
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of EUV induced depolymerization and side reactions of polyphthalaldehydes for dry develop EUV resist applications†
Bilal A.
Naqvi
 *ab,
Jared
Schwartz
c,
Anthony
Engler
d,
Paul A.
Kohl
c,
Stefan
De Gendt
ab and
Danilo
De Simone
b
*ab,
Jared
Schwartz
c,
Anthony
Engler
d,
Paul A.
Kohl
c,
Stefan
De Gendt
ab and
Danilo
De Simone
b
aDepartment of Chemistry, KU Leuven, 3001 Leuven, Belgium. E-mail: bilalabbas.naqvi@kuleuven.be
bimec, Kapeldreef 75, 3001 Leuven, Belgium
cSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, USA
dCain Department of Chemical Engineering, Louisiana State University, Baton Rouge, LA, USA
First published on 11th March 2025
Abstract
With the emergence of extreme ultraviolet (EUV) lithography, poly(phthalaldehyde) (PPA) has regained attention as a potential photoresist four decades after its initial introduction to the field of lithography in the 1980s. The main advantage of PPA is its chemical amplification via spontaneous depolymerization upon exposure to external stimuli such as EUV photons, offering an alternative to acid amplification to increase sensitivity for chain scission resists. Chain scission depolymerization results in the formation of small molecular fragments and monomers that can vaporize making PPA a potential candidate for a dry-developed resist. Dry development, unlike conventional liquid development, prevents the pattern collapse of critical nanometer features by eliminating surface tension forces. However, single-component, end-capped PPA was found to be insensitive to EUV exposure. In this study, cyclic PPA and a copolymer of phthalaldehyde and propanal demonstrated significantly higher EUV sensitivity. However, complete and dry removal was not achieved under the tested conditions. The reason for incomplete film removal via dry development was investigated using spectroscopic and surface analysis techniques including grazing angle Fourier-transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (NMR), and time of flight-secondary ion mass spectroscopy (TOF-SIMS). In this report, the main challenges of dry-develop PPA-based EUV resists are discussed.
1. Introduction
To match the scaling needs for miniaturization of integrated circuits, extreme ultraviolet (EUV) lithography was introduced in the last decade for high-volume chip fabrication. EUV can print 12 nm lines, and is intended to be used in manufacturing 7 nm node and beyond devices.1 The advent of the high numerical aperture (NA) EUV tool will push the boundaries of resolution to a sub 8 nm critical dimension (CD).2 The limiting factor in unlocking the full scaling capabilities of EUV lithography scanners is believed to be the lack of suitable photoresists. The performance of photoresists is gauged by the resolution, line edge roughness (LER), and sensitivity (RLS) tradeoff.3,4 For matching high-volume manufacturing, high performing and sensitive resists (<20 mJ cm−2) are needed.5 As feature sizes shrink, LER requirements become increasingly stringent. Some sources suggest the benchmarking LER criteria to be less than 10% of the CD for optimal photoresist performance.5The conventional photoresist used in high volume manufacturing is a chemically amplified resist (CAR) which is comprised of a polymer matrix, a photo acid generator (PAG) and a quencher. The solubility switch in CARs, both in DUV and EUV lithography, occurs through acid generation, acid diffusion, and acid catalyzed deprotection reactions in the polymer resin. In EUV lithography, the reactions for acid generation are driven by low energy secondary electrons (∼10 eV)6 which are generated by direct ionization of the polymer matrix by EUV photons (92 eV). These secondary electrons also induce other reactions such as bond scission and cross-linking.7,8 CARs are not designed to cater to these additional reactions and some of these additional crosslinking reactions are also unwanted in positive tone CARs.
Another challenge in lithography is stochastic failures. Stochastic failures arise primarily due to two reasons. Firstly, from the photon shot noise; the EUV photon (13.5 nm or 92 eV) possesses roughly 14 times higher energy than the ArF DUV photon (193 nm or 6 eV), which means there are far fewer photons in an EUV exposure pixel at the same energy dose compared to energetically equivalent DUV exposures. The photon shot noise effect, an intrinsic limitation of EUV lithography, is aggravated due to the use of thinner film photoresists. Secondly, the chemical inhomogeneity in multi-component CARs also leads to stochastics in acid generation, diffusion, and the deprotection reactions.9 Novel resist platforms with different reaction mechanisms may improve chemical stochastics. With increasing resolution, the pattern collapse is also becoming a concern as the aspect ratio of the patterns is increasing.10
To overcome these hurdles in EUV lithography, many polymeric resist platforms were developed as an alternative to CARs. One of the alternate approaches was the use of a single chain polymeric resist where the cation of the acid is bound to the polymer. This resist utilizes both acid amplification, and electron generated radical formation and cross-linking to achieve a solubility switch.8,11 Other types of simpler polymeric resists are bond scission-based resists such as PMMA; however, they suffer from low sensitivity and resolution.12 Another polymer which has been investigated recently is poly(phthalaldehyde) (PPA). PPA is a thermodynamically metastable polymer with a low ceiling temperature (ca. −36 °C).13,14 The end-capped and cyclic PPA are kinetically stabilized such that they can be used above room temperature in a metastable state.15 The application of an external stimulus such as heat, acid, or radiation to PPA can cleave polymer backbone bonds, which results in spontaneous depolymerization of the chain forming primarily o-phthalaldehyde monomers.13,16,17
Previously, Ito and Willson studied PPA as a photoresist for DUV and electron-beam lithography.16 They studied the performance of PPA by attaching different end-caps as well as mixing PAGs, as acid can also induce depolymerization in PPA, with the polymer to trigger the spontaneous depolymerization via backbone scission. Moreover, the depolymerization of PPA resulted in the formation of relatively high vapor pressure monomers, leading to the “spontaneous development” of the film during exposure.16 This feature makes PPA a potential dry-develop resist.13,18
There is a resurgence in interest in PPA as a photoresist for EUV lithography mainly due to the following reasons: the simpler single component structure of PPA would be beneficial for overcoming chemical stochastics. The amplified scission reaction leading to depolymerization of PPA is also beneficial for overcoming stochastics while simultaneously improving the sensitivity to EUV. Lastly, the depolymerization of PPA results in the formation of highly volatile monomers, which facilitates liquid-free development. This process can occur either spontaneously by sublimation or through external methods of dry development, such as convection.19
However, end-capped PPA was reported to be relatively insensitive to EUV. A dose of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mJ cm−2 was insufficient to clear 30 nm films of the material.20 In other reports, PAG-tethered PPA showed better sensitivity.21 Nonetheless, the addition of small molecule PAGs and the use of liquid developers are likely to suffer from common RLS tradeoff issues.
