
Flower-shaped cobalt oxide nano-structures as an efficient, flexible and stable electrocatalyst for the oxygen evolution reaction†
C. K.
Ranaweera‡
a,
C.
Zhang‡
a,
S.
Bhoyate
a,
P. K.
Kahol
b,
M.
Ghimire
c,
S. R.
Mishra
c,
Felio
Perez
d,
Bipin Kumar
Gupta
 e and
Ram K.
Gupta
*a
e and
Ram K.
Gupta
*a
aDepartment of Chemistry, Pittsburg State University, Pittsburg, KS 66762, USA. E-mail: ramguptamsu@gmail.com
bDepartment of Physics, Pittsburg State University, Pittsburg, KS 66762, USA
cDepartment of Physics, The University of Memphis, Memphis, TN 38152, USA
dIntegrated Microscopy Center, The University of Memphis, Memphis, TN 38152, USA
eCSIR – National Physical Laboratory, Dr K.S. Krishnan Road, New Delhi 110012, India
First published on 28th March 2017
Abstract
The industrial application of water splitting for oxygen evolution requires low cost, high performance and stable electrocatalysts which can operate at low overpotential. Here, we develop a high performance and stable electrocatalyst for the oxygen evolution reaction (OER) using earth abundant materials. A binder free approach for the synthesis of flower-shaped cobalt oxide (Co3O4) composed of nanosheets showed high OER catalytic activity. The Co3O4 electrode requires a low overpotential of 356 mV to achieve a current density of 10 mA cm−2 with a low onset potential of 284 mV. The electrode showed outstanding flexibility and stability. The catalytic activity of the Co3O4 electrode was very stable up to the 2000th cycle of the polarization study. The high catalytic activity and structural stability arise due to efficient and fast charge transportation through the nanosheets of Co3O4 which are in direct contact with the conducting nickel of the electrode. The porous structure of Co3O4 allows easy access of the electrolyte and escape of generated oxygen without damaging the structure. Collectively, the flower-shaped nanostructured Co3O4 electrode can be used as a flexible and high performance electrode for the OER in an industrial setup.
The oxygen evolution reaction (OER) is a key step in many electrochemical processes, such as water electrolysis, solar to energy generation and metal–air batteries.1–4 Among these processes, water electrolysis is attracting considerable research interest as it can provide hydrogen and oxygen as fuels in a sustainable way. However, oxygen evolution through water electrolysis requires a high overpotential, which is due to slow kinetics and the involvement of several steps.5,6 Therefore, efficient electrocatalysts need to be developed which can accelerate the kinetics and lower the overpotential. To date, IrO2 and RuO2 are considered to be the ultimate electrocatalysts for the oxygen evolution reaction; however their applications are limited due to high cost and limited availability of the precious metals.7–9
Recently, several non-precious metal-based electrocatalysts have been synthesized and used to reduce the oxygen overpotential in water splitting processes. Among the various non-precious metal based electrocatalysts, transition metal based materials and perovskites have been considered to be most promising.10–20 Multi-shelled hollow structures have been synthesized via various techniques for energy applications.21–27 Lv et al. synthesized hollow mesoporous NiCo2O4 nanocages and studied their electrochemical properties for the oxygen evolution reaction.28 The hollow nanostructured NiCo2O4 showed an overpotential of about 0.34 V at a current density of 10 mA cm−2 with a Tafel slope of 75 mV per decade. Further, Chen et al. improved the catalytic properties of NiCo2O4 by growing a hierarchically porous nitrogen-doped graphene (PNG)–NiCo2O4 composite.29 The OER kinetics were also observed to improve as Tafel slopes of 156, 249 and 254 mV per decade were reported for PNG–NiCo2O4, NG–NiCo2O4 and PNG, respectively. Tang et al. used stainless steel containing Fe, Ni, Cr and Mn for OER catalysts.30 They grew in situ thin films of Fe(Ni)OOH by immersing stainless steel in an alkaline oxidant solution containing NaOH and (NH4)2S2O8. The electrode showed an overpotential of 300 mV at 10 mA cm−2 with a low Tafel slope of 34 mV per decade. Other groups have also used stainless steel for water oxidation due to the presence of Fe, Ni, Cr in it.31–35
