
Control of aggregation-induced emission versus fluorescence aggregation-caused quenching by bond existence at a single site in boron pyridinoiminate complexes†
Madoka
Yamaguchi
,
Shunichiro
Ito
,
Amane
Hirose
,
Kazuo
Tanaka
 * and
Yoshiki
Chujo
*
* and
Yoshiki
Chujo
*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: kazuo123@chujo.synchem.kyoto-u.ac.jp; chujo@chujo.synchem.kyoto-u.ac.jp
First published on 3rd April 2017
Abstract
This manuscript reports the control of the luminescence properties of organoboron complexes between fluorescence aggregation-caused quenching (ACQ) and aggregation-induced emission (AIE) with or without a chemical bond at a single site in the pyridinoiminate skeleton. Novel boron complexes with (FBPI) and without (BPI) the fused structure in the ligand moiety were designed and synthesized. From the optical measurements, it was demonstrated that their solution- and solid-state emission behaviors oppositely varied with the presence of the fused structures. FBPI showed critical ACQ in a poor solvent. In contrast, BPI presented AIE behaviors. Additionally, from further evaluation of the solid-state emissive properties, it was shown that both boron complexes had crystallization-induced emission enhancement (CIEE) properties. Finally, it was demonstrated that reversible regulation of the emission intensity by external stimuli such as heating and solvent vapor fuming was accomplished with BPI based on the CIEE properties.
Introduction
Most of the luminescence properties in conventional organic dyes are spoiled in the condensed state due to aggregation-caused quenching (ACQ). To overcome ACQ and achieve bright solid-state emissive materials, the photochemistry in the class of aggregation-induced emission (AIE)-active molecules, which can present emission only in the aggregated state, has attracted attention.1–3 Since the first report on AIE-active molecules,4 many researchers have dedicated much effort to exploring new AIE-active dyes5–10 and to understanding the molecular mechanism of their AIE behaviors for the application of AIE-active molecules as optically-functional materials.11–15 Owing to their huge efforts, so far, various types of AIE-active molecules have been found, and in most cases, AIE properties were explained by the restriction of intramolecular rotation (RIR) mechanism, where suppression of intramolecular motions should be responsible for the enhancement of emission intensity with aggregate formation.1–3 Moreover, it was revealed that AIE-active molecules are versatile especially for preparing stimuli-responsive solid-state emissive materials. Based on sensitive responses in the optical properties of AIE-active materials toward environmental factors or external stimuli, various types of luminescent chromism such as thermo-, vapo- and mechanochromism have been demonstrated.16–18 Thus, the development of new AIE-active dyes and exploration of their properties should be of great significance not only to comprehend AIE behaviors at the molecular level but also to produce solid-state emissive materials with tunable functions.By employing “element-blocks” which are defined as a minimum functional unit composed of heteroatoms, highly-functional polymeric materials can be fabricated with unique properties originating from each element.19 From this standpoint in material design, boron complexes are promising “element-blocks” especially for obtaining luminescent materials.20,21 For example, boron dipyrromethene (BODIPY) is known to be one of the conventional boron complexes having superior optical properties such as large light-absorption ability, intense luminescence and sharp spectra.22 Indeed, it has been reported from recent studies that BODIPY-containing polymers are applicable as an advanced optical material in various applications such as near infrared-emissive dyes,23,24 cell-imaging,25 and light-harvesting antennae.26 In another instance, it has been shown that boron diketonates and their derivatives27 presented various interesting properties such as room-temperature phosphorescence,28 dual-emissive properties, applications as an oxygen sensor29–31 and reversible mechanochromic luminescence.32,33 By incorporating boron diketonate into polymers, bright emissive materials were readily obtained.34 Furthermore, a series of solid-state emissive materials with stimuli-responsiveness have been developed.35–39 We found that the resulting ketoiminate and diiminate complexes had AIE properties by replacing oxygen with nitrogen in ACQ-active boron diketonate.40,41 By using these complexes as an AIE-inducible “element-block”, a series of AIE-active materials such as solid-state emissive conjugated polymers,42,43 film-type chemical sensors,44 stimuli-responsive chromic hydrogels,45 and mechanochromic luminescent materials46–48 have been developed. Thus, further exploration of luminescent boron “element-blocks” and discovery of their superior functions are a topic with high relevance for developing highly-functional optical materials.
From theoretical investigation, it was implied that large structural relaxation might occur in the excited state of AIE-active boron complexes.49 During the structural relaxation process, the probability of excitation decay increased and very slight emission could be induced in the solution. Whereas, in the aggregation state, it is likely that conformational alteration in the excited state should be highly suppressed by structural restriction. In addition, disturbance of intermolecular interaction at the boron moiety by intramolecular distortion or steric hindrance could contribute to the suppression of ACQ. As a result, solid-state emission was obtained from the boron complexes with the nitrogen-substituted ligands.41,42 From this mechanistic speculation, it was supposed that AIE and ACQ behaviors can be switched at the same molecular skeleton by regulating the degree of structural flexibility around boron. To prove the validity of this idea, we designed and synthesized new boron complexes with or without the fused structure in the ligand moiety. In this report, their opposite behaviors of emission intensity changes from solution to aggregation are described. Moreover, regulation of the solid-state emissive properties of the synthesized boron complex is also mentioned. This is the first example, to the best of our knowledge, to demonstrate the control of AIE and ACQ properties with the presence of a chemical bond at a single site in a boron complex.
