
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShorter might be better: oligo(oxazoline)s for thermoresponsive polymersomes†
Léa
Daoud
a,
Laura Vasilica
Arsenie
a,
Belkacem Tarek
Benkhaled
b,
Kilian
Caillaud
c,
Mona
Semsarilar
 b,
Luc
Picton
c,
Didier
Le Cerf
c and
Vincent
Lapinte
*a
b,
Luc
Picton
c,
Didier
Le Cerf
c and
Vincent
Lapinte
*a
aICGM, University of Montpellier, CNRS, ENSCM, 34090 Montpellier, France. E-mail: vincent.lapinte@umontpellier.fr
bIEM, UMR 5635, University of Montpellier, CNRS, ENSCM, 34096 Montpellier, France
cUniversity of Rouen Normandie, INSA Rouen Normandie, CNRS, PBS Laboratory, UMR 6270, Rouen, France
First published on 27th August 2024
Abstract
Amphiphilic poly(2-oxazoline)s offer various opportunities for a number of applications where polymer self-assembly in water is required. Oligomers are particularly interesting for their chain mobility since they can increase the membrane permeability similar to bio-inspired liposomes. Here, the synthesis of various triblock oligomers (oligoOx) was investigated using only oxazoline monomers with hydrophilic 2-methyl-2-oxazoline (MeOx) and hydrophobic 2-butyl-2-oxazoline (BuOx). OligoOx with various MeOx/BuOx ratios were self-assembled in PBS by direct dissolution or film rehydration and evaluated using DLS and TEM. The formed large vesicles were thermoresponsive with reversible swelling/deswelling at a cloud point of 30 °C. These oligomeric architectures could open a new path for the preparation of stable and permeable polymersomes mimicking cellular compartments.
Introduction
Thermoresponsive polymers have been largely investigated in life sciences.1–11 They exhibit changes in chain conformation in response to temperature. The temperature-dependent solubility, known as the cloud point (CP), is a common characteristic of low critical solution temperature (LCST) and upper critical solution temperature (UCST) derived systems.4–11 For LCST based systems, below CP, the free polymer chains are presented in solution while the chains aggregate upon heating above the CP, whereas the opposite phenomenon is observed for UCST systems.4,5 For example, in the case of LCST systems, these transitions around CP are attributed to the replacement of interactions between the polymer and solvent at low temperature with the interactions between polymer chains at high temperature.In recent years, particular attention has been devoted to the development of polymersomes.12–16 Inspired by liposomes, polymersomes could be used for cellular internalization and drug encapsulation. Although liposomes have ultra-thin membranes (around 3–5 nm), polymersomes are endowed with thicker membranes spanning from 10 to 50 nm, making them more robust, resistant, and often more advantageous in terms of stability.13 However, unlike liposomes, polymersomes’ membranes are impermeable.13
According to some studies, using thermoresponsive polymers for the preparation of polymersomes could result in the formation of responsive drug delivery membranes.12–16 This occurs since the thermoresponsive polymer chains forming the polymersomes undergo swelling/deswelling transitions in response to temperature changes.
However, these systems are made of polymers with molar masses of up to 10 kDa (middle and long chain lengths), while the use of oligomers (below 1 kDa) has received very little attention.1–16 Oligomers may offer better control over chain interactions during self-assembly, potentially allowing for precise shaping.17,18
Motivated by the potential of using oligomers in the design of discrete thermoresponsive self-assembled systems, we focused on using poly(oxazoline)s. Polyoxazolines (POx) have many advantages, such as being non-toxic and biocompatible as well as exhibiting thermoresponsive behavior depending on their pendant R group.19–42 Another significant advantage of POx is that the hydrophobic/hydrophilic balance can be efficiently adjusted via designing co-polymers with hydrophobic and hydrophilic segments. This allows the formation of well-defined self-assembled architectures such as micelles, polymersomes, rods etc.43–66 Herein, most of the examples predominantly involve diblock POx with hydrophobic and hydrophilic blocks.43–49,56,62–66 Generally, most thermoresponsive polyoxazoline based assemblies are based on polymers derived from 2-isopropyl-2-oxazoline (iPrOx), explored alone and/or in association with 2-methyl-2-oxazoline (MeOx) or 2-ethyl-2-oxazoline (EtOx), with all the three structures providing short side chains, which ensures molecular mobility and results in the formation of different shapes, including polymersomes.67–71 However, oxazoline co-monomers with longer side chains (up to 3) were less employed to develop thermoresponsive polymersomes.66,72 For example, Daubian et al. prepared polymersome membranes with a mean width of around 50 nm by using amphiphilic diblock copolymers made of hydrophilic PEO and hydrophobic polyoxazoline containing a nine carbon atom branched chain (poly(ethylene oxide)-b-poly(2-(3-ethylheptyl)-2-oxazoline) (PEO-b-PEHOx)).66 However, the resulting morphologies were obtained by the solvent-switching method without exhibiting LCST behavior.
Moreover, when examining the type of POx architecture involved in polymersome formation, very few examples were found to deal with ABA and/or BAB triblock architectures.46,51,59,61,65,73–75 Krumm et al. prepared vesicles from triblock polyoxazolines (Mn ranging between 8400 g mol−1 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) composed of internal 2-phenyl-2-oxazoline (PheOx) hydrophobic blocks and hydrophilic 2-methyl-2-oxazoline (MeOx) segments.73 Spulber et al. developed nanoreactors from polymersomes composed of amphiphilic block copolymers based on PMeOx-b-PDMS-b-PMeOx.74 Similar results were also reported by Belluati and coll.75 Nevertheless, these nano-objects were made with long PDMS chains (up to 54 units) while the length of PMeOx blocks varied between 5 and 10 monomer units.75
000 g mol−1) composed of internal 2-phenyl-2-oxazoline (PheOx) hydrophobic blocks and hydrophilic 2-methyl-2-oxazoline (MeOx) segments.73 Spulber et al. developed nanoreactors from polymersomes composed of amphiphilic block copolymers based on PMeOx-b-PDMS-b-PMeOx.74 Similar results were also reported by Belluati and coll.75 Nevertheless, these nano-objects were made with long PDMS chains (up to 54 units) while the length of PMeOx blocks varied between 5 and 10 monomer units.75
Synthesizing POx oligomers is a major challenge. Research and reports in this area are quite limited,59,61 underscoring that one of the primary challenges during the synthesis of oligo(oxazoline)s is the purification step. In addition, these few examples address the synthesis of homo-oligomers,59,61 as the development of co-oligomers has not yet been reported to the best of our knowledge.
In this work, we synthesized ω-aminated amphiphilic oligomers of oxazoline copolymers with hydrophobic crystalline 2-butyl-2-oxazoline (BuOx) and hydrophilic 2-methyl-2-oxazoline (MeOx), allowing us to achieve statistical and block architectures. Homopolymers of MeOx and BuOx represent boundary cases with opposite behaviors in terms of hydrophilicity, with PMeOx being highly hydrophilic, while PBuOx is highly hydrophobic. However, incorporating these MeOx and BuOx co-monomers into co-oligomers enables adjustments of CP within a more practical and desirable range.25 These copolymers were obtained by two-step synthesis consisting of the quenching of living oxazolinium species with an excess of potassium phthalimide, followed by the conversion of the phthalimide group into the amino group by the addition of hydrazine. Compared to commonly reported ω-aminated POx synthesised by a reaction between ammonia and the living oxazolinium species, which results in only 80% amino end functionalization,40 this route is more advantageous since it enables the quantitative assessment of end-capping efficiency.41 Moreover, we investigated the self-assembly and the thermoresponsive behavior of the oligomers in physiological buffer (pH 7.4). The comprehensive analysis conducted provides valuable insights into the tailored oxazoline oligomers for specific applications.
Experimental
Materials
2-Methyl-2-oxazoline (MeOx, 97%), 2-n-butyl-2-oxazoline (BuOx, 99%), 1,6-hexanediol (HD, 99%), triethylamine (TEA, 97%), hydrazine monohydrate (95%), anhydrous acetonitrile (99.9%) and p-toluenesulfonyl chloride (TsCl, 96%) were acquired from Sigma Aldrich. Potassium phthalimide (99%) was bought from Thermo Fischer. Diethyl ether (99% purity) was obtained from Honeywell. Ethanol (98%) and hydrochloric acid (solution 37% w/w) were purchased from Sigma Aldrich and used without further purification. Dimethylformamide (DMF, 99.8%) was acquired from Fisher Scientific. Deuterated chloroform (CDCl3) was provided by Sigma Aldrich. Phosphate buffer saline (PBS) solution (pH 7.4) was purchased from Merck.Instruments
1H-NMR and 13C-NMR spectra were recorded on a 400 MHz Bruker Aspect spectrometer. DOSY experiments were performed using a 600 MHz Bruker spectrometer. CDCl3 was used as a deuterated solvent. The chemical shifts were given in parts per million (ppm) and the chemical shifts of protons were relative to tetramethylsilane (TMS) at δ = 0. The samples were prepared by dissolving 10 mg of the polymer in 0.5 mL of CDCl3.Size exclusion chromatography (SEC) analyses were performed on a PLGPC 50 Plus instrument thermostated at 50 °C and equipped with a RI differential refractometer detector, one 8 μm Polar Gel-M pre-column (7.5 × 50 mm), two 8 μm Polar Gel-M columns (7.5 × 300 mm) and a Varian model 410 autosampler. SEC was performed in DMF containing 0.1 wt% LiCl, with a flow rate of 0.8 mL min−1 at 50 °C. The samples were prepared by dissolving 10 mg of the oligomer in 1.5 mL of DMF and then they were filtered using TE36 Whatman PTFE-supported membrane filter paper (0.45 μm, 47 mm diameter) before the injection. The data were calibrated using polymethylmethacrylate (PMMA) standards.
The FTIR (Fourier transform infra-red) spectra were recorded with a PerkinElmer Spectrum spectrometer equipped with an ATR (attenuated total reflectance) cell with a ZnSe crystal. The wavelength range studied is 4000 cm−1 to 650 cm−1.
MALDI-TOF measurements were recorded in reflection mode using R-cyano-4-hydroxycinnamic acid (HCCA) as a matrix with NaI. The polymer was dissolved in acetonitrile at a concentration of 10 mg mL−1.
Polymerisations were performed in a micro-wave Anton Paar 300 mono-wave reactor.
The thermoresponsive behaviour of the oligomers in solution was evaluated using dynamic light scattering (DLS). The instrument employed for this purpose was a Malvern Instruments Zetasizer Nano ZS single-angle device. The DLS system was equipped with a He–Ne (helium–neon) laser source with a wavelength of 633 nm and a power of 35 mW. The detector of the instrument was positioned at an angle of 173°. By utilizing DLS, the scattering of light by the sample under investigation was analyzed, providing information about the size and dynamics of the particles or aggregates present in the solution. Herein, DLS was employed to assess the changes in particle size and the aggregation behavior of the oligomer system in solution as a function of temperature. The use of a specific laser source and detector angle allowed for accurate and precise measurements of the scattering phenomenon, enabling the determination of the thermo-responsive behavior in solution of the oligomers under investigation. PBS was filtered before use. The measurement interval was selected between 10 °C and 50 °C. First, the samples were equilibrated for 10 min. Subsequently, the temperature was increased by 2 °C followed by 5 min of equilibration at the given temperature and the acquisition of scattering data for 3 min. This process was repeated until the samples reached 50 °C. For the reversibility tests of thermal changes, the sample was cooled back to 10 °C (2 °C min−1) and equilibrated for 10 min before the acquisition of DLS data. DLS measurements have been done in triplicate, with the final reported values being presented as mean values of the three measurements.
