
Construction of a novel INHIBIT logic gate through a fine-tuned assembly of anthryl fluorophores via selective anion recognition and host–guest interactions†
Lei Zhang*a,
Yilong Hea,
Na Zhangb,
Daosheng Liua,
Jiao Hana and
Weitao Gongc
aCollege of Chemistry, Chemical Engineering and Environmental Engineering, Liaoning Shihua University, No. 1 Dandong Road, Fushun 113001, Liaoning, China. E-mail: lnpuzhanglei@163.com; Tel: +86-024-56861711
bDepartment of Chemical Engineering, Fushun Vocational Technology Institute, Fushun 113122, Liaoning, China
cState Key Laboratory of Fine Chemicals, School of Chemical Engineering, School of Chemistry, Dalian University of Technology, Dalian 116024, P. R. China
First published on 18th December 2015
Abstract
A novel ligand (AAP) based on an anthryl fluorophore was rationally designed and synthesized. The presence of H2PO4− (Pi) could induce the effective assembly of the ligand which leads to a strong excimer emission, while β-cyclodextrin (β-CD) disassembled the Pi–ligand complex through a host–guest interaction with terminal adamantane groups. This disassembly causes a considerable decrement in emissive intensity as well as a clear blue-shift in emissive wavelength. By manipulating the assembly and disassembly of anthryl fluorophores with Pi and β-CD, a novel INHIBIT logic gate was constructed using Pi and β-CD as the chemical inputs and fluorescence emission as the output.
1. Introduction
A molecular logic gate, mimicking the real functions of a semiconductor logic gate in modern computing, extends information processing to the molecular level and thus has attracted much attention in the field of supramolecular chemistry.1–7 Since the discovery of the first AND gate in solution by A. P. de Silva,8 molecular logic gates have experienced rapid growth and various types of logic gates, such as NAND, OR, XNOR, XOR, NOR, NOT and INHIBIT, have been established.9–15 Among them, the INHIBIT gate involves a combination of AND and NOT logic functions and deserves increasing attention due to its non-commutative, in which the output signal is inhibited by only one powerful input.16–19 The operation of INHIBIT gate is usually derived from some switchable molecular systems, especially by changing the optical properties. Despite the striking number of INHIBIT gates developed in recent years, their design mechanisms are controlled by intramolecular charge transfer (ICT),20,21 photo-induced electron transfer (PET)22 and fluorescent resonance energy transfer (FRET).23 In this sense, development of INHIBIT logic gate with new design mechanisms is an attractive challenge.Anthryl fluorophore was used to obtain novel multi-responsive fluorescent assembly and dis-assembly phenomena.24–26 If one can tune the assembly properties of anthryl moiety by introducing different inputs, novel logic gates could be constructed. Until now, however, few relative reports were found with respect to logic gates built on the assembly of anthryl fluorophore to the best of our knowledge.
Anion-induced self-assembly has attracted much attention recently, since it provides a new and efficient strategy to build functional supramolecular structures. Beer and co-workers reported a novel anion-templating strategy for the construction of rotaxanes and catenanes as efficient anions sensors.27–29 Wu and co-workers presented the anion-induced assembly process to build novel double and triple helical supramolecular structures.30,31 In our previous work, a series of selective chemosensors containing anthryl fluorophore towards H2PO4− anions in polar solvents, even in aqueous solution were achieved.32–34 In that case, the high selectivity is derived from the subtle control of the assembly of anthryl moiety by ligand structure, anions and solvent polarity.
In this work, a new ligand AAP was designed and synthesized as a potential INHIBIT logic gate. It is composed of two amide-pyridinium moieties as binding sites and an anthryl fluorophore as the signaling part. Adamantane group was introduced at the two terminals for controlling the assembly behavior via potential host–guest interaction with β-CD. The detailed design rationale is exhibited in Fig. 1 and it is based on the following hypotheses: (1) under the role of Pi, the ligand AAP could assemble to form Pi–AAP complex according to our previous reports. In this case, two anthracene groups are located closer, which would lead to a different emissive wavelength compared with the pure ligand, (2) terminal adamantane moieties are known to be easily incorporated into the cavity of β-CD by host–guest interactions. If the formation of Pi–AAP complex could be disturbed by this host–guest interaction between adamantane and β-CD, the assembly behavior of anthryl fluorophore might change accordingly giving rise to differences in fluorescence property. In a word, taking advantage of Pi anion and β-CD as two different chemical inputs and fluorescence emission as the output, a potential INHIBIT logic gate will be achieved.
 | ||
| Fig. 1 Schematic representation of the proposed Pi-induced assembly and β-CD induced dis-assembly processes. | ||
2. Experimental
2.1. Material and characterization
All the chemicals were purchased from commercial sources and used without further purification unless mentioned. Acetonitrile was pre-dried by shaking with type 4A molecular sieves, and then distilled over calcium hydride before use. Other solvents were used as received. 1H and 13C NMR spectra were recorded on a Bruker Av400 NMR spectrometer at 400 MHz and 126 MHz, respectively. Chemical shifts are given in parts per million (ppm) relative to tetramethylsilane (TMS). High resolution mass spectra (HRMS) were recorded on a Waters GC-TOF and LC/Q-TOF mass spectrometer. Fluorescence spectra were obtained from a Jasco FP-8300 spectrofluorometer. UV-Vis absorption spectra were all taken on a Hitachi UV-4100 spectrophotometer. Quartz cells with 1 cm path length were used.2.2. Synthetic procedures
The synthetic route of AAP was shown in Scheme 1. | ||
| Scheme 1 Synthetic route of AAP. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3OH = 5:3) to give a yellow solid. Then the yellow solid was dissolved in DMF. After dropwise addition of excess saturated aqueous KPF6 solution, 50 mL H2O was added in one portion and a light yellow precipitate was formed. The precipitate was filtered and washed with water repeatedly. After drying in vacuum, AAP (0.77 g, 58%) was obtained as a yellow powder which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.15 (s, 2H), 9.68 (s, 2H), 8.61 (d, J = 8.6 Hz, 2H), 8.56 (dd, J = 6.9, 3.2 Hz, 4H), 8.37 (d, J = 6.1 Hz, 2H), 7.96 (dd, J = 8.6, 6.1 Hz, 2H), 7.77 (dd, J = 6.9, 3.1 Hz, 4H), 7.06 (s, 4H), 2.03 (s, 6H), 1.89 (s, 12H), 1.70 (d, J = 6.5 Hz, 12H). 13C NMR (126 MHz, DMSO-d6) δ 177.18, 139.63, 137.86, 135.01, 134.80, 131.42, 130.56, 128.23, 128.15, 127.13, 126.01, 124.63, 56.21, 41.27, 37.85, 36.35, 35.75, 27.35, 26.82. ESI-MS calcd for C48H52N4O22+: 716.4090 [M]2+; found: 358.2043 (m/2z).
:CH3OH = 5:3) to give a yellow solid. Then the yellow solid was dissolved in DMF. After dropwise addition of excess saturated aqueous KPF6 solution, 50 mL H2O was added in one portion and a light yellow precipitate was formed. The precipitate was filtered and washed with water repeatedly. After drying in vacuum, AAP (0.77 g, 58%) was obtained as a yellow powder which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.15 (s, 2H), 9.68 (s, 2H), 8.61 (d, J = 8.6 Hz, 2H), 8.56 (dd, J = 6.9, 3.2 Hz, 4H), 8.37 (d, J = 6.1 Hz, 2H), 7.96 (dd, J = 8.6, 6.1 Hz, 2H), 7.77 (dd, J = 6.9, 3.1 Hz, 4H), 7.06 (s, 4H), 2.03 (s, 6H), 1.89 (s, 12H), 1.70 (d, J = 6.5 Hz, 12H). 13C NMR (126 MHz, DMSO-d6) δ 177.18, 139.63, 137.86, 135.01, 134.80, 131.42, 130.56, 128.23, 128.15, 127.13, 126.01, 124.63, 56.21, 41.27, 37.85, 36.35, 35.75, 27.35, 26.82. ESI-MS calcd for C48H52N4O22+: 716.4090 [M]2+; found: 358.2043 (m/2z).3. Results and discussion
To verify our conjecture, the sensing ability of ligand AAP towards different anions was first studied to figure out the selectivity of anion-induced assembly properties of AAP. Pure AAP in CH3CN exhibited almost no fluorescence owing to the quenching effect of photo-induced electron transfer (PET) from anthryl moiety to pyridinium.33 Among various anions tested, only Pi induced clear fluorescence change, while others such as I−, F−, OH−, CN−, Cl−, Br−, NO3− and AcO−, showed neglectable changes (Fig. 2a), indicating that AAP can be used as the selective Pi sensing agent. This result is in accordance with our previous results. Compared with that of pure ligand, the Pi–AAP complex exhibited strong green fluorescence centered at 500 nm, which is the well-known excimer emission of anthryl fluorophore,32 confirming the anion-induced assembly of AAP. | ||
| Fig. 2 (a) Fluorescence spectra of AAP (5.0 μM) upon addition of various anions (5 equiv.) in CH3CN (λex = 360 nm). (b) Absorption spectra of AAP (5.0 μM) upon addition of various amounts of Pi in CH3CN. | ||
UV-Vis absorption experiment provides further evidence about Pi-induced self-assembly of ligand AAP, by showing the apparent red-shift in absorption of anthryl fluorophore upon titration of Pi into ligand AAP. As reported, the longer wavelength absorption band compared with the pure AAP indicated the formation of association of anthryl fluorophore in ground state (Fig. 2b). The clear red-shift of the absorption bands of anthracene indicates the formation of J-aggregate.36–38 The 1H NMR spectra before and after the addition of Pi into AAP were also recorded and shown in Fig. 3. Protons of the anthryl moiety showed clear shifting towards higher magnetic field after the addition of 0.5 equiv. Pi. This result further demonstrates the binding of Pi with AAP. Fig. S5† shows the fluorescent titration data for AAP with Pi. The Job plot for the binding between AAP and Pi showed a 1:1 stoichiometry (Fig. S6†). From the fluorescence titration, the binding constant and detection limit of AAP toward Pi was calculated to be (5.3 ± 0.1) × 105 M−1 and 1.842 × 10−7 M respectively.
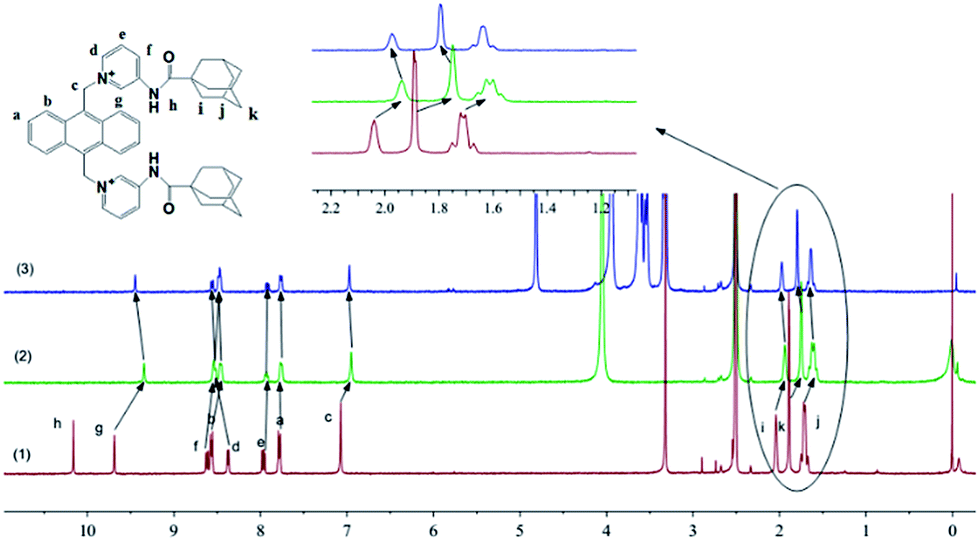 | ||
| Fig. 3 1H NMR spectra of (1) AAP; (2) after addition of Pi (0.5 equiv.); (3) then after addition of β-CD (0.5 equiv.). | ||
Once confirming the formation of Pi–AAP complex, which showed elegant assembled excimer emission of anthryl fluorophore, our attention transferred to how to dis-assemble or disturb this Pi–AAP complex aiming to change the fluorescence. Considering the selective binding between adamantane moiety and β-CD as previously reported, the fluorescence change due to the addition of β-CD into Pi–AAP complex was therefore investigated. Upon titration with β-CD, as shown in Fig. 4a, the excimer emission coming from Pi–AAP complex decreased. In contrast, a new fluorescence emission centered at about 440 nm increased, which is the classical monomer emission of anthryl fluorophore. Accordingly, a clear fluorescence color change from green to blue was observed (Fig. 4a inset). This result indicated the dis-assembly behavior of Pi–AAP complex after the addition of β-CD. In UV-Vis absorption titration between Pi–AAP complex and β-CD, the apparent blue-shift of anthryl fluorophore further evidences the β-CD induced dis-association of Pi–AAP complex in ground state (Fig. 4b). The occurrence of dis-assembly Pi–AAP complex was due to the host–guest interaction between adamantane and β-CD. The 1H NMR investigation of Pi–AAP complex solution before and after addition of β-CD evidences the formation host–guest complex. Signals of methylene protons on adamantyl group showed downfield shift after addition of β-CD (Fig. 3 inset).39
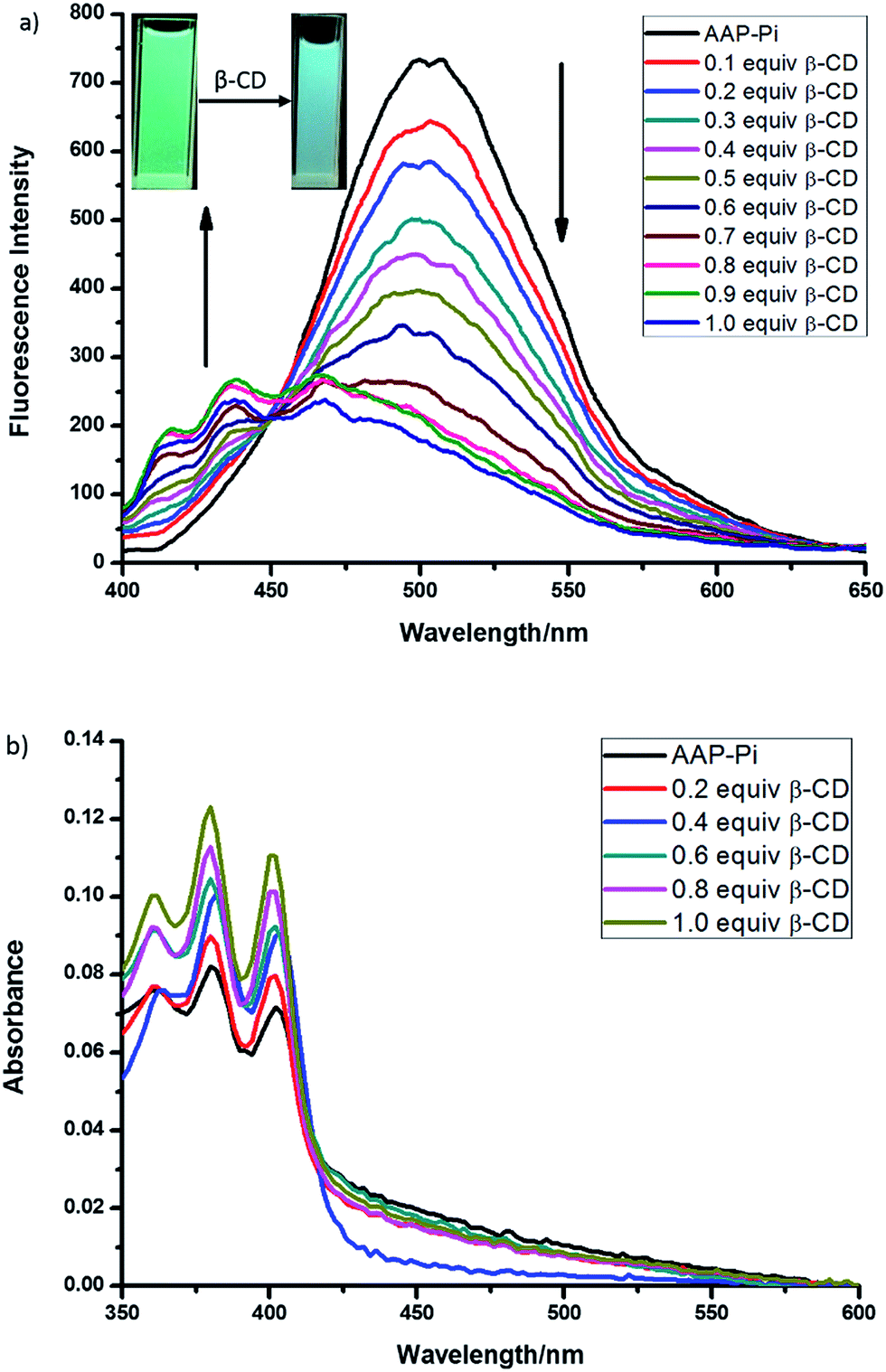 | ||
| Fig. 4 (a) Fluorescence spectra of AAP–Pi complex (5.0 μM) upon addition of various amounts of β-CD in CH3CN (λex = 360 nm). (b) Absorption spectra of AAP–Pi complex (5.0 μM) upon addition of various amounts of β-CD in CH3CN. | ||
Changes in fluorescence due to reverse addition of β-CD and Pi to AAP were also studied. Some interesting results obtained were shown in Fig. 5. When β-CD was first introduced into pure AAP solution instead of Pi, still rather weak fluorescence of anthryl fluorophore was obtained together with a subtle wavelength change. This result means the presence of host–guest interaction between adamantane and β-CD. More interestingly, when Pi was added into the AAP–β-CD complex solution, Pi-induced excimer emission was not found, enhancement of monomer emission was alone observed (Fig. 5b). This result indicated that the Pi-induced assembly of ligand AAP was inhibited by the formation of AAP–β-CD complex. As for the enhancement of monomer emission, it might be due to the block of photo-induced electron transfer (PET) from anthryl moiety to pyridinium by the interaction with Pi anions.
