
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly and ambipolar charge transport in columnar phases of polynuclear gold isocyano–triphenylene complexes†
Estela
de Domingo
a,
Gregorio
García
 a,
Emiliano
Tritto
a,
Bertrand
Donnio
*b,
Asmita
Shah
c,
Dharmendra Pratap
Singh
*c and
Silverio
Coco
*a
a,
Emiliano
Tritto
a,
Bertrand
Donnio
*b,
Asmita
Shah
c,
Dharmendra Pratap
Singh
*c and
Silverio
Coco
*a
aIU CINQUIMA/Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, 47071 Valladolid, Castilla y León, Spain. E-mail: silverio.coco@uva.es
bInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504 (CNRS-Université de Strasbourg), F-67034 Strasbourg Cedex 2, France. E-mail: bertrand.donnio@ipcms.unistra.fr
cUnité de Dynamique et Structure des Matériaux Moléculaires (UDSMM), Université du Littoral Côte d’Opale (ULCO), 62228 Calais cedex, France. E-mail: dharmendra.singh@univ-littoral.fr
First published on 19th March 2025
Abstract
An uncommon approach to the synthesis of ambipolar semiconductors based on di- and tri-nuclear gold isocyano–triphenylene complexes of the formula [(AuX)n(CN–C6H4–O–(CH2)6)n–TriPh] (n = 2, 3; X = Cl, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ph) is described. Although mesomorphism has only been obtained with chloro derivatives, the trialkynyl complex has turned out to be a precursor of gold nanoparticles. The chloro complexes self-assemble in lamello-columnar phases, whose supramolecular organizations were confirmed by SAXS/WAXS experiments. Both the tri(chloro-gold) and the di(chloro-gold) complexes display high ambipolar charge transport along the columnar stacking direction, either in the mesophase (trichloro derivative) or in the crystalline solid state (dichloro complex). The analysis of ambipolar charge transport in the chloro-gold compounds has been performed using the time-of-flight (ToF) technique. The dichloro compound exhibits an ambipolar charge carrier mobility of the order of 10−4 cm2 V−1 s−1, whereas the trinuclear compound displays an ambipolar charge carrier mobility of the order of 10−3 cm2 V−1 s−1, which is attributed to the addition of a supplementary peripheral –NC–Au–Cl complex unit, offering a drift field to the charge carriers and a lower optical bandgap. Quantum chemical calculations show that the introduction of an additional –NC–Au–Cl fragment to the dinuclear complex to give the trinuclear derivative promotes a cofacial stacking of the molecules, which increases the mobility of the charge carriers of the system. Due to their ambipolar charge carrier mobility, the poly-nuclear gold isocyano–triphenylene complexes demonstrate their potential for use in organic electronics and optoelectronic devices.
C–Ph) is described. Although mesomorphism has only been obtained with chloro derivatives, the trialkynyl complex has turned out to be a precursor of gold nanoparticles. The chloro complexes self-assemble in lamello-columnar phases, whose supramolecular organizations were confirmed by SAXS/WAXS experiments. Both the tri(chloro-gold) and the di(chloro-gold) complexes display high ambipolar charge transport along the columnar stacking direction, either in the mesophase (trichloro derivative) or in the crystalline solid state (dichloro complex). The analysis of ambipolar charge transport in the chloro-gold compounds has been performed using the time-of-flight (ToF) technique. The dichloro compound exhibits an ambipolar charge carrier mobility of the order of 10−4 cm2 V−1 s−1, whereas the trinuclear compound displays an ambipolar charge carrier mobility of the order of 10−3 cm2 V−1 s−1, which is attributed to the addition of a supplementary peripheral –NC–Au–Cl complex unit, offering a drift field to the charge carriers and a lower optical bandgap. Quantum chemical calculations show that the introduction of an additional –NC–Au–Cl fragment to the dinuclear complex to give the trinuclear derivative promotes a cofacial stacking of the molecules, which increases the mobility of the charge carriers of the system. Due to their ambipolar charge carrier mobility, the poly-nuclear gold isocyano–triphenylene complexes demonstrate their potential for use in organic electronics and optoelectronic devices.