Transmission electron microscopy (TEM) images were acquired using a JEOL 1400 P + 120 kV. 10 μL of polymer solutions at 1 g L−1 were deposited onto a carbon coated grid (Formvar/carbon 200 mesh, copper) for 30 seconds and then blotted with filter paper to remove excess solution. Then, the sample loaded grid was stained with 10 μL of 1% ammonium molybdate solution for 15 seconds before this solution was removed with filter paper. The grid was allowed to dry for 15 min in a fume hood. For the study at 50 °C, the polymer solutions at 1 g L−1 were incubated at 50 °C by using an oil bath for heating for 1 h. The samples were quickly loaded onto the grid using the protocol described above.
DSC thermograms were recorded using a Q2000 (TA Instruments) apparatus in a sealed pan under a nitrogen flow (50 mL min−1). An empty pan was used as a reference. The samples were analyzed from 0 to 140 °C with 10 °C min−1 heating and 10 °C min−1 cooling between the first and second runs.
Methods
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C of aromatic tosylate, m), 133 (CC–CH3 of aromatic tosylate, z), 130 and 128 (CC of aromatic tosylate, c and d), 70.28 (S–O–CH2– of alkyl linker, b), 28.87 (S–O–CH2–CH2–CH2– of alkyl linker, y and y′), 24.95 (S–O–CH2–CH2–CH2– of alkyl linker, j and j′), 21.65 (CH3 of tosylate backbone, e).
C–C of aromatic tosylate, m), 133 (CC–CH3 of aromatic tosylate, z), 130 and 128 (CC of aromatic tosylate, c and d), 70.28 (S–O–CH2– of alkyl linker, b), 28.87 (S–O–CH2–CH2–CH2– of alkyl linker, y and y′), 24.95 (S–O–CH2–CH2–CH2– of alkyl linker, j and j′), 21.65 (CH3 of tosylate backbone, e).
The reaction mixture was filtered and the solvent was removed. The residue was dissolved in chloroform and washed twice with a saturated solution of NaHCO3 and once with brine. The organic phase was concentrated under reduced pressure. Yield: 60%. 1H NMR (CDCl3), δ (ppm), oligoMeOx: 7.7 (8H, He), 3.2–3.5 (CH2 backbone of oligoMeOx, Hb and terminal CH2 groups of HDOTs, Ha′), 2.1–2.3 (CH3 backbone of oligoMeOx, Hc), 1.2–1.6 (8H, Ha). OligoBuOx: 1H NMR (CDCl3), Fig. S3.†δ (ppm): 7.7 (8H, He), 3.2–3.5 (CH2 backbone of oligoBuOx, Hb and terminal CH2 groups of HDOTs, Ha′), 2.2–2.4 (vicinal CH2 of CO from oligoBuOx, Hc), 1.5–1.8 (CH2 of the pendant oligoBuOx chain, Hd and Hf, CH2 alkyl chains of HDOTs, 8H, Ha), 0.9–1.0 (terminal CH3 of the oligoBuOx backbone, Hg).
O from oligoBuOx, Hc), 1.5–1.8 (CH2 of the pendant oligoBuOx chain, Hd and Hf, CH2 alkyl chains of HDOTs, 8H, Ha), 0.9–1.0 (terminal CH3 of the oligoBuOx backbone, Hg).
For all syntheses, the total co-monomer-to-initiator ratio of M/I was equal to 10 while preparing different compositions (from 20% to 50% BuOx). The total co-monomer concentration was adjusted to 4 M. The solution was heated in a microwave reactor at 140 °C and 300 W. After cooling the resulting solution to room temperature, a 10-fold excess of potassium phthalimide (5.6 g, 30.5 mmol, 10 equiv.) was added and the reaction mixture was stirred overnight at 70 °C. The reaction mixture was filtered and the solvent was removed. For statistical co-oligomers, the resulting residue was dissolved in chloroform and washed twice with a saturated solution of NaHCO3 and once with brine. The organic phase was concentrated under reduced pressure. However, block and reverse block co-oligomers showed some issues during the organic/aqueous separation step. For this reason, the residue obtained after filtration in the case of block and reverse block architectures was purified by using a different method: the filtrate was concentrated and precipitated twice in cold diethyl ether. The resulting product was dried over vacuum. Yield: 65%. 1H NMR (CDCl3), δ (ppm): 7.7 (8H, He), 3.2–3.5 (CH2 backbone of oligoMeOx and oligoBuOx, Hb and terminal CH2 groups of HDOTs, Ha′), 2.2–2.4 (vicinal CH2 of CO from oligoBuOx, Hd), 2.1–2.3 (CH3 backbone of oligoMeOx, Hc), 1.2–1.8 (CH2 of the pendant oligoBuOx chain, Hf and Hi, CH2 alkyl chains of HDOTs, 8H, Ha), 0.8–0.9 (terminal CH3 of the oligoBuOx backbone, Hg).
O from oligoBuOx, Hd), 2.1–2.3 (CH3 backbone of oligoMeOx, Hc), 1.2–1.8 (CH2 of the pendant oligoBuOx chain, Hf and Hi, CH2 alkyl chains of HDOTs, 8H, Ha), 0.8–0.9 (terminal CH3 of the oligoBuOx backbone, Hg).
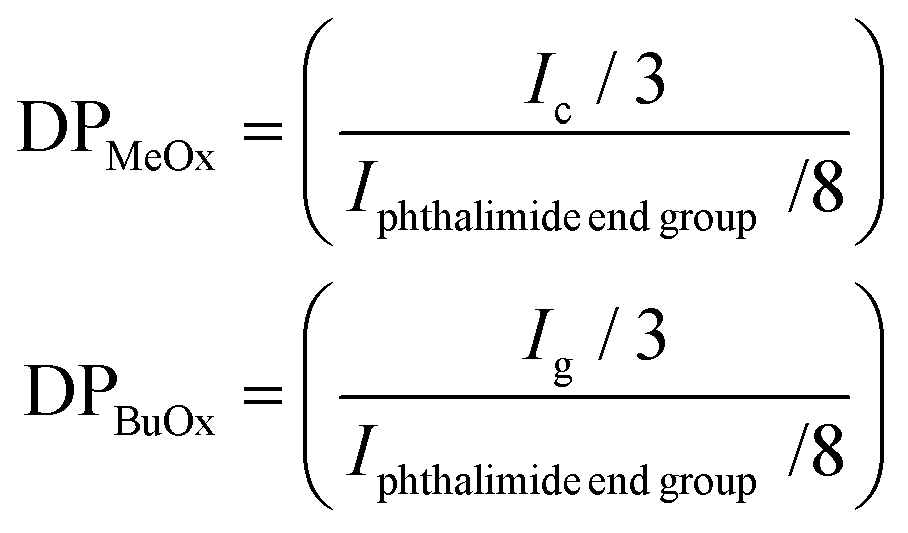 | (1) |
For statistical (S), block (B) and reverse block (RB) co-oligomers, the total number of methyl oxazoline and butyl oxazoline units was calculated as described previously, while the total degree of polymerisation was calculated as the sum between DPMeOx and DPBuOx, as stated in eqn (2):
| DPtot = DPMeOx + DPBuOx | (2) |
For S, B and RB co-oligomers, the molar % of MeOx (or BuOx) units was calculated by dividing the number of MeOx (or BuOx) repeating motifs (DPMeOx or DPBuOx) by the total degree of polymerisation (DPtot), and the result is expressed as a percentage according to eqn (3):
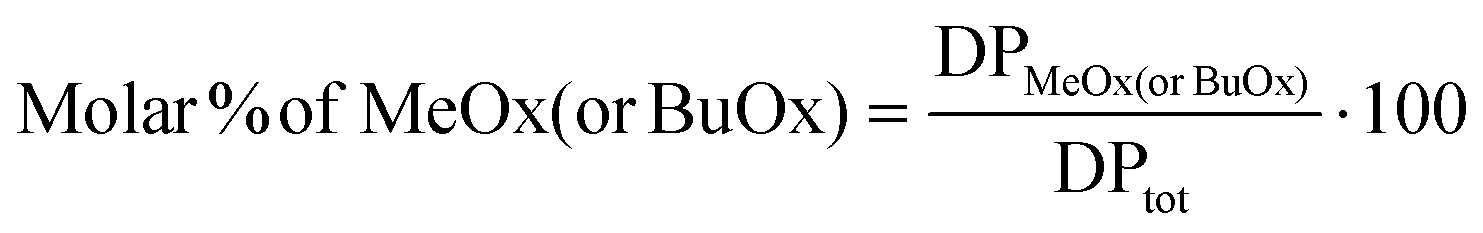 | (3) |
For oligoMeOx and oligoBuOx homo-oligomers, experimental Mn was calculated as the sum between the total DPtot multiplied by the molar mass of the repetitive unit and adding the molar mass of chain extremities (Mext) as well as the molar mass of the internal six-carbon linker (Mint), according to eqn (4). A similar procedure was applied in the case of co-oligomers by following eqn (5):
| For homo-oligomers: Mn = DPtot·MMeOx (or BuOx) + Mext | (4) |
| For co-oligomers: Mn = (DPMeOx·MMeOx) + (DPBuOx·MBuOx) + Mext | (5) |
The degree of functionalisation (DF) with phthalimide-end groups was calculated by comparing the value of integral signals of aromatic protons of phthalimide end-groups (at 7.6 ppm) with that of methylene protons from HDOTs in the proximity of oligoOx (noted as a′ at around 3.2 ppm or around 3.6 ppm, each of them integrating to 2) (Fig. S3†). The obtained value was divided by the theoretical value of the integral signals of phthalimide protons corresponding to theoretical 100% functionalisation (equal to 8) and multiplied by 100 to obtain the final DF (%) according to eqn (6):
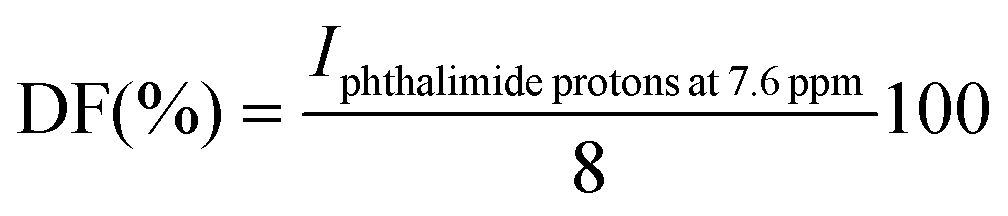 | (6) |
Results and discussion
Synthesis of oligoOx
ω-Aminated oligoOx were synthesized in two steps. First, a bifunctional six-carbon chain ditosylate initiator was used in the CROP of MeOx (BuOx), and the living oxazolinium species were quenched with an excess of potassium phthalimide. The six carbon-chain internal linker of the ditosylate initiator induces structural flexibility in the resulting oligoOx, which is an important characteristic of self-assemblies requiring a sharp convolution of the central hydrophobic block. The end-capping efficiency with phthalimide groups was evaluated by determination of the degree of functionalisation (DF) by 1H-NMR. This was done by comparing the value of the integral signals of methylene protons from the difunctional initiator in the proximity of oligoOx (noted as a′ at around 3.2 ppm) with the value of the integral signals of aromatic protons of phthalimide end-groups (at 7.6 ppm) (Fig. S3†). The obtained value of the integral signals of phthalimide was then divided by the theoretical value (equal to 8) and multiplied by 100 to obtain the final DF (%), as described in eqn (6). As shown in Table 1, the first phthalimide protection step resulted in the DF between 89% and 96%, which is in accordance with the literature.41| Code | DPn th | DPn expa | %BuOx/%MeOx expb |