Results and discussion
According to the previous reports on the AIE properties of boron ketoiminates and diiminates, it was proposed that intramolecular motions involving the boron-coordinating nitrogen atom should play a critical role in their AIE behaviors.40,41,49 Therefore, we presumed that regulation of structural flexibility at the nitrogen could drastically influence the optical properties. Based on this idea, boron complexes BPI and FBPI were designed and synthesized (Scheme 1). The ligands for BPI and FBPI were prepared through imine formation and the coupling reaction with imidoyl chloride and lithiated methylpyridine, respectively. By treating these ligands with boron trifluoride-diethyl etherate, boron complexes were obtained. The products were characterized by 1H, 13C, and 11B NMR spectroscopies and mass measurements and elemental analyses (Fig. S1 and S2, ESI†). From these data, it was confirmed that the products had designed structures. Both products showed good solubility in common organic solvents such as chloroform, dichloromethane, tetrahydrofuran and acetonitrile, and decomposition was hardly observed in the air and during analytical procedures. Therefore, it was concluded that both boron complexes had high enough stability for evaluating the series of optical properties.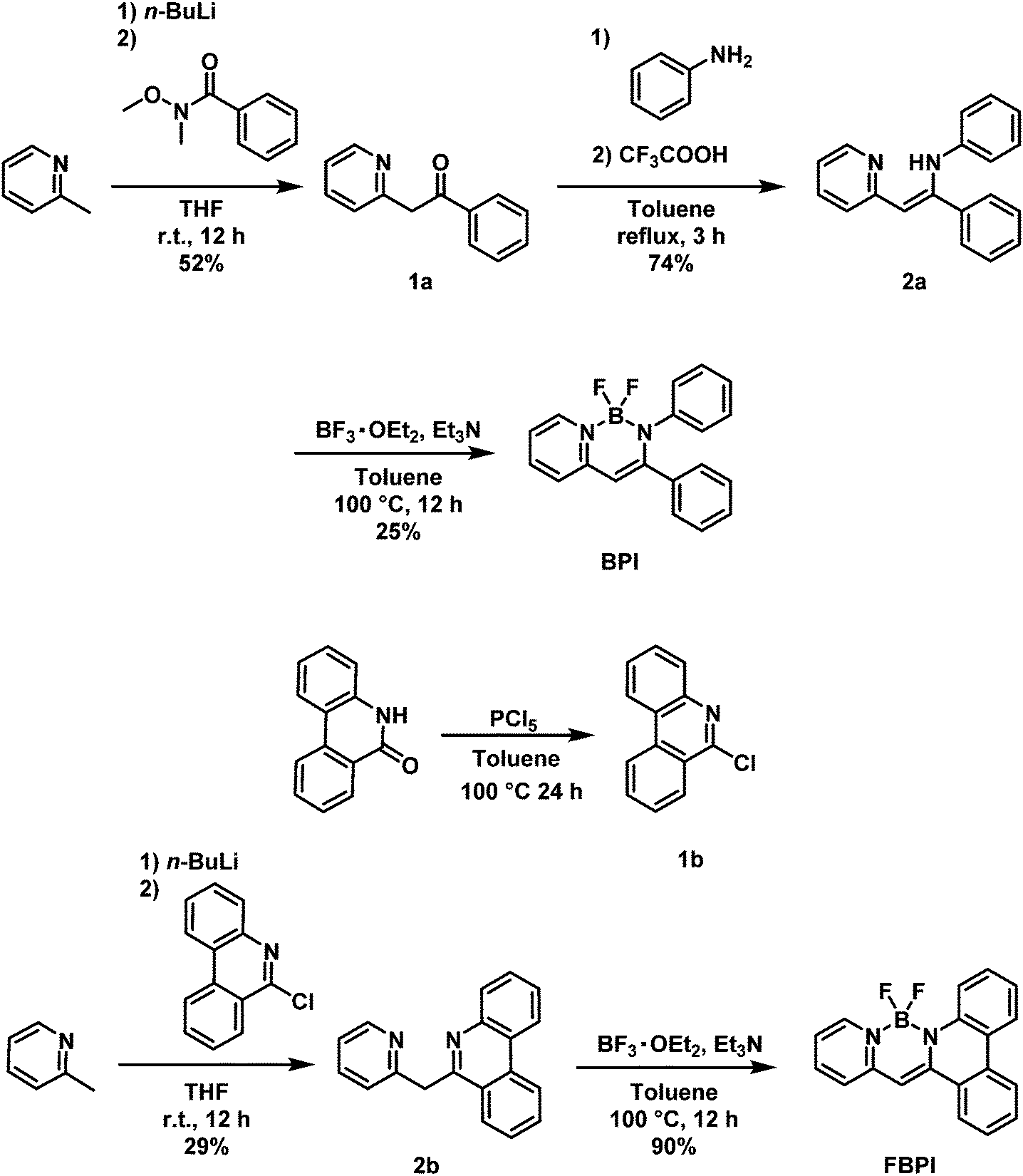 | ||
| Scheme 1 Synthesis of boron complexes. | ||
The optical properties of BPI and FBPI in the solution state were investigated (Table 1). In the UV-vis light absorption spectra in CHCl3 (1.0 × 10−5 M), large absorption bands attributable to π–π* transition were observed from both compounds (Fig. 1). FBPI showed a larger absorption band in the longer wavelength region than BPI. This result means that FBPI had smaller band gap energy than BPI. To determine the energy levels of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of both complexes, cyclic voltammetry (CV) was carried out with the solution samples (Fig. S3 and Table S1, ESI†). From the onset values of redox waves, energy levels were estimated. Accordingly, it was shown that FBPI possessed higher HOMO and lower LUMO levels than BPI. These data clearly indicate that FBPI had a narrower band gap than BPI. To theoretically support these optical and electrochemical properties of boron complexes, quantum chemical calculations using the density functional theory (DFT) method at the B3LYP/6-31+G(d,p) level were performed (Fig. S4, ESI†).50 Corresponding to the experimental data, FBPI presented a narrower band gap originating from higher HOMO and lower LUMO levels. It is likely that the π-conjugated system should be extended through the higher planar fused structure in FBPI. Then, a narrower band gap was obtained.
