
Diazopyridine–Ni(II) complexes exhibiting intra-chain ferromagnetic interaction after irradiation: formation of magnetic gel†
Koya
Mori
a,
Rumi
Eguchi
a,
Satoru
Karasawa
*ab and
Noboru
Koga
*a
aGraduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, 812-8582 Japan. E-mail: karasawa@fc.phar.kyushu-u.ac.jp; koga@fc.phar.kyushu-u.ac.jp; Fax: +81-92-642-6545; Tel: +81-92-642-6593
bPRESTO, Japan Science and Technology Agency, Kawaguchi, 332-0012 Japan
First published on 5th August 2015
Abstract
Diazopyridine–nickel complexes, 1 and 2, were obtained by mixing di(4-pyridyl)diazomethane, D1py2, with Ni(R-hfpip)2; hfpip = 4-(4-R-phenylimino)-1,1,1,5,5,5-hexafluoro-2-pentanone and R = n-hexyl (1) and n-octyl (2), in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The discrete reference complex [Ni(n-hexyl-hfpip)(4NOpy)2] (3) was also prepared. In the tube inversion test and the rheological measurements, the sample (20 mM) of 1 and 2 dissolved in EtOH–20% CH2Cl2 solution showed gelation behavior at −4 °C. In a scanning transmission electron microscopy (STEM) image of the xerogel sample, fibers with widths of 500–700 nm were observed. The Vis-NIR spectrum of the gel sample for 1 showed the typical broad absorption at ∼995 nm due to the octahedral Ni(II) complex, suggesting that the pyridine ligands in D1py2 coordinated to the Ni(II) ion to form the chain structure. Complex 3 showed ferromagnetic interaction with J/kB = 26.7 K between the aminoxyl and the Ni(II) through the pyridine ring. Similarly, the thermal profile of χmol values after irradiation of the gel sample of 1 showed a ferromagnetic interaction between the generated triplet carbenes and the Ni(II) ions and provided a magnetic chain structure. The magnetic correlation length was estimated to be 4–5 units from the maximum value of χmolT (12.7 cm3 K mol−1 at 7 K).
:1 ratio. The discrete reference complex [Ni(n-hexyl-hfpip)(4NOpy)2] (3) was also prepared. In the tube inversion test and the rheological measurements, the sample (20 mM) of 1 and 2 dissolved in EtOH–20% CH2Cl2 solution showed gelation behavior at −4 °C. In a scanning transmission electron microscopy (STEM) image of the xerogel sample, fibers with widths of 500–700 nm were observed. The Vis-NIR spectrum of the gel sample for 1 showed the typical broad absorption at ∼995 nm due to the octahedral Ni(II) complex, suggesting that the pyridine ligands in D1py2 coordinated to the Ni(II) ion to form the chain structure. Complex 3 showed ferromagnetic interaction with J/kB = 26.7 K between the aminoxyl and the Ni(II) through the pyridine ring. Similarly, the thermal profile of χmol values after irradiation of the gel sample of 1 showed a ferromagnetic interaction between the generated triplet carbenes and the Ni(II) ions and provided a magnetic chain structure. The magnetic correlation length was estimated to be 4–5 units from the maximum value of χmolT (12.7 cm3 K mol−1 at 7 K).
1. Introduction
Quantum magnets, having large magnetic anisotropy (D and E), such as single-molecule magnets (SMMs),1 single-chain magnets (SCMs),2 and single-ion magnets (SIMs)3 have been extensively studied in the field of molecule-based magnets. Their application to molecular memory, magnetic devices, and quantum computers is anticipated.4 We have been studying a heterospin system5 consisting of organic spins and 3d transition metal ions in the field of molecule-based magnets.6 In this heterospin system, the organic spins of aminoxyl, 4NOpy,7 and triplet carbene generated by photolysis of diazo groups, D1py,8D1py2,9 and D2py2,10 were used. By using the latter spin source, the mono- and dinuclear octahedral cobalt complexes were reported to show SMM behavior after irradiation in solid and frozen solution conditions.11 The obtained SMMs had large activation barriers and hysteresis loops of magnetization with large coercive forces. To develop the field of molecule-based magnets, the construction of new functional materials having magnetic properties is interesting and significant. This time, our heterospin systems were combined with gel, being soft materials and the construction of the magnetic gel exhibiting intense magnetic interactions was studied. Gels have been widely studied and used in various fields such as tissue engineering,12 cell batteries,13 photo devices,14 and environmental materials.15 Recently, not only general gels made of polymers and organic molecules16 but also metallogels17 containing metal ions and/or metal complexes have been developed and examined. The crystals and powders naturally exhibited the magnetic properties of the solid state. In contrast, the gel potentially has both properties of the solid state and solution state, which are shown as the values of G′ and G′′ in rheological measurement.16,17 Therefore, the magnetic properties of gel should exhibit both the properties of the solid and solution state. Furthermore, the gel and the crystals (powders) favor the structures that are kinetically stable and thermodynamically stable, respectively. Accordingly, we can investigate the magnetic properties of a kinetically stable structure of the materials by the gel samples.We have also studied the gelation behavior of aromatic compounds having the urea unit, alkyl chain, and oligoethyleneglycol units.18 Generally, the structure of gels obtained from polymers19 and supra-molecules20 is a 3-D network consisting of 1-D fibers. Accordingly, we applied our heterospin chain as the main framework of the gel structure and used a combination of D1py2 and M(R-hfpip)2; R-hfpip = 4-(4-R-phenylimino)-1,1,1,5,5,5-hexafluoropentanone. The combination of D1py2 and Co(Br-hfpip)2 was reported to form chain complexes of [Co(Br-hfpip)2D1py2]n showing SCM like magnetic behavior.21 For gel formation, first, the alkyl-chain substituents, n-hexyl and n-octyl, were attached to the hfipip ligand. The complexes obtained from D1py2 and Ni(R-hfpip)2; R = n-hexyl and n-octyl, were expected to form a metallogel and a magnetic gel. Herein, we describe the formation of magnetic gel, its morphology, and its magnetic behavior before and after irradiation (Scheme 1).
 | ||
| Scheme 1 Formula of pyridine derivatives carrying diazo and aminoxyl, and M(R-hfpip)2 complexes. | ||
2. Experimental
2.1. Materials
Unless otherwise stated, the preparative reactions were carried out under a high purity dry nitrogen atmosphere. Diethylether, tetrahydrofuran (THF), and 2-methyltetrahydrofuran (MTHF), were distilled from sodium benzophenone ketyl. Dichloromethane and acetonitrile were distilled by CaH2. D1py2 and 4NOpy were prepared in a manner similar to the procedure reported previously.5a:AcOEt (40:1) as the eluent to separately afford the yellow oil in 84% yield (4.8 g, 13.1 mmol). 1H NMR (500 MHz, CDCl3); δ 11.9 (s, 1H), 7.22 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.01 (s, 1H), 2.63 (t, J = 7.8 Hz, 2H), 1.62 (quin, J = 7.8 Hz, 2H), 1.36–1.27 (m, 6H), 0.88 (t, J = 6.5 Hz, 3H). FAB-MS; 367.1 [M]+. Anal. Calcd for C17H19F6NO, C 55.59, H 5.21, N 3.81, found C 55.29, H 4.99, N 3.91.
:AcOEt (40:1) as the eluent to separately afford the yellow oil in 76% yield. 1H NMR (500 MHz, CDCl3); δ 11.9 (s, 1H), 7.22 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.01 (s, 1H), 2.63 (t, J = 7.8 Hz, 2H), 1.62 (quin, J = 7.8 Hz, 2H), 1.36–1.27 (m, 10H), 0.88 (t, J = 6.5 Hz, 3H). FAB-MS; 395.0 [M]+. Anal. Calcd for C19H23F6NO, C 57.72, H 5.86, N 3.54, found C 57.79, H 5.89, N 3.61.
