
Nano-cuprous oxide catalyzed one-pot synthesis of a carbazole-based STAT3 inhibitor: a facile approach via intramolecular C–N bond formation reactions†
C. P. Baburajeeva,
Chakrabhavi Dhananjaya Mohanb,
Govindagouda S. Patilc,
Shobith Rangappad,
Vijay Pandeye,
Anusha Sebastiana,
Julian E. Fuchsf,
Andreas Benderf,
Peter E. Lobiee,
Basappa*a and
Kanchugarakoppal S. Rangappa*b
aLaboratory of Chemical Biology, Department of Chemistry, Bangalore University, Central College Campus, Palace Road, Bangalore 560001, India. E-mail: salundibasappa@gmail.com
bDepartment of Studies in Chemistry, University of Mysore, Manasagangotri, Mysore 570006, India. E-mail: rangappaks@yahoo.com
cBal pharma Ltd, No 61-B, Bommasandra, Anekal Taluk, Bangalore 560099, India
dFrontier Research Center for Post-Genome Science and Technology, Hokkaido University, Sapporo 060-0808, Japan
eCancer Science Institute of Singapore, National University of Singapore, Singapore 117599
fCentre for Molecular Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK
First published on 29th March 2016
Abstract
In this study, we report the one-pot synthesis of substituted carbazole derivatives using nano cuprous oxide as a catalyst via intramolecular C–N bond forming reactions. Among the synthesized carbazoles, 3′-((3-acetyl-6-chloro-9H-carbazol-9-yl)methyl)-[1,1′-biphenyl]-2-carbonitrile (ACB) was identified as a lead antiproliferative agent against lung cancer cell lines A549 and LLC with an IC50 of 13.6 and 16.4 μM respectively. Furthermore, we found that the lead compound suppresses the constitutive phosphorylation of STAT3 (Tyr-705) in A549, HCC-2279 and H1975 cells. We analyzed the levels of phospho-STAT3 and LSD1 in the nuclear extract of ACB treated HCC-2279 cells to evaluate the transcriptional activity of STAT3. We found the downregulation of phospho-STAT3 without any change in the expression of LSD1 indicating that ACB downregulates the transcriptional activity of STAT3. Molecular docking analysis revealed that ACB makes a favorable interaction with Arg-609 and Ser-613 in the pTyr site of the SH2 domain of STAT3.
Introduction
Signal transducer and activator of transcription (STAT) proteins were originally identified as inducible transcription factors in the context of cytokine signaling two decades ago.1,2 STAT1, 2, 3, 4, 5a, 5b and 6 are the seven members of the STAT family and STAT3 is being persistently activated in several types of cancers.3,4 In an unstimulated cell, STAT3 remains as a monomer in the cytoplasm and the stimulation of upstream receptors with various ligands including cytokines and growth factors leads to the phosphorylation of STAT3 (Y705) following dimerization, and translocation into nucleus to transcribe STAT3 target genes.5,6 Various proliferative, angiogenic, survival, pro-inflammatory, antiapoptotic, metastatic proteins including, cyclin D1, HIF-1α, VEGF, Bcl-2, MMPs, COX-2 and RANTES are regulated by JAK-STAT3 signaling contributing to tumorigenesis.7–9 Studies have revealed that, blockade of STAT3 signaling by natural/synthetic small molecules and RNA interference significantly decrease the cell survival and antiapoptosis of cancer cells.10,11Carbazole was initially isolated in 1872 by Graebe and Glazer.12 The natural occurrence and medicinal properties of carbazole was demonstrated after the discovery of murrayanine, a carbazole derived alkaloid from Murraya koenigii Spreng in 1964.13,14 Since then several natural sources have been identified for the presence of carbazole derivatives including blue-green algae, Aspergillus species and Actinomadura species. Furthermore, carbazoles have been shown to induce their anticancer effect in various cancer models by inhibiting wide array of targets.15–19 BC3EE2,9B and KPYB10602 are the synthetic carbazole derivatives which have been reported to possess good antioncogenic property in several types of cancer cells. BC3EE2,9B was reported to upregulate autophagy and synergistically sensitizes human glioblastoma cells to temozolomide.20 KPYB10602, a lactam-fused carbazole derivative and inhibitor of kinesin spindle protein, induced anticancer activity in human ovarian cancer cells and in vivo tumor model.21 Furthermore, various novel carbazole derivatives were reported to downregulate the phosphorylation of STAT3 (ref. 22 and 23) and effectively inhibiting the in vivo growth of human TNBC and lung tumor xenografts.24 Therefore, carbazole has been identified as a privileged scaffold in medicinal chemistry for the development of therapeutics against cancer.
Several research groups have reported the synthesis of carbazoles using various synthetic strategies.25–27 The development of mild and efficient protocol for the preparation of carbazoles remains as a challenge for researchers in synthetic organic chemistry. In continuation of our effort to develop synthetic small molecules and explore their pharmacological properties,28–32 herein, we report the one-pot synthesis of substituted carbazole derivatives using nano cuprous oxide as a catalyst via intramolecular C–N bond forming reactions. The advantage of this method lies in the fact that N-alkylation and C–N bond formation takes simultaneously and eventually resulted in one-pot synthesis of title compounds. In addition, we identified 3′-((3-acetyl-6-chloro-9H-carbazol-9-yl)methyl)-[1,1′-biphenyl]-2-carbonitrile (ACB) as lead cytotoxic agent against lung cancer cells. Further, ACB was demonstrated to induce cytotoxicity in cancer cells by modulating the transcriptional activity of STAT3 and molecular docking analysis revealed that ACB makes favorable interaction with Arg-609 and Ser-613 in pTyr site of SH2 domain of STAT3.