| λ abs [nm] | ε [M−1 cm−1] | λ PL [nm] | Stokes shiftd [cm−1] | Φ PL | τ [ns] | |
|---|---|---|---|---|---|---|
| a Molar absorption coefficients at the peak top wavelength. b Excited at λabs. c Excited at 453 nm. d Calculated by 1/λabs × 107 − 1/λPL × 107. e Determined as an absolute value. f Excited at 375 nm. g Not detectable due to too weak emission. | ||||||
| BPI | 406 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
464b | 3079 | 0.01 | n.d.g |
| FBPI | 483 | 53900 |
491c | 337 | 0.69 | 4.01 (100%) |
| 453 | 35800 |
522c | 4.00 (100%) | |||
 | ||
| Fig. 1 UV-vis absorption and PL spectra of (a) BPI and (b) FBPI in CHCl3 (1.0 × 10−5 M). Photographs of the solutions of BPI and FBPI in CHCl3 (1.0 × 10−5 M) under visible (c) and UV (d) irradiation. | ||
Next, to examine the electronic structures in the excited state, photoluminescence (PL) spectra were measured with the solution samples (1.0 × 10−5 M in CHCl3) containing boron complexes (Fig. 1 and Table 1). Because of the small Stokes shift of FBPI, we recorded an emission spectrum with the excitation light at the absorption maximum in the shorter wavelength region (453 nm). Interestingly, distinctly different luminescent behaviors were observed. The luminescence quantum yield of BPI was very small (Φ = 0.01), while FBPI showed efficient emission (Φ = 0.68). Furthermore, FBPI showed an emission band in the longer wavelength region with a much smaller Stokes shift than BPI. Additionally, clear structured emission was observed from FBPI. Because of the enhanced molecular rigidity of FBPI by introducing the fused ring structure compared to BPI, molecular motions should be suppressed in the excited state, resulting in the observation of vibrational peaks from FBPI. These results suggest that the electronic structures of the boron complexes should be significantly different in the excited state.
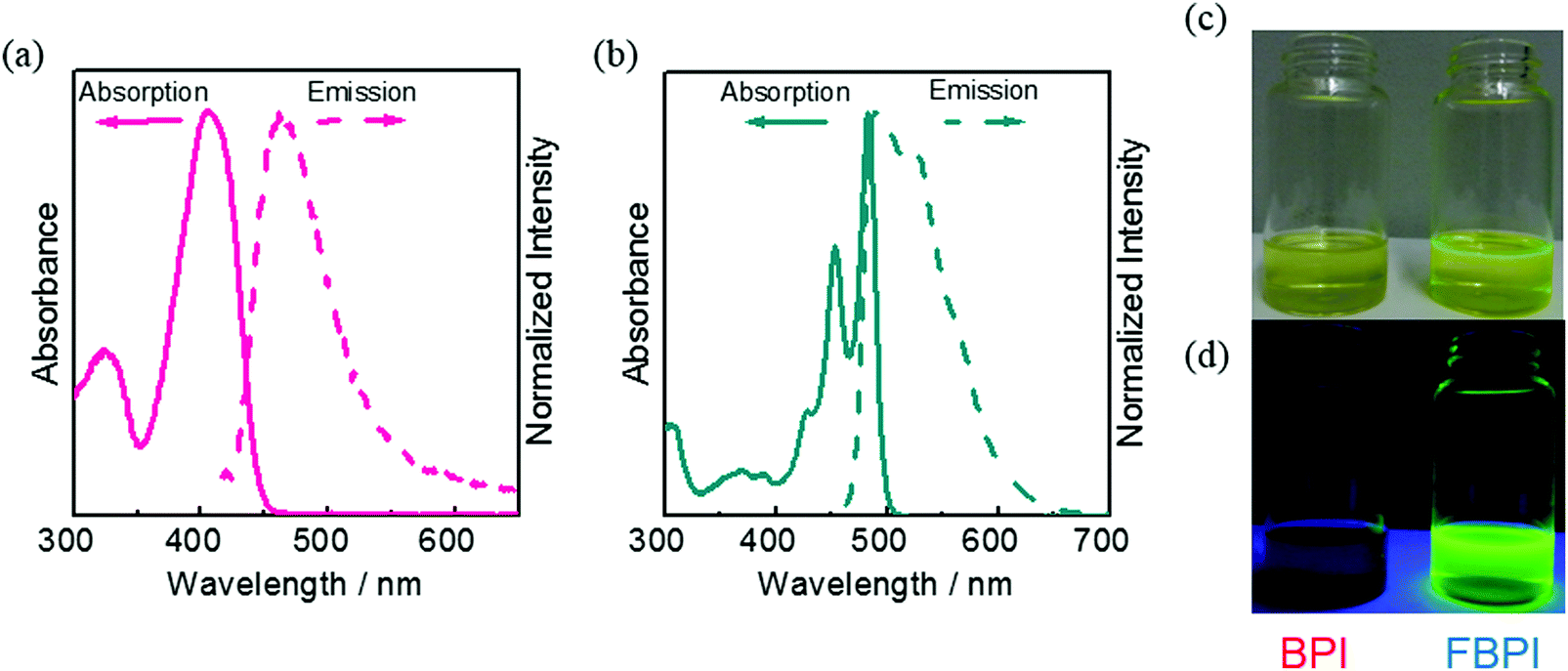
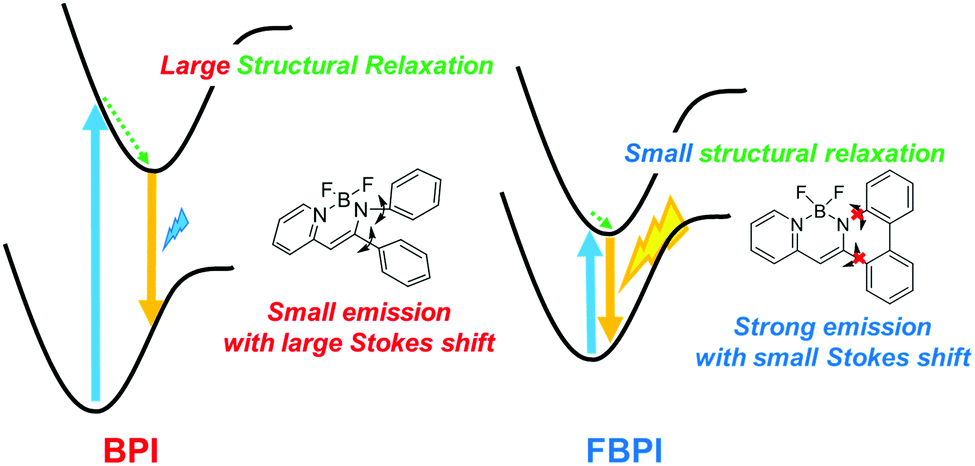
To understand the differences in emission properties between BPI and FBPI in the solution state, quantum chemical calculations were executed. Optimized structures of the boron complexes in the ground and excited states were estimated using DFT and time-dependent DFT (TD-DFT) methods at the B3LYP/6-31+G(d,p) level, respectively (Fig. 2). It was found that the molecular conformation of BPI in the S1 state was distinctly different from that in the S0 state. This result means that twisting at the phenyl ring and at the ligand site of BPI should proceed during structural relaxation in the excited state. In contrast, subtle differences were observed in the optimized structures of FBPI between the S1 and S0 states. It is reasonable that structural alteration should be highly restricted because of the rigid fused structure in FBPI. The significant difference in the Stokes shift in the PL spectra can be explained by these differences in the degree of structural relaxation in the excited state. Because of slight structural alteration in FBPI before and after relaxation in the excited state, the emission band is capable of appearing near the position of the absorption band. On the other hand, the emission band of BPI should be detected in the longer wavelength region because of relatively-larger structural relaxation in the excited state.