Introduction
Columnar liquid crystals (CLCs) are a representative class of self-assembled materials with a great variety of potential applications as soft organic semiconductors with unidirectional charge carrier transport properties.1–15 Triphenylene derivatives are among the most studied CLCs for applications in electronics, optoelectronics and LEDs.16,17 Their properties, including charge mobility along the columnar stacking direction, can be modulated through tailoring of the triphenylene core with a variety of functional groups,18–22 among which are metal–organic complexes.23–29 The functionalization of triphenylene molecules with moieties prone to self-organization in domains at the nanoscale sufficiently far from the aromatic core, for example, at the end of peripheral chains, has become a useful strategy for developing mesophases with segregated columns of different nature.30,31 This columnar segregation allows for a finer modulation of electronic properties such as ambipolar charge transport32–34 and photoconduction.35 In the case of triphenylene molecules functionalized with a metal group, interesting and unique hybrid inorganic/organic columnar mesophases, whose structures contain columnar packings of triphenylene moieties and independent stacks of metal fragments, have been reported.29,36–42 In these mesophases, the effect of the inorganic columns on the optical and charge mobility properties of the material is remarkable. For example, using platinum-containing moieties able to establish intermolecular metallophilic interactions, highly stabilized phosphorescent mesophases with high hole mobility along the columnar stack have been obtained.37 The high charge carrier mobilities of these mesophases are not restricted to the presence of metallophilic interactions but most likely to the ability of the metal containing moieties to self-organize into large columnar domains. Hence, in triphenylene molecules bearing Salen-type oxovanadium(IV) complexes,38 which self-assemble into one-dimensional structures via M⋯O interactions, high charge mobilities have also been found. In addition, an extraordinary enhancement of the hole mobility upon metal coordination to the free triphenylene Schiff bases is observed. This reveals the clear contribution of the metal–organic groups to increasing the conductivity in these systems and the high potential of metal–organic triphenylene molecules as semiconducting soft materials.Studies on the influence of metal–organic moieties on the thermal, optical, and conduction properties of mesomorphic triphenylene derivatives have been restricted to monometallic systems. However, the triphenylene moiety has many functionalization possibilities, particularly at the peripheral 2,3,6,7,10,11-positions, which represents an attractive platform for the design of polydentate ligands.43 Although neither is mesomorphic, there have already been a few reports of trimetallic derivatives of triphenylene with Zn(II),44 Cu(II),45 and Au(I),46 as well as some triphenylene-based metal–organic frameworks.47
On these grounds, we focus our attention on functionalizing triphenylene systems with two or three metal complexes, with the aim not only of preparing polymetallic liquid crystals, but also to study the effect of incorporating various metal groups on the charge mobility of the material. In this work, we report a new series of mesomorphic di- and tri-nuclear gold isocyano–triphenylene complexes of the formula [(AuX)n(CN–C6H4–O–(CH2)6)n–TriPh] (n = 2, 3; X = Cl, CC–Ph). The chloro complexes display high ambipolar charge transport along the columnar stacking direction, either in the mesophase or in the crystalline solid state. Interestingly, the charge carrier mobility (using ToF) increases ten-fold when going from the dinuclear derivative to the trinuclear complex. The trialkynyl gold derivative (12) is not a liquid crystal but undergoes extensive decomposition upon heating, yielding gold nanoparticles.
The study of charge transport in triphenylene-based columnar mesogens is of great interest as these supramolecular systems have demonstrated their utility in fabricating various optoelectronic devices. For example, the charge carrier mobility for holes in the hexa-alkoxytriphenylenes (HATn) varies from 10−5 to 0.01 cm2 V−1 s−1 depending on the molecular ordering and columnar phases exhibited by the materials; however, the addition of any electron-deficient moiety can lead to changes in molecular geometry and, thus, charge carrier mobility.48–50 In general, one of the disadvantages of the HATn system is the absence of ambipolar charge mobility, which could not be realized under typical experimental conditions; therefore, efforts must be made to obtain ambipolar charge mobility in triphenylene-based columnar systems, which has been investigated herein.
Additionally, the observed mesophase charge mobility in these systems has been observed to be temperature-invariant,51,52 which has also been investigated in this study.
Results and discussion
Synthesis and characterization
The previously unreported di- and tri-isocyanide triphenylene ligands were synthesized following standard methods, as shown in Scheme 1. | ||
| Scheme 1 Synthesis of the di- and tri-isocyanide triphenylene ligands (7, 8) and their di- and tri-nuclear gold complexes (9–12). | ||
The gold complexes were prepared by the reaction of the free isocyanide with the corresponding gold precursor. They were isolated as white solids. Full synthetic description, C, H, and N analyses, yields, MALDI-TOF mass spectra, and relevant IR and NMR characterization data are given in the experimental details (ESI,† Fig. S1–S25).
The UV-vis absorption and fluorescence spectra of the free isocyanide and the metal complexes in dichloromethane solution are all very similar (Fig. 1, and Table S1, ESI†), displaying a spectral pattern typical of 2,3,6,7,10,11-hexaalkoxytriphenylenes, and related monometallic isocyanide complexes.37 In the compounds described here, the triphenylene and the gold complexes are not electronically connected due to the use of an alkyl chain as a linker between them. Thus, as observed experimentally, a significant influence on its electronic transitions is not expected. Analogous results have been reported for related triphenylene mononuclear compounds.36 In the solid state, all the compounds display emission spectra similar to those recorded in dichloromethane solution, but their intensity decreases noticeably as described for related systems.37,41
 | ||
| Fig. 1 UV-vis absorption (a) and normalized luminescence emission spectra (b) of the di- and tri-isocyanide ligands and their gold complexes in dichloromethane solution (10−5 M) at 298 K. | ||
Thermal behaviour and self-organization properties
The mesomorphic properties of the free isocyanides (7, 8) and their gold(I) complexes (9–12) have been studied by polarised optical microscopy (POM), differential scanning calorimetry (DSC, Fig. S26–S31, ESI†), and small- and wide-angle X-ray scattering (S/WAXS, Fig. 4 and Fig. S32–S34, ESI†). The optical, thermal and thermodynamic data are collected in Table 1. The phase assignment in Table 1 is based on the results of the S/WAXS studies.| Compound | Transitiona | T (°C)b | ΔHb (kJ mol−1) |
|---|---|---|---|
| a Cr, crystal phase; CrLamCol, lamello-columnar crystal (crystalline phase with a rectangular sublattice); LamColrec: lamellar phase with a rectangular superlattice (lamello-columnar mesophase); I, isotropic liquid; mesophase assignment based on S/WAXS studies. b Data collected from the first and second heating, and first cooling DSC cycles. The transition temperatures are given as peak onsets. | |||
| (CN)2–TriPh (7) | Cr → I | 57 | 84.1 |
| (CN)3–TriPh (8) | Cr → I | 52 | 35.7 |
| [(AuCl)2((CN)2–TriPh)] (9) | Cr1 → Cr2 | 44 | 3.0 |
| Cr2 → Cr3 | 68 | 5.5 | |
| Cr3 → Cr4 | 77 | −3.7 | |
| Cr4 → I | 81 | 20.1 | |
| I → CrLamCol | 54 | 12.5 | |
| CrLamCol → I | 57 | 14.5 | |
| [(AuCC–Ph)2((CN)2–TriPh)] (10) | Cr → I | 18 | 4.1 |
| [(AuCl)3((CN)3–TriPh)] (11) | Cr → I | 112 | 52.5 |
| I → LamColrec | 49 | 16.9 | |
| LamColrec → I | 55 | 16.4 | |
| [(AuCC–Ph)3((CN)3–TriPh)] (12) | Cr → I(dec) | 142 | 28.5 |
Upon heating, the free isocyanide ligands (7, 8), the alkynyl derivatives and the dinuclear chloro gold complex (9) melt directly into an isotropic liquid and are not mesomorphic. In contrast, the trinuclear gold compound (11) shows a monotropic thermotropic liquid crystal behaviour. The texture observed by POM on cooling from the isotropic liquid (Fig. 2) is compatible with the occurrence of columnar mesophases, which was confirmed by small-angle X-ray diffraction studies.
