
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2-based polycarbonates from biobased cyclic terpenes with end-of-life usage potential†
Philipp
Holzmüller
a,
Jasmin
Preis
b and
Holger
Frey
 *a
*a
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de
bPSS Polymer Standards Service GmbH, In der Dalheimer Wiese 5, 55120 Mainz, Germany
First published on 27th August 2024
Abstract
Biobased polymers have garnered increasing attention in recent years, aiming at more sustainable materials. This study focuses on the synthesis of polycarbonates sourced from cyclic terpenoid-based monomers and CO2, representing polymers derived from a biobased feedstock. Menthyl, thymyl, and carvacryl glycidyl ethers, synthesized from menthol, thymol, and carvacrol and epichlorohydrin were copolymerized with CO2 using catalytic systems such as (R,R)-(salcy)-Co(III)Cl (Co(Salen)Cl) and bis(triphenylphosphine)-iminium chloride ([PPN]Cl) or triethylborane (TEB)/[PPN]Cl. Moderate to high molar mass polymers (up to 60 kg mol−1) were obtained with low dispersities (Mw/Mn < 1.24) via solvent-free bulk copolymerization. Despite the sterically demanding nature of these monomers, the cobalt-based catalyst system exhibited high monomer conversion, polymer selectivity, and carbonate linkage content. The resulting polycarbonates exhibited glass transition temperatures (Tg) ranging from 41 to 58 °C, when the polymer backbone consisted solely of polycarbonate linkages. However, with decreasing polycarbonate linkage content, the Tg value dropped to 0 °C for the menthol based polycarbonate. The aromatic side chain polycarbonates displayed not only the highest Tg values, but also the highest thermal stability, with T5% reaching 260 °C. The thymol-based polycarbonate exhibited a Young's modulus (E) of 645 ± 43 MPa and an elongation at break (ε) of 5 ± 2%, as determined by tensile testing. All three biobased polymers underwent complete degradation under strong basic conditions (5 M KOH) within 30 hours, yielding their respective diols and CO2, thus offering potential for end-of-life usage. CO2 generated by thermal decomposition can be recycled for copolymerization, while the diols could find application for other purposes.
Introduction
The United Nations (UN) launched the Sustainable Development Goals (SDGs) initiative in 2015, aiming at universal guidelines for achieving fair and responsible development while preserving the well-being of both humanity and ecosystems.1 This initiative comprises a targeted agenda to be achieved by 2030, consisting of 17 goals and 169 targets focused on promoting economic growth, environmental conservation, social equity, and human welfare.2 Biobased polymers emerge as significant contributors to achieving the SDGs, particularly objectives related to “Responsible Consumption and Production”, “Clean Water and Sanitation”, and “Life on Land”.3 Established polycarbonates, derived from bisphenol A (BPA), are widely used as thermoplastic polymers renowned for their toughness, impact resistance, and optical clarity, making them prevalent in medical, optical, and electronic applications.4 However, polycarbonates based on BPA and phosgene pose toxicity risks due to the release of hazardous volatile compounds, particularly when utilized in food containers.5 An alternative approach to produce more sustainable polycarbonates is the utilization of CO2 and epoxides, employing ring opening copolymerization (ROCOP).6,7 For instance, poly(propylene carbonate) (PPC), obtained by copolymerizing CO2 and propylene oxide (PO), represents a polycarbonate with high CO2 content and is produced on industrial-scale.8 Additionally, this polycarbonate route offers the possibility to use the greenhouse gas CO2 as C1 building block, contributing to carbon capture and usage.9 However, the synthesis of PO and therefore also PPC still relies on fossil resources. Consequently, there is a growing interest in developing nature-based polycarbonates relying on biobased epoxides and CO2, offering a more sustainable alternative to achieve the SDGs.Various natural sources such as phenylpropanoids,10 sugars,11 castor oil,12 fatty acids,13 and notably terpenes14 have been utilized for CO2 based polycarbonate synthesis. One prominent example is poly(limonene oxide) carbonate (PLimC), derived from limonene oxide (LO) and CO2. Several authors have suggested scalability of PLimC production and its suitability for applications like gas separation membranes due to its permeability and film-forming properties.15–17 Moreover, PLimC can be post-modified to achieve and tailor a variety of polymer properties, including antibacterial, sea water solubility, or rubber-like properties.17 However, a limitation arises as only a few catalysts, primarily zinc-based compounds, have shown efficacy in polymerizing limonene oxide. Consequently, other structurally demanding terpene-based polycarbonates have been explored, utilizing monomers such as menth-2-ene oxide18 and pinene oxide.19
These terpene-based monomers face challenges in terms of specific catalysts requirements and epoxide synthesis, often requiring multistep syntheses. For instance, in the case of limonene oxide (LO) synthesis for ROCOP, selective trans-LO synthesis is usually necessary. Recently, long-chain terpene-based polycarbonates derived from citronellyl glycidyl ether20 and geranyl glycidyl ether21 have been published by our group. The monomers were synthesized in one-step reactions from the respective alcohols and epichlorohydrin (ECH) and require simple purification via distillation. Traditionally, ECH is produced through a multi-step process involving propylene and chlorine, which is not considered highly sustainable due to the use of chlorine.22,23 However, ECH can also be derived from triacylglycerol via the Epicerol process.23–25 The bio-sourcing of ECH from glycerol is particularly sustainable when glycerol is obtained as a byproduct from biodiesel production.26 While this synthesis route for ECH is more sustainable, ECH itself must still be handled with care and within closed environments.