 | ||
| Fig. 2 Optimized structures of boron complexes in the (a) S0 and (b) S1 state calculated using the DFT and TD-DFT methods at the B3LYP/6-31G+(d,p) level, respectively. | ||
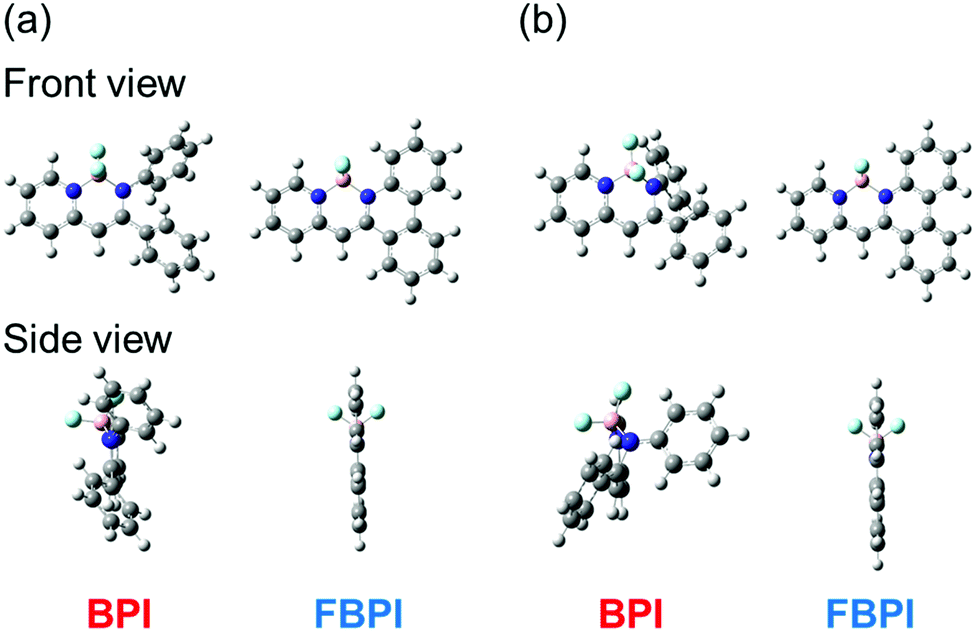
PL spectra were measured in 2-methyltetrahydrofuran (2-Me-THF) at 77 K (1.0 × 10−5 M, Fig. S5 and Table S2, ESI†) to investigate the influence of suppression of molecular motion and structural relaxation on optical properties. Under frozen conditions, both complexes showed larger emission bands in the shorter wavelength regions than those under room-temperature conditions. It should be emphasized that BPI presented bright emission, indicating that intramolecular motion should be responsible for excitation decay peculiarly in BPI. Additionally, boron complexes showed phosphorescence at low temperatures (Fig. S6 and Table S3, ESI†). In previous studies, it was reported that boron diketonates and diiminates showed phosphorescence.28,43 According to these molecules, it is implied that the existence of the lone-pair electrons on pyridine or imine groups could be responsible for phosphorescence. From these results, a plausible model of emission in the solution state is proposed (Fig. 3). In BPI, non-radiative decay frequently occurs during large structural relaxation in the excited state. Therefore, low emission intensity should be induced. On the other hand, owing to the rigid fused structure of FBPI, non-radiative decay should be effectively suppressed. Hence, efficient emission can be obtained with a small Stokes shift.
 | ||
| Fig. 3 Plausible emission mechanisms of BPI and FBPI in the solution state. | ||
The optical properties of boron complexes in the condensed state were investigated. Initially, the intensity ratios were monitored in acetonitrile by adding H2O as a poor solvent (Fig. 4 and Fig. S7, ESI†). To form aggregation in both samples, we performed experiments with a relatively higher concentration (1.0 × 10−4 M) than those for evaluating the optical properties. Similar emission tendencies to those from the solution samples were obtained upon addition of H2O up to 70 vol%. Then, by increasing the water content (H2O ≥ 80 vol%), white turbidity appeared in both samples. From this water content, the emission behaviors in both samples oppositely changed. From the sample containing BPI, significant emission enhancement was observed by increasing the water content in the sample. In contrast, the emission intensity of FBPI drastically decreased in the aggregated state. These data including the optical properties in the solution state clearly indicate that BPI is an AIE-active molecule, whereas FBPI shows ACQ similarly to common organic dyes.