1: To a solution (1 ml) of Hex-hfpip (800 mg, 2.2 mmol) in MeOH was added aqueous solution (3 ml) of NaOH (90 mg, 2.3 mmol) and the solution was stirred vigorously for 5 min. To the solution was added aqueous solution (2 ml) of NiCl2·6H2O and subsequently stirred vigorously for 30 min. After filtration, the residue was dissolved by CH2Cl2 and dried over MgSO4. The solvent was evaporated to afford Ni(Hex-hfpip)2 (869 mg, 1.1 mmol) as dark green oil. The nickel complex was used for the next step without further purification. The solution (1 ml) of D1py2 (74 mg, 0.38 mmmol) in CH2Cl2 was mixed with the solution (1 ml) of Ni(Hex-hfpip)2 (300 mg, 0.38 mmol l) in CH2Cl2 and then MeOH (8 ml) was added. The resulting yellow precipitate was collected by a suction filtration and was washed with MeOH. 1 was obtained as yellow powder in 34% yield (125 mg, 0.13 mmol). IR (KBr); ν(N2); 2063 cm−1. M.P. (decomp.): 185 °C. Anal. Calcd for C45H44F12N6NiO2·0.5CH2Cl2, C 53.06, H 4.40, N 8.16, found C 53.13, H 4.44, N 8.27.
2: To a solution (1 ml) of Oct-hfpip (869 mg, 2.2 mmol) in MeOH was added aqueous solution (3 ml) and NaOH (90 mg, 2.3 mmol). The solution was stirred vigorously for 5 min, aqueous solution (2 ml) of NiCl2·6H2O (259 mg, 1.1 mmol) was added, and then subsequently stirred vigorously for 30 min. After filtration, the residue was dissolved in CH2Cl2 and dried over MgSO4. The solvent was evaporated to afford Ni(Oct-hfpip)2 (844 mg) as dark green oil. The obtained nickel complex was used for the next step without further purification. The solution (1 ml) of D1py2 (69 mg, 0.36 mmol) in CH2Cl2 was mixed with the solution (1 ml) of Ni(Oct-hfpip)2 (300 mg, 0.36 mmol) in CH2Cl2 and then MeOH (8 ml) was added. The resulting yellow precipitate was collected by a suction filtration and was washed with MeOH. 2 was obtained as yellow powder in 50% yield (198 mg, 0.18 mmol). IR (KBr); ν(N2); 2063 cm−1. M.P. (decomp.): 143 °C. Anal. Calcd for C49H52F12N6NiO2, C 56.39, H 5.02, N 8.05, found C 56.29, H 4.99, N 7.91.
3: To a solution (10 ml) of hydroxylamine of 4NOHpy (70 mg, 0.42 mmol) in CH2Cl2 was added Ag2O (0.7 g, 3 mmol) and the solution was stirred for 10 min. After removal of Ag2O by filtration, the filtrate containing 4NOpy was added to the solution (10 ml) of Ni(Hex-hfpip)2 (170 mg, 0.21 mmol) in CH3CN. The solution was condensed by reduced pressure and left at −30 °C. 3 was obtained as red blocks in 10% yield (27.0 mg, 0.022 mmol). M.P.; 112–116 °C. Anal. Calcd for C52H62F12N6NiO4, C 55.68, H 5.57, N 7.49, found C 55.73, H 5.65, N 7.47.
2.2. General methods
Infrared spectra were recorded on a JASCO 420 FT-IR spectrometer. 1H NMR spectra were measured on a Bruker Biospin AVANCE III 500 Fourier transform spectrometer using CDCl3 as the solvent and referenced to TMS. FAB mass spectra (FAB MS) by using 4-nitorobenzylalcohol (NBA) as a matrix were recorded on a JEOL JMS-SX102 spectrometer. UV-Vis infrared spectra were recorded on a JASCO V570 spectrometer. Scanning electron microscopy (SEM) was carried out on a Shimadzu Superscan SSX-550. Melting points and phase transition temperatures were determined by DSC. Powder X-ray diffraction (PXRD) measurements were performed by using a Bruker SAXS NANOSTAR in a 2θ range of 2–10° at 23 °C. Rheological measurements were performed by an Anton Parr MCR 301 rheometer. Elemental analyses were performed at the Analytical Center of the Faculty of Science in Kyushu University.2.3. Single crystal X-ray diffraction (SXRD)
Suitable single crystals were glued onto a glass fiber using epoxy resin. X-ray diffraction data were collected on a Rigaku RAXIS-RAPID diffractometer with graphite monochromated CuKα radiation (λ = 1.54187 Å) for 3. Reflections were collected at 123 ± 1 K. The molecular structures were solved by direct methods (SHELXL 97).22 The refinements were converged using the full-matrix least squares method from the Crystal Structure software package23 to give P21/c (no. 14) for 3. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were refined isotropically. Crystallographic data collection and structural refinement information for 3 are listed in Table S1.† Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication no. CCDC 1406724.2.4. SQUID measurements
Direct current (dc) magnetic susceptibility data were obtained with a Quantum Design MPMS2 SQUID magneto/susceptometer, and corrected for the magnetization of the sample holder, capsule, and cellophane tape, and/or diamagnetic contributions to the samples, which were estimated from Pascal constants.6A hand-made irradiation system5a was used for photolysis of diazo chromophore inside the SQUID apparatus. For the effective photolysis of the diazo-nickel complex with its irradiation system, less than 1 mg of powder was used and 10–20 mM concentrations for the gel and frozen solution were used. In that case, absolute M values were small, therefore, obtained M values before and after irradiation measurements below 10 and 50 K were used as valid values. To avoid raising the temperature and decomposing the sample by local heating, the cooling value in SQUID apparatus, which is able to control the flow amounts of the helium gas for cooling, was maintained to open during irradiation.
3. Results and discussion
3.1. Preparation