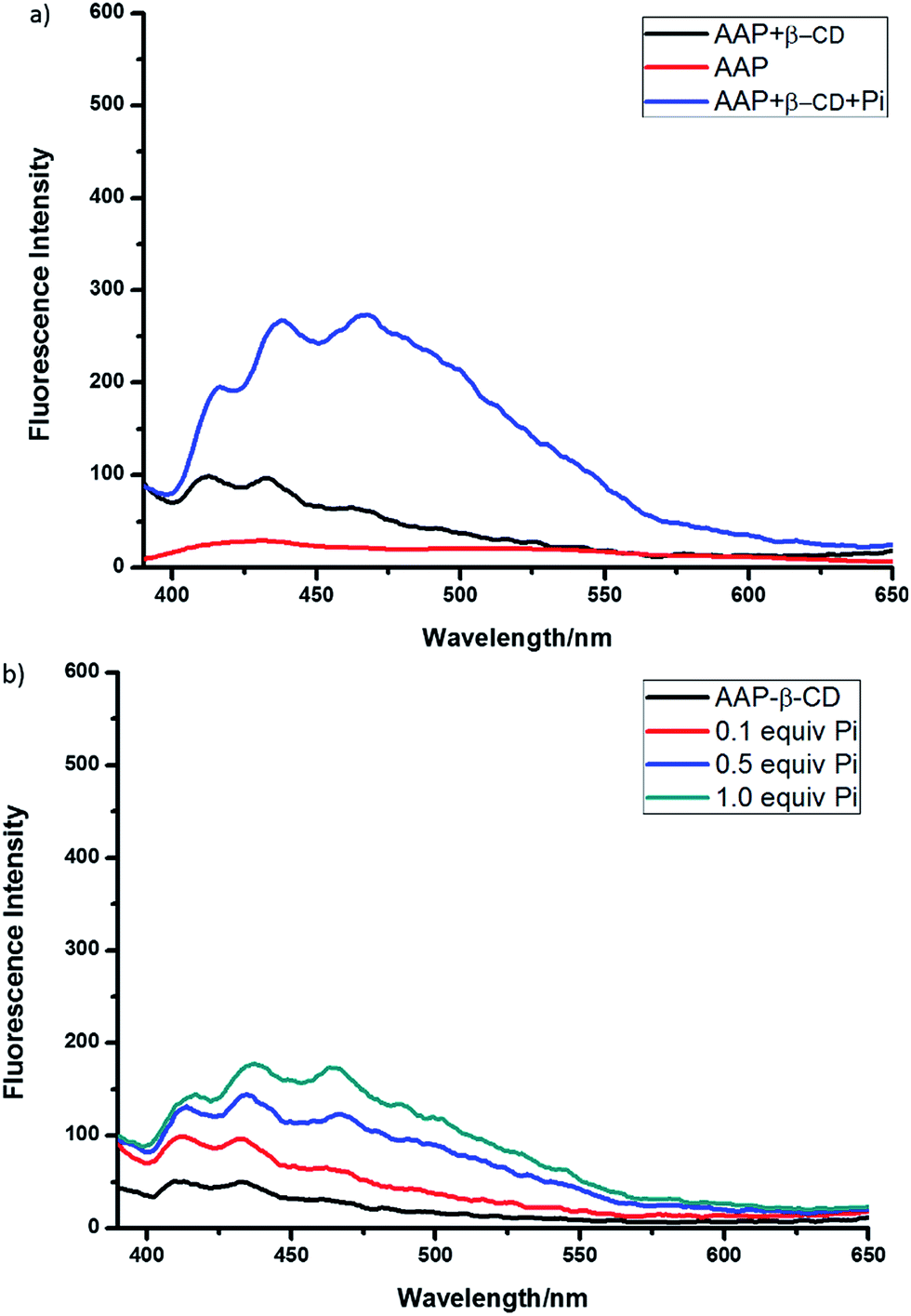 | ||
| Fig. 5 (a) Fluorescence spectra of AAP (5.0 μM) upon addition of β-CD (1 equiv.) and Pi (1 equiv.) in CH3CN (λex = 360 nm). (b) Fluorescence spectra of AAP–β-CD complex (5.0 μM) upon addition of different amounts of Pi in CH3CN (λex = 360 nm). | ||
These results obtained above demonstrated clearly that the assembly phenomena of anthryl fluorophore in ligand AAP could be tuned by introducing Pi and β-CD as two chemical inputs. Pi could induce the assembly of AAP to give excimer emission, and β-CD could inhibit this Pi-induced assembly through the host–guest interaction. Accordingly, this system correlates very well with an INHIBIT logic gate as shown in Fig. 6. In considering the INHIBIT logic gate properties of AAP, in our system, Pi and β-CD act as two chemical inputs, while the excimer emission at 500 nm acts as the output. The output is “0” and the gate is “OFF” when (1) both the Pi and β-CD are absent, (2) β-CD alone is present, (3) both the Pi and β-CD are present. Correspondingly if the output is “1” and the gate is “ON” only when Pi alone is present. Thus, a new INHIBIT gate is constructed by controlling the assembly phenomena of anthryl fluorophore by inputting different chemical stimuli.
 | ||
| Fig. 6 Output and the corresponding truth tables for the INHIBIT gates (λex = 360 nm) of AAP using Pi (input 1) and β-CD (input 2) as inputs. | ||
4. Conclusions
In summary, a potential INHIBIT logic gate based on an anthryl fluorophore was rationally designed and synthesized. Under the role of H2PO4−, anthryl fluorophore could assemble to give apparent excimer fluorescent emission, while β-CD could inhibit this Pi-induced assembly process via host–guest interaction with adamantane moieties. Based on this phenomena, a novel INHIBIT gate was constructed. This is the first example of INHIBIT gate that combines anion-induced assembly and host–guest triggered inhibition. Further work will be focused on the exploration of the detailed structure–property relationship for the construction of novel logic gates.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 21206016) and the Fundamental Research Funds for the Central Universities (DUT14LK08) and the Science Research General Foundation of Liaoning Education Department (L2014157).Notes and references
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2010, 39, 174–188 RSC.
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC.
- K. He, Y. Li, B. Xiang, P. Zhao, Y. Hu, Y. Huang, W. Li, Z. Nie and S. Yao, Chem. Sci., 2015, 6, 3556–3564 RSC.
- A. J. M. Huxley, M. Schroeder, H. Q. N. Gunaratne and A. P. de Silva, Angew. Chem., Int. Ed., 2014, 53, 3622–3625 CrossRef CAS PubMed.
- S. Kou, H. N. Lee, D. van Noort, K. M. K. Swamy, S. H. Kim, J. H. Soh, K. M. Lee, S. W. Nam, J. Yoon and S. Park, Angew. Chem., Int. Ed., 2008, 47, 872–876 CrossRef CAS PubMed.
- J. Ling, B. Daly, V. A. D. Silverson and A. P. de Silva, Chem. Commun., 2015, 51, 8403–8409 RSC.
- A. Sreejith and A. Ajayaghosh, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2012, 51, 47–56 Search PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- D. C. Magri, M. C. Fava and C. J. Mallia, Chem. Commun., 2014, 50, 1009–1011 RSC.
- E. T. Ecik, A. Atilgan, R. Guliyev, T. B. Uyar, A. Gumus and E. U. Akkaya, Dalton Trans., 2014, 43, 67–70 RSC.
- D. C. Magri, G. J. Brown, G. D. McClean and A. P. de Silva, J. Am. Chem. Soc., 2006, 128, 4950–4951 CrossRef CAS PubMed.
- C. P. Collier, E. W. Wong, M. Belohradský, F. M. Raymo, J. F. Stoddart, P. J. Kuekes, R. S. Williams and J. R. Heath, Science, 1999, 285, 391–394 CrossRef CAS PubMed.
- A. P. de Silva, I. M. Dixon, H. Q. N. Gunaratne, T. Gunnlaugsson, P. R. S. Maxwell and T. E. Rice, J. Am. Chem. Soc., 1999, 121, 1393–1394 CrossRef CAS.
- A. R. Chowdhury, P. Ghosh, B. G. Roy, S. K. Mukhopadhyay, P. Mitra and P. Banerjee, RSC Adv., 2015, 5, 62017–62023 RSC.
- P. Ghosh, B. G. Roy, S. Jana, S. K. Mukhopadhyay and P. Banerjee, Phys. Chem. Chem. Phys., 2015, 17, 20288–20295 RSC.
- K. C. Chang, I. H. Su, Y. Y. Wang and W. S. Chung, Eur. J. Org. Chem., 2010, 4700–4704 CrossRef CAS.