Experimental section
Materials and method
All chemicals used were purchased from Sigma-Aldrich. 1H and 13C NMR spectra were recorded on a Bruker WH-200 (400 MHz) spectrometer in CDCl3 or DMSO-d6 as solvent, using TMS as an internal standard and chemical shifts are expressed as ppm. High resolution mass spectra were determined on a WATERS Q-TOF Premier-HAB213 instrument, mass spectra were determined on a Agilent LC-MS and the elemental analyses were carried out using an Elemental Vario Cube CHNS rapid Analyzer. The progress of the reaction was monitored by TLC pre-coated silica gel G plates. Lung cancer cell lines were cultured as described previously.33 pSTAT3 (Y705), STAT3, β-actin antibodies and LSD1 (sc-271720) were purchased from Santa Cruz, USA.Typical procedure for the synthesis of 1-(2′-amino-5′,6-dichloro-[1,1′-biphenyl]-3-yl)ethanone (1a)
To a solution of 1-(3-bromo-4-chlorophenyl)ethanone (700 mg, 0.3 mmol) in 10 ml of distilled water in a sealed tube, (2-(2-amino-5-chlorophenyl)-4,5,5-trimethyl-1,3,2-dioxaborolan-4-yl)methylium (0.32 mmol), SCS-Bi2O3 (0.24 mmol), TBAB (0.15 mmol) and [1,1′bis(diphenylphosphino)ferrocene]dichloropalladium(II) (0.009 mmol) was added and the reaction mixture was heated to 110 °C for the required time. The reaction was cooled to room temperature and filtered to remove the base and catalyst. The product was extracted from the water layer by thrice in ethyl acetate (5 ml), dried with magnesium sulfate, filtered, and concentrated in vacuum. The crude product was purified by recrystallization using ethanol. The yield of the obtained product (1a) was 85% (714 mg).Typical procedure for the synthesis of 2′,5-dichloro-5′-ethyl-[1,1′-biphenyl]-2-amine (1b)
To a mixture of (1a) (500 mg, 3 mmol) in tetrahydrofuran (5 ml), sodiumborohydride (NaBH4) (6 mmol), boron trifluoride diethyletherate (BF3 etherate) (6 mmol) was added at 10–15 °C. The reaction mass was stirred at room temperature for 10–12 h and completion of the reaction was monitored by TLC. 1 ml of dil. suphuric acid was added and the pH is adjusted to 7.5 using 50% sodium hydroxide solution. The product (1b) is extracted with dichloromethane, dried and concentrated. The yield of the obtained product (1b) was 89% (426 mg).Typical procedure for the synthesis of 1-(2′-amino-5′,6-dichloro-[1,1′-biphenyl]-3-yl)ethanol (1c)
To a solution of 1a (500 mg, 3 mmol) in ethanol (10 ml), 14 N sodium hydroxide (5 ml) NaBH4 (6 mmol) was added lot-wise at 10–15 °C. The resulting mixture was stirred at room temperature for 10–12 h and completion of the reaction was monitored by TLC. Upon the completion of reaction, excess NaBH4 was removed by dil. hydrochloric acid and the solvent was concentrated under reduced pressure. The residue was diluted with water (8 ml) then extracted with dichloromethane, dried and concentrated to obtain solid product (1c) with the yield of 90% (453 mg).General procedure for copper-catalyzed intramolecular C–N bond-formation reaction
A reaction flask was charged with substrate 1a–c (1 mmol), 2a–e (1 mmol), DMEDA (0.1 mmol), Cu2O (8 mol%), and K2CO3 (3 mmol) followed by reagent grade DMF (5 ml). The reaction mixture was heated to 150 °C for 15 h. After completion of the reaction, mixture is cooled to room temperature, quenched with water and diluted with ethyl acetate (10 ml) and the aqueous layer was extracted with ethyl acetate (3 ml). The combined organic layer was washed with water (5 ml), dried over anhydrous sodium sulfate and the solvent was removed in vacuum. The crude product was purified using silica gel column chromatography. All the new compounds exhibited spectral properties consistent with the assigned structures and were fully characterized by their spectroscopic data (1H, IR, mass, elemental and 13C NMR analysis).Pharmacology
MTT assay
The antiproliferative effect of the compounds synthesized against A549 and LLC cells was determined by the MTT dye uptake method as described previously.34,35 Briefly, cancer cells (2.5 × 104 ml−1) were incubated in triplicate in a 96-well plate, in the presence of varying compound concentrations at a volume of 0.2 ml, for different time intervals at 37 °C. Thereafter, 20 μl MTT solution (5 mg ml−1 in PBS) was added to each well. After 2 h incubation at 37 °C, a 0.1 ml lysis buffer (20% SDS, 50% dimethylformamide) was added; incubation was performed for 1 h at 37 °C, and the optical density (OD) at 570 nm was measured by plate reader.Western blotting
Western blotting analysis was performed as described earlier.36,37 For detection of phospho-proteins, ACB treated whole-cell extracts were lysed in lysis buffer (20 mM Tris (pH 7.4), 250 mM NaCl, 2 mM EDTA (pH 8.0), 0.1% Triton X-100, 0.01 mg ml−1 aprotinin, 0.005 mg ml−1 leupeptin, 0.4 mM PMSF, and 4 mM NaVO4). Lysates were then spun at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min to remove insoluble material and resolved on SDS-gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with 5% non-fat milk, and probed with anti-phospho-STAT3/STAT3 antibodies (1:1000) overnight at 4 °C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence.
000 rpm for 10 min to remove insoluble material and resolved on SDS-gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with 5% non-fat milk, and probed with anti-phospho-STAT3/STAT3 antibodies (1:1000) overnight at 4 °C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence.
Molecular docking analysis
We followed a dual strategy to analyze structure–activity relationships (SAR) of the 15 carbazole compounds. We performed a ligand-based analysis of SAR trends for carbazole core substituents (R groups) as well as N-substitutions (R1 groups) inspired by matched molecular pair analysis.38 We extracted matching substituents and identified enriched chemical groups amongst compounds of higher activity.On the other hand, we attempted to rationalize the SAR trends by docking the carbazole compounds to the SH2 domain of STAT3. Therefore, we extracted the SH2 domain from the crystal structure of the STAT3 homo-dimer (PDB: 1BG1)39 and prepared the structure for docking using protonate 3D as implemented in Molecular Operating Environment (MOE, version 2014.0901).40 Since the pTyr binding site has been described as a hot-spot for small molecule binding,41 we chose the pTyr as center for in silico docking experiments. We deleted bound protein residues in that region and docked the carbazole series after establishing successful redocking of pTyr with an RMSD < 2 Å using MOE's default settings. Resulting docking poses were visualized using PyMOL (Schrodinger LLC, version 1.6.0.0).
