
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct exfoliation of graphite into graphene in aqueous solutions of amphiphilic peptides†
Meiwen
Cao
a,
Ningning
Wang
a,
Lei
Wang
a,
Yu
Zhang
a,
Yucan
Chen
a,
Zilong
Xie
a,
Zongyi
Li
b,
Elias
Pambou
b,
Ruiheng
Li
b,
Cuixia
Chen
a,
Fang
Pan
b,
Hai
Xu
*a,
Jeffery
Penny
c,
John R. P.
Webster
d and
Jian R.
Lu
*b
aState Key Laboratory of Heavy Oil Processing and Centre for Bioengineering and Biotechnology, China University of Petroleum (East China), 66 Changjiang West Road, Qingdao 266580, China. E-mail: xuh@upc.edu.cn; Tel: +86-532-86981569
bBiological Physics Laboratory, School of Physics and Astronomy, University of Manchester, Schuster Building, Manchester M13 9PL, UK. E-mail: j.lu@manchester.ac.uk; Tel: +44 (0)161-2003926
cManchester Pharmacy School, University of Manchester, Stopford Building, Manchester M13 9PT, UK
dSTFC ISIS Facility, Rutherford Appleton Laboratory, Didcot OX11 0QX, UK
First published on 18th November 2015
Abstract
Different amphiphilic peptides were used to mediate the direct exfoliation of graphite into few-layered graphene flakes in aqueous solutions. Charge was found to be an important parameter in determining their graphite exfoliating efficiency. The anionic molecules were more favorable than the cationic ones leading to a higher efficiency. The gemini-type peptide IleIleIleCys–CysIleIleIle (I3C–CI3) exhibited the highest efficiency, which might be attributed to its specific physicochemical properties and interactions with graphene sheets. I3C–CI3 adsorbed onto the graphene surface as either monomers or self-assembled nanoaggregates. These adsorbed species increased both electrostatic and steric repulsions between the graphene/I3C–CI3 composites. More interestingly, the graphene/I3C–CI3 composites showed a reversible pH-dependent dispersion/aggregation. This behavior resulted from the pH-sensitive protonation of the peptide molecules and was rarely found in the graphene dispersions exfoliated by traditional surfactants. Moreover, the graphene/I3C–CI3 dispersion was used to fabricate free-standing macroscopic composite films that contained different nanostructures. The study expands the library of available agents for direct graphite exfoliation to produce graphene sheets. Employing peptide molecules as graphene exfoliating and stabilizing agents avoids the use of toxic reagents, which may allow fabrication of functional composite materials for biocompatible applications.
1 Introduction
Since the first experimental isolation by Geim and coworkers in 2004,1 graphene has attracted great interest due to its fundamental and practical significance. Graphene monolayers are comprised of sp2-bonded carbon atoms with a two-dimensional lattice. They have high surface areas and show unusual properties in the electronic,2 thermal,3,4 and mechanical aspects,5,6 and hence single- and few-layer graphene materials are highly promising for technological uses including electronic devices, energy generation and storage, reinforced composite materials, and chemical and biological materials.7,8A variety of approaches have been utilized to produce graphene materials. Micromechanical cleavage,1 epitaxial growth,9–11 and some special methods12 can yield single-layer graphene sheets of high quality. However, these methods are unsuitable for large scale production due to the drawbacks of time consumption and low output. Reduction of graphene oxide (GO) produced from the aggressive oxidation of graphite is a powerful approach that can yield large quantities of graphene sheets.13–15 However, the reduced GO still contains a significant amount of defects and the structure and original properties of graphene are never fully restored, resulting in restrictions for their applications. Compared with the above methods, sonication assisted liquid-phase graphene exfoliation has some obvious advantages.16–23 This method directly exfoliates pristine graphite to graphene in organic solvents or surfactant solutions, which features ease of processing and up-scalable production. More importantly, the graphene obtained by this method has a very low density of defects and the liquid phase colloidal dispersions readily allow the subsequent synthesis of coatings and composite materials.24,25
Graphite is a hydrophobic material with stable interlayer adhesion that makes its direct exfoliation into graphene difficult. Several particular organic solvents such as N,N-dimethylformamide,19N-methylpyrrolidone,17,26,27 and ortho-dichlorobenzene20,27 have been demonstrated to be effective for graphite exfoliation. These solvents are nonpolar and have surface energy close to that of graphene (e.g., having surface tension values of ∼40 mJ m−2) so as to ensure efficient graphite exfoliation.17 However, these solvents have their own disadvantages. Their high boiling points and toxicity to multiple organs limit their viability for real manipulation and application.28 Referring to these aspects, water is the best choice for the dispersal of graphene because it not only allows easy composite formation but also has non-toxicity.29 However, water has a surface tension of 72.8 mJ m−2 at 20 °C,30 which is too high for direct graphite exfoliation. In this case, suitable stabilizing agents are needed for graphene exfoliation in water, for example, polymers and surfactants.16,22,31–33
Researchers have devoted considerable efforts to the production of graphene directly from pristine graphite in water/surfactant solutions. Coleman and coworkers have produced single- and few-layer graphene sheets using various types of surfactants.16,18,34 They found that the dispersed graphene concentration depended on either the zeta potential of the surfactant-coated flakes (in the case of ionic surfactants) or the magnitude of the steric potential barrier stabilizing the flakes (in the case of non-ionic surfactants).34 Guardia et al. paid particular attention to the use of non-ionic surfactants as graphene dispersing agents and showed that some non-ionic surfactants could lead to graphene concentrations up to about 1 mg mL−1.35 Vadukumpully et al. produced graphene flakes from graphite using cationic surfactant cetyl trimethyl ammonium bromide (CTAB) as a stabilizer.36 An et al. obtained a stable graphene dispersion in water by functionalization of graphene with 1-pyrenecarboxylic acid through a non-covalent π–π stacking mechanism.37 Green et al. firstly obtained graphene dispersions using the planar surfactant sodium cholate, and then isolated graphene flakes with controlled thicknesses using density gradient ultracentrifugation.38 These studies pioneered the study of surfactant-assisted graphite exfoliation and probed into the exfoliating mechanisms, which are crucial for further graphene processing and prospective applications.
Recently, one of the focuses of graphene studies is its bio-applications, for example, biosensors, thermal therapy, drug carriers, and so on.7,39 For such applications, biocompatible graphene dispersing systems are highly needed.40 However, many surfactants used for graphene exfoliation in water are unsuitable largely due to their possible toxicity and low biocompatibility. Some amphiphilic peptide molecules, for example, short surfactant-like peptides and lipopeptides, are similar to traditional surfactants in both molecular structures and physiochemical properties, and furthermore, they usually show better biocompatibility. Therefore, we are interested in establishing whether such molecules can help to exfoliate graphite to graphene efficiently in water. In the present study, we have compared the efficiency of 18 amphiphilic peptides in graphite exfoliation. Our studies revealed that the anionic molecules were more favorable than the cationic ones, demonstrating relatively high efficiency. Peptide IleIleIleCys–CysIleIleIle (I3C–CI3) exhibited the highest efficiency, possibly due to its specific physicochemical properties and geometry as well as interactions with graphene sheets. In addition, the graphene/I3C–CI3 composites showed a reversible pH-dependent dispersion/aggregation behaviour. These findings have expanded the library of available agents for direct graphene exfoliation and may allow the scalable graphene production for biocompatible composite materials.