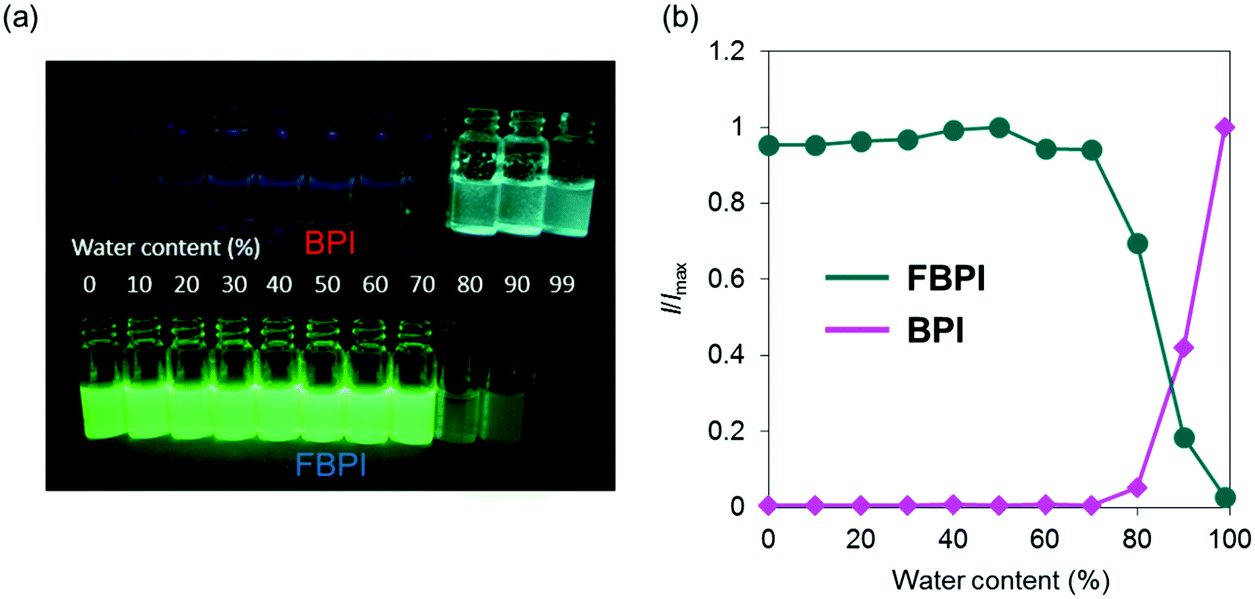 | ||
| Fig. 4 (a) Photographs of BPI and FBPI under UV irradiation with variable water contents in acetonitrile (1.0 × 10−4 M). (b) Changes in emission-intensity ratios of BPI and FBPI by increasing water contents in the samples. | ||
To gather further information on the opposite behaviors, AIE versus ACQ in the boron complexes, the emission properties were examined by changing intermolecular interaction in the solid state (Table 2). The optical properties were evaluated in the crystalline state (Fig. 5). From the PL spectra in the crystalline state, it was observed that FBPI showed an emission band in the longer wavelength region than BPI. This trend was also found in the solution state. On the other hand, different behaviors were obtained in luminescence quantum yield measurements. BPI showed more efficient emission (ΦPL = 0.58) in the crystalline state than in the solution state (ΦPL = 0.01). In contrast, FBPI provided less efficient emission (ΦPL = 0.07) in the crystalline state than in the solution state (ΦPL = 0.69). To investigate the emission behaviors in the crystalline state in detail, single-crystal structure analyses were carried out. The most impressive point is that BPI and FBPI formed dimer structures in the crystalline state. Face-to-face π–π (av. 3.35 Å) and CH–F interactions (2.52 Å) were observed in the BPI dimer (Fig. S8 and Table S4, ESI†). Meanwhile, only face-to-face π–π interaction (av. 3.33 Å) was detected in the FBPI dimer (Fig. S9 and Table S5, ESI†). Additionally, from the comparison of π-stacking areas between the dimers, less significant overlapping at the molecular surface was observed in the BPI dimer (Fig. 6). Only a side part in BPI seemed to be slightly overlapped. In contrast, overlapping at the surface of π-conjugated rings was obviously found in the FBPI dimer. The difference in emission quantum yields between BPI and FBPI in the crystalline state can be reasonably explained by these structural features in these dimers. In the crystal packing of FBPI, intermolecular interaction should occur via π-stacking, resulting in ACQ. Therefore, a critical decrease in the emission quantum yield was observed in the crystalline state. In contrast, because of the small overlap in the dimer, BPI can avoid ACQ. Furthermore, excitation decay induced by structural relaxation would be suppressed in the crystalline state. Thus, efficient emission can be obtained from the crystalline sample of BPI.