Results and discussion
Chemistry
We achieved the synthesis of substrate 1-(2′-amino-5′,6-dichloro-[1,1′-biphenyl]-3-yl)ethanone (1a), by the Suzuki coupling reaction as recently developed in our laboratory from commercially available (2-(2-amino-5-chlorophenyl)-4,5,5-trimethyl-1,3,2-dioxaborolan-4-yl)methylium and 1-(3-bromo-4-chlorophenyl) ethanone.42 The resulting product (1a) was readily reduced to corresponding 2′,5-dichloro-5′-ethyl-[1,1′-biphenyl]-2-amine (1b) and 1-(2′-amino-5′,6-dichloro-[1,1′-biphenyl]-3-yl)ethanol (1c) as depicted in Scheme 1(i–iii). | ||
| Scheme 1 Schematic representation for the preparation of title compounds. (i) SCS-Bi2O3, Pd(pdppf)Cl2, water, 110 °C, TBAB; (ii) NaBH4/BF3 ethyl etherate, THF, 12 h; (iii) NaBH4/NaOH, ethanol, reflux, 10 h; (iv) R1Br (2a–e), K2CO3/nano Cu2O (8 mol%)/DMEDA, DMF/150 °C, 15 h; R = COCH3, CH2CH3, CHOHCH3; R1 = benzyl, 2,6-dichlorobenzyl, 4-iodobenzyl, bromo ethane, 4-bromomethyl-2-cyanobiphenyl. | ||
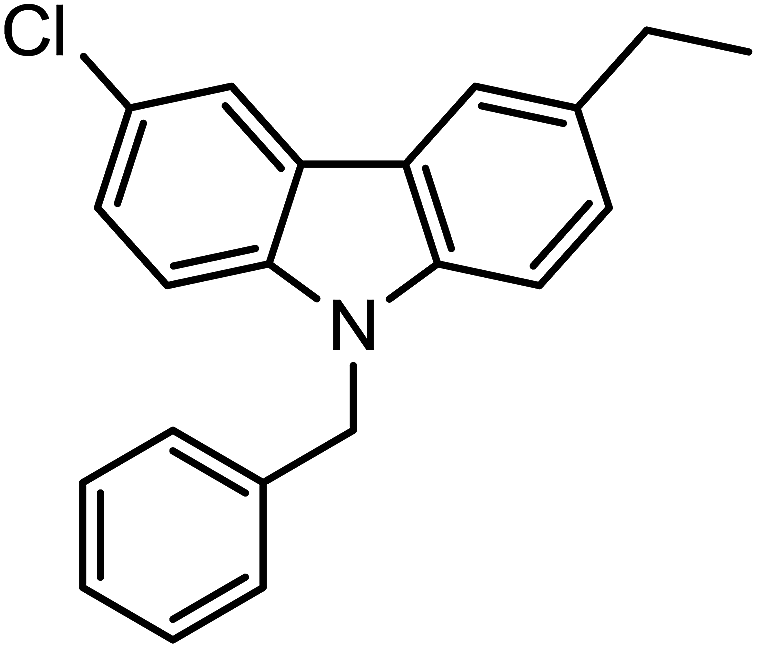
In the next step, we developed an efficient protocol for the direct one-pot synthesis of N-substituted carbazoles via N-alkylation followed by C–N bond forming coupling reaction using Cu catalyst in the presence of various ligands. The conversion of substrate 1-(2′-amino-5′,6-dichloro-[1,1′-biphenyl]-3-yl)ethanone (1a) to the corresponding N-substituted carbazole derivative 1-(9-benzyl-6-chloro-9H-carbazol-3-yl)ethanone (3a) was carried out by treating 1a with benzylbromide (2a), Cu catalyst and a base (Scheme 2 and Table 1). The preliminary analysis suggested that the use of combination of 8 mol% Cu2O and 10 mol% N,N′-dimethylethane-1,2-diamine (DMEDA) in presence of K2CO3 at 150 °C in DMF resulted in efficient conversion to 3a. We observed the incomplete conversion when 5 mol% Cu2O was used despite of longer reaction time and high temperature. Interestingly, it was found that nano Cu2O accelerated the reaction with lesser reaction time and better yield (Table 1, entry 12) which can be attributed to availability of large surface area for reaction.
 | ||
| Scheme 2 Schematic representation of synthesis of 1,9-substituted-6-chlorocarbazoles via N-alkylation followed by C–N bond forming coupling reaction using Cu catalyst in presence of ligand. | ||
| Entry | 8 mol% catalyst | Ligand | Base | Temp/time | Yielda |
|---|---|---|---|---|---|
| a The reaction was carried out in DMF solvent. | |||||
| 1 | CuO | DMEDA | KO′Bu | 150 °C/20 h | 31% |
| 2 | Cu(OAc)2 | DMEDA | KO′Bu | 150 °C/20 h | NR |
| 3 | CuI | DMEDA | KO′Bu | 150 °C/20 h | NR |
| 4 | Cu2O | DMEDA | KO′Bu | 150 °C/20 h | 47% |
| 5 | Nano CuO | DMEDA | KO′Bu | 150 °C/20 h | 42% |
| 6 | Nano Cu2O | DMEDA | KO′Bu | 150 °C/20 h | 56% |
| 7 | CuO | DMEDA | K2CO3 | 150 °C/20 h | 32% |
| 8 | Cu(OAc)2 | DMEDA | K2CO3 | 150 °C/20 h | NR |
| 9 | CuI | DMEDA | K2CO3 | 150 °C/20 h | NR |
| 10 | Cu2O | DMEDA | K2CO3 | 150 °C/20 h | 55% |
| 11 | Nano CuO | DMEDA | K2CO3 | 150 °C/20 h | 43% |
| 12 | Nano Cu2O | DMEDA | K2CO3 | 150 °C/15 h | 79% |
| 13 | Nano Cu2O | — | K2CO3 | 150 °C/15 h | NR |
| 14 | Nano Cu2O | 1,10-Phenanthroline | K2CO3 | 150 °C/15 h | 79% |
| 15 | Nano Cu2O | Xantphos | K2CO3 | 150 °C/15 h | 74% |
| 16 | Nano Cu2O | Ethylenediamine | K2CO3 | 150 °C/15 h | 71% |
We observed no reaction in the absence of the ligand (Table 1, entry 13) whereas, the use of bidentate ligands such as 1,10-phenanthroline, xantphos and ethylene diamine resulted in the good yield of the product (Table 1, entry 14–16). These results encouraged us to examine the generality of one-pot method for the synthesis of series of novel N-substituted carbazoles (Scheme 1(iv)). This protocol was applied successfully on carbonyl (1a), alkane (1b) and hydroxyl (1c) substituents with reasonable yields as shown in Table 2.
| Entry | Amines | Alkyl halides | Productc | Yieldb (%) | M.P (°C) | IC50 (μM) | |
|---|---|---|---|---|---|---|---|
| A549 | LLC | ||||||
| a Reaction condition: K2CO3/nano Cu2O (8 mol%)/DMEDA, DMF/150 °C, 15 h.b Isolated yields.c All the synthesized compounds exhibited spectral properties consistent with the assigned structures and were fully characterized by their spectroscopic data (1H, IR, LC MS, elemental and 13C NMR, HRMS analysis). | |||||||
| 3a | 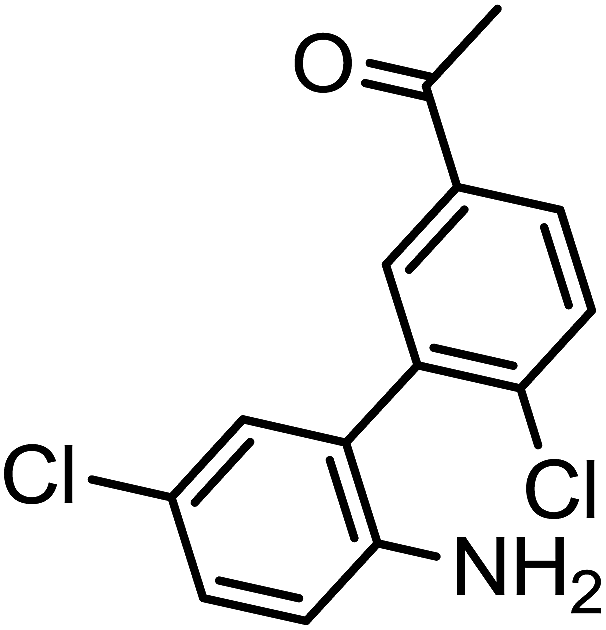 |
 |
 |
79 | 118–119 | 38.2 ± 2.7 | 35.2 ± 4.3 |
| 3b |  |
 |
74 | 185–186 | 17.4 ± 0.1 | 19.1 ± 0.2 | |
| 3c |  |
 |
68 | 162–163 | 18.2 ± 2.7 | 21.6 ± 4.2 | |
| 3d |  |
 |
79 | 122–123 | 22.1 ± 2.2 | 24.7 ± 3.1 | |
| 3e |  |
 |
78 | 144–145 | 13.6 ± 1.1 | 16.4 ± 2.3 | |
| 3f | 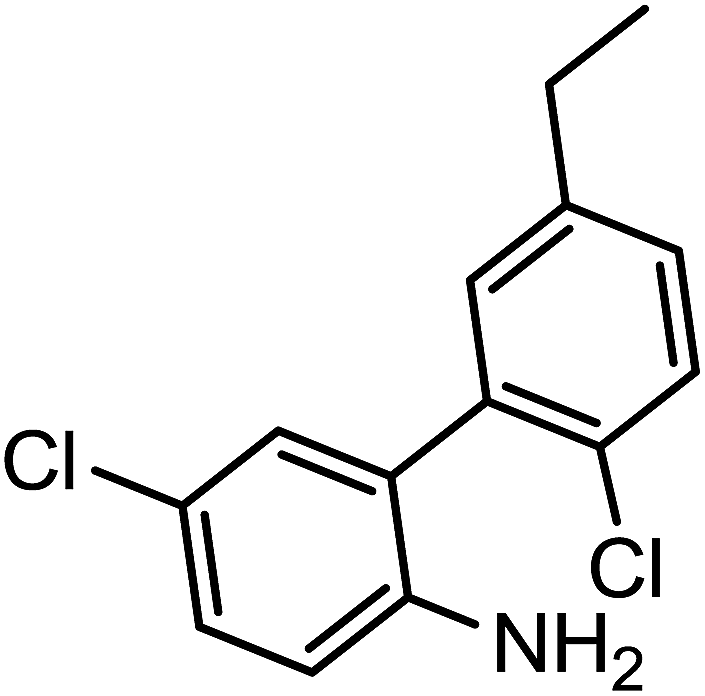 |
 |
 |
79 | 124–125 | 48 ± 4.1 | >50 |
| 3g | 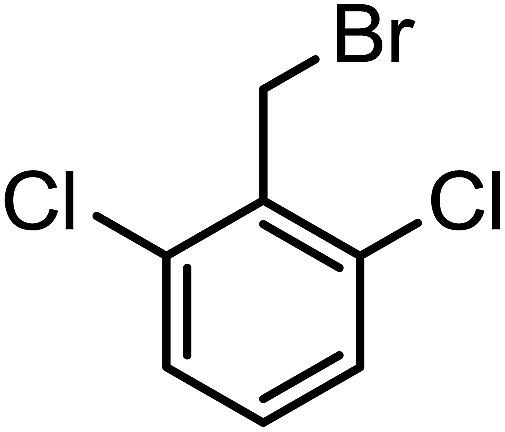 |
 |
71 | 165–166 | >50 | >50 | |
| 3h | 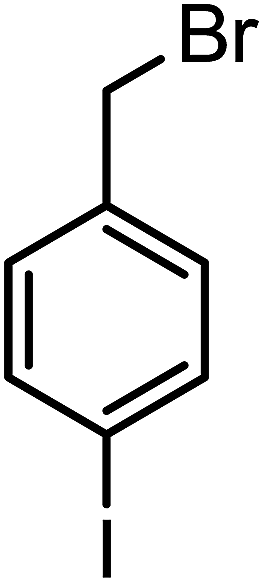 |
 |
69 | 143–144 | >50 | >50 | |
| 3i |  |
 |
78 | 128–129 | >50 | >50 | |
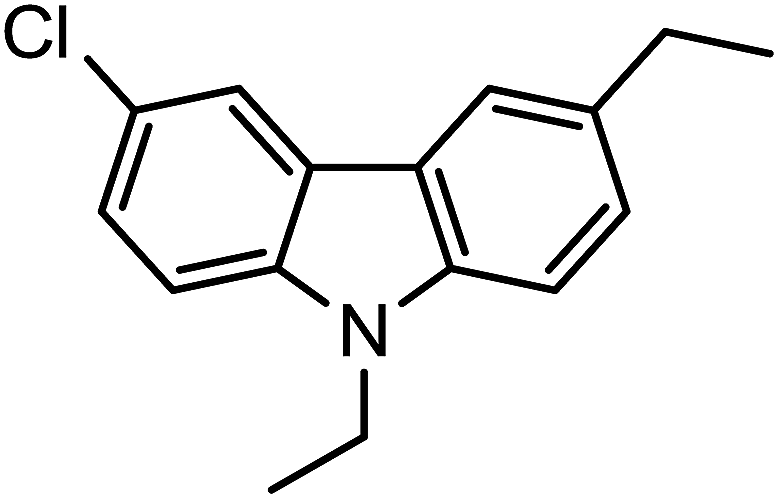
| 3j | 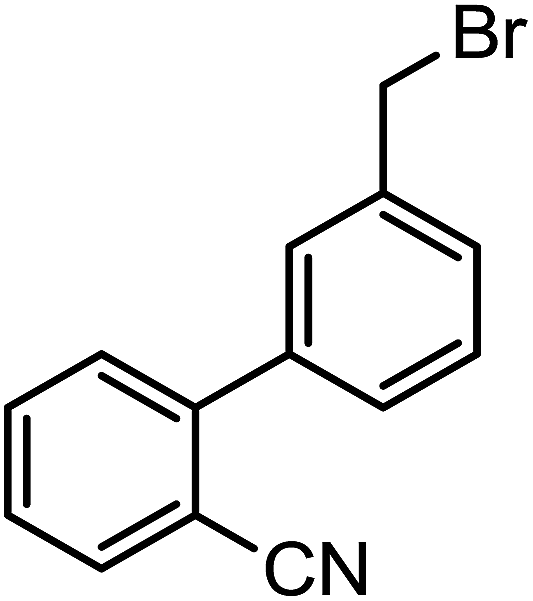 |
 |
77 | 140–141 | 17.2 ± 1.3 | 19.2 ± 2.7 | |
| 3k |  |
 |
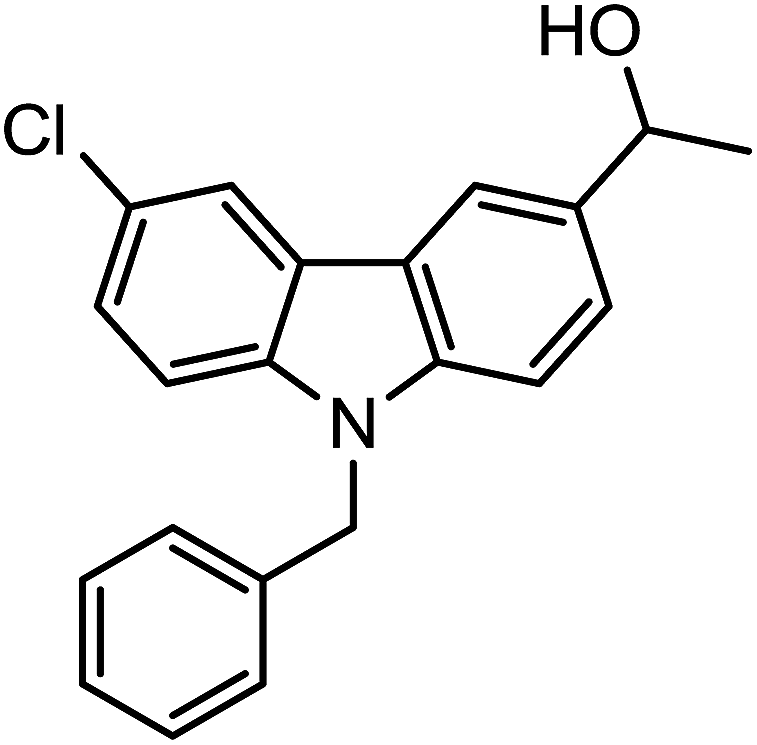 |
79 | 125–126 | 23.1 ± 4.1 | 22.1 ± 1.3 |
| 3l | 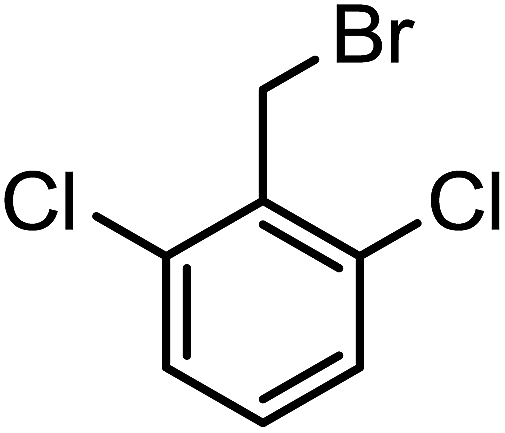 |
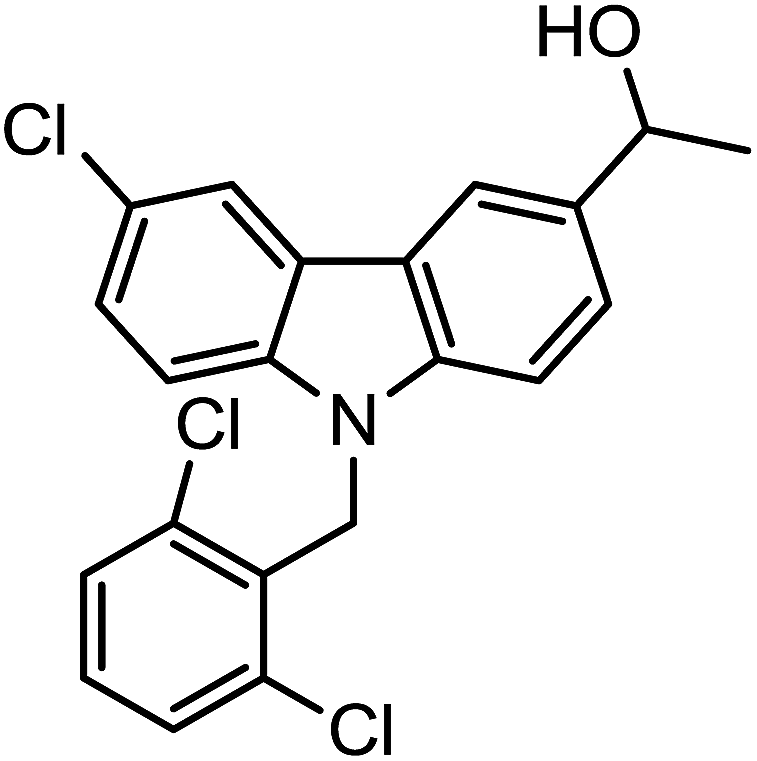 |
72 | 141–142 | 35.6 ± 3.6 | 32.4 ± 2.1 | |
| 3m |  |
 |
70 | 119–120 | 26.4 ± 1.7 | 38.2 ± 3.2 | |
| 3n |  |
 |
74 | 116–117 | >50 | >50 | |
| 3o |  |
 |
77 | 132–133 | 18.6 ± 1.8 | 21.7 ± 2.7 | |
Mechanism
On the basis of previous mechanistic studies in copper catalyzed intramolecular C–N bond formation,43 we propose the possible concerted mechanism for the conversion of 2-amino-2′-chloro biphenyls to N-substituted carbazoles as shown in Scheme 3. | ||
| Scheme 3 Possible concerted mechanism for the conversion of 2-amino-2′-chloro biphenyls to 1,9-substituted-6-chlorocarbazole. | ||
As it is clear from the Scheme 3 that initially a Cu–L complex was formed which in turn coordinates with secondary amine by intramolecular oxidative addition to form complex A. The complex A further reacts with base to form Cu–N bond to form complex B. Reductive elimination step regenerates the copper catalyst and formation of the desired product C. Our findings suggested that secondary amines were more liable for cyclisation compare to primary amines25,44 wherein poor ring formation was observed in the reaction carried out without using benzylbromide (2a) (Scheme 2), which apparently support N-benzylation occurs before the coordination of the copper complex with amine nitrogen.