000 mJ cm−2 was insufficient to clear 30 nm films of the material.20 In other reports, PAG-tethered PPA showed better sensitivity.21 Nonetheless, the addition of small molecule PAGs and the use of liquid developers are likely to suffer from common RLS tradeoff issues.
In this study, single component cyclic PPA (c-PPA) was used, and it was found that the material is extremely sensitive to EUV. Additionally, a copolymer of PPA and propanal was also tested as aliphatic aldehydes have higher vapor pressures than aromatic aldehydes15 which can promote film removal after depolymerization. Post exposure reactions were studied in the context of spontaneous development. Despite the material's high sensitivity to EUV radiation, approximately 25% of the film remained on the wafer even at doses as high as 250 mJ cm−2. This anomaly in the thickness contrast curve indicates that additional reactions are taking place, producing non-volatile byproducts that limit the film's dry development. This saturation of the thickness loss was also seen previously;1,20 however, a detailed analysis of this behavior was not reported. In this paper, an in-depth analysis was carried out to understand the underlying reason for this saturation of thickness loss, despite the high EUV sensitivity of c-PPA and PA-25. Different spectroscopy techniques including Fourier-transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (NMR), and time of flight-secondary ion mass spectroscopy (TOF-SIMS) were used to understand this phenomenon.
2. Materials and methods
PPA samples were polymerized using methods described in ref. 22. Briefly, o-phthalaldehyde (>99.7%, purchased from TCI and used as-received) was charged into a glass round-bottom flask, which had been thoroughly cleaned and dried overnight in a 150 °C oven, and dissolved in anhydrous DCM (purchased from EMD Millipore) to a concentration of 0.746 M. A dilute catalyst solution was prepared in a separate, dry 20 mL vial with boron trifluoride diethyl etherate (ca. 48% BF3, purchased from Acros Organics) and anhydrous DCM. A small volume of this catalyst solution was taken up in a syringe to ensure a molar ratio of monomer-to-catalyst of 500:1. These preparations took place in a N2-rich glovebox before sealing the flask with rubber septa. Polymerizations were chilled to −78 °C with the use of dry ice and acetone before injecting the catalyst solution into the flask. After >1 h a small volume of pyridine (99%, Alfa Aesar) in THF (ACS grade, BDH Chemicals) was injected to quench the catalyst and mixed for >10 min. The polymerization solution was then precipitated dropwise into a volumetric excess of vigorously stirred MeOH (BDH Chemicals) and stirred for >1 h before separating the resulting white powder via vacuum filtration. The synthesis of PA25 used the same procedure and initial monomer concentration, but fed at a molar ratio of 2:1 propanal-to-phthalaldehyde. c-PPA has a Mn of 75 kg mol−1 and Đ = 2.03, and the co-polymer is composed of 25 mol% propanal, abbreviated as PA-25 (Fig. 1a), with a Mn = 35.0 kg mol−1 and Đ = 2.31. The molecular structures of cPPA and PA-25 are shown in Fig. 1a.
 | ||
| Fig. 1 Molecular structure of materials used in this study. (a) Molecular structure of cyclic PPA and the co-polymer of phthalaldehyde and propanal – PA-25 (also cyclic in nature). (b) The molecular structure of “l-Br PPA” and “l-PPA” and (c) the molecular structure of the model CAR. The EUV induced thickness contrast curves of these materials were sourced from references to compare them with those of c-PPA and PA-25. | ||
In Fig. 1b, the two variants of linear end capped PPA, namely “l-Br PPA” and “l-PPA”, are shown. The EUV-induced thickness contrast curves (Fig. 2) for these PPA derivatives (Fig. 1b), sourced from ref. 1 and 20, were compared with those of c-PPA and PA-25. The EUV induced thickness contrast curves of c-PPA and PA-25 were also compared with those of a model CAR (Fig. 3a), sourced from ref. 7. The structure of this model CAR is shown in Fig. 1c.