| λ PL (crystal) [nm] | λ PL (amorphous) [nm] | Φ PL (crystal) | Φ PL (amorphous) | τ (crystal) [ns] | τ (amorphous) [ns] | |||
|---|---|---|---|---|---|---|---|---|
| a Excited at λabs in CHCl3 (1 × 10−5 M). b Excited at 453 nm. c Determined as an absolute value. d Excited at 375 nm. e Not detectable due to too weak emission. | ||||||||
| BPI | 497a | 491a | 0.58 | 0.04 | 2.45 (28%) | 4.87 (72%) | 0.84 (37%) | 3.13 (63%) |
| FBPI | 560b | 616b | 0.07 | >0.01 | 2.57 (41%) | 5.89 (59%) | n.d.e | |
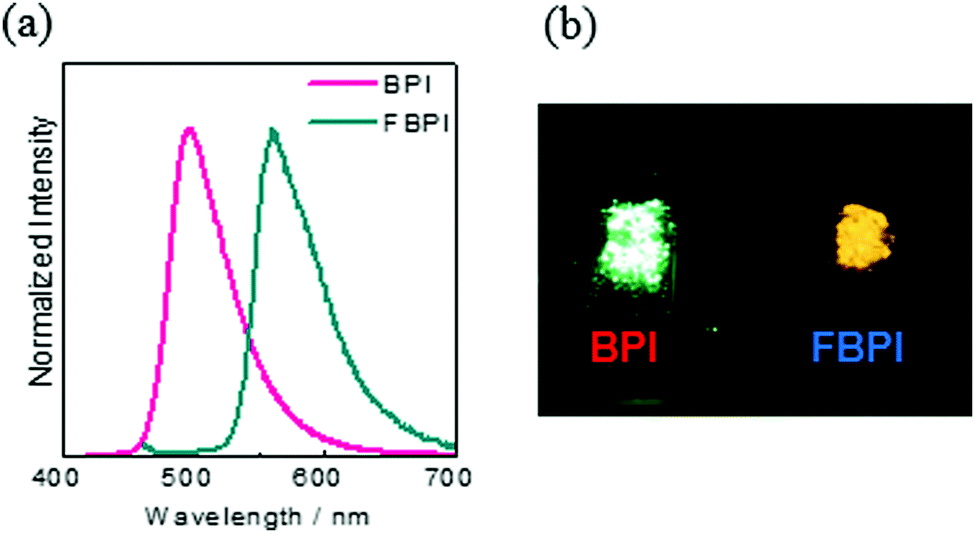 | ||
| Fig. 5 (a) PL spectra of BPI and FBPI in the crystalline state. (b) Photograph of BPI and FBPI in the crystalline state under UV irradiation. | ||
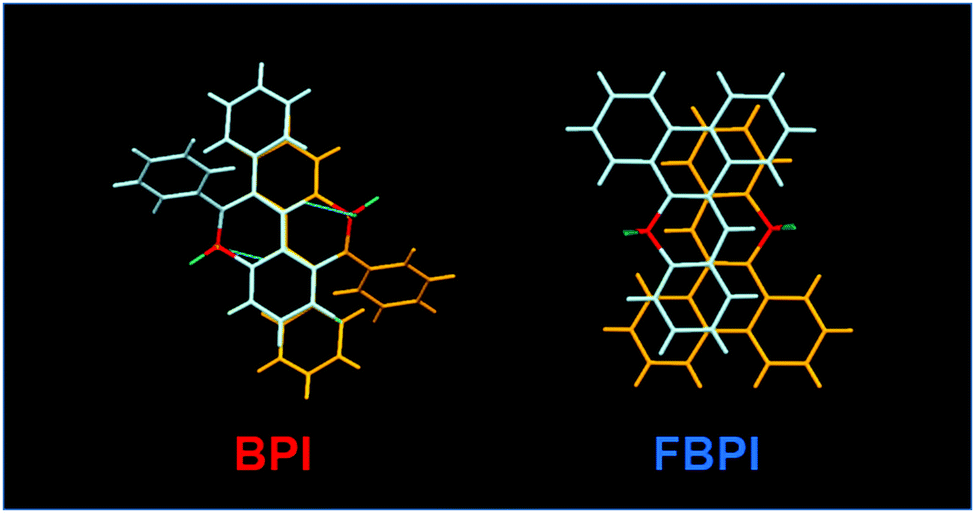 | ||
| Fig. 6 Capped-stick drawings of BPI and FBPI dimer structures. | ||
The thermal properties were surveyed with thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. S10, S11 and Table S6, ESI†). Accordingly, decomposition temperatures with 5 wt% weight loss were obtained at 256 °C and 312 °C from BPI and FBPI, respectively. The melting temperatures of the boron complexes were detected below each decomposition temperature (BPI: 141 °C, FBPI: 205 °C). Based on these thermal data, amorphous samples were prepared by rapidly cooling the melting sample in a refrigerator at −20 °C to investigate the optical properties in the amorphous state. To check the formation of the amorphous states, powder X-ray diffraction (XRD) was performed (Fig. S12, ESI†). Since no sharp diffraction patterns were detected from either sample after cooling, it was confirmed that homogeneous amorphous samples were produced.
The PL spectra of the boron complexes in the crystalline and amorphous samples were compared (Fig. S13, S14 and Table S2, ESI†). Both complexes exhibited stronger emission in the crystalline state than in the amorphous state. These results indicate that both complexes have crystallization-induced emission enhancement (CIEE) properties.41 Compared with the PL spectra in the crystalline state, the blue-shifted emission band was obtained from the amorphous sample of BPI, meanwhile the amorphous FBPI presented the red-shifted emission band. Taking account of the X-ray crystallography data that boron complexes formed the dimer structures in the crystal packing, it is implied that intermolecular interaction might be reduced in the amorphous BPI sample because of intrinsic steric structures. In contrast, the degree of π-stacking should be enhanced in the random distribution of planar FBPI molecules. Thus, peak shifts were observed in the PL spectra with the amorphous samples.