M
n NMRc (g mol−1) |
DFNMRd (%) |
M
n SEC
(g mol−1) |
Đ |
F
phil
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| For oligoMeOx and oligoBuOx, ** refers to the global DPn (theoretical th and experimental exp); for S, B and RB co-oligomers, * refers to the DPn (theoretical th and experimental exp) calculated for each repetitive unit of BuOx/MeOx.a Determined by 1H-NMR and according to eqn (1) and (2).b Determined by 1H-NMR and according to eqn (3).c Determined by 1H-NMR and according to eqn (4) and (5).d Determined by 1H-NMR and according to eqn (6).e Determined by SEC in DMF, calibrated by using PMMA standards.f Fphil: hydrophilic fraction, calculated by 1H-NMR as: Mn (hydrophilic part)/Mn (oligomer), where Mn (hydrophilic part) = (DPexp of MeOx × MMeOx) + 2 × Mamino group; DPexp of MeOx: experimental degree of polymerisation of MeOx; MMeOx: molecular weight of MeOx; Mamino group: molecular weight of the amino group. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Homo-oligomers | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| OligoMeOx | **10 | **12 | 0/100 | 1380 | 92 | 1500 | 1.08 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| OligoBuOx | **10 | **10 | 100/0 | 1380 | 95 | 1280 | 1.10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Statistical (S) co-oligomers | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S1 | *2/8 | *1.6/8.4 | 16/84 | 1010 | 93 | 1140 | 1.36 | 0.71 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S2 | *3/7 | *2.4/7.6 | 24/76 | 1170 | 94 | 1070 | 1.21 | 0.55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S3 | *4/6 | *5.0/5.0 | 50/50 | 1310 | 96 | 1260 | 1.37 | 0.32 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block (B) co-oligomers | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B1 | *2/8 | *1.0/9.0 | 10/90 | 1080 | 89 | 2490 | 1.20 | 0.70 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B2 | *3/7 | *3.5/7.5 | 32/68 | 1170 | 93 | 1270 | 1.13 | 0.54 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B3 | *4/6 | *4.0/6.0 | 40/60 | 1160 | 92 | 1060 | 1.39 | 0.44 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reverse block (RB) co-oligomers | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RB1 | *2/8 | *1.0/9.0 | 10/90 | 1010 | 90 | 1600 | 1.26 | 0.75 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RB2 | *3/7 | *2.8/7.2 | 28/72 | 1083 | 91 | 1800 | 1.16 | 0.56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RB3 | *4/6 | *3.8/6.2 | 38/62 | 1125 | 94 | 1360 | 1.07 | 0.47 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Then, hydrazine was added to convert phthalimide functions into amino groups. This strategy was employed to functionalize homo-oligomers and co-oligomers with a target degree of polymerisation (DPn) of 10 (Fig. 1 and Table 1). However, purification of amino-containing oligoOx was challenging. Inspired by the literature reports showing that typically, due to their low molecular weight, POx oligomers are isolated through liquid/liquid extraction and/or consecutive precipitation steps,59,61 the first strategy of purification involves chloroform/aqueous solution extraction of the final oligomer. Nevertheless, the extraction step results in low yields because the hydrophilic oligoOx is soluble in both aqueous and organic chloroform solvents. Thus, the second alternative strategy involves the purification of the product through repeated precipitation–filtration cycles, which improved the final yield of the resulting oligomers to 60%. However, as the oligomers undergo slow precipitation, this method was time-consuming. 1H-NMR spectroscopy was used to follow the deprotection of phthalimide groups into amino functions, confirmed by the complete disappearance of the signal characteristic of the aromatic phthalimide units at 7.76 ppm (Fig. 1A, B and D). In addition, it is important to note that 1H-NMR spectra indicated the absence of proton signals in the 4.2 ppm–4.8 ppm range (which are characteristic of free non-terminated oxazolinium species). The absence of these signals demonstrates the efficiency of the end-capping steps. FTIR spectroscopy confirmed the effective transformation of phthalimide functions into amino groups, as evidenced by the absence of the typical band at 1710 cm−1 specific to the stretching vibrations of the CO group of phthalimide (Fig. 1C). After deprotection, as observed in Fig. 1D, 1H-NMR spectroscopy showed the presence of all signals corresponding to the resulting oligomers. For example, in the case of oligoMeOx, the protons of the six-carbon internal linker were observed at around 1.1–1.6 ppm (labeled a), while the protons of the oligomer backbone were located at around 2–2.3 ppm (labeled c, methyl protons) and at around 3.2–3.4 ppm (labeled b, methylene protons). When analysing oligoBuOx, well-defined signals were observed at 0.9 ppm (corresponding to the terminal methyl group of the BuOx backbone labeled g), as well as the methylene protons of the pendant group between 1.3 and 1.5 ppm (e, f) or 2.3 ppm (d), together with those of the oligomer backbone (b, a, a′). The integrals of these signals were used further to determine the total degree of polymerization (DP). For example, in the case of oligoMeOx, the number of methyl oxazoline units was calculated considering the integral values of the methyl protons of the oligomer backbone (Hc, between 2.1 and 2.3 ppm), compared to the integral values of the aliphatic carbon linker (Ha, 1.1–1.6 ppm). Additional comprehensive analysis of oligoOx structures was performed using MALDI-TOF (Fig. S6 and S7†), showing a coherent correlation between the calculated Mn and those experimentally determined.
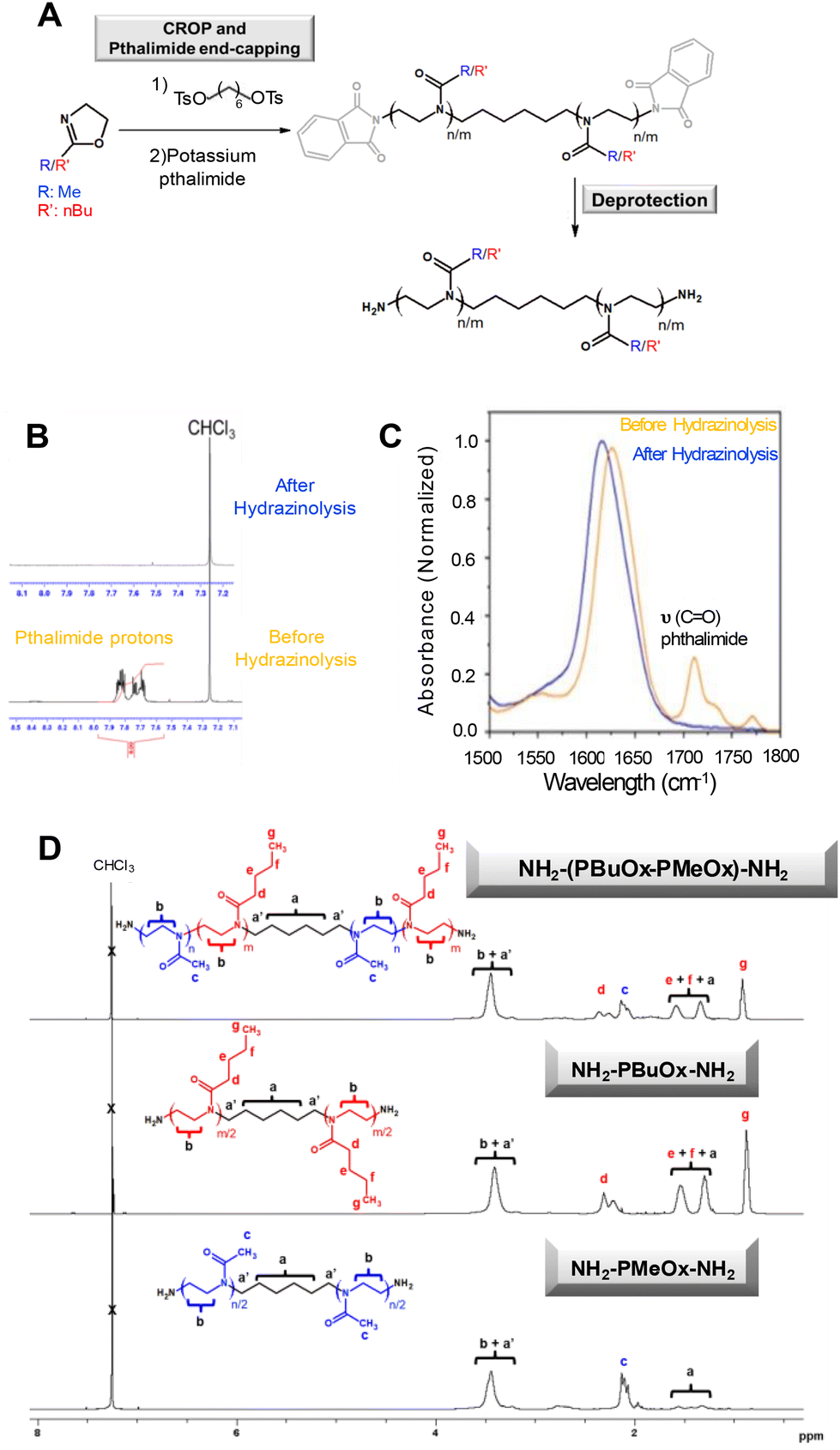 | ||
| Fig. 1 (A) Two-step synthesis of amphiphilic oligoOx: CROP (anhydrous acetonitrile, microwave, 300 W, 140 °C) and termination by using a phthalimide ending agent (70 °C, overnight); deprotection by using hydrazine (ethanol, reflux, overnight); the deprotection step was followed by (B) 1H-NMR and (C) FTIR spectroscopy; (D) 1H-NMR spectra of oligoOx ended with amino groups. | ||
Statistical, block and reverse block oligoOx
