
Cyclometallation of the NNN pincer ligand in complexes of platinum†
Jessica C.
DeMott
,
John R.
Dekarske‡
,
Billy J.
McCulloch
and
Oleg V.
Ozerov
*
Department of Chemistry, Texas A&M University, College Station, TX 77842, USA. E-mail: ozerov@chem.tamu.edu
First published on 28th August 2015
Abstract
Platinum complexes of the diimino/carbazolyl NNN pincer ligand have been prepared and characterized. Cyclometallation of the mesityl substituent was observed to occur unexpectedly easily in (NNN)PtOTf, which also catalyzed cyclometallation in (NNN)PtMe.
Introduction
Pincer complexes have gained in popularity in part because they offer a robust and well-defined framework for study of elementary reactions at transition metal centers.1 Our group, among many others, has been particularly attracted to pincer ligands based on the diarylamido central unit with ortho-positioned side donors (Chart 1).2–5 They provide a semi-rigid framework for binding transition metals while the amido donor may display redox6 or acid/base7,8 non-innocence. Carbazole can be regarded as a special diarylamine with greatly reduced basicity of the amido and increased rigidity of the underlying framework.9 We recently explored10 Pd complexes of the bis(imino)carbazolyl (NNN) pincer ligand pioneered by Gibson et al.11 Here we report the synthesis of NNN complexes of Pt, and observations of unexpectedly facile cyclometallation.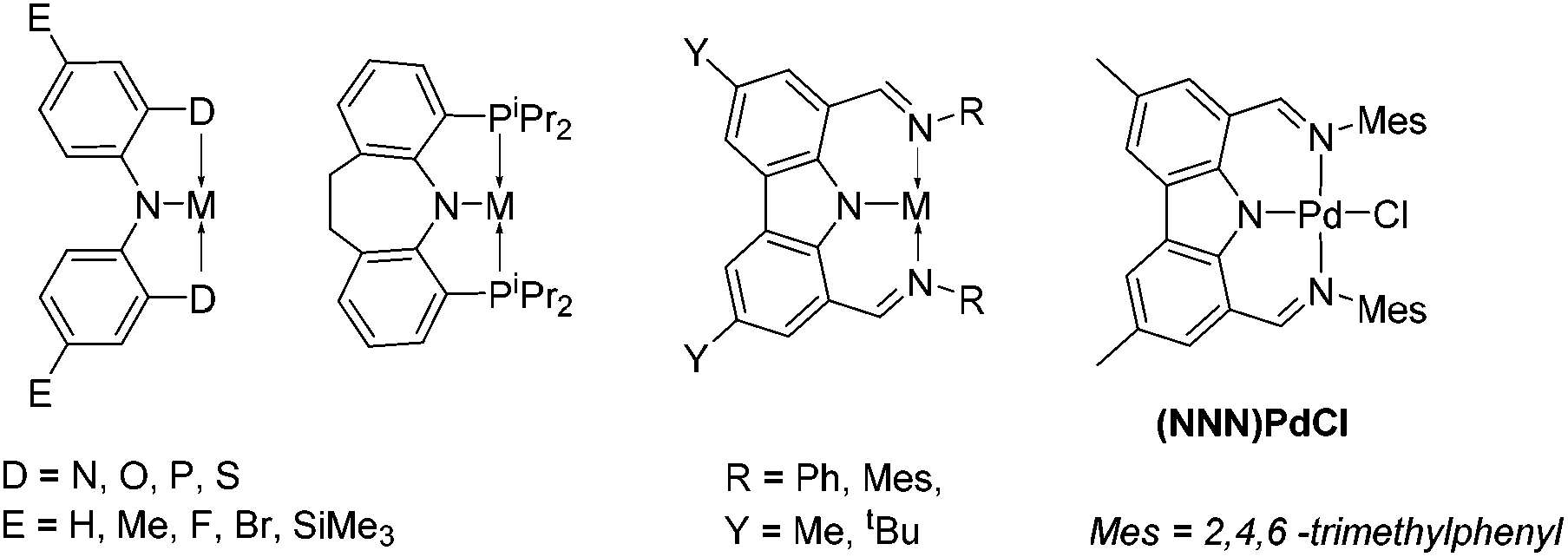 | ||
| Chart 1 Examples of diarylamido based pincer complexes. | ||
Results and discussion
Thermolysis of (NNN)Me with (MeCN)2PtCl2 in mesitylene for 24 h at reflux led to the formation of (NNN)PtCl (1) that was isolated in 41% yield (Scheme 1). Although it required considerably more forcing conditions, this preparation mimicked the synthesis of (NNN)PdCl,10 and also the related syntheses of PNP pincer complexes of group 10 metals by N-Me cleavage.12 Similarly to (NNN)PdCl, a quasi-reversible oxidation was observed for 1 by cyclic voltammetry. The formal potential for this primarily ligand-based10,13 redox event for 1 (0.65 V vs. Fc/Fc+) is a few hundredths of a volt more negative than for (NNN)PdCl (0.69 V), as was seen for other Pd–Pt comparisons with diarylamido-based pincer ligands.13 The synthesis of (NNN)PtMe (2) was accomplished by treatment of 1 with MeMgCl in toluene, followed by heating the mixture at 80 °C for 20 h, with a 74% isolated yield. 