Carbazole derivatives suppresses the proliferation of A549 and LLC cells
We evaluated all the synthesized carbazole derivatives for their antiproliferative activity against A549 and LLC cells. Among the tested carbazoles, compounds 3e (ACB) and 3j showed significant antiproliferative effect (IC50 < 20 μM) against A549 & LLC cells. ACB was identified as the lead compound with IC50 of 13.6 and 16.4 μM against A549 and LLC cells respectively. Paclitaxel was used as the positive control which displayed the IC50 of 0.0044 μM against A549 cells.ACB downregulates the constitutive STAT3 phosphorylation in lung cancer cells
Phosphorylation of STAT3 at Y705 causes the dimerization and translocation into nucleus to relay the oncogenic signals by expressing the genes involved in proliferation, antiapoptosis, angiogenesis and tumor evasion.5 Carbazoles are known for their inhibitory activity against the phosphorylation of STAT3 (Tyr-705), in turn inducing the antiproliferative effect in cancer cells.24 Therefore, we treated A549, HCC-2279 and H1975 cells with 10 μM of ACB for 6 h and investigated the effect of lead compound on the persistent activation of STAT3 by western blot analysis using antibodies which recognize phosphorylation of STAT3 at Tyr-705. ACB significantly downregulated the STAT3 phosphorylation in all the tested cell lines (Fig. 1A). The levels of total STAT3 and β-actin remain unchanged indicating the inhibitory efficacy of carbazoles on STAT3 phosphorylation.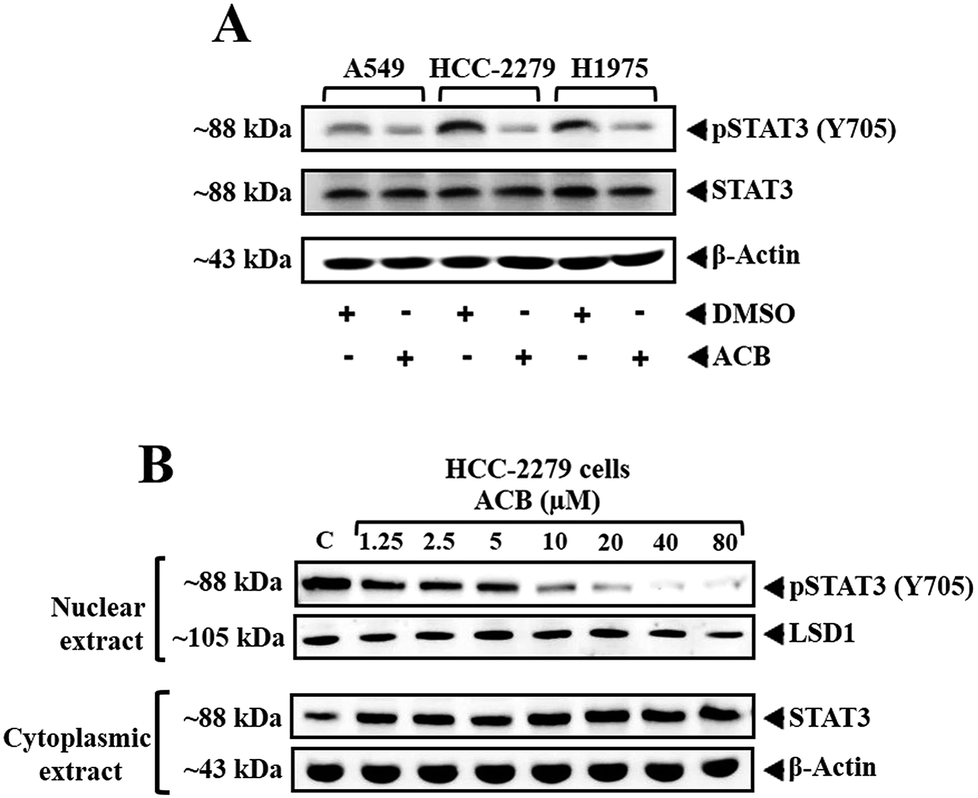 | ||
| Fig. 1 ACB downregulates phospho-STAT3 in lung cancer cells. (A) A549, HCC-2279 and H1975 cells (5 × 105 ml−1) were treated with the indicated concentrations (0, 1.25, 2.5, 5, 10, 20, 40 and 80 μM) of ACB for 6 h, after which whole-cell extract was prepared and resolved on SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed for phospho-STAT3, and the same blot was stripped and reprobed with STAT3 antibody to verify equal protein loading. (B) HCC-2279 cells were treated with different doses of ACB (0, 1.25, 2.5, 5, 10, 20, 40 and 80 μM) and nuclear and cytoplasmic extract was prepared. Nuclear extract was resolved on SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed for phospho-STAT3 and LSD1. Cytoplasmic extract was also resolved on SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed for STAT3 and β-actin antibody to verify equal protein loading. | ||
Given the role of STAT3 phosphorylation in translocation to nucleus and DNA binding, we further investigated the levels of phospho-STAT3 and lysine demethylase (LSD1) in the nuclear extract of HCC-2279 cells treated with different doses of ACB (0, 1.25, 2.5, 5, 10, 20, 40 and 80 μM). ACB downregulated the nuclear pool of phospho-STAT3 indicating that ACB inhibits the phosphorylation of STAT3, thereby translocation into nucleus and in turn reduces its DNA binding ability (Fig. 1B). LSD1 was used as nuclear marker and loading control for nuclear protein. The levels of LSD1, cytoplasmic STAT3 and β-actin remain unchanged.
In silico interaction studies of carbazoles that targets STAT3 in lung cancer cells
Matched pair analysis identified the acetyl group on the core carbazole as most promising substituent at the R position. In four of five matched molecular series, the acetyl group is associated with highest cellular activities. An ethyl substituent is associated with poor solubility of the corresponding compounds, especially in connection with chlorine substitutions on the R1 group. On the other hand, we investigated N-substituents (R1 position) and found the 3-(2-cyano-phenyl)-benzyl substitution as decoration with highest activity in two out of three matched molecular series. Thus, ACB represents the optimum candidate molecule for further development from a matched pairs perspective.On the other hand, we attempted to rationalize the SAR trends by docking the carbazoles to the SH2 domain of STAT3. Therefore, we extracted the SH2 domain from the crystal structure of the STAT3 homo-dimer (PDB: 1BG1)39 and chose the pTyr as center for in silico docking experiments. Interestingly, the 3-(2-cyano-phenyl)-benzyl substituted compounds such as ACB, 3j and 3o showed distinct behavior in molecular docking apart from conserved shape fit and hydrophobic interactions. In general, the R groups are buried in the pTyr binding site and thereby form hydrogen bonds via the acetyl and hydroxyethyl group (see Fig. 2). This explains, the higher activity of acetyl and ethyl decoration over ethyl which lacks the acceptor functionality resembling the pTyr in binding to the side-chain of Arg-609. Here, 3-(2-cyano-phenyl)-benzyl substituted compounds are predicted to interact with Arg-609 and Ser-613 in the pTyr site via the cyano group. This also leads to a change in the SAR for this particular series as expected after a reorientation of the binding mode.45 Since the cyano groups replaces the R-group, the SAR for R substitutions is flatter and shows little activity difference for acetyl-3′-((3-acetyl-6-chloro-9H-carbazol-9-yl)methyl)-[1,1′-biphenyl]-2-carbonitrile (ACB), hydroxyethyl (compound 3o), and ethyl decoration (compound 3j).