PMeOx is widely recognised in the literature for its hydrophilicity.29 On the other hand, PBuOx exhibits greater hydrophobicity which decreases the cloud point of thermoresponsive polymers.29 To modulate self-assembly and thermoresponsive properties, various amphiphilic oligoOx incorporating both MeOx and BuOx in different MeOx/BuOx ratios were synthesized. Different compositions (i.e., ranging from 10 to 50% molar BuOx, corresponding to hydrophilic fractions Fphil from 0.7 to 0.3) were prepared (Fig. 2 and Table 1).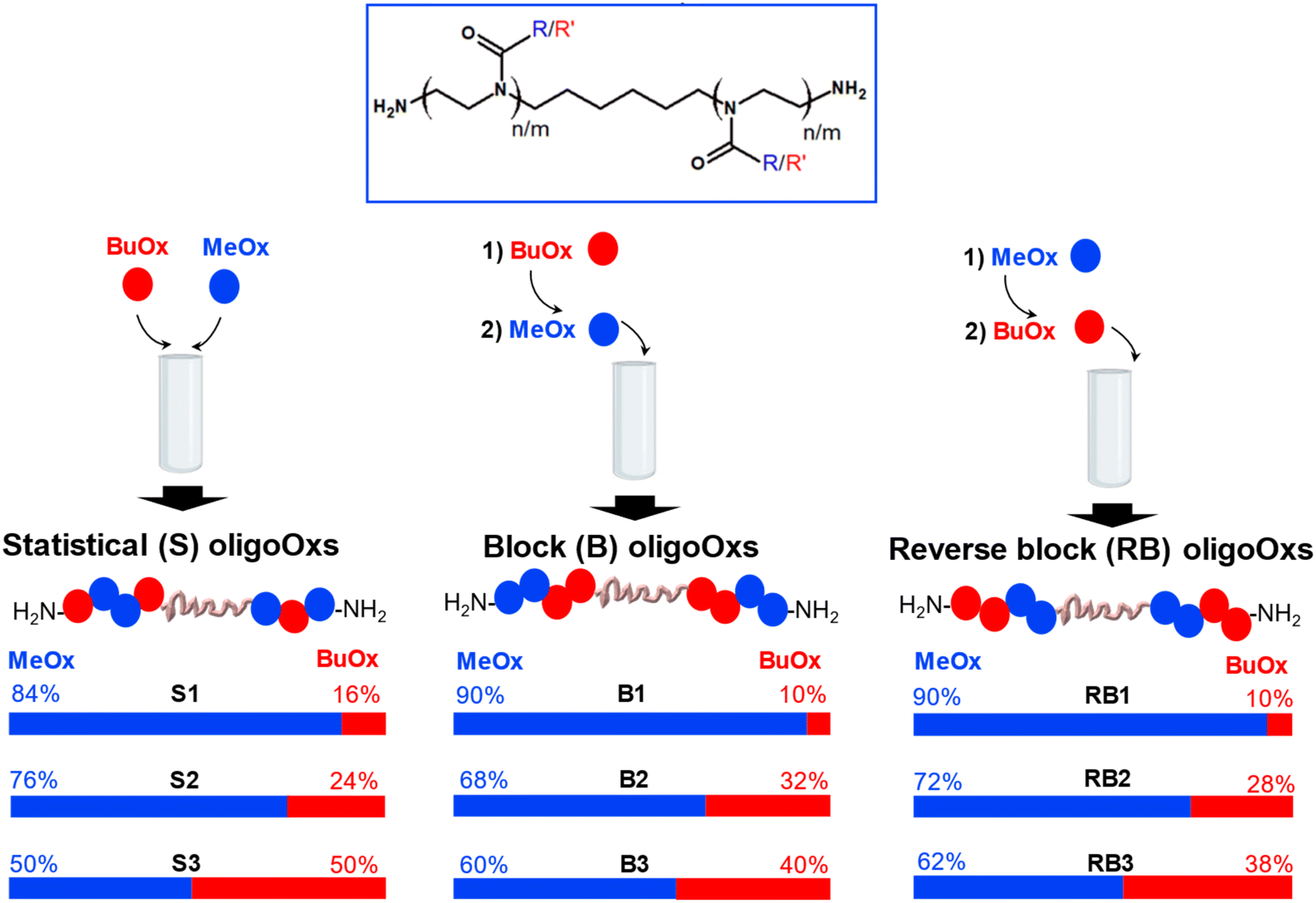 | ||
| Fig. 2 General design of different architectures of amphiphilic oligoOxs: statistical (S), block (B) and reverse block (RB) as a function of the order of MeOx and BuOx monomer addition. | ||
All the oligoOx were characterised by 1H-NMR spectroscopy (Fig. 1D and Table 1), DOSY (Fig. S4†) and SEC (Fig. S5†). For instance, 1H-NMR was used for determining the degree of polymerization (DP), which in the cases of statistical (S), block (B) and reverse-block (RB) oligoOx was calculated as the sum of the methyl-2-oxazoline units DPMeOx and butyl-2-oxazoline units DPBuOx respectively. Overall, these results revealed that all oligomers had a composition close to the targeted ones, as demonstrated by similar experimental (DPexp) and theoretical (DPth) DP values (Table 1). In addition, DOSY NMR is a highly sensitive technique that was used to provide evidence of block copolymer formation. If the block copolymerisation is successful, the correlation spots characterizing the 1H-NMR signals of participating blocks are characterized by only a single, distinct diffusion coefficient.76
DOSY-NMR experiments (Fig. S4†) were performed in CDCl3 for three oligoOx structures (oligoMeOx, oligoBuOx, and the B2 block co-oligomer) providing the same total DP of 10. The three oligomers provided well-aligned spots belonging to the hydrogen atoms of the polyoxazoline backbone and the six-carbon chain linker, which was proved by a unique horizontal line corresponding to the logarithmic value of the diffusion coefficient (logD). In addition, this characterization method is suitable for samples providing diffusion coefficients (D) of 10−12 ≤ D ≤ 2 × 10−3 m2 s−1.77 Respectively, the D was 7.95 × 10−10 m2 s−1 for oligoMeOx (Fig. S4A†), 1.99 × 10−10 m2 s−1 for oligoBuOx (Fig. S4B†), and 3.98 × 10−10 m2 s−1 for co-oligoOx (B2) (Fig. S4C†), which falls between those of oligoMeOx and oligoBuOx. All values were found to be in the corresponding range according to the literature.77 It is important to highlight that the unique diffusion coefficient obtained for the B2 co-oligomer proved that the copolymerization occurred successfully without any residual compounds obtained through the CROP process.
Furthermore, SEC analysis revealed monomodal distribution and dispersities below 1.4 (Fig. S5† and Table 1). The slight broadening of the SEC traces may be related to the bifunctional nature of oligomers, potentially causing inter-chain aggregation due to their proximity, impacting the final dispersity. Recently, similar behavior was reported for multifunctional polyoxazolines prepared using tri/tetrafunctional triflate initiators with star-like architectures, showing rather broad dispersities by SEC, which were explained by polymer aggregation (due to polyfunctionality).78,79 In another work, polypropylene glycol (PPG) ditosylate initiators were used to prepare difunctional polymethyloxazolines with the PPG backbone, with broad dispersities explained similarly.79 Moreover, it is important to emphasize that the chosen protocol (simultaneous addition of MeOx and BuOx for S oligomers, the first addition of BuOx for B oligomers, or the first addition of highly reactive MeOx for RB oligomers) does not affect the formation of relatively well-defined oligomers, as proved by similar experimental Mn values obtained by 1H-NMR, as well as by the consistent hydrophilic fractions (Fphil) obtained for all the three investigated co-oligomers.
Investigation of self-assembly properties
Self-assembled oligomers were prepared by direct dissolution of oligoOx powders in PBS buffer solution (pH 7.4) at a concentration of 1 g L−1, with the resulting solutions being investigated by using TEM and DLS. This concentration was chosen for further experiments since higher concentrations led to undesired aggregation phenomena observed by DLS (data not shown).
First, control experiments were performed on statistical oligoOx designed with hydrophilic MeOx and hydrophobic BuOx units (S). As expected, no self-assembly was noticed as a consequence of limited hierarchical control over the arrangement of hydrophilic and hydrophobic units (Fig. S11†). Otherwise, we investigated the self-assembly of block oligomers.
B1 (with the lowest % of BuOx of 10%, Fphil: 0.70) did not self-assemble in particular morphologies due to its high hydrophilicity that hinders self-assembly (Table 2 and Fig. 3). The increase of hydrophobicity in B2 (32% BuOx, Fphil: 0.54) led to the formation of organised large vesicles with a polymersome-like morphology. TEM and DLS analyses indicated a similar size (219 nm by TEM versus 252 nm by DLS) and a slight polydisperse distribution (PDI: 0.2 by DLS) (Table 2, Fig. 3 and Fig. S8A–F†).
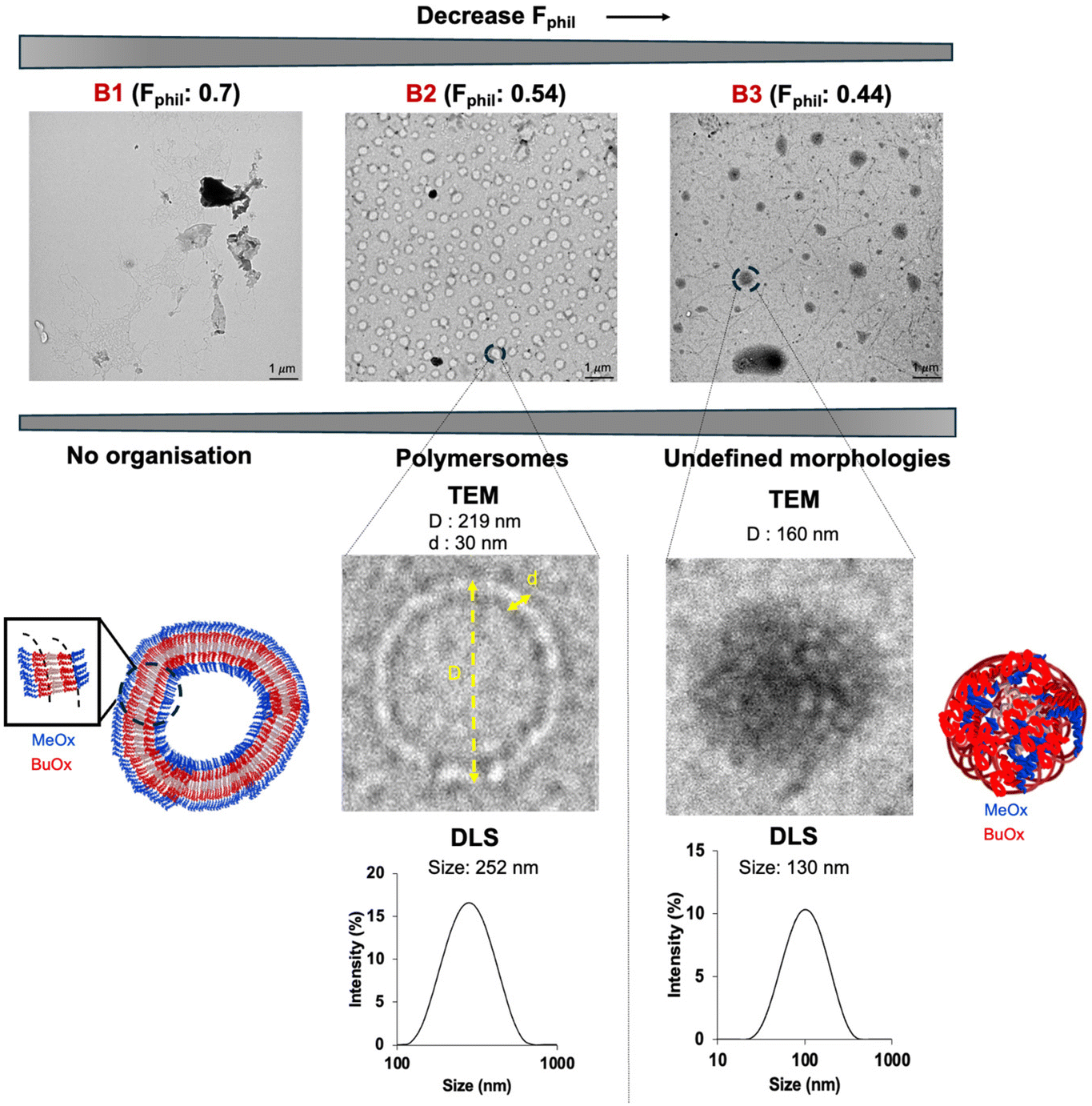 | ||
| Fig. 3 Effect of the Fphil ratio on the self-assembly of block oligoOx (B). Scale bar: 1 μm. | ||
| Code | F phil | Self-assembly at 25 °C | Self-assembly at 50 °C | ||||
|---|---|---|---|---|---|---|---|
| Size (nm) | Size (nm) | ||||||
| TEM | DLS | Morphology | TEM | DLS | Morphology | ||
| Method I: direct dissolution | |||||||
| B1 | 0.70 | N.D. | <50 | Aggregates | N.D. | N.D. | Aggregates |
| B2 | 0.54 | 219 | 252 | Polymersomes | 350 | 360 | Polymersomes |
| B3 | 0.44 | 160 | 130 | Globular/spherical | 110 | 127 | Undefined |
| RB1 | 0.75 | N.D. | >500 | Aggregates | N.D. | >500 | Aggregates |
| RB2 | 0.56 | ||||||
| RB3 | 0.47 | ||||||
| Method II: film rehydration | |||||||
| B2 | 0.54 | 111 | 101 | Polymersomes | 257 | 290 | Polymersomes |
| RB2 | 0.56 | 210 | 180 | Spheres | N.D. | >500 | Aggregates |
These polymersomes had a membrane thickness of around 30 nm, which is six times larger than that of a usual liposome (around 5 nm).13
Interestingly, similar phenomena have been reported in studies of polymersomes made from polyoxazoline containing long side alkyl chains, where solvent impurities coming from the solvent switch method were implicated.66,72 However, the presence of solvent impurities was excluded in our case, as shown by 1H-NMR data (Fig. 1). Thus, we presume that these values are a consequence of the fact that the layer's organization is not uniform, with some regions composed of hydrophobic BuOx stacked chains, while in other parts, hydrophilic MeOx segments are inserted. This imperfect arrangement might potentially retain water molecules inside, thus leading to an overestimation of the membrane thickness.