However, these long-chain terpene-based polymers exhibit Tg values below −29 °C, making them unsuitable for usage as potential thermoplastic materials like PLimC. Nonetheless, the class of terpenes offers a wide range of structures, comprising more than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 variants with linear, cyclic, or multicyclic configurations.27 From a polymer point of view, cyclic terpenes are of particular interest as their molecular structure is rather rigid. This is expected to result in polymers with potentially enhanced glass temperatures, rendering them suitable for applications as engineering plastics.28 Some of the simplest cyclic terpenoid structures suitable for monomer synthesis with ECH include carvacrol, thymol, and menthol. They can be extracted from oregano,29 thyme,30 and mint,31 respectively, but can also be produced in bioreactors.32 These terpenoids find extensive application in the pharmaceutical industry, particularly due to their antioxidant, antiseptic, antibacterial, antifungal, and antimicrobial properties.33 In polymer chemistry, carvacrol, thymol, and menthol have been utilized especially for polymer blends in which these terpenoids enhanced the bioactivity/bioresistance of the polymers or the polymers acted as host material to slowly release these terpenoids.34 However, carvacrol, thymol, and menthol derivatives have also been polymerized to polyamides,35 polyesters,36 polyacrylates,37 polybenzoxazines,38 polyphenols,39 with menthol being the only one used for polycarbonate synthesis recently.18
000 variants with linear, cyclic, or multicyclic configurations.27 From a polymer point of view, cyclic terpenes are of particular interest as their molecular structure is rather rigid. This is expected to result in polymers with potentially enhanced glass temperatures, rendering them suitable for applications as engineering plastics.28 Some of the simplest cyclic terpenoid structures suitable for monomer synthesis with ECH include carvacrol, thymol, and menthol. They can be extracted from oregano,29 thyme,30 and mint,31 respectively, but can also be produced in bioreactors.32 These terpenoids find extensive application in the pharmaceutical industry, particularly due to their antioxidant, antiseptic, antibacterial, antifungal, and antimicrobial properties.33 In polymer chemistry, carvacrol, thymol, and menthol have been utilized especially for polymer blends in which these terpenoids enhanced the bioactivity/bioresistance of the polymers or the polymers acted as host material to slowly release these terpenoids.34 However, carvacrol, thymol, and menthol derivatives have also been polymerized to polyamides,35 polyesters,36 polyacrylates,37 polybenzoxazines,38 polyphenols,39 with menthol being the only one used for polycarbonate synthesis recently.18
Another issue to consider when discussing biobased polycarbonates in the context of fulfilling the SDGs is their end-of-life usage. In recent years, there has been increasing attention given to the degradation and recycling of polymers to enable a circular economy.40 Various strategies have been explored in the field of CO2-based polycarbonates to achieve circularity.41 For instance, polycarbonates can undergo solvolysis under acidic or basic conditions yielding in the corresponding diols and CO2.42 In such cases, the toxicity and potential reuse options of the resulting molecules need to be carefully evaluated. Additionally, CO2-based polycarbonates can be degraded to their respective cyclic carbonates via catalysis,43 which can then be repurposed for applications such as green solvents or electrolytes in batteries.44 Chemical recycling to monomers (CRM) for CO2-based polycarbonates has also garnered increasing attention in recent years due to its potential for full circularity.45,46 Thereby, the focus is on the straightforward and efficient recovery of the respective epoxide and CO2 using a catalytic system.47 For terpene-based polycarbonates, only studies for poly(limonene oxide) carbonate (PLimC) have been conducted, aiming at achieving CRM and demonstrating degradability in anaerobic environments.48
In this study, we present the copolymerization of different biobased cyclic terpenyl glycidyl ethers with CO2 to produce polycarbonates. Our main focus was on extending the platform of terpene-based polycarbonates. We successfully synthesized a range of copolymers based on carvacryl glycidyl ether (CarGE), thymyl glycidyl ether (ThyGE) and menthyl glycidyl ether (MeGE) (see Scheme 1). Different parameters like catalyst loading, catalytic system, solvents and chain transfer agents were varied to investigate the effect on the copolymerization with CO2. The resulting polymers were analysed regarding their material properties like glass temperature, thermal stability and mechanical performance. We also investigated polymer degradation under basic conditions and the possible usage of the degradation products.