Among other transition metal based electrocatalysts, Co3O4 shows promising behaviour for OER.6 The catalytic activities of crystalline and amorphous Co3O4 films were compared, and it was observed that amorphous Co3O4 films are more catalytically active than crystalline Co3O4 films.6 The crystalline and amorphous Co3O4 films showed Tafel slopes of 49 and 36 mV per decade, respectively. It was observed that the exchange current density also depends on the nature of the films. Exchange current densities of 2.0 × 10−10 and 5.4 × 10−12 A cm−2 were observed for the crystalline and amorphous Co3O4 films, respectively. The effect of the size of Co3O4 on the catalytic activity for the OER was also examined.36 It was also observed that the catalytic properties depend on the size of the synthesized Co3O4. A current density of 10 mA cm−2 was observed at overpotentials of 328, 363 and 382 mV for anodes loaded with 1 mg cm−2 of 5.9, 21.1 and 46.9 nm sized Co3O4 nanoparticles, respectively. The observed OER activities were directly correlated with the surface area of the Co3O4 nanoparticles. The electrocatalytic activity of Co3O4 was improved by synthesizing Co3O4/NiCo2O4 double-shelled nanocage structures.10 An overpotential of 410 and 340 mV was required for Co3O4 and Co3O4/NiCo2O4 double-shelled nanocages, respectively, to provide a current density of 10 mA cm−2. The OER kinetics of Co3O4 were also improved by making Co3O4/NiCo2O4 double-shelled nanocages as they showed Tafel slopes of 110 and 88 mV per decade, respectively. In general, the electrocatalytic activities of the synthesized materials were tested by loading the materials over a current collector using a polymer binder. These polymeric binders increase the contact resistance and block the active site of the catalyst and thus reduce the catalytic activities.37 Schäfer et al. used various ways to oxidize different types of stainless steel in a binder free approach for OER applications.33,38–40 For example, they oxidized the surface of steel S235 by Cl2 gas and observed its catalytic activity. They reported an overpotential of 462 mV (at 1 mA cm−2) at pH 7, which decreased to 347 mV (at 1 mA cm−2) at pH 13. They also used a facile anodization process to convert AISI Ni42 steel into a bifunctional (OER and HER) electrocatalyst. The performance of the prepared catalyst was very stable even under standard industrial operation conditions. Lu et al. used a binder free approach to synthesize Co3O4 and studied the catalytic activities of the deposited Co3O4 film.41 They observed a Tafel slope of 74 mV per decade, which was better than those of Co3O4 (110 mV per decade) and Co3O4/NiCo2O4 (88 mV per decade) double-shelled nanocages tested using a polymer binder.10
Herein, we report a facile binder free approach to synthesize flower-shaped Co3O4 nanostructures which are composed of nanosheets for the oxygen evolution reaction (details in the ESI†). The binder free Co3O4 electrode showed a low overpotential of 356 mV to gain a current density of 10 mA cm−2 in an alkaline medium. Furthermore, stability and flexibility tests confirmed no obvious decay in the current density of the Co3O4 electrode during the OER process. The porous nanosheets of Co3O4 over nickel foam allow fast electron and ion transportation through the nanosheets to the electrode with excellent structural stability, and thus high catalytic performance.