Overall, this behaviour is favourably influenced by the equibalanced hydrophilic weight fraction (Fphil: 0.54), which allows the self-assembly of many polymers into well-defined nano-objects, especially polymersomes, as reported in the literature.12–18 We note in Fig. S8† the singular tortuosity of the polymersome membrane. This phenomenon is rarely reported in the literature and can be attributed to the flexibility of the polyoxazoline backbone which ensures local folding of the membrane.12–18 The increase in the hydrophobic ratio of BuOx in B3 (40%, Fphil: 0.44) revealed a transition into morphologies with undefined shapes, even in nano-sized objects (160 nm by TEM, 130 nm & PDI: 0.19 by DLS) (Table 2, Fig. 3 and Fig. S9A–E†). This result was quite surprising since B3 provides quite a close hydrophilic weight fraction as B2, which might be theoretically suitable for polymersome formation according to the literature.12–18 For B3, another kind of self-assembly with undefined shapes organized into undefined globular-like morphologies was also observed, as shown in Fig. S9A.† This might come from the slight increase of %BuOx, driving an increase in crystallinity, as additionally supported by DSC thermograms showing a shift of about 21 °C of the melting point (Fig. S10A and B†). Briefly, the self-organisation of block oligoOx into polymersomes is driven by the triblock BuOx structure with a compromise between its low Tg (characteristic of a flexible polymer chain) and its crystalline nature. This is further supported by the TEM image of the formulation of control diblock oligoOx (noted as CB2) in PBS. CB2 provides the same BuOx/MeOx and DP as B2, prepared with the commercial tosylate initiator. As expected, no particular organisation into polymersomes was observed (Fig. S11C†), which emphasizes the importance of triblock oligomer architecture for organisation into polymersome-like morphologies.
However, as the crystallinity increases along with the increased amount of BuOx, the oligomer chains become less flexible, which could impede their organization into polymersomes.
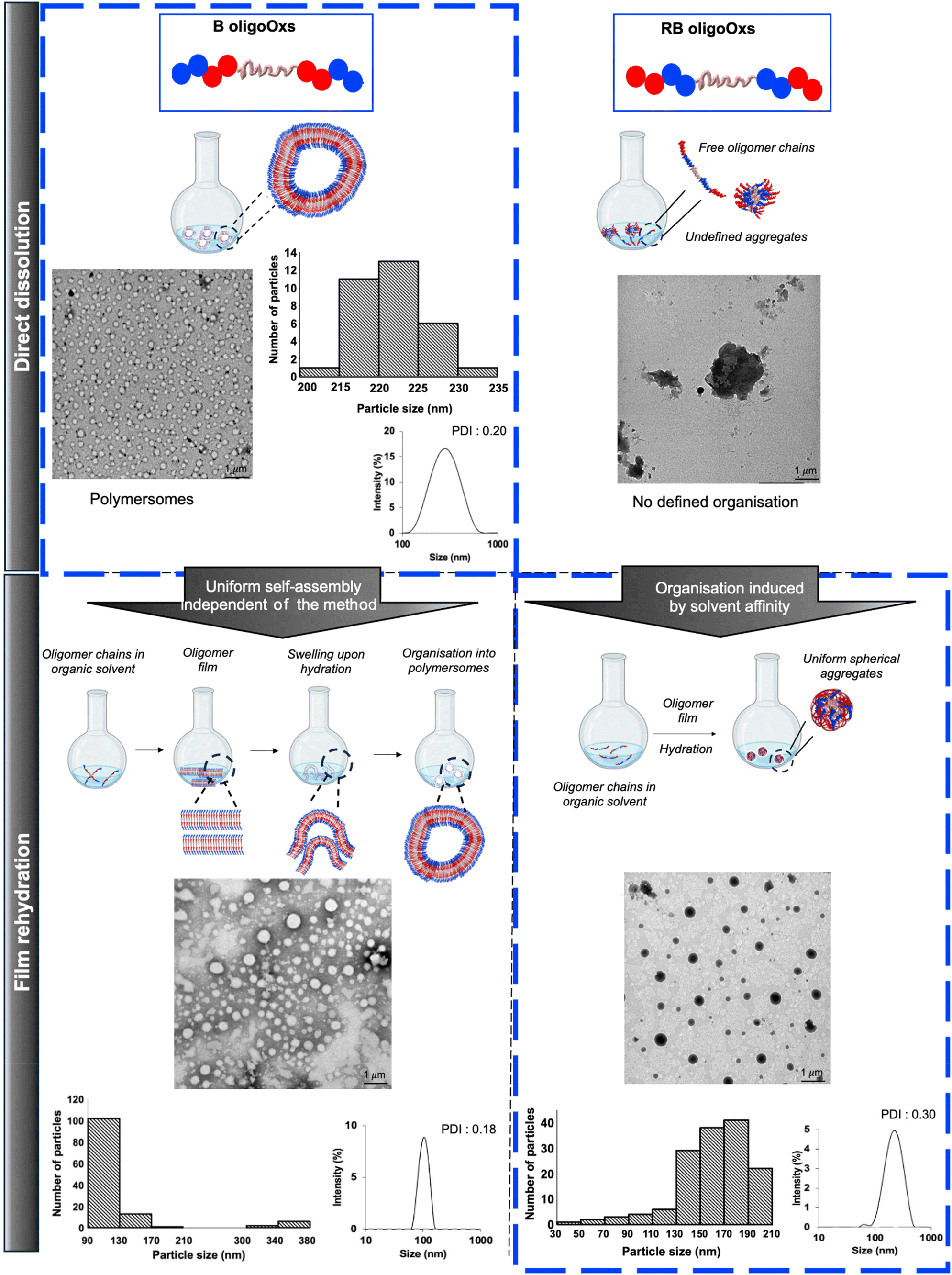 | ||
| Fig. 4 Effect of oligoOx architectures B2 and RB2 and the preparation method on the self-assembly at 25 °C. Scale bar: 1 μm. The contrast differences are assigned to the preference of the contrasting agent towards the imaging of hydrophilic compounds. | ||
Although the direct dissolution method in PBS is highly practical, avoiding organic solvents for oligomer solubilization, other methods of self-assembly of polymers in solution are usually employed including the co-solvent process, film rehydration, etc. There is no universal method to precisely predict the formation of a particular self-assembled morphology. Consequently, B2 and RB2 formulations were also prepared by film rehydration (Fig. 4). B2 self-assembled with similar vesicle morphologies to those obtained by the direct dissolution method (Table 2, Fig. 4 and Fig. S13†) with relatively well-defined polymer vesicles shown by DLS (101 nm & PDI: 0.18). TEM analysis (Fig. 4) of this formulation revealed a mean distribution of around 110 nm and a secondary distribution of around 380 nm, while the membrane thickness was around 12 nm. These results in terms of size were slightly different from those obtained when B2 was directly dissolved in PBS (i.e. showing a uniform mean size of around 250 nm). This difference could be related to the film rehydration method used herein which, despite being straightforward, has some drawbacks such as the lack of control over the resulting particle sizes. This issue, which has been reported in the literature,80 could thus explain the difference in the size of the resulting particles. Moreover, for self-assembled RB2 architectures prepared via the film rehydration method, the morphology switched from undefined organization into nanosized, spherical aggregates (210 nm by TEM; 180 nm & PDI: 0.30 by DLS) (Fig. 4 and Fig. S12†). In addition, contrasting differences between B2 and RB2 could be observed in TEM and might be attributed to the hydrophilic nature of the contrast agent, potentially favoring the imaging of hydrophilic compounds. In the case of RB, the hydrophilic MeOx blocks are located internally, while the hydrophobic blocks are positioned at the extremities of the structure. This suggests that the contrast agent preferentially images the hydrophilic core of RB.
Overall, these results highlight the importance of block architecture in gaining structural stability driven by internal hydrophobic BuOx segments (in particular at equibalanced Fphil) that promote the self-assembly into polymersomes, regardless of the method employed for the preparation of the co-oligomers. Finally, the direct dissolution process was preferred for the self-assembly of ABA amphiphilic triblock oligoOx B in PBS, while the film rehydration process was more suitable to self-assemble the BAB amphiphilic triblock oligoOx RB.