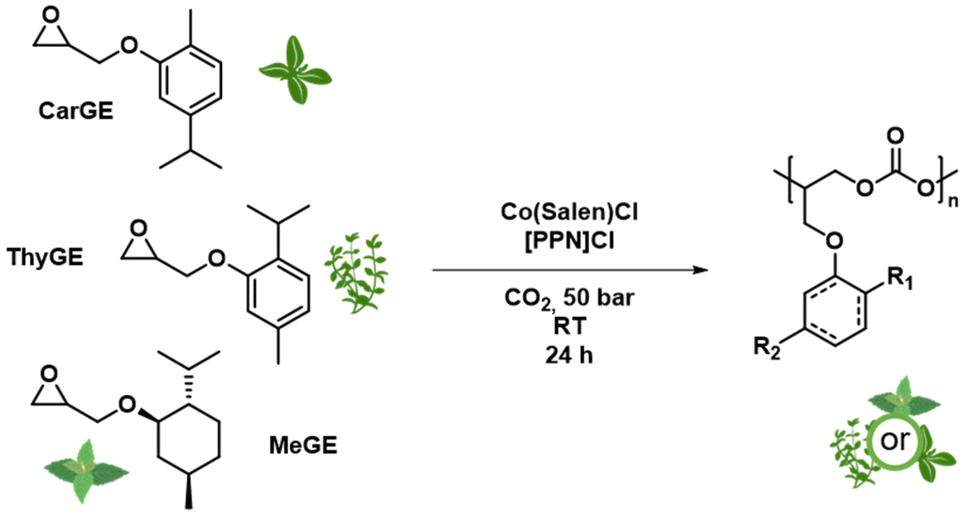 | ||
| Scheme 1 Synthesis of cyclic terpenoid-based polycarbonates. | ||
Results and discussion
Monomer synthesis
The terpenoid-based epoxides investigated in this study were prepared using a Williamson ether synthesis approach. This method involves reaction of the respective terpenoid alcohol with ECH. We adapted a method known for similar epoxide structures to achieve high yields of up to 89% of the phenyl-based terpenoids.49 The menthol-based epoxide, synthesized according to patent literature, was obtained in yields up to 74%.50To the best of our knowledge, none of the resulting epoxides – CarGE, ThyGE, or MeGE – has been previously utilized in any polymerization process. Demonstrating scalability, we synthesized all epoxide monomers on a 50 g scale and efficiently purified them via vacuum distillation (see ESI† for detailed methods). ECH can be obtained from triacylglycerol via the Epicerol process (see ESI Schemes S1 and S2†).23–25 To ensure the absence of protic impurities in the monomer, all epoxides were treated with iodomethane and sodium hydride, achieving quantitative yields. This method not only prevents catalyst deactivation, but also minimizes additional polymer initiation by alcohols, ultimately enhancing the polymers’ molar masses and dispersities.15 The successful synthesis of all terpenoid-based epoxides was verified by 1H and 13C NMR spectroscopy (see ESI Fig. S1–S6†). The 1H NMR spectrum of CarGE (Fig. 1) illustrates the characteristic epoxide signals highlighted in blue. Comparison of the 1H NMR peaks of the bulky side chain (in green) reveals a marginal variation among the alcohol, monomer, and polymer. However, specific peaks for epoxide and polycarbonate can be identified.