1 and 2 displayed the expected C2v symmetry in their 1H and 13C NMR spectra. The Pt-bound Me group in 2 gave rise to singlet resonances in the 1H NMR (δ 0.32 ppm) and 13C{1H} NMR (−10.5 ppm) spectra with the expected satellite peaks from coupling to 195Pt (2JPt–H = 70 Hz, 1JPt–C = 772 Hz).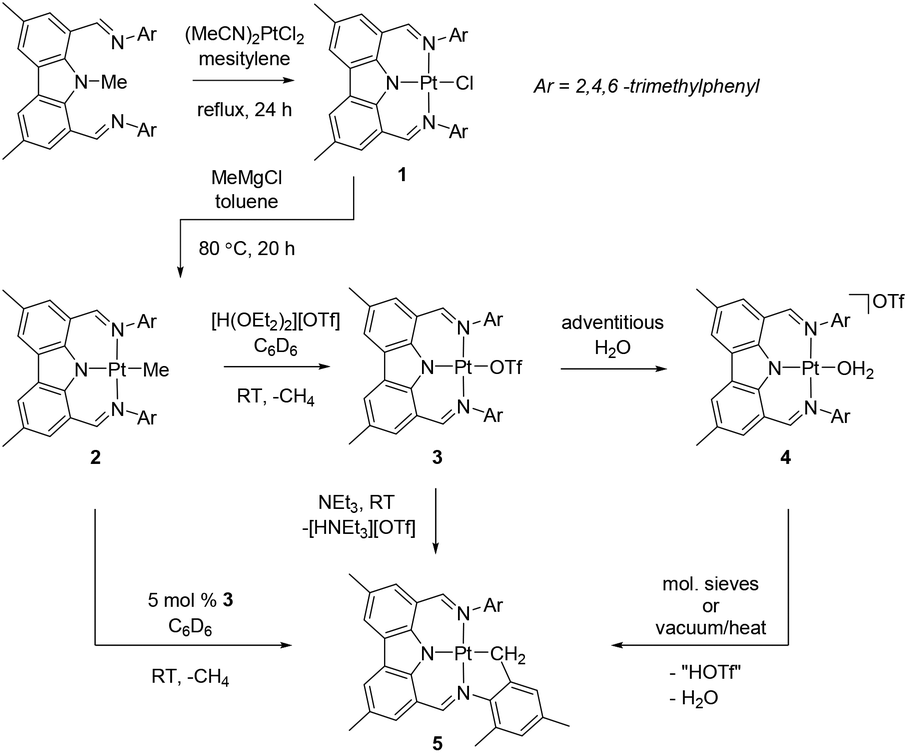 | ||
| Scheme 1 Syntheses and reactivity of (NNN)Pt complexes. | ||
A suspension of 2 in benzene rapidly reacted with one equiv. of triflic acid (in an ether solution) to lose methane and produce 3. When the reaction was performed under thoroughly dry conditions, 89% yield of pure 3 was isolated. It was noticed that in some reactions an impurity was present that was identified as the water adduct [(NNN)Pt–OH2][OTf] 4. We previously noted an analogous water adduct formation in the synthesis of the Pd analog (NNN)PdOTf.10 However, unlike in the Pd case, molecular sieves or application of vacuum were not effective at removing water from 4 and (re)generating 3. A small sample of the pure water adduct 4 was isolated following passing crude 3 through silica gel. The 1H NMR signal corresponding to the coordinated water molecule was observed at 4.86 ppm in C6D6. We were able to obtain two single crystals of 4 with different cocrystallized molecules: one contained a molecule of benzene in the asymmetric unit, while the other contained half of a disordered molecule of fluorobenzene, a molecule of triflic acid, and solvent-accesible voids modeled with SQUEEZE plausibly containing two water molecules each (Fig. 1 and ESI†). Triflic acid appears to be hydrogen bonded to the triflate anion of 4. The presence of HOTf was confirmed by its observation in the NMR spectrum of the dissolved crystals from this batch. The geometries of the central 4 cations in both structures are very similar. The corresponding Pt–N and Pt–O distances are statistically indistinguishable while the angles about Pt define a nearly perfect square planar environment with only 0–2° differences between the two structures. Similarly to the structure of (NNN)PdCl, the NNN ligand is nearly perfectly planar and pairwise π-stacking of the carbazole units is evident in the solid-state structures of 4.