 | ||
| Fig. 2 Optical polarizing microscopy photographs (crossed polarizers) on cooling from the isotropic phase of [Au3Cl3((CN)3–TriPh)] (11) at 48 °C. | ||
The compounds described here do not decompose when they reach the isotropic state, except for the trinuclear alkynyl complex (12) that undergoes extensive decomposition upon reaching the clearing temperature. The decomposition leads to the formation of a dark red product, whose UV-vis spectrum in dichloromethane solution shows surface plasmon absorption at 530 nm (Fig. S35, ESI†), characteristic of gold nanoparticles.53 Transmission electron micrographs showed spherical nanoparticles with an average diameter of 17.3 ± 0.5 nm with a broad particle size distribution (Fig. 3). This thermal instability has been studied in alkynylisocyanide gold mesogens without the triphenylene group and is associated with the thermal lability of the gold–isocyanide bond.54
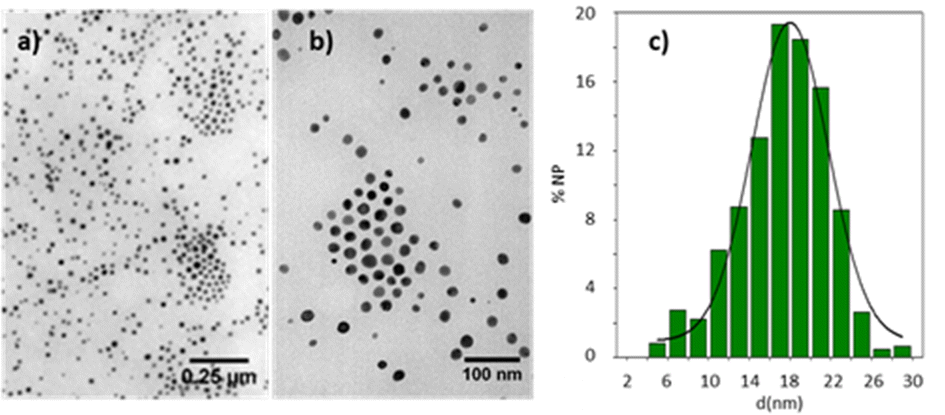 | ||
| Fig. 3 TEM pictures (a) and (b) and histogram (c) of nanoparticle size distribution obtained by thermal decomposition of 12 at 133 °C. | ||
S/WAXS studies
As just described, the tri(chloro-gold) complex (11) is mesomorphic, but only after having been heated into the isotropic liquid (above ca. 115 °C) and then cooled down to ambient temperature (Fig. S30, ESI†). The phase that is formed after a large supercooling (ca. 60 °C) and clears at ca. 58 °C on subsequent heating is thus transient and monotropic. The lamello-columnar mesophase was confirmed by SAXS/WAXS measurements performed at several temperatures. The X-ray pattern recorded at 20 °C, after being cooled from the isotropic liquid, displays a broad signal in the wide-angle range, corresponding to the average lateral distance between molten aliphatic chains with a maximum at ca. 4.11 Å (hch) and several sharp reflections in the small-angle range (Fig. 4).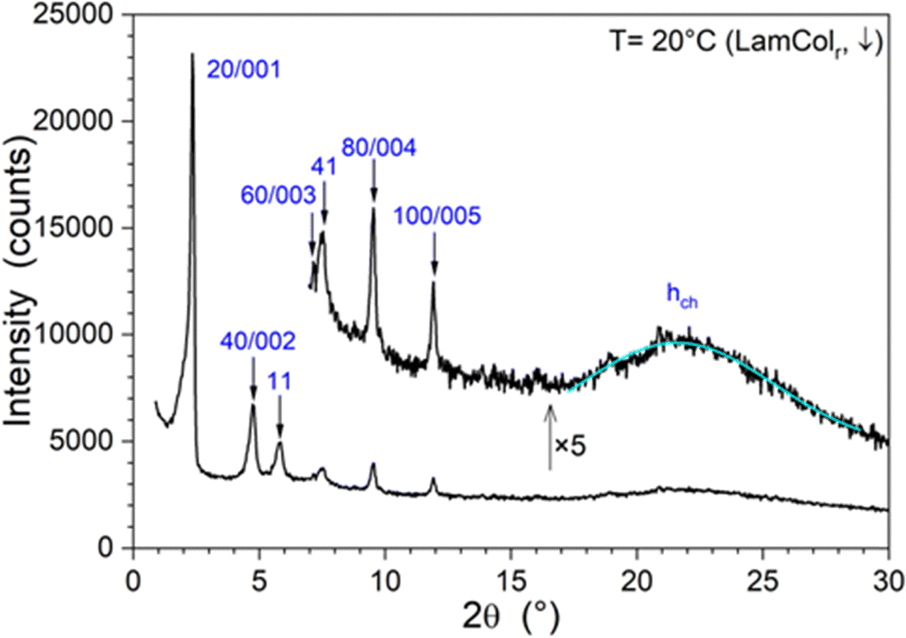 | ||
| Fig. 4 Representative S/WAXS pattern of 11 at 20 °C after cooling from the isotropic liquid (both lamellar and columnar indexations are shown). Additional patterns of 11 are shown in Fig. S29 (ESI†). | ||
The peaks were indexed within a 2D rectangular lattice of p2gg planar symmetry (hk with h + k = 2n + 1, Table S2, ESI†), with the parameters a = 74.1 Å and b = 15.6 Å (Table 2).