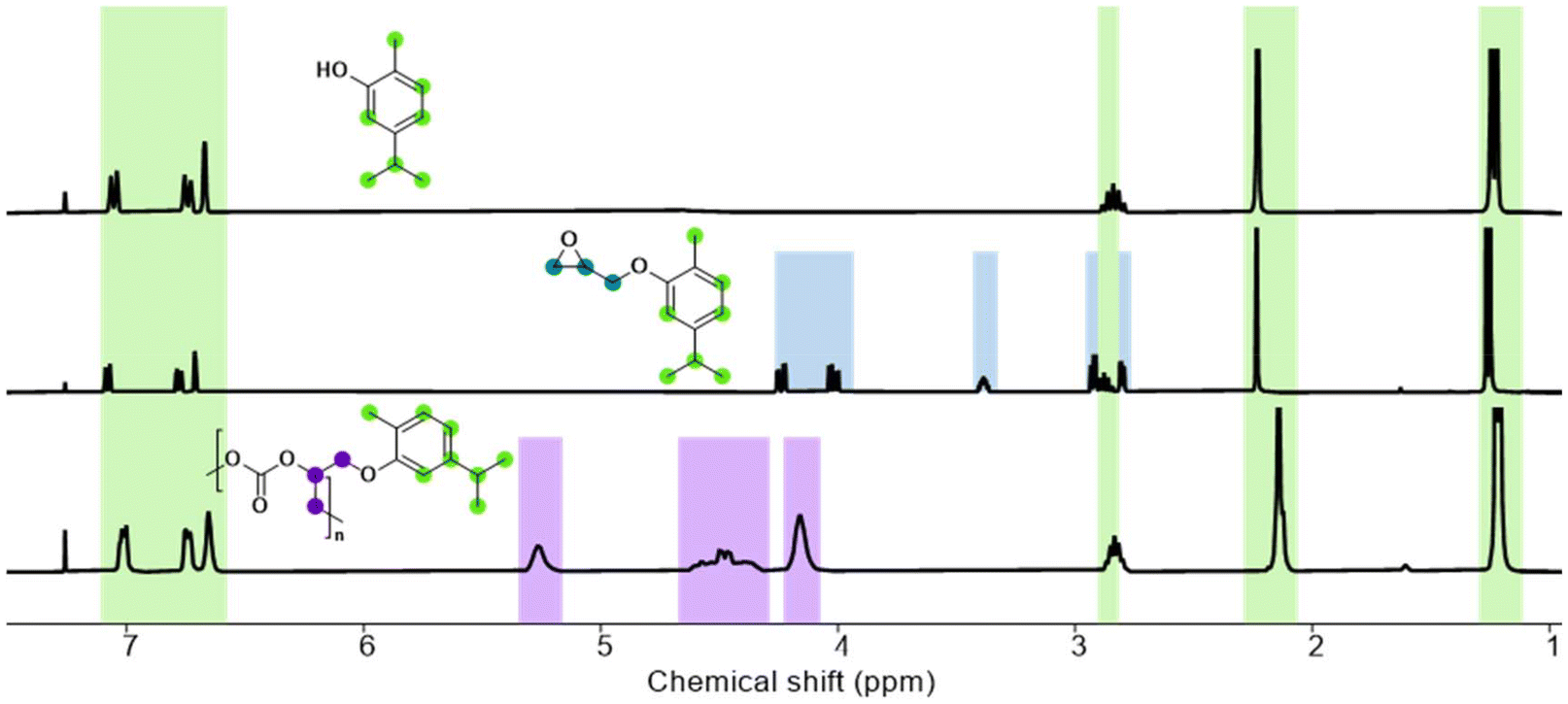 | ||
| Fig. 1 1H NMR spectra of carvacrol, CarGE and PCarGEC, measured in CDCl3. | ||
Copolymerization of CarGE with CO2
CarGE was chosen for in-depth copolymerization analysis due to a less sterically demanding structure compared to the other monomers. CarGE and CO2 were copolymerized in bulk using Co(Salen)Cl and [PPN]Cl as catalysts, employing 50 bar at room temperature for 24 hours, see Table 1 (detailed procedure in the ESI†). The catalytic system was chosen because of its known high tolerance for sterically demanding monomers like cyclohexene oxide and other glycidyl ethers.51 50 bar CO2 pressure was chosen as standard operating pressure because this binary catalyst demonstrates best performance under high CO2 pressure.10 The copolymerization was stopped by releasing the excess CO2, and the success of the copolymerization was primarily assessed via1H NMR spectra determining the selectivity towards polymer formation (see ESI Fig. S20†). In the case of CarGE copolymerization, small traces of cyclic carbonates were identified, indicating a selectivity towards the formation of polycarbonate despite its demanding side chain compared to monomers like propylene oxide (PO). The noticeable side chain structure does not facilitate backbiting of the chain end, resulting in no observable differences compared to PO.52 The polymers were purified by precipitation in methanol, reaching yields up to 86%. Verification of the successful polycarbonate synthesis was achieved by 1H NMR and 13C NMR characterization of the isolated polymer, in which the typical proton signals of the CO2/epoxide based polycarbonate backbone were identified at 5.20 and 4.50 ppm (see ESI Fig. S7 and S8†). Furthermore the selectivity towards polycarbonate linkages was investigated via1H NMR characterization to ensure a low content of polyether defects. The Co(Salen)Cl catalyst showed high selectivity for polycarbonate linkages, consistently yielding polycarbonate linkages exceeding 99%. Optimization of the monomer:initiator:catalyst ratio was pivotal in achieving high molar masses of the resulting PCarGEC while minimizing the catalyst loading. Entries 1–7 in Table 1 illustrate that the highest Mn can be achieved with a monomer:initiator:catalyst ratio of 2000:2:2, resulting in a Mn of 59.5 kg mol−1 with a dispersity (Đ) of 1.15, determined via size exclusion chromatography with THF as an eluent and polystyrene standards (SEC, THF, PS). The ratio of catalyst to co-catalyst (initiator) was always maintained at 1:1, based on investigations for this system that revealed this ratio performs best.53 Despite efforts to reduce the water content in the reaction mixture (use of small reactor volumes and CO2 storage over molecular sieve), bimodality in SEC curves persisted due to residual water traces in CO2 (see Fig. 2). Furthermore, with lower initiator concentrations this bimodality was more dominant, which is due to increasing water/initiator ratios. Similar results were obtained for the recently published long chain terpenoid based polycarbonates by our group.21 In general, the molar mass can be adjusted by varying the ratio of monomer, catalyst and initiator. Moreover, the addition of chain transfer agents (CTAs), e.g. 1,4-benzenedimethanol (BDM), to the reaction mixture allows to reach lower molar masses without higher catalyst loadings (see Table 1, entry 8). This is a common strategy for fine-tuning molar masses in polycarbonate synthesis.7,54 Herein, BDM, acting as a bifunctional initiator, induces a shift in the SEC curve, transitioning from a monofunctional-dominated bimodal curve to a strongly bimodal curve (see ESI Fig. S28†). SEC (THF, PS) was chosen as the standard analytical method for the polycarbonates, commonly used by most polycarbonate research groups. However, due to the significant structural differences between the CarGE based polycarbonate structure and the polystyrene standards used for SEC calibration, additional absolute molar mass determination was conducted. This was achieved by SEC with universal calibration in combination with intrinsic viscosity measurements as a well-known method.55 Table S4† shows a discrepancy between the two absolute and relative methods, ranging between a 12–18% deviation, where the relative method consistently underestimates the molar mass. Therefore, the absolute molar mass for all CarGE-based polycarbonates is assumed to be higher than determined via SEC (THF, PS). When the catalyst loading is insufficient, both conversion and selectivity decrease within the 24 hour reaction time, resulting in lower molar masses as evidenced in Table 1, entry 7. For consistent comparison between various copolymerizations and their catalyst loadings, a fixed 24 hour time slot was consistently set. However, when reducing the reaction time to 6 hours for the monomer:initiator:catalyst ratio of 2000:2:2, 70% conversion was already achieved (see ESI Table S3†). Toluene was added as a solvent to the reaction mixture (see Table 1, entry 9) with the intention of improving bimodality/dispersity, as demonstrated in the literature.18 After opening the reactor, the reaction mixture was slightly viscous but not solid, unlike the case without toluene. Comparing entries 4 and 9 in Table 1, no improvement in dispersity was observed, and molar mass, conversion, and polycarbonate linkages remained stable.