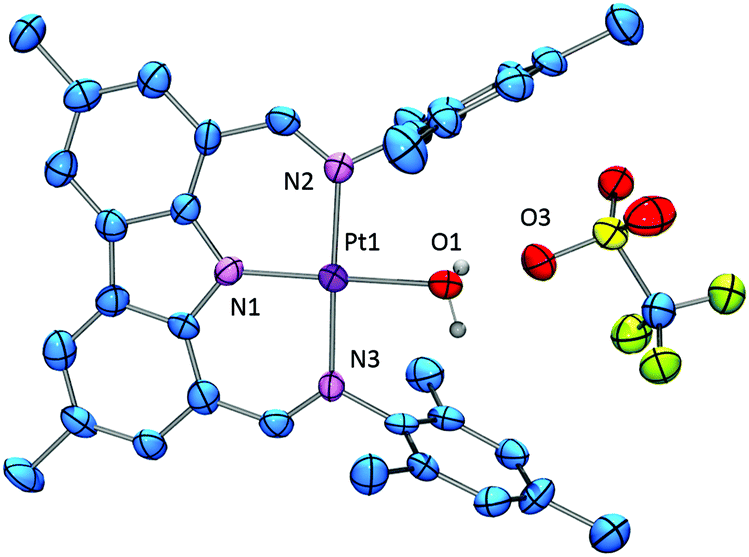 | ||
| Fig. 1 ORTEP drawing (50% probability ellipsoids) of 4. Omitted for clarity: H atoms and C6D6. Selected distance (Å) and angles (°) follow: Pt1–O1, 2.078(6); Pt1–N1, 1.933(6); Pt1–N2, 2.018(6); Pt1–N3, 2.023(6); N2–Pt1–N3, 177.1(2); N1–Pt1–O1, 175.7(2); N1–Pt1–N2, 91.0(3); N1–Pt1–N3, 90.5(3); N2–Pt1–O1, 87.9(2); N3–Pt1–O1, 90.8(2). | ||
Treatment of 3/4 mixtures with molecular sieves or heating under vacuum led to the appearance of a new Pt product in the mixture, which was identified as cyclometallated (NNN)Pt–CH2–C6H2(CH3)2 (5). 5 arises from cyclometallation of one of the mesityl substituents with a formal loss of triflic acid from 3. We were able to prepare pure 5 by treatment of 3 with Et3N in benzene at ambient temperature. The NMR spectra of 5 are consistent with partial loss of symmetry associated with cyclometallation. The equivalent protons of the Pt-bound CH2 group resonate at δ 2.35 ppm in C6D6. The 195Pt NMR chemical shifts14 of 2 (−2483 ppm) and 5 (−2589 ppm) are quite similar, and distinct from those of 1 (−1849 ppm) and 3 (−1493 ppm). Interestingly, we found that 3 serves as the catalyst for cyclometallation of 2. The latter is indefinitely stable in C6D6 solutions at ambient temperature, but addition of 5% 3 led to the appearance of 15% of 5 and free methane within 5 min (and 80% conversion to 5 after 18 h). Since 3 loses HOTf under vacuum and to a weak base such as water, it seems plausible that 2 can serve as the base to promote cyclometallation of 3 to 5 by accepting the formal equivalent of HOTf, evolving methane and regenerating 3.
The solid-state structure of 5 was determined by X-ray diffraction methods (Fig. 2). The coordination environment about Pt is approximately square-planar and is defined by the now tetradentate cyclometallated NNN ligand. With the exception of the intact mesityl group, this molecule possesses a remarkably flat extended conjugated system about the Pt center. Not surprisingly, molecules of 5 in the solid state display extensive π-stacking (Fig. 2).
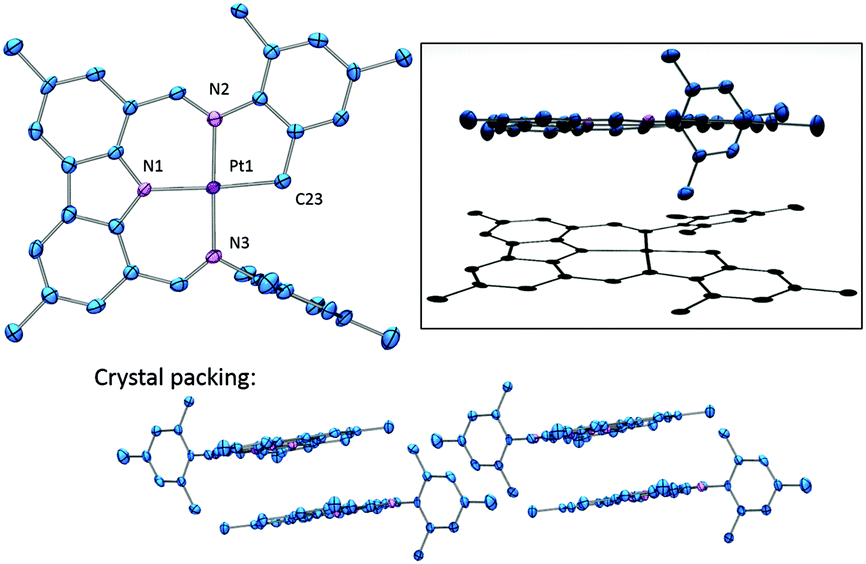 | ||
| Fig. 2 ORTEP drawing (50% probability ellipsoids) of 5 with crystal packing and Pov-ray generated figure depicting side-view and shadow. Omitted for clarity: H atoms. Selected distance (Å) and angles (°) follow: Pt1–C23, 2.027(5); Pt1–N1, 2.007(4); Pt–N2, 2.011(4); Pt–N3, 2.012(4); N2–Pt1–N3, 178.37(15); N1–Pt1–C23, 176.43(16); N1–Pt1–N2, 92.89(16); N1–Pt1–N3, 88.70(16); N2–Pt1–C23, 83.59(18); N3–Pt1–C23, 94.82(18). | ||
Mesityl group is a common substituent on a nitrogen atom in polydentate ligands. Cyclometallation of an N-mesityl moiety is well documented in the chemistry of early transition,15 lanthanide,16 and main group17 metals. However, it is quite rare for late metals18 and the existing examples have not been formed by electrophilic cyclometallation observed in 3. On the other hand, cyclometallation of the NNN ligand in this work can be related to the literature examples of intermolecular C–H activation by PtII–OTf complexes of amido-based pincer ligands (Scheme 2). In those examples from Peters,19 Liang,20 and our own work,8 activation of aromatic solvents proceeded sluggishly at elevated temperatures in contrast to the rapid ambient-temperature C–H activation of 3. While the intramolecular nature of the reaction in 3 cannot be underestimated, it is worth pointing out that the examples in Scheme 2 were judged to proceed by a dissociative mechanism. This loss of triflate may be more facile in the NNN system as evidenced by the ready displacement of it by water (for both Pt and Pd), which is not observed for the corresponding PNP complexes. The steric bulk projected by the mesityl groups of the NNN ligand towards the non-pincer coordination site at the square planar metal center may be favouring coordination of smaller ligands (e.g., water). We hypothesize that evolution of the NNN ligand aimed at preventing cyclometallation while preserving strong steric pressure on the reactive site may lead to exciting intermolecular reactivity, and this will be a subject of future studies.