Based on the CIEE properties, reversible control of optical properties was performed by using external stimuli as a trigger (Fig. 7 and Fig. S15, Table S7, ESI†). The amorphous sample coated on quartz glass was placed in saturated CH2Cl2 vapor or heated on a hot plate at 100 °C. Bathochromic shifts and increases in emission intensity were observed from the amorphous BPI samples. From XRD monitoring, it was indicated that a phase transition from amorphous to crystalline was induced by both external stimuli (Fig. S16, ESI†). The crystallized sample recovered to the amorphous state upon melting, followed by rapid cooling. In particular, these cycles were repeatable many times with both external stimuli. From these results, it is proposed that BPI could have large potential for applications in chemosensors for heat and volatile organic molecules. Although the same procedure was applied to the FBPI sample, significant changes were hardly observed. It was assumed that the amorphous–crystal transition could proceed smoothly triggered by external stimuli because of weak intermolecular interaction in the amorphous samples of BPI. On the other hand, due to intrinsic strong π–π interaction and electronically-stabilized distributions in the amorphous state of FBPI according to the PL spectra, phase transition could be disturbed. Therefore, sensitivity in luminescent chromism toward external stimuli varied in the boron complexes.
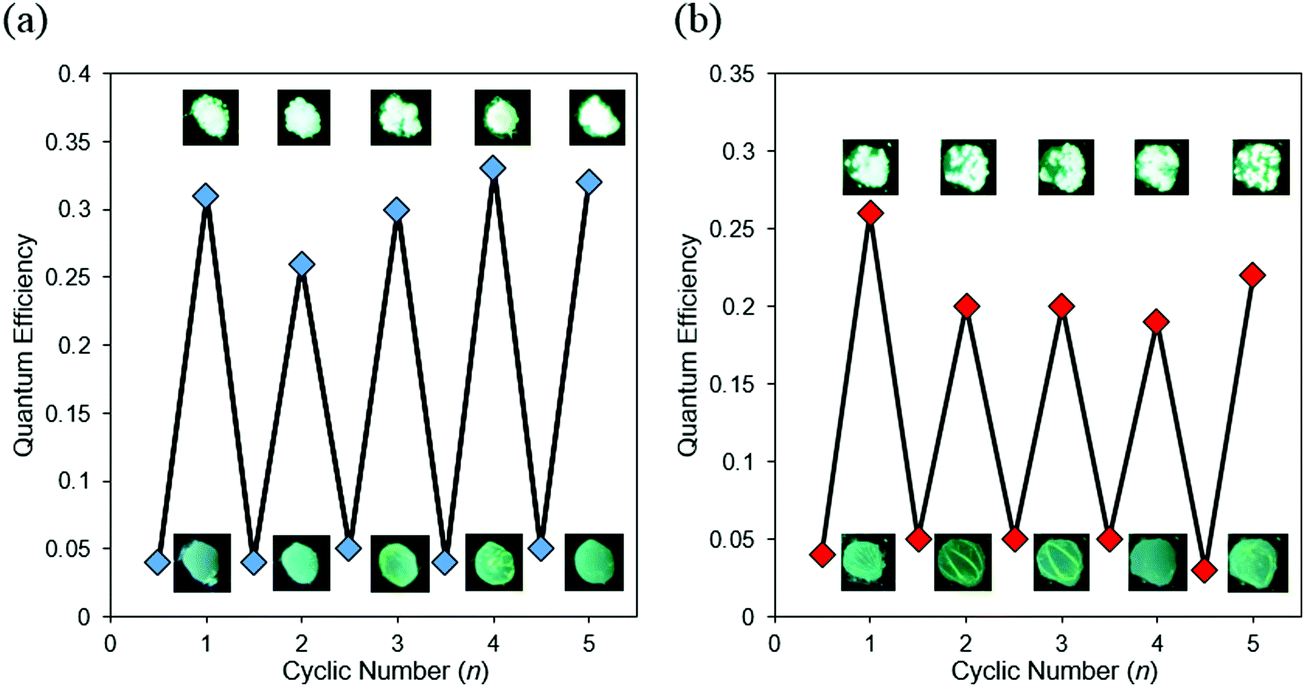 | ||
| Fig. 7 Repeated switching of BPI between amorphous and crystalline states by (a) fuming-melting and (b) heating-melting cycles. | ||
To evaluate the influence of intermolecular interaction in the solid state on emission quenching, the optical properties were monitored in a poly(methyl methacrylate) (PMMA) matrix (Fig. S17 and Table S8, ESI†). It was proposed that each boron complex would be isolated in the matrix, and intermolecular interaction should be restricted. Indeed, significant emission bands with similar emission efficiencies (BPI: ΦPL = 0.31; FBPI: ΦPL = 0.35) were detected from both film samples. These data clearly indicate that intermolecular interaction induces ACQ in the solid state of the boron complexes, especially FBPI.
Conclusion
By employing the pyridinoiminate skeleton, it was illustrated that an ACQ-presenting luminescent dye or an AIE-active molecule can be, respectively, prepared in the presence or absence of a chemical bond at a single site in the boron complex. From the series of mechanistic studies, it was suggested that molecular flexibility in the excited state and planarity of the complex were critically changed by the existence of the chemical bond. Therefore, opposite behaviors between luminescent quenching and enhancement could be observed in aggregate formation. This research includes two significant issues: Firstly, new optically-functional “element-blocks”, boron pyridinoiminates, were obtained in this research. Both boron complexes would be individually useful in each scene and application. FBPI and its derivatives are promising as scaffolds for fabricating functional materials with advanced luminescence properties. Because of its high planarity, which is advantageous for constructing robust π-conjugated systems, multi-functions and stimuli-responsiveness are potentially inducible by connecting with other functional units. The phosphorescence of FBPI might be applicable for developing chromism sensors. Furthermore, it can be expected that BPI is a versatile AIE-inducible “element-block”. Based on environment-sensitive optical properties in the solid state as demonstrated here, various stimuli-responsive materials can be proposed. Secondly, the photochemistry of BPI could be feasible for designing a new series of AIE-active materials based on theoretical presumption. Improvement of structural flexibility around the boron moiety could be a valid strategy for realizing AIE behaviors according to preprogrammed designs. Thus, it can be said that our findings described here could contribute not only to comprehending luminescence properties in boron-containing compounds but also to extending application fields of organic luminescent dyes.Acknowledgements
This work was partially supported by the Research Institute for Production Development (for K. T.) and a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks (No. 2401)” (JSPS KAKENHI Grant Number JP24102013).References
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- A. Fukazawa, Y. Ichihashi and S. Yamaguchi, New J. Chem., 2010, 34, 1537–1540 RSC.