The phase purity and crystallinity of the synthesized cobalt oxide were studied using X-ray diffraction analysis (Fig. 1a). All the diffraction peaks observed in the synthesized sample match with those of the cubic spinel phase of Co3O4 (JCPDS: 74-2120). No peaks other than those due to Co3O4 were observed, suggesting the phase purity and high crystallinity of the synthesized cobalt oxide. The lattice parameter of the synthesized Co3O4 was calculated using the most intense peak (311), and it was found to be 0.808 nm. The observed lattice parameter of Co3O4 matches well with its theoretical value of 0.808 nm. The chemical composition of the synthesized cobalt oxide was further examined using X-ray photoelectron spectroscopy. Fig. 1b shows the survey-scan spectrum of the cobalt oxide. The deconvolution of the complex Co 2p spectrum (Fig. 1c) confirms the existence of two chemically distinct species: Co2+ and Co3+. The peaks at around 794.8 and 779.6 eV correspond to the Co 2p1/2 and Co 2p3/2 spin–orbit peaks of Co3O4. The presence of Co3O4 can be further confirmed by the O 1s XPS spectrum (Fig. 1d). The peak at 529.7 eV is corresponding to the lattice oxygen in the spinel Co3O4.42 The other observed peaks are associated with the oxygen of OH− and the H2O adsorbed onto the surface of Co3O4.43,44
 | ||
| Fig. 1 (a) XRD powder pattern, (b) XPS survey scan spectrum, (c) XPS spectra of Co 2p, and (d) XPS spectra of O 1s of the synthesized Co3O4. | ||
Fig. 2 shows the SEM images of Co3O4 at various magnifications. Co3O4 densely grows on the nickel foam with a flower-shaped morphology. The higher magnification images show that these flower-shaped structures are made of nanosheets. These individual flower-shaped nanostructures are very porous in nature, which will allow easy access of the electrolyte to the entire surface of the Co3O4. These porous nanosheets in direct contact with the nickel foam will also allow fast electron and ion transportation through the nanosheets to the electrode with excellent structural stability and thus could provide high catalytic activity.
 | ||
| Fig. 2 SEM images of synthesized Co3O4 at various magnifications. | ||
The electrocatalytic activities of Co3O4 were studied in alkaline solution for the oxygen evolution reaction. Fig. 3a shows the OER polarization curves of the nickel foam, platinum foil and Co3O4 grown over nickel foam. The Co3O4 electrode showed the lowest overpotential and the highest current density compared with those of nickel foam and platinum. The onset potentials for Co3O4, nickel foam and platinum were found to be 284, 327 and 328 mV, respectively (Fig. S1, ESI†). The overpotentials of 356, 441 and 459 mV were observed at 10 mA cm−2 for Co3O4, nickel foam and platinum, respectively. The Tafel slopes of Co3O4, nickel foam and platinum in 1 M NaOH were calculated to be 68, 142 and 220 mV per decade, respectively. As seen in Fig. 3b, the slope for the Co3O4 electrode remains linear even at higher current densities, indicating fast electron and mass transfer between the surface of Co3O4 and the electrolyte.45 Jeon et al. observed an overpotential of 377 mV at 10 mA cm−2 for a chemical solution deposited Co3O4 thin film.46 Chou et al. synthesized different phases of cobalt oxide such as CoO and Co3O4 for OER applications.47 Overpotentials of 495 and 496 mV at 10 mA cm−2 for CoO and Co3O4, respectively, were reported. Mesoporous Co3O4 has been synthesized using porous silica as a hard template via a nanocasting route as a catalyst for OER activity.48 They observed that the aging temperature of the silica hard template for Co3O4 affects the overpotential. They observed overpotentials from 525 to 636 mV at 10 mA cm−2 for Co3O4 with different aging temperatures. The electrochemical activities of a series of Co3O4 structures and their nanocomposites were investigated as oxygen evolution catalysts.49 It was observed that among the ordered mesoporous composites, Co3O4–CuCo2O4 showed the best performance as an OER catalyst. Co3O4 and Co3O4–CuCo2O4 showed overpotentials of 528 and 498 mV at 10 mA cm−2, respectively.