2 Experimental section
2.1 Materials
Peptide molecules (Fig. 1) were synthesized following the methods described elsewhere41–43 or purchased from GL Biochem (Shanghai) Ltd. Pristine graphite was purchased from Jinpeng (Qingdao) Ltd. Other chemicals were purchased from Sigma-Aldrich and used as received. Milli-Q water (18 MΩ cm) was used for all the experiments.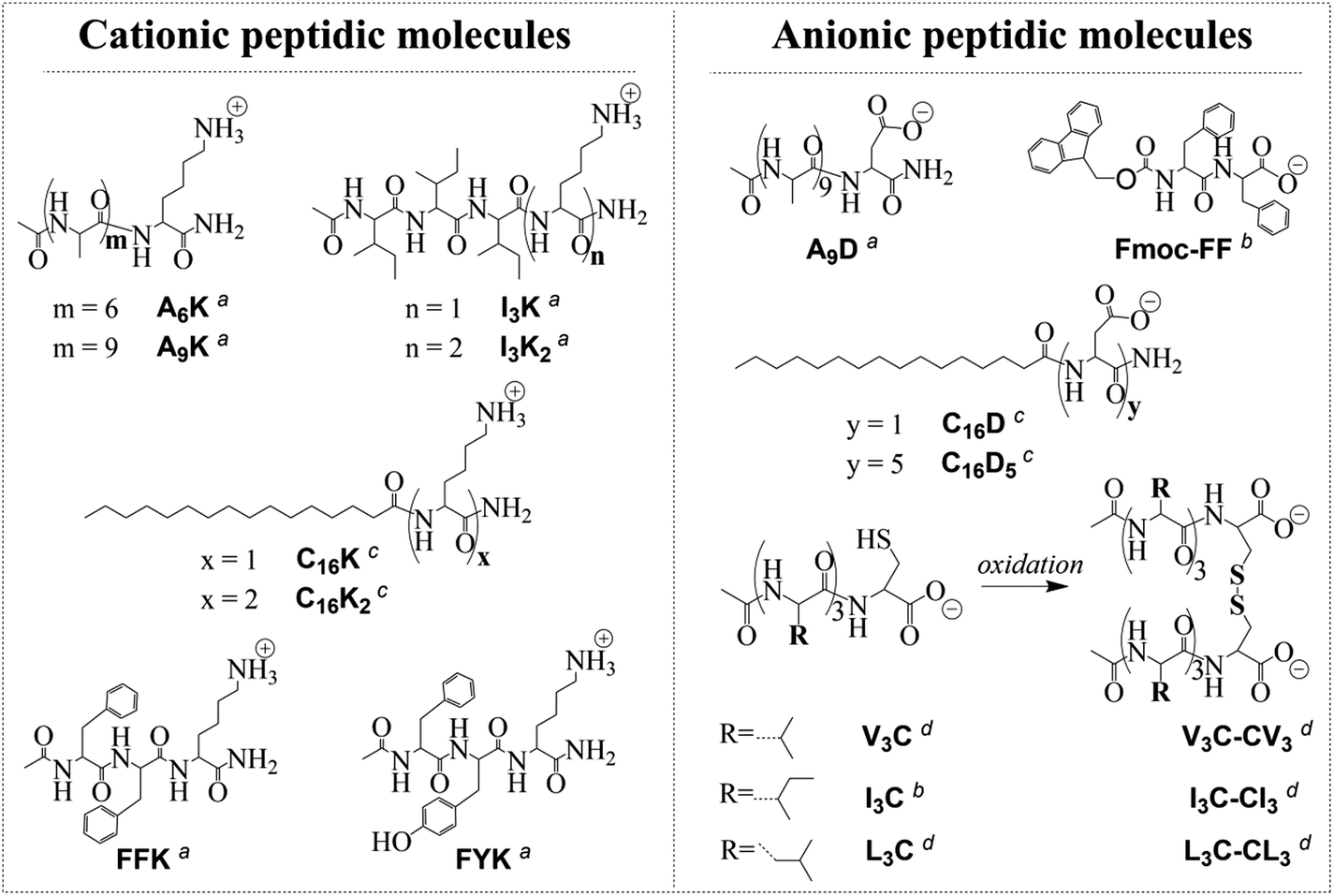 | ||
| Fig. 1 Molecular structures of the peptide molecules used for graphite exfoliation in water. These molecules can be divided into several categories according to different classification criteria: (1) cationic molecules and anionic molecules by charge (e.g., A9K and A9D), (2) non-aromatic molecules and aromatic molecules by aromaticity (e.g., A9K and FFK), (3) peptides and lipopeptides by possession or lack of a long alkyl chain (e.g., A9D and C16D), (4) single-chain molecules and gemini molecules by topological structuring via disulfide bonding (e.g., I3C and I3C–CI3). a,c,d Synthesized following the methods described in ref. 41–43, respectively. b Purchased from GL Biochem (Shanghai) Ltd and used as received. | ||
2.2 Graphite exfoliation
10.0 mg of peptide molecules and 50.0 mg of graphite were added into an Eppendorf tube with 20 mL of water. Specifically, for peptides V3C, I3C, and L3C with a functional thiol group, dithiothreitol (DTT) was added to the suspension at a molar ratio of DTT/peptide of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, in an attempt to avoid disulfide bond formation by oxidation. The suspension pH was adjusted to ∼10 for anionic molecules using dilute NaOH and to 3–4 for cationic molecules using dilute HCl to make sure that the molecules were fully charged. The suspension was then subjected to sonication in an ultrasound clean bath (KQ-200KDE System, 40 kHz) for a total of 24 h. The bath temperature was controlled to be below 65 °C during sonication. Following sonication, the suspension was centrifuged at different speeds to sediment un-exfoliated graphite particles or thick flakes, and the top supernatant containing few-layer graphene flakes was collected as the final graphene dispersion.
:1, in an attempt to avoid disulfide bond formation by oxidation. The suspension pH was adjusted to ∼10 for anionic molecules using dilute NaOH and to 3–4 for cationic molecules using dilute HCl to make sure that the molecules were fully charged. The suspension was then subjected to sonication in an ultrasound clean bath (KQ-200KDE System, 40 kHz) for a total of 24 h. The bath temperature was controlled to be below 65 °C during sonication. Following sonication, the suspension was centrifuged at different speeds to sediment un-exfoliated graphite particles or thick flakes, and the top supernatant containing few-layer graphene flakes was collected as the final graphene dispersion.