 | ||
| Fig. 2 SEC traces of PCarGEC presented in Table 1 with different catalyst loading ratios (THF, PS, RI detector). Mn of the respective polymers is shown in the legend. | ||
| Entry | Catalyst | [m]0:[i]0:[cat]0a |
Conv.b (%) | Select.c (%) | PCd (%) | M n (Đ) (kg mol−1) | T g (°C) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: Co(Salen)Cl or TEB as a catalyst and [PPN]Cl as an initiator, monomer (1 mL), 50 bar CO2, r.t. (reaction with TEB at 60 °C), 24 hours.a [m]0 = monomer equivalents, [i]0 = initiator equivalents, [cat]0 = catalyst equivalents.b Determined via1H NMR spectroscopy from the non-purified reaction mixture after opening the reactor; conv. = epoxide conversion determined via comparison of the relative integrals in the 1H NMR spectrum of PC, CC, PE, and monomer.c Determined via1H NMR spectrum from the non-purified reaction mixture after opening the reactor; select. = polymer selectivity determined via comparison of the relative integrals in the 1H NMR spectrum for PC against CC.d Determined via comparison of the relative integrals in the 1H NMR spectrum for PC against PE.e Determined via SEC (THF, PS) and RI detector.f BDM was added as a CTA. The total concentration of [PPN]Cl and BDM is indicated as [i]0. [PPN]Cl and Co(Salen)Cl have the same ratio.g 0.3 mL toluene was added as solvent to the reaction mixture. | |||||||
| 1 | Co(Salen)Cl | 2000:8:8 |
100 | 92 | >99 | 20.5 (1.13) | 48 |
| 2 | Co(Salen)Cl | 2000:5:5 |
100 | 93 | >99 | 35.8 (1.12) | 48 |
| 3 | Co(Salen)Cl | 2000:4:4 |
100 | 94 | >99 | 43.8 (1.10) | 48 |
| 4 | Co(Salen)Cl | 2000:3:3 |
99 | 93 | >99 | 50.0 (1.13) | 49 |
| 5 | Co(Salen)Cl | 2000:2.5:2.5 |
98 | 93 | >99 | 53.1 (1.13) | 49 |
| 6 | Co(Salen)Cl | 2000:2:2 |
97 | 94 | >99 | 59.5 (1.15) | 49 |
| 7 | Co(Salen)Cl | 2000:1:1 |
38 | 86 | >99 | 33.1 (1.15) | 49 |
| 8 | Co(Salen)Clf | 2000:14:4 |
100 | 81 | >99 | 20.5 (1.12) | 47 |
| 9 | Co(Salen)Clg | 2000:3:3 |
96 | 92 | >99 | 51.0 (1.12) | 48 |
| 10 | TEB | 2000:2:4 |
35 | 0 | — | — | — |
| 11 | TEB | 2000:2:8 |
26 | 0 | — | — | — |
| 12 | TEB | 2000:2:16 |
62 | 22 | 80 | 11.9 (1.25) | 37 |
| 13 | TEB | 2000:2:24 |
94 | 78 | 97 | 38.9 (1.13) | 47 |
Independent of the chosen reaction conditions, the 13C NMR spectra exhibit three peaks for the carbonate carbon, with one peak being notably dominant (see ESI Fig. S19†). This dominance indicates the prevalent formation of head-to-tail linkages, a characteristic behaviour of the Co(Salen)Cl/[PPN]Cl catalyst system.56 To confirm the predominance of polycarbonate linkages in PCarGEC, MALDI-ToF characterization was employed, where the repeating unit matches the sum of epoxide units and CO2 (see ESI Fig. S22†). All PCarGEC polymers show a glass temperature (Tg) between 47 and 49 °C, with a slightly higher Tg with higher molar masses. The Co(Salen)Cl/[PPN]Cl catalytic system poses challenges due to cobalt's environmental impact due to its mining.57 In search of sustainable alternatives, attention has shifted to the metal-free catalyst triethylborane (TEB) in combination with the initiator [PPN]Cl.58 In initial trials using this system to prepare PCarGEC, the epoxide:[PPN]Cl:TEB ratio was found to be pivotal for selective polycarbonate copolymerization. Entries 10–13 in Table 1 illustrate the copolymerization of CarGE with CO2 by TEB catalysis, underscoring the critical role of selecting appropriate epoxide:[PPN]Cl:TEB ratios. The importance of these ratios was already reported in literature for the copolymerization of phenyl glycidyl ether with CO2 to achieve the respective polycarbonate.59 Here, insufficient TEB loading in the reaction mixture results in very low conversion and the formation of cyclic carbonates. With an increased TEB amount (Table 1, entry 12), conversion improves, but selectivity and content of polycarbonate linkages remain low. Only with a substantial TEB surplus, almost full conversion is achieved. Good selectivity, and high content of polycarbonate linkages are obtained, although not as high as with the cobalt-based system. Comparing the PCarGEC results with the copolymerization results of phenyl glycidyl ether and CO2, the more demanding structure of CarGE shows no worsening influence on polymerization selectivity.59 A low ether linkage content in the polymer already affects the Tg by decreasing it, as evidenced in Table 1, entry 13. Higher content of ether linkages increases this effect (Table 1, entry 12). A reaction temperature of 60 °C was necessary for all TEB catalysed copolymerizations, while lower temperatures did not induce any reaction. Regardless of the catalytic system and Mn of the presented polymers, all PCarGECs exhibited long-term stability at room temperature or in the refrigerator without any autonomous post-polymerization modifications or degradation.