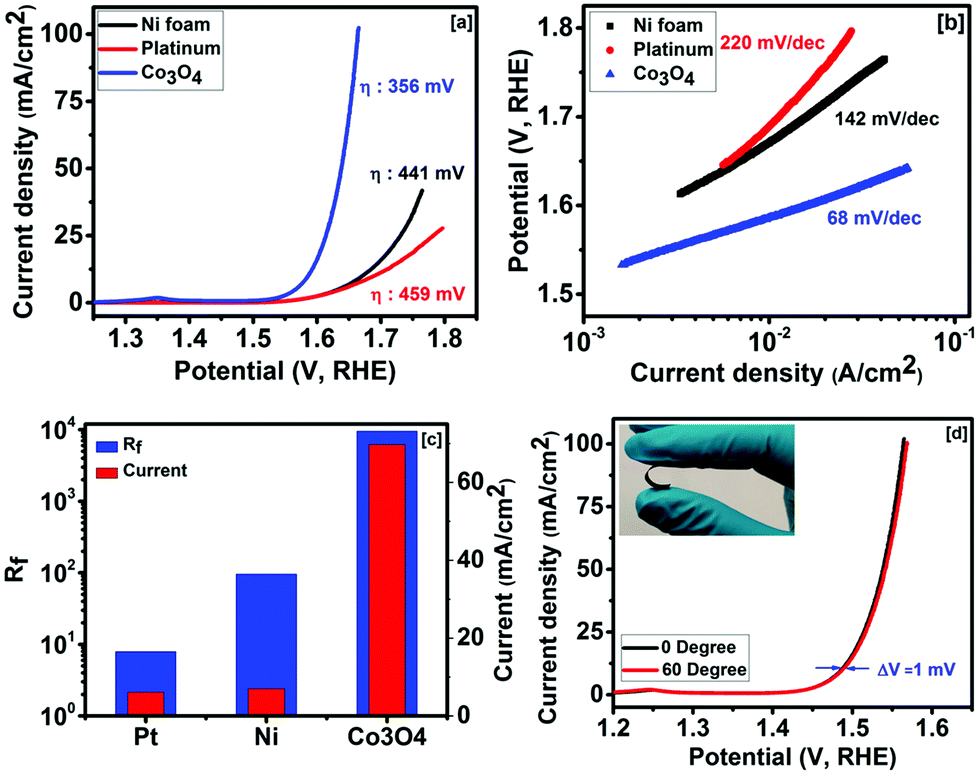 | ||
| Fig. 3 (a) OER polarization curves, (b) Tafel slope, (c) roughness factor and current density (at 1.65 V, RHE) for platinum, nickel foam and Co3O4, and (d) flexibility testing of the Co3O4 electrode (inset of (d) shows flexibility of Co3O4 electrode). | ||
Our synthesized Co3O4 showed an overpotential of 356 mV at 10 mA cm−2 and a Tafel slope of 68 mV per decade. The better performance of our Co3O4 electrode could be due to the highly porous and nanostructured morphology, which allows easy access of the electrolyte to the entire surface of Co3O4 and easy escape of the generated oxygen from the electrode. In addition, the direct growth of Co3O4 on nickel foam reduces the series resistance and the binder free approach does not block the active sites for the OER reaction. The specific surface area and roughness factor of nickel foam, platinum foil and Co3O4 grown over nickel foam were estimated using cyclic voltammetry measurements in non-faradaic regions. The CV curves show typical rectangular curves with no oxidation or reduction peaks, suggesting regular features of an electrical double layer capacitor (Fig. S2(a), ESI†). A linear relationship was observed between the current density and the scan rate of the samples as seen in Fig. S2(b) (ESI†). The slope of these curves provides the value of the double layer capacitance [i = C(dE/dt)]. Double layer capacitances of 0.48, 5.75 and 573 mF cm−2 were calculated for platinum, nickel foam and the Co3O4 electrode, respectively. The roughness factor (Rf) was determined using the following equation:41,50
| Rf = Cdl/Cdl* | (1) |
The effect of bending on the OER current density of Co3O4 was studied to investigate its applicability as a flexible electrode. The linear sweep voltammogram curves of the Co3O4 electrode at 0° and 60° bending show almost identical behaviors (Fig. 3d). As seen in Fig. 3d, a difference of 1 mV in overpotential at 10 mA cm−2 was observed between 0° and 60° bending. The long term stability of the Co3O4 electrode was studied. Fig. 4a shows the linear sweep voltammetry curves of cobalt oxide at various cycles. The performance of the Co3O4 electrode was very stable up to the 2000th cycle. A small difference (∼4 mV) in overpotential at 10 mA cm−2 was observed between the 2000th cycle and the first cycle. Our results suggest that the Co3O4 electrode could be used as a high performance and flexible OER electrode. The stability of the Co3O4 electrode was further examined using chronoamperometry. As seen in Fig. 4b, the current density was almost constant for over 20 h of study. Our results confirmed that the method adopted in this work could be used to fabricate highly efficient and durable electrocatalysts for the oxygen evolution reaction in the electrolysis of water.