 | ||
| Fig. 2 Molecular docking of carbazole compounds to the STAT3 SH2 domain: (A) the binding site of pTyr-705 (sticks in elemental colors, carbon in white) in the SH2 domain (PDB: 1BG1) is associated with polar interactions with Ser-613 and Arg-609 (both shown as lines). (B) Compound 3c is predicted to position the acetyl function in the pTyr site forming hydrogen bonding to Arg-609. (C) ACB shows a switch in binding mode compared to compound 3c and places a cyano function in the pTyr site. In both binding modes carbazole show shape complementarity to the binding site and form van der Waals contacts to the peptide binding groove. | ||
Conclusion
STAT3 is identified as one of the attractive molecular therapeutic targets for the designing of small molecule inhibitors because of its persistent activation in more than 20 types of cancers including various solid and liquid tumors. We therefore for the first time implemented the one-pot synthesis of carbazole derivatives using nano-cuprous oxide as a catalyst via intramolecular C–N bond forming reactions. Initial investigation on the antiproliferative activity of carbazoles on lung cancer cells projected ACB as the lead structure. Further examination revealed that, ACB suppresses the phosphorylation of STAT3 thereby interfering with its transcriptional activity. Analysis of phospho-STAT3 in whole-cell and nuclear extracts clearly demonstrated the mechanism of action of ACB. The results of the STAT3 inhibition readout are closely correlated with antiproliferative activity of ACB. In silico analysis substantially supported our in vitro experimental evidences. Previous reports have suggested that, carbazole inhibits STAT3 phosphorylation by modulating protein-tyrosine phosphatase PTPN6 and it requires further investigation to explore the possible involvement of phosphatases in ACB mediated STAT3 inhibition in lung cancer cells.Acknowledgements
This research was supported by University Grants Commission (41-257-2012-SR), Vision Group Science and Technology, Department of Science and Technology (No. SR/FT/LS-142/2012) to Basappa. KSR thanks DST-JSPS (DST/INT/JAP/P-79/09), DST Indo-Korea [INT/Indo-Korea/122/2011-12] for financial support and Institution of Excellence, University of Mysore for instrumentation facility. CDM thanks the University of Mysore for Department of Science and Technology-Promotion of University Research and Scientific Excellence (DST-PURSE) for Research Associate fellowship.References
- A. Subramaniam, M. K. Shanmugam, T. H. Ong, F. Li, E. Perumal, L. Chen, S. Vali, T. Abbasi, S. Kapoor, K. S. Ahn, A. P. Kumar, K. M. Hui and G. Sethi, Br. J. Pharmacol., 2013, 170, 807–821 CrossRef CAS PubMed.
- J. J. O'Shea, M. Gadina and Y. Kanno, J. Immunol., 2011, 187, 5475–5478 CrossRef PubMed.
- C. N. Revanna, Basappa, V. Srinivasa, L. Fi, K. S. Siveen, X. Dai, S. N. Swamy, D. G. Bhadregowda, G. Sethi, K. Mantelingu, A. Bender and K. S. Rangappa, MedChemComm, 2014, 5, 32–40 RSC.
- P. Rajendran, F. Li, K. A. Manu, M. K. Shanmugam, S. Y. Loo, A. P. Kumar and G. Sethi, Br. J. Pharmacol., 2011, 163, 283–298 CrossRef CAS PubMed.
- C. D. Mohan, H. Bharathkumar, K. C. Bulusu, V. Pandey, S. Rangappa, J. E. Fuchs, M. K. Shanmugam, X. Dai, F. Li, A. Deivasigamani, K. M. Hui, A. P. Kumar, P. E. Lobie, A. Bender, Basappa, G. Sethi and K. S. Rangappa, J. Biol. Chem., 2014, 289, 34296–34307 CrossRef CAS PubMed.
- J. Zhang, K. S. Ahn, C. Kim, M. K. Shanmugam, K. S. Siveen, F. Arfuso, R. P. Samy, A. Deivasigamani, L. H. Lim, L. Wang, B. C. Goh, A. P. Kumar, K. M. Hui and G. Sethi, Antioxid. Redox Signaling, 2015 DOI:10.1089/ars.2015.6418.
- K. S. Siveen, S. Sikka, R. Surana, X. Dai, J. Zhang, A. P. Kumar, B. K. Tan, G. Sethi and A. Bishayee, Biochim. Biophys. Acta, 2014, 1845, 136–154 CAS.
- R. L. Carpenter and H.-W. Lo, Cancers, 2014, 6, 897–925 CrossRef CAS PubMed.
- A. Jarnicki, T. Putoczki and M. Ernst, Cell Div., 2010, 5, 1–15 CrossRef PubMed.
- Y. Liu, P. K. Li, C. Li and J. Lin, J. Biol. Chem., 2010, 285, 27429–27439 CrossRef CAS PubMed.
- L. Konnikova, M. Kotecki, M. M. Kruger and B. H. Cochran, BMC Cancer, 2003, 3, 23 CrossRef PubMed.
- H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef.
- D. P. Chakraborty, B. K. Barman and P. K. Bose, Tetrahedron, 1965, 21, 681–685 CrossRef CAS.
- K. C. Das, D. P. Chakraborty and P. K. Bose, Experientia, 1965, 21, 340 CrossRef CAS PubMed.
- A. Gluszynska, Eur. J. Med. Chem., 2015, 94, 405–426 CrossRef CAS PubMed.
- M. S. Shaikh, R. Karpoormath, N. Thapliyal, R. A. Rane, M. B. Palkar, A. M. Faya, H. M. Patel, W. S. Alwan, K. Jain and G. A. Hampannavar, Anti-Cancer Agents Med. Chem., 2015, 15, 1049–1065 CrossRef CAS PubMed.
- P. Padmaja, G. Koteswara Rao, A. Indrasena, B. V. Subba Reddy, N. Patel, A. B. Shaik, N. Reddy, P. K. Dubey and M. P. Bhadra, Org. Biomol. Chem., 2015, 13, 1404–1414 CAS.