Copolymerization of ThyGE and MeGE with CO2
We selected the thymol-based glycidyl ether ThyGE, a constitutional isomer of CarGE, for copolymerization to investigate the impact of the methyl/isopropyl group on the phenyl ring (Scheme 1). Additionally, we investigated the menthol-based glycidyl ether MeGE to see the effect of the phenyl ring in comparison to the cyclohexanyl ring. Furthermore, Wambach et al. recently reported the menthol-based polycarbonate poly(menth-2-ene carbonate) (PMen2C) as a terpene-based polymer.18The herein presented menthol-based glycidyl ether induces higher chain flexibility compared to menth-2-ene oxide. In Table 2, the copolymerization of ThyGE or MeGE with CO2 leads to results comparable to CarGE when using Co(Salen)Cl as a catalyst: mostly full conversion, high polymer selectivity, and high content of polycarbonate linkages. Mn up to 60 kg mol−1 with moderate dispersities was achieved. Similar to PCarGEC, these polymers display bimodality due to traces of water acting as initiator (see ESI Fig. S29†). Notably, PThyGEC and PMeGEC also showed an underestimation (13–18%) of the molar mass via SEC (THF, PS) (see ESI Table S4†). Both polycarbonate structures are confirmed by 1H and 13C NMR spectroscopy (see ESI Fig. S9–S12†) and MALDI-ToF characterization (see ESI Fig. S23 and S24†). After 6 hours of copolymerization, ThyGE and MeGE reached monomer conversions of 81% and 89%, respectively (see ESI Table S3†). The addition of toluene as a solvent to the reaction mixture does also not improve dispersities (see ESI Tables S1 and S2†). Mn for both polymers can also be tuned by the addition of BDM (see ESI Tables S1 and S2†). The 13C NMR indicates a slightly higher head-to-tail selectivity in both cases compared to PCarGEC (see ESI Fig. S19†). PThyGEC shows a Tg of up to 58 °C, while the Tg for PMeGEC reaches only 41 °C. MeGE and ThyGE are also copolymerizable with CO2 by TEB catalysis. While the copolymerization of ThyGE shows similar results regarding polymer selectivity and content of carbonate linkages compared to CarGE (see Table S1†), MeGE afforded only polyether structures without any polycarbonate linkages with insufficient TEB loading (see Table S2†). Adjusting the TEB loading enabled an increase in polycarbonate linkages, albeit only up to a maximum of 57%. The content of polycarbonate linkages notably impacts the Tg values in the MeGE based polycarbonate, resulting in a Tg range from 0 to 41 °C with 0% or 100% content of polycarbonate linkages in the final polymer (see ESI Fig. S30†). Overall, the Co(Salen)Cl catalytic system showed superior polymerization performance considering both polymer selectivity and content of polycarbonate linkages compared to the TEB catalytic system for the monomers MeGE and ThyGE.