2.3 Transmission electron microscopy (TEM)
TEM images were obtained on a JEM 1400Plus (JEOL) electron microscope operated at 120 kV. Two methods were used for TEM sample preparation. In Method 1, a drop of the peptide-treated graphene dispersion was cast onto a copper grid (200 mesh) covered with a carbon film and allowed to dry in air. In Method 2, after casting the graphene dispersion onto the copper grid, the sample was negatively stained with 2% uranyl acetate.2.4 Atomic force microscopy (AFM)
AFM measurements were performed on a MultiMode Nanoscope IVa AFM (Digital Instruments, Santa Barbara, CA) in tapping mode under ambient conditions. TESP silicon probes (Veeco, Santa Barbara, CA) were used with a nominal spring constant of 42 N m−1. For sample preparation, a drop of 10–15 μL of the peptide-treated graphene dispersion was pipetted onto a freshly cleaved mica surface. After adsorption for 10–30 s, the sample was dried with a nitrogen stream. Tapping mode images were recorded as 512 × 512 pixel images. Image analysis was carried out using the Digital Instruments Nanoscope software (version V530r3sr3).2.5 Scanning electron microscopy (SEM)
The paper-like graphene/I3C–CI3 composite film was prepared by vacuum filtration of the peptide-treated graphene dispersion through a nylon filter membrane of 0.22 μm pore size. After vacuum drying, the film was coated with a thin platinum layer and then characterized using a JEOL JSM-840 instrument operated at 15 kV.2.6 UV-Vis measurements
UV-Vis absorption spectra of the peptide-treated graphene dispersion were recorded on a UV-vis spectrophotometer (UV-1700 Phama Spec, Shimadzu) at ambient temperature. The spectra were measured in the 200–800 nm wavelength range with a cuvette of 1 cm path-length. Absorption by peptide molecules was negligible, except for those dispersions stabilized by molecules containing aromatic moieties in their structure (e.g., FFK, FYK, and Fmoc-FF). However, in all cases the absorbance of peptide molecules approached zero at a wavelength of more than 450 nm. This allowed use of the absorbance of the dispersion at 660 nm as an estimate of the relative concentration of suspended graphene.2.7 Raman spectra
Raman spectra were recorded on a Horiba Jobin-Yvon LabRam instrument at a laser excitation wavelength of 532 nm. Note that the peptide-treated graphene dispersion was filtered to form a thin film for Raman measurements.2.8 Zeta potential (ζ) and dynamic light scattering (DLS) measurements
The ζ measurements were performed at 25 °C using a Malvern Nano-ZS instrument (ZEN3600, Malvern Instruments, Worcestershire, UK) equipped with a 4 mW He–Ne laser at a wavelength of 633 nm. A clear disposable capillary cell (DTS1060C) was used. Sizes of the graphene/I3C–CI3 composites were measured through DLS and the intensity weighted hydrodynamic size distributions were presented.2.9 X-ray photoelectron spectroscopy (XPS)
XPS measurements of the pristine graphite and the graphene/I3C–CI3 composite film were performed on the Thermo Scientific ESCALab 250Xi using 200 W monochromated Al Kα radiation. The 500 μm X-ray spot was used for XPS analysis. The hydrocarbon C1s line at 284.8 eV from adventitious carbon was used for energy referencing.3 Results and discussion
The relative efficiency of different peptide molecules in graphite exfoliation and stabilization can be directly estimated from visual observation of the treated dispersions. The inset photos in Fig. 2 show several representative dispersions after sonication and centrifugation. The black or grey color of the dispersions signifies the presence of a large amount of suspended graphitic species, especially the I3C- and I3C–CI3-treated samples that were dark in appearance. These dispersions were observed to be stable for at least 1 month with practically no signs of floating or precipitated materials, indicating high stability. According to the Beer–Lambert Law, A = αCl (α: the absorption coefficient, C: the concentration, l: the cell pathlength), the A/l (absorbance per unit-cell-length) is proportional to the concentration. Here the A/l values of the dispersions at 660 nm were measured and taken as a quantitative measurement of the concentration of the suspended species, which can reflect the relative exfoliating and dispersing ability of different peptide molecules (Fig. 2). The results showed that nearly all the anionic molecules (except L3C and L3C–CL3) gave A/l values of above 20 m−1, while all the cationic molecules gave A/l values lower than 10 m−1. This indicated that for peptide molecules the negative charges were more beneficial than the positive charges for improving the efficiency in graphite exfoliation and suspension. There were little differences between the A/l values of non-aromatic compared to aromatic molecules and of peptides compared to lipopeptides. Therefore, aromaticity and acylation of the peptide molecules did not change their graphite exfoliation efficiency significantly. For the single chain peptides of V3C, I3C, and L3C, there was little difference with their corresponding gemini-type molecules in exfoliating efficiency. The reason may be that although DTT was added to the solutions, the long sonication process with increased temperature would oxidize a considerable amount of thiol groups to form disulfide bonds and turn the single-chain molecules into the gemini molecules.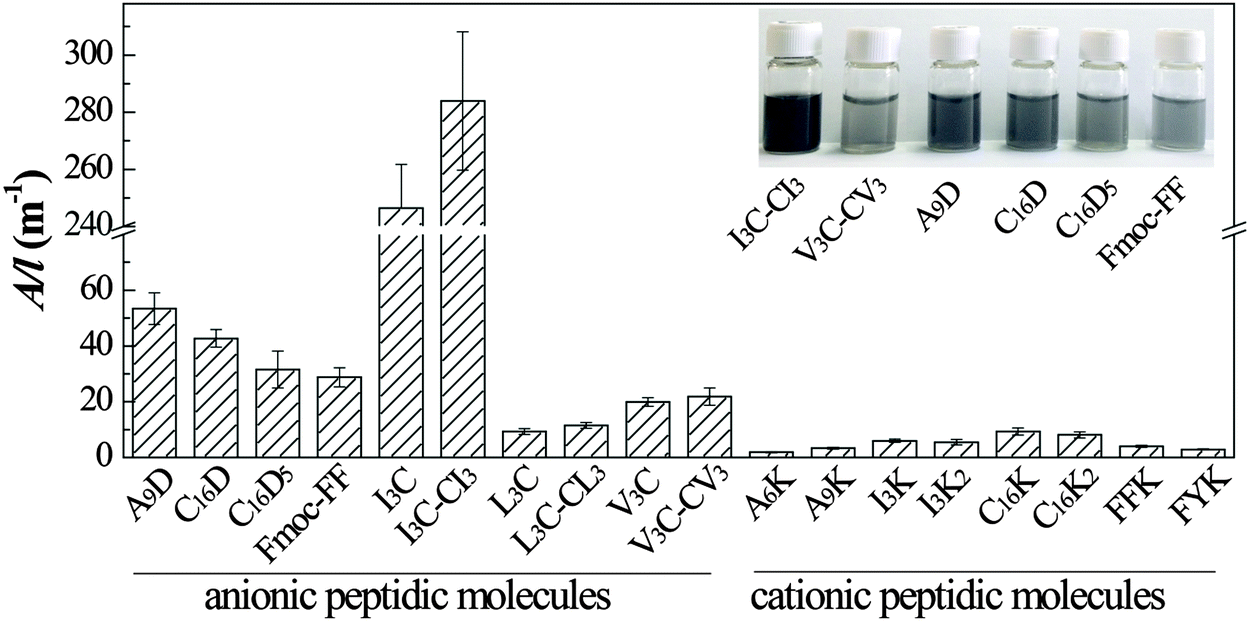 | ||