 | ||
| Fig. 2 Comparison of thickness contrast after EUV exposure for different PPA derivatives: l-Br PPA (green – with PEB and IPA development) & l-PPA (black without any development step) adapted from ref. 1 and 20. cPPA (red) and PA-25 (violet) used in this study are at least two orders of magnitude more sensitive than other two derivatives. | ||
 | ||
| Fig. 3 (a) Comparison of thickness contrast curves after EUV exposure; cPPA and PA-25 (without any development step) to a model CAR after application of PEB and wet development (taken from ref. 7). (b) Optical image of EUV exposure points on the PA-25 coated wafer, the spot with exposure shows a decrease in thickness after spontaneous development after EUV exposure. | ||
2.1. EUV exposure and outgassing analysis
Polymers were dissolved in anisole (2 wt%) and filtered through a 0.2 μm filter. The solution was spin-coated on a blank 200 mm Si wafer followed by a post application bake at 80 °C for 60 s to obtain 20 nm thick films. The coated wafers were then exposed to EUV radiation in a custom designed EUV tool (more details on the setup are given here23) with subsequent increasing dose at different spots. The film thickness of the exposed spots was measured using a KLA-Tencor SCD-100 ellipsometer. Another wafer was coated for outgassing analysis, which was exposed to EUV and the mass spectra of outgassing species were recorded using an in situ mass spectrometer (more details on the setup are given here24). For TOF-SIMS and FTIR, silicon coupons (1.5 cm2) coated with PA-25 were exposed to EUV radiation using a raster pattern to ensure uniform sample coverage with EUV dose.2.2. FTIR spectroscopy
PA-25 coated Si coupons (2 × 2 cm) were EUV exposed in a raster pattern with different doses. The attenuated total internal reflection (ATR) FTIR setup using the Nicolet™ iS50 FTIR Spectrometer and Harrick VariGATR™ grazing angle accessory was used to obtain the IR spectra. A germanium crystal was used to clamp the sample to enhance the sensitivity. The incidence angle was set at 65°. A liquid nitrogen-cooled mercury cadmium telluride (MCT) detector was used. The background spectrum collected was that of the atmosphere. Subsequently base-line correction was then employed. Each spectrum was analyzed to identify general changes in the chemical composition before and after varying EUV exposure dose.2.3. TOF-SIMS
PA-25 samples, exposed with EUV in a raster pattern with different exposure doses, were used for conducting TOF-SIMS analysis. TOF-SIMS was conducted using an ION-TOF GmbH TOF-SIMS with a dual beam configuration. The sputter ion source consisted of a gas cluster ion beam (GCIB) using Ar2000+ at 5 keV and 30 pA, which was rastered over an area of 300 μm2 at an angle of 45°. The GCIB was chosen as the sputter ion source to preserve the molecular information of the different organic components in the layer, that is, to minimize the sputter-induced damage and fragmentation. The analysis used a Bi1+ beam at an energy of 15 keV and a current of approximately 0.5 pA. The beam scanned over an area of 150 × 150 μm. The instrument was set to spectrometry mode for high mass resolution.3. Results and discussion
3.1. Thickness contrast curve
The EUV sensitivity of the polymers was evaluated by measuring the thickness contrast curves after exposure. Fig. 2 shows the comparison of thickness contrast curves of c-PPA, PA-25, l-Br PPA and l-PPA. Both c-PPA and PA-25 are more sensitive as compared to previously reported l-Br PPA1 and l-PPA19 (structure shown in Fig. 1b). The contrast curves for c-PPA, PA-25 and l-PPA were recorded after EUV exposure without any development step (i.e. spontaneous development in a vacuum during exposure), while the l-Br PPA was developed by applying a post exposure bake and isopropanol (IPA).There is a postulation that EUV can induce bond scission either on the main chain or at the end-caps, similar to what was reported for acid induced bond scission in end capped PPA,25 although the exact mechanism is not clear. In some cases, it was reported that acetal linkages in PPA are not sensitive to EUV and adding EUV ‘sensitive end-caps’ improves the sensitivity of PPA.1 However, we noticed that the c-PPA, which is a homopolymer lacking endcaps, is extremely sensitive to EUV indicating that the acetal bonds were susceptible to chain scission during EUV exposure. The contrast curve decreases sharply at first but starts to plateau after reaching ∼50% thickness, which is also more clearly seen in Fig. 3a by comparing the thickness contrast curve of cPPA and PA-25 to that of the model CAR. cPPA, PA-25 and the model CAR lose 50% of the original thickness after a dose of ∼20 mJ cm−2. Afterwards, there is a complete solubility switch in the CAR after the wet development step. It is also worth noting that the model CAR is almost unaffected at very low doses. In contrast, there is a sudden decrease in the thickness of cPPA and PA-25 even at lower doses (without any wet development step), which indicates the start of depolymerization. The trend in thickness loss for cPPA and PA-25 is the same at lower doses (<50 mJ cm−2). The slightly higher thickness loss in PA-25 at higher doses indicates that the presence of a small amount of propanal in the resist (25 mol%, 12.6 wt%) promotes material removal after exposure due to an increase in vapor pressure. Fig. 3b shows the EUV exposure spots on a PA-25 thin film. The contrast shows loss of thickness at the exposure point. These images were taken right after exposure without any development step.
The saturation of the curves at higher dose was also seen in previous PPA reports, which was attributed to the reattachment of monomers to the reactive anionic chain ends.1 However, a detailed analysis is needed to investigate the remnants on the wafer which can also then point towards challenges to be solved in dry development of PPA for EUV lithography. The presence of materials on the wafer, even after a dose of >200 mJ cm−1,2 requires more characterization to better understand the reason of incomplete film removal (despite the materials’ high sensitivity to EUV and a decrease in sudden film loss after EUV exposure without any development step). In this report, different spectroscopy techniques were used to find the underlying cause to these remnants on the wafers. For further characterization, the PA-25 polymer was used to understand the nature of the remaining substance on the wafer as the film removal was slightly better in it as compared to cPPA.