| Compounds | Parameters | |
|---|---|---|
| a a rec, brec, Arec: rectangular lattice parameters and area; Vmol: molecular volume; ρ: molecular density; Nmol: number of molecules (columns)/per lattice unit (Nmol = NCol); hmol: molecular thickness, hmol = Nmol × Vmol/Arec; dLamCol, dLam: lamellar periodicity; ΣLam: molecular area, ΣLam = Nmol × Vmol/dLamCol., σLam: mesogen area; Nmes, number of mesogens (–PhNCAuCl), σLam = ΣLam/Nmes = brec × hmol/Nmes). See also Fig. 5 and Fig. S31 (ESI) for a graphical representation of these parameters. | ||
| (9) | CrLamCol (crystalline phase with a p2mg rectangular sublattice) | |
| V mol = 2625 Å3 (ρ = 1.18 g cm−3) | ||
| CrColrec | CrLam | |
| a rec = 81.22 Å, brec = 24.52 Å | d LamCol = 81.22 Å | |
| A rec = 1991.5 Å2 | d Lam = 40.61 Å | |
| N mol = 4 | Σ Lam = 129.28 Å2 | |
| h mol = 5.27 Å | N mes = 2 | |
| σ Lam = 32.31 Å2 | ||
| (11) | LamColrec-p2gg | |
| V mol = 2690 Å3 (ρ = 1.315 g cm−3) | ||
| Colrec | Lam | |
| a rec = 74.09 Å, brec = 15.57 Å | d LamCol = 74.09 Å | |
| A rec = 1153.6 Å2 | d Lam = 37.20 Å | |
| N mol = 2 | Σ Lam = 72.61 Å2 | |
| h mol = 4.66 Å | N mes = 3 | |
| σ Lam = 24.20 Å2 | ||
The pattern further reveals a strong layering of the molecules, as most of the reflections could also be indexed as 00l reflections of a lamellar system (Table S2, ESI†); thus, the phase can be assigned as lamello-columnar with a rectangular symmetry. Making the reasonable assumption that both the triphenylene parts and the metallic fragments segregate in the volume since they have incompatible molecular shapes (i.e. discs vs. rods),37–41 the supramolecular organisation in the lamello-columnar phase results from the superposition of two sublayers of different nature separated by the aliphatic continuum (spacers and peripheral aliphatic chains): one of the sublayers would consist of a single row of columns, with the latter resulting from the stacking of the triphenylene cores, and the other one would accommodate the peripheral phenyl(isonitrile)gold(I) chloride protomesogens (Fig. 5). The ratio between the molecular volume (Vmol) and the lattice area (Arec), scaled by the number of molecules per lattice, Nmol, according to the formula, hmol = [Vmol × Nmol]/[Arec], simultaneously provides values for Nmol and hmol, the lattice thickness (Table 2), after successive trial and error attempts, consistent with the symmetry elements of the planar group and the various geometrical constraints. This calculation shows that two complexes can be organized within such a rectangular lattice, with a thickness hmol of about 4.66 Å, close to hch. The non-centrosymmetric lattice, deduced from the reflection rules, results from both the slight out-of-plane tilt of the triphenylenes within the columns, resulting in the columns having an ellipsoid cross-section instead of a cylindrical one,55 and the alternate orientation of the transversal long elliptic axis along the a-axis. The other sublayer is filled by the side-by-side antiparallel arrangement of 3 phenyl(isonitrile)gold(I) chloride fragments distributed in a way that the sum of their transverse area (ΣLam = 3 × σLam) is compensated by that of one triphenylene transverse cross-section in the mesophases (Fig. 5).
