Polyethylenimine-impregnated siliceous mesocellular foam particles as high capacity CO2 adsorbents
Junqi
Zhao†
a,
Fritz
Simeon†
a,
Yujun
Wang
b,
Guangsheng
Luo
b and
T. Alan
Hatton
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: tahatton@mit.edu; Fax: 617.253.8723; Tel: 617.253.4588
bDepartment of Chemical Engineering, Tsinghua University, 1 Tsinghua Yuan Street, Beijing, 100084, China
First published on 25th April 2012
Abstract
Siliceous mesostructured cellular foams (MCF) impregnated with polyethylenimine (PEI) of various molecular weights and structures were evaluated as CO2 adsorbents. The MCF solid support consisted of a well-defined interconnected three-dimensional mesoporous structure with large cell diameter of 30.3 nm and large window diameter of 11.3 nm, filled with polyethylenimine up to 70 weight percent or about 22.3% nitrogen atom by weight of the adsorbents. While other mesoporous solid supports lost their porosity after PEI impregnation, our MCF solid support maintained its pore volume over the range of 1.12 to 1.64 cm3 g−1. The importance of the porosity of PEI-impregnated MCF adsorbents for high capacity CO2 adsorbents was demonstrated. The highest CO2 sorption capacity (180.6 mg-CO2/g-adsorbent or 393.6 mg-CO2/g-PEI at 75 °C) was obtained for silica supports loaded with 50 weight percent branched PEI with average molecular weight of 600 g mol−1. Under dry atmospheric CO2 gas, this adsorbent reached the theoretical CO2 capacity of 0.50 mole-CO2 per mole-nitrogen within less than about 8 min, making this adsorbent one of the most effective CO2 adsorbents reported. Repeated multiple sorption cycles demonstrated good stability of this adsorbent for CO2 capture. The initial sorption kinetics determined the overall CO2 sorption capacity, which was limited by the formation of a carbamate layer as a result of the CO2–PEI complexation that due to inhibition of CO2 diffusion; the kinetics of “ionic” gelation of the impregnated PEI by CO2 controlled the overall performance of the CO2 adsorbents. At 75 °C, the operating temperature favored the molecular mobility of PEI and unrestricted diffusion of CO2 to allow the theoretical CO2 capacity of the PEI to be attained. Lower temperatures limited the mobilities of PEI and CO2 and the kinetics of “ionic” gel formation dominated, causing a lowered overall performance of the CO2 adsorbents. Overall, this study points to the importance of interconnected porous channel networks to optimize the performance of PEI-impregnated mesoporous silica particles.
Introduction
Anthropogenic greenhouse gas carbon dioxide (CO2) in the Earth's atmosphere has been cited as a primary cause of global climate change and threatens global public health and welfare. Since the Industrial Revolution in the 1700's, fossil fuels have become the primary energy source and driving force for sustainable global economic growth, and it is likely that human dependence on fossil fuels will continue for the rest of this century. Excessive anthropogenic fossil fuel consumption is responsible for a rising atmospheric CO2 concentration, which is currently at a level of 392 ppm. To mitigate the CO2 emission problem and to minimize the global threat offered by CO2 while sustaining economic growth, major transformation through “clean fossil-fuel” development is required. Carbon capture and sequestration (CCS) technology is an effective and important part of CO2 emission abatement strategies, with the major CCS efforts to date focusing on the removal of CO2 directly from large-scale carbon emitters and storing it in secure geologic reservoirs. Although the existing carbon capture technologies are feasible technically, their implementation is still a challenging task due to their energy demands, capital investment and operational costs, and environmental impact.Liquid amine scrubbing has been the chemical absorption process most investigated over the past three decades for CO2 capture.1–3 In this continuous scrubbing process, dilute CO2 is separated from a gas mixture through chemical sorption by a liquid amine solution. At lower temperatures, the acid gas undergoes a spontaneous exothermic acid–base reaction generating stable carbamate or carbonate salts. In the desorber units, the reaction products are broken down by stripping with steam at higher temperature, regenerating the amine sorbents and producing a concentrated CO2 stream. Considerable quantities of steam and heat are required to elevate the temperature of the solution to favor release of the CO2. Substantial parasitic energy losses result from the need to use excess steam and heat in order to meet the kinetic requirements of the process. Increasing the concentration of amine solutions will decrease the parasitic energy loss and reduce the overall CO2 capture costs;4 however, contact of the concentrated scrubbing solutions with iron steel accelerates corrosion rates with their consequent detrimental effects on the boilers of the desorption units.
Adsorption processes for CO2 scrubbing have recently gained more attention as promising methods to lower parasitic energy losses.5–19 Ideally, high concentrations of amines can be incorporated within solid inorganic supports with lower corrosion issues due to the negligible contact between the amine solution and surfaces other than those of the solid inorganic supports. Porous inorganic materials such as zeolites and silicates are ideal solid supports since they provide high pore volumes and large surface areas for optimizing the CO2 capture capacity per total weight of adsorbent. Furthermore, amine-inorganic hybrid adsorbents offer the potential to lower the parasitic energy loss because the heat capacity of inorganic supports is lower than that of water (Csilica = 0.8 kJ·kg−1·K−1versus CH2O = 4.2 kJ·kg−1·K−1). These mesoporous silica supports have unique properties such as high porosity, uniform pore dimensions, and large surface area, easy for various post-chemical modifications.20–29
Impregnation of porous inorganic solid supports with amine molecules can be through either chemical conjugation30–38 or physical adsorption.39–47 Chemical conjugation improves the chemical stability and prevents the leaching of the amines; however, only low amine concentrations can be loaded on the porous supports. Amine conjugation through the polymerization of aziridine on mesoporous silica surfaces was demonstrated by Hicks et al.,48 who obtained stable CO2 adsorbents containing about 10% nitrogen atom by weight. Physical impregnation of mesoporous silica supports is an easy method to obtain high concentration of dispersed amines. Polyethylenimine (PEI) is the most commonly used macromolecule for CO2 adsorbents because of its high amine content, about 33% nitrogen by weight. The hydroxyl groups on the silica surface provide strong interactions with the amino groups of PEI facilitating the distribution of the polymer throughout the pore space. This dispersion of PEI molecules within mesoporous silica supports substantially increases their CO2 sorption capacity and improves the CO2 sorption kinetics relative to those observed with pure PEI.