3.2. EUV induced outgassing analysis
The mass spectra of the outgassing species collected during the EUV exposure of PA-25 show expected peaks indicative of fragmentation of poly(phthalaldehyde) and propanal (Fig. 4). The peak at 134 m/z is attributed to the phthalaldehyde monomer. Other prominent peaks arising from fragmentation and rearrangement of phthalaldehydes are benzofuran (m/z 118), benzaldehyde (m/z 105), benzene (C6H5+m/z 77), and C4H3+ (m/z 51). The expected peaks from propanal are also observed at 58 m/z while those at 39 m/z (C3H3+) can be attributed to fragmentation of both propanal and phthalaldehyde. The strongest peak is 29 m/z arising from the CHO+ fragment of both monomers. The presence of 28 m/z could be a mixture of C2H4+ from propanal and CO (CO could be released during transformation of phthalaldehyde (m/z 134) to benzofuran (m/z 118) and from propanal fragmentation as well). The relatively higher intensity of benzene in the mass spectra observed here differs slightly from previously reported EUV-induced poly(phthalaldehyde) fragmentation patterns.20 Although the overall fragmentation patterns are generally consistent with each other, the findings reported in the next section may provide additional reasons for the benzene peak. The mass spectrum of the outgassed species from EUV exposure suggests that the polymer depolymerizes to its constituent monomers, phthalaldehyde and propanal. These monomers can then sublime leaving a patterned substrate to serve as a potentially dry-develop resist. The depolymerization reaction of PPA is shown in Fig. 5.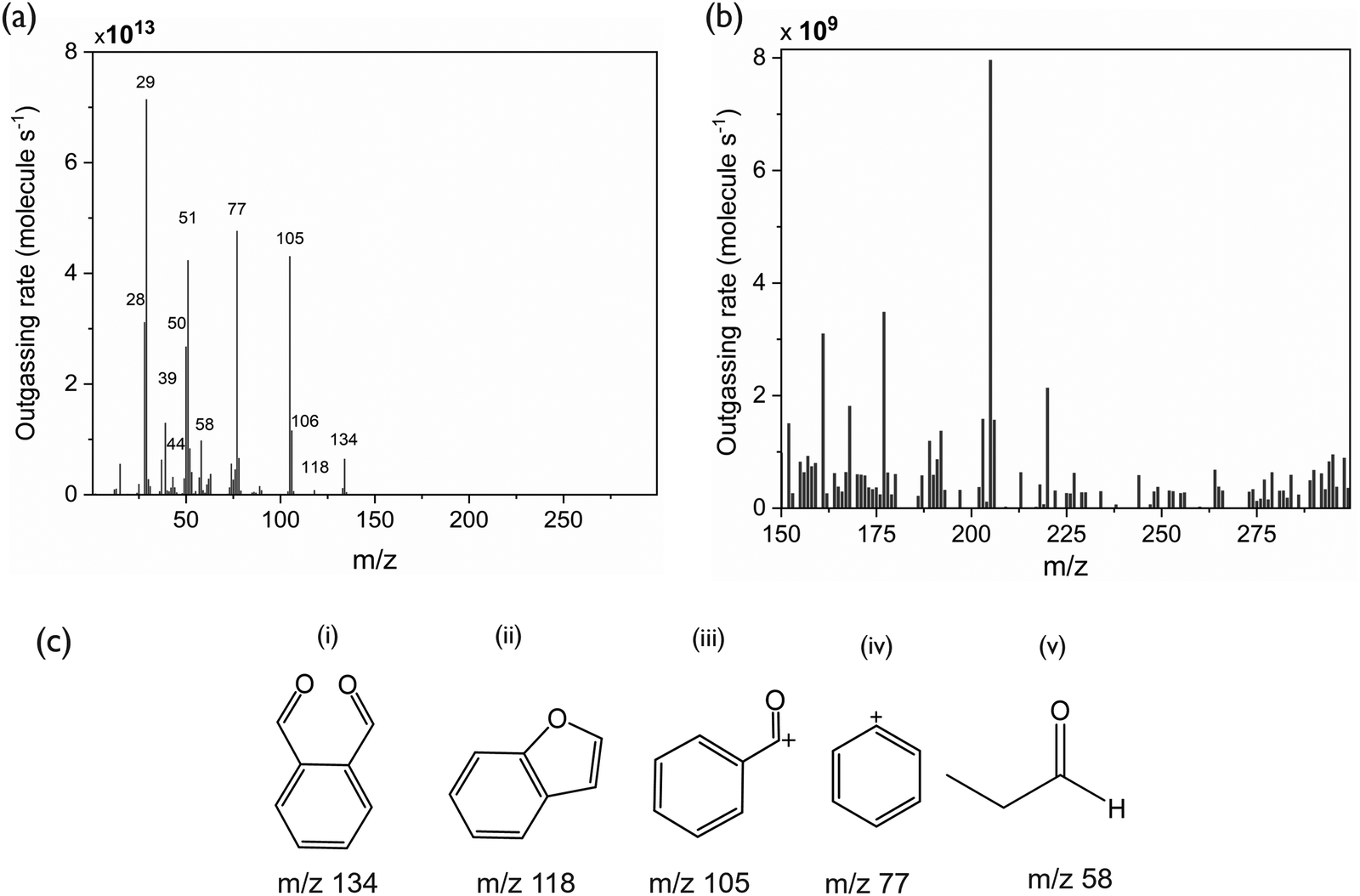 | ||
| Fig. 4 (a) Mass spectra of outgassing species during EUV exposure of PA-25. (b) Zoomed-in view of peaks present at higher m/z (>150). (c) Molecular structure of major fragments of phthaldehyde and propanal. | ||
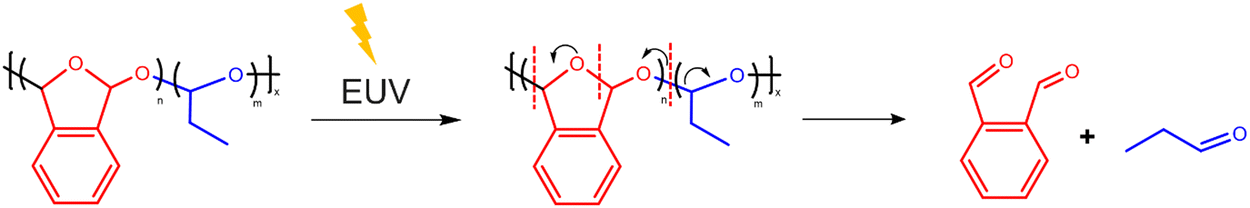 | ||
| Fig. 5 Proposed EUV induced depolymerization of PA-25. | ||
Interestingly, there are some peaks arising at higher m/z ranges (Fig. 4b). The higher m/z (>150) peaks have four orders of magnitude lower intensity than the peaks at lower m/z. Nonetheless, these peaks indicate the formation of larger fragments than expected from depolymerization, during or after the EUV exposure of PA-25.
An additional exposed PA-25 (250 mJ cm−2) sample was subjected to a wet development step with isopropanol (IPA) (Fig. S1, ESI†), which serves as a good solvent for the monomers but a poor solvent for PA-25. In the wet developed samples, the saturation trend continued, and the effect of wet development was minimal. The thickness similarities between wet and dry developed films (exposed at higher doses) suggests the remaining residue is both non-volatile and insoluble in IPA. This further strengthened our hypothesis of additional reactions apart from depolymerization. Apart from IPA, acetone was also tried for wet development, however acetone was too harsh and dissolved un-exposed polymer as well. In a previous report,20 a mixture of IPA and Anisole was used. However, this mixture didn’t have good selectivity between exposed and un-exposed parts making it unsuitable as a developer.