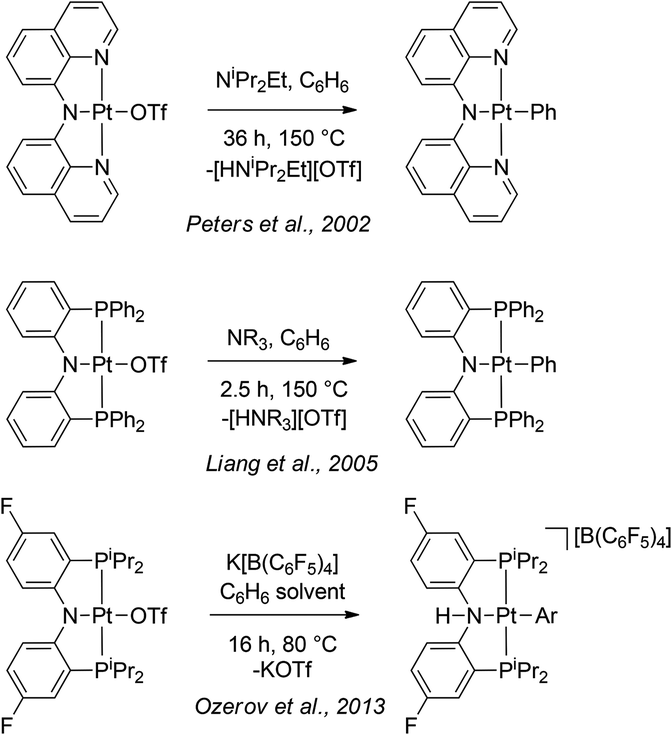 | ||
| Scheme 2 C–H activation of benzene by (XNX)Pt–OTf pincer complexes. | ||
Conclusions
In summary, a series of Pt complexes supported by a bis(imino)carbazolyl pincer ligand possessing mesityl sidearms has been synthesized. Comparison to the previously reported (NNN)PdX complexes10 demonstrates very similar metrics, redox potentials, and facile displacement of triflate by water at the metal center. This behavior is in contrast to that observed for analogous (PNP)MOTf complexes and is believed to result from the alternative steric profile and extended steric bulk of the mesityl groups. Surprisingly, weak bases including 2 in the presence of a catalytic amount of 3 were observed to consume an equivalent of HOTf to form the cyclometallated mesityl complex 5 through facile intramolecular C–H activation at room temperature.Experimental section
General considerations and physical methods
Unless otherwise noted, all manipulations and reactions were performed under argon, using standard glovebox and Schlenk line techniques. Toluene, triethylamine, pentane, and C6D6 were dried over NaK/Ph2CO/18-crown-6, distilled and stored over molecular sieves in an Ar-filled glovebox. Diethyl ether was dried and deoxygenated (by purging) using a solvent purification system and stored over molecular sieves in an Ar-filled glovebox. Fluorobenzene and CH2Cl2 were dried over CaH2, distilled, and stored over molecular sieves in an Ar-filled glovebox.NMR spectra were recorded on a Varian Inova 300 spectrometer (1H NMR, 300.0 MHz, 13C NMR, 75.4 MHz, 19F NMR, 282.2 MHz, 31P NMR, 121.4 MHz), a Varian Inova 400 spectrometer (1H NMR, 399.5 MHz, 31P NMR, 161.7 MHz, 13C NMR, 100.5 MHz, 195Pt NMR, 85.5 MHz) and a Varian Inova 500 spectrometer (1H NMR, 499.7 MHz, 13C NMR, 125.7 MHz, 19F NMR, 470.1 MHz, 31P NMR, 202.3 MHz) in noted solvents. Chemical shifts are given in δ (ppm). 19F NMR spectra were referenced externally using CF3COOH at δ −78.5 ppm. 31P NMR spectra were referenced externally with 85% phosphoric acid at δ 0. 195Pt NMR spectra were referenced externally with K2PtCl4 aqueous solution at δ −1620. 1H NMR and 13C NMR spectra were referenced using the solvent signals. Elemental analyses were performed by CALI, Inc. (Parsippany, NJ).