| Fig. 2 The A/l values of the peptide-treated graphene dispersions that were obtained through centrifugation at 5000 rpm for 10 min after sonication in peptide solutions. The inset shows the photographs of several typical dispersions with relatively high exfoliation efficiency. | ||
UV-Vis and Raman spectra were recorded to determine the species in the dispersions and the results are shown in Fig. 3. The peptide molecules I3C, I3C–CI3, C16D, C16D5, and A9D all demonstrated a UV-vis absorbance peak at 267–269 nm (Fig. 3a). This peak is characteristic of graphene flakes and can be attributed to the π–π* transition of polyaromatic systems.44–46 The UV-vis spectrum of Fmoc-FF seemed to be a little different from the others by giving a blue-shifted peak at 263 nm, which possibly arises from the combined contribution of the aromatic rings of Fmoc and Phe moieties and the polyaromatic system of graphene. Raman spectra were used to determine the quality of the starting pristine graphite and the produced graphene flakes (Fig. 3b). Two prominent Raman bands for graphitic materials, a defect-induced D band at 1345 cm−1 and a G band of in-plane vibration of sp2 carbon at 1570 cm−1, were observed.47,48 The D/G intensity ratio for graphite powder was 0.11, indicating that the starting material possessed a high structural quality (very low defect content). On the other hand, for the sample treated with I3C–CI3, the D/G ratio was 0.27, being close to the range of 0.26–0.6 which is typical for graphene flakes stabilized by surfactants.18 The increased D intensity could be attributed to an increased fraction of defects within the graphene flakes, mainly from the graphene edges caused by sonication cutting rather than point defects on the basal plane.16,47,49,50
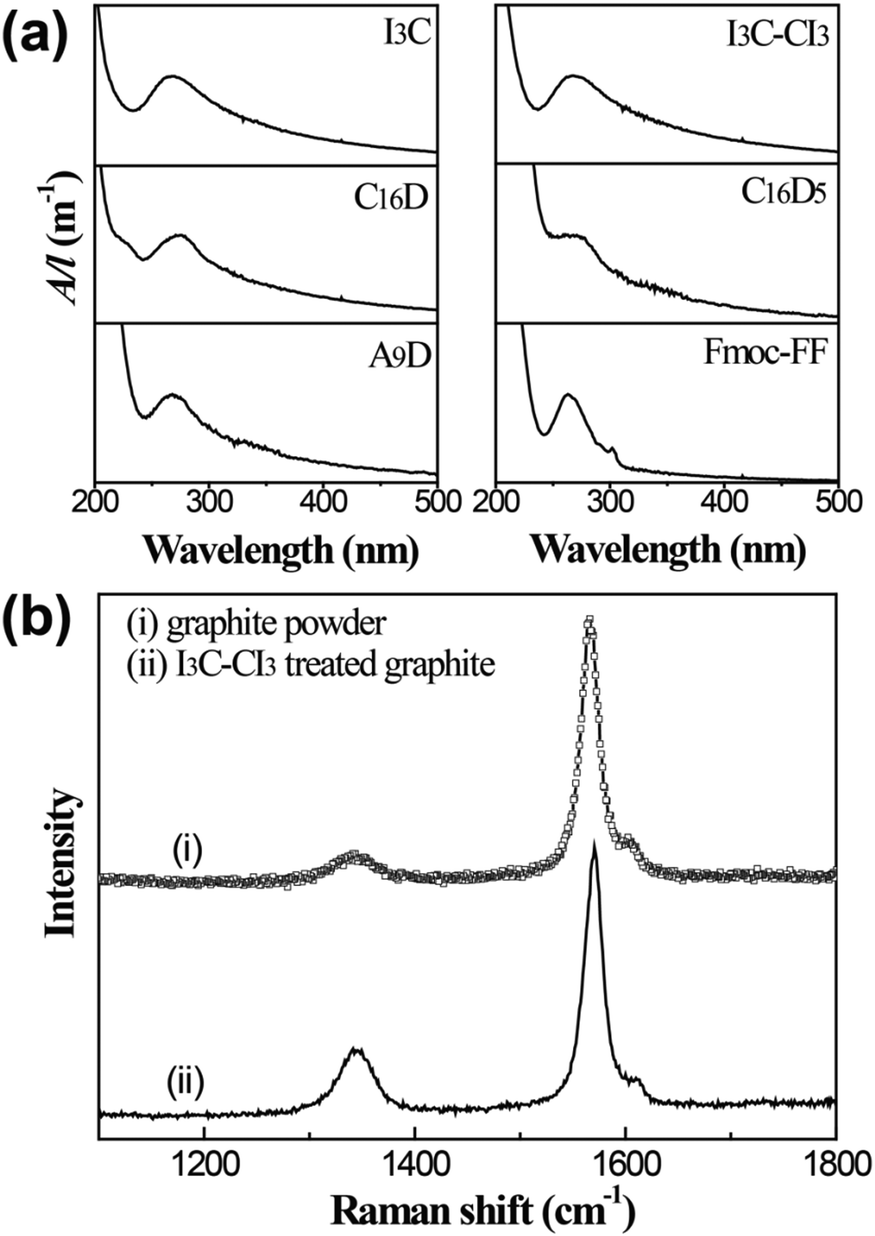 | ||
| Fig. 3 (a) Typical UV-Vis spectra of the peptide-treated graphene dispersions that were obtained through centrifugation at 5000 rpm for 10 min after sonication in peptide solutions. (b) Raman spectra of the starting pristine graphite and the I3C–CI3-treated sample. | ||
The amphiphilic peptide molecules used here have charged amine (–NH3+)/carboxyl (–COO−) groups as the hydrophilic heads and alkyl chains/repeated hydrophobic amino residues as the hydrophobic tails (Fig. 1). The structural characteristic makes these molecules similar to traditional surfactants in terms of their amphiphilic nature.41,51 It is thus suggested that the role of peptide molecules in graphite exfoliation is similar to that of traditional surfactants.34 The molecules bind to the exposed graphitic surface by the hydrophobic segments via hydrophobic interactions. The charged –NH3+/–COO− groups are exposed outward (as verified by the zeta potential measurement) to separate the graphitic layers and suspend them well in aqueous solution.37 The lower efficiency of the cationic molecules in comparison with that of the anionic molecules may be ascribed to the cation–π interaction. It has been well documented that the binding of simple cations to aromatic systems is quite substantial and may be interpreted by an electrostatic model.52,53 Here the positive –NH3+ groups of the cationic peptide molecules could bind to the polyaromatic graphitic surface via the cation–π interaction. This interaction would reduce the electrostatic repulsion between the graphene/peptide composites and lead to a decreased exfoliating efficiency. Additionally, although an aromatic component has been proposed to improve the molecular binding with graphene surface via π–π interactions and thus to facilitate graphene stabilization,54,55 there was no indication that the aromatic peptide molecules (e.g. FFK and Fmoc-FF) gave higher exfoliating efficiency in the present case.
The I3C–CI3-treated graphene dispersion was taken as a representative sample for further detailed investigations. The dispersion was firstly separated into several aliquots and each was centrifuged at a chosen speed for 90 min. The supernatants were then removed and their absorbance was measured. As shown in Fig. 4a, their A/l values at 660 nm were relatively large, above 250 m−1 at centrifugation speeds of lower than 1500 rpm. A sharp decrease of the A/l value was observed when the centrifugation speed was increased to above 1500 rpm. Then over the speed range of 5000–8000 rpm the A/l value reached a plateau at approximately 50 m−1. The centrifugation-induced decrease in the A/l value is likely due to the removal of graphitic particles and/or larger graphene flakes from the dispersion.38 However, the A/l value was still ∼50 m−1 even at a higher centrifugation speed of 8000 rpm, indicating a significant suspending power of the I3C–CI3 molecule toward graphene. By taking the absorption coefficient at 660 nm of α = 1390 mL mg−1 m−1,16 the graphene concentration was calculated to be approximately 0.03 mg mL−1 for the dispersion centrifuged at 8000 rpm for 90 min. Of note is that when higher centrifugation speeds were applied, the sizes of the dispersed graphene flakes decreased, as verified by the DLS size measurements (Fig. 4b).
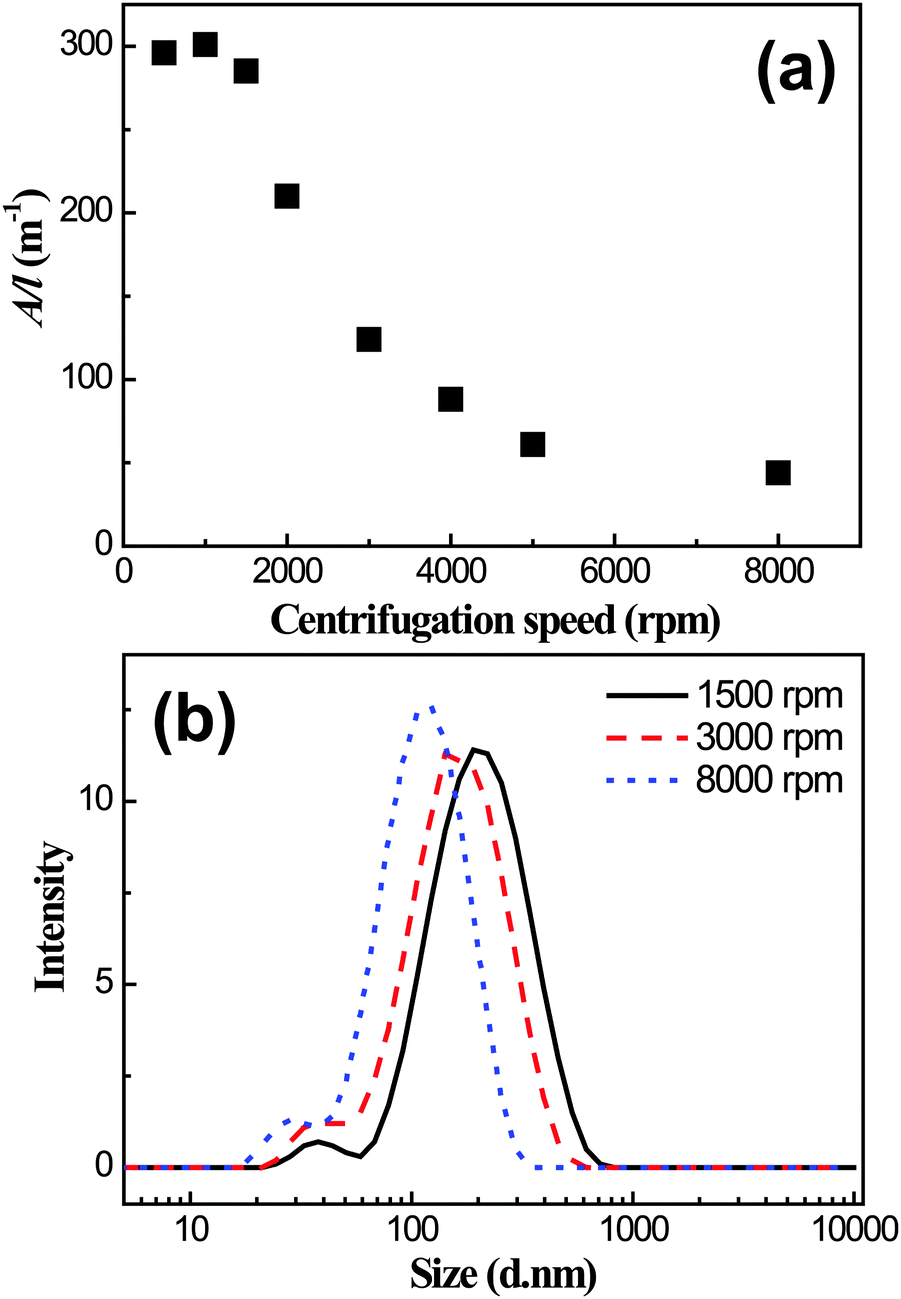 | ||
| Fig. 4 (a) The A/l values at 660 nm for the I3C–CI3-treated graphene dispersions after centrifugation at different speeds for 90 min. (b) Size distributions of the dispersed graphene flakes in solutions centrifuged at different speeds as measured by DLS. | ||
TEM measurements were performed to characterize the suspended species in the I3C–CI3-treated graphene dispersion, as shown in Fig. 5. For both the unstained sample and the negatively stained one, many two-dimensional objects with lateral dimensions of several hundred nanometers were observed, which correspond to graphene flakes. These findings clearly demonstrated the successful exfoliation of graphite into graphene. In addition to graphene flakes, considerable nanorods and nanofibrils were also observed to coexist, especially in the TEM image of the negatively stained sample (Fig. 5b, the image contrast for the nanorods and nanofibrils was significantly enhanced by negative staining). These nanorods and nanofibrils are most likely to be the self-assembled aggregates of I3C–CI3.43
 | ||
| Fig. 5 TEM images of (a) the unstained and (b) the negatively stained samples showing the suspended flake species in the I3C–CI3-treated graphene dispersion after centrifugation at 5000 rpm for 10 min. | ||
The notably high graphite exfoliation efficiency of I3C–CI3 is ascribed to the complexation between the I3C–CI3 molecules and aggregates and graphene sheets. It is reasonable that the I3C–CI3 molecules adsorb on the graphene surface in two ways, monomer adsorption and aggregate adsorption. The adsorbed I3C–CI3 species all expose the dissociated carboxyl groups outward, resulting in the formation of an electrostatic double-layer distribution and thereby stabilizing the graphene/I3C–CI3 composites against aggregation. Besides, being larger in size the adsorbed I3C–CI3 aggregates can introduce steric repulsion into the system, which will also help improve the graphene suspending capability.34 Our previous study has indicated that I3C–CI3 had relatively high self-assembling ability due to its specific molecular geometry as well as the high hydrophobicity and strong β-sheet forming propensity of the constituent Ile residue,43 which can account significantly for its higher graphite exfoliation efficiency relative to other peptide molecules.
The thickness and the number of layers per graphene flake could be estimated by edge counting (Fig. 6).16,26,48 The statistics showed that >90% of the graphene flakes had a thickness of less than 10 layers and the averaged thickness was 5–6 layers per flake (Fig. 6f). The thickness distribution indicated incomplete exfoliation; however, this was typical for graphene exfoliated using solvents or surfactants.18,23,26,34,48 Note that the sample used for the TEM measurements was treated at 5000 rpm for 10 min, a moderate centrifugation speed and time, and if larger speeds were applied, a lower thickness distribution for the graphene sheets could be obtained.38 Moreover, the presence of corrugation, scrolling, and agglomeration on the edge of graphene, which is typical for the mechanically stirred system, might also interfere with the thickness analysis. The results from tapping mode AFM measurements (Fig. 6e) also confirmed the presence of graphene flakes. Folding of the graphene flakes could be observed and the cross-section analysis indicated that most of the graphene flakes were below 8 nm. The I3C–CI3 rods and fibrils with a height of ∼5.0 nm could also be observed in the AFM image, some of which were bound to the graphene flakes. All these AFM findings were consistent with the TEM observation.
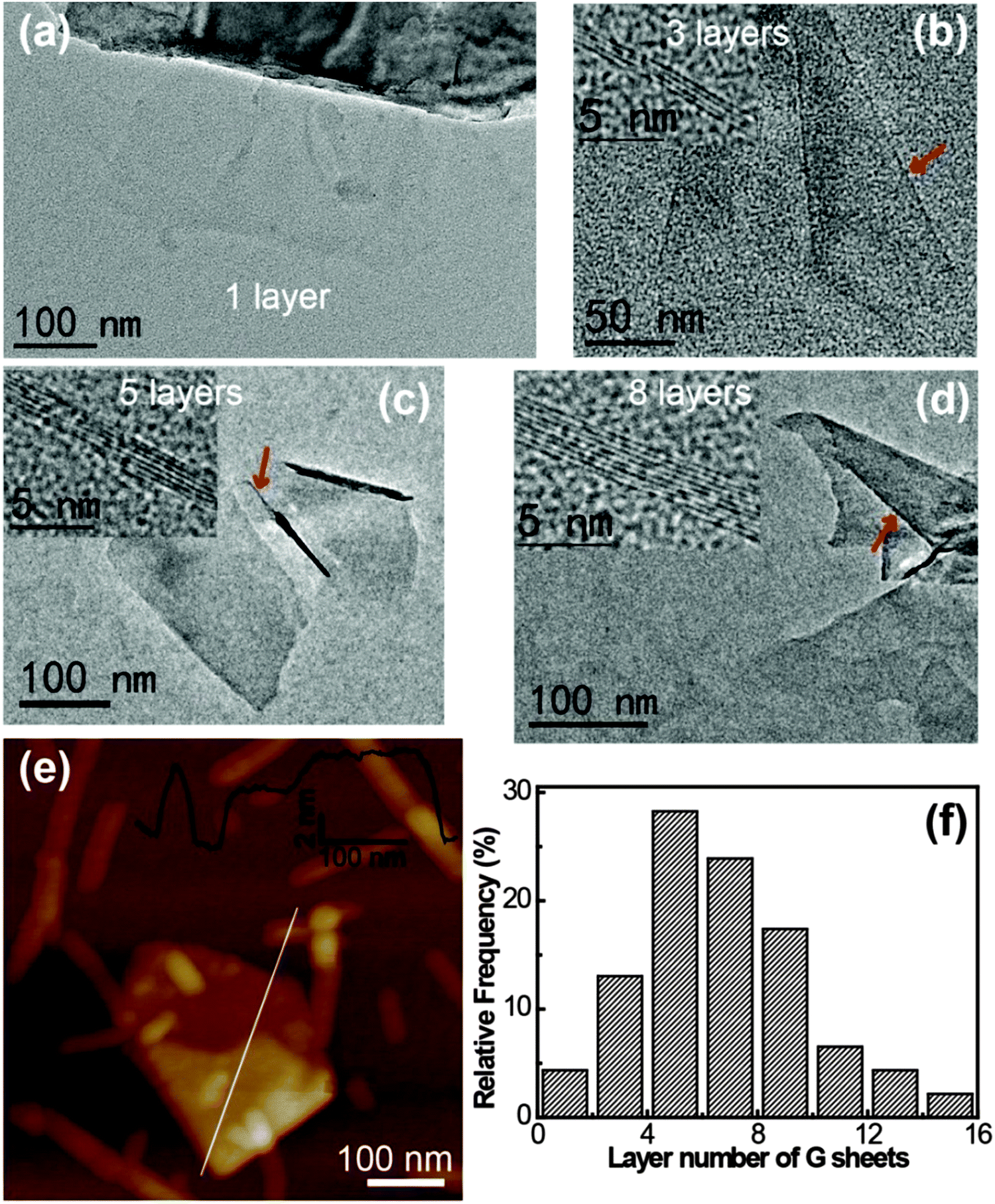 | ||
| Fig. 6 TEM images of (a) a single-layer graphene sheet, (b) a tri-layer graphene sheet, (c) a five-layer graphene sheet, and (d) an eight-layer graphene sheet. (e) AFM image of a graphene sheet by drop casting onto freshly cleaved mica. (f) Statistical analysis of flake size. Histograms showing flake thickness, expressed as the number of graphene monolayers per flake. | ||
Several other representative peptides, A9D, C16D, and Fmoc-FF that have relatively high graphite exfoliation efficiency, were also investigated by TEM and DLS to probe into the different dispersing properties of these molecules (Fig. S1, ESI†). The results showed that they could produce graphene sheets in the size range of 100–1000 nm. Interestingly, in the case of A9D, many plate-like aggregates could be found, which were believed to be A9D self-assembled structures. Some of these plates stacked onto graphene sheets. And in the case of Fmoc-FF, many tapes formed by Fmoc-FF could also be found to bind with graphene sheets. These results clearly show that complexation between peptide aggregates and graphene sheets is a characteristic feature of peptide-mediated graphite exfoliation.
Since the protonation state of carboxyl groups is pH-sensitive, we wondered whether the behavior of graphene/I3C–CI3 composites in the dispersion could be controlled by pH adjustment. The pH values of the graphene/I3C–CI3 dispersions were adjusted between 2 and 10 using 1.0 M HCl or NaOH solutions. After 30 min of incubation, the photographs of the dispersions at different pH values were taken and the absorbance of the supernatants at 660 nm was measured as shown in Fig. 7a and b, respectively. Interestingly, the graphene/I3C–CI3 species in the dispersions showed a pH-dependent dispersibility. The dispersions were homogeneous and transparent with an absorbance of approximately 0.8 when the pH value was 5 and above, indicating that the graphene/I3C–CI3 composites were stably suspended in the dispersions. And at these pHs the composite size was around 350 ± 200 nm from the DLS results (Fig. S2A, ESI†). As the pH value dropped below 5, substantial precipitation was observed at the bottom of the dispersions, and absorbance of the supernatants decreased significantly to 0.1 and below, indicating a poor dispersibility. The pH-responsiveness of the dispersions was further assessed by ζ potential monitoring, a method widely employed to quantify electrostatic repulsions and colloidal stability.56,57 As graphene sheets themselves are nonpolar, a ζ value close to zero is expected; however, when coated with peptide I3C–CI3, the graphene–peptide composites are expected to have a nonzero ζ value due to the ionization of the –COOH groups. Fig. 7c shows variation of the ζ potential with pH. The ζ potential of the dispersion gave a transition point at a pH of around 5. At a pH of more than 5, the ζ potential averaged at approximately −42 mV, a value that can support a stable suspension, whilst at a pH of less than 5, the absolute ζ potential values were in the range of −30 to +10 mV, values that are accepted to be unable to support a stable colloidal dispersion.57 In a previous study by Skaltsas et al., a pH dependent dispersion of the graphene/block copolymer system in NMP/H2O mixed solution has been reported.27
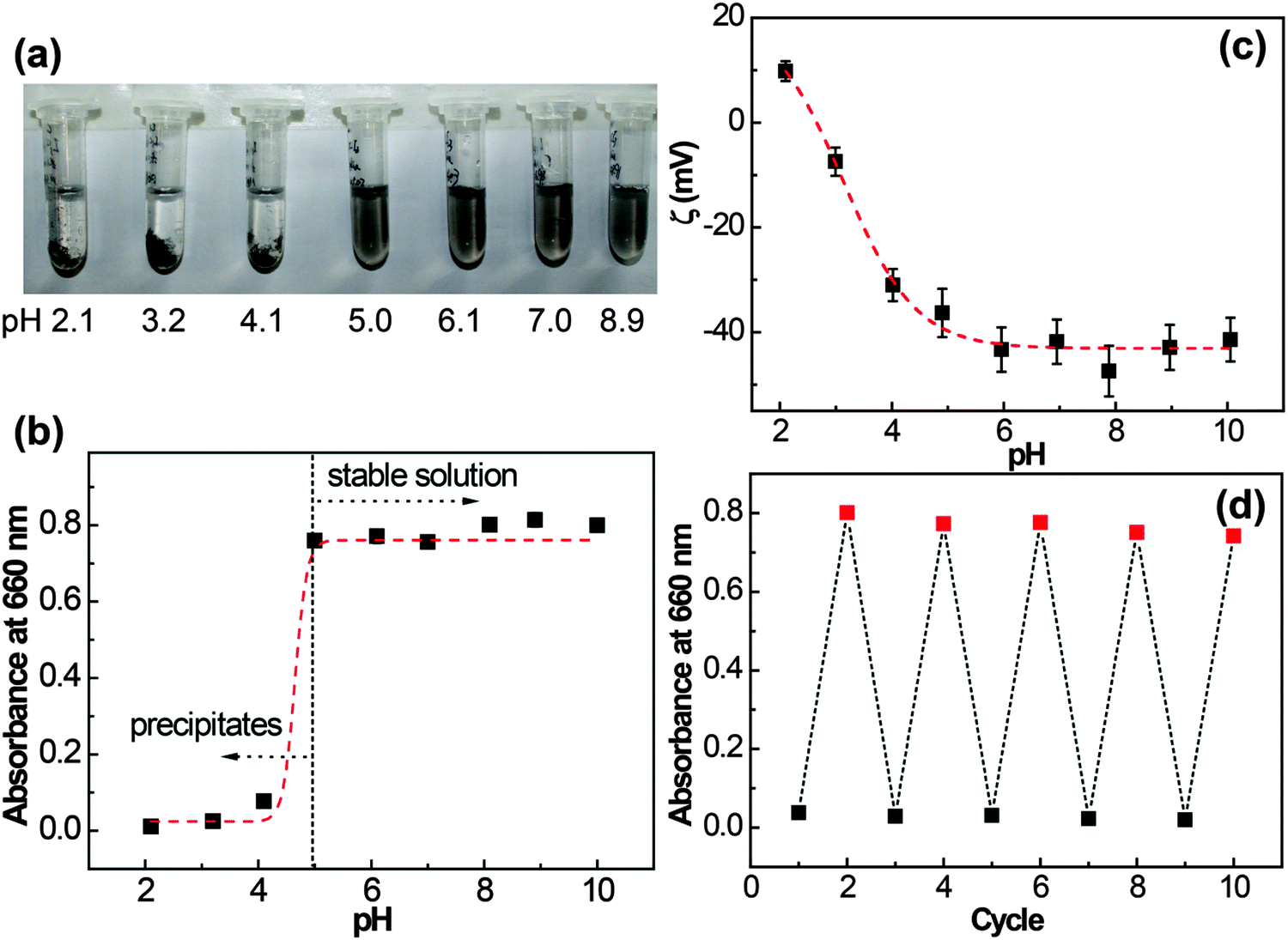 | ||
| Fig. 7 The I3C–CI3-treated graphene dispersion was first centrifuged at 5000 rpm for 10 min, diluted 10 times (to aid visualization by eye) and then the pH was adjusted to indicated values. The resulting dispersions were used to obtain (a) photographs of the graphene dispersions at different pH values, (b) absorbance of their upper supernatants, (c) ζ potential values of the dispersions at different pH values and (d) absorbance of a dispersion by alternatively adjusting pH between 3 (black solid square) and 10 (red solid square). | ||
Moreover, the dispersion/aggregation of the graphene/I3C–CI3 composites could be repeatedly switched by reversibly changing the pH between basic (pH 3) and acidic (pH 10) conditions, as shown by the associated changes in absorbance at 660 nm (Fig. 7d) and the DLS results for the dispersions at pH 10 (Fig. S2B, ESI†). At higher pH, the –COOH groups of I3C–CI3 were deprotonated and the graphene/I3C–CI3 composites were stably dispersed due to inter-sheet electrostatic and steric repulsions. With a reduction in pH, the –COOH groups became protonated and the inter-sheet electrostatic repulsion was minimized, while intermolecular association (hydrogen bonding and/or hydrophobic interactions) became dominant, so the graphene/I3C–CI3 composites became aggregated. Therefore, the protonation/deprotonation of I3C–CI3 molecules can be viewed as an engine for the dispersion/aggregation of the graphene/I3C–CI3 composites. The pH-responsiveness is a great advantage of peptide molecules in comparison with traditional surfactants, which can be used to conduct the reversible manipulation of graphene dispersibility with pH as a stimulus.