The structural properties of mesoporous silica supports impact the performance of the CO2 adsorbents. Amorphous silica supports with a random array of pore dimensions are not suitable because their entire pore volume may not be accessible to amine molecules, whereas ordered mesoporous silica supports provide large surface areas and well-defined porous structures that make them excellent candidates as solid supports. The total pore volume and pore size of mesoporous silica supports impact the performance of the CO2 adsorbents. Franchi et al.40 synthesized pore-expanded MCM-41 (PE-MCM-41) with a total pore volume of 2.03 cm3 g−1, about twice that of the standard MCM-41. The large pore volume accommodates a larger quantity of amine resulting in higher CO2 adsorption capacity compared with that of MCM-41. Son et al.42 demonstrated that the CO2 sorption capacity and the CO2 sorption kinetics are controlled by the pore size of the silica supports. By developing a series of mesoporous silica supports impregnated with PEI (MCM-41, MCM-48, SBA-15, SBA-16 and KIT-6 with corresponding average pore diameters of 2.8 nm, 3.1 nm, 4.1 nm, 5.5 nm and 6.5 nm, respectively), they showed that the CO2 adsorption capacity and the time required for each adsorbent to reach 70% of its equilibrium capacity increased with increasing average pore diameter of the mesoporous silica samples, with the best performance observed using the PEI-impregnated KIT-6 adsorbent. The high surface areas of mesoporous silica supports provide open and accessible sites for the stabilization of a large number of amine molecules; however, an excessively large surface area lowers the theoretical amine efficiency for CO2 capture since the amino groups interacting with the surfaces of solid supports are no longer available for complexation with CO2. Franchi et al.40 reported that the amine efficiency of diethanolamine (DEA)-impregnated MCM-41 (MCM-41 BET surface area of 1138 m2 g−1) is about 0.37 mole CO2 per mole amine, less than the ideal dry CO2 case of 0.50 mole CO2 per mole amine. From previous investigations, we learn that, in order to develop a high capacity CO2 adsorbent, mesoporous inorganic solid supports should have high pore density, large pore diameter and adequately large surface area.
Of the previously developed mesoporous silica supports, mesocellular foam (MCF) silica seems to be the most suitable candidate for high capacity CO2 adsorbents. These porous silicas are known to have large pore sizes (20–50 nm), high pore volumes (up to 2.6 cm3 g−1), adequate large surface areas (500–1000 m2 g−1), and robust frameworks.49,50 MCFs are composed of uniform spherical unit cells that interconnect by windows with a narrow size distribution. These three-dimensional porous networks provide not only large surface areas but also highly-interconnected channels that provide open structures for ease of CO2 sorption. In the present study, we synthesized siliceous MCF particles as support materials for PEI impregnation. Various molecular weights and molecular structures of PEI were impregnated physically into the porous MCFs and their performance in CO2 capture was assessed. We also addressed the influence of the sorption conditions and PEI loading on the CO2 capture performance. This study demonstrates the importance of interconnected porous channel networks in optimizing the performance of PEI-impregnated mesoporous silica particles.
Results and discussion
Siliceous mesostructured cellular foams (MCF) impregnated with PEI
Fig. 1 shows TEM and SEM images of empty (Fig. 1(a) and 1(d)) and PEI-impregnated (Fig. 1(b), 1(c), 1(e) and 1(f)) MCFs. The TEM image shows typical MCF solid supports with interconnected three-dimensional porous network silica structures. The pore structures of these supports were characterized by nitrogen adsorption-desorption isotherms at 77 K. Fig. 2 (black line) shows the nitrogen sorption isotherm for blank MCFs, which typically show type IV isotherms with hysteresis adsorption-desorption loops. The Barrett-Joyner-Halenda (BJH) pore analysis method was used to determine the cell diameter from the N2 adsorption isotherm profile, while the window diameter was determined from the N2 desorption isotherm profile. The overall surface area of the MCFs was determined using the Brunauer-Emmett-Teller (BET) surface area analysis method. The results are compared with those of previously reported mesoporous silica particles (Table 1). Our mesoporous silica had unique three-dimensional pore structures with ultra-large cell-like pores, about 30.3 nm on average, interconnected by windows with an average opening of 11.3 nm. This MCF silica possessed an ultra-high pore volume of 3.14 cm3 g−1, and a relatively large BET surface area of 628 m2 g−1, significantly better than the corresponding values reported in the literature for mesostructured porous silica supported CO2 adsorbents. The overall geometric features of the synthesized MCFs make them promising as candidate materials for high capacity CO2 adsorbents. | ||
| Fig. 1 TEM and SEM micrographs of PEI-impregnated MCFs. TEM images: (a) MCF, (b) MCF-PEI(600b)-50, and (c) MCF-PEI(600b)-70. SEM images: (d) MCF, (e) MCF-PEI(600b)-50, and (f) MCF-PEI(600b)-70. | ||
 | ||
| Fig. 2 (a) Nitrogen adsorption-desorption isotherms at 77 K, (b) BJH pore geometric analysis of window diameter of MCF silica adsorbents, and (c) BJH pore geometric analysis of cell diameter of MCF silica adsorbents. | ||
Polyethylenimine (PEI) was impregnated into the synthesized MCFs using a “wet impregnation” method, in which the MCFs were physically mixed with PEI solutions in methanol.43 The various PEI-impregnated MCFs are designated as MCF-PEI(X)-Y, where X denotes the molecular weight (grams per mol) and the molecular structure of PEI (b for branched-PEI and l for linear-PEI), and Y denotes the PEI weight percentage. Fig. 1(b), 1(c), 1(e) and 1(f) show the electron micrograph images of PEI-impregnated MCF particles. No change in particle structure was observed following PEI impregnation, and the TEM images suggested that the pore sizes of the PEI-impregnated MCFs were similar to those of the blank MCFs. The pore properties of the PEI-impregnated MCF particles were characterized by nitrogen adsorption-desorption isotherms at 77 K, as summarized in Table 2. Using the BJH pore analysis approach, we inferred that there were no structural changes, at least in terms of the cell and window diameters, in the MCFs after PEI impregnation. The cell diameters of PEI-impregnated MCFs were in the range of 29.4 to 31.2 nm, while the window diameters were in the range of 10.8 to 11.3 nm.