D1py2 and 4NOpy were prepared by the procedure reported previously. R-Hfpip; 4-(4-R-phenylimino)-1,1,1,5,5,5-hexafluoropentanone (R = n-hexyl and n-octyl) were prepared by the condensation reaction of 1,1,1,5,5,5,-hexafluoropentane-2,4-dione, hfac, with the corresponding para substituted aniline in the presence of the dehydration catalyst, montmorillonite K10. Ni(R-hfpip)2 were prepared by the reaction of R-hfpip and NiCl2·6H2O under basic conditions. D1py2 was mixed with Ni(R-hfpip)2 to afford 1 (R = n-hexyl) and 2 (R = n-octyl) as yellowish powders. To understand the magnetic properties between the nickel ion and the organic spin through pyridine rings, the stable radical, 4NOpy was used in place of D1py2 and the discrete nickel complex, Ni(Hex-hfpip)2(4NOpy)2, was prepared as a reference complex, 3. The reference complex 3 was prepared in a manner similar to the procedure for 1 and was obtained as a single crystal of red block.3.2. Crystal structure
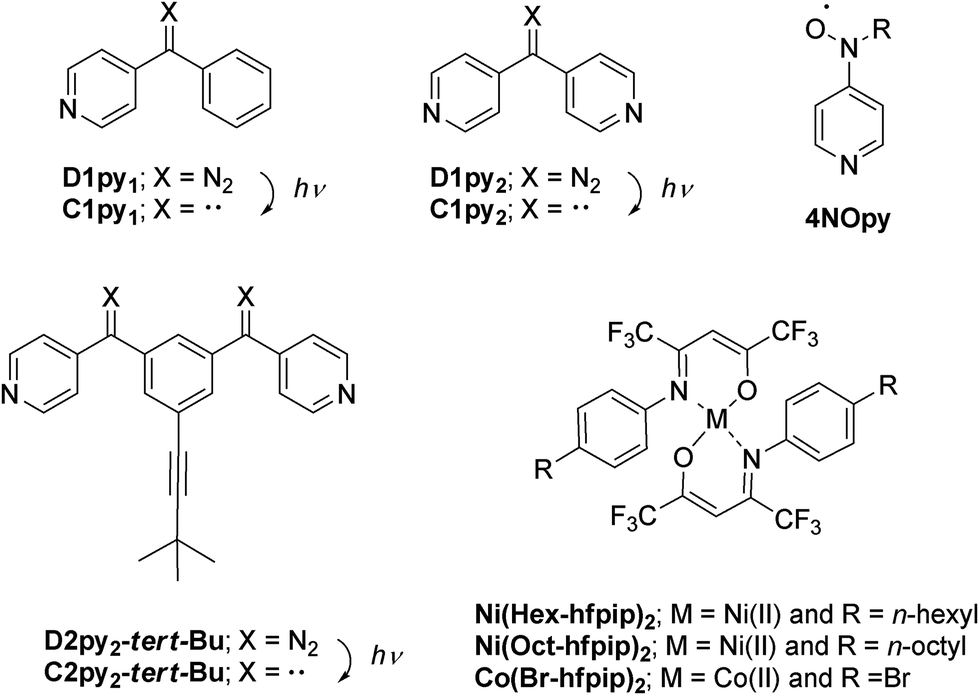
The crystal structure of single crystals for 3 was analyzed by X-ray crystallography. The measurement was performed by Cu radiation at 123 K and the diffraction data were analyzed by SHELXL 97 software.22 Discrete heterospin nickel complex 3 was crystallized in the space group of P21/c (no 14) with Z = 4 (Z′ = 1). Crystallographic data collection and structural refinement information for 3 is shown in Table S1.†In 3, the pyridine rings of two 4NOpy coordinated to the Ni(II) ion in cis configuration. The molecular structure was a compressed octahedron, in which the bond distances between the Ni(II) ion and the O atoms of hfpip were short. The molecular structure and the crystal packing with short contacts of 3 are shown in Fig. 1, and the full structure of 3 is shown in Fig. S1.†
 | ||
| Fig. 1 Molecular structure (a) and crystal packing (b) of 3 with a ball and stick model; color code: Ni, (green), N (blue), O (red), and C (gray). Red dotted lines in (b) indicate the short distance of 3.194(3) Å between NO and C(β) in the Py ring. Phenyl, tert-Bu, CF3, and hexyl group and H atoms are omitted for the sake of clarity. | ||
The bond distances around the Ni(II) ion were rNi–Nimi = 2.127(3) and 2.123(3) Å with the N atoms of hfpip, rNi–Npy = 2.154(3) and 2.160(3) Å with the N atoms of pyridine rings, and rNi–o = 1.992(3) and 2.003(3) Å with the O atoms of hfpip. The dihedral angles between the XY plane defined by the four N atoms coordinating to the Ni(II) ion and two pyridine rings were 71.57(0.06)° and 77.94(0.06)°. The dihedral angles between the pyridine ring and the NO plane were 2.74(0.16) and 22.42(0.19)°. Those bond lengths and the dihedral angles suggested that the magnetic interaction might take place effectively between the Ni(II) ion and the aminoxyl through pyridine rings.5a Selected bond lengths and dihedral angles of 3 are listed in Table 1.
| Bond lengths of coordination site in Ni ion/Å | |
| r Ni–Npy | 2.154(3) and 2.160(3) |
| r Ni–Nimi | 2.123(3) and 2.127(3) |
| r Ni–O | 1.992(3) and 2.003(3) |
| Dihedral angles between the XY plane and pyridine rings/° | |
| XY–py | 71.57(0.06) and 77.94(0.06) |
| Dihedral angles between pyridine and NO/° | |
| py–NO | 2.74(0.16) and 22.42(0.19) |
In crystal packing, intermolecular short contacts (red dotted line in Fig. 1) were observed and the distance, rO–C, between the O atom of the aminoxyl center and the C(β) atom at the position of β of the pyridine ring was 3.194(3) Å. This short contact is often observed in heterospin systems using aminoxyl as an organic spin.5a The observed short contacts might cause the intermolecular magnetic interaction in the cryogenic temperatures.
3.3. Gelation test and rheological measurements for 1 and 2
To confirm gel formation, the gel tests for 1 and 2 were carried out. The complexes were dissolved in 13 solvents and then 20 mM solution samples were prepared. Each sample was kept at −4 °C and the solutions after 24 h were visually confirmed (Table 2). For 1 having the n-hexyl group, the solubility for alcohols (entries 1–3 in Table 2) and aliphatic hydrocarbon (entries 12–14) solvents was poor and the precipitates appeared immediately after dissolving in the solvents. In contrast, the solubility in halogenated (entries 8 and 9), ethereal (entries 6, 7 and 11), and aprotic polar (entries 4, 5 and 10) solvents was high and the solutions were maintained even at −4 °C. Therefore, the mixed solvents of CH2Cl2 and EtOH, which were good and medium/poor solvents, respectively, were used as the solvent for 1. In EtOH solutions containing 20–40% CH2Cl2 (20–40% CH2Cl2/EtOH, entries 15–17), increase in the solvent viscosity was observed. By the tube inversion test,24 the sample dissolved in 20% CH2Cl2/EtOH (entry 16) became a transparent gel at −4 °C and the gel was maintained for more than 6 h at r.t. (Fig. 2). In contrast, 11 mM samples dissolved in 20% CH2Cl2/EtOH showed no gelation behavior. Furthermore, 2 having the n-octyl group showed similar solubility to 1 against 13 solvents and the gel accompanied with the powder was obtained under 20% CH2Cl2/EtOH (entry 16) condition. | ||
| Fig. 2 Photographs of “sol” (left) and “gel” (right) samples for 1 at rt: solutions (20 mM) dissolved in 20% CH2Cl2/EtOH at rt. | ||
| Entry | Solvents | 1 | 2 |
|---|---|---|---|
| S, S(h), I, P, G, and WG denote solution, solution by heating, insoluble, precipitation, gel, and weak gel, respectively. | |||
| 1 | MeOH | S(h) → P | S(h) → P |
| 2 | EtOH | S(h) → P | S(h) → P |
| 3 | BuOH | I | I |
| 4 | MeCN | I | I |
| 5 | Acetone | S | S |
| 6 | MTHF | S | S |
| 7 | 1,4-Dioxane | S | S |
| 8 | CH2Cl2 | S | S |
| 9 | CHCl3 | S | S |
| 10 | AcOEt | S | S → P |
| 11 | Et2O | S | S |
| 12 | n-Hexane | I | I |
| 13 | c-Hexane | S(h) | S(h) → P |
| 14 | Toluene | S(h) | S(h) → P + WG |
| 15 | 10% CH2Cl2/EtOH | S → P | S → P |
| 16 | 20% CH2Cl2/EtOH | G | S → P + G |
| 17 | 40% CH2Cl2/EtOH | WG | S |
Photographs of the gel and sol sample for 1 are shown in Fig. 2 and the tests for the gelation in various solvents are summarized in Table 2.