3.3. FTIR spectroscopy
FTIR spectroscopy was performed to study the nature of these remnants remaining on the substrate. Fig. 6 shows the FTIR spectra of PA-25, unexposed and with increasing EUV dose. Peaks representing the polymer backbone, particularly the C–O stretching in saturated ethers at 1190 cm−1, decrease with increasing EUV dose. This indicates a breaking of the polymer backbone bonds. The peak at 1690 cm−1 in the unexposed sample is ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of aromatic aldehyde, arising from the residual phthalaldehyde monomer. However, it diminishes upon EUV exposure and a new peak emerges at 1775 cm−1 after EUV exposure. This peak is more indicative of CO stretching in aromatic esters. The presence of the CO peak at 1775 cm−1 suggests the possibility of other concurrent reactions during EUV exposure resulting in a low vapor pressure ester. The 1H-NMR spectra (Fig. S2 and S3, ESI†) also point to the presence of an aromatic ester in the EUV exposed samples, as well as aldehyde peaks that that are inconsistent with propanal or phthalaldehyde.
O stretching of aromatic aldehyde, arising from the residual phthalaldehyde monomer. However, it diminishes upon EUV exposure and a new peak emerges at 1775 cm−1 after EUV exposure. This peak is more indicative of CO stretching in aromatic esters. The presence of the CO peak at 1775 cm−1 suggests the possibility of other concurrent reactions during EUV exposure resulting in a low vapor pressure ester. The 1H-NMR spectra (Fig. S2 and S3, ESI†) also point to the presence of an aromatic ester in the EUV exposed samples, as well as aldehyde peaks that that are inconsistent with propanal or phthalaldehyde.
 | ||
| Fig. 6 (a) FTIR spectra of unexposed and exposed PA-25. The green region represents C–C and C–O peaks which are representative of the polymer back bone. (b) Zoomed view of the peaks of interest. The peak highlighted with purple (1690 cm−1), present in the un-exposed sample only, represents aromatic CO stretching coming from leftover monomer from the synthesis. The peaks highlighted in red (1775) cm−1 also represent CO stretching. This peak may be a mixed signal from an aromatic ester or an aromatic carbonate. | ||
3.4. Hypothesized EUV induced reaction of the phthalaldehyde monomer
Based on the above discussion, it can be concluded that EUV-induced reactions in PA-25 can lead to the formation of esters. These esters could be of varying chain lengths and chemical structures. For further chemical characterization using TOF-SIMS, some structures were specified based upon previous reports, results of NMR (Fig. S3, ESI†) and FTIR analysis (Fig. 6). This facilitated the identification of the specific m/z range to focus on during TOF-SIMS analysis. The details of this reaction scheme are given in the following paragraph.The energetic EUV photon can ionize the polymer leading to formation ions and neutral radicals. These neutral radicals are highly reactive species and can undergo a variety of reactions. Previous reports on deep UV photolysis of phthalaldehyde also mention the formation of ketene-enol intermediates and radicals, which eventually leads to the formation of non-PPA type dimers.26 Some reports suggested the formation of a ketene and its equivalent bi-radical through intramolecular hydrogen abstraction.26,27 Other reports mention the formation of radicals by intermolecular hydrogen transfer in photo-excited phthalaldehydes and other aromatic aldehydes.28,29 Despite varied reaction pathways, all reports mention the formation of phthalide and isomeric mixtures of dimers. For simplicity, the hydrogen abstraction reaction, subsequent formation of radicals and in turn dimers were the focus of additional analysis.