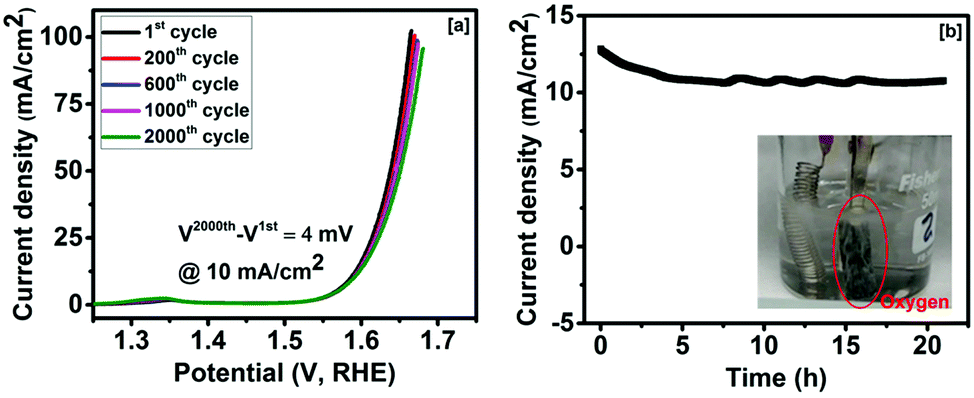 | ||
| Fig. 4 (a) Linear sweep voltammetry curves and (b) time-dependent current density curve for the synthesized Co3O4 (inset of (b) shows oxygen evolution during experiment). | ||
The performance of the Co3O4 electrode was also studied using electrochemical impedance spectroscopy. Fig. S3 (ESI†) shows the Nyquist plots of the Co3O4 electrode at different potentials. The behavior of the Nyquist plots depends on the potential. At lower potential, the Nyquist plot shows a depressed semicircle near the origin (at high frequency), followed by a straight line at higher impedance (at low frequency). The straight line in the low frequency region starts to convert into a semicircle with an increase in the potential. It is further noted that the semicircle in the high frequency region is almost independent of the potential, whereas the semicircle in the low frequency region depends on the overpotential. The semicircle at low frequency is related to the OER process as its diameter reduces with increasing overpotential, which is due to a faster reaction at high overpotential. EIS measurements of the Co3O4 electrode before and after the long-term stability test were performed. Fig. S4(a) (ESI†) shows the Nyquist plot of the Co3O4 electrode before the stability test. The EIS data fit well with the equivalent circuit given in the inset of Fig. S4(a) (ESI†), where Rs is the electrolyte resistance, Cdl is the double layer capacitance at the Co3O4/electrolyte interface, Rct, Cpc and R1 are related to the OER process, and Cox and Rox could be due to additional contribution from the underlying Co3O4 passive film.51 The Nyquist plots before and after the stability test are shown in Fig. S4(b) (ESI†). A small increase in the impedance of the Co3O4 electrode was seen after the stability test (inset of Fig. S4(b) (ESI†)). A small increment in Rs, Rct, R1 and Rox was observed after the stability test (Table S1, ESI†). The electrochemical investigations on the Co3O4 electrode confirm that our binder free approach can be used to fabricate low-cost, high performance, durable and flexible electrodes for the oxygen evolution reaction which can operate at low overpotentials.