| Entry | Monomer | [m]0:[i]0:[cat]0a |
Conv.b (%) | Select.c (%) | PCd (%) | M n (Đ) (kg mol−1) | T g (°C) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: Co(Salen)Cl as a catalyst and [PPN]Cl as an initiator, monomer (1 mL), 50 bar CO2, r.t., 24 hours.a [m]0 = monomer equivalents, [i]0 = initiator equivalents, [cat]0 = catalyst equivalents.b Determined via1H NMR spectroscopy from the non-purified reaction mixture after opening the reactor; conv. = epoxide conversion determined via comparison of the relative integrals in the 1H NMR spectrum of PC, CC, PE, and monomer.c Determined via1H NMR spectrum from the non-purified reaction mixture after opening the reactor; select. = polymer selectivity determined via comparison of the relative integrals in the 1H NMR spectrum for PC against CC.d Determined via comparison of the relative integrals in the 1H NMR spectrum for PC against PE.e Determined via SEC (THF, PS) and RI detector. | |||||||
| 1 | ThyGE | 2000:8:8 |
100 | 93 | >99 | 27.9 (1.10) | 55 |
| 2 | ThyGE | 2000:4:4 |
100 | 94 | >99 | 48.0 (1.18) | 57 |
| 3 | ThyGE | 2000:3:3 |
99 | 95 | >99 | 60.0 (1.24) | 58 |
| 4 | MeGE | 2000:8:8 |
100 | 95 | >99 | 21.5 (1.11) | 39 |
| 5 | MeGE | 2000:4:4 |
100 | 98 | >99 | 27.2 (1.11) | 40 |
| 6 | MeGE | 2000:2:2 |
95 | 98 | >99 | 29.6 (1.12) | 41 |
Polymer properties
The three presented terpene-based polycarbonates exhibit similar structures, of which the structures based on thymol and carvacrol are even constitutional isomers. While the copolymerization performance of each monomer is similar, they show significant differences in their polymer properties. When examining similar molar masses, PThyGEC displays the highest Tg at 58 °C, PMeGEC shows the lowest Tg at 41 °C, while PCarGEC ranged in between with 49 °C (see Fig. 3A). One key observation is the impact of π–π stacking on the Tg for PCarGEC and PThyGEC, whereas polycarbonates derived from MeGE possess lower Tg values due to absence of this additional interaction. According to literature, the presence of π–π stacking leads to an increase of the Tg.28 | ||
| Fig. 3 (A) DSC measurements and (B) TGA measurements of the three different terpenoid-based copolymers. | ||
Surprisingly, the Tg difference between PThyGEC and PCarGEC is almost 10 K, despite the only distinction being the position of the methyl/isopropyl group at the phenyl ring. This suggests that PThyGEC might have a more tightly packed chain structure, while PCarGEC's isopropyl chains contribute to plasticizing the polymer structure. The position of the isopropyl/methyl group in PCarGEC and PThyGEC significantly influences the glass transition. Nonetheless, all three polymers exhibit lower Tg values compared to polycarbonates where the cyclic structure is part of the polymer backbone, such as poly(cyclohexene oxide) carbonate (PCHC) (Tg = 118 °C), PLimC (Tg = 130 °C), or PMen2C (Tg = 144 °C).14 The presence of the “glycidyl ether spacer” obviously results in a less rigid polymer structure, leading to lower Tg. This effect is particularly noticeable when comparing PMeGEC with PMen2C, where the Tg difference exceeds 100 °C. In addition, the thermal degradation for each polymer shows differences (see Fig. 3B). While PThyGEC and PCarGEC exhibit nearly identical T5% values at 260 °C, PMeGEC exhibits a lower T5% at 221 °C when comparing similar molar masses. When comparing the T5% values of pure menthol and thymol, thymol shows also a higher T5%.60 In comparison to PLimC (T5% = 229 °C), PCarGEC and PThyGEC exhibit higher thermal stability, but are surpassed by PCHC (T5% = 283 °C), and PMen2C (T5% = 308 °C).18
Tensile testing of PCarGEC, PThyGEC, and PMeGEC samples has also been carried out. All polymers showed promising film-forming properties when solvent casting from chloroform and annealing at 100 °C. However, specific challenges arose for PMeGEC: the film was non-processible at lower temperatures due to high brittleness, while at room temperature, it became sticky (see ESI Fig. S33A†). This characteristic results from its Tg close to room temperature. PCarGEC showed successful film formation, but exhibited significant brittleness when punched into the dog bone shape, causing the film to shutter (see ESI Fig. S33B†). Consequently, conducting tensile testing was not feasible. PThyGEC displayed good film-forming properties (see ESI Fig. S33C†). Upon manual bending, the resulting films exhibited high flexibility (see ESI Fig. S34†). Comparing all three polycarbonates, it becomes evident that not only the Tg value plays a crucial role, but also the side chains.
All PThyGEC tensile tests were conducted from the reuse of the same polymer batch (after tensile testing, dissolving polymer and subsequent film generation), highlighting its reprocessability. The films show no direct failure after reaching their stress maximum displaying some elastic deformation. This phenomenon may stem from the slow flow of polymer chains, influenced by weak chain entanglements and interactions. The films demonstrate a Young's modulus (E) of 645 ± 43 MPa and an elongation at break (ε) of 5 ± 2%, both at ambient temperature. The elongation at break falls within a similar range as that of PCHC.61 Comparing the Young's modulus, PThyGEC falls in a similar range as PPC.61 However, the higher Tg of PThyGEC compared to PPC (Tg = 20–40 °C) is a significant advantage. In contrast to PCHC, PThyGEