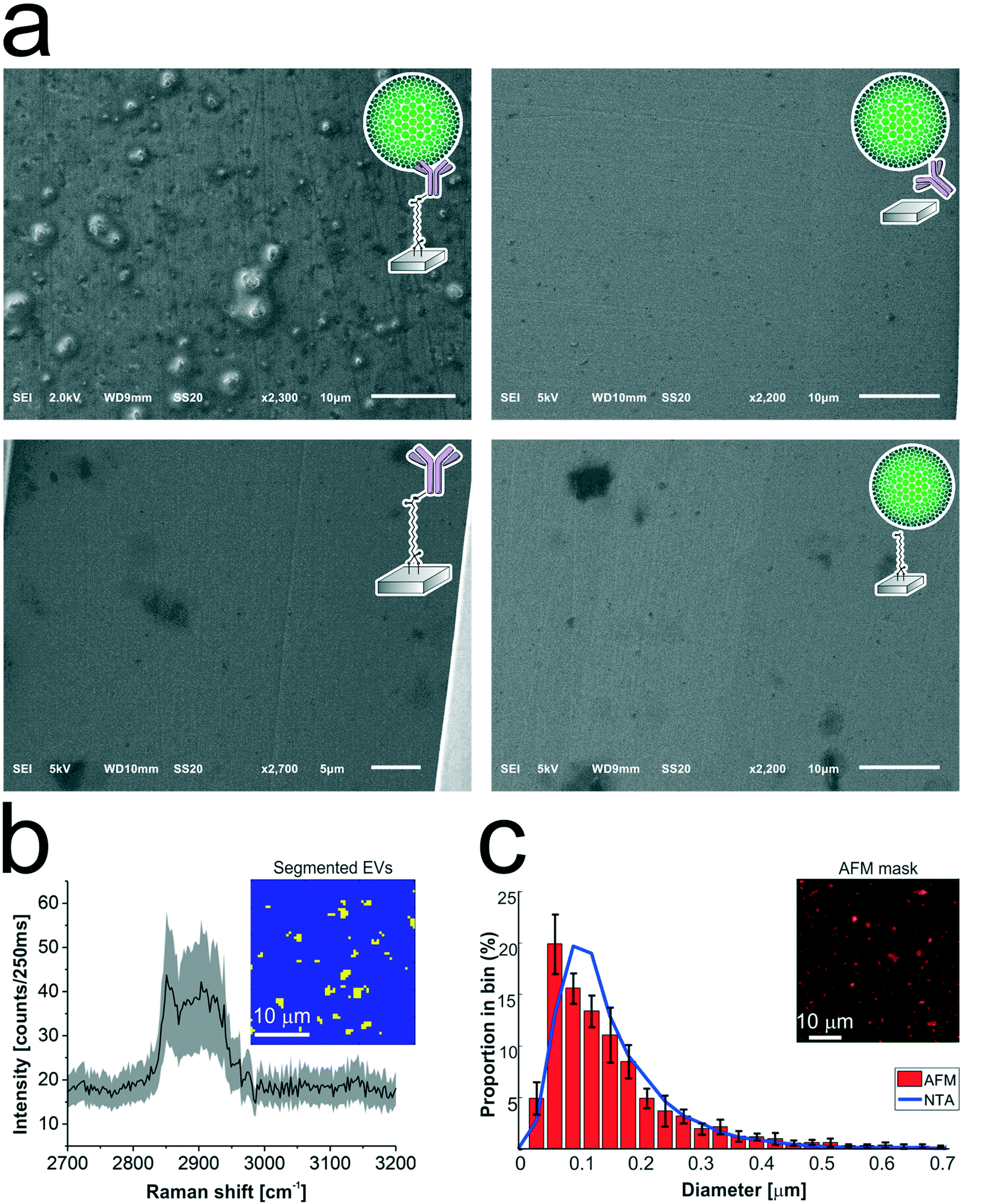 | ||
| Fig. 5 Analysis of the tdEVs captured on anti-EpCAM functionalized stainless steel substrates. a) SEM imaging revealing the specific capture of tdEVs (obtained from LNCaP cells) on anti-EpCAM functionalized stainless steel substrates (top left), while no object was captured on negative control samples (top right; without carbodiimide/NHS activation; bottom left; without functionalization with anti-EpCAM antibodies; and bottom right, without exposure to tdEV samples). b) Mean Raman spectrum (black line) and standard deviation (shaded area) of all EVs segmented from the ROI presented in the inset (30 μm × 30 μm and 64 × 64 pixels). The Raman spectrum (Raman shift range 2700–3200 cm−1) shows a lipid–protein band (2850–2950 cm−1) with a characteristic peak at 2851 cm−1, which corresponds to the CH2 symmetric stretch of lipids. In the inset, yellow pixels correspond to EVs and blue to the background. c) Size distribution of the surface-immobilized LNCaP-derived EVs determined by AFM (red histogram), and of the same sample in suspension before its immobilization on the surface as determined by NTA (blue line). Histogram: bin width 30 nm, error bars corresponding to the standard deviation (n = 5). Inset: Mask used for counting EVs on the AFM image, showing all objects detected with a height greater than 25 nm. | ||
As a next step, the same substrates, after capture of the tdEVs, were analyzed using Raman spectroscopy and AFM imaging. Hyperspectral Raman images were acquired on 30 μm × 30 μm ROIs (64 × 64 pixels), and analyzed using PCA in the spectral region between 2700 to 3200 cm−1 that contains the most intense peaks. EVs were identified as regions with pixels of high intensity values in certain scores. Such pixels were next segmented and used not only as a mask to identify the locations corresponding to EVs in all the images, as depicted in the inset of Fig. 5b, but also to compute a mean Raman spectrum for EVs, as presented in Fig. 5b. This spectrum comprises a characteristic lipid–protein band at 2850–2950 cm−1, and a clear peak at 2851 cm−1 that corresponds to the CH2 symmetric stretch of lipids.60
Finally, AFM images were used for quantitative analysis of the captured EVs. In five considered areas of 50 μm × 50 μm, a total of 5.4 × 103 tdEVs were detected, which corresponds to a surface density of 4.3 × 105 tdEVs mm−2. These objects presented a size range of 54 to 3840 nm and an average diameter of 101 ± 111 nm, and Fig. 5c shows the particle distribution up to 0.7 μm, since most of the particles were found in the 0–0.7 μm range. Noteworthy, the particle size distribution and average size as determined by AFM were overall in good agreement with data obtained using nanoparticle tracking analysis (NTA), with yet a slight shift in the size distribution i.e., 167 ± 91 nm (see ESI 4† and Fig. 5c, blue line). The difference observed can be accounted for by the lower detection limit of the latter technique, or by shrinking of the EVs due to dehydration before multi-modal analysis.51,61
Multi-modal analysis using SEM, Raman and AFM imaging
Finally, tdEVs captured on functionalized stainless steel substrates were analyzed successively using SEM, Raman and AFM imaging for their multi-modal characterization, and this last series of analysis was performed after fabrication of navigation markers on the stainless steel substrates. To identify interesting ROIs, the samples were first analyzed with SEM at a low resolution. It should be noted that for this first step of SEM imaging, the ROIs were not extensively exposed to the electron beam to avoid electron beam-induced deposition of amorphous carbon,62 which would hinder later analysis by Raman spectroscopy. Next, the distance between the measured location and the nearest set of markers was noted so as to find back the same region in the different imaging techniques. Following this, the same ROIs on the surface were successively imaged using hyperspectral Raman spectroscopy and SEM to characterize the EV size and morphology. Similarly, and with the help of the navigation markers, the ROIs were traced back and imaged with AFM. A key element in this multi-modal analytical process is the presence of navigation markers: due to their varying pitch, size and shape, each location in the sample region, as defined by the microchannel in the last PDMS device, can be matched to a unique combination of markers to assign spatial reference points to a ROI. This reference enables to retrieve objects of interest after transferring the stainless steel substrates between different instruments.
Fig. 6 presents the images of this multi-modal analysis, for the individual techniques as well as overlaid images. Noteworthy, a very good correlation exists between the images acquired with the individual techniques, with similar patterns observed in all 3 techniques (Fig. 6c, f, and i). Surprisingly, SEM imaging (Fig. 6a) revealed the presence of two types of particles, which could easily be distinguished based on their morphology and size: on one hand, small and elongated objects with an aspect ratio of approximately 1:7, and, on the other hand, compact, solidified crystalline particles with irregular shapes and well-defined edges, and with a height comparable to their lateral size. The larger particles were identified as cyanoacrylate glue particles, while the smaller particles were captured tdEVs, which was confirmed by Raman imaging (vide infra). Glue particles are presumably created upon release of the last PDMS device used to fabricate the navigation markers. In SEM, tdEVs present a much lower contrast than glue particles due to differences in molecular density.
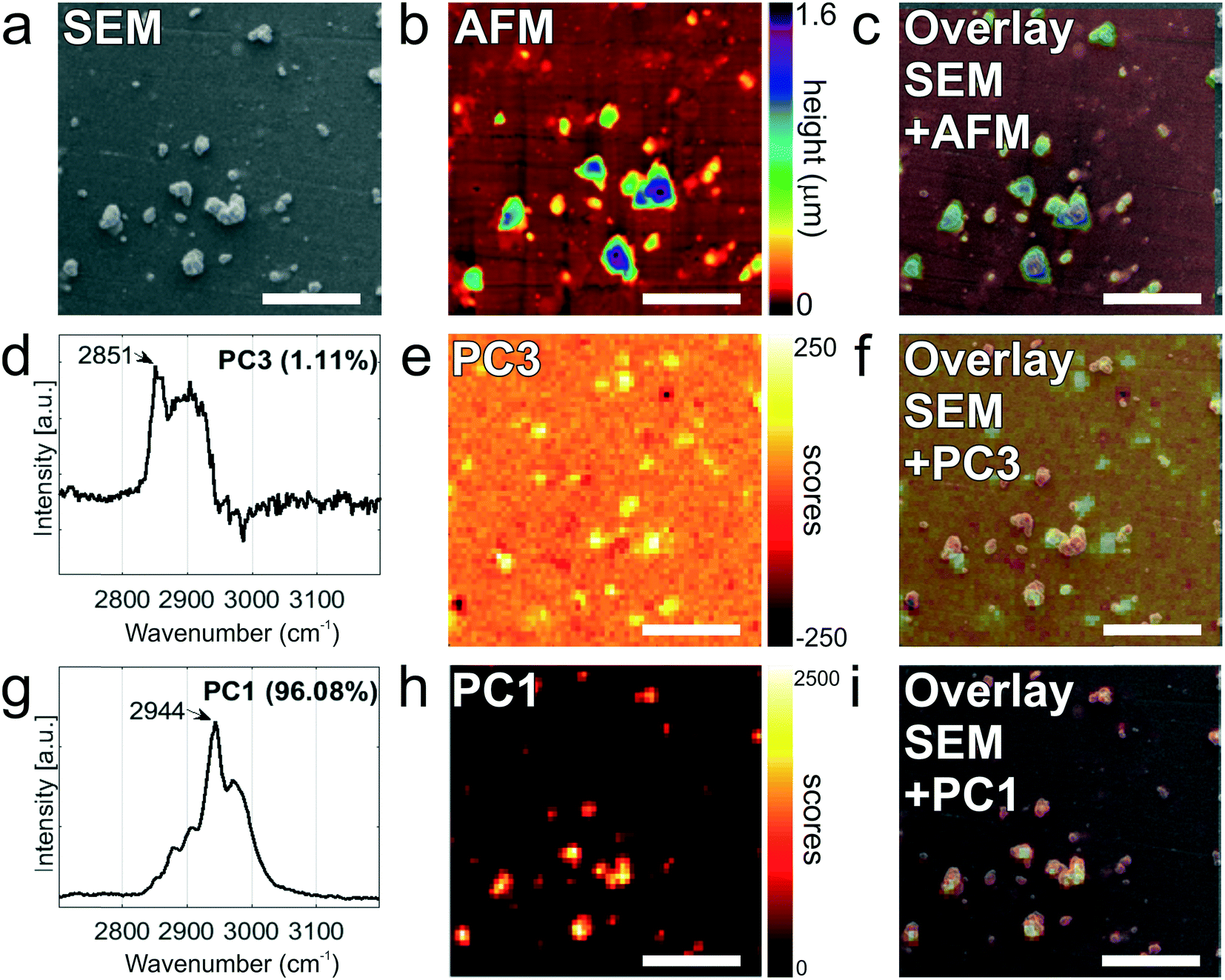 | ||
| Fig. 6 Multi-modal analysis of LnCAP-derived EVs on anti-EpCAM functionalized stainless steel substrates. a) SEM image of a selected ROI (30 μm × 30 μm). b) Corresponding AFM height image of the same ROI. c) AFM-SEM overlaid image. d) Raman spectrum in the 2700–3200 cm−1 region of the PC3 displaying a characteristic lipid–protein band (2850–2950 cm−1) specific to EVs, with a characteristic peak at 2851 cm−1, which corresponds to the CH2 symmetric stretch of lipids. e) Raman image of scores on PC3 (1.11%) showing the position of EVs in the Raman image. f) Overlay image showing excellent correspondence between the PC3 image of scores (h) with the SEM image (a). g) Raman spectrum in the 2700–3200 cm−1 region corresponding to the PC1 displaying a v(CH) stretching region of the cyanoacrylate glue with a characteristic peak at 2944 cm−1. h) Raman image of scores on PC1 (96.08%) showing the position of the glue particles in the Raman image. i) Overlay image showing excellent correspondence between the PC1 image of scores on PC1 (e) with the SEM image presented in (a). | ||
Multivariate analysis of the Raman data by PCA performed in the high frequency region (2700 and 3200 cm−1) as before confirms the presence of distinct populations of objects on the stainless steel substrates, whose Raman profile was distinct enough, as observed from the loading vectors PC3 (for tdEVs) and PC1 (for glue particles), respectively, in Fig. 6d and g. Measurements were conducted here on single EVs captured on the surface, and the signal-to-noise ratio was better in the high frequency region, which was therefore solely considered for data analysis. Yet, it allowed distinguishing tdEVs from other particles. As before, a clear Raman peak was found in the PC3 (Fig. 6d) loading at 2851 cm−1, which corresponds to the CH2 symmetric stretch of lipids, and a lipid–protein band between 2850 and 2950 cm−1, which is characteristic of EVs. These bands were absent in the PC1 loading, and are indeed not expected for cyanoacrylate (glue) particles. In contrast, the PC1 loading (Fig. 6g) presented a CH stretching region with a prominent peak at 2944 cm−1 and a CN stretching region with a peak at around 2247 cm−1, which are both characteristic of cyanoacrylate glue.63 It is worth noticing that the optical contrast of the images of scores is superior to the electron contrast in the SEM images, which clearly highlights the added value of correlative SEM-Raman imaging. The Raman images of the PC3 and PC1 scores in Fig. 6e and h reveal the respective distribution of the EVs and glue particles.
As a last analytical modality, AFM was employed to characterize the objects captured on the surface. Although AFM is typically slower than SEM, its resolution is higher, which allows detecting both more and smaller particles compared to the two other techniques. The resolution of particles below 0.5 μm in SEM is complicated by the low contrast in absence of gold coating of the sample, whereas if became apparent only from AFM analysis that the majority of particles is in fact smaller than 120 nm (see Fig. 5c). Moreover, AFM provides quasi-3D morphological information, which is of great interest to characterize the height of the captured EVs. Given also the low contrast in SEM due to the low acceleration voltages, the AFM data is altogether more suitable for studying the size distribution of captured EVs (Fig. 5c). Fig. 6b presents an AFM image corresponding to the previously discussed Raman and SEM images, and Fig. 6c an overlay image of the AFM and SEM data, showing good agreement between the data acquired by both techniques. In future studies, AFM could also be considered to examine the mechanical properties of the captured EVs (e.g., by nanoindentation64).
Altogether, data acquired by AFM are fully in line with both Raman and SEM data, and they all demonstrate the specific capture of tdEVs by the covalently bound antibodies on the stainless steel substrates.
Conclusion
We reported here a platform for the selective capture of tumor derived EVs (tdEVs) followed by their multi-modal analysis using SEM, Raman and AFM imaging to correlate size, morphological and chemical information at the individual EV level. Stainless steel, selected here for its suitability for all three imaging techniques, was first chemically modified with a CDPA monolayer onto which anti-EpCAM antibodies targeting tumor-derived EVs were immobilized. IRRAS and XPS characterization of the CDPA-functionalized surfaces revealed a densely packed and well-ordered monolayer, and XPS confirmed proper immobilization of the antibodies. Furthermore, EVs isolated from LNCaP prostate cancer cell lines were successfully captured on the antibody-conjugated stainless steel substrates, and successively analyzed using Raman spectroscopy, SEM and AFM. The integration of navigation markers on the stainless steel substrates after EV capture was instrumental here to track back individual EVs between the different analytical techniques. However, their fabrication using cyanoacrylate injected in patterned PDMS channels resulted in the creation of glue particles, which were detected together with the EVs. In future work, therefore, such navigation markers should be machined in the substrate and not onto the substrate to alleviate these contamination issues. Nonetheless, good agreement was found between the three techniques considered here, with excellent overlay of the images acquired by the individual modalities.As a proof of concept, in this paper, tdEVs isolated from cancerous cell lines were captured and analyzed. As a next step, the same platform will be challenged with more complex samples, such as blood samples, after implementation of anti-fouling moieties, e.g., based on polyethylene glycol. Furthermore, the proposed multi-modal approach can easily be expanded in the future to other optical (e.g., confocal fluorescence microscopy or infrared spectroscopy), electron (e.g., energy-dispersive X-ray spectroscopy) and probe (e.g., force spectroscopy) microscopy techniques as well as other analysis techniques, e.g., surface plasmon resonance (SPR) and mass spectroscopy (MS).
Conflicts of interest
C. Otto declares a potential conflict of interest as a managing director of the company Hybriscan Technologies B.V. (http://www.hybriscan.com).Acknowledgements
The authors would like to thank Naoual Ouazzani Chahdi for her experimental contribution. This work is part of the Perspectief Program Cancer ID with project numbers 14193 and 14196, which is funded by the Netherlands Organization for Scientific Research (NWO) and co-financed by JEOL Europe B.V., Hybriscan Technologies B.V., Aquamarijn Micro Filtration B.V. and Lionix International B.V.References
- M. Poudineh, E. H. Sargent, K. Pantel and S. O. Kelley, Nat. Biomed. Eng., 2018, 2, 72–84 CrossRef CAS PubMed.
- R. Vaidyanathan, R. H. Soon, P. Zhang, K. Jiang and C. T. Lim, Lab Chip, 2018, 11–34 Search PubMed.
- C. Alix-Panabières and K. Pantel, Clin. Chem., 2013, 59, 110–118 CrossRef PubMed.
- F. A. W. Coumans, S. T. Ligthart, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2012, 18, 5711–5718 CrossRef PubMed.
- F. Coumans, G. Van Dalum and L. W. M. M. Terstappen, Cytometry, Part A, 2018, 1197–1201 CrossRef PubMed.
- A. Nanou, F. A. W. Coumans, G. van Dalum, L. L. Zeune, D. Dolling, W. Onstenk, M. Crespo, M. S. Fontes, P. Rescigno, G. Fowler, P. Flohr, C. Brune, S. Sleijfer, J. S. de Bono and L. W. M. M. Terstappen, Oncotarget, 2018, 9, 19283–19293 CrossRef PubMed.
- M. Yáñez-Mó, P. R. M. Siljander, Z. Andreu, A. B. Zavec, F. E. Borràs, E. I. Buzas, K. Buzas, E. Casal, F. Cappello, J. Carvalho, E. Colás, A. Cordeiro-Da Silva, S. Fais, J. M. Falcon-Perez, I. M. Ghobrial, B. Giebel, M. Gimona, M. Graner, I. Gursel, M. Gursel, N. H. H. Heegaard, A. Hendrix, P. Kierulf, K. Kokubun, M. Kosanovic, V. Kralj-Iglic, E. M. Krämer-Albers, S. Laitinen, C. Lässer, T. Lener, E. Ligeti, A. Line, G. Lipps, A. Llorente, J. Lötvall, M. Manček-Keber, A. Marcilla, M. Mittelbrunn, I. Nazarenko, E. N. M. Nolte-’t Hoen, T. A. Nyman, L. O'Driscoll, M. Olivan, C. Oliveira, É. Pállinger, H. A. Del Portillo, J. Reventós, M. Rigau, E. Rohde, M. Sammar, F. Sánchez-Madrid, N. Santarém, K. Schallmoser, M. S. Ostenfeld, W. Stoorvogel, R. Stukelj, S. G. Van Der Grein, M. H. Vasconcelos, M. H. M. Wauben and O. De Wever, J. Extracell. Vesicles, 2015, 4, 1–60 Search PubMed.
- V. Sunkara, H.-K. Woo and Y.-K. Cho, Analyst, 2016, 141, 371–381 RSC.
- A. Liga, A. D. B. Vliegenthart, W. Oosthuyzen, J. W. Dear and M. Kersaudy-Kerhoas, Lab Chip, 2015, 15, 2388–2394 RSC.
- B. György, T. G. Szabó, M. Pásztói, Z. Pál, P. Misják, B. Aradi, V. László, É. Pállinger, E. Pap, Á. Kittel, G. Nagy, A. Falus and E. I. Buzás, Cell. Mol. Life Sci., 2011, 68, 2667–2688 CrossRef.
- R. Xu, D. W. Greening, H. Zhu, N. Takahashi and R. J. Simpson, J. Clin. Invest., 2016, 126, 1152–1162 CrossRef PubMed.
- E. Reátegui, K. E. Van Der Vos, C. P. Lai, M. Zeinali, N. A. Atai, B. Aldikacti, F. P. Floyd, A. Khankhel, V. Thapar, F. H. Hochberg, L. V. Sequist, B. V. Nahed, B. Carter, M. Toner, L. Balaj, D. Ting, X. O. Breakefield and S. L. Stott, Nat. Commun., 2018, 9, 2018 CrossRef PubMed.
- Q. Zhu, M. Heon, Z. Zhao and M. He, Lab Chip, 2018, 18, 1690–1703 RSC.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS.
- H. Im, H. Shao, Y. Il Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- K. W. Witwer, E. I. Buzás, L. T. Bemis, A. Bora, C. Lässer, J. Lötvall, E. N. Nolte-’t Hoen, M. G. Piper, S. Sivaraman, J. Skog, C. Théry, M. H. Wauben and F. Hochberg, J. Extracell. Vesicles, 2013, 2, 2013 Search PubMed.
- Y. Yuana, A. N. Böing, A. E. Grootemaat, E. van der Pol, C. M. Hau, P. Cizmar, E. Buhr, A. Sturk and R. Nieuwland, J. Extracell. Vesicles, 2015, 4, 2015 Search PubMed.
- B. J. Tauro, D. W. Greening, R. A. Mathias, H. Ji, S. Mathivanan, A. M. Scott and R. J. Simpson, Methods, 2012, 56, 293–304 CrossRef CAS PubMed.
- Y. Yoshioka, N. Kosaka, Y. Konishi, H. Ohta, H. Okamoto, H. Sonoda, R. Nonaka, H. Yamamoto, H. Ishii, M. Mori, K. Furuta, T. Nakajima, H. Hayashi, H. Sugisaki, H. Higashimoto, T. Kato, F. Takeshita and T. Ochiya, Nat. Commun., 2014, 5, 3591 CrossRef PubMed.
- E. Willms, C. Cabañas, I. Mäger, M. J. A. Wood and P. Vader, Front. Immunol., 2018, 9, 2018 CrossRef PubMed.
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Lab Chip, 2014, 14, 1891–1900 RSC.
- N. Koliha, Y. Wiencek, U. Heider, C. Jüngst, N. Kladt, S. Krauthäuser, I. C. D. Johnston, A. Bosio, A. Schauss and S. Wild, J. Extracell. Vesicles, 2016, 5, 2016 Search PubMed.
- S. Yadav, K. Boriachek, M. N. Islam, R. Lobb, A. Möller, M. M. Hill, M. S. Al Hossain, N. T. Nguyen and M. J. A. Shiddiky, ChemElectroChem, 2017, 4, 967–971 CrossRef CAS.
- S. C. Guo, S. C. Tao and H. Dawn, J. Extracell. Vesicles, 2018, 7, 1–16 Search PubMed.
- F. A. W. Coumans, A. R. Brisson, E. I. Buzas, F. Dignat-George, E. E. E. Drees, S. El-Andaloussi, C. Emanueli, A. Gasecka, A. Hendrix, A. F. Hill, R. Lacroix, Y. Lee, T. G. Van Leeuwen, N. Mackman, I. Mäger, J. P. Nolan, E. Van Der Pol, D. M. Pegtel, S. Sahoo, P. R. M. Siljander, G. Sturk, O. De Wever and R. Nieuwland, Circ. Res., 2017, 120, 1632–1648 CrossRef CAS PubMed.
- H. Im, K. Lee, R. Weissleder, H. Lee and C. M. Castro, Lab Chip, 2017, 17, 2892–2898 RSC.
- J. C. Contreras-Naranjo, H. J. Wu and V. M. Ugaz, Lab Chip, 2017, 17, 3558–3577 RSC.
- C. L. Hisey, K. D. P. Dorayappan, D. E. Cohn, K. Selvendiran and D. J. Hansford, Lab Chip, 2018, 18, 3144–3153 RSC.
- T. G. Kormelink, G. J. A. Arkesteijn, F. A. Nauwelaers, G. van den Engh, E. N. M. Nolte-’t Hoen and M. H. M. Wauben, Cytometry, Part A, 2016, 89, 135–147 CrossRef CAS PubMed.
- Y. Yuana, T. H. Oosterkamp, S. Bahatyrova, B. Ashcroft, P. Garcia Rodriguez, R. M. Bertina and S. Osanto, J. Thromb. Haemostasis, 2010, 8, 315–323 CrossRef CAS PubMed.
- E. van der Pol, A. Sturk, T. van Leeuwen, R. Nieuwland, F. Coumans, F. Mobarrez, G. Arkesteijn, M. Wauben, P. R. M. Siljander, V. Sánchez-López, R. Otero-Candelera, L. A. Ramón, S. Dolz, V. Vila, N. Mackman, J. Geddings, F. Mullier, N. Bailly, J. Y. Han, H. C. Kwaan, I. M. Weiss, E. I. Buzás, E. Pállinger, P. Harrison, J. Kraan, B. D. Hedley, A. LazoLangner, A. Enjeti, P. J. Norris, C. Paris, S. Susen, A. Bonnefoy, I. Delorme, W. L. Chandler, C. Hau, H. C. D. Aass, D. Connor, X. Wu, R. Dragovic, L. M. Uotila, R. Lacroix and S. Robert, J. Thromb. Haemostasis, 2018, 16, 1236–1245 CrossRef CAS PubMed.
- P. Zhang, J. Crow, D. J. Lella, X. Zhou, G. Samuel, A. K. Godwin and Y. Zeng, Lab Chip, 2018, 18, 3790–3801 RSC.
- P. Zhang, M. He and Y. Zeng, Lab Chip, 2016, 16, 3033–3042 RSC.
- N. Sebaihi, B. De Boeck, Y. Yuana, R. Nieuwland and J. Pétry, Meas. Sci. Technol., 2017, 28, 8pp CrossRef.
- W. Lee, A. Nanou, L. Rikkert, F. A. W. Coumans, C. Otto, L. W. M. M. Terstappen and H. L. Offerhaus, Anal. Chem., 2018, 90, 11290–11296 CrossRef CAS PubMed.
- V. Shpacovitch and R. Hergenröder, Anal. Chim. Acta, 2018, 1005, 1–15 CrossRef CAS PubMed.
- S. Obeid, A. Ceroi, G. Mourey, P. Saas, C. Elie-Caille and W. Boireau, Biosens. Bioelectron., 2017, 93, 250–259 CrossRef CAS PubMed.
- G. Pocsfalvi, C. Stanly, A. Vilasi, I. Fiume, G. Capasso, L. Turiák, E. I. Buzas and K. Vékey, Mass Spectrom. Rev., 2015, 35, 3–21 CrossRef PubMed.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed.