- D. Sarkar, A. Pramanik, S. Jana, P. Karmakar and T. K. Mondal, Sens. Actuators, B, 2015, 209, 138–146 CrossRef CAS.
- S. Mardanya, S. Karmakar, S. Das and S. Baitalik, Sens. Actuators, B, 2015, 209, 701–713 CrossRef.
- A. R. Chowdhury, P. Ghosh, B. G. Roy, S. K. Mukhopadhyay, N. C. Murmu and P. Banerjee, Sens. Actuators, B, 2015, 220, 347–355 CrossRef CAS.
- V. Luxami and S. Kumar, Dalton Trans., 2012, 41, 4588–4593 RSC.
- A. Coskun, E. Deniz and E. U. Akkaya, Org. Lett., 2005, 7, 5187–5189 CrossRef CAS PubMed.
- S. H. Lee, J. Y. Kim, S. K. Kim, J. H. Lee and J. S. Kim, Tetrahedron, 2004, 60, 5171–5176 CrossRef CAS.
- M. Yuan, W. Zhou, X. Liu, M. Zhu, J. Li, X. Yin, H. Zheng, Z. Zuo, C. Ouyang, H. Liu, Y. Li and D. Zhu, J. Org. Chem., 2008, 73, 5008–5014 CrossRef CAS PubMed.
- S. Malkondu, D. Turhan and A. Kocak, Tetrahedron Lett., 2015, 56, 162–167 CrossRef CAS.
- D. Zhai, W. Xu, L. Zhang and Y. T. Chang, Chem. Soc. Rev., 2014, 43, 2402–2411 RSC.
- Y. J. Huang, W. J. Ouyang, X. Wu, Z. Li, J. S. Fossey, T. D. James and Y. B. Jiang, J. Am. Chem. Soc., 2013, 135, 1700–1703 CrossRef CAS PubMed.
- M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11463–11466 CrossRef CAS PubMed.
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC.
- B. R. Mullaney, A. L. Thompson and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11642–11646 CrossRef.
- C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q. S. Li and X. J. Yang, Chem. Commun., 2010, 46, 5376–5378 RSC.
- J. Zhao, D. Yang, Y. Zhao, X. J. Yang, Y. Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2014, 53, 6632–6636 CrossRef CAS PubMed.
- W. T. Gong, D. Na, L. Fang, H. Mehdi and G. L. Ning, Org. Biomol. Chem., 2015, 13, 1979–1982 CAS.
- W. T. Gong, Q. L. Zhang, F. R. Wang, B. Gao, Y. Lin and G. L. Ning, Org. Biomol. Chem., 2012, 10, 7578–7583 CAS.
- W. T. Gong, S. Bao, F. R. Wang, J. W. Ye, G. L. Ning and K. Hiratani, Tetrahedron Lett., 2011, 52, 630–634 CrossRef CAS.
- V. A. Ermokhin, N. A. Klenova and P. P. Purygin, Pharm. Chem. J., 2008, 42, 175–176 CrossRef CAS.
- Y. Molard, D. M. Bassani, J. P. Desvergne, P. N. Horton, M. B. Hursthouse and J. H. R. Tucker, Angew. Chem., Int. Ed., 2005, 44, 1072–1075 CrossRef CAS PubMed.
- H. K. Cho, D. H. Lee and J. I. Hong, Chem. Commun., 2005, 1690–1692 RSC.
- F. Fages, J. P. Desvergne, H. B. Laurent, J. M. Lehn, Y. Barrans, P. Marsau, M. Meyer and A. M. A. Gary, J. Org. Chem., 1994, 59, 5264–5271 CrossRef CAS.
- K. Ohga, Y. Takashima, H. Takahashi, Y. Kawaguchi, H. Yamaguchi and A. Harada, Macromolecules, 2005, 38, 5897–5904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, characterization of ligand AAP, including 1H NMR, 13C NMR and HR-MS. See DOI: 10.1039/c5ra20120a |
| This journal is © The Royal Society of Chemistry 2016 |