Synthesis of Pt complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 (2.17 g, 6.23 mmol) was added to the 250 mL Schlenk flask. Reaction was put under reflux in an oil bath at 180 °C for 24 h. Reaction changed color from yellow to brown-orange after approximately an hour sitting in the oil bath. Volatiles were removed by vacuum. The resulting oily brown residue was filtered through Celite and silica gel using toluene. Volatiles were removed under vacuum which yielded an orange powder. Solid was filtered off and washed with cold pentane (3 × 10 mL). Yield: 1.8 g, 41%. 1H NMR (300 MHz, C6D6): δ 8.16 (s, 2H, Ar–H), 8.15 (s, 2H, N
21 (2.17 g, 6.23 mmol) was added to the 250 mL Schlenk flask. Reaction was put under reflux in an oil bath at 180 °C for 24 h. Reaction changed color from yellow to brown-orange after approximately an hour sitting in the oil bath. Volatiles were removed by vacuum. The resulting oily brown residue was filtered through Celite and silica gel using toluene. Volatiles were removed under vacuum which yielded an orange powder. Solid was filtered off and washed with cold pentane (3 × 10 mL). Yield: 1.8 g, 41%. 1H NMR (300 MHz, C6D6): δ 8.16 (s, 2H, Ar–H), 8.15 (s, 2H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.10 (s, 2H, Ar–H), 6.84 (s, 4H, Ar–H), 2.45 (s, 6H, Ar–CH3), 2.32 (s, 12H, Ar–CH3), 2.18 (s, 6H, Ar–CH3). 13C{1H} NMR (75.4 MHz, CDCl3): δ 157.8 (CN), 151.8 (CAr), 136.2 (CAr), 135.7 (CAr), 131.3 (CAr), 130.9 (CAr), 128.4 (CAr), 128.0 (CAr), 125.6 (CAr), 117.1 (CAr), 21.5 (Ar–CH3), 21.0 (Ar–CH3), 18.5 (Ar–CH3). 195Pt NMR (85.7 MHz, CDCl3): δ −1849. Elem. An. Found (Calculated) for C34H34N3PtCl: C, 57.07 (57.10); H, 4.73 (4.79)%.
CH), 8.33 (s, 2H, Ar–H), 7.26 (s, 2H, Ar–H), 6.83 (s, 4H, Ar–H), 2.53 (s, 6H, Ar–CH3), 2.24 (s, 12H, Ar–CH3), 2.19 (s, 6H, Ar–CH3), 0.32 (s, 3H, JPt–H = 69.6 Hz, Pt–CH3). 1H NMR (300 MHz, CD2Cl2): δ 8.79 (s, 2H, JPt–H = 98 Hz, NCH), 8.56 (s, 2H, Ar–H), 7.73 (s, 2H, Ar–H), 6.93 (s, 4H, Ar–H), 2.72 (s, 6H, Ar–CH3), 2.32 (s, 6H, Ar–CH3), 2.13 (s, 12H, Ar–CH3), −0.39 (s, 3H, JPt–H = 70.5 Hz, Pt–CH3). 13C{1H} NMR (100.5 MHz, CDCl3): 158.0 (CN), 151.2 (CAr), 138.77 (CAr), 135.5 (CAr), 131.0 (CAr), 130.8 (CAr), 128.4 (CAr), 127.4 (CAr), 127.0 (CAr), 117.6 (CAr), 21.7 (Ar–CH3), 20.9 (Ar–CH3), 18.1 (Ar–CH3), −10.5 (s, JPt–C = 772 Hz, Pt–Me). 195Pt NMR (85.7 MHz, CD2Cl2): δ −2483. Elem. An. Found (Calculated) for C35H37N3Pt: C, 60.53 (60.51); H, 5.46 (5.37)%.
CH), 7.00 (s, 2H, Ar–H), 6.92 (s, 4H, Ar–H), 2.50 (s, 12H, Ar–CH3), 2.41 (s, 6H, Ar–CH3), 2.22 (s, 6H, Ar–CH3). 13C{1H} NMR (75.4 MHz, C6D6): 160.7, 149.4, 136.7, 135.9, 132.4, 131.8, 129.4, 125.5, 117.0, 21.1, 21.0, 19.5, –CF3 signal not resolved. 19F NMR (282.2 MHz, C6D6): δ −77.78. 195Pt NMR (85.7 MHz, C6D6): δ −1493. Elem. An. Found (Calculated) for C35H34F3N3O3SPt: C, 50.57 (50.72); H, 4.18 (4.14)%.
CH), 7.12 (s, 2H, Ar–H), 7.07 (s, 4H, Ar–H), 4.86 (br, 2H, Pt–OH2), 2.45 (s, 6H, Ar–CH3), 2.35 (s, 6H, Ar–CH3), 2.20 (s, 12H, Ar–CH3). 13C{1H} NMR (75.4 MHz, C6D6): 158.6 (CN), 145.9 (CAr), 138.8 (CAr), 135.8 (CAr), 132.0 (CAr), 130.8 (CAr), 129.4 (CAr), 128.8 (CAr), 125.9 (CAr), 117.1 (CAr), 21.12 (Ar–CH3), 21.0 (Ar–CH3), 18.0 (Ar–CH3), CF3 signal not resolved. 19F NMR (282.2 MHz, C6D6): δ −78.70. Elem. An. Found (Calculated) for C35H36F3N3O4SPt: C, 49.43 (49.64); H, 4.44 (4.29)%.