- H. Imoto, K. Kizaki, S. Watase, K. Matsukawa and K. Naka, Chem. – Eur. J., 2015, 21, 12105–12111 CrossRef CAS PubMed.
- H. Imoto, K. Nohmi, K. Kizaki, S. Watase, K. Matsukawa, S. Yamamoto, M. Mitsuishi and K. Naka, RSC Adv., 2015, 5, 94344–94350 RSC.
- H. Eguchi, H. Nishiyama, S. Inagi and I. Tomita, Asian J. Org. Chem., 2016, 6, 165–168 CrossRef.
- N. Komiya, T. Muraoka, M. Iida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054–16061 CrossRef CAS PubMed.
- M. Shimada, M. Tsuchiya, R. Sakamoto, Y. Yamanoi, E. Nishibori, K. Sugimoto and H. Nishihara, Angew. Chem., Int. Ed., 2016, 55, 3022–3026 CrossRef CAS PubMed.
- J. Luo, K. Song, F. l. Gub and Q. Miao, Chem. Sci., 2011, 2, 2029–2034 RSC.
- J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
- A. Prlj, N. Došlić and C. Corminboeuf, Phys. Chem. Chem. Phys., 2016, 18, 11606–11609 RSC.
- F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 14492–14497 CrossRef CAS PubMed.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- L. Yan, Y. Zhang, B. Xu and W. Tian, Nanoscale, 2016, 8, 2471–2487 RSC.
- X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC.
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 633–643 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235–1255 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., 2008, 120, 1202–1219 CrossRef.
- X. Ma, X. Mao, S. Zhang, X. Huang, Y. Cheng and C. Zhu, Polym. Chem., 2013, 4, 520–527 RSC.
- R. Yoshii, A. Nagai and Y. Chujo, J. Polym. Sci., Part A-1: Polym. Chem., 2010, 48, 5348–5356 CrossRef CAS.
- T. Mosaiab, I. In and S. Y. Park, Macromol. Rapid Commun., 2013, 34, 1408–1415 CrossRef CAS PubMed.
- H. Yeo, K. Tanaka and Y. Chujo, Macromolecules, 2013, 46, 2599–2605 CrossRef CAS.
- K. Tanaka and Y. Chujo, NPG Asia Mater., 2015, 7, e223 CrossRef CAS.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed.
- J. S-Kosicka, C. A. DeRosa, W. A. Morris, Z. Fan and C. L. Fraser, Macromolecules, 2014, 47, 3736–3746 CrossRef PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. A. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- G. M. Palmer, A. N. Fontanella, G. Zhang, G. Hanna, C. L. Fraser and M. W. Dewhirst, J. Biomed. Opt., 2010, 15, 066021 Search PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- F. Ito and T. Sagawa, RSC Adv., 2013, 3, 19785–19788 RSC.
- K. Tanaka, K. Tamashima, A. Nagai, T. Okawa and Y. Chujo, Macromolecules, 2013, 46, 2969–2975 CrossRef CAS.
- D. Zhao, G. Li, D. Wu, X. Qin, P. Neuhaus, Y. Cheng, S. Yang, Z. Lu, X. Pu, C. Long and J. You, Angew. Chem., 2013, 125, 13921–13925 CrossRef.
- S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647–2662 RSC.
- A. Sakai, E. Ohta, Y. Yoshimoto, M. Tanaka, Y. Matsui, K. Mizuno and H. Ikeda, Chem. – Eur. J., 2015, 21, 18128–18137 CrossRef CAS PubMed.
- M. Tanaka, E. Ohta, A. Sakai, Y. Yoshimoto, K. Mizuno and H. Ikeda, Tetrahedron Lett., 2013, 54, 4380–4384 CrossRef CAS.
- M. Tanaka, S. Muraoka, Y. Matsui, E. Ohta, A. Sakai, T. Ogaki, Y. Yoshimoto, K. Mizuno and H. Ikeda, ChemPhotoChem DOI:10.1002/cptc.201600028.
- R. Yoshii, A. Nagai, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2013, 19, 506–4512 CrossRef PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2014, 20, 8320–8324 CrossRef CAS PubMed.
- R. Yoshii, K. Tanaka and Y. Chujo, Macromolecules, 2014, 47, 2268–2278 CrossRef CAS.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed.
- A. Hirose, K. Tanaka, R. Yoshii and Y. Chujo, Polym. Chem., 2015, 6, 5590–5595 RSC.
- K. Suenaga, R. Yoshii, K. Tanaka and Y. Chujo, Macromol. Chem. Phys., 2016, 217, 414–417 CrossRef CAS.
- R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2015, 21, 7231–7237 CrossRef CAS PubMed.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 3, 5314–5319 RSC.
- K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2017, 23, 1409–1414 CrossRef CAS PubMed.
- K. Tanaka, K. Nishino, S. Ito, H. Yamane, K. Suenaga, K. Hashimoto and Y. Chujo, Faraday Discuss., 2017, 196, 31–42 RSC.
- M. J. Frish, et al., Gaussian 09 (Revision D.01), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1533275 and 1533277. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00076f |
| This journal is © the Partner Organisations 2017 |