In summary, we have used a facile method to synthesise a binder free Co3O4 nanostructured electrode. The as-synthesized Co3O4 nanostructure electrode was tested for the oxygen evolution reaction in alkaline solution. Co3O4 showed a highly promising electrocatalytic behaviour. In alkaline solution, the Co3O4 electrode showed a low overpotential of 356 mV (at 10 mA cm−2) with great electrochemical stability and flexibility. The Co3O4 electrode showed almost identical oxygen evolution curves on bending (1 mV difference in overpotential before and after bending). The high catalytic activity of the Co3O4 electrode was due to the highly porous structure and the direct contact of the Co3O4 nanosheets with the nickel foam. Thus, it is anticipated that this binder free approach can be used to synthesize low cost and high performance electrocatalysts for industrial water splitting processes.
Author contributions
RKG conceived the project and designed the experiments. RKG also performed some structural and electrochemical characterizations, interpreted and analyzed the data, and prepared the manuscript. CR, CZ and SB synthesized and performed some electrochemical characterizations. MG and SM recorded the SEM images. FP performed the XPS measurements. All the authors reviewed and commented on the manuscript.Competing financial interests
The authors declare no competing financial interests. Requests for materials should be addressed to RKG.Acknowledgements
Dr Ram K. Gupta expresses his sincere acknowledgment to the Polymer Chemistry Initiative, Pittsburg State University for providing financial and research support. SM thanks funding from FIT-DRONES at the University of Memphis.References
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- Y.-C. Lu, Z. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- T. J. Meyer, Nature, 2008, 451, 778–779 CrossRef CAS PubMed.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- S. Cherevko, T. Reier, A. R. Zeradjanin, Z. Pawolek, P. Strasser and K. J. J. Mayrhofer, Electrochem. Commun., 2014, 48, 81–85 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- J. Yu, Y. Zhong, W. Zhou and Z. Shao, J. Power Sources, 2017, 338, 26–33 CrossRef CAS.
- X. Wang, Y. Zheng, J. Yuan, J. Shen, A.-J. Wang, L. Niu and S. Huang, Electrochim. Acta, 2016, 212, 890–897 CrossRef CAS.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- Y. Xu, A. Tsou, Y. Fu, J. Wang, J.-H. Tian and R. Yang, Electrochim. Acta, 2015, 174, 551–556 CrossRef CAS.
- X. Xu, Y. Pan, W. Zhou, Y. Chen, Z. Zhang and Z. Shao, Electrochim. Acta, 2016, 219, 553–559 CrossRef CAS.
- J. Wang, Y. Fu, Y. Xu, J. Wu, J.-H. Tian and R. Yang, Int. J. Hydrogen Energy, 2016, 41, 8847–8854 CrossRef CAS.
- C. Jin, X. Cao, L. Zhang, C. Zhang and R. Yang, J. Power Sources, 2013, 241, 225–230 CrossRef CAS.
- K. Elumeeva, J. Masa, J. Sierau, F. Tietz, M. Muhler and W. Schuhmann, Electrochim. Acta, 2016, 208, 25–32 CrossRef CAS.
- D. S. Bick, A. Kindsmüller, G. Staikov, F. Gunkel, D. Müller, T. Schneller, R. Waser and I. Valov, Electrochim. Acta, 2016, 218, 156–162 CrossRef CAS.
- H. A. Bandal, A. R. Jadhav, A. A. Chaugule, W. J. Chung and H. Kim, Electrochim. Acta, 2016, 222, 1316–1325 CrossRef CAS.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- J. Wang, H. Tang, H. Ren, R. Yu, J. Qi, D. Mao, H. Zhao and D. Wang, Adv. Sci., 2014, 1, 1400011 CrossRef PubMed.
- J. Zhang, H. Ren, J. Wang, J. Qi, R. Yu, D. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 CAS.
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef CAS PubMed.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Materials Chemistry Frontiers, 2017, 1, 414–430 RSC.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- X. Lv, Y. Zhu, H. Jiang, X. Yang, Y. Liu, Y. Su, J. Huang, Y. Yao and C. Li, Dalton Trans., 2015, 44, 4148–4154 RSC.
- S. Chen and S.-Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed.
- D. Tang, O. Mabayoje, Y. Lai, Y. Liu and C. B. Mullins, ChemistrySelect, 2017, 2, 2230–2234 CrossRef CAS.
- F. Yu, F. Li and L. Sun, Int. J. Hydrogen Energy, 2016, 41, 5230–5233 CrossRef CAS.