CH), 8.43 (s, 1H, JPt–H = 90 Hz, NCH), 8.37 (s, 1H, Ar–H), 8.31 (s, 1H, Ar–H), 7.50 (s, 1H, Ar–H), 7.34 (s, 1H, Ar–H), 7.00 (s, 1H, Ar–H), 6.96 (s, 2H, Ar–H), 6.52 (s, 1H, Ar–H), 2.95 (s, 2H, JPt–H = 66 Hz, Pt–CH2–Ar), 2.56 (s, 3H, Ar–CH3), 2.53 (s, 3H, Ar–CH3), 2.35 (s, 3H, Ar–CH3), 2.32 (s, 3H, Ar–CH3), 2.28 (s, 6H, Ar–CH3), 2.09 (d, 3H, Ar–CH3). 1H NMR (300 MHz, CD2Cl2): δ 9.68 (s, 1H, JPt–H = 90 Hz, NCH), 8.77 (s, 1H, JPt–H = 90 Hz, NCH), 8.58 (s, 1H, Ar–H), 8.53 (s, 1H, Ar–H), 7.90 (s, 1H, Ar–H), 7.78 (s, 1H, Ar–H), 7.06 (s, 2H, Ar–H), 6.93 (s, 1H, Ar–H), 6.76 (s, 1H, Ar–H), 2.85 (s, 3H, Ar–CH3), 2.76 (s, 3H, Ar–CH3), 2.74 (s, 3H, Ar–CH3), 2.46 (s, 3H, Ar–CH3), 2.35 (s, 2H, coupling to Pt obscured, Pt–CH2–Ar), 2.27 (s, 3H, Ar–CH3), 2.19 (d, 6H, Ar–CH3). 13C{1H} NMR (75.4 MHz, CD2Cl2): δ 157.3, 155.1, 152.4, 151.4, 151.2, 138.2, 138.0, 136.2, 135.9, 131.9, 131.0, 130.9, 130.4, 129.6, 128.6, 128.0, 127.8, 127.7, 127.7, 126.7, 126.3, 118.8, 118.1, 24.5, 21.7, 21.0, 20.4, 17.9, 16.4. 195Pt NMR (85.7 MHz, C6D6): δ −2589. Elem. An. Found (Calculated) for C34H33N3Pt: C, 60.28 (60.17); H, 4.87 (4.90)%.
CH), 7.10 (s, 2H, Ar–H), 6.84 (s, 4H, Ar–H), 2.45 (s, 6H, Ar–CH3), 2.32 (s, 12H, Ar–CH3), 2.18 (s, 6H, Ar–CH3). 13C{1H} NMR (75.4 MHz, CDCl3): δ 157.8 (CN), 151.8 (CAr), 136.2 (CAr), 135.7 (CAr), 131.3 (CAr), 130.9 (CAr), 128.4 (CAr), 128.0 (CAr), 125.6 (CAr), 117.1 (CAr), 21.5 (Ar–CH3), 21.0 (Ar–CH3), 18.5 (Ar–CH3). 195Pt NMR (85.7 MHz, CDCl3): δ −1849. Elem. An. Found (Calculated) for C34H34N3PtCl: C, 57.07 (57.10); H, 4.73 (4.79)%.
CH), 8.33 (s, 2H, Ar–H), 7.26 (s, 2H, Ar–H), 6.83 (s, 4H, Ar–H), 2.53 (s, 6H, Ar–CH3), 2.24 (s, 12H, Ar–CH3), 2.19 (s, 6H, Ar–CH3), 0.32 (s, 3H, JPt–H = 69.6 Hz, Pt–CH3). 1H NMR (300 MHz, CD2Cl2): δ 8.79 (s, 2H, JPt–H = 98 Hz, NCH), 8.56 (s, 2H, Ar–H), 7.73 (s, 2H, Ar–H), 6.93 (s, 4H, Ar–H), 2.72 (s, 6H, Ar–CH3), 2.32 (s, 6H, Ar–CH3), 2.13 (s, 12H, Ar–CH3), −0.39 (s, 3H, JPt–H = 70.5 Hz, Pt–CH3). 13C{1H} NMR (100.5 MHz, CDCl3): 158.0 (CN), 151.2 (CAr), 138.77 (CAr), 135.5 (CAr), 131.0 (CAr), 130.8 (CAr), 128.4 (CAr), 127.4 (CAr), 127.0 (CAr), 117.6 (CAr), 21.7 (Ar–CH3), 20.9 (Ar–CH3), 18.1 (Ar–CH3), −10.5 (s, JPt–C = 772 Hz, Pt–Me). 195Pt NMR (85.7 MHz, CD2Cl2): δ −2483. Elem. An. Found (Calculated) for C35H37N3Pt: C, 60.53 (60.51); H, 5.46 (5.37)%.
CH), 7.00 (s, 2H, Ar–H), 6.92 (s, 4H, Ar–H), 2.50 (s, 12H, Ar–CH3), 2.41 (s, 6H, Ar–CH3), 2.22 (s, 6H, Ar–CH3). 13C{1H} NMR (75.4 MHz, C6D6): 160.7, 149.4, 136.7, 135.9, 132.4, 131.8, 129.4, 125.5, 117.0, 21.1, 21.0, 19.5, –CF3 signal not resolved. 19F NMR (282.2 MHz, C6D6): δ −77.78. 195Pt NMR (85.7 MHz, C6D6): δ −1493. Elem. An. Found (Calculated) for C35H34F3N3O3SPt: C, 50.57 (50.72); H, 4.18 (4.14)%.