Suspension of graphene in water in large quantities facilitates processing the graphene dispersions into macroscopic materials that are required in many cases. Here, we demonstrated the preparation of free-standing, paper-like graphene/I3C–CI3 composite films by simple filtration of the dispersions. The films could be handled without breaking (Fig. 8a), indicating their high mechanical strength and flexibility. The SEM characterization showed the detailed film morphology (Fig. 8b). The graphene flakes were generally homogeneous in size and they assembled into a close-packed, overlapped arrangement. From the highly magnified image shown in the inset of Fig. 8b, the graphene flakes showed a bumped rough surface, which is likely caused by the surface-bound I3C–CI3 nanoaggregates.
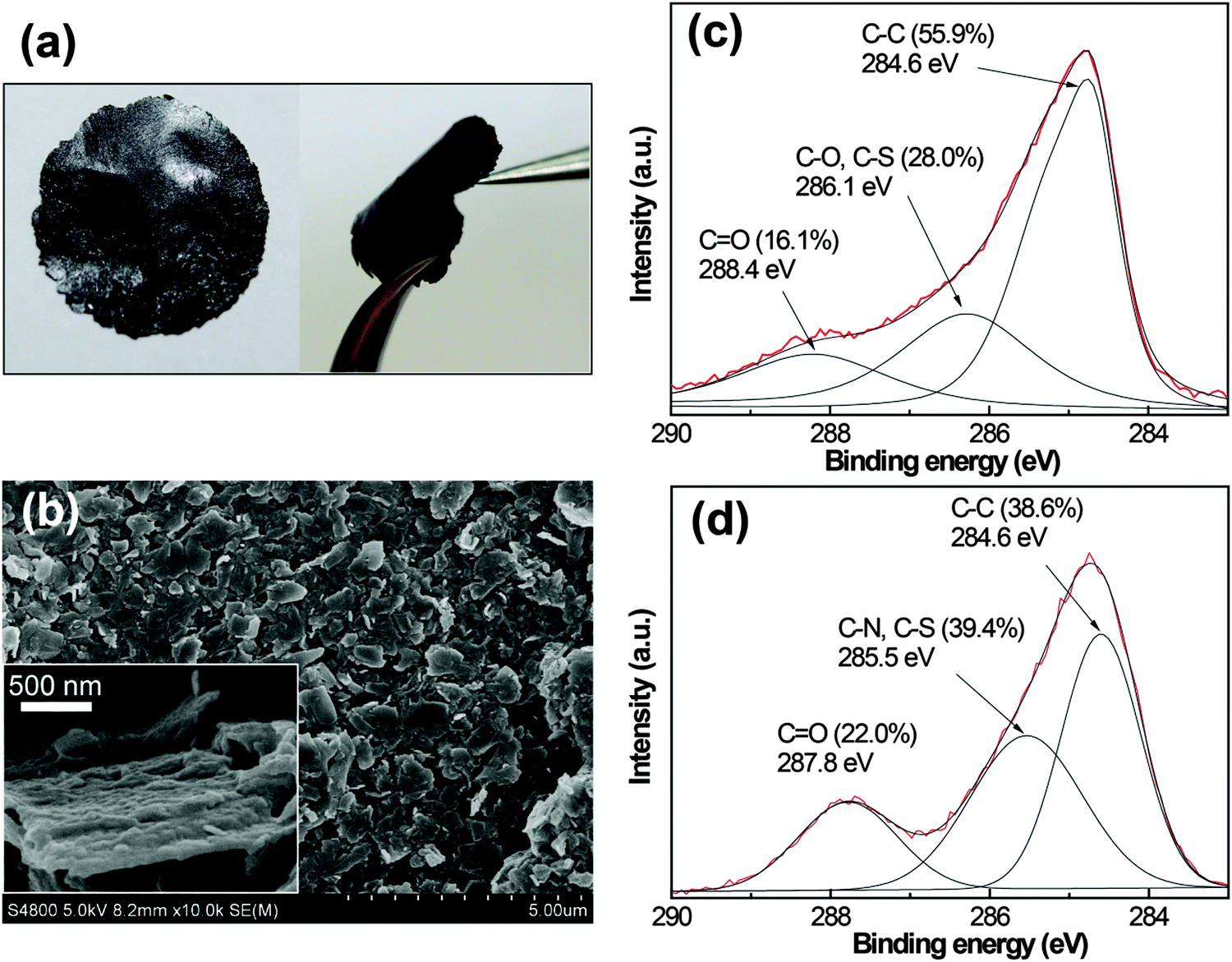 | ||
| Fig. 8 (a) The photographs and (b) the SEM images (inset, a magnified image) of the graphene/I3C–CI3 composite film prepared by simple filtration of a dispersion through a nylon filter membrane of 0.22 μm pore size. Note that the dispersion was obtained by centrifugation at 5000 rpm for 10 min. XPS profiles (C1s spectra) of (c) the graphene/I3C–CI3 composite and (d) the I3C–CI3 powder. | ||
XPS is a powerful tool to detect the levels of C for carbon-based materials. It has been shown that the graphene exfoliated from pristine graphite contained very few defects and the C1s spectra exhibited a main band centered at 284.6 eV associated with the graphitic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C component.16,35 For the C1s spectra of the graphene/I3C–CI3 composite, however, the signal in the binding energy range of 285–290 eV was significant (Fig. 8c). The spectra can be deconvoluted into three peaks centered at 284.6, 286.1, and 288.4 eV, respectively, and the latter two are most likely to be related to the oxygenated functionalities. By comparing with the C1s spectra of I3C–CI3 itself (Fig. 8d), the peak at ∼286 eV was ascribed to C–O, C–S, and C–N species while the peak at ∼288 eV was ascribed to CO species, respectively. These signals are mainly from the I3C–CI3 molecules, though very little might come from the carboxyl and epoxy groups of graphene defects. Obviously, a high amount of I3C–CI3 was integrated in the composite film, consistent with the SEM observation. Furthermore, the weight ratio of I3C–CI3 in the composite film was assessed by thermogravimetric analysis (Fig. S3, ESI†) to be about 53%.
C component.16,35 For the C1s spectra of the graphene/I3C–CI3 composite, however, the signal in the binding energy range of 285–290 eV was significant (Fig. 8c). The spectra can be deconvoluted into three peaks centered at 284.6, 286.1, and 288.4 eV, respectively, and the latter two are most likely to be related to the oxygenated functionalities. By comparing with the C1s spectra of I3C–CI3 itself (Fig. 8d), the peak at ∼286 eV was ascribed to C–O, C–S, and C–N species while the peak at ∼288 eV was ascribed to CO species, respectively. These signals are mainly from the I3C–CI3 molecules, though very little might come from the carboxyl and epoxy groups of graphene defects. Obviously, a high amount of I3C–CI3 was integrated in the composite film, consistent with the SEM observation. Furthermore, the weight ratio of I3C–CI3 in the composite film was assessed by thermogravimetric analysis (Fig. S3, ESI†) to be about 53%.