 | ||
| Fig. 5 Schematic representations (2D projection) of the LamColrec mesophase of compound 11 and of the 2D sublattice of the CrLamCol crystalline phase of 9, with respectively 2 and 4 molecules per lattice: black and blue elliptic cross-sections of ribbons of the columns formed by the stacking of the triphenylene mesogenic fragments (slightly titled), arranged in a rectangular lattice/sublattice (red lines) with a p2gg (left) or p2mg (right) plane group, respectively; black and blue rectangles, sublayers formed by the metallic protomesogenic units (–Ph–NC–Au–Cl). The rest of the lattice is occupied by the aliphatic chains (not shown). See also Fig. S31 (ESI†). | ||
The di(chloro-gold) complex (9) is however not mesomorphic and displays a rather complicated thermal behaviour. During the first heating, two crystal-to-crystal phase transitions are detected at ca. 50.5 °C and 71.6 °C, respectively, immediately followed by a recrystallisation event at 76.5 °C, and complete clearing at 84 °C (Fig. S28, ESI†). On cooling, the isotropic-to-crystal phase transition is strongly supercooled (ca. 30 °C). The following cycles are much simpler and consist of a single transition between a crystalline phase and an isotropic liquid at 59 °C (and ca. 53 °C on cooling). The SAXS/WAXS pattern, recorded at 20 °C on cooling from the isotropic liquid (Fig. S32, ESI†), displays many sharp reflections over the entire investigated angular range, along with a broad wide-angle scattering signal, emerging from lateral interactions between partially molten chains (hch). Such a typical pattern reveals a crystal-like, three-dimensional long-range organization, coexisting with partially molten chain segments (this type of phase can also be described as a soft crystalline phase). Similar to the tri(chloro-gold) complex, the di(chloro-gold) complex displays a strong lamellar structure and an in-plane rectangular sublattice, but the p2mg planar symmetry (a = 81.22 Å, b = 24.52 Å) of a more complex 3D structure could be deduced from SAXS/WAXS (the 3D structure, which could be indistinctly of monoclinic, quadratic or orthorhombic nature, could not be solved at this stage). The overall lamello-columnar supramolecular organisation is similar to the above, in that it consists of the alternation of sublayers of columns formed by the stacking of the triphenylene cores and of phenyl(isonitrile)gold(I) chloride protomesogens separated by the aliphatic continuum (Fig. 5). Due to the molecular difference, and using the same geometrical approach, four complexes can be organized within such a lattice, with a thickness hmol of about 5.27 Å this time. One sublayer would consist of the lateral arrangement of triphenylene columns in a single row, with alternate orientation of their long transversal long axis with respect to both a- and b-parameters of the rectangular lattice, whereas the other sublayer is filled by 4 phenyl(isocyanide)gold(I) chloride fragments in a way that their transverse area (ΣLam = 4 × σLam) is compensated by the transverse cross-sections of two triphenylene moieties in the crystal 3D lattice. The expansion of the lattice from trigold to digold complexes may be due to the extra chain with respect to the former compound, which requires more space and thus the increase of the number of molecules per lattice.
Charge transport properties
The ambipolar charge transport properties were analysed by determining the hole and electron mobilities in compounds 9 and 11 using the time-of-flight (ToF) technique. Firstly, the compounds were filled in a 20 μm homeotropically anchored ITO-coated cells. Due to the strong π–π stacking capability of the triphenylene core, both compounds preferably adopt homeotropic orientation, i.e. the columnar axis remains perpendicular to the substrate. The compounds were irradiated using a Nd:YAG pulse laser with an excitation wavelength of 355 nm. A suitable bias voltage (positive for holes and negative for electrons) was applied to observe the transient photocurrent curves for the positive and negative charge carriers (i.e. holes and electrons, respectively). After estimating the transit time at different temperatures, the electron and hole mobilities were determined by using the equation: μe,h(T) = d2/V·τ, where d is the thickness of the cell, V is the applied positive or negative bias voltage and τ is the transit time for charge carriers at a particular temperature as described in the previous articles.56,57The transient photocurrent curves of both compounds are presented in Fig. 6, which evinced the dispersive nature of charge propagation corresponding to electrons and holes. Usually, the dispersive nature of the photocurrent curves could be due to several factors such as the bulk nature of molecules, chemical or ionic impurities, the presence of shallow traps and defects in the materials, etc.;58–60 however, in our case, the dispersive nature is probably attributed to the presence of a partially ionic moiety (–NC–Au–Cl) at the terminals of both compounds causing an ionic hindrance in the electronic charge transport (Fig. 6a and b). It is also observed that the magnitude of photocurrent also depends on the laser intensity. The observed photocurrent is directly proportional to the generated charge carriers, i.e. I(t) = dQ/dt; however, the generation of charge carriers remarkably depends on the production of excitons and subsequent dissociation into electrons and holes after the absorption of incident photonic energy via laser irradiation on the material. Following the quantum mechanical approach, the total surface charge density received at the bottom electrode will be JBE = Ne/A, where N is the number of generated charge carriers and A is the surface area of the bottom electrode. It is therefore evident from Fig. 6c that the magnitude of photocurrent increases with increasing intensity of laser irradiation.