The hypothesized reaction scheme is shown in Fig. 7. First, upon excitation, the phthalaldehyde monomer (1) forms two radicals; hydrogen radical and (2). The intermolecular hydrogen abstraction leads to the generation of a mixture of phthalaldehyde (2) and a hydroxy-benzyl radical (3). (2) can then rearrange to a phthalide radical (4). Second, the phthalide radical (4) reacts with the phthalaldehyde monomer (1) via hydrogen abstraction giving an epimeric mixture of a hemiacetal which can be oxidized to di-hydrobisphthalide (5). (5) is a known photo-induced decomposition product of the phthalaldehyde monomer.26,29 However, dilute monomer solutions were used for the analysis in the past for photodecomposition study, whereas in the present case the concentration of phthalaldehyde is much higher. Thus, the reaction possibilities are also higher. Finally, due to the abundance of radicals, the additional products numbered (6), (7) and (8) are proposed (Fig. 7). Any of these proposed structures could feasibly produce the aromatic carbonyl peaks observed in the FTIR (Fig. 7) and NMR spectra. It is important to note that only representative products were proposed and selected for the ease of detection in the further analysis performed below. Nonetheless, the abundance of free radicals may result in higher molecular weight molecules or oligomeric species that are structurally distinct from PPA oligomers. It has also been pointed out that the chain length of these products may be longer than the predicted structures.29
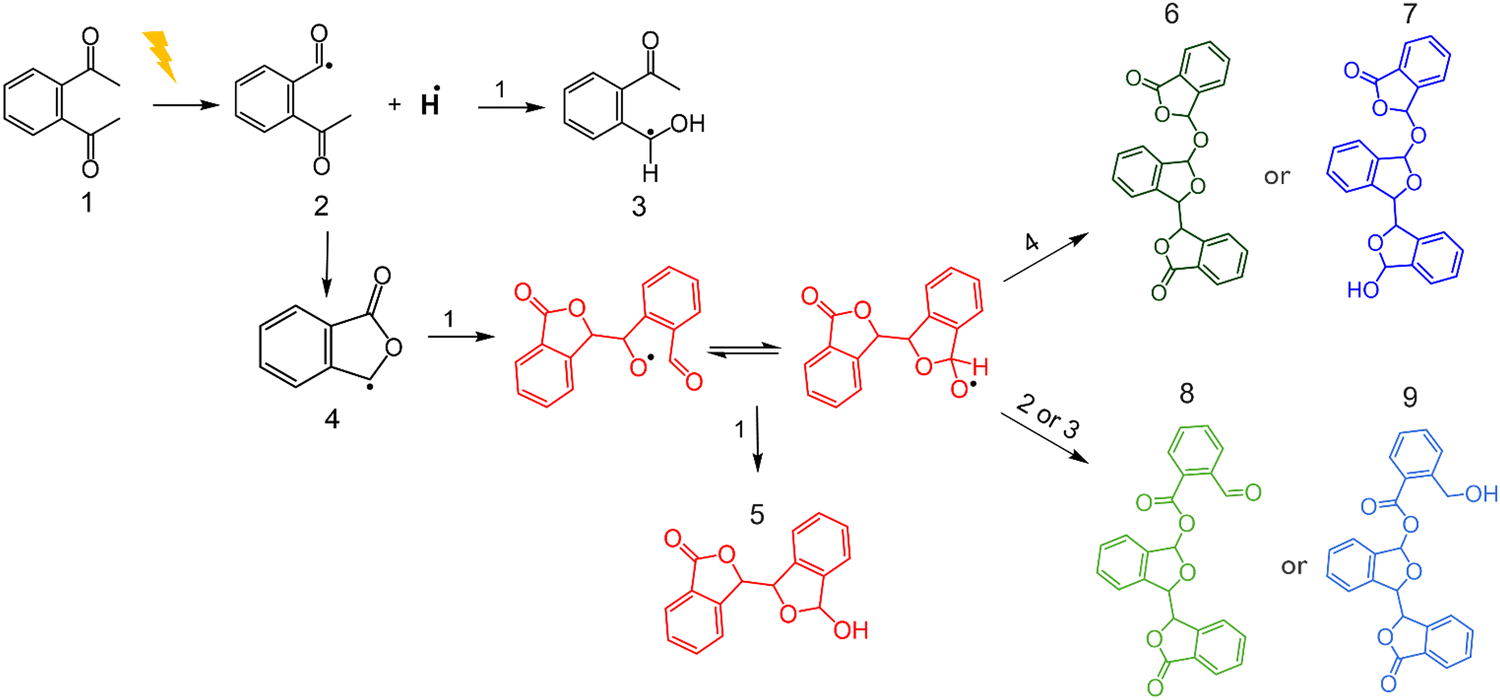 | ||
| Fig. 7 Proposed reaction scheme of phthalaldehyde radical generation and cross-linking. | ||
3.5. TOF-SIMS
A useful technique to study the chemical structure of molecules in the solid phase is TOF-SIMS. The irradiation of a surface with primary ions creates a collision cascade of secondary ions, radicals, and photons within a shallow thickness on the surface (∼10 Å). Due to the impact of low energy secondary ions, secondary particles generated farther from the impact site are generally a molecular fragment of larger organic compounds. Therefore, TOF-SIMS allows the detection of the molecular structures of compounds near the surface of a substrate while causing minimal damage to the original chemical structure.To verify the presence of the chemical species shown in Fig. 7, TOF-SIMS was employed. Unexposed PA-25 was used as a reference sample and five EUV exposed samples were used in TOF-SIMS analysis to identify the remaining compounds on the substrates. Three samples were exposed at relatively lower exposure doses of 5, 7.5 and 10 mJ cm−2 and two other samples were exposed to higher doses of 50 and 100 mJ cm−2. Fig. 8 shows the analyzed data from the mass spectra obtained by TOF-SIMS. The m/z signal from the phthalaldehyde monomer and its fragments (m/z ≤ 134) is detected in unexposed and exposed samples. However, the peaks from the heavier fragments (m/z > 134), as predicted in Fig. 7, are present in exposed samples only. This affirms our hypothesis that additional reactions during EUV exposure leads to the formation of heavier fragments which cannot sublime under high vacuum conditions. The signal from the monomer and its fragments (m/z ≤ 134) in the unexposed sample could be coming either from leftover monomer from the synthesis (as it is also seen in the FTIR spectra in Fig. 6) or due to the depolymerization caused due to the impact of the analysis beam of TOF-SIMS. The intensity ratio between the signal from the monomer and its fragments (m/z ≤ 134) in the unexposed sample is almost similar to what was observed in EUV induced outgassing (the highest peak is m/z 77). Moreover, upon EUV exposure, the overall signal intensities of the monomer and its fragments (m/z ≤ 134) increase significantly—by several orders of magnitude—compared to the unexposed sample, indicating the depolymerization of polyphthalaldehyde. However, in the exposed samples the intensity ratio between the monomer and its fragments (m/z ≤ 134) changes, with the highest peak now observed at m/z 118. This change in ratio in the exposed samples indicates that the monomer is not the only source of these signals (m/z ≤ 134). The chemical species formed after EUV exposure, like those shown in Fig. 7, may also be contributing to these signals. In the exposed samples the intensity of all m/z decreases with the increase in EUV dose. This indicates that heavier fragments (m/z > 134) are further reacting thus reducing the intensities at m/z 268, 400 and 402. The decrease in intensity of monomeric fragments (m/z ≤ 134) also points to the formation of heavier or more stable products. The full mass spectrum is given in the ESI† (Fig. S4). There are several peaks which indicate a variety of molecular structures and chain lengths. Moreover, some of the peaks are highest in the 10 mJ cm−2 exposed samples, which could be fragments of longer and relatively stable molecule chains formed after EUV exposure.