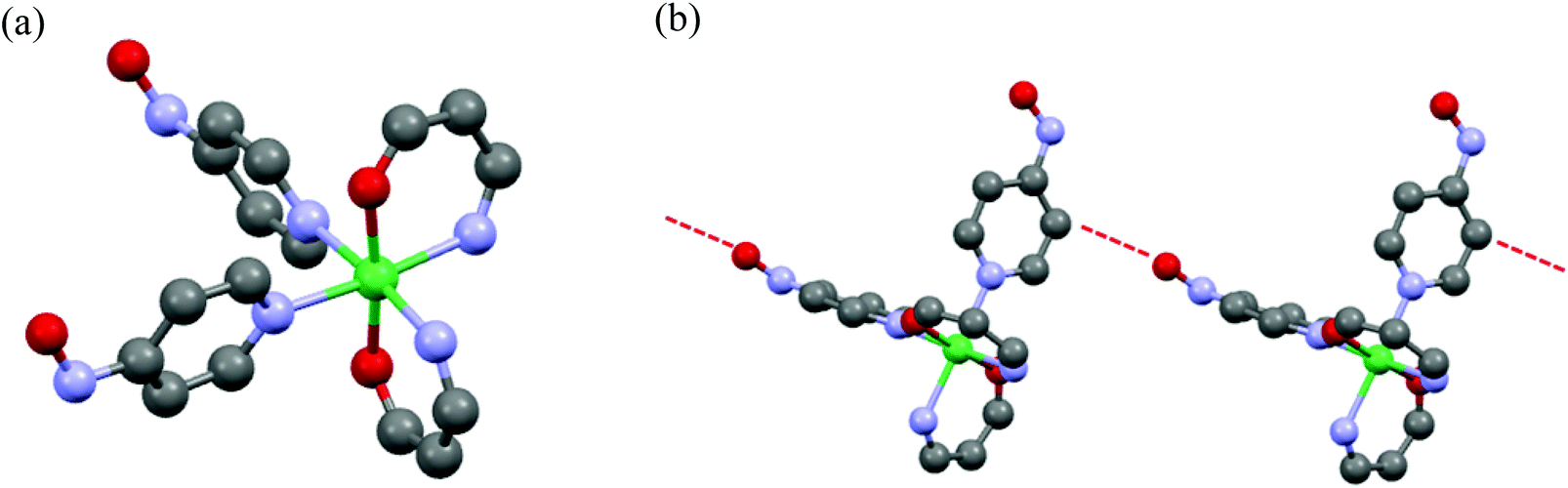
Rheological measurements of the obtained gel samples for 1 (entries 16 and 17) and 2 (entry 16) were carried out at 23 °C.25 The gel samples were prepared in parallel-type plates with a diameter of 50 mm and set in a rheometer. The strain (γ/%) dependence of the values of elastic modulus (G′) and viscous modulus (G′′) are shown in Fig. 3 for 1 and S2† for 2. According to the measurements, the oscillating frequency of 1 Hz was used under sweeping strain, γ, from 0.01 to 1000%. The G′ and G′′ indicate the ability by which a deformed material changed its original geometry back and the tendency of a material to flow, respectively. Therefore those rheological parameters provide the gel properties for the hardness and the softness from the value of G′ in a low γ region, and the stability and the strength by mechanical stress from an inflection point of the G′ value by changing the mechanical strain. The typical gel samples exhibit a G′ value independent of strain until the formation is broken and furthermore the G′ value exceeds the G′′ by about one order of magnitude.26
 | ||
| Fig. 3 Strain dependence of G′(filled circles or squares) and G′′ (empty circles and square) of the gel 1 obtained according to the conditions in entry 16 (a) and entry 17 (b). The dotted lines parallel to the horizontal axes and the dotted arrows indicate the plateau regions of G′ values in low γ regions and inflection points of G′ values upon increasing the strain. | ||
In the gel sample of 1 (entry 16), the G′ values in low γ regions were plateaus around 1.2–1.8 × 10 kPa. In this region, the G′ values were one order of magnitude larger than one of the G′′, indicating that the gel behaves as an elastic solid. On increasing the strain, an inflection point of the G′ value was observed at γ = 2.5% and the magnitude of G′′ values exceeded G′ values above 20% strain (G′ < G′′), indicating that the gel (entry 16) behaves as a viscous solution and the transformation from gel to sol took place at that point. In contrast, in the weak gel sample of 1 (entry 17), the G′ values showed a plateau around 6.6–7.1 kPa in a low γ region and an inflection point was observed at 0.63% strain. Furthermore, the transformation took place at 10% strain. When compared with the properties of those gels, the gel (entry 16) had larger G′ values in the low γ region and exhibited an inflection point at stronger strain, rather than the weak gel (entry 17). These results indicated that the potential properties of the gel were different and the gel obtained from the conditions in entry 16 had more hardness and strength against mechanical strain compared to the weak gel (entry 17). In the gel of 2 (entry 16), the observed gel behavior, in which the G′ values showed the plateau around 1.0–1.2 × 10 kPa in the low γ region, and an inflection point was observed at 1.59% strain, was consistent with that of the gel of 1 (entry 16). G′ and G′′ vs. γ plots of 1 and 2 are shown in Fig. 3 and S2,† respectively.
3.4. Vis-NIR spectra
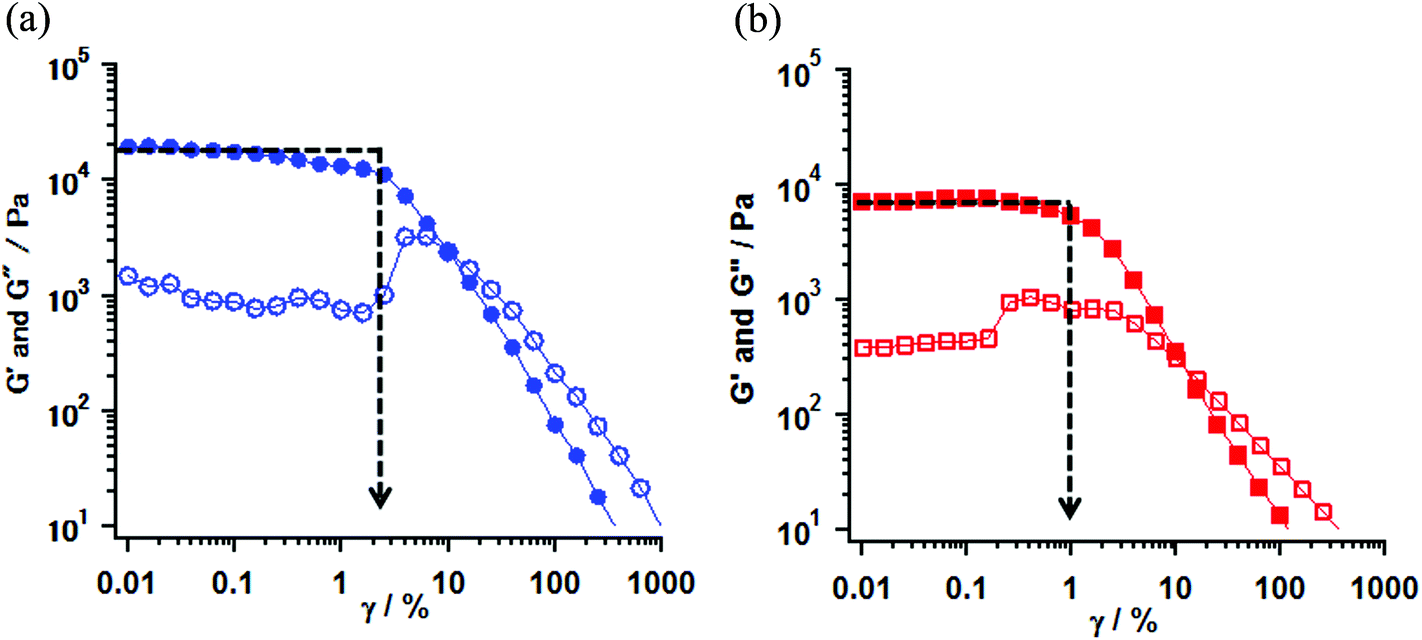
To confirm the coordination structure of 1 as the gel, 15 mM solution in MTHF, and powder samples, the visible-near infrared (Vis-NIR) spectra in the range of 500–1200 nm were examined. The gel obtained from the solution (20 mM) of 1 in 20% CH2Cl2/EtOH was used as a sample for the measurement. The 15 mM solution in MTHF showed no gelation behavior. In three samples, a set of shoulder and round maxima at λmax = ∼610 (16.4 × 103) and ∼995 (10.1 × 103) nm (cm−1), which were characteristic to the octahedron in the Ni(II) complex and were assigned as 3A2g → 3T1g and 3A2g → 3T2g, respectively,27 were observed. The octahedral structure by the coordination of D1py2 to the Ni(II) ion might suggest that the obtained Ni complexes consisted of a chain or a discrete cyclic structure. Vis-NIR spectra in the powder and the gel sample of 1 are shown in Fig. 4 and the spectrum for the 15 mM solution of 1 in MTHF is shown in Fig. S3.†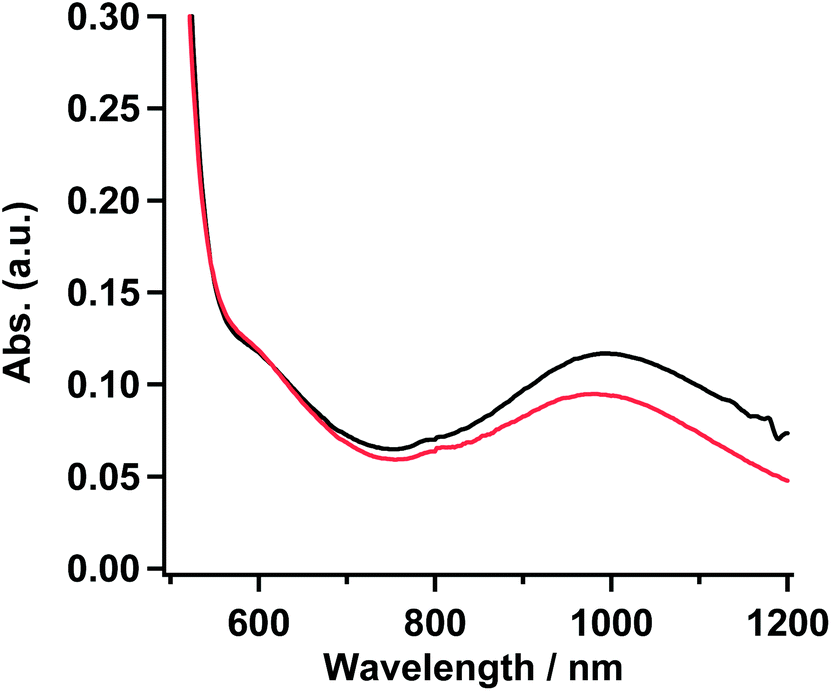 | ||
| Fig. 4 Absorption spectra of 1 in powder (red) and gel sample (black) obtained from the 20% CH2Cl2/EtOH solution. | ||
3.5. Morphology of the gel
To reveal the morphology of 1, the scanning electron microscopy (SEM) observations of the samples of the xerogel and the powder were carried out. The xerogel sample was prepared by freeze-drying the gel of 1. The SEM images for the xerogel and the powder exhibited the fibers of 500–600 nanometer width and the crystalline solid of micrometer size, respectively. The fibers observed in the gel might suggest that 1 had the chain structure formed by alternately binding D1py2 and Ni(Hex-hfpip)2 in the molecular level. The morphologies of 1 in the xerogel and the powder are shown in Fig. 5. In addition, counts vs. width of fiber obtained from the SEM image are summarized in Fig. S4.† | ||
| Fig. 5 SEM images of xerogel (a) and powder (b) samples for 1. White lines are scale bars. | ||
3.6. PXRD measurement
To deduce the molecular structure of the xerogel samples, PXRD measurements of xerogels for 1 and 2 were carried out in the 2θ range of 2–10° at 23 °C. Furthermore the distances obtained from PXRD peaks were compared to the SXRD results for the analogue cobalt chain complex, [Co(Br-hfpip)2D1py2]n, reported previously (Fig. S1′†).21 In the crystal structure of [Co(Br-hfpip)2D1py2]n, two pyridine rings in D1py2 coordinated to the Co ion in trans configuration and the distances of one pitch, one unit, and the width of the chain complex, which corresponded to A, B and C in Fig. S1′,† were 22.0, 10.0, and 13.7 Å, respectively.The xerogel samples obtained from freeze-drying gel of 1 (entry 16) and 2 after removal of powder (entry 16) were used. According to the results of PXRD measurements, the same patterns between 1 and 2, in which two intense peaks in small angle regions and some broadening peaks in small and wide angle regions, were clearly observed. The two intense peaks at 3.81 and 5.74° for 1 and 3.70 and 5.63° for 2 corresponded to the distances of 22.7 and 15.2 Å for 1 and 23.4 and 15.5 Å for 2, respectively. When compared with the distances obtained from SXRD results of the analogue [Co(Br-hfpip)2D1py2]n, the distances corresponded to the former and latter peaks were consistent with those of one pitch within the chain and the width of the chain, respectively (A and C in Fig. S1′†). Furthermore, the weak broadening peaks at 9.0 (9.6) and 8.8° (9.8 Å) for 1 and 2 in PXRD were observed at similar distances to a complex unit (B in Fig. S1′†) in a chain. Accordingly, the resulting PXRD patterns indicated that each xerogel of 1 and 2 formed similar chain structures, in which coordination geometry of the pyridine rings in D1py2 might be trans configuration such as the analogue [Co(Br-hfpip)2D1py2]n (Fig. 6).
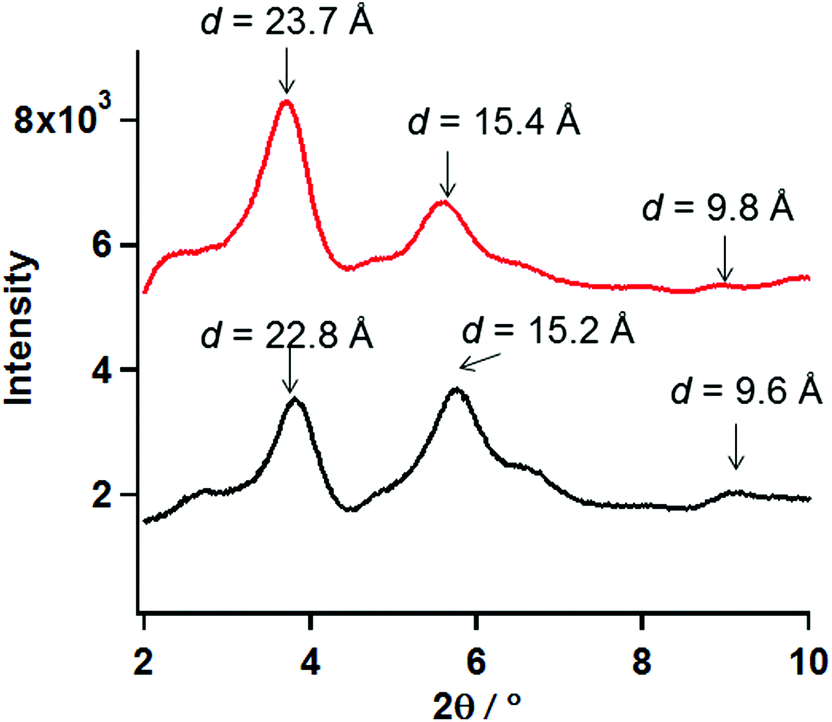 | ||
| Fig. 6 PXRD patterns of the xerogel samples of 1 (black) and 2 (red) in the 2θ range of 2–10°at 23 °C. | ||
3.7. Magnetic properties
To understand the magnetic interaction between the nickel(II) ion and the organic spin quantitatively, the magnetic measurements for the reference complex 3 together with 1 were performed by using a SQUID magneto/susceptometer.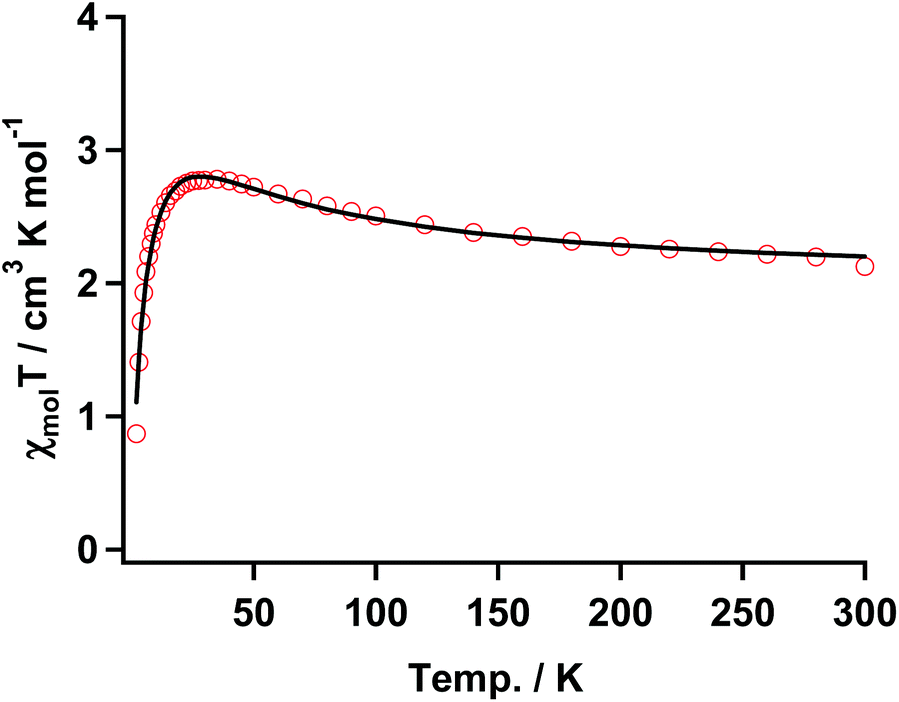 | ||
| Fig. 7 χ mol T vs. T plot of 3 with the fitting result (solid black line) according to the three spin model in eqn (1). The parameters are shown in the text. | ||
The temperature dependence of χmol values was applied to fitting of a linear three spin model: H = −J(SNiS1 + SNiS2), and the theoretical eqn (1), where J/kB, N, kB, μB, θ denote the magnetic coupling parameter, Avogadro number, Boltzman constant, Bohr magneton, and Weiss constant, respectively, fitted the experimental data. The values obtained by fitting were g = 2.14, J/kB = 26.7 K, and θ = −4.21 K, respectively, indicating that ferromagnetic interaction between Ni(II) and aminoxyl through the pyridine rings took place and the ground state of 3 was S = 4/2. In contrast, the obtained negative θ value indicated that intermolecular interaction of 3 antiferromagnetically took place. The fitting result is shown in Fig. 7 as a solid line.
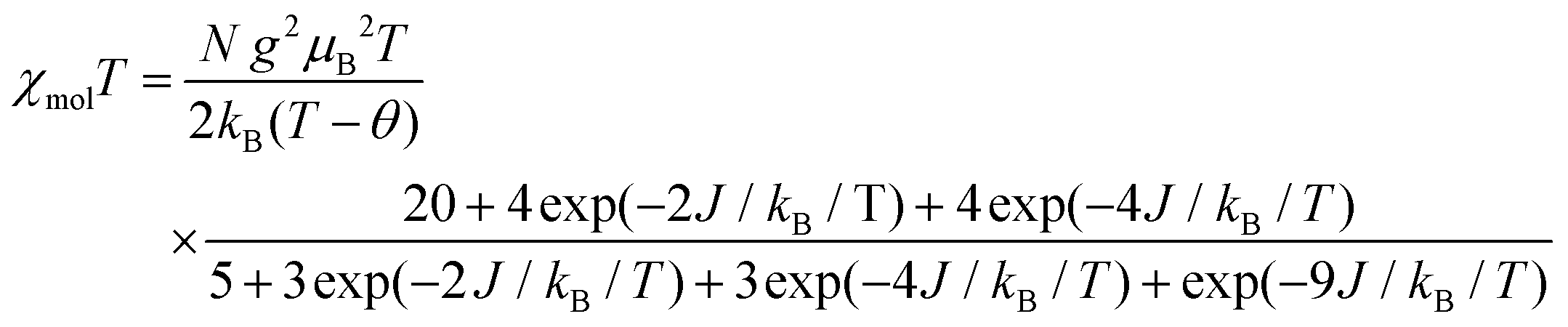 | (1) |
The thermal profile in the χmolT vs. T plot for the discrete complex of 3 indicated that aminoxyl ferromagnetically interacted with the Ni(II) ion of Ni(Hex-hfpip)2 through the pyridine rings. This result indicated that the magnetic coupling of the carbene generated by photolysis of 1 with the Ni(II) ion might be ferromagnetic.
The magnetic susceptibility measurements before and after irradiation were performed in the temperature range of 50–2 K under conditions similar to that for 3. The χmolT vs. T plots for the samples of 20 mM gel, 15 mM frozen solution, and powder of 1 before and after irradiation are summarized in Fig. 8.
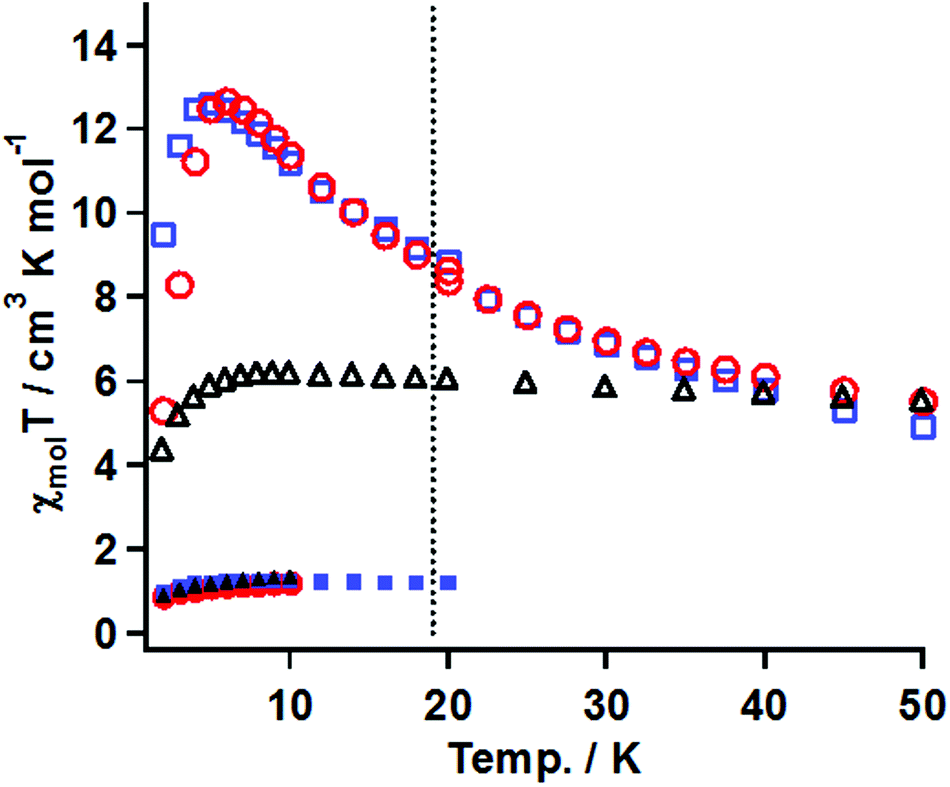 | ||
| Fig. 8 χ mol T vs. T plots before (filled marks) and after (blank marks) irradiation of 1 for samples of 20 mM gel (red circles), powder (blue squares), and 15 mM frozen solution (black triangles). Thermal profiles of the gel and powder samples were carried out under 5 kOe (50–20 K) and 0.5 kOe (below 18 K), respectively. A dotted delimiter line was put on 19 K. | ||
(A) Gel sample. The χmolT value before irradiation was 1.1 cm3 K mol−1 at 20 K, which was consistent with the magnetically isolated Ni(II) spin value when g = 2.0. On cooling, the χmolT value was constant until 2 K, suggesting that the ground state was S = 2/2. After irradiation for 33 h, the χmol values were collected at a constant field of 5 kOe in the temperature range of 50–20 K and at 0.5 kOe in 18–2 K. The χmolT value at 50 K was 5.5 cm3 K mol−1. This value was much larger than the magnetically isolated value of 2 cm3 K mol−1, which was calculated from S = 2/2 for the Ni(II) ion and S = 2/2 for triplet carbene, and the value of 3 cm3 K mol−1 calculated by a ferromagnetic Ni(II)-carbene unit with S = 4/2. The χmolT value at 50 K indicated that the ferromagnetic interaction between the triplet carbene and the Ni(II) ion took place to form a species with a high-spin ground state. On cooling, the χmolT value gradually increased and reached a maximum of 12.7 cm3 K mol−1 at 6 K. The observed increase of the χmolT values indicated that the correlation length of the magnetic chain became longer with decreasing temperature. The magnetic correlation length, n, was roughly estimated from eqn (2) and (3).29 In the ln(χmolT) vs. T−1 plot (Fig. S6†), the data from 50–15 K were fitted by a linear line model, giving the straight line and the value of Δξ = 13.5 K from the slope, which was the energy to create a domain wall. In the Heisenberg limit (|D| ≪|J|), the value of Δξ was correlated to 4ST2 √|JD| and kBT ln(2n) represented in eqn (2). Those parameters of J, D, ST, kB, T* and n denote the magnitude of magnetic interaction and zero-field splitting value within the Ni- triplet carbene complex, spin quantum number of one unit (S = 4/2 in this case), Boltzmann constant, the maxima value of χmolT and the correlation length, respectively. When using the Δξ value of 13.5 K and the T* value of 6 K in eq. (2), obtained from the χmolT vs. T plot (Fig. 8), it resulted in the value of n = 5.
| Δξ = kBT* ln(2n) = 4S2√|JD| | (2) |
| χnT/Ceff ≈ n | (3) |
In eqn (3), an effective Curie constant, Ceff, in 1 after irradiation was roughly deduced to the value of 3 cm3 K mol−1 calculated by a ferromagnetic Ni(II)-carbene unit with S = 4/2. From the maximum value of 12.7 cm3 K mol−1 at 6 K in χmolT vs. T plot, the n value, correlation length, was estimated to be 4 units. From the results of eqn (2) and (3), accordingly, n value was estimated to be 4–5 units. Below 5 K, the χmolT values steeply decreased due to intermolecular antiferromagnetic interaction.
(B) In the frozen sample solution sample. Before irradiation, the thermal profile of χmolT values was similar to those for the gel sample. After irradiation for 19 h, the χmol values were collected under conditions similar to that for the gel sample. The χmolT value at 50 K was 5.5 cm3 K mol−1, which was similar to the gel sample. On cooling, interestingly, the χmolT values were nearly constant until 6 K and then decreased below 6 K. The constant χmolT values in the range 50–6 K and their values of 5.5–6 cm3 K mol−1 might suggest that ferromagnetic species with a high-spin ground state formed and its S values was S = 6/2, respectively.
(C) Powder. The thermal profiles before and after irradiation for 20 h were similar to those of the gel sample. The maximum value after irradiation of χmolT was 12.6 cm3 K mol−1 at 5 K corresponding to the correlation length of 4–5 units, which was comparable with the value of the gel sample.
3.8. Molecular and magnetic structures of 1 in gel, solution, and powder samples
Vis-NIR spectra of the gel, the solution, and the powder samples of 1 showed a set of shoulder and round absorption maxima at λmax = ∼610(16.4 × 103) and ∼995(10.1 × 103) nm (cm−1) due to the octahedral Ni(II) complex, suggesting that the pyridine rings in D1py2 coordinated to the Ni(II) ions to form a chain and a cyclic structure. In magnetic measurements after irradiation of gel and frozen solution samples, the χmolT values at 50 K for both samples were similar at 5.5 cm3 K mol−1. This value of 5.5 cm3 K mol−1 was larger than 3.0 cm3 K mol−1 for the ferromagnetic unit (carbene-Ni(II) ion) with S = 4/2, indicating that the generated carbenes ferromagnetically interacted with the Ni(II) ions to form the high-spin species. The reference complex [Ni(Hex-hfpip)2(4NOpy)2] also showed ferromagnetic interaction with J/kB = 26.7 K between the aminoxyl and the Ni(II) through the pyridine ring. The thermal profiles of the χmolT values in the temperature range of 50–7 K showed different magnetic behaviors. In the χmolT vs. T plots, the χmolT values for the gel sample gradually increased with decreasing temperature, while those for the frozen solution sample were nearly constant. The observed increase of the χmolT values for the gel sample indicated that the correlation length of the magnetic chain became longer with decrease in temperature. In contrast, the observed temperature-independent thermal profile of the χmolT values for the frozen solution sample might suggest the formation of discrete high-spin species having a cyclic structure. The powder sample showing the similar magnetic behavior to that for the gel might have a chain structure. The magnetic behaviors observed in the gel and powder samples were similar to that for the copper chain complex, [Cu(hfac)2(D1py2)]n reported previously.9b Plausible molecular and magnetic structures of 1 in the gel and the frozen solution are depicted in Fig. 9.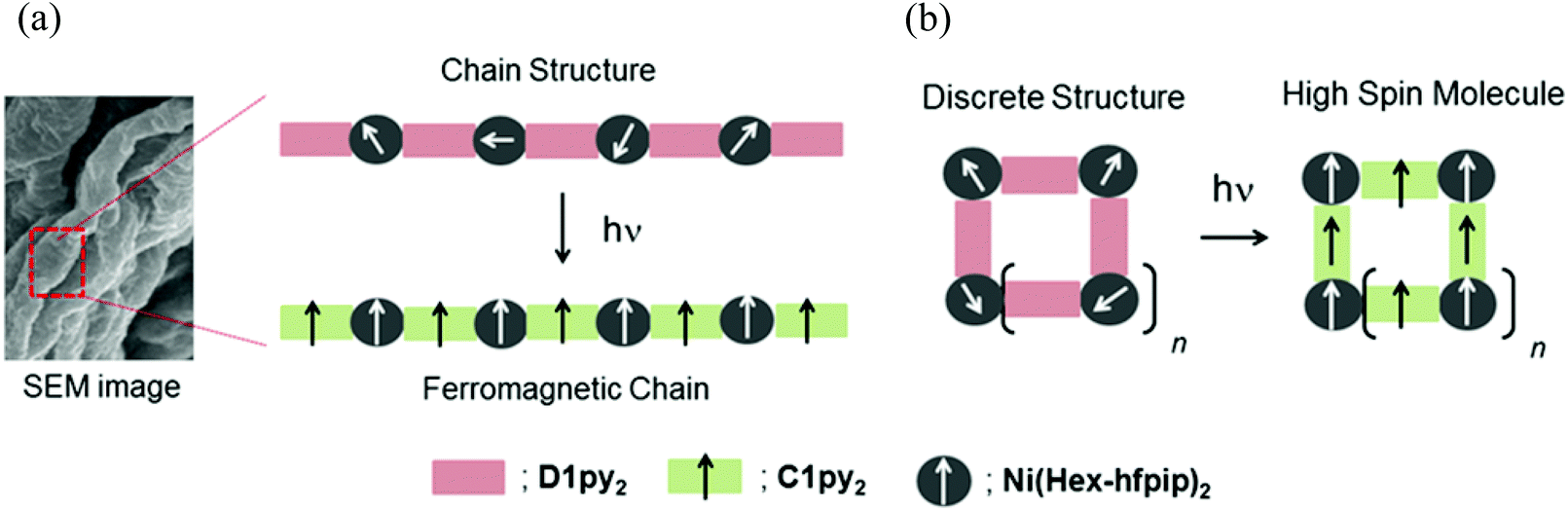 | ||
| Fig. 9 Plausible molecular and magnetic structures in the gel (a) and the frozen solution (b) of 1. | ||
4. Conclusion
The octahedral Ni(II) complexes consisting of D1py2 and Ni(R-hfpip)2; R = n-hexyl (1) and n-octyl (2), were prepared. In the gelation test and the rheological measurements, 1 and 2 were found to become a gel in 20 mM solution of 20% CH2Cl2/EtOH at −4 °C. In the SEM image of the xerogel obtained from freeze- drying the gel sample, fibers with the width of ca. 500–600 nm were observed. The resulting gel exhibited magnetic behavior of the ferromagnetic chain after irradiation. No example of a gel showing such strong magnetic interaction has been reported. Attempts to construct the magnetic gel showing the magnetic behavior of single-chain magnets (SCMs) by using anisotropic transition metal ions are in progress.Acknowledgements
This work was partially supported by Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. S. K. appreciates Grants-in-Aid for Scientific Research (B)(2)(no. 25288038) from the Japan Society for the Promotion of Science (JSPS), and PRESTO program on Molecular Technology from Japan Science Technology Agency (JST).References
- (a) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, New York, 2006 Search PubMed; (b) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66 CrossRef CAS.
- (a) Z.-X. Wang, X. Zhang, Y.-Z. Zhang, M.-X. Li, H. Zhao, M. Andruh and K. R. Dunbar, Angew. Chem., Int. Ed., 2014, 53, 11567 CrossRef CAS; (b) R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837 CrossRef PubMed; (c) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS.
- (a) K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952 CrossRef CAS PubMed; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed.
- (a) M. Jenkins, T. Hümmer, M. J. Martínez-Pérez, J. García-Ripoll, D. Zueco and F. Luis, New J. Phys., 2013, 15, 095007 CrossRef; (b) G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251 CrossRef CAS.
- (a) N. Koga and S. Karasawa, Bull. Chem. Soc. Jpn., 2005, 78, 1384 CrossRef CAS; (b) I.-R. Jeon, J. G. Park, D. J. Xiao and T. D. Harris, J. Am. Chem. Soc., 2013, 135, 16845 CrossRef CAS PubMed; (c) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538 CrossRef CAS.
- O. Kahn, Molecular Magnetism, Wiley-VCH Publishers, Weinheim, 1993 Search PubMed.
- (a) S. Kanegawa, S. Karasawa, M. Nakano and N. Koga, Chem. Commun., 2004, 3590 Search PubMed; (b) M. Kitano, Y. Ishimaru, K. Inoue, N. Koga and H. Iwamura, Inorg. Chem., 1994, 33, 6012 CrossRef CAS.
- (a) S. Karasawa and N. Koga, Inorg. Chem., 2011, 50, 5186 CrossRef CAS; (b) S. Karasawa, D. Yoshihara, N. Watanabe, M. Nakano and N. Koga, Dalton Trans., 2008, 1418 RSC; (c) S. Karasawa, G. Zhou, H. Morikawa and N. Koga, J. Am. Chem. Soc., 2003, 125, 13676 CrossRef CAS PubMed.
- (a) S. Karasawa, Y. Sano, T. Akita, N. Koga, T. Itoh, H. Iwamura, P. Rabu and M. Drillon, J. Am. Chem. Soc., 1998, 120, 10080 CrossRef CAS; (b) Y. Sano, M. Tanaka, N. Koga, K. Matsuda, H. Iwamura, P. Rabu and M. Drillon, J. Am. Chem. Soc., 1997, 119, 8246 CrossRef CAS.
- (a) D. Yoshihara, S. Karasawa and N. Koga, Inorg. Chim. Acta, 2015, 428, 57 CrossRef CAS PubMed; (b) D. Yoshihara, S. Karasawa and N. Koga, Polyhedron, 2011, 30, 3211 CrossRef CAS PubMed; (c) D. Yoshihara, S. Karasawa and N. Koga, J. Am. Chem. Soc., 2008, 130, 10460 CrossRef CAS PubMed.
- (a) S. Karasawa, K. Nakano, D. Yoshihara, N. Yamamoto, J. Tanokashira, T. Yoshizaki, Y. Inagaki and N. Koga, Inorg. Chem., 2014, 53, 5447 CrossRef CAS PubMed; (b) S. Karasawa, K. Nakano, J. Tanokashira, N. Yamamoto, T. Yoshizaki and N. Koga, Dalton Trans., 2012, 13656 RSC; (c) S. Karasawa, D. Yoshihara, N. Watanabe, M. Nakano and N. Koga, Dalton Trans., 2008, 1418 RSC; (d) H. Tobinaga, M. Suehiro, T. Itoh, G. Zhou, S. Karasawa and N. Koga, Polyhedron, 2007, 26, 1905 CrossRef CAS PubMed.
- (a) N. Annabi, K. Tsang, S. M. Mithieux, M. Nikkhah, A. Ameri, A. Khademhosseini and A. S. Weiss, Adv. Funct. Mater., 2013, 23, 4950 CrossRef CAS PubMed; (b) H. C. Fiegel, C. Lange, U. Kneser, W. Lambrecht, A. R. Zander, X. Rogiers and D. Kluth, J. Cell. Mol. Med., 2006, 10, 577 CrossRef CAS PubMed.
- (a) J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS; (b) P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddn, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402 CrossRef CAS PubMed.
- Y.-H. Kim, J.-S. Heo, T.-H. Kim, S. Park, M.-H. Yoon, J. Kim, M. S. Oh, G.-R. Yi, Y.-Y. Noh and S. K. Park, Nature, 2012, 489, 128 CrossRef CAS PubMed.
- T. Ono, T. Sugimoto, S. Shinkai and K. Sada, Nat. Mater., 2007, 6, 429 CrossRef CAS PubMed.
- (a) Y. Wu, Y. Hirai, Y. Tsunobuchi, H. Tokoro, H. Eimura, M. Yoshio, S. Ohkoshi and T. Kato, Chem. Sci., 2012, 3, 3007 RSC; (b) M. Shirakawa, N. Fujita, H. Shimakoshi, Y. Hisaeda and S. Shinkai, Tetrahedron, 2006, 62, 2016 CrossRef CAS.
- (a) G. Nandi, H. M. Titi, R. Thakuria and I. Goldberg, Cryst. Growth Des., 2014, 14, 2714 CrossRef CAS; (b) J. Park, J. H. Lee, J. Jaworski, S. Shinkai and J. H. Jung, Inorg. Chem., 2014, 53, 7181 CrossRef CAS; (c) C. Gonçalves, Y. Lalatonne, L. Melro, G. Badino, M. F. Ferreira, L. David, C. F. G. C. Geraldes, L. Motte, J. A. Martins and F. M. Gama, J. Mater. Chem. B, 2013, 1, 5853 RSC; (d) M. Takauchi, S. Tanaka and S. Shinkai, Chem. Commun., 2005, 5539 RSC.
- H. Hayashi, K. Ohkubo, S. Karasawa and N. Koga, Langmuir, 2011, 27, 12709 CrossRef CAS PubMed.
- (a) H. Kamata, Y. Akagi, Y. Kayasuga-Karita, U.-i. Chung and T. Sakai, Science, 2014, 343, 873 CrossRef CAS; (b) K. Takemura, H. Ajiro, T. fujiwara and M. Akashi, RSC Adv., 2014, 4, 63 RSC.
- (a) H. Komatsu, S. Matsumoto, S. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5580 CrossRef CAS PubMed; (b) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS PubMed; (c) S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58 CrossRef CAS PubMed; (d) J. H. Jung, S. Shinkai and T. Shimizu, Chem. – Eur. J., 2002, 8, 2684 CrossRef CAS.
- S. Karasawa and N. Koga, Inorg. Chem., 2011, 50, 2055 CrossRef CAS PubMed.
- SHELX 97: G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- Crystal Structure 3.5.1: Crystal Structure Analysis Package, Rigaku and Rigaku/MSC, 2000–2003, 9009 New Trails Dr. The Woodlands, TX 77381, USA.
- X.-D. Xu, J. Zhang, L.-J. Chen, X.-L. Zhao, D.-X. Wang and H.-B. Yang, Chem. – Eur. J., 2012, 18, 1659 CrossRef CAS.
- K. Matsumoto, A. Shundo, M. Ohno, S. Fujita, K. Saruhashi, N. Miyachi, K. Miyaji and K. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 2192 RSC.
- (a) S. Bhattacharya, S. Sengupta, S. Bala, A. Goswami, S. Ganguly and R. Mondal, Cryst. Growth Des., 2014, 14, 2366 CrossRef CAS; (b) J. H. Lee, S. Kang, J. Y. Lee, J. Jaworski and J. H. Jung, Chem. – Eur. J., 2013, 19, 16665 CrossRef CAS PubMed.
- (a) M. Arakawa, N. Suzuki, S. Kishi, M. Hasegawa, K. Satoh, E. Horn and Y. Fukuda, Bull. Chem. Soc. Jpn., 2008, 81, 127 CrossRef CAS; (b) K. Miyamoto, M. Sakamoto, C. Tanaka, E. Horn and Y. Fukuda, Bull. Chem. Soc. Jpn., 2005, 78, 1061 CrossRef CAS.
- (a) P. B. Sczaniecki and J. Lesniak, J. Magn. Reson., 1982, 46, 185 CAS; (b) J. Reedijik and B. Nieuwenhuijse, Recl. Trav. Chim. Pays-Bas, 1972, 91, 533 CrossRef PubMed.
- (a) H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420 CrossRef CAS PubMed; (b) M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K. Sugiura, M. Yamashita, C. Coulon and R. Clérac, J. Am. Chem. Soc., 2005, 127, 3090 CrossRef CAS PubMed; (c) C. Coulon, R. Clérac, R. Lecrem, W. Wernsdorfer and H. Miyasaka, Phys. Rev. B: Condens. Matter, 2004, 69, 132408 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional X-ray structure of 3. Rheological measurement of the gel for 2. Vis-NIR spectrum in a 15 mM MTHF solution sample of 1. M vs. irradiation time plots. ln(χmolT) vs. T plot after irradiation of the gel for 1. CCDC 1406724 for (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00109a |
| This journal is © the Partner Organisations 2015 |