 | ||
| Fig. 6 Ambipolar transient photocurrent curves for (a) 9, (b) 11 and (c) 11 with two different laser intensities, i.e. I3 ≈ 3% (equivalent to 3 mW) and I4 ≈ 4% (equivalent to 4 mW). | ||
After extracting the transit time of charge carriers from the photocurrent curves, the ambipolar charge mobility has been calculated for the electrons and holes, which is plotted in Fig. 7a and b, respectively, for both compounds 9 and 11. Compound 9 showed an ambipolar charge carrier mobility of the order of 10–4 cm2 V−1 s−1 which is almost temperature invariant; however, the ambipolar charge carrier mobility has been found to be superior for compound 11. The charge carrier mobility has been found of the order of 10−3 cm2 V−1 s−1, in which electron mobility (μe) is superior to hole mobility (μh). This increase in mobility in compound 11 is analogous to its LUMO level energy and optical bandgap (∼3.43 eV, Table S3, ESI†), which is lower than that for compound 9 (∼3.46 eV, Table S3, ESI†). The observed mobility in 9 and 11 is in good agreement with the previously reported triphenylene-based compounds.15,54,55
 | ||
| Fig. 7 Temperature dependence of ambipolar charge carrier mobility for compounds (a) 9 and (b) 11. μe and μh, respectively, represent the electron and hole mobilities. | ||
Quantum chemical calculations
According to previous works on charge transport in discotic liquid crystals, including triphenylene derivatives, charge-carrier mobility is highly dependent on molecular arrangement within the columnar stacking. This arrangement is mainly determined by three key structural parameters: π-stacking, lateral displacement, and azimuthal angle (τ), the last of these being the most critical factor.61,62 Therefore, to shed light on the difference in the charge transport values of the studied complexes, quantum chemical calculations were performed on dimer models to describe the relative disposition between two adjacent molecules within the columnar structures, (Fig. 8). The quantum chemical calculations have been carried out keeping the inter-disc distances at the experimental values (5.27 Å for 9 and 4.66 Å for 11). See the ESI† for more details and Fig. S37. | ||
| Fig. 8 Relative energy (ΔE) evolution as a function of the azimuthal angle (τ) between triphenylene cores. Intermolecular π-stacking distances have been fixed according to experimental data from X-ray diffraction studies. ΔE values were calculated with respect to the minimum energy situation for each complex (τ = 45° and 60° for the di(chloro-gold) and tri(chloro-gold) complexes, respectively). Representations of two stacked molecules of di(chloro-gold) and tri(chloro-gold) complexes (9 and 11, respectively, where both molecules are highlighted in different colours) corresponding to configurations with minimal energy as a function of the azimuthal angle (τ) are also displayed. H atoms are omitted. | ||
For the di(chloro-gold) complex (9), the most stable arrangements were found for triphenylene cores with azimuthal angles of 0°, 45°, 70°, 125°, and 165°, which correspond to three staggered configurations (45°, 70°, 165°) and two eclipsed arrangements (0°, 125°). The lowest energy arrangement is for the staggered configuration with τ = 45°, but the relative energies of the other four minima are 4.1 kcal mol−1 for τ = 0° (eclipsed), 3.7 kcal mol−1 for τ = 70° (staggered), and 2.2 kcal mol−1 for both τ = 165° (staggered) and τ = 125° (eclipsed). Consequently, the predominant contribution to the description of the molecular stacking in this compound is due to staggered configurations. These results are consistent with previous studies indicating that alternating π-stacking geometries generally yield the most favourable arrangements.60 For the tri(chloro-gold) complex (11), five minima were also identified. Three of them involve staggered configurations (τ = 40°, 60°, and 160°), while two exhibit eclipsed arrangements (τ = 0° and 125°). The minimal energy corresponds to the alternating arrangement between the triphenylene cores with τ = 60°. However, in this case the other four configurations (two staggered and two eclipsed) show very low energy differences (between 0.5 kcal mol−1 and 1.2 kcal mol−1) compared to the minima.
In short, for both the di(chloro-gold) and tri(chloro-gold) complexes, the relative disposition of molecules within the columnar structure can be described as a combination of alternating and eclipsed configurations between the aromatic cores, with alternating arrangements being the predominant contribution. This finding aligns with previous studies on triphenylene-related systems.60 However, in our case, the presence of the gold substituent also allows the eclipsed configurations to be easily accessible, especially in the tri(chloro-gold) complex, where the contribution of the eclipsed configurations is comparable to that of the staggered arrangements. This circumstance allows cofacial stackings that enhance charge transport along the column.61 Therefore, the lower energy difference between alternating and eclipsed dispositions in the tri(chloro-gold) complex allows eclipsed structures to play a more significant role in describing the supramolecular structure within the column, thereby improving charge transport properties.
Conclusions
This study shows that the chemical polyfunctionalization of triphenylene mesogens at the end of the alkoxy substituents with –NC–Au–X (Cl, CC–Ph) synthons constitutes a simple strategy to design semiconducting columnar phases with high ambipolar charge mobility. As the number of organometallic groups increases from two to three, the intermolecular interactions become stronger. Consequently, the transition temperatures increase, and the appearance of columnar mesophases is favored for the chloro complexes. In contrast, the trialkynyl derivative undergoes extensive decomposition upon heating, leading to the formation of spherical gold nanoparticles.
The charge carrier mobility of this system was studied on the di- and tri(chloro-gold) compounds. The di(chloro-gold) compound (9) shows an ambipolar charge carrier mobility of 10−4 cm2 V−1 s−1. The introduction of a third organometallic fragment into the system results in an increase in charge mobility by one order of magnitude. This increase has been associated with the greater tendency of 11 to stack co-facially, which enhances charge transport along the column. This trend is also consistent with the LUMO level energy and optical bandgap (3.43 eV) of 11, which are found to be lower than those of the di(chloro-gold) compound 9. These results evidence the potential use of poly-nuclear gold isocyano–triphenylene complexes in organic electronics and optoelectronic devices because of their appreciable ambipolar charge carrier mobility and self-assembled supramolecular nature.
Author contributions
All authors have contributed equally to this paper.Data availability
The authors declare that the data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the University of Valladolid. We thank Prof. Pablo Espinet for helpful discussions and Dr Rubén Chico for his help in the nanoparticle characterization. E.D. thanks MEDC for her FPU contract. BD thanks CNRS and the University of Strasbourg for support and Dr Benoît Heinrich for performing the S/WAXS measurements. DPS is thankful to ULCO for financial assistance via BQR, BQI, and POLE MTE projects. This research has made use of the high-performance computing resources of the Castilla y León Supercomputing Center (SCAYLE, www.scayle.es), financed by the European Regional Development Fund (ERDF). The authors thankfully acknowledge the use of the computer resources at Lusitania II and the technical support provided by Cénits-COMPUTAEX.Notes and references
- B. R. Kaafarani, Chem. Mater., 2011, 23, 378–396 Search PubMed.