- H. Schäfer, S. M. Beladi-Mousavi, L. Walder, J. Wollschläger, O. Kuschel, S. Ichilmann, S. Sadaf, M. Steinhart, K. Küpper and L. Schneider, ACS Catal., 2015, 5, 2671–2680 CrossRef.
- H. Schafer, S. Sadaf, L. Walder, K. Kuepper, S. Dinklage, J. Wollschlager, L. Schneider, M. Steinhart, J. Hardege and D. Daum, Energy Environ. Sci., 2015, 8, 2685–2697 Search PubMed.
- F. Moureaux, P. Stevens, G. Toussaint and M. Chatenet, J. Power Sources, 2013, 229, 123–132 CrossRef CAS.
- J. S. Chen, J. Ren, M. Shalom, T. Fellinger and M. Antonietti, ACS Appl. Mater. Interfaces, 2016, 8, 5509–5516 CAS.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CAS.
- Y. Luo, J. Jiang, W. Zhou, H. Yang, J. Luo, X. Qi, H. Zhang, D. Y. W. Yu, C. M. Li and T. Yu, J. Mater. Chem., 2012, 22, 8634–8640 RSC.
- H. Schafer, D. M. Chevrier, K. Kuepper, P. Zhang, J. Wollschlaeger, D. Daum, M. Steinhart, C. He, U. Krupp, K. Muller-Buschbaum, J. Stangl and M. Schmidt, Energy Environ. Sci., 2016, 9, 2609–2622 Search PubMed.
- H. Schäfer, K. Küpper, J. Wollschläger, N. Kashaev, J. Hardege, L. Walder, S. Mohsen Beladi-Mousavi, B. Hartmann-Azanza, M. Steinhart, S. Sadaf and F. Dorn, ChemSusChem, 2015, 8, 3099–3110 CrossRef PubMed.
- H. Schäfer, D. M. Chevrier, P. Zhang, J. Stangl, K. Müller-Buschbaum, J. D. Hardege, K. Kuepper, J. Wollschläger, U. Krupp, S. Dühnen, M. Steinhart, L. Walder, S. Sadaf and M. Schmidt, Adv. Funct. Mater., 2016, 26, 6402–6417 CrossRef.
- B. Lu, D. Cao, P. Wang, G. Wang and Y. Gao, Int. J. Hydrogen Energy, 2011, 36, 72–78 CrossRef CAS.
- Y. Tan, Q. Gao, C. Yang, K. Yang, W. Tian and L. Zhu, Sci. Rep., 2015, 5, 12382 CrossRef CAS PubMed.
- S. Xiong, J. S. Chen, X. W. Lou and H. C. Zeng, Adv. Funct. Mater., 2012, 22, 861–871 CrossRef CAS.
- C. Shang, S. Dong, P. Hu, J. Guan, D. Xiao, X. Chen, L. Zhang, L. Gu, G. Cui and L. Chen, Sci. Rep., 2015, 5, 8335 CrossRef CAS PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- H. S. Jeon, M. S. Jee, H. Kim, S. J. Ahn, Y. J. Hwang and B. K. Min, ACS Appl. Mater. Interfaces, 2015, 7, 24550–24555 CAS.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- H. Tüysüz, Y. J. Hwang, S. B. Khan, A. M. Asiri and P. Yang, Nano Res., 2013, 6, 47–54 CrossRef.
- T. Grewe, X. Deng, C. Weidenthaler, F. Schüth and H. Tüysüz, Chem. Mater., 2013, 25, 4926–4935 CrossRef CAS.
- L. M. Da Silva, L. A. De Faria and J. F. C. Boodts, Electrochim. Acta, 2001, 47, 395–403 CrossRef CAS.
- E. B. Castro, C. A. Gervasi and J. R. Vilche, J. Appl. Electrochem., 1998, 28, 835–841 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization and some additional figures. See DOI: 10.1039/c7qm00108h |
| ‡ C. K. Ranaweera and C. Zhang contributed equally. |
| This journal is © the Partner Organisations 2017 |