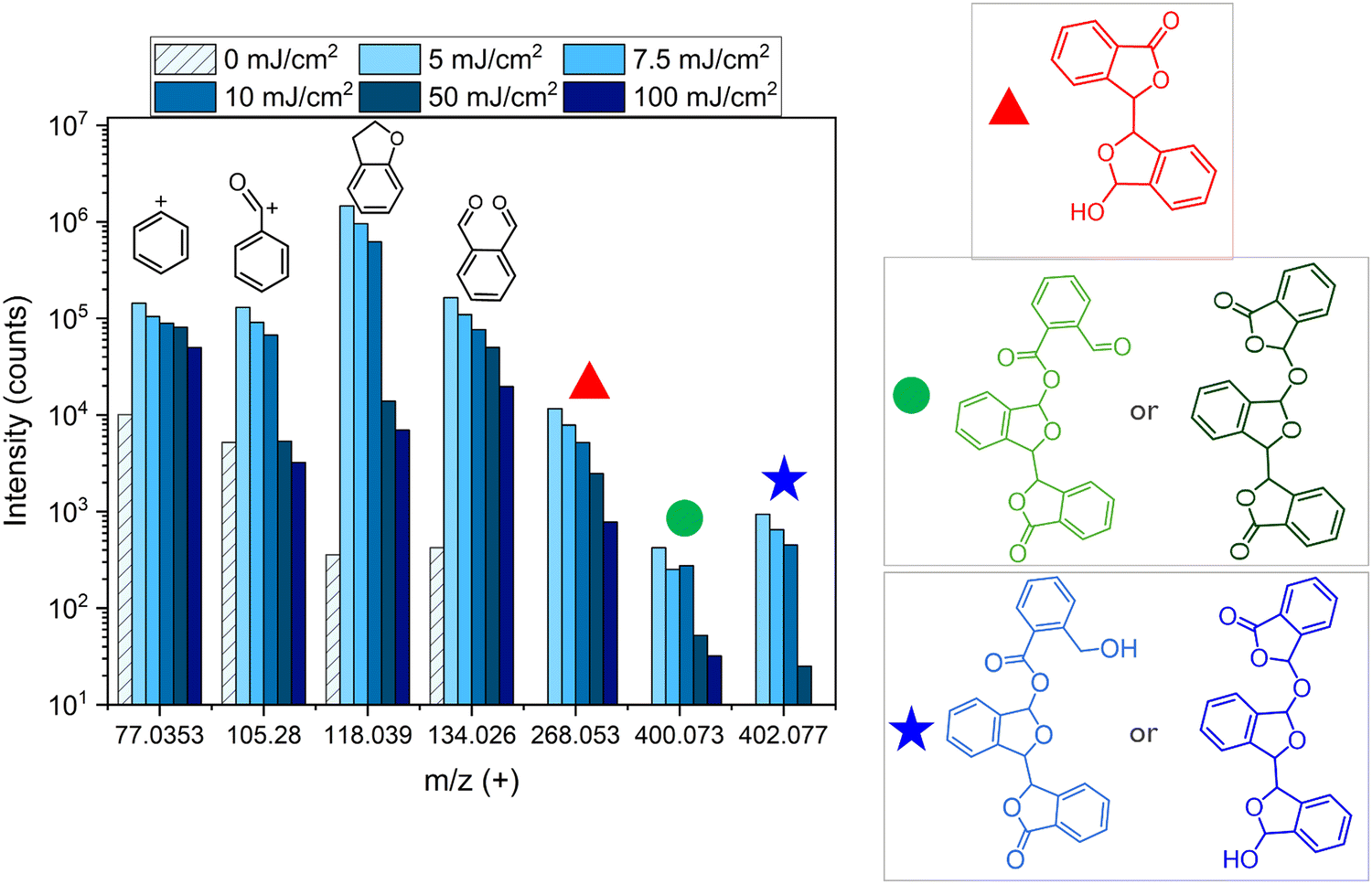 | ||
| Fig. 8 Mass spectra of un-exposed and EUV exposed PA-25 obtained in positive mode using TOF-SIMS. It shows the peak of phthalaldehyde and its fragments. In addition to that, the peaks at 268 m/z, 400 m/z and 402 m/z correspond to the heavier low volatile species formed after EUV exposure. | ||
The chemical characterization and contrast curves point towards additional EUV-induced reaction and byproduct formation along with the usual depolymerization of PA-25. The tone reversal phenomena is also seen in other EUV resists such as PMMA and CAR at higher doses.7,12,30 However, it only becomes evident at higher doses, although the high energy EUV photon can cause bond cleavage and radical formation readily. At lower doses, chain scission is dominant, however at higher doses, the chain scission becomes the limiting step due to the consumption of polymer back chains. Also, the increase in the number of free radicals at higher doses leads to a pronounced tone reversal effect.31 In the case of PA-25, due to its higher sensitivity, it depolymerizes faster after a small EUV dose followed by additional reactions with further EUV exposure. The products of these additional reactions were traced to be aromatic esters using FTIR, NMR and TOF-SIMS spectroscopies. The source of the formation of this aromatic ester can be traced back to the formation of phthalides. Phthalides can form in two ways: either as a radical chain end during depolymerization or directly from the phthalaldehyde monomer following depolymerization. Both pathways produce aromatic esters, which were insensitive to EUV exposure and remain on the wafer even after higher exposure doses. Therefore, it can be postulated that depolymerization and cross-linking were probably in-balance with each other until the point where the thickness contrast curves flatten. As the dose increased, the amount of polymer decreased, and cross-linking became the dominant reaction pathway.
4. Conclusion
In this study, we showed that c-PPA and PA-25 are extremely sensitive to EUV exposures. The instantaneous decrease in thickness after EUV exposure showed spontaneous depolymerization and development of PA-25. However, at higher doses, the thickness contrast curve flattens out with almost 25% of thickness remaining after an exposure of 200 mJ cm−2. Wet development with IPA also did not completely remove the remaining film. EUV-induced outgassing analysis revealed that upon EUV exposure, cPPA and its co-polymer PA25 undergo depolymerization into its constituent monomers. However, residual thickness after high dose exposures and the observation of the FTIR peak at 1775 cm−1 (belonging to aromatic ester) suggested the occurrence of additional reactions in addition to depolymerization. TOF-SIMS analysis confirmed the formation of larger fragments arising from undesirable side reactions of the phthalaldehyde monomer, which explains the saturation observed in the thickness contrast curve at higher EUV doses. Overall, the cPPA and PA-25 proved to be sensitive to EUV. In contrast to CARs, it shows amplification of the chemical reaction due to spontaneous depolymerization. After depolymerization, the phthalaldehyde monomer underwent additional reactions forming non-volatile molecules. These additional reactions are attributed to the formation of radical chain ends or radical monomers. This radical formation can be further investigated using radical quenchers such as DMPO and TEMPO as a follow up work. These radical quenchers may also inhibit the formation of side products, if they proceed via a radical mechanism. This may be a subject of future reports. Nonetheless, it is shown in this paper that these larger fragments did not evaporate and were not soluble in IPA either. The abundance of side reactions and products along with chain scission in PPA upon EUV irradiation limits its use as a single component EUV resist.Author contributions
Conceptualization: all authors conceived the concept and plan for the research. Investigation: BN, JS and AE planned and conducted the experiments for data collection. Data analysis: BN, JS and AE conducted the data analysis. Writing – original draft: BN wrote the original draft, and all authors then reviewed and edited the draft. Supervision: DD, SDG and PAK jointly supervised the research.Data availability
The data provided in this article are original. Analysis based on the data is shown in the article and the ESI.† Raw data can be made available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Alexis Franquet and Rita Tilmann at the MCA department of imec for performing TOF-SIMS analysis. The authors (ACE and PAK) gratefully acknowledge the financial support from the Laboratory Directed Research and Development program at Sandia National Laboratories under contract DE-NA-0003525. Any subjective views or opinions expressed in the paper do not necessarily represent the views of the U.S. Department of Energy (DOE) or the US. Government.References
- J. Deng, S. Bailey, R. Ai, A. Delmonico, G. Denbeaux, S. Jiang and C. K. Ober, ACS Macro Lett., 2022, 1049–1054 Search PubMed.