- W. Pisula, M. Zorn, J. C. Chang, K. Müllen and R. Zentel, Macromol. Rapid Commun., 2009, 30, 1179–1202 CrossRef CAS.
- T. Kato, T. Yasuda and Y. Kamikawa, Chem. Commun., 2009, 729–739 RSC.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 Search PubMed.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 Search PubMed.
- M. Oukachmih, P. Destruel, L. Seguy, G. Ablart, P. Jolinat, S. Archambeau, M. Mabiala, S. Fouet and H. Bock, Sol. Energy Mater. Sol. Cells, 2005, 85, 535–543 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkotter, K. Müllen, E. Moons, R. Friend and J. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS.
- Y. Wang, J. Shi, J. Chen, W. Zhu and E. Baranoff, J. Mater. Chem. C, 2015, 3, 7993–8005 RSC.
- R. J. Bushby, S. M. Kelly and M. O’Neill, Liquid crystalline semiconductors: materials, properties and applications, Springer, Dordrecht, Netherlands, 2013 Search PubMed.
- T. Wohrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef.
- R. Termine and A. Golemme, Int. J. Mol. Sci., 2021, 22, 877 CrossRef CAS PubMed.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- R. Uttam, A. Khare, S. Kumar and R. Dhar, Liq. Cryst., 2021, 49, 523–542 CrossRef.
- M. Vadivel, S. Singh, D. P. Singh, V. A. Raghunathan and S. Kumar, J. Phys. Chem. B, 2021, 125, 10364–10372 CAS.
- W. C. Chen, Y. H. Zhang, H. F. Zheng, H. T. Li, F. L. Guo, G. Ni, M. L. Ma, C. W. Shi, R. Ghadari and L. H. Hu, Chem. Commun., 2020, 56, 1879–1882 Search PubMed.
- S. Kumar, Liq. Cryst., 2020, 47, 1195–1203 CrossRef CAS.
- N. Boden, R. J. Bushby and A. N. Cammidge, Liq. Cryst., 1995, 18, 673–676 CAS.
- N. Boden, R. J. Bushby, A. N. Cammidge and G. Headdock, J. Mater. Chem., 1995, 5, 2275–2281 RSC.
- S. Kumar, M. Manickam, V. S. K. Balagurusamy and H. Schonherr, Liq. Cryst., 1999, 26, 1455–1456 CAS.
- R. J. Bushby, N. Boden, C. A. Kilner, O. R. Lozman, Z. Lu, Q. Liu and M. A. Thornton-Pett, J. Mater. Chem., 2003, 13, 470–474 RSC.
- N. Boden, R. J. Bushby, Z. B. Lu and A. N. Cammidge, Liq. Cryst., 1999, 26, 495–499 CrossRef CAS.
- S. Kumar and S. K. Varshney, Liq. Cryst., 2001, 28, 161–163 CrossRef CAS.
- J. L. Schulte, S. Laschat, R. Schulte-Ladbeck, V. von Arnim, A. Schneider and H. Finkelmann, J. Organomet. Chem., 1998, 552, 171–176 CrossRef CAS.
- A. N. Cammidge and H. Gopee, Chem. Commun., 2002, 966–967 RSC.
- B. Mohr, G. Wegner and K. Ohta, Chem. Commun., 1995, 995–996 RSC.
- F. Yang, X. Bai, H. Guo and C. Li, Tetrahedron Lett., 2013, 54, 409–413 CrossRef CAS.
- J. Shi, Y. Wang, M. Xiao, P. Zhong, Y. Liu, H. Tan, M. Zhu and W. Zhu, Tetrahedron, 2015, 71, 463–469 CrossRef CAS.
- R. Chico, C. Domínguez, B. Donnio, B. Heinrich, S. Coco and P. Espinet, Cryst. Growth Des., 2016, 16, 6984 CrossRef CAS.
- S. P. Gupta, M. Gupta and S. K. Pal, ChemistrySelect, 2017, 2, 6070–6077 CAS.
- E. Beltrán, M. Garzoni, B. Feringán, A. Vancheri, J. Barberá, J. L. Serrano, G. M. Pavan, R. Giménez and T. Sierra, Chem. Commun., 2015, 51, 1811–1814 Search PubMed.
- B. Feringán, P. Romero, J. L. Serrano, C. L. Folcia, J. Etxebarria, J. Ortega, R. Termine, A. Golemme, R. Giménez and T. Sierra, J. Am. Chem. Soc., 2016, 138, 12511–12518 Search PubMed.
- H. Hayashi, W. Nihashi, T. Umeyama, Y. Matano, S. Seki, Y. Shimizu and H. Imahori, J. Am. Chem. Soc., 2011, 133, 10736–10739 CAS.
- K.-Q. Zhao, L.-L. An, X.-B. Zhang, W.-H. Yu, P. Hu, B.-Q. Wang, J. Xu, Q.-D. Zeng, H. Monobe, Y. Shimizu, B. Heinrich and B. Donnio, Chem. – Eur. J., 2015, 21, 10379–10390 CAS.
- T. Umeyama, N. Tezuka, F. Kawashima, S. Seki, Y. Matano, Y. Nakao, T. Shishido, M. Nishi, K. Hirao, H. Lehtivuori, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Angew. Chem., Int. Ed., 2011, 50, 4615–4619 CAS.
