New second-order nonlinear optical (NLO) hyperbranched polymers containing isolation chromophore moieties derived from one-pot “A2 + B4” approach via Suzuki coupling reaction†
Wenbo
Wu
a,
Lijin
Huang
a,
Li
Xiao
a,
Qi
Huang
a,
Runli
Tang
a,
Cheng
Ye
b,
Jingui
Qin
a and
Zhen
Li
*a
aDepartment of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: lizhen@whu.edu.cn; lichemlab@163.com; Fax: (+86) 027 68755363
bInstitute of Chemistry, The Chinese Academy of Sciences, Beijing 100080, China
First published on 14th May 2012
Abstract
In this paper, a facile synthetic route was designed to prepare two new hyperbranched polymers HP1 and HP2 useful as second-order nonlinear optical (NLO) materials through a one-pot “A2 + B4” approach via a simple Suzuki coupling reaction. Thanks to the effect of the isolation chromophores to the main chromophore and the three-dimensional spatial isolation effect of the hyperbranched structure, these hyperbranched polymers demonstrated high second-harmonic generation coefficients (up to 73.6 pm V−1) and good optical transparency (with the maximum absorption as 466 nm in film).
Introduction
In the last few decades, considerable attention has been paid to organic/polymeric second-order nonlinear optical (NLO) materials due to their huge potential applications in photonics and many advantages over conventional inorganic crystalline materials, such as large nonlinearity, good processability, ultrafast response time and superior chemical flexibility.1 For the development of NLO materials, one of the major problems encountered is how to efficiently translate the large β values of the organic chromophores into high macroscopic NLO activities of polymers, since the strong intermolecular dipole–dipole interactions among the chromophore moieties in the polymeric system make the poling-induced noncentrosymmetric alignment of chromophores a daunting task.2 Fortunately, an elaborate three-dimensional (3D) structure was reported to minimize the dipole–dipole interactions by locking a chromophore inside the dendron shell and, thus, enhance the macroscopic NLO activities by applying the site-isolation principle.3 Many novel series of NLO dendrimers and hyperbranched polymers were prepared as typical examples.4 According to this principle, from 2006, our group designed and synthesized some NLO dendrimers and hyperbranched polymers through a different approach according to the concept of a “suitable isolation group”5 and our obtained results also demonstrated that the introduction of the NLO chromophore moieties to the system of dendrimers or hyperbranched polymers would lead to novel NLO polymers with very large macroscopic NLO effects.6 Usually, dendrimers require highly complicated and repetitive synthetic routes, resulting in the production of dendrimers being relatively costly.6h,j In contrast to dendrimers, hyperbranched polymers are of special interest as their easy synthetic accessibility, typically by one pot syntheses, allows their production in large quantities and their application on an industrial scale.7 Although the structure of hyperbranched polymers are imperfect and still contain linear units, they inherit the properties of dendrimers, such as good solubility, low viscosity and multifunctionality at end groups.7,8On the other hand, very recently, different from previous literatures that usually used non-polar groups as the isolation group according to the site-isolation principle, we used one chromophore with a lower μβ value as the isolation group for another chromophore group with a high μβ value. The result showed that introduction of isolation chromophores into NLO materials will improve the optical transparency and poling efficiency. Thus, we designed and synthesized a new series of NLO hyperbranched polymers HPS1 and HPS2 (Chart S1†) derived from an AB4-type monomer containing two chromophores with a regular structure by adjusting the amount of catalyst, solvent and reaction time, also using the end-capped groups.9 In HPS1 and HPS2, the nitro-based azo chromophore was used as the main chromophore with its β value of 19.8 × 10−30 esu, while the sulfonyl-based azo chromophore was used as an isolation chromophore with a β value of 13.0 × 10−30 esu.10 Thanks to the introduction of the isolation chromophore and the unique three-dimensional architecture, the obtained hyperbranched polymers exhibited very good NLO activities (with d33 values of 117.6 and 167.4 pm V−1, respectively) and optical transparency (the λmax of HPS1 in film was only 458 nm). The detailed data is shown in Table S1 in the ESI†.
However, there was no literature concerning hyperbranched NLO polymers containing isolation chromophores, and the AB4 approach to synthesize HPS1 and HPS2 reported by our group was a little difficult, limiting their large-scale application in the real world.9 Thus, design of a simple way to prepare these hyperbranched NLO polymers containing isolation chromophores was necessary. In this paper, we designed two new hyperbranched NLO polymers HP1 and HP2 (Scheme 1), similar to HPS1 and HPS2 reported before. The two polymers could be synthesized easily through an “A2 + B4” approach via a Suzuki coupling reaction, and demonstrated a good NLO coefficient and outstanding optical transparency. What’s more, this is the first time that the Suzuki coupling reaction has been applied to the synthesis of a hyperbranched NLO polymer to the best of our knowledge. Herein, we would like to report the synthesis, characterization and optical nonlinearities of these two polymers in detail.
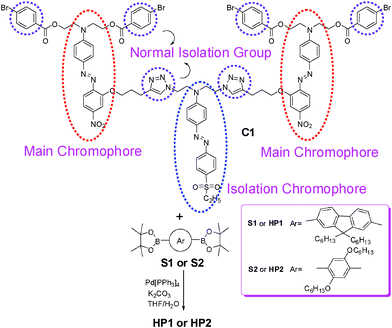 | ||
| Scheme 1 The synthesis of hyperbranched polymers HP1 and HP2. | ||
Results and discussion
Synthesis
The B2-type monomers boronic ester S1, S2 and the nitro-based chromophore C3 containing one yne group and two bromine groups as main chromophore was synthesized in our previous work,6e,11 while the synthetic pathway of the other chromophores is presented in Scheme 2. The sulfonyl-based chromophore C2 containing two azido groups as isolation chromophores was prepared under a normal azo-coupling reaction between the donor S3 and the acceptor S4 according to our previous work.6c,f,g From 2001, the Cu(I)-catalyzed cycloaddition between azides and alkynes to yield triazoles, which is often referred to as the “click reaction”, has aroused much interest among researchers because of its remarkable features, such as nearly quantitative yields, mild reaction conditions, broad tolerance toward functional groups, low susceptibility to side reactions and simple product isolation.12 In this work, under a typical “click chemistry” reaction between chromophores C2 and C3, the A4-type monomer C1 with the regular ABA structure was prepared easily in excellent yield (98%). And it is worth pointing out that the formed triazoles in the reaction could be utilized to act as good isolation groups6f–j between the main chromophore (the nitro-based one) and the isolation chromophore (the sulfonyl-based one). | ||
| Scheme 2 The synthesis of A4-type monomer C1. | ||
The polymerization procedure proceeded smoothly through the Suzuki coupling reaction using Pd(PPh3)4 as a catalyst, K2CO3 as a base and a mixture of THF/H2O (10/1, v/v) as the solvent. The copolymerization of A4 and B2 monomers might lead to the formation of gelation rapidly and many parameters could affect the final products in the “A2 + B4” polymerization, such as temperature, reaction time, concentrations of the monomers and so on.13 According to our previous work, to synthesize hyperbranched polymers also via a one-pot Suzuki reaction through an “A2 + B4” approach,13 the concentration of the monomer C1 was fixed at 8 × 10−3 mmol mL−1 to avoid the possible formation of gelation. However, the hyperbranched polymers obtained were not soluble in any solvent if the reaction times were long. Thus, the reaction must be stopped before the formation of gelation and the reaction time was also different (Table 1) due to their different copolymer units. In fact, once a little insoluble solid yielded, the reactions were stopped by the addition of methanol. Thus, their gelation points should be very close to that reaction time. At last, the obtained hyperbranched polymers were readily soluble in common polar organic solvents, such as CH2Cl2, CHCl3, THF, DMF and DMSO, and their solutions could be easily spin-coated into thin solid films. Therefore, it was convenient to test their NLO and other properties based on the solutions and thin films.
| No. | Reaction time (h) | Yield (%) | M w a (×103) | M w/Mna | T g b (°C) | T d c (°C) |
|---|---|---|---|---|---|---|
| a Determined by GPC in THF on the basis of a polystyrene calibration. b Glass transition temperature (Tg) of polymers detected by the DSC analyses under argon at a heating rate of 10 °C min−1. c The 5% weight loss temperature of polymers detected by the TGA analyses under nitrogen at a heating rate of 10 °C min−1. | ||||||
| HP1 | 8 | 73.3 | 7.51 | 1.78 | 143 | 275 |
| HP2 | 24 | 51.1 | 11.39 | 1.54 | 158 | 260 |
Characterizations
Spectroscopic techniques were employed to characterize the structures of the products and all of them gave satisfactory analysis data corresponding to their expected structures (see Figs 1 and S1–S7 in ESI†). Fig. 1 showed the FT-IR spectra of the hyperbranched polymers HP1, HP2 and the A4-type monomer C1. The absorption bands associated with the nitro groups and sulfonyl ones were at about 1515, 1338 and 1140 cm−1, respectively, and all of the peaks were present in the IR spectra of both the monomer and polymers, indicating the chromophore was stable during the Suzuki polymerization procedure. | ||
| Fig. 1 The FT-IR spectra of hyperbranched polymers HP1, HP2 and their corresponding monomer C1. | ||
In the 1H NMR spectra of the hyperbranched polymers HP1 and HP2, no unexpected resonance peaks were observed. The chemical shifts were consistent with the proposed polymer structure, as demonstrated in Scheme 1, however, showing an inclination of signal broadening due to polymerization. Fig. S3† is the 1H NMR spectra of HP1. Compared with the spectra of the monomer C1 (Fig. S1 in ESI†), it was easily seen that there were some new peaks at 2.1–2.4 (CCH in fluorene) and so on, indicating the success of polymerization. The same phenomenon could also be found in the 1H NMR spectra of HP2.
The molecular weights of HP1 and HP2, determined by gel permeation chromatography (GPC) using THF as the solvent and mono-disperse polystyrene as the calibration standard, were around 104 g mol−1, with polydispersity indexes (PDI) of 1.78 and 1.54, respectively (Table 1), showing the successful polymerization. However, it is still necessary to point out that the GPC analysis using linear polystyrenes as calibration standards often underestimate the molecular weights of hyperbranched polymers, which were 3D branched structures as mentioned before.14 Thus, the true molecular weights of the these hyperbranched polymers should be higher than those given in Table 1.
As shown in Fig. 2, these polymers were thermolytically resistant, with the 5% weight loss temperature (Td) as high as 275 and 260 °C, respectively. As the chemical structures of HP1 and HP2 were analogous, their Td was also nearly the same. Their glass transition temperatures (Tg) were also investigated using a differential scanning calorimeter (DSC) with the results summarized in Table 1. The Tgs of HP1 and HP2 were 143 and 158 °C, respectively. HP2 exhibited a higher glass transition temperature than HP1, possibly due to its slightly higher molecular weight.
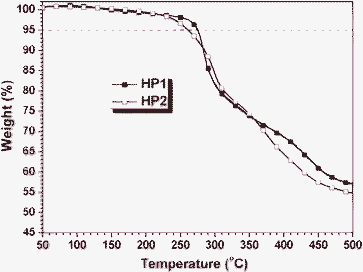 | ||
| Fig. 2 TGA thermograms of hyperbranched polymers, measured in nitrogen at a heating rate of 10 °C min−1. | ||
UV-vis spectra and effects of isolation chromophore
The UV-vis absorption spectra of the polymers in different solvents are demonstrated in Figs 3 and S8–S12†, while the maximum absorption wavelengths for the π–π* transition of the azo moieties in them are shown in Table 2. In comparison with the UV-vis absorption spectrum of C1, except the peaks for the π–π* transition of the azo chromophores, there was another peak around 330 nm in the spectra of the polymers, which could be associated with the conjugative rings of their copolymer units, again indicating the success of polymerization. | ||
| Fig. 3 UV-Vis spectra of hyperbranched polymers HP1, HP2 and their corresponding monomer C1 in THF (0.02 mg mL−1). | ||
In our previous work, it was obviously observed that the maximum absorption wavelength of the chromophore moieties of polymers constructed by two different chromophore moieties with the regular AB structure was red-shifted in comparison with that of the corresponding chromophore.9 The same phenomenon was observed in HPS1 and HPS2; there was an obvious red-shift in the UV-vis spectra of HPS2 compared to the corresponding chromophore. In this work, the λmaxs of HP1 and HP2 were also red-shifted in comparison with their corresponding A4-type chromophore C1, which should be caused by the effect of the isolation chromophore. However, the change of the maximum absorption was not so large, just about 6 nm red-shifted, which might indicate that the effect of the isolation chromophore was as strong as our previous work. The reason might be as follows. In our previous work,9 it was confirmed that just in the orderly AB structure, the chromophore with lower μβ value would act as the isolation chromophore to enhance the poling efficiency of NLO polymers, which means that the distance between the two chromophores could not be too far. But in this work, it was a little different: due to the present of the copolymer unit, there were large isolation groups between the two nitro chromophores, as shown in Fig. 4. On the other hand, the chromophore moieties in hyperbranched polymers might exhibit a blue-shifted maximum absorption in comparison with those of the free chromophores in the same solvent, due to the branched structure and site-isolation effects according to the literature and our previous work.6c,f,g These two reasons made the maximum absorption wavelength of these two polymers red-shifted not so large. Anyhow, the effect of the isolation chromophore could also be observed in this work.
 | ||
| Fig. 4 The sketch map of chromophores and isolation groups in the hyperbranched polymers HP1 and HP2. | ||
NLO Properties
To evaluate the NLO activity of the polymers, their poled thin films were prepared. The most convenient technique to study the second-order NLO activity is to investigate the second harmonic generation (SHG) processes characterized by d33, an SHG coefficient. The method for the calculation of the SHG coefficients (d33) for the poled films has been reported in our previous papers.15 From the experimental data, the d33 values of HP1 and HP2 were calculated to be 40.0 and 73.6 pm V−1, respectively, at 1064 nm fundamental wavelength (Table 3). To check the reproducibility, we repeated the measurements three times and got the same results.| No. | T e a (°C) | l s b (μm) | d 33 c (pm V−1) | d 33(∞)d (pm V−1) | Φ e | N f |
|---|---|---|---|---|---|---|
| a The best poling temperature. b Film thickness. c Second harmonic generation (SHG) coefficient. d The non-resonant d33 values calculated using the approximate two-level model. e Order parameter Φ = 1 − A1/A0, A1 and A0 are the absorbance of the polymer film after and before corona poling, respectively. f The loading density of the effective chromophore moieties, which was the ratios of the molecular weights of all of the chromophores (main chromophore and isolation chromophore) and the molecular weights of hyperbranched polymers. | ||||||
| HP1 | 145 | 0.27 | 40.0 | 7.5 | 0.11 | 0.40 |
| HP2 | 153 | 0.30 | 73.6 | 13.8 | 0.13 | 0.43 |
Here, the obtained NLO results were encouraging, and the polymers demonstrated relatively high d33 values, because of the effect of the isolation chromophore and the 3D topological structure of hyperbranched polymers. However, their NLO coefficient was lower than HPS1 and HPS2, which were synthesized through an AB4 approach via a “click chemistry” reaction. That was understandable. First of all, the hyperbranched polymers derived from AB4 monomers with the controlled structure more like dendrimers, have a better 3D topological structure,6c,f,g,9 and the void-rich topological structure could minimize the optical loss in the NLO process. On the other hand, as mentioned above, there were isolation groups between two nitro chromophores, as shown in Fig. 4. However, the isolation group here was too large and would decrease the loading density of the effective chromophore moieties to a large degree. It is well-known that in theory, under identical experimental conditions, the d33 value was proportional to the density of the chromophore moieties.5a So the d33 values of HP1 and HP2 were lower than HPS1 and HPS2, as a result of their lower density of the chromophore moieties (around 0.4 vs. around 0.6). Since HP2 (0.43) has a slightly higher loading density of the chromophore moieties than HP1 (0.40), its d33 value was also higher. At the same time, as we mentioned before, the shorter distance means a higher effect of the two types of chromophores. As the bulk of the phenyl ring was much smaller than the fluorene rings, the effect of the isolation chromophore in HP2 might be higher in HP1, and the NLO coefficient of HP2 was higher than that of HP1.
On the other hand, these phenomena could be explained by the concept of “suitable isolation groups” proposed in our previous work and it is confirmed that this concept could be used in hyperbranched polymers. In this work, there were two isolation groups around each sulfonyl-based chromophore, while there were three isolation groups around each nitro-based chromophore. According to our previous work on the suitable isolation groups of these two chromophores, the isolation group between two nitro-based chromophores was too large and the bulk of the phenyl ring or triazole rings was enough.6c,f–i Thus, in this work, the larger the B2-type unit, the lower the d33 value. HP2 showed a higher d33 value than HP1. In HPS1 and HPS2, the isolation groups around the chromophores were all phenyl rings or triazole rings, so they had the higher d33 values than the hyperbranched polymers in this work.
Anyway, HP1 and HP2 still demonstrated a high NLO coefficient due to the effect of isolation chromophores and the 3D topological structure of hyperbranched polymers, in comparison with some of our previous polymers that only contained normal isolation groups.5 As there might be some resonant enhancement due to the absorption of the chromophore moieties at 532 nm, the NLO properties of the dendrimers should be smaller, as shown in Table 3 (d33(∞)). As mentioned in the introduction, introduction of isolation chromophores into NLO materials would improve the optical transparency. HP1 and HP2 demonstrated good optical transparency with the maximum absorption wavelength of them in film at 466 nm. Thus, the d33(∞) values of HP1 and HP2 were still high (7.5 and 13.8 pm V−1), giving them potential for applications in optical fields.
To further explore the alignment of the chromophore moieties in the polymers, the order parameters (Φ) (Table 3) were also measured and calculated from the change of the UV-vis spectra of their thin films before and after poling under an electric field, according to the equation described in Table 3 (footnote e). Fig. 5 shows the UV-vis spectra of the film of hyperbranched polymers HP1 and HP2 before and after poling. The tested Φ values were in good accordance with their d33 values, further confirming our idea.
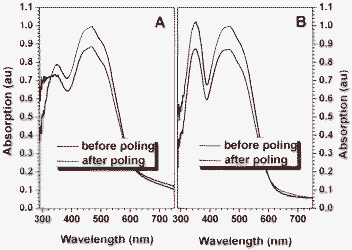 | ||
| Fig. 5 Absorption spectra of the film of HP1 (A) and HP2 (B) before and after poling. | ||
The dynamic thermal stabilities of the NLO activities of the polymers were investigated by depoling experiments, in which the real-time decays of their SHG signals were monitored as the poled films were heated from 35 to 145 °C in air at a rate of 4 °C min−1. Fig. 6 shows the decay of the SHG coefficient of HP1 and HP2 as a function of temperature; the onset temperatures for decays were found to be higher than 100 °C. The results indicated that the long-term temporal stability of the polymers was relatively good.
 | ||
| Fig. 6 Decay curves of the SHG coefficients of hyperbranched polymers HP1 and HP2 as a function of the temperature. | ||
Experimental section
Materials and instrumentations
Tetrahydrofuran (THF) was dried over and distilled from K–Na alloy under an atmosphere of dry nitrogen. Chromophore C3 and the monomers boronic ester S1 and S2 were synthesized as in our previous work and the sulfonyl-based chromophore C2 was prepared under a normal azo-coupling reaction between the donor S3 and the acceptor S4 according to our previous work. All other reagents were used as received.1H NMR spectra were measured on a Varian Mercury300 spectrometer using tetramethylsilane (TMS; δ = 0 ppm) as the internal standard. The Fourier transform infrared (FTIR) spectra were recorded on a PerkinElmer-2 spectrometer in the region of 3000–400 cm−1. UV-visible spectra were obtained using a Shimadzu UV-2550 spectrometer. Elemental analyses (EA) were performed by a CARLOERBA-1106 microelemental analyzer. Matrix-assisted laser desorption ionization time-of-flight mass spectra were measured on a Voyager-DE-STR MALDI-TOF mass spectrometer (MALDI-TOF MS; ABI, American) equipped with a 337 nm nitrogen laser and a 1.2 m linear flight path in positive ion mode. Gel permeation chromatography (GPC) was used to determine the molecular weights of polymers. GPC analysis was performed on a Waters HPLC system equipped with a 2690D separation module and a 2410 refractive index detector. Polystyrene standards were used as calibration standards for GPC, DMF was used as an eluent and the flow rate was 1.0 mL min−1. Thermal analysis was performed on a NETZSCH STA449C thermal analyzer at a heating rate of 10 °C min−1 in nitrogen at a flow rate of 50 cm3 min−1 for thermogravimetric analysis (TGA) and the thermal transitions of the polymers. The thickness of the films was measured with an Ambios Technology XP-2 profilometer.
Synthesis of the monomer C1
The sulfonyl-based chromophore C2 (47 mg, 0.11 mmol), the nitro-based chromophore C3 (184 mg, 0.24 mmol), CuSO4·5H2O (10 mol%), NaHCO3 (20 mol%) and ascorbic acid (20 mol%) were dissolved in THF (3 mL)/H2O (0.5 mL) under nitrogen in a Schlenk flask. The mixture was stirred at room temperature for 3 h, then extracted with chloroform and washed with brine. The organic layer was dried over anhydrous magnesium sulfate and purified by column chromatography using THF/chloroform (1/2) as the eluent to afford red solid chromophore C1 (220 mg, 98%). IR (KBr), υ (cm−1): 1723 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1511, 1341 (–NO2), 1120 (–SO2). 1H NMR (300 MHz, CDCl3, 298 K) δ (TMS, ppm): 1.30 (t, J = 7.2 Hz, 3H, –CH3), 2.24 (m, 4H, –CH2–), 2.96 (t, J = 7.2 Hz, 4H, –CCH2–), 3.16 (q, J = 7.2 Hz, 2H, –SCH2–), 3.72 (s, br, 4H, –NCH2–), 3.91 (t, J = 6.0 Hz, 8H, –NCH2–), 4.14 (t, J = 6.0 Hz, 4H, –NCH2–), 4.38 (s, br, 4H, –OCH2–), 4.55 (t, J = 6.0 Hz, 8H, –COOCH2–), 6.57 (d, J = 4.2 Hz, 4H, ArH), 6.94 (d, J = 4.5 Hz, 8H, ArH), 7.23 (s, 2H, CCH), 7.53–7.65 (m, 4H, ArH), 7.77–7.98 (m, 16H, ArH). 13C NMR (75 MHz, CDCl3, 298 K) δ (ppm): 21.55, 22.32, 23.64, 25.91, 26.26, 28.17, 28.35, 28.94, 29.73, 42.73, 46.94, 49.59, 50.82, 55.09, 61.47, 67.29, 68.24, 107.59, 108.86, 110.12, 111.39, 111.65, 116.18, 117.15, 120.80, 122.15, 122.68, 122.95, 125.71, 125.95, 128.24, 129.33, 133.06, 138.56, 138.89, 144.24, 144.62, 145.59, 146.54, 146.89, 147.92, 150.57, 154.80, 166.18. MALDI-TOF MS: calcd for (C88H81N17O16SBr4): m/z [M + H]+: 1984.3; found: m/z 1984.1. C88H81N17O16SBr4 (EA) (%, found/calcd): C, 53.03/53.26; H, 4.17/4.11; N, 11.65/12.00.
O), 1511, 1341 (–NO2), 1120 (–SO2). 1H NMR (300 MHz, CDCl3, 298 K) δ (TMS, ppm): 1.30 (t, J = 7.2 Hz, 3H, –CH3), 2.24 (m, 4H, –CH2–), 2.96 (t, J = 7.2 Hz, 4H, –CCH2–), 3.16 (q, J = 7.2 Hz, 2H, –SCH2–), 3.72 (s, br, 4H, –NCH2–), 3.91 (t, J = 6.0 Hz, 8H, –NCH2–), 4.14 (t, J = 6.0 Hz, 4H, –NCH2–), 4.38 (s, br, 4H, –OCH2–), 4.55 (t, J = 6.0 Hz, 8H, –COOCH2–), 6.57 (d, J = 4.2 Hz, 4H, ArH), 6.94 (d, J = 4.5 Hz, 8H, ArH), 7.23 (s, 2H, CCH), 7.53–7.65 (m, 4H, ArH), 7.77–7.98 (m, 16H, ArH). 13C NMR (75 MHz, CDCl3, 298 K) δ (ppm): 21.55, 22.32, 23.64, 25.91, 26.26, 28.17, 28.35, 28.94, 29.73, 42.73, 46.94, 49.59, 50.82, 55.09, 61.47, 67.29, 68.24, 107.59, 108.86, 110.12, 111.39, 111.65, 116.18, 117.15, 120.80, 122.15, 122.68, 122.95, 125.71, 125.95, 128.24, 129.33, 133.06, 138.56, 138.89, 144.24, 144.62, 145.59, 146.54, 146.89, 147.92, 150.57, 154.80, 166.18. MALDI-TOF MS: calcd for (C88H81N17O16SBr4): m/z [M + H]+: 1984.3; found: m/z 1984.1. C88H81N17O16SBr4 (EA) (%, found/calcd): C, 53.03/53.26; H, 4.17/4.11; N, 11.65/12.00.
Synthesis of HP1
A mixture of chromophore C1 (79.6 mg, 0.04 mmol), the boronic ester S1 (46.9 mg, 0.08 mmol), potassium carbonate (220.8 mg, 1.6 mmol), THF (5 mL)/water (0.5 mL) and Pd(PPh3)4 (1.5 mg) was carefully degassed and charged with argon. Then, the reaction mixture was stirred at 60 °C for 8 h. Before gelation, a lot of methanol was poured into the mixture, then filtered. The obtained solid was dissolved in THF and the insoluble solid was filtered out. After removal of the solvent, the residue was further purified by several precipitations from THF into methanol and the obtained solid was then washed with a lot of acetone and dried in a vacuum at 40 °C to a constant weight. The resultant polymer was obtained as a red powder (68.0 mg, 73.3%). Mw = 7510, Mw/Mn = 1.78 (GPC, polystyrene calibration). IR (KBr), υ (cm−1): 1724 (CO), 1518, 1336 (–NO2), 1120 (–SO2). 1H NMR (300 MHz, CDCl3, 298 K) δ (ppm): 0.4–0.8 (–CH3), 0.8–1.4 (–CH2–), 1.6–2.1 (–CH2–), 2.1–2.4 (–CCH2–), 2.8–3.0 (–CH2–), 3.0–3.2 (–SCH2–), 3.6–4.2 (–NCH2– and –OCH2–), 4.2–4.8 (–OCH2– and –COOCH2–), 6.4–6.7 (ArH), 6.8–7.2 (ArH), 7.2–8.2 (ArH and CCH). 13C NMR (75 MHz, CDCl3, 298 K) δ (ppm): 7.41, 13.95, 21.79, 22.48, 23.73, 28.35, 29.57, 31.36, 40.31, 47.23, 49.87, 50.66, 55.41, 61.88, 68,57, 109.24, 111.96, 112.43, 116.52, 117.50, 120.40, 121.55, 122.36, 126.15, 126.98, 127.15, 128.05, 128.45, 129.21, 130.17, 131.09, 131.83, 138.41, 138.83, 140.71, 144.65, 144.99, 146.02, 146,34, 147.17, 148.26, 149.24, 150.83, 151.92, 155.12, 155.74, 165.74, 166.41.
Synthesis of HP2
A mixture of chromophore C1 (79.6 mg, 0.04 mmol), the boronic ester S1 (42.4 mg, 0.08 mmol), potassium carbonate (220.8 mg, 1.6 mmol), THF (5 mL)/water (0.5 mL) and Pd(PPh3)4 (1.5 mg) was carefully degassed and charged with argon. Then, the reaction mixture was stirred at 60 °C for 24 h. Before gelation, a lot of methanol was poured into the mixture, then filtered. The obtained solid was dissolved in THF and the insoluble solid was filtered out. After removal of the solvent, the residue was further purified by several precipitations from THF into methanol and the obtained solid was then washed with a lot of acetone and dried in a vacuum at 40 °C to a constant weight. The resultant polymer was obtained as a red powder (45.0 mg, 51.1%). Mw = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 390, Mw/Mn = 1.54 (GPC, polystyrene calibration). IR (KBr), υ (cm−1): 1724 (CO), 1518, 1336 (–NO2), 1120 (–SO2). 1H NMR (300 MHz, CDCl3, 298 K) δ (ppm): 0.6–1.0 (–CH3), 1.0–1.8 (–CH2–), 1.8–2.4 (–CH2–), 2.8–3.0 (–CH2–), 3.1–3.2 (–SCH2–), 3.4–4.2 (–NCH2–), 4.2–4.4 (–OCH2–), 4.4–4.8 (–OCH2– and –COOCH2–), 5.1–5.2 (–OCH2–), 6.4–6.6 (ArH), 6.8–7.2 (ArH), 7.2–8.2 (ArH and CCH). 13C NMR (75 MHz, CDCl3, 298 K) δ (ppm): 7.20, 13.75, 13.89, 21.52, 22.27, 22.45, 23.19, 25.40, 26.12, 27.14, 28.07, 28.87, 29.11, 29.44, 31.09, 31.67, 31.93, 46.84, 50.41, 61.36, 68.30, 69.21, 108.81, 111.63, 112.02, 115.51, 116.24, 122.65, 124.75, 125.87, 127.72, 128.49, 129.08, 129.39, 129.79, 131.77, 134.96, 143.12, 144.73, 149.99, 166.25.
390, Mw/Mn = 1.54 (GPC, polystyrene calibration). IR (KBr), υ (cm−1): 1724 (CO), 1518, 1336 (–NO2), 1120 (–SO2). 1H NMR (300 MHz, CDCl3, 298 K) δ (ppm): 0.6–1.0 (–CH3), 1.0–1.8 (–CH2–), 1.8–2.4 (–CH2–), 2.8–3.0 (–CH2–), 3.1–3.2 (–SCH2–), 3.4–4.2 (–NCH2–), 4.2–4.4 (–OCH2–), 4.4–4.8 (–OCH2– and –COOCH2–), 5.1–5.2 (–OCH2–), 6.4–6.6 (ArH), 6.8–7.2 (ArH), 7.2–8.2 (ArH and CCH). 13C NMR (75 MHz, CDCl3, 298 K) δ (ppm): 7.20, 13.75, 13.89, 21.52, 22.27, 22.45, 23.19, 25.40, 26.12, 27.14, 28.07, 28.87, 29.11, 29.44, 31.09, 31.67, 31.93, 46.84, 50.41, 61.36, 68.30, 69.21, 108.81, 111.63, 112.02, 115.51, 116.24, 122.65, 124.75, 125.87, 127.72, 128.49, 129.08, 129.39, 129.79, 131.77, 134.96, 143.12, 144.73, 149.99, 166.25.
Preparation of polymer thin films
The polymers were dissolved in THF (concentration ∼4 wt%) and the solutions were filtered through syringe filters. Polymer films were spin coated onto indium-tin-oxide (ITO)-coated glass substrates, which were cleaned by N,N-dimethylformide, acetone, distilled water and THF sequentially in an ultrasonic bath before use. Residual solvent was removed by heating the films in a vacuum oven at 40 °C.NLO measurement of poled films
The second-order optical nonlinearity of the polymers was determined by an in situ second harmonic generation (SHG) experiment using a closed temperature-controlled oven with optical windows and three needle electrodes. The films were kept at 45° to the incident beam and poled inside the oven, and the SHG intensity was monitored simultaneously. Poling conditions were as follows: temperature, different for each polymer (Table 1); voltage, 7.5 kV at the needle point; gap distance, 0.8 cm. The SHG measurements were carried out with a Nd:YAG laser operating at a 10 Hz repetition rate and an 8 ns pulse width at 1064 nm. A Y-cut quartz crystal served as the reference.Conclusions
In summary, we have successfully synthesized two new NLO hyperbranched polymers containing isolation chromophore moieties through one-pot A2 + B4 Suzuki coupling polymerization for the first time. The two polymers were soluble in organic solvents and demonstrated good macroscopic NLO effects due to the effect of isolation chromophores to the main chromophore and the three-dimensional spatial isolation effect of the hyperbranched structure. Coupled with the good optical transparency and high temporal stability of the polymers, they might be promising candidates for practical NLO applications.Acknowledgements
We are grateful to the National Science Foundation of China (no. 21034006) for financial support.References
- (a) D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31 CrossRef CAS; (b) Y. Bai, N. Song, J. P. Gao, X. Sun, X. Wang, G. Yu and Z. Y. Wang, J. Am. Chem. Soc., 2005, 127, 2060 CrossRef CAS; (c) T. J. Marks and M. A. Ratner, Angew. Chem., Int. Ed., 1995, 34, 155 CrossRef CAS; (d) D. Yu, A. Gharavi and L. P. Yu, J. Am. Chem. Soc., 1995, 117, 11680 CrossRef CAS; (e) S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS; (f) M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401 CrossRef CAS; (g) Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson, W. H. Steier and L. R. Dalton, Science, 2000, 288, 119 CrossRef CAS; (h) C. V. Mclaughlin, M. Hayden, B. Polishak, S. Huang, J. D. Luo, T.-D. Kim and A. K. Y. Jen, Appl. Phys. Lett., 2008, 92, 151107 CrossRef; (i) L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25 CrossRef CAS; (j) J. D. Luo, X. H. Zhou and A. K. Y. Jen, J. Mater. Chem., 2009, 19, 7410 RSC; (k) J. D. Luo, S. Huang, Z. W. Shi, B. M. Polishak, X. H. Zhou and A. K. Y. Jen, Chem. Mater., 2011, 23, 544 CrossRef CAS.
- (a) B. H. Robinson and L. R. Dalton, J. Phys. Chem. A, 2000, 104, 4785 CrossRef CAS; (b) B. H. Robinson, L. R. Dalton, H. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorova, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. K.-Y. Jen, Chem. Phys., 1999, 245, 35 CrossRef CAS.
- (a) J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4782 CrossRef; (b) J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 Search PubMed; (c) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS.
- (a) M. J. Cho, D. H. Choia, P. A. Sullivan, A. J.-P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013 CrossRef CAS; (b) Y. V. Pereverzev, K. N. Gunnerson, O. V. Prezhdo, P. A. Sullivan, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, A. K.-Y. Jen and L. R. Dalton, J. Phys. Chem. C, 2008, 112, 4355 CrossRef CAS; (c) H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K. -Y. Jen, Adv. Funct. Mater., 2002, 12, 565 CrossRef CAS; (d) W. Shi, J. Luo, S. Huang, X.-H. Zhou, T.-D. Kim, Y.-J. Cheng, B. M. Polishak, T. R. Younkin, B. A. Block and A. K.-Y. Jen, Chem. Mater., 2008, 20, 6372 CrossRef.
- (a) Z. Li, Q. Li and J. Qin, Polym. Chem., 2011, 2, 2723 RSC; (b) Z. Li, Q. Zeng, Z. Li, S. Dong, Z. Zhu, Q. Li, C. Ye, C. Di, Y. Liu and J. Qin, Macromolecules, 2006, 39, 8544 CrossRef CAS; (c) Z. Li, Z. Li, C. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951 CrossRef CAS; (d) Q. Zeng, Z. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634 CrossRef CAS; (e) Q. Li, Z. Li, F. Zeng, W. Gong, Z. Li, Z. Zhu, Q. Zeng, S. Yu, C. Ye and J. Qin, J. Phys. Chem. B, 2007, 111, 508 CrossRef CAS; (f) Z. Li, P. Li, S. Dong, Z. Zhu, Q. Li, Q. Zeng, Z. Li, C. Ye and J. Qin, Polymer, 2007, 48, 3650 CrossRef CAS; (g) Z. Li, S. Dong, G. Yu, Z. Li, Y. Liu, C. Ye and J. Qin, Polymer, 2007, 48, 5520 CrossRef CAS; (h) Z. Li, S. Dong, P. Li, Z. Li, C. Ye and J. Qin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2983 CrossRef CAS.
- (a) Z. Li, A. Qin, J. W. Y. Lam, Y. Dong, Y. Dong, C. Ye, I. D. Williams and B. Z. Tang, Macromolecules, 2006, 39, 1436 CrossRef CAS; (b) Z. Zhu, Z. Li, Y. Tan, Z. Li, Q. Li, Q. Zeng, C. Ye and J. Qin, Polymer, 2006, 47, 7881 CrossRef CAS; (c) Z. Li, G. Yu, P. Hu, C. Ye, Y. Liu, J. Qin and Z. Li, Macromolecules, 2009, 42, 1589 CrossRef CAS; (d) Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2010, 1, 78 RSC; (e) Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Macromol. Chem. Phys., 2010, 211, 916 CrossRef CAS; (f) W. Wu, Y. Fu, C. Wang, C. Ye, J. Qin and Z. Li, Chem.–Asian J., 2011, 6, 2787 CrossRef CAS; (g) Z. Li, W. Wu, G. Qiu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1977 CrossRef CAS; (h) Z. Li, G. Yu, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 3864 CrossRef CAS; (i) Z. Li, W. Wu, Q. Li, G. Yu, L. Xiao, Y. Liu, C. Ye, J. Qin and Z. Li, Angew. Chem., 2010, 122, 2823 CrossRef ; Angew. Chem. Int. Ed. 2010, 49, 2763.
- (a) Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS; (b) A. Sunder, J. Heinemann and H. Frey, Chem.–Eur. J., 2000, 6, 2499 CrossRef CAS; (c) M. Häuβer and B. Z. Tang, Adv. Polym. Sci., 2007, 209, 1 CrossRef; (d) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS; (e) M. Scholl, Z. Kadlecova and H.-A. Klok, Prog. Polym. Sci., 2009, 34, 24 CrossRef CAS.
- (a) L. Zhi, J. Wu, J. Li, M. Stepputat, U. Kolb and K. Müllen, Adv. Mater., 2005, 17, 1492 CrossRef CAS; (b) C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS; (c) C. J. Hawker, Adv. Polym. Sci., 1999, 147, 113 CrossRef CAS; (d) D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS.
- W. Wu, C. Ye, G. Yu, Y. Liu, J. Qin and Z. Li, Chem.–Eur. J., 2012, 18, 4426 CrossRef CAS.
- A. Ulman, C. S. Willand, W. Kohler, D. R. Robello, D. J. Williams and L. Handley, J. Am. Chem. Soc., 1990, 112, 7083 CrossRef CAS.
- Z. Li, P. Hu, G. Yu, W. Zhang, Z. Jiang, Y. Liu, C. Ye, J. Qin and Z. Li, Phys. Chem. Chem. Phys., 2009, 11, 1220 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708 CrossRef ; Angew. Chem. Int. Ed. 2002, 41, 2596; (b) B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 15020 CrossRef CAS; (c) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS; (d) J. F. Lutz, Angew. Chem., 2007, 119, 1036 CrossRef ; Angew. Chem., Int. Ed. 2007, 46, 1018; (e) C. Ornelas, J. R. Aranzaes, E. Cloutet, S. Alves and D. Astruc, Angew. Chem., 2007, 119, 890 CrossRef ; Angew. Chem., Int. Ed. 2007, 46, 872; (f) A. Qin, C. K. W. Jim, W. Lu, J. W. Y. Lam, M. Häussler, Y. Dong, H. H. Y. Sung, I. D. Williams, G. K. L. Wong and B. Z. Tang, Macromolecules, 2007, 40, 230 Search PubMed; (g) H. Gao, G. Louche, B. S. Sumerlin, N. Jahed, P. Golas and K. Matyjaszewski, Macromolecules, 2005, 38, 8979 CrossRef CAS; (h) B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540 CrossRef CAS.
- (a) Z. Li, Y. Liu, G. Yu, Y. G. Wen, Y. Guo, L. Ji, J. Qin and Z. Li, Adv. Funct. Mater., 2009, 19, 2677 CrossRef CAS; (b) W. Wu, S. Ye, G. Yu, Y. Liu, J. Qin and Z. Li, Macromol. Rapid Commun., 2012, 33, 164 CrossRef CAS.
- (a) Z. Muchtar, M. Schappacher and A. Deffieux, Macromolecules, 2001, 34, 7595 CrossRef CAS; (b) M. W. Weimer, J. M. J. Fréchet and I. Gitsov, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 955 CrossRef CAS; (c) Y. Kim and O. W. Webster, Macromolecules, 1992, 25, 5561 CrossRef CAS.
- (a) Z. Li, J. Qin, S. Li, C. Ye, J. Luo and Y. Cao, Macromolecules, 2002, 35, 9232 CrossRef CAS; (b) Z. Li, C. Huang, J. Hua, J. Qin, Z. Yang and C. Ye, Macromolecules, 2004, 37, 371 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: The synthetic route to HPS1 and HPS2, NMR spectra, TOF mass spectra of chromophore C1, NMR spectra and UV-vis spectra of hyperbranched polymers HP1 and HP2. See DOI: 10.1039/c2ra20255g/ |
| This journal is © The Royal Society of Chemistry 2012 |