4 Conclusions
The large-scale direct exfoliation of graphite into few-layered (<10 layers) graphene flakes in aqueous dispersions was successfully realized with the help of peptide molecules. The exfoliating efficiency of different molecules was evaluated. Charge was found to be important in determining the exfoliating efficiency and the anionic peptide molecules were found to be more favorable than the cationic ones leading to relatively high efficiency. Specifically, peptide I3C–CI3 exhibited the highest efficiency possibly due to its specific physicochemical properties and interactions with graphene sheets. Interestingly, the resulting graphene/I3C–CI3 composites showed a reversible pH-dependent suspension/aggregation. Such characteristics related closely to the intrinsic nature of peptide molecules and are rarely found in the graphene dispersions exfoliated by traditional surfactants.34–37 Moreover, the graphene/peptide dispersion could be used to conveniently fabricate macroscopic materials with different compositions and nanostructures.The study expanded the library of available agents for direct graphite exfoliation to produce graphene. Because peptide molecules usually have high biocompatibility, employing peptide molecules as graphene exfoliating and stabilizing agents avoids the use of toxic reagents and allows the scalable graphene production for biocompatible materials.37 The second feature of peptide molecules is their stimuli-responsiveness, and the strategy of using this feature to control the behaviors of graphene in solutions may open a window for broadening the applications of graphene,58 for example, to construct stimuli-responsive graphene-based chemical/biological sensors. Thirdly, some peptide molecules are capable of self-assembling into distinct nanostructures such as fibrils, tubes, ribbons, and vesicles,59 which provides good opportunities for the integration of specific functionalities of peptide nanostructures and graphene sheets (e.g. the three-dimensional network of the peptide fibrils and the mechanical reinforcement of graphene), and allows for the fabrication of novel functional composite materials.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21473255, 21003160) and the Fundamental Research Funds for the Central Universities (14CX02055A). We also thank the funding support from UK Engineering and Physical Sciences Research Council (EPSRC) and Innovative UK under KTP008143 and the Graphene Grand Challenges by the University of Manchester.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- H. Y. Mao, S. Laurent, W. Chen, O. Akhavan, M. Imani, A. A. Ashkarran and M. Mahmoudi, Chem. Rev., 2013, 113, 3407–3424 CrossRef CAS PubMed.
- H.-P. Cong, J.-F. Chen and S.-H. Yu, Chem. Soc. Rev., 2014, 43, 7295–7325 RSC.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- P. W. Sutter, J.-I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406–411 CrossRef CAS PubMed.
- J. Coraux, A. T. N'Diaye, C. Busse and T. Michely, Nano Lett., 2008, 8, 565–570 CrossRef CAS PubMed.
- W. Zhang, J. Cui, C.-A. Tao, Y. Wu, Z. Li, L. Ma, Y. Wen and G. Li, Angew. Chem., 2009, 121, 5978–5982 CrossRef.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155–3162 CrossRef CAS PubMed.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- C. E. Hamilton, J. R. Lomeda, Z. Sun, J. M. Tour and A. R. Barron, Nano Lett., 2009, 9, 3460–3462 CrossRef CAS PubMed.
- X. Wang, P. F. Fulvio, G. A. Baker, G. M. Veith, R. R. Unocic, S. M. Mahurin, M. Chi and S. Dai, Chem. Commun., 2010, 46, 4487–4489 RSC.
- A. Ciesielski and P. Samori, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- W. Du, X. Jiang and L. Zhu, J. Mater. Chem. A, 2013, 1, 10592–10606 CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- U. Khan, H. Porwal, A. O'Neill, K. Nawaz, P. May and J. N. Coleman, Langmuir, 2011, 27, 9077–9082 CrossRef CAS PubMed.
- T. Skaltsas, N. Karousis, H. Yan, C. Wang, S. Pispas and N. Tagmatarchis, J. Mater. Chem., 2012, 22, 21507–21512 RSC.
- A. O'Neill, U. Khan, P. N. Nirmalraj, J. Boland and J. N. Coleman, J. Phys. Chem. C, 2011, 115, 5422–5428 Search PubMed.
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis, A. K. Stubos and C. Trapalis, Solid State Commun., 2009, 149, 2172–2176 CrossRef CAS.
- T. Skaltsas, S. Pispas and N. Tagmatarchis, Chem. – Eur. J., 2013, 19, 9286–9290 CrossRef CAS PubMed.
- T. Skaltsas, N. Karousis, S. Pispas and N. Tagmatarchis, Nanotechnology, 2014, 25, 445404 CrossRef CAS PubMed.
- R. J. Smith, M. Lotya and J. N. Coleman, New J. Phys., 2010, 12, 125008 CrossRef.
- L. Guardia, M. J. Fernández-Merino, J. I. Paredes, P. Solís-Fernández, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 2011, 49, 1653–1662 CrossRef CAS.
- S. Vadukumpully, J. Paul and S. Valiyaveettil, Carbon, 2009, 47, 3288–3294 CrossRef CAS.
- X. An, T. Simmons, R. Shah, C. Wolfe, K. M. Lewis, M. Washington, S. K. Nayak, S. Talapatra and S. Kar, Nano Lett., 2010, 10, 4295–4301 CrossRef CAS PubMed.
- A. A. Green and M. C. Hersam, Nano Lett., 2009, 9, 4031–4036 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- H. Xu, J. Wang, S. Han, J. Wang, D. Yu, H. Zhang, D. Xia, X. Zhao, T. A. Waigh and J. R. Lu, Langmuir, 2008, 25, 4115–4123 CrossRef.
- D. H. Jia, K. Tao, J. Q. Wang, C. D. Wang, X. B. Zhao, M. Yaseen, H. Xu, G. H. Que, J. R. P. Webster and J. R. Lu, Soft Matter, 2011, 7, 1777–1788 RSC.
- M. Cao, C. Cao, P. Zhou, N. Wang, D. Wang, J. Wang, D. Xia and H. Xu, Colloids Surf., A, 2015, 469, 263–270 CrossRef CAS.
- Y. Zhu, D. K. James and J. M. Tour, Adv. Mater., 2012, 24, 4924–4955 CrossRef CAS PubMed.
- T. Kim, H. Lee, J. Kim and K. S. Suh, ACS Nano, 2010, 4, 1612–1618 CrossRef CAS PubMed.
- A. S. Douglas and M. Donald, Principles of Instrumental Analysis, Brooks/Cole, Thompson Learning, New York, 1998 Search PubMed.
- J. N. Coleman, Acc. Chem. Res., 2012, 46, 14–22 CrossRef PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- C. Casiraghi, A. Hartschuh, H. Qian, S. Piscanec, C. Georgi, A. Fasoli, K. S. Novoselov, D. M. Basko and A. C. Ferrari, Nano Lett., 2009, 9, 1433–1441 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- S. Han, S. Cao, Y. Wang, J. Wang, D. Xia, H. Xu, X. Zhao and J. R. Lu, Chem. – Eur. J., 2011, 17, 13095–13102 CrossRef CAS PubMed.
- S. Mecozzi, A. P. West and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10566–10571 CrossRef CAS.
- N. S. Scrutton and A. R. C. Raine, Biochem. J., 1996, 319, 1–8 CrossRef CAS PubMed.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed.
- V. Nicolosi, H. Cathcart, A. R. Dalton, D. Aherne, G. R. Dieckmann and J. N. Coleman, Biomacromolecules, 2008, 9, 598–602 CrossRef CAS PubMed.
- R. W. O'Brien, B. R. Midmore, A. Lamb and R. J. Hunter, Faraday Discuss. Chem. Soc., 1990, 90, 301–312 RSC.
- Z. Sun, X. Huang, F. Liu, X. Yang, C. Rosler, R. A. Fischer, M. Muhler and W. Schuhmann, Chem. Commun., 2014, 50, 10382–10385 RSC.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of graphene dispersions treated by other peptides, more DLS results, and TGA results of the composite film. See DOI: 10.1039/c5tb02065d |
| This journal is © The Royal Society of Chemistry 2016 |