- R. Chico, E. de Domingo, C. Domínguez, B. Donnio, B. Heinrich, R. Termine, A. Golemme, S. Coco and P. Espinet, Chem. Mater., 2017, 29, 7587–7595 CAS.
- V. Conejo-Rodrıguez, B. Donnio, B. Heinrich, R. Termine, A. Golemme and P. Espinet, J. Mater. Chem. C, 2023, 11, 1435–1447 Search PubMed.
- M. Barcenilla, M. J. Baena, B. Donnio, B. Heinrich, L. Gutiérrez, S. Coco and P. Espinet, J. Mater. Chem. C, 2022, 10, 9222–9231 CAS.
- E. de Domingo, C. L. Folcia, J. Ortega, J. Etxebarria, R. Termine, A. Golemme, S. Coco and P. Espinet, Inorg. Chem., 2020, 59, 10482–10491 CAS.
- E. Tritto, R. Chico, G. Sanz-Enguita, C. L. Folcia, J. Ortega, S. Coco and P. Espinet, Inorg. Chem., 2014, 53, 3449 CAS.
- E. Tritto, R. Chico, J. Ortega, C. L. Folcia, J. Etxebarria, S. Coco and P. Espinet, J. Mater. Chem. C, 2015, 3, 9385 RSC.
- A. B. Miguel-Coello, M. Bardají, S. Coco, B. Donnio, B. Heinrich and P. Espinet, Inorg. Chem., 2018, 57, 4359–4369 CrossRef CAS PubMed.
- D. Sonet and B. Bibal, Tetrahedron Lett., 2019, 60, 872–884 CrossRef CAS.
- V. Bhalla, H. Arora, A. Dhir and M. Kumar, Chem. Commun., 2012, 48, 4722–4724 RSC.
- V. Bhalla, H. Singh and M. Kumar, Dalton Trans., 2012, 41, 11413–11418 RSC.
- S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2013, 52, 7009–7013 CAS.
- N. Contreras-Pereda, S. Pané, J. Puigmartí-Luis and D. Ruiz-Molina, Coord. Chem. Rev., 2022, 460, 214459 CAS.
- H. Lino, J.-I. Hanna, R. J. Bushby, B. Movaghar, B. J. Whitaker and M. J. Cook, Appl. Phys. Lett., 2005, 87, 132102 Search PubMed.
- A. Shah, D. P. Singh, B. Duponchel, F. Krasisnski, A. Daoudi, S. Kumar and R. Douali, J. Mol. Liq., 2021, 342, 117353 CAS.
- P. Mahesh, A. Shah, K. Swamynathan, D. P. Singh, R. Douali and S. Kumar, J. Mater. Chem. C, 2020, 8, 9252–9261 CAS.
- T. Kreouzis, K. J. Donovan, N. Boden, R. J. Bushby, O. R. Lozman and Q. Liu, J. Chem. Phys., 2001, 114, 1797–1802 CAS.
- D. Markovitsi, A. Germain, P. Millié, P. Lécuyer, L. K. Gallos, P. Argyrakis, H. Bengs and H. Ringsdorf, J. Phys. Chem., 1995, 99, 1005–1017 CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CAS.
- R. Chico, E. Castillejos, P. Serp, S. Coco and P. Espinet, Inorg. Chem., 2011, 50, 8654–8662 CAS.
- B. Donnio, B. Heinrich, H. Allouchi, J. Kain, S. Diele, D. Guillon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 15258–15268 CAS.
- I. Bala, W.-Y. Yang, S. P. Gupta, J. De, R. A. Kumar Yaday, D. P. Singh, D. K. Dubey, J.-H. Jou, R. Douali and S. Kumar Pal, J. Mater. Chem. C, 2019, 7, 5724–5738 CAS.
- A. Shah, B. Duponchel, A. Gowda, S. Kumar, M. Becuwe, C. Davoisne, C. Legrand, R. Douali and D. P. Singh, New J. Chem., 2020, 44, 14872–14878 CAS.
- S. Dhingra, S. P. Gupta, A. Shah, D. P. Singh and S. Kumar Pal, Chem. Commun., 2024, 60, 2922–2925 RSC.
- A. Shah, V. K. Vishwakarma, N. Lhouvum, A. A. Sudhakar, P. Kumar, A. K. Srivastava, F. Dubois, T. Chomchok, N. Chattham and D. P. Singh, J. Mol. Liq., 2024, 393, 123535 CrossRef CAS.
- M. Gupta, A. Krishna KM, S. Sony, S. Dhingra, A. Shah and D. P. Singh, Chem. Commun., 2023, 59, 10652–10655 RSC.
- V. Lemaur, D. A. da Silva Filho, V. Coropceanu, M. Lehmann, Y. Geerts, J. Piris, M. G. Debije, A. M. van de Craats, K. Senthilkumar, L. D. A. Siebbeles, J. M. Warman, J.-L. Brédas and J. Corni, J. Am. Chem. Soc., 2004, 126, 3271–3279 CrossRef CAS PubMed.
- X. Feng, V. Marcon, W. Pisula, M. R. Hansen, J. Kirkpatrick, F. Grozema, D. Andrienko, K. Kremer and K. Müllen, Nat. Mater., 2009, 8, 421–426 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, full details of synthetic methods, spectroscopic and analytical data for the new compounds, MALDI-TOF mass spectra, DSC thermograms not included in the text and X-ray diffraction patterns not included in the text. See DOI: https://doi.org/10.1039/d5tc00575b |
| This journal is © The Royal Society of Chemistry 2025 |