- A. Erdmann, H. Mesilhy, P. Evanschitzky, V. Philipsen, F. Timmermans and M. Bauer, J. Micro/Nanolithogr., MEMS, MOEMS, 2020, 19, 1–16 Search PubMed.
- 2015 International Technology Roadmap for Semiconductors (ITRS), ITRS Roadmap, 2015, p. 65.
- G. M. Gallatin, P. Naulleau and R. Brainard, Adv. Resist. Mater. Process Technol., 2007, 6519, 651911 Search PubMed.
- D. De Simone, P. Vanelderen and G. Vandenberghe, J. Photopolym. Sci. Technol., 2017, 30, 613–617 CAS.
- D. L. Goldfarb, A. Afzali-Ardakani and M. Glodde, Adv. Patterning Mater. Process., 2016, 9779, 97790A Search PubMed.
- I. Pollentier, Y. Vesters, A. Rathore, P. Vanelderen, J. Petersen, D. De Simone and G. Vandenberghe, Proc. SPIE Adv. Patterning Mater. Process., 2018, 105860C, 11 Search PubMed.
- B. A. Naqvi, S. Enomoto, K. Machida, Y. Takata, T. Kozawa, Y. Muroya, S. De Gendt and D. De Simone, Chem. Mater., 2024, 36(3), 1459–1471 CAS.
- T. Kozawa and S. Tagawa, Jpn. J. Appl. Phys., 2010, 49, 06GF02 Search PubMed.
- S. Heo, S. Baek, M. Gupta, H. S. Suh, K. Kato, S. Takeda, W. Shibayama and R. Sakamoto, J. Micro/Nanopatterning, Mater., Metrol., 2024, 23, 1–9 Search PubMed.
- S. Enomoto, T. Yoshino, K. Machida and T. Kozawa, Jpn. J. Appl. Phys., 2019, 58, 056504 CAS.
- A. Rathore, I. Pollentier, H. Singh, R. Fallica, D. De Simone and S. De Gendt, J. Mater. Chem. C, 2020, 8, 5958 CAS.
- A. Engler, C. K. Lo and P. A. Kohl, Polym. Adv. Technol., 2021, 32, 2142–2150 CrossRef CAS.
- J. P. Lutz, O. Davydovich, M. D. Hannigan, J. S. Moore, P. M. Zimmerman and A. J. McNeil, J. Am. Chem. Soc., 2019, 141, 14544–14548 CrossRef CAS PubMed.
- O. Phillips, J. M. Schwartz, A. Engler, G. Gourdin and P. A. Kohl, Proc. – Electron. Compon. Technol. Conf., 2017, 772–779 Search PubMed.
- H. Ito and C. G. Willson, Polym. Eng. Sci., 1983, 23, 1012–1018 CAS.
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 6342–6360 CAS.
- J. Lopez Ninantay, A. Engler, J. Schwartz and P. A. Kohl, ECS J. Solid State Sci. Technol., 2024, 13, 054004 CAS.
- A. Engler, C. Tobin, C. K. Lo and P. A. Kohl, J. Mater. Res., 2020, 35, 2917–2924 CAS.
- A. Rathore, I. Pollentier, S. S. Kumar, D. De Simone and S. De Gendt, J. Micro/Nanopatterning, Mater., Metrol., 2021, 20, 1–8 Search PubMed.
- J. Deng, S. Bailey, S. Jiang and C. K. Ober, J. Am. Chem. Soc., 2022, 144, 19508–19520 CAS.
- A. Engler, O. Phillips, R. C. Miller, C. Tobin and P. A. Kohl, Macromolecules, 2019, 52, 4020–4029 CAS.
- I. Pollentier, G. Aksenov, A.-M. Goethals, R. Gronheid, R. Jonckheere and M. Leeson, Altern. Lithogr. Technol., 2009, 7271, 727146 Search PubMed.
- I. Pollentier, A.-M. Goethals, R. Gronheid, J. Steinhoff and J. Van Dijk, Extreme Ultraviolet Lithogr., 2010, 7636, 76361W Search PubMed.
- M. Tsuda, M. Hata, R. I. E. Nishida and S. Oikawa, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 77–89 CAS.
- J. C. Scaiano, M. V. Encinas and M. V. George, J. Chem. Soc., Perkin Trans. 2, 1980, 724–730 CAS.
- J. C. Netto-Ferreira and J. C. Scaiano, Can. J. Chem., 1993, 71, 1209 CAS.
- M. Cocivera and A. M. Trozzolo, J. Am. Chem. Soc., 1970, 92, 1772–1774 CAS.
- D. A. Harrison, R. N. Schwartz and J. Kagan, J. Am. Chem. Soc., 1970, 92, 5793–5795 CAS.
- T. H. Fedynyshyn, R. B. Goodman, A. Cabral, C. Tarrio and T. B. Lucatorto, Adv. Resist Mater. Process. Technol., 2010, 7639, 76390A Search PubMed.
- D. J. Carbaugh, S. Kaya and F. Rahman, Mater. Res. Express, 2019, 6, 045308 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc05250a |
| This journal is © The Royal Society of Chemistry 2025 |