- I. Tatischeff, E. Larquet, J. M. Falcón-Pérez, P.-Y. Turpin and S. G. Kruglik, J. Extracell. Vesicles, 2012, 1, 19179 CrossRef PubMed.
- C. Krafft, K. Wilhelm, A. Eremin, S. Nestel, N. von Bubnoff, W. Schultze-Seemann, J. Popp and I. Nazarenko, Nanomedicine, 2017, 13, 835–841 CrossRef CAS PubMed.
- N. Eswaran, V. A. Sundaram, K. A. Rao and S. T. Balasundaram, 3 Biotech, 2018, 8, 1–6 CrossRef PubMed.
- K. A. Kondratov, T. A. Petrova, V. Yu Mikhailovskii, A. N. Ivanova, A. A. Kostareva and A. V. Fedorov, Cell tissue biol., 2017, 11, 181–190 CrossRef.
- F. J. Timmermans and C. Otto, Rev. Sci. Instrum., 2015, 86, 2015 CrossRef PubMed.
- A. Enciso-Martinez, F. J. Timmermans, A. Nanou, L. W. M. M. Terstappen and C. Otto, Analyst, 2018, 143, 4495–4502 RSC.
- S. Sharma, H. I. Rasool, V. Palanisamy, C. Mathisen, M. Schmidt, D. T. Wong and J. K. Gimzewski, ACS Nano, 2010, 4, 1921–1926 CrossRef CAS PubMed.
- M. Kosian, M. M. J. Smulders and H. Zuilhof, Langmuir, 2016, 32, 1047–1057 CrossRef CAS PubMed.
- A. Raman, M. Dubey, I. Gouzman and E. S. Gawalt, Langmuir, 2006, 22, 6469–6472 CrossRef CAS PubMed.
- G. Tizazu, A. M. Adawi, G. J. Leggett and D. G. Lidzey, Langmuir, 2009, 25, 10746–10753 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, 2nd edn, 2013 Search PubMed.
- E. van der Pol, F. A. W. Coumans, A. E. Grootemaat, C. Gardiner, I. L. Sargent, P. Harrison, A. Sturk, T. G. van Leeuwen and R. Nieuwland, J. Thromb. Haemostasis, 2014, 12, 1182–1192 CrossRef CAS PubMed.
- H. Abdi and L. J. Williams, Wiley Interdiscip. Rev. Comput. Stat., 2010, 2, 433–459 CrossRef.
- J. Ter Maat, R. Regeling, C. J. Ingham, C. A. G. M. Weijers, M. Giesbers, W. M. De Vos and H. Zuilhof, Langmuir, 2011, 27, 13606–13617 CrossRef CAS PubMed.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS.
- R. Maoz and J. Sagiv, J. Colloid Interface Sci., 1984, 100, 465–496 CrossRef CAS.
- A. Debrassi, E. Roeven, S. Thijssen, L. Scheres, W. M. De Vos, T. Wennekes and H. Zuilhof, Langmuir, 2015, 31, 5633–5644 CrossRef CAS PubMed.
- R. Arnold, W. Azzam, A. Terfort and C. Wöll, Langmuir, 2002, 18, 3980–3992 CrossRef CAS.
- C. D. Bain and G. M. Whitesides, J. Phys. Chem., 1989, 93, 1670–1673 CrossRef CAS.
- P. E. Laibinis, C. D. Bain and G. M. Whitesides, J. Phys. Chem., 1991, 95, 7017–7021 CrossRef CAS.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493–541 CrossRef CAS.
- E. Van Der Pol, A. G. Hoekstra, A. Sturk, C. Otto, T. G. Van Leeuwen and R. Nieuwland, J. Thromb. Haemostasis, 2010, 8, 2596–2607 CrossRef CAS PubMed.
- F. J. Timmermans, L. Chang, H. A. G. M. van Wolferen, A. T. M. Lenferink and C. Otto, Opt. Lett., 2017, 42, 1337–1340 CrossRef CAS PubMed.
- H. G. M. Edwards and J. S. Day, J. Raman Spectrosc., 2004, 35, 555–560 CrossRef CAS.
- B. W. H. Roos and G. J. L. Wuite, Adv. Mater., 2009, 1187–1192 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00081j |
| ‡ These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2019 |