| Sample | S BET (m2 g−1) | V t (cm3 g−1) | D BJHA (nm) | D BJHD (nm) |
|---|---|---|---|---|
| MCF | 628 | 3.14 | 30.3 | 11.3 |
| SBA-15 | 954 | 1.16 | 7.6 | 5.6 |
| MCF-PEI(600b)-15 | 388 | 2.29 | 30.1 | 10.9 |
| MCF-PEI(600b)-30 | 304 | 1.97 | 31.2 | 10.8 |
| MCF-PEI(600b)-50 | 199 | 1.30 | 30.8 | 11.1 |
| MCF-PEI(600b)-70 | 20 | 0.13 | — | — |
| MCF-PEI(25000b)-50 | 51 | 1.12 | 29.4 | 10.9 |
| MCF-PEI(25000l)-50 | 215 | 1.64 | 30.8 | 11.3 |
| SBA-15-PEI(25000b)-50 | 25 | 0.06 | 4.9 | 3.2 |
Dispersion of PEI within the internal pores of MCFs was expected to lower the BET surface area and the pore volume of the adsorbents. At a loading of 15 wt% of PEI(600b), the BET surface area of PEI-impregnated MCFs was 388 m2 g−1, about half the BET surface area of the blank MCF. Increasing the PEI content to 30 wt% and 50 wt% further decreased the measured BET surface areas to 304 m2 g−1 and 199 m2 g−1, respectively. When the mesoporous MCF silica was impregnated with 70 wt% PEI(600b), we observed a significant decrease in BET surface area to 20 m2 g−1 indicating the mesoporous MCF was almost completely filled by PEI. Similarly, the pore volume of the adsorbents decreased with PEI impregnation, as summarized in Table 2. Interestingly, our results show that because of its unique pore geometry, MCF-PEI(600b)-50 still possesses significant porosity with a pore volume of 1.30 cm3 g−1. Ma et al.51 developed PEI-impregnated MCM-41 and SBA-15 adsorbents for CO2 capture, and found that the pore volume of MCM-41 decreased from 1.15 cm3 g−1 to 0.03 cm3 g−1 after 50 wt% PEI impregnation, while the pore volume of SBA-15 decreased from 1.31 cm3 g−1 to 0.20 cm3 g−1. In this study, impregnating MCFs with 50 wt% of high molecular weight PEI, PEI(25000b) and PEI(25000l), still resulted in highly porous CO2 adsorbents, with pore volumes of 1.12 cm3 g−1 and 1.64 cm3 g−1, respectively. The high porosity of PEI-impregnated MCFs should improve the CO2 sorption kinetics of these adsorbents.
CO2 sorption capacity of MCFs and PEI-impregnated MCFs
The CO2 sorption capacities of MCFs, SBA-15 and their PEI-impregnated versions were measured using a Rubotherm suspension microbalance to monitor CO2 adsorption uptake under various experimental conditions. Instead of isothermal equilibrium CO2 adsorption capacities, we report here working capacities since these values are more relevant to evaluation of the performance of CO2 adsorbents. The CO2 adsorption uptake was monitored for 96 min following exposure to a sudden change in CO2 concentration under isothermal conditions.Using the same method as used for the MCFs, we impregnated the internal pores of SBA-15 with 50-wt% of either PEI(600b) or PEI(25000b). The nitrogen sorption profiles of SBA-15 and PEI-impregnated SBA-15 at 77 K are shown in Fig. 2, and the pore properties are summarized in Table 2. Mesoporous SBA-15 had a larger BET surface area than mesoporous MCFs, about 954 m2 g−1, but a smaller pore diameter of 7.6 nm. The total pore volume of SBA-15 was about three-times lower than that of mesoporous MCFs. PEI(25000b)-impregnated SBA-15 had a smaller BET surface area, 25.4 m2 g−1, and a lower total pore volume, 0.06 cm3 g−1, than did the blank SBA-15. At a loading of 50 wt% PEI(25000b) on SBA-15, most of the mesopores were filled with PEI resulting in a significant decrease in both BET surface area and pore volume. When SBA-15 was impregnated with 50 wt% PEI(600b), no nitrogen gas adsorption was observed indicating a complete filling of the pores, in agreement with the results of Ma et al.51 The small pore diameters of SBA-15 limited the pore diffusion of branched PEI through the pores, and possibly led to clogging, preventing further impregnation by the PEI molecules.
Fig. 3 shows the CO2 adsorption capacities of PEI-impregnated MCF silica and PEI-impregnated SBA-15, as summarized in Table 3. The empty MCF showed a low CO2 adsorption capacity of 10.6 mg-CO2/g-adsorbent, while that of the empty SBA-15 was slightly higher at 23.5 mg-CO2/g-adsorbent. The CO2 adsorption capacity of empty mesoporous silica supports correlated directly with their BET surface areas. Mesoporous SBA-15 supports with a BET surface area of 1229 m2 g−1 showed double the CO2 adsorption capacity of the mesoporous MCF supports that had a surface area of only 628 m2 g−1. The strong correlation between the surface area and the CO2 sorption capacity of the solid supports indicated that CO2 adsorbed as a monolayer on the surfaces of these mesoporous silica particles.
 | ||
| Fig. 3 Adsorption and desorption curves for PEI(600b) and PEI(25000b)-impregnated MCFs and SBA-15 at 75 °C. | ||
| Sample | Sorption T/°C | Adsorption capacity | Desorption efficiency (%) | Time taken to reach a specific percentage of the individual CO2 adsorption capacity (min) | ||||
|---|---|---|---|---|---|---|---|---|
| mg-CO2/g-adsorbent | mg-CO2/g-PEI | 50% | 70% | 90% | 95% | |||
| MCFs | 75 | 10.6 | — | 107.5 | — | — | — | — |
| SBA-15 | 75 | 23.5 | — | 103.1 | — | — | — | — |
| MCF-PEI(600b)-15 | 75 | 33.3 | 222.0 | 100.0 | 5.5 | 5.5 | 6.9 | 9.6 |
| MCF-PEI(600b)-30 | 75 | 102.3 | 341.0 | 99.1 | 5.5 | 5.5 | 5.5 | 6.9 |
| MCF-PEI(600b)-50 | 50 | 113.4 | 226.8 | 62.1 | 5.5 | 6.9 | 20.6 | 34.3 |
| MCF-PEI(600b)-50 | 65 | 148.6 | 297.2 | 96.9 | 5.5 | 5.5 | 10.9 | 23.3 |
| MCF-PEI(600b)-50 | 75 | 180.6 | 361.2 | 100.7 | 5.5 | 5.5 | 8.2 | 16.5 |
| MCF-PEI(600b)-50 | 100 | 150.2 | 300.4 | 89.5 | — | — | — | — |
| MCF-PEI(600b)-70 | 75 | 192.6 | 275.1 | 100.2 | 5.5 | 6.9 | 26.1 | 52.1 |
| MCF-PEI(25000b)-50 | 75 | 161.3 | 322.6 | 99.4 | 5.5 | 5.5 | 11.0 | 19.2 |
| MCF-PEI(25000l)-50 | 75 | 112.3 | 224.6 | 101.5 | — | — | — | — |
| SBA-15-PEI(600b)-50 | 75 | 116.4 | 232.8 | 98.5 | 5.5 | 8.2 | 48 | 71.3 |
| SBA-15-PEI(25000b)-50 | 75 | 107.8 | 215.6 | 100.3 | 5.5 | 12.3 | 39.8 | 52.1 |
| PEI(600b) | 75 | — | 111.0 | 62.0 | 6.9 | 17.8 | 46.6 | 57.6 |
| PEI(25000b) | 75 | — | 62.6 | 93.7 | — | — | — | — |
Impregnation of mesoporous silica solid supports with PEI increased substantially their CO2 sorption capacities. PEI-impregnated MCFs showed higher CO2 adsorption capacity and faster CO2 sorption rates than did PEI-impregnated SBA-15. The CO2 adsorption capacity of MCF-PEI(600b)-50 was 180.6 mg-CO2/g-adsorbent corresponding to 361.2 mg-CO2/g-PEI, and 90% CO2 sorption capacity was attained within 8 min. The CO2 adsorption capacity of SBA15-PEI(600b)-50 was 116.4 mg-CO2/g-adsorbent, corresponding to 232.8 mg-CO2/g-PEI; 90% CO2 adsorption capacity was reached after 48 min. With high molecular weight PEI, 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, MCF-PEI(25000b)-50 showed a CO2 adsorption capacity of 322.6 mg-CO2/g-PEI, whereas SBA15-PEI(25000b)-50 had a CO2 adsorption capacity of 215.6 mg-CO2/g-PEI. Although the SBA15-PEI(25000b)-50 reached 90% CO2 adsorption capacity more quickly than did SBA15-PEI(600b)-50, in about 39 min, its CO2 adsorption rate was still significantly slower than that observed with MCF-PEI(25000b)-50, for which the adsorption time was about 11 min.
000 g mol−1, MCF-PEI(25000b)-50 showed a CO2 adsorption capacity of 322.6 mg-CO2/g-PEI, whereas SBA15-PEI(25000b)-50 had a CO2 adsorption capacity of 215.6 mg-CO2/g-PEI. Although the SBA15-PEI(25000b)-50 reached 90% CO2 adsorption capacity more quickly than did SBA15-PEI(600b)-50, in about 39 min, its CO2 adsorption rate was still significantly slower than that observed with MCF-PEI(25000b)-50, for which the adsorption time was about 11 min.
We postulate that the superiority of PEI-impregnated MCFs is due to the large pore diameter and the three-dimensional interconnected porous channel networks that prevent clogging of the pores and maintain the porosity of the adsorbents after PEI impregnation. Evidence for the important role of the porosity of PEI-impregnated adsorbents in determining their CO2 adsorption capacities and sorption kinetics is provided by our results. MCF-PEI(25000b)-50, with a pore volume of 1.12 cm3 g−1, showed lower CO2 sorption capacity and slower CO2 sorption kinetics than did MCF-PEI(600b)-50 with its pore volume of 1.30 cm3 g−1. Similarly, SBA15-PEI(600b)-50 with completely filled pores showed lower CO2 adsorption capacity and slower CO2 sorption kinetics than did SBA15-PEI(25000b)-50. Table 4 summarizes the performance of a number of PEI-impregnated mesoporous inorganic substrates reported previously. Our PEI-impregnated MCFs show the highest adsorption capacities in terms of CO2 per gram of PEI under the experimental conditions reported in the literature to date.
| Support | S BET (m2 g−1) | V t (cm3 g−1) | D P (nm) | PEI loading amount (wt%) | Test conditions | Method | Adsorption capacity | Ref | |
|---|---|---|---|---|---|---|---|---|---|
| mg-CO2/g-adsorbent | mg-CO2/g-PEI | ||||||||
| a Results from our study. | |||||||||
| MCM-41 | 1,486 | 1.00 | 2.75 | 50 | Pure CO2 stream; 100 mL min−1; 1 atm; 75 °C | TGA | 112 | 224 | 42 |
| MCM-48 | 1,162 | 1.17 | 3.1 | 50 | Pure CO2 stream; 20 mL min−1; 1 atm; 75 °C | TGA | 119 | 238 | 41 |
| SBA-15 | 753 | 0.94 | 5.5 | 50 | TGA | 127 | 254 | ||
| SBA-16 | 736 | 0.75 | 4.1 | 50 | TGA | 129 | 258 | ||
| KIT-6 | 895 | 1.22 | 6.0 | 50 | TGA | 135 | 270 | ||
| HMS | 561 | 1.44 | 9.8 | 60 | Pure CO2 stream; 30 mL min−1; 1 atm; 75 °C | TGA | 184 | 307 | 55 |
| MCF | 628 | 3.14 | 30.3 | 50 | Pure CO2 stream; 100 mL min−1; 1 atm; 75 °C | Rubotherm suspension balance | 181 | 362 | a |
| 11.3 | |||||||||
Thermal Gravimetric Analysis (TGA) under nitrogen indicated that PEI-impregnated MCFs are thermally stable for application as CO2 adsorbents over a wide temperature range (Fig. 4). No significant change in sample mass was observed when temperature was ramped between room temperature and 200 °C regardless of the PEI molecular weight and molecular structure. Between 250 and 400 °C a more rapid decrease in mass occurred indicating a degradation of the impregnated PEI molecules within the CO2 adsorbents. The sample was heated to 800 °C, and above 400 °C no further change in sample mass was observed indicating the strong thermal stability of the mesoporous MCF silica support. We further evaluated multiple cycles of CO2 adsorption-desorption by the PEI-impregnated MCF adsorbent at 75 °C, as shown in Fig. 5 for MCF-PEI(600b)-50. The performance of this adsorbent was excellent over the five adsorption-desorption cycles shown, with no significant changes in adsorption capacity and kinetics.
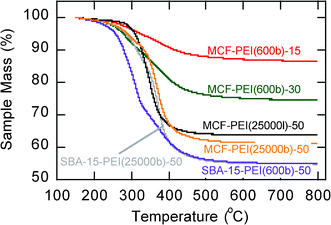 | ||
| Fig. 4 Thermal gravimetric analysis (TGA) profiles for the PEI-impregnated MCF adsorbents. | ||
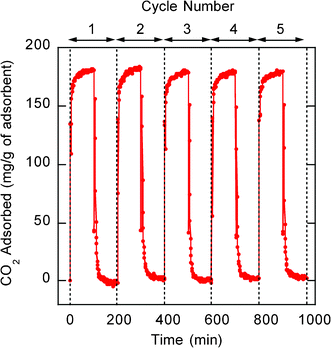 | ||
| Fig. 5 Multiple adsorption-desorption cycles with MCF-PEI(600b)-50 at 75 °C. | ||
Effects of PEI molecular weight, molecular structure and loading on CO2 sorption performance
The synergistic effects of polymeric amines and inorganic solid supports on the CO2 adsorption capacity have spurred the recent interest in exploring this hybrid material for developing high capacity CO2 adsorbents.52 Inorganic mesoporous solid supports generally have low CO2 adsorption capacity; similarly, pure polyamines show relatively lower CO2 sorption capacity than do polymer-impregnated inorganic hybrid adsorbents. In particular, pure PEI molecules, either branched or linear, and containing a mixture of primary, secondary and tertiary amines, have lower CO2 adsorption working capacity than predicted based on their composition. The primary and secondary amines interact strongly with CO2 through the formation of zwitterionic intermediates to form carbamate salts. Under dry conditions, the tertiary amine does not undergo complexation with CO2; its normal role is to catalyze the hydration of CO2 in the presence of water molecules to form carbonate salts. In the absence of water, the primary and secondary amines of PEI react with CO2 with a molar ratio of 2 to 1 (two moles of amino groups per mole of CO2). In the presence of water, however, the primary, secondary and tertiary amines are all reactive toward CO2 to form carbonate salts with a molar ratio of 1 to 1 (one mole of amine per mole of CO2). In this study, we explored three different PEIs, PEI(600b), PEI(25000b) and PEI(25000l). The branched PEI(600b) and PEI(25000b) contain primary, secondary and tertiary amino groups in the ratio of 42:33:25;53 whereas, the linear PEI(25000l) contains only secondary amines. We evaluated the adsorption working capacity of these polymers under dry CO2 conditions, where, the theoretical CO2 adsorption capacity of branched PEI is 382 mg-CO2/g-PEI(b) and that of the linear PEI is 500 mg-CO2/g-PEI(l).
Table 3 summarizes the measured CO2 adsorption working capacity of PEI. The low molecular weight branched PEI(600b) has a higher CO2 adsorption working capacity than does the high molecular weight branched PEI(25000b), i.e. 111 mg-CO2/g-PEI(600b) versus 62.6 mg-CO2/g-PEI(25000b). CO2 adsorption working capacities of these polymers were still below the theoretical limit, primarily because of the slow observed CO2 adsorption kinetics with pure PEI samples. Fig. 6(a) (grey line) shows the development of the mass change profile of PEI(600b) at 75 °C under dry CO2 with time. When the PEI molecules were dispersed within MCF solid supports, we observed a substantial increase in their CO2 sorption capacities (Fig. 6(b) and Table 3).
 | ||
| Fig. 6 (a) Adsorption and desorption curves for PEI(600b)-impregnated MCFs with different PEI loading amounts at 75 °C. (b) Mass of CO2 adsorbed by PEI(600b)-impregnated MCFs with different PEI loading amounts at 75 °C. | ||
At 75 °C, impregnation of PEI(600b) within mesoporous MCF particles provided a significant increase in the adsorption capacity of the adsorbent. Adsorbents with higher PEI contents showed higher CO2 adsorption capacity per gram of adsorbents (Fig. 6(b)). At 50 wt% PEI loading, the CO2 adsorption capacity was 180.6 mg-CO2/g-adsorbent corresponding to 361.2 mg-CO2/g-PEI(600b). Therefore, dispersing PEI(600b) within the mesoporous MCFs supports increased the CO2 adsorption capacity of PEI to close to its theoretical limit of 382 mg-CO2/g-PEI(b). Under these experimental conditions, it is one of the best performing CO2 adsorbents reported in literature to date.
A further increase in the PEI(600b) content to 70 wt% resulted in an additional, slight increase in the CO2 adsorption capacity of the adsorbent. Relative to the mass of PEI(600b), however, the capacity decreased to 275.1 mg-CO2/g-PEI(600b), corresponding to 71% of the theoretical CO2 adsorption capacity. This decrease at high PEI content may be attributed to the overall evolution of the structure of the adsorbents with changes in PEI content. For the MCF impregnated with 70 wt% PEI(600b), analysis of the nitrogen adsorption-desorption isotherms at 77 K showed a significant decrease in the BET surface area to 20 m2 g−1 and also a decrease in pore volume to 0.13 cm3 g−1, significantly lower than the respective values of 198.8 m2 g−1 and 1.30 cm3 g−1 reported above for MCF-PEI(600b)-50. This analysis suggested that the porosity of PEI-impregnated MCF supports plays an important role in determining the CO2 adsorption capacity, and that completely filling the mesoporous inorganic supports lowers dramatically the PEI efficiency of the amine-inorganic hybrid adsorbents. Although the CO2 adsorption capacity of MCF-PEI(600b)-70 was higher than that of pure PEI(600b), the CO2 sorption kinetics in MCF-PEI(600b)-70 particles were similar to those of pure PEI(600b) (Fig. 6), which were slower than the CO2 adsorption kinetics observed with MCF-PEI(600b)-50. Therefore, our results emphasize the importance of the porosity of PEI-impregnated mesoporous inorganic adsorbents in ensuring higher capacities and faster kinetics of CO2 adsorption.
We also evaluated the influence of PEI molecular weight and structure on the performance of 50 wt% PEI-impregnated MCF adsorbents as summarized in Table 3. As with PEI(600b), impregnation of the mesoporous MCF supports with PEI(25000b) improved the CO2 adsorption capacity of the polymers, from 62.6 to 322.6 mg-CO2/g-PEI(25000b), demonstrating the superiority of mesoporous MCFs over other silica supports as excellent platforms for amine-impregnated CO2 adsorbents. Similarly, a low CO2 adsorption capacity of linear PEI(25000l), i.e. 10 mg-CO2/g-PEI(25000l), is significantly increased to 224.6 mg-CO2/g-PEI(25000l) when this polymer is dispersed within MCF solid supports.
Effect of sorption temperature on the CO2 sorption performance of MCF-PEI(600b)-50
The adsorption of CO2 by PEI-impregnated MCF adsorbents was driven mainly by exothermic chemical reaction between the acidic CO2 molecules and the basic amino groups within the PEI molecules, and the equilibrium CO2 adsorption capacity should be lower at higher operating temperatures. Here we evaluated the effect of operating temperature on the performance of the MCF-PEI(600b)-50 adsorbent. Isothermal CO2 sorption capacities of MCF-PEI(600b)-50 at various operating temperatures between 50 °C and 100 °C were characterized using the Rubotherm suspension microbalance. The CO2 adsorption and desorption profiles on MCF-PEI(600b)-50 at various operating temperatures are shown in Fig. 7, and the results are summarized in Table 3. The CO2 adsorption capacity is reported as the working capacity of the adsorbents, i.e. the CO2 uptake after 96 min exposure to pure CO2 gas at atmospheric pressure. | ||
| Fig. 7 Adsorption and desorption curves for MCF-PEI(600b)-50 at different temperatures. | ||
At 50 °C, the MCF-PEI(600b)-50 CO2 adsorption capacity was found to be 113.4 mg-CO2/g-adsorbent. Increasing the operating temperature to 65 °C, interestingly, increased the CO2 adsorption capacity to 148.6 mg-CO2/g-adsorbent. At 75 °C, the adsorbent showed a CO2 adsorption capacity of 180.6 mg-CO2/g-adsorbent, reaching the theoretical CO2 adsorption capacity for branched PEI under dry CO2 conditions. At 50 °C, it took about 21 min for the adsorbent to reach 90% of its 96 min capacity. Increasing operating temperatures to 65 °C and 75 °C shortened the time required to reach 90% of the CO2 adsorption capacity to about 11 and 8 min, respectively. The adsorption profiles for MCF-PEI(600b)-50 at 50 °C, 65 °C and 75 °C, showed an initial rapid uptake of CO2, up to 30 min, following which the amount of CO2 adsorbed increased more slowly at a rate of 0.09 mg-CO2/g-adsorbent per minute, suggesting that the CO2 working capacity had already been attained within 30 min. An increasing trend of CO2 adsorption capacity with an increase in operating temperature is counter-intuitive for exothermic adsorption. Yue et al.54 explained this commonly observed trend with amine-impregnated silicas as reflecting a strongly diffusion-controlled process. Wang et al.55 proposed a more detailed mechanism for CO2 adsorption on PEI-impregnated mesoporous solid supports, suggesting that gas adsorption includes two rate regimes, i.e., rapid sorption over surface layers of PEI and diffusion and sorption inside the bulk multi layers of PEI. The first step is controlled by isothermal equilibrium of CO2 adsorption, while the second step is controlled by the diffusion. We believe that this model does not depict the complete mechanism for CO2 adsorption within PEI-impregnated silica adsorbents. If the difference in CO2 adsorption capacity was due mainly to diffusion-controlled processes, we should observe faster CO2 adsorption rates after 30 min at 75 °C than at 50 °C because of the higher CO2 molecular diffusion rates at higher temperatures.
As with the Wang et al.55 model, we postulate that the CO2 sorption on PEI-impregnated mesoporous solid supports includes two sorption regimes, chemisorption on PEI surface layers followed by bulk diffusion of CO2 within the multiple layers of PEI. The initial step of CO2 sorption is critical in determining the working capacity of the adsorbents, but, we believe, the formation of a carbamate “ionic” gel through the complexation of CO2 and PEI inhibits further CO2 diffusion into the gel, and that the operating conditions for CO2 adsorption control the kinetics of formation of this gel layer. To understand the detailed mechanistic aspects of CO2 adsorption on PEI-impregnated MCFs adsorbents, we performed the following experiment. The CO2 adsorption on MCFs-PEI(600b)-50 adsorbent was carried out at 50 °C for 96 min, following which the operating temperature was increased to 75 °C and the adsorption was further carried out for another 96 min. The temperature profiles and the CO2 sorption capacity at various times are depicted in Fig. 8. The CO2 sorption capacity reached 113.4 mg-CO2/g-adsorbent after the first 96 min. Increasing the operating temperature subsequently to 75 °C resulted in only a slight increase in CO2 sorption capacity to 123.0 mg-CO2/g-adsorbent after the next 96 min. No obvious additional mass change was observed after 45 min adsorption at 75 °C, suggesting that the CO2 adsorbent had reached its maximum working capacity. This capacity was significantly lower than the CO2 sorption capacity observed when the experiment was performed at 75 °C right from the beginning, for which the CO2 sorption capacity was 180.6 mg-CO2/g-adsorbent.
 | ||
| Fig. 8 (Black line) Mass profile of CO2 adsorbed per gram of adsorbent when the sample was equilibrated with CO2 at 50 °C and then the temperature was increased to 75 °C. (Green line) Adsorption curve for MCF-PEI(600b)-50 at 75 °C. The dotted lines indicate the CO2 working capacities (mg-CO2 per gram-adsorbent) at each of these temperatures. | ||
Chemical reaction of CO2 with PEI produces carbamate through a two-step reaction. The acid–base reaction of one molecule of CO2 and one molecule of amine forms a zwitterionic intermediate that is then deprotonated by another amine molecule to form the final carbamate product. The formation of carbamate essentially creates “ionic” crosslinks within the surface and the bulk PEI layers as observed with other amine sorbents. Fig. 9 illustrates the proposed process. CO2 adsorption may also cause swelling of the PEI films so that they eventually fill the MCF pores and inhibit the CO2 diffusion. We believe, however, that the formation of “ionic” crosslinks is the dominant factor in limiting the CO2 transport because typical polymeric swelling would increase the total volume by less than 10 percent under atmospheric CO2.56 Amundsen et al.,57 for instance, reported that the densities and dynamic viscosities of monoethanolamine solutions increase significantly with increasing CO2 loading at various temperatures ranging from 25 to 80 °C. The formation of carbamates with PEI increases significantly the density and viscosity of the PEI layer and inhibits further CO2 diffusion. At higher temperature, PEI has a lower viscosity and higher molecular mobility. When the CO2 adsorption was performed at 75 °C, the higher PEI mobility and CO2 diffusivity allowed the formation of multiple layers of carbamate between CO2 and PEI. The lower binding constant between CO2 and amino groups at higher temperature, and the lower cross-linking efficiency, further allowed deeper CO2 penetration, with the formation of a thicker carbamate layer. At the lower temperature of 50 °C, the lower mobility of PEI and the lower CO2 diffusivity, coupled with the stronger binding constant and hence more strongly gelled polymers, decreased the number of PEI-CO2 carbamate layers. This formation of the PEI-CO2 carbamate layer inhibited further CO2 diffusion significantly. This process explains the difference in isothermal CO2 sorption capacity at 75 °C when the experiment was performed only at 75 °C, and when the experiment was performed at 50 °C and then elevated to 75 °C. Since the formation of these PEI-CO2 carbamate layers inhibits the CO2 sorption processes, for high CO2 capacity, the adsorbents should have high porosity after PEI impregnation to ensure CO2 penetration into the deepest pores of the particles.
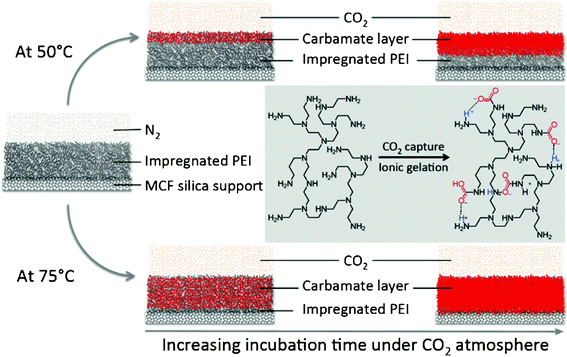 | ||
| Fig. 9 Schematic illustration of PEI-impregnated MCF adsorbents during CO2 capture and release. The initial step of CO2 sorption is critical in determining the working capacity of the adsorbents. The formation of a carbamate “ionic” gel through the complexation of CO2 and PEI inhibits further CO2 diffusion into the gel; and that the operating conditions for CO2 adsorption control the kinetics of formation of this gel layer. | ||
Conclusion
We have developed PEI-impregnated MCF adsorbents for enhanced CO2 capture performance in which siliceous mesocellular foam solid supports provide high pore volumes and pore sizes to accommodate high concentrations of PEI molecules. Under dry atmospheric CO2 gas, these adsorbents reached their theoretical CO2 capacity of 0.50 mole-CO2 per mole-amine within less than about 8 min, making them some of the most effective CO2 adsorbents reported. Repeated multiple sorption cycles demonstrated good stability of this adsorbent for CO2 capture. Our study pointed to the importance of initial sorption kinetics in determining the overall dynamic CO2 sorption capacities observed in both this work and in other reported studies. We propose that a gelled carbamate layer formed through complexation of the CO2 and PEI inhibits the CO2 diffusion, and that the kinetics of “ionic” gelation of impregnated PEI by CO2 adsorption controls the overall performance of these CO2 adsorbents. Highly porous adsorbents are necessary for high capacity CO2 sorption. The MCF silica maintained its porosity even at high weight percentage of PEI. Generally, our study points to the importance of interconnected porous channel networks in optimizing the performance of PEI-impregnated mesoporous silica particles.Experimental
Chemicals
Pluronic® P123 (poly (ethylene oxide)-block-poly (propylene oxide)-block-poly (ethylene oxide), EO20-PO70-EO20, Mw = 5800) was purchased from BASF Corporation. Mesitylene (≥99.0%, TMB) was produced by Fluka Analytical. Ethanol (pure) and methanol (≥99.9%) were purchased from Koptec and OmniSolv, respectively. Tetraethyl orthosilicate (98%, TEOS), hydrochloric acid (37%, HCl), ammonium fluoride (≥99.99%, NH4F), and polyethylenimine (Mw = 25000, branched, liquid, PEI(25000b)) were all purchased from Sigma-Aldrich Co. Polyethylenimine (Mw = 600, branched, liquid, PEI(600b)) and polyethylenimine (Mw = 25000, linear, solid, PEI(25000l)) were produced by Alfa Aesar.
Preparation of PEI-impregnated MCFs and SBA-15 adsorbents
Pure silica MCFs were prepared by a hydrothermal method developed by Schmidt-Winkel that used TEOS as the silica source, P123 as the template, and TMB as the swelling agent.58 In this process, 0.8 g of P123 was dissolved in 30 mL of HCl aqueous solution at a concentration of 1.6 M at ambient temperature; 2.0 mL of TMB was then added and the resulting solution was stirred vigorously at 39 °C for 2 h. Then 1.84 mL of TEOS was dropped in and the mixture was stirred for 24 h; 1.0 mL of NH4F aqueous solution with a concentration of 1 wt% was added and the resulting mixture was transferred into an autoclave and kept at 100 °C for 30 h. The white solid obtained was filtered, washed with deionized water and ethanol, dried at 100 °C for 12 h, and calcined at 550 °C for 6 h. SBA-15 was prepared without adding TMB or NH4F, and the other operations were the same as those with the MCFs.MCF- or SBA-15-supported PEI was prepared by a wet impregnation method. In a typical preparation, the desired amount of PEI was dissolved in 1.5 mL of methanol under stirring for about 15 min, after which 0.1 g of calcined MCFs or SBA-15 was added to the solution. The resultant slurry was stirred continuously for about 30 min, and then dried at 70 °C for 16 h under 700 mm Hg vacuum. The as-prepared adsorbent was denoted as MCF-PEI(X)-Y, where X denotes the molecular weight (grams per mol) and the molecular structure of PEI (b for branched-PEI and l for linear-PEI), and Y denotes the PEI weight percentage.
Characterization of the adsorbents
The N2 adsorption/desorption was carried out on a Quantachrome Autosorb 1 automated adsorption apparatus. The sample was outgassed at 75 °C for 24 h using a high vacuum line prior to adsorption. The BET surface area was determined from the linear part of the BET plot (P/P0 = 0.10–0.30) from the adsorption branch. The total pore volume was determined as the volume of liquid nitrogen adsorbed at a relative pressure of 0.995. The average pore size was obtained from the adsorption branch. The BJH adsorption/desorption pore size was taken as the peak of the pore size distribution as calculated from the adsorption/desorption branch using the BJH method. For TG analyses, the samples were heated at a heating rate of 10 °C min−1 up to 800 °C.Adsorption and desorption measurements
The weight changes of the adsorbents were followed to determine the performances of the adsorbents using a magnetic suspension balance (Rubotherm). In a typical adsorption/desorption process, about 10 mg of the adsorbent was placed in a small sample container, heated to 100 °C in N2 atmosphere at a flow of 100 mL min−1, and held at that temperature for about 30 min until no weight change was observed. The temperature was then adjusted to the operating temperature, and the gas was switched from N2 to 99.8% bone-dry CO2 at a flow rate of 100 mL min−1 to perform adsorption. After that, the gas was switched to 99.995% pure N2 at a flow rate of 100 mL min−1 to perform desorption at the same temperature. In our experiment, adsorption of CO2 on the PEI-impregnated silica was found to follow a quick adsorption phase followed by a slow approach to equilibrium, as has been observed in several literature reports of amine-impregnated silica adsorbents.42,43,59,60 Thus, it is more practical to refer to the “working capacity” in reporting the amine-impregnated silica adsorbent performance, because it is more efficient to shorten the length of the adsorption cycle in an industrial process that to allow equilibration to occur. In this study, the period for each of the adsorption and desorption cycles was 96 min, and working adsorption capacity and working desorption efficiency were used to evaluate the PEI-impregnated silica adsorbent performance. The working adsorption capacities and working desorption efficiency are calculated as follows: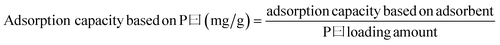 | (1) |
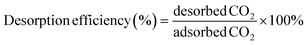 | (2) |
Acknowledgements
We acknowledge the Tsinghua-Cambridge-MIT Alliance (TCMA) for Low Carbon Energy for funding.References
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467–4475 CrossRef CAS.
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- C. L. Leci, Energy Convers. Manage., 1997, 38, S45–S50 CrossRef CAS.
- J. C. Abanades and D. Alvarez, Energy Fuels, 2003, 17, 308–315 CrossRef CAS.
- L. Bastin, P. S. Barcia, E. J. Hurtado, J. A. C. Silva, A. E. Rodrigues and B. Chen, J. Phys. Chem. C, 2008, 112, 1575–1581 CAS.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339–8344 CrossRef CAS.
- E. Diaz, E. Munoz, A. Vega and S. Ordonez, Ind. Eng. Chem. Res., 2008, 47, 412–418 CrossRef CAS.
- L. P. Ding, S. K. Bhatia and F. Liu, Chem. Eng. Sci., 2002, 57, 3909–3928 CrossRef CAS.
- T. C. Drage, A. Arenillas, K. M. Smith, C. Pevida, S. Piippo and C. E. Snape, Fuel, 2007, 86, 22–31 CrossRef CAS.
- N. D. Hutson, S. A. Speakman and E. A. Payzant, Chem. Mater., 2004, 16, 4135–4143 CrossRef CAS.
- M. B. Kim, Y. S. Bae, D. K. Choi and C. H. Lee, Ind. Eng. Chem. Res., 2006, 45, 5050–5058 CrossRef CAS.
- H. Lu, E. P. Reddy and P. G. Smirniotis, Ind. Eng. Chem. Res., 2006, 45, 3944–3949 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS.
- R. V. Siriwardane, M. S. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279–284 CrossRef CAS.
- P. Sun, J. R. Grace, C. J. Lim and E. J. Anthony, AIChE J., 2007, 53, 2432–2442 CrossRef CAS.
- K. S. Walton, M. B. Abney and M. D. LeVan, Microporous Mesoporous Mater., 2006, 91, 78–84 CrossRef CAS.
- Z. Yong, V. Mata and A. E. Rodriguez, Ind. Eng. Chem. Res., 2001, 40, 204–209 CrossRef CAS.
- Z. Yong and A. E. Rodrigues, Energy Convers. Manage., 2002, 43, 1865–1876 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- X. A. Diao, Y. J. Wang, J. Q. Zhao and S. L. Zhu, Chin. J. Chem. Eng., 2010, 18, 493–499 CrossRef CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159–162 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- J. Q. Zhao, Y. J. Wang, G. S. Luo and S. L. Zhu, Bioresour. Technol., 2010, 101, 7211–7217 CrossRef CAS.
- J. Q. Zhao, Y. J. Wang, G. S. Luo and S. L. Zhu, Particuology, 2011, 9, 56–62 CrossRef CAS.
- J. Q. Zhao, Y. J. Wang, G. S. Luo and S. L. Zhu, Bioresour. Technol., 2011, 102, 529–535 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446–458 CrossRef CAS.
- N. Hiyoshi, K. Yogo and T. Yashima, Chem. Lett., 2004, 33, 510–511 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Ind. Eng. Chem. Res., 2005, 44, 3702–3708 CrossRef CAS.
- S. Kim, J. Ida, V. V. Guliants and J. Y. S. Lin, J. Phys. Chem. B, 2005, 109, 6287–6293 CrossRef CAS.
- C. Knofel, J. Descarpentries, A. Benzaouia, V. Zelenak, S. Mornet, P. L. Llewellyn and V. Hornebecq, Microporous Mesoporous Mater., 2007, 99, 79–85 CrossRef.
- G. P. Knowles, S. W. Delaney and A. L. Chaffee, Ind. Eng. Chem. Res., 2006, 45, 2626–2633 CrossRef CAS.
- Z. Liang, B. Fadhel, C. J. Schneider and A. L. Chaffee, Microporous Mesoporous Mater., 2008, 111, 536–543 CrossRef CAS.
- J. W. Wei, J. J. Shi, H. Pan, W. Zhao, Q. Ye and Y. Shi, Microporous Mesoporous Mater., 2008, 116, 394–399 CrossRef CAS.
- F. Zheng, D. N. Tran, B. J. Busche, G. E. Fryxell, R. S. Addleman, T. S. Zemanian and C. L. Aardahl, Ind. Eng. Chem. Res., 2005, 44, 3099–3105 CrossRef CAS.
- T. C. Drage, A. Arenillas, K. M. Smith and C. E. Snape, Microporous Mesoporous Mater., 2008, 116, 504–512 CrossRef CAS.
- R. S. Franchi, P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2005, 44, 8007–8013 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514–1520 CrossRef CAS.
- W. J. Son, J. S. Choi and W. S. Ahn, Microporous Mesoporous Mater., 2008, 113, 31–40 CrossRef CAS.
- X. C. Xu, C. S. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- X. C. Xu, C. S. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Microporous Mesoporous Mater., 2003, 62, 29–45 CrossRef CAS.
- X. C. Xu, C. S. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113–8119 CrossRef CAS.
- X. C. Xu, C. S. Song, B. G. Miller and A. W. Scaroni, Fuel Process. Technol., 2005, 86, 1457–1472 CrossRef CAS.
- M. B. Yue, L. B. Sun, Y. Cao, Z. J. Wang, Y. Wang, Q. Yu and J. H. Zhu, Microporous Mesoporous Mater., 2008, 114, 74–81 CrossRef CAS.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS.
- Y. Han, S. S. Lee and J. Y. Ying, Chem. Mater., 2007, 19, 2292–2298 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, D. Y. Zhao, P. D. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254–255 CrossRef CAS.
- X. L. Ma, X. X. Wang and C. S. Song, J. Am. Chem. Soc., 2009, 131, 5777–5783 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS.
- A. von Harpe, H. Petersen, Y. X. Li and T. Kissel, J. Controlled Release, 2000, 69, 309–322 CrossRef CAS.
- M. B. Yue, Y. Chun, Y. Cao, X. Dong and J. H. Zhu, Adv. Funct. Mater., 2006, 16, 1717–1722 CrossRef CAS.
- X. X. Wang, V. Schwartz, J. C. Clark, X. L. Ma, S. H. Overbury, X. C. Xu and C. S. Song, J. Phys. Chem. C, 2009, 113, 7260–7268 CAS.
- J. D. Wind, S. M. Sirard, D. R. Paul, P. F. Green, K. P. Johnston and W. J. Koros, Macromolecules, 2003, 36, 6433–6441 CrossRef CAS.
- T. G. Amundsen, L. E. Oi and D. A. Eimer, J. Chem. Eng. Data, 2009, 54, 3096–3100 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 121, 254–255 CrossRef.
- C. Chen, W. J. Son, K. S. You, J. W. Ahn and W. S. Ahn, Chem. Eng. J., 2010, 161, 46–52 CrossRef CAS.
- G. G. Qi, Y. B. Wang, L. Estevez, X. N. Duan, N. Anako, A. H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 CAS.
Footnote |
| † Authors contributions: these authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2012 |