- F. Sha, Y. Tao, C. Y. Tang, F. Zhang and X. Y. Wu, J. Org. Chem., 2015, 80, 8122–8133 CrossRef CAS PubMed.
- A. Caruso, D. Iacopetta, F. Puoci, A. R. Cappello, C. Saturnino and M. S. Sinicropi, Mini-Rev. Med. Chem., 2016, 16, 630–643 CAS.
- C. L. Chen, L. Cen, J. Kohout, B. Hutzen and C. Chan, Mol. Cancer, 2008, 7, 78 CrossRef PubMed.
- M. Takenaga, Y. Yamamoto, T. Takeuchi, Y. Ohta, Y. Tokura, A. Hamaguchi, D. Asai, H. Nakashima, S. Oishi and N. Fujii, Biochem. Biophys. Res. Commun., 2015, 463, 222–228 CrossRef CAS PubMed.
- A. Botta, E. Sirignano, A. Popolo, C. Saturnino, S. Terracciano, A. Foglia, M. S. Sinicropi, P. Longo and S. Di Micco, Mol. Inf., 2015, 34, 689–697 CrossRef CAS.
- C. Saturnino, C. Palladino, M. Napoli, M. S. Sinicropi, A. Botta, M. Sala, A. Carcereri de Prati, E. Novellino and H. Suzuki, Eur. J. Med. Chem., 2013, 60, 112–119 CrossRef CAS PubMed.
- S. Hou, Y. W. Yi, H. J. Kang, L. Zhang, H. J. Kim, Y. Kong, Y. Liu, K. Wang, H. S. Kong, S. Grindrod, I. Bae and M. L. Brown, J. Med. Chem., 2014, 57, 6342–6353 CrossRef CAS PubMed.
- W. D. Guerra, R. A. Rossi, A. B. Pierini and S. M. Barolo, J. Org. Chem., 2015, 80, 928–941 CrossRef CAS PubMed.
- F. Xiao, Y. Liao, M. Wu and G.-J. Deng, Green Chem., 2012, 14, 3277–3280 RSC.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892–2895 CrossRef CAS PubMed.
- C. P. Baburajeev, C. Dhananjaya Mohan, H. Ananda, S. Rangappa, J. E. Fuchs, S. Jagadish, K. Sivaraman Siveen, A. Chinnathambi, S. Ali Alharbi, M. E. Zayed, J. Zhang, F. Li, G. Sethi, K. S. Girish, A. Bender, Basappa and K. S. Rangappa, Sci. Rep., 2015, 5, 14195 CrossRef CAS PubMed.
- K. S. Rakesh, S. Jagadish, A. C. Vinayaka, M. Hemshekhar, M. Paul, R. M. Thushara, M. S. Sundaram, T. R. Swaroop, C. D. Mohan, Basappa, M. P. Sadashiva, K. Kemparaju, K. S. Girish and K. S. Rangappa, PLoS One, 2014, 9, e107182 Search PubMed.
- R. Roopashree, C. D. Mohan, T. R. Swaroop, S. Jagadish, B. Raghava, K. S. Balaji, S. Jayarama, Basappa and K. S. Rangappa, Bioorg. Med. Chem. Lett., 2015, 25, 2589–2593 CrossRef CAS PubMed.
- N. C. Anilkumar, M. S. Sundaram, C. D. Mohan, S. Rangappa, K. C. Bulusu, J. E. Fuchs, K. S. Girish, A. Bender, Basappa and K. S. Rangappa, PLoS One, 2015, 10, e0131896 Search PubMed.
- S. Anusha, C. D. Mohan, H. Ananda, C. P. Baburajeev, S. Rangappa, J. Mathai, J. E. Fuchs, F. Li, M. K. Shanmugam, A. Bender, G. Sethi, Basappa and K. S. Rangappa, Bioorg. Med. Chem. Lett., 2016, 26, 1056–1060 CrossRef CAS PubMed.
- X. N. Wang, S. J. Wang, V. Pandey, P. Chen, Q. Li, Z. S. Wu, Q. Wu and P. E. Lobie, Medicine, 2015, 94, e860 CrossRef CAS PubMed.
- H. Bharathkumar, C. D. Mohan, H. Ananda, J. E. Fuchs, F. Li, S. Rangappa, M. Surender, K. C. Bulusu, K. S. Girish, G. Sethi, A. Bender, Basappa and K. S. Rangappa, Bioorg. Med. Chem. Lett., 2015, 25, 1804–1807 CrossRef CAS PubMed.
- N. Ashwini, M. Garg, C. D. Mohan, J. E. Fuchs, S. Rangappa, S. Anusha, T. R. Swaroop, K. S. Rakesh, D. Kanojia, V. Madan, A. Bender, H. P. Koeffler, Basappa and K. S. Rangappa, Bioorg. Med. Chem., 2015, 23, 6157–6165 CrossRef CAS PubMed.
- H. K. Keerthy, C. D. Mohan, K. Sivaraman Siveen, J. E. Fuchs, S. Rangappa, M. S. Sundaram, F. Li, K. S. Girish, G. Sethi, Basappa, A. Bender and K. S. Rangappa, J. Biol. Chem., 2014, 289, 31879–31890 CrossRef CAS PubMed.
- V. Pandey, Z. S. Wu, M. Zhang, R. Li, J. Zhang, T. Zhu and P. E. Lobie, Breast Cancer Res., 2014, 16, 429 CrossRef PubMed.
- C. Kramer, J. E. Fuchs, S. Whitebread, P. Gedeck and K. R. Liedl, J. Med. Chem., 2014, 57, 3786–3802 CrossRef CAS PubMed.
- S. Becker, B. Groner and C. W. Muller, Nature, 1998, 394, 145–151 CrossRef CAS PubMed.
- P. Labute, Proteins, 2009, 75, 187–205 CrossRef CAS PubMed.
- I. H. Park and C. Li, J. Mol. Recognit., 2011, 24, 254–265 CrossRef CAS PubMed.
- S. Anusha, B. S. Anandakumar, C. D. Mohan, G. P. Nagabhushana, B. S. Priya, K. S. Rangappa, Basappa and C. G. T. Chandrappa, RSC Adv., 2014, 4, 52181–52188 CAS.
- J. Peng, M. Ye, C. Zong, F. Hu, L. Feng, X. Wang, Y. Wang and C. Chen, J. Org. Chem., 2011, 76, 716–719 CrossRef CAS PubMed.
- C. T. Brain and J. T. Steer, J. Org. Chem., 2003, 68, 6814–6816 CrossRef CAS PubMed.
- C. Kramer, J. E. Fuchs and K. R. Liedl, J. Chem. Inf. Model., 2015, 55, 483–494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01906d |
| This journal is © The Royal Society of Chemistry 2016 |