CH), 7.12 (s, 2H, Ar–H), 7.07 (s, 4H, Ar–H), 4.86 (br, 2H, Pt–OH2), 2.45 (s, 6H, Ar–CH3), 2.35 (s, 6H, Ar–CH3), 2.20 (s, 12H, Ar–CH3). 13C{1H} NMR (75.4 MHz, C6D6): 158.6 (CN), 145.9 (CAr), 138.8 (CAr), 135.8 (CAr), 132.0 (CAr), 130.8 (CAr), 129.4 (CAr), 128.8 (CAr), 125.9 (CAr), 117.1 (CAr), 21.12 (Ar–CH3), 21.0 (Ar–CH3), 18.0 (Ar–CH3), CF3 signal not resolved. 19F NMR (282.2 MHz, C6D6): δ −78.70. Elem. An. Found (Calculated) for C35H36F3N3O4SPt: C, 49.43 (49.64); H, 4.44 (4.29)%.
CH), 8.43 (s, 1H, JPt–H = 90 Hz, NCH), 8.37 (s, 1H, Ar–H), 8.31 (s, 1H, Ar–H), 7.50 (s, 1H, Ar–H), 7.34 (s, 1H, Ar–H), 7.00 (s, 1H, Ar–H), 6.96 (s, 2H, Ar–H), 6.52 (s, 1H, Ar–H), 2.95 (s, 2H, JPt–H = 66 Hz, Pt–CH2–Ar), 2.56 (s, 3H, Ar–CH3), 2.53 (s, 3H, Ar–CH3), 2.35 (s, 3H, Ar–CH3), 2.32 (s, 3H, Ar–CH3), 2.28 (s, 6H, Ar–CH3), 2.09 (d, 3H, Ar–CH3). 1H NMR (300 MHz, CD2Cl2): δ 9.68 (s, 1H, JPt–H = 90 Hz, NCH), 8.77 (s, 1H, JPt–H = 90 Hz, NCH), 8.58 (s, 1H, Ar–H), 8.53 (s, 1H, Ar–H), 7.90 (s, 1H, Ar–H), 7.78 (s, 1H, Ar–H), 7.06 (s, 2H, Ar–H), 6.93 (s, 1H, Ar–H), 6.76 (s, 1H, Ar–H), 2.85 (s, 3H, Ar–CH3), 2.76 (s, 3H, Ar–CH3), 2.74 (s, 3H, Ar–CH3), 2.46 (s, 3H, Ar–CH3), 2.35 (s, 2H, coupling to Pt obscured, Pt–CH2–Ar), 2.27 (s, 3H, Ar–CH3), 2.19 (d, 6H, Ar–CH3). 13C{1H} NMR (75.4 MHz, CD2Cl2): δ 157.3, 155.1, 152.4, 151.4, 151.2, 138.2, 138.0, 136.2, 135.9, 131.9, 131.0, 130.9, 130.4, 129.6, 128.6, 128.0, 127.8, 127.7, 127.7, 126.7, 126.3, 118.8, 118.1, 24.5, 21.7, 21.0, 20.4, 17.9, 16.4. 195Pt NMR (85.7 MHz, C6D6): δ −2589. Elem. An. Found (Calculated) for C34H33N3Pt: C, 60.28 (60.17); H, 4.87 (4.90)%.
Reactivity studies
Acknowledgements
We are thankful to the US National Science Foundation (grant CHE-1062840 for the REU site at TAMU and grant CHE-1300299 to O.V.O.) and the Welch Foundation (grant A-1717 to O.V.O.) for support of this research. We are also grateful to Ms. Linda Redd for editorial assistance, to Allen Lunsford for assistance with cyclic voltammetry measurements, and to Prof. Andrei Vedernikov for insightful discussions.Notes and references
- For information on the chemistry of pincer ligands in general, see: (a) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (b) The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. Jensen, Elsevier, Amsterdam, 2007 Search PubMed; (c) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed.
- (a) L.-C. Liang, Coord. Chem. Rev., 2006, 250, 1152 CrossRef CAS PubMed; (b) O. V. Ozerov, in The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. Jensen, Elsevier, Amsterdam, 2007, pp. 287–309 Search PubMed; (c) D. J. Mindiola, Acc. Chem. Res., 2006, 39, 813 CrossRef CAS PubMed.
- (a) L. Fan, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 326 CrossRef CAS; (b) A. M. Winter, K. Eichele, H.-G. Mack, S. Potuznik, H. A. Mayer and W. C. Kaska, J. Organomet. Chem., 2003, 682, 149 CrossRef CAS; (c) L.-C. Liang, J.-M. Lin and C.-H. Hung, Organometallics, 2003, 22, 3007 CrossRef CAS.
- (a) S. B. Harkins and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 2030 CrossRef CAS PubMed; (b) E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 11161 CrossRef CAS PubMed; (c) M. T. Whited and R. H. Grubbs, Acc. Chem. Res., 2009, 42, 1607 CrossRef CAS PubMed; (d) T. Cantat, C. R. Graves, B. L. Scott and J. L. Kiplinger, Angew. Chem., Int. Ed., 2009, 48, 3681 CrossRef CAS PubMed.
- (a) S. B. Harkins and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 2885 CrossRef CAS PubMed; (b) Z. Csok, O. Vechorkin, S. B. Harkins, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2008, 130, 8156 CrossRef CAS PubMed.
- (a) A. T. Radosevich, J. G. Melnick, S. A. Stoian, D. Bacciu, C.-H. Chen, B. M. Foxman, O. V. Ozerov and D. G. Nocera, Inorg. Chem., 2009, 48, 9214 CrossRef CAS PubMed; (b) D. Adhikari, S. Mossin, F. Basuli, J. C. Huffman, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2008, 130, 3676 CrossRef CAS PubMed; (c) V. N. Madhira, P. Ren, O. Vechorkin, Z. Hu and D. A. Vicic, Dalton Trans., 2012, 41, 7915 RSC; (d) J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916 CrossRef CAS PubMed.
- (a) C.-I Lee, N. A. Hirscher, J. Zhou, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2015, 34, 3099 CrossRef CAS; (b) L. C. Gregor, C.-H. Chen, C. M. Fafard, L. Fan, C. Guo, B. M. Foxman, D. G. Gusev and O. V. Ozerov, Dalton Trans., 2010, 39, 3195 RSC.
- J. C. DeMott, N. Bhuvanesh and O. V. Ozerov, Chem. Sci., 2013, 4, 642 RSC.
- (a) I. Kieltsch, G. G. Dubinina, C. Hamacher, A. Kaiser, J. Torres-Nieto, J. M. Hutchison, A. Klein, Y. Budnikova and D. A. Vicic, Organometallics, 2010, 29, 1451 CrossRef CAS; (b) T. Inagaki, L. T. Phong, A. Furuta, J. Ito and H. Nishiyama, Chem. – Eur. J., 2010, 16, 3090 CrossRef CAS PubMed; (c) S.-F. Lu, D.-M. Du, S.-W. Zhang and J. Xu, Tetrahedron: Asymmetry, 2004, 15, 3433 CrossRef CAS PubMed; (d) S. Wanniarachchi, B. J. Liddle, J. Toussaint, S. V. Lindeman, B. Bennett and J. R. Gardinier, Dalton Trans., 2010, 39, 3167 RSC; (e) M. S. Mudadu, A. N. Singh and R. P. Thummel, J. Org. Chem., 2008, 73, 6513 CrossRef CAS PubMed.
- A. M. Hollas, W. Gu, N. Bhuvanesh and O. V. Ozerov, Inorg. Chem., 2011, 50, 3673 CrossRef CAS PubMed.
- (a) J. A. Gaunt, V. C. Gibson, A. Haynes, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Organometallics, 2004, 23, 1015 CrossRef CAS; (b) V. C. Gibson, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 2718 RSC; (c) G. J. P. Britovsek, V. C. Gibson, O. D. Hoarau, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Inorg. Chem., 2003, 42, 3454 CrossRef CAS PubMed; (d) J. M. Barbe, B. Habermeyer, T. Khoury, C. P. Gros, P. Richard, P. Chen and K. M. Kadish, Inorg. Chem., 2010, 49, 8929 CrossRef CAS PubMed.
- L. Fan, L. Yang, C. Guo, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 4778 CrossRef CAS; (a) O. V. Ozerov, C. Guo, L. Fan and B. M. Foxman, Organometallics, 2004, 23, 5573 CrossRef CAS.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916 CrossRef CAS PubMed.
- J. R. L. Priqueler, I. S. Butler and F. D. Rochon, Appl. Spectrosc. Rev., 2006, 41, 185 CrossRef CAS PubMed.
- (a) K. D. Conroy, W. E. Piers and M. Parvez, J. Organomet. Chem., 2008, 693, 834 CrossRef CAS PubMed; (b) P. Oulie, C. Freund, N. Saffon, B. Martin-Vaca, L. Maron and D. Bourissou, Organometallics, 2007, 26, 6793 CrossRef CAS; (c) L. P. Spencer and M. D. Fryzuk, J. Organomet. Chem., 2005, 690, 5788 CrossRef CAS PubMed; (d) Y. Schrodi, R. R. Schrock and P. J. Bonitatebus, Jr., Organometallics, 2001, 20, 3560 CrossRef CAS.
- S. Hamidi, L. N. Jende, H. M. Dietrich, C. Maichle-Mössmer, K. W. Törnroos, G. B. Deacon, P. C. Junk and R. Anwander, Organometallics, 2013, 32, 1209 CrossRef CAS.
- (a) S. Horchler, E. Parisini, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, J. Chem. Soc., Dalton Trans.: Inorg. Chem., 1997, 2761 RSC; (b) S. Schulz, L. Haeming, R. Herbst-Irmer, H. W. Roesky and G. M. Sheldrick, Angew. Chem., Int. Ed. Engl., 1994, 33, 969 CrossRef PubMed; (c) K. M. Waggoner and P. P. Power, J. Am. Chem. Soc., 1991, 113, 3385 CrossRef CAS.
- (a) S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2013, 135, 6018 CrossRef CAS PubMed; (b) E. Kwan Huang, W.-M. Cheung, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Organometallics, 2013, 32, 733 CrossRef CAS; (c) E. R. King, G. T. Sazama and T. A. Betley, J. Am. Chem. Soc., 2012, 134, 17858 CrossRef CAS PubMed.
- S. B. Harkins and J. C. Peters, Organometallics, 2002, 21, 1753 CrossRef CAS.
- L.-C. Liang, J.-M. Lin and W.-Y. Lee, Chem. Commun., 2005, 2462 RSC.
- L. R. Falvello, R. Garde, E. M. Miqueleiz, M. Tomás and E. P. Urriolabeitia, Inorg. Chim. Acta, 1997, 264, 297 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, X-ray crystallographic data. CCDC 1405023 (4·HOTf·C6H5F), 1405024 (4·C6D6), and 1405025 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00102a |
| ‡ J.R.D. was an NSF-REU participant from Hampden-Sydney College in 2013. Current address: Department of Chemistry, University of Virginia, Charlottesville, VA 22904, USA. |
| This journal is © the Partner Organisations 2015 |