 | ||
| Fig. 5 (A) Thermal transition temperature of block oligoOxs vs. pure PMeOx and PBuOx; (B) The temperature effect on the disaggregation of self-assembled B3 oligomers into smaller nano-objects. All measurements were recorded for a concentration of 1 mg mL−1 in PBS. Scale bar: 1 μm. | ||
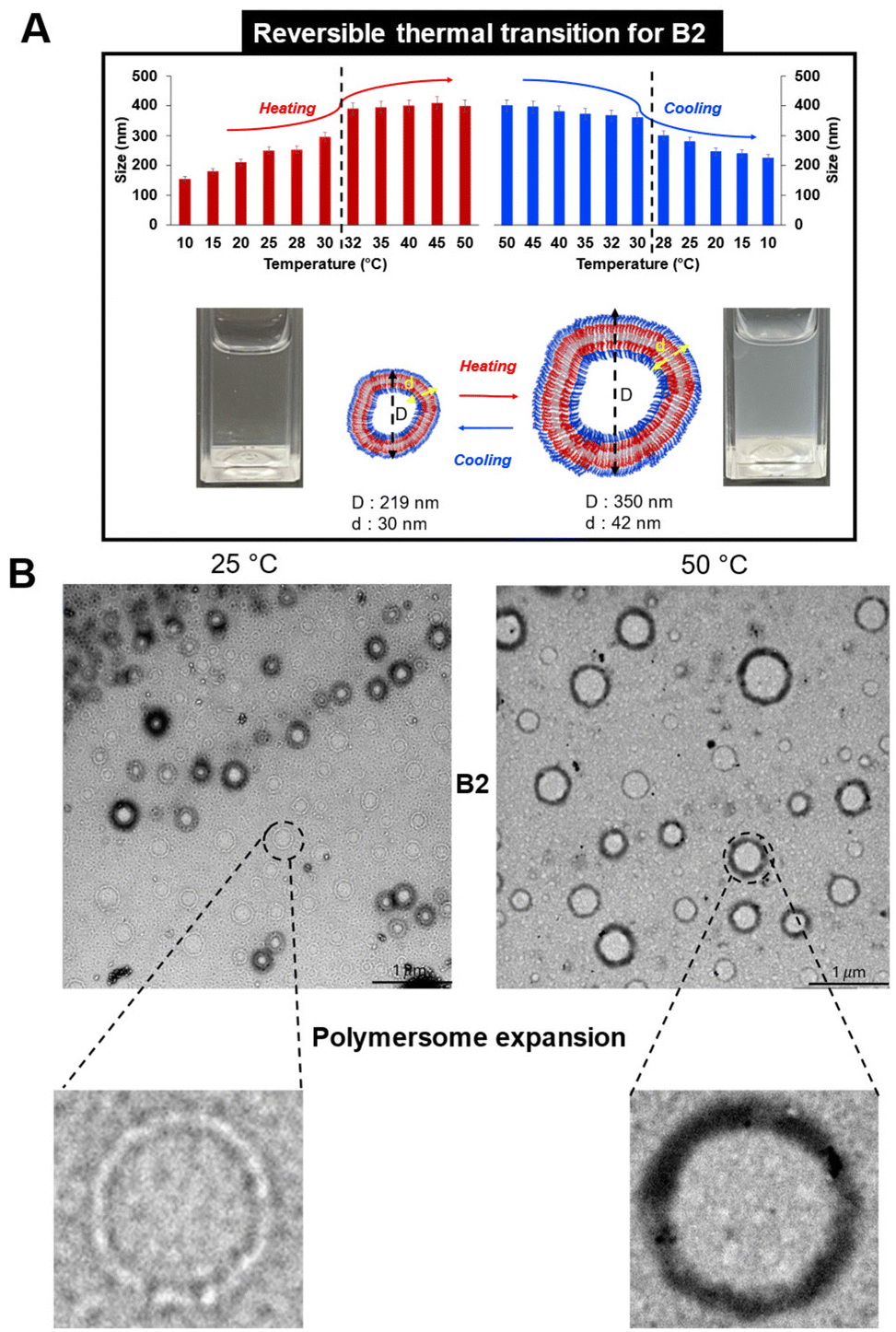 | ||
| Fig. 6 (A) Reversible polymersome shape transition for B2 evaluated by DLS and (B) TEM. All measurements were recorded for a concentration of 1 mg mL−1 in PBS. Scale bar: 1 μm. | ||
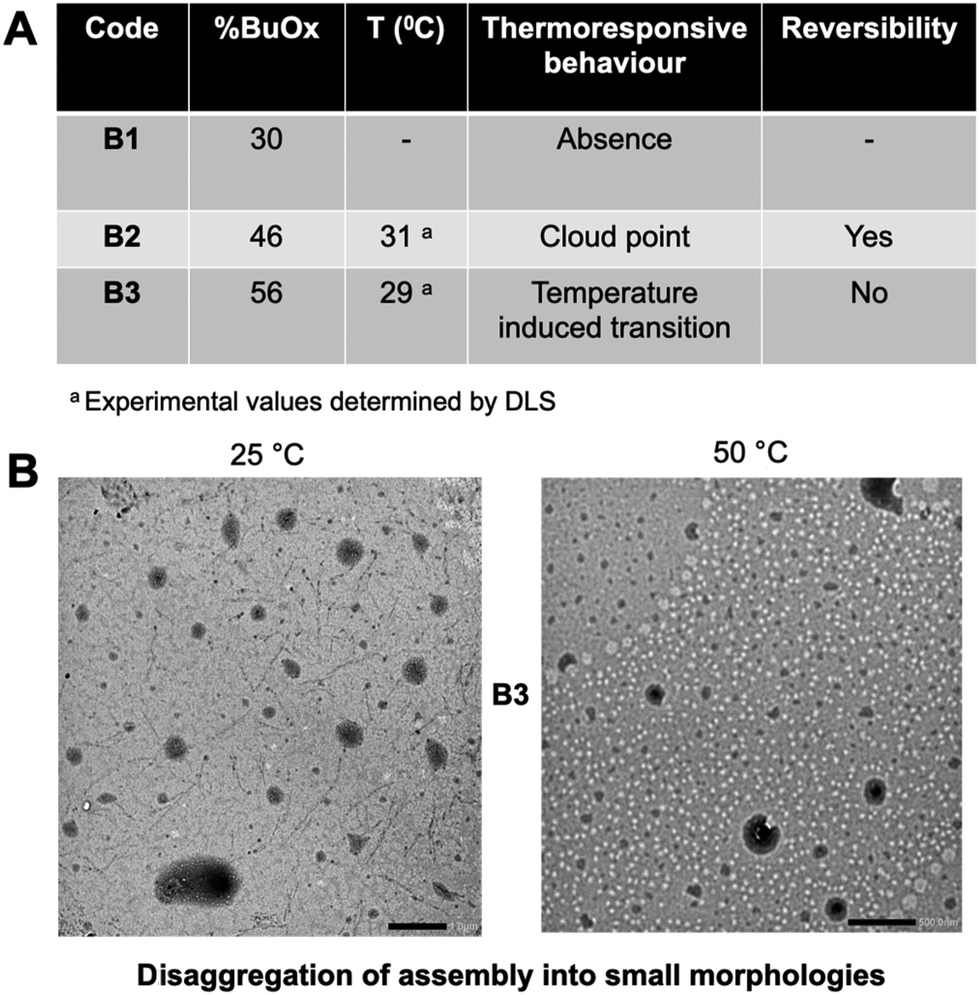
A possible explanation of this behavior might be related to the use of a flexible oligomer backbone (–CH2–CH2–N– repeat units of oxazoline and an internal six-carbon chain linker). This feature might allow an easier reorganization into self-assembled structures with expanded/swelled conformation, even at higher temperatures. This flexibility is more restricted in the case of long polymer chains above their CP since the effect of hydrophobic interactions is stronger, promoting the shape collapse/deswelling.
The reversibility of the thermo-responsiveness of B2 was also studied by DLS during successive heating and cooling cycles between 10 and 50 °C (Fig. 6A). In addition, B2 showed a reversible CP transition of around 30 °C, as proved by a decrease in particle size upon cooling followed by returning to a clear solution. In the case of B3, the lower Fphil ratio led to a slight size decrease of around 29 °C, as detected by DLS (Fig. S14†) and TEM (Fig. 5B). A possible explanation is that an excess of hydrophobicity (the lowest Fphil of 0.44 for B3) may lead to the destabilization of the oligomer assembly at higher temperatures, prompting chain reorganization into smaller nano-assemblies. However, the temperature variation in solution was irreversible for the B3 formulation.
Moreover, the thermoresponsive behavior of RB formulations prepared by the direct dissolution method was investigated (Fig. S11 and S15†). RB1 (10% BuOx) did not present a response in temperature changes, which was expected due to the high hydrophilicity of the system, as in the case of B1 (Fig. S15†). However, no size transition with temperature was observed for RB2 (28% BuOx), which is attributed to the hydrophilic internal MeOx segments that might hinder the aggregation (Fig. S11†). Whereas in the case of RB3 (prepared with the highest amount of BuOx, 40%), the formulation presented a very polydisperse profile, as proved by DLS traces at both temperatures (25 °C and 50 °C), indicating uncontrolled aggregation (Fig. S15†).
Conclusions
Oligomers, being short in length, provide heightened structural mobility allowing precise control over chain interactions and acting as the driving force for the self-assembly of stimuli-responsive polymers.A preliminary challenge concerns the synthesis of controlled amphiphilic oligomers, which is not always straightforward in the case of polyoxazoline chemistry, which is usually employed to obtain macromolecules with higher DPs. This work emphasizes amphiphilic low molecular weight oligoOx, synthesized in two steps using hydrophilic MeOx and hydrophobic BuOx monomers. The versatility of POx chemistry was demonstrated with direct access to ABA, BAB amphiphilic triblock oligoOx and statistical oligoOx using one pot or successive addition of monomers, with, in all cases, control of molecular mass. The MeOx/BuOx ratio of 0.5 revealed, as expected, a promising trade-off for the well-defined self-organisation of oligomers in solution. As previously reported in the literature, the solubility of oligoOx, the crystallization ability and the process of self-assembly drive the oligoOx self-organisation.
OligoOx B2 spontaneously reorganized themselves into large polymersomes with tortuous membranes. In contrast to polymersomes based on high molecular weight polymers already reported in the literature, polymersomes of oligoOx B2 swelled upon heating and reversibly de-swelled upon cooling. These thermoresponsive polymersomes exhibited a reversible cloud point of around 30 °C. Otherwise, reverse BAB triblock oligoOx RB2 self-assembled into spherical nano-objects driven by the crystallization of BuOx, which changed when heated but without a detectable CP temperature.
As far as we know, this is the first report on large unilamelar vesicles based on oligooxazolines. This is a promising path to prepare polymersomes with flexible membranes, which is the limitation of classical polymersomes in comparison with liposomes, opening avenues towards new nanostructured systems used in biological applications.
Author contributions
LD: experiments and writing the manuscript; LVA: experiments, conceptualization, writing the manuscript, review and editing; BTB: experiments and writing the manuscript; KC: writing the manuscript; MS, LP, and DC: manuscript correction and funding; VL: manuscript correction, review and editing, supervision, and funding. All authors approved the final form of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project is supported by the Chimie Balard Cirimat Carnot Institute through the ANR program No. 16 CARN 0008-01 and the I2C Carnot Institute through the ANR program No. 20 CARN 0036-0.References
- R. Liu, M. Fraylich and B. R. Saunders, Thermoresponsive copolymers: from fundamental studies to applications, Colloid Polym. Sci., 2009, 287, 627–643, DOI:10.1007/s00396-009-2028-x.
- M. A. Ward and T. K. Georgiou, Thermoresponsive Polymers for Biomedical Applications, Polymers, 2011, 3, 1215–1242, DOI:10.3390/polym3031215.
- D. Roy, W. A. Brooks and B. S. Summerlin, New directions in thermoresponsive polymers, Chem. Soc. Rev., 2013, 42, 7214–7243, 10.1039/C3CS35499G.
- M. I. Gibson and R. K. O'Reilly, To aggregate, or not to aggregate? considerations in the design and application of polymeric thermally-responsive nanoparticles, Chem. Soc. Rev., 2013, 42, 7204–7213, 10.1039/C3CS60035A.
- S. Hocine and M.-H. Li, Thermoresponsive self-assembled polymer colloids in water, Soft Matter, 2013, 9, 5839–5861, 10.1039/C3SM50428J.
- S. Strandman and X. X. Zhu, Thermo-responsive block copolymers with multiple phase transition temperatures in aqueous solutions, Prog. Polym. Sci., 2015, 42, 154–176, DOI:10.1016/j.progpolymsci.2014.10.008.
- Y.-J. Kim and Y. T. Matsunaga, Thermo-responsive polymers and their application as smart biomaterials, J. Mater. Chem. B, 2017, 5, 4307–4321, 10.1039/C7TB00157F.
- M. Sponchioni, U. C. Palmiero and D. Moscatelli, Thermo-responsive polymers: Applications of smart materials in drug delivery and tissue engineering, Mater. Sci. Eng., C, 2019, 102, 589–605, DOI:10.1016/j.msec.2019.04.069.
- P. Zarrintaj, M. Jouyandeh, M. R. Ganjali, B. S. Hadavand, M. Mozafari, S. S. Sheiko, M. Vatankhah-Varnoosfaderani, T. J. Gutiérrez and M. R. Saeb, Thermo-sensitive polymers in medicine: A review, Eur. Polym. J., 2019, 117, 402–423, DOI:10.1016/j.eurpolymj.2019.05.024.
- Y. Kotsuchibashi, Recent advances in multi-temperature-responsive polymeric materials, Polym. J., 2020, 52, 681–689, DOI:10.1038/s41428-020-0330-0.
- D. J. Phillips and M. I. Gibson, Towards being genuinely smart: ‘isothermally responsive’ polymers as versatile, programmable scaffolds for biologically-adaptable materials, Polym. Chem., 2015, 6, 1033–1043, 10.1039/C4PY01539H.
- P. A. Beales, S. Khan, S. P. Muench and L. J. C. Jeuken, Durable vesicles for reconstitution of membrane proteins in biotechnology, Biochem. Soc. Trans., 2017, 45, 15–26, DOI:10.1042/BST20160019.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Liposomes and polymersomes: a comparative review towards cell mimicking, Chem. Soc. Rev., 2018, 47, 8572–8612, 10.1039/C8CS00162F.
- Y. Zhu, S. Cao, M. Huo, J. C. M. van Hest and H. Che, Recent advances in permeable polymersomes: fabrication, responsiveness and applications, Chem. Sci., 2023, 14, 7411–7438, 10.1039/D3SC01707A.
- S. Rottet, S. Iqbal, P. A. Beales, A. Lin, J. Lee, M. Rug, C. Scott and R. Callaghan, Characterisation of hybrid polymersome vesicles containing the efflux pumps NaAtm1 or P-Glycoprotein, Polymers, 2020, 12, 1049–1071, DOI:10.3390/polym12051049.
- P. L. W. Welzen, S. W. Martinez Ciriano, S. Cao, A. F. Mason, I. A. B. Welzen-Pijpers and J. C. M. van Hest, Reversibly self-assembled pH-responsive PEG-p(CL-g-TMC) polymersomes, J. Polym. Sci., 2021, 59, 1241–1252, DOI:10.1002/pol.20200871.
- B. Van Genabeek, B. A. G. Lamers, C. J. Hawker, E. W. Meijer, W. R. Gutekunst and B. V. K. J. Schmidt, Properties and applications of precision oligomer materials; where organic and polymer chemistry join forces, J. Polym. Sci., 2021, 59(5), 373–403, DOI:10.1002/pol.20200862.
- L. Xu, L. Yang and S. Lei, Self-assembly of conjugated oligomers and polymers at the interface: structure and properties, Nanoscale, 2012, 4, 4399–4415, 10.1039/C2NR30122A.
- R. Hoogenboom, Poly(2-oxazoline)s: A Polymer Class with Numerous Potential Applications, Angew. Chem., Int. Ed., 2009, 48(43), 7978–7994, DOI:10.1002/anie.200901607.
- B. Guillerm, S. Monge, V. Lapinte and J.-J. Robin, How to Modulate the Chemical Structure of Polyoxazolines by Appropriate Functionalization, Macromol. Rapid Commun., 2012, 33(19), 1600–1612, DOI:10.1002/marc.201200266.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Poly(2-oxazoline)s as polymer therapeutics, Macromol. Rapid Commun., 2012, 33(19), 1613–1631, DOI:10.1002/marc.201200354.
- O. Sedlacek, B. D. Monnery, S. K. Filippov, R. Hoogenboom and M. Hruby, Poly(2-Oxazoline)s – Are They More Advantageous for Biomedical Applications Than Other Polymers?, Macromol. Rapid Commun., 2012, 33(19), 1648–1662, DOI:10.1002/marc.201200453.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Poly(2-oxazoline)s based biomaterials: A comprehensive and critical update, Biomaterials., 2018, 178, 204–280, DOI:10.1016/j.biomaterials.2018.05.022.
- M. M. Bloksma, R. M. Paulus, H. P. C. van Kuringen, F. van der Woerdt, H. M. L. Lambermont-Thijs, U. S. Schubert and R. Hoogenboom, Thermoresponsive Poly(2-oxazine)s, Macromol. Rapid Commun., 2012, 33, 92–96, DOI:10.1002/marc.201100587.
- C. Weber, R. Hoogenboom and U. S. Schubert, Temperature responsive bio-compatible polymers based on poly(ethylene oxide) and poly(2-oxazoline)s, Prog. Polym. Sci., 2012, 37(5), 686–714, DOI:10.1016/j.progpolymsci.2011.10.002.
- A. Pospisilova, S. K. Filippov, A. Bogomolova, S. Turner, O. Sedlacek, N. Matushkin, Z. Cernochova, P. Stepanek and M. Hruby, Glycogen-graft-poly(2-alkyl-2-oxazolines) – the new versatile biopolymer-based thermoresponsive macromolecular toolbox, RSC Adv., 2014, 4, 61580–61588, 10.1039/C4RA10315G.
- R. Konefał, J. Spěváček and P. Černoch, Thermoresponsive poly(2-oxazoline) homopolymers and copolymers in aqueous solutions studied by NMR spectroscopy and dynamic light scattering, Eur. Pol. J., 2018, 100, 241–252, DOI:10.1016/j.eurpolymj.2018.01.019.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. van Lankvelt and U. S. Schubert, Microwave-assisted synthesis and properties of a series of poly(2-alkyl-2-oxazoline)s, Des. Monomers Polym., 2012, 8(6), 659–671, DOI:10.1163/156855505774597704.
- R. Hoogenboom and H. Schlaad, Thermoresponsive poly(2-oxazoline)s, polypeptoids, and polypeptides, Polym. Chem., 2017, 8, 24–40, 10.1039/C6PY01320A.
- L. Loukotová, R. Konefał, K. Venclíková, D. Machová, O. Janoušková, M. Rabyk, M. Netopilík, E. Mázl Chánová, P. Štěpánek and M. Hrubý, Hybrid thermoresponsive graft constructs of fungal polysaccharide β-glucan: Physico-chemical and immunomodulatory properties, Eur. Polym. J., 2018, 106, 118–127, DOI:10.1016/j.eurpolymj.2018.07.004.
- S. Huber, N. Hutter and R. Jordan, Effect of end group polarity upon the lower critical solution temperature of poly(2-isopropyl-2-oxazoline), Colloid Polym. Sci., 2008, 286, 653–1661, DOI:10.1007/s00396-008-1942-7.
- S. Huber and R. Jordan, Modulation of the lower critical solution temperature of 2-Alkyl-2-oxazoline copolymers, Colloid Polym. Sci., 2008, 286, 395–402, DOI:10.1007/s00396-007-1781-y.
- C. Diehl and H. Schlaad, Thermo-responsive polyoxazolines with widely tuneable LCST, Macromol. Biosci., 2009, 9(2), 157–161, DOI:10.1002/mabi.200800213.
- R. Hoogenboom, H. M. L. Lambermont-Thijs, M. J. H. C. Jochems, S. Hoeppener, C. Guerlain, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Schizophrenic gradient copolymer: switching and reversing poly(2-oxazoline) micelles based on UCST and subtle solvent changes, Soft Matter, 2009, 5, 3590–3592, 10.1039/B912491H.
- G. Le Fer, C. Amiel and G. Volet, Copolymers based on azidopentyl-2-oxazoline: Synthesis, characterization and LCST behavior, Eur. Polym. J., 2015, 71, 523–533, DOI:10.1016/j.eurpolymj.2015.08.028.
- M. M. Lübtow, M. Mrlik, L. Hahn, A. Altmann, M. Beudert, T. Lühmann and R. Luxenhofer, Temperature-Dependent Rheological and Viscoelastic Investigation of a Poly(2-methyl-2-oxazoline)-b-poly(2-iso-butyl-2-oxazoline)-b-poly(2-methyl-2-oxazoline)-Based Thermogelling Hydrogel, J. Funct. Biomater., 2019, 10(3), 36–52, DOI:10.3390/jfb10030036.
- M. Madau, G. Morandi, C. Rihouey, V. Lapinte, H. Oulyadi, D. Le Cerf, V. Dulong and L. Picton, A mild and straightforward one-pot hyaluronic acid functionalization through termination of poly-(2-alkyl-2-oxazoline), Polymer, 2021, 230, 124059, DOI:10.1016/j.polymer.2021.124059.
- M. Madau, G. Morandi, C. Rihouey, V. Lapinte, H. Oulyadi, D. Le Cerf, V. Dulong and L. Picton, Thermo-responsive hydrogels from hyaluronic acid functionalized with poly(2-alkyl-2-oxazoline) copolymers with tuneable transition temperature, Polymer, 2022, 244, 124643, DOI:10.1016/j.polymer.2022.124643.
- G. Bretel, D.-T. Tran, G. Morandi, V. Lapinte, S. Marais and L. Hespel, Synthesis of an original oxazoline based monomer containing a photosensitive azobenzene moiety and investigation of the behavior in solid state, in aqueous and vapor media of a photo and thermo-responsive polyoxazoline copolymer, Polymer, 2023, 271, 125812, DOI:10.1016/j.polymer.2023.125812.
- S. Weydert, S. Zurcher, S. Tanner, N. Zhang, R. Ritter, T. Peter, M. J. Aebersold, G. Thompson-Steckel, C. Forro, M. Rottmar, F. Stauffer, I. A. Valassina, G. Morgese, E. M. Benetti, S. Tosatti and J. Voros, Easy to Apply Polyoxazoline-Based Coating for Precise and Long-Term Control of Neural Patterns, Langmuir, 2017, 33(35), 8594–8605, DOI:10.1021/acs.langmuir.7b01437.
- L. Tauhardt, M. Frant, D. Pretzel, M. Hartlieb, C. Bucher, G. Hildebrand, B. Schroter, C. Weber, K. Kempe, M. Gottschaldt, K. Liefeith and U. S. Schubert, Amine-end functionalised poly(2-ethyl-2-oxazoline) as promising coating material for antifouling applications, J. Mater. Chem. B., 2014, 2, 4883–4893, 10.1039/c4tb00193a.
- T. Brossier, B. T. Benkhaled, M. Colpaert, G. Volpi, O. Guillaume, S. Blanquer and V. Lapinte, Polyoxazoline hydrogels fabricated by stereolithography, Biomater. Sci., 2022, 10, 2681–2691, 10.1039/d2bm00138a.
- C. Legros, M.-C. De Pauw-Gillet, K. C. Tam and D. Taton, Lecommandoux, S. Crystallisation-driven self-assembly of poly(2-isopropyl-2-oxazoline)-block-poly(2-methyl-2-oxazoline) above the LCST, Soft Matter, 2015, 11, 3354–3359, 10.1039/C5SM00313J.
- M. Hijazi, M. Schmidt, H. Xia, J. Storkmann, R. Plothe, D. Dos Santos, U. Bednarzick, C. Krumm and J. C. Tiller, Investigations on the thermoresponsive behavior of copoly(2-oxazoline)s in water, Polymer, 2019, 175, 294–301, DOI:10.1016/j.polymer.2019.05.040.
- D. Pizzi, J. Humphries, J. P. Morrow, N. L. Fletcher, C. A. Bell, K. J. Thurecht and K. Kempe, Poly(2-oxazoline) macromonomers as building blocks for functional and biocompatible polymer architectures, Eur. Polym. J., 2019, 121, 109258–109299, DOI:10.1016/j.eurpolymj.2019.109258.
- M. Sahn, C. Weber and U. S. Schubert, Poly(2-oxazoline)-Containing Triblock Copolymers: Synthesis and Applications, Polym. Rev., 2019, 59(2), 240–279, DOI:10.1080/15583724.2018.1496930.
- D. Babuka, K. Kolouchova, L. Loukotova, O. Sedlacek, O. Groborz, A. Skarkova, A. Zhigunov, E. Pavlova, R. Hoogenboom, M. Hruby and P. Stepanek, Self-Assembly, Drug Encapsulation, and, Cellular Uptake of Block and Gradient Copolymers of 2-Methyl-2-oxazine and 2-n-Propyl/butyl-2-oxazoline, Macromolecules, 2021, 54(23), 10667–10681, DOI:10.1021/acs.macromol.1c01794.
- Ł. Otulakowski, M. Kasprów, A. Strzelecka, A. Dworak and B. Trzebicka, Thermal Behaviour of Common Thermoresponsive Polymers in Phosphate Buffer and in Its Salt Solutions, Polymers, 2021, 13, 90–104, DOI:10.3390/polym13010090.
- M. Hruby, S. K. Filippov, J. Panek, M. Novakova, H. Mackova, J. Kucka, D. Vetvicka and K. Ulbrich, Polyoxazoline thermoresponsive micelles as radionuclide delivery systems, Macromol. Biosci., 2010, 10(8), 916–924, DOI:10.1002/mabi.201000034.
- R. Chapman, P. J. M. Bouten, R. Hoogenboom, K. A. Jolliffe and S. Perrier, Thermoresponsive cyclic peptide – poly(2-ethyl-2-oxazoline) conjugate nanotubes, Chem. Commun., 2013, 49, 6522–6524, 10.1039/C3CC42327A.
- Y. Jung, J.-H. Kim and W.-D. Jang, Linear and cyclic poly(2-isopropyl-2-oxazoline)s for fine control of thermoresponsiveness, Eur. Polym. J., 2017, 88, 605–612, DOI:10.1016/j.eurpolymj.2016.09.003.
- A. Amirova, S. Rodchenko, Z. Makhmudova, G. Cherkaev, S. Milenin, E. Tatarinova, M. Kurlykin, A. Tenkovtsev and A. Filippov, Synthesis, Characterization, and Investigation of Thermosensitive Star-Shaped Poly(2-isopropyl-2-oxazolines) Based on Carbosilane Dendrimers, Macromol. Chem. Phys., 2017, 218, 1600387–1600398, DOI:10.1002/macp.201600387.
- Y. Chu, H. Li, H. Huang, H. Zhou, Y. Chen, B. Andreas, L. Liu and Y. Chen, Uni-molecular nanoparticles of poly(2-oxazoline) showing tunable thermoresponsive behaviors, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 174–183, DOI:10.1002/pola.28889.
- T. Sezonenko, X.-P. Qiu, F. M. Winnik and T. Sato, Dehydration, Micellization, and Phase Separation of Thermosensitive Polyoxazoline Star Block Copolymers in Aqueous Solution, Macromolecules, 2019, 52(3), 935–944, DOI:10.1021/acs.macromol.8b02528.
- M. N. Leiske, Poly(2-oxazoline)-derived star-shaped polymers as potential materials for biomedical applications: A review, Eur. Polym. J., 2023, 185, 111832, DOI:10.1016/j.eurpolymj.2023.111832.
- J. Zhang, B. Farias-Mancilla, M. Destarac, U. S. Schubert, D. J. Keddie, C. Guerrero-Sanchez and S. Harrisson, Asymmetric Copolymers: Synthesis, Properties, and Applications of Gradient and Other Partially Segregated Copolymers, Macromol. Rapid Commun., 2018, 1800357–1800379, DOI:10.1002/marc.201800357.
- R. J. R. Peters, I. Louzao and J. C. M. van Hest, From polymeric nanoreactors to artificial organelles, Chem. Sci., 2012, 3, 335–343, 10.1039/c2sc00803c.
- G. Delaittre, Telechelic poly(2-oxazoline)s, Eur. Polym. J., 2019, 121, 109281–109306, DOI:10.1016/j.eurpolymj.2019.109281.
- L. Korchia, C. Bouilhac, J.-J. Robin and V. Lapinte, Amphiphilic photo-induced triblock polyoxazoline through coumarin dimerization: Efficient synthetic tool for nanoparticles, Eur. Polym. J., 2017, 88, 636–644, DOI:10.1016/j.eurpolymj.2016.09.024.
- L. Korchia, C. Bouilhac, V. Lapinte, C. Travelet, R. Borsali and J.-J. Robin, Photodimerization as an alternative to photocrosslinking of nanoparticles : proof of concept with amphiphilic linear polyoxazoline bearing coumarin unit, Polym. Chem., 2015, 6, 6029–6040, 10.1039/c5py00834d.
- L. Korchia, C. Bouilhac, A. Aubert, J.-J. Robin and V. Lapinte, Light-switchable nanoparticles based on amphiphilic diblock, triblock and heterograft polyoxazoline, RSC Adv., 2017, 7, 42690–42699, 10.1039/c7ra07094b.
- L. Korchia, V. Lapinte, C. Travelet, R. Borsali, J.-J. Robin and C. Bouilhac, UV-responsive amphiphilic graft copolymers based on coumarin and polyoxazoline, Soft Matter, 2017, 13, 4507–4520, 10.1039/c7sm00682a.
- A. Oudin, J. Chauvin, L. Gibot, M.-P. Rols, S. Balor, D. Goudounèche, B. Payré, B. Lonetti, P. Vicendo, A.-F. Mingotaud and V. Lapinte, Amphiphilic polymers based on polyoxazoline as relevant nanovectors for photodynamic therapy, J. Mater. Chem. B, 2019, 7, 4973–4983, 10.1039/c9tb00118b.
- R. Obeid, E. Maltseva, A. F. Thunemann, F. Tanaka and F. M. Winnik, Temperature response of self-assembled micelles of telechelic hydrophobically modified poly(2-alkyl-2-oxazoline)s in water, Macromolecules, 2009, 42, 2204–2214, DOI:10.1021/ma802592f.
- N. Xue, X. Hou, X.-P. Qiu, X. Song, Q. Feng and X. Liu, Synthesis and solution properties of telechelic poly(2-isopropyl-2-oxazoline) bearing perfluoro end groups, Polym. Chem., 2023, 14, 37–46, 10.1039/d2py00815g.
- D. Daubian, J. Gaitzsch and W. Meier, Synthesis and complex self-assembly of amphiphilic block copolymers with a branched hydrophobic poly(2-oxazoline) into multicompartment micelles, pseudo-vesicles and yolk/shell nanoparticles, Polym. Chem., 2020, 11, 1237–1249, 10.1039/c9py01559k.
- C. Turan, I. Terzioglu, D. Gundogdu and I. Erel-Goktepe, Synthesis of poly(2-isopropyl-2-oxazoline)-b-poly(2-phenyl-2-oxazoline)-b-poly(2-isopropyl-2-oxazoline) and its self-assembly into polymersomes: Temperature-dependent aqueous solution behavior, Mater. Today Commun., 2023, 25, 106094–106110, DOI:10.1016/j.mtcomm.2023.106094.
- A. Zahoranová and R. Luxenhofer, Poly(2-oxazoline)- and Poly(2-oxazine)-Based Self-Assemblies, Polyplexes, and Drug Nanoformulations—An Update, Adv. Healthcare Mater., 2021, 10, 2001382–2001409, DOI:10.1002/adhm.202001382.
- T. Kirila, A. Smirnova and M. Kurlykin, et al., Self-organization in aqueous solutions of thermosensitive star-shaped and linear gradient copolymers of 2-ethyl-2-oxazoline and 2-isopropyl-2-oxazoline, Colloid Polym. Sci., 2020, 298, 535–546, DOI:10.1007/s00396-020-04638-z.
- A. I. Amirova, O. V. Golub and T. U. Kirila, et al., The effect of arm number and solution concentration on phase separation of thermosensitive poly(2-isopropyl-2-oxazoline) stars in aqueous solutions, Colloid Polym. Sci., 2016, 294, 947–956, DOI:10.1007/s00396-016-3853-3.
- T. Y. Kirila, M. P. Kurlykin and A. V. Tenkovtsev, et al., Behavior of a Thermosensitive Star-Shaped Polymer with Polyethyloxazoline-block-Polyisopropyloxazoline Copolymer Arms, Polym. Sci., Ser. A, 2018, 60, 249–259, DOI:10.1134/S0965545X18030069.
- J. Lefley, R. Terracciano, Z. Varanaraja, J. Beament and C. R. Becer, Self-Assembly Behavior of Amphiphilic Poly(2-ethyl-2-oxazoline)-b-poly(2-isostearyl-2-oxazoline) Block Copolymers, Macromolecules, 2024, 57(12), 5881–5891, DOI:10.1021/acs.macromol.4c00833.
- C. Krumm, C. P. Fik, M. Meuris, G. J. Dropalla, H. Geltenpoth, A. Sickmann and J. C. Tiller, Well-defined amphiphilic poly(2-oxazoline) ABA-triblock copolymers and their aggregation behaviour in aqueous solution, Macromol. Rapid Commun., 2012, 33, 1677–1682, DOI:10.1002/marc.201200192.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, Photoreaction of a hydroxyalkyphenone with the membrane of polymersomes: a versatile method to generate semipermeable nanoreactors, J. Am. Chem. Soc., 2013, 135(24), 9204–9212, DOI:10.1021/ja404175x.
- A. Belluati, V. Mikhalevich, S. Y. Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, How do the properties of amphiphilic polymer membranes influence the functional insertion of peptide pores?, Biomacromolecules, 2020, 21(2), 701–715, DOI:10.1021/acs.biomac.9b01416.
- Y. Bakkour, V. Darcos, S. Li and J. Coudane, Diffusion ordered spectroscopy (DOSY) as a powerful tool for amphiphilic block copolymer characterization and for critical micelle concentration (CMC) determination, Polym. Chem., 2012, 3, 2006–2010, 10.1039/C2PY20054F.
- C. Bouilhac, C. Travelet, A. Graillot, S. Monge, R. Borsali and J.-J. Robin, Synthesis of fatty phosphonic acid based polymethacrylamide by RAFT polymerization and self-assembly in solution, Polym. Chem., 2014, 5, 2756–2767, 10.1039/C3PY01512B.
- L. Plet, G. Delecourt, M. Hanafi, N. Pantoustier, G. Pembouong, P. Midoux, V. Bennevault and P. Guégan, Controlled star poly(2-oxazoline)s: Synthesis, characterization, Eur. Polym. J., 2020, 122, 109323–109335, DOI:10.1016/j.eurpolymj.2019.109323.
- B. Brissault, A. Kichler, C. Leborgne, N. Jarroux, H. Cheradame and C. Guis, Amphiphilic Poly[(propylene glycol)-block,-(2-methyl-2-oxazoline)] Copolymers for Gene Transfer in Skeletal Muscle, ChemMedChem, 2007, 2, 1202–1207, DOI:10.1002/cmdc.200700068.
- M. Leitgeb, Z. Knez and M. Primožič, Sustainable technologies for liposome preparation, J. Supercrit. Fluids, 2020, 165, 104984–105001, DOI:10.1016/j.supflu.2020.104984.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00564c |
| This journal is © The Royal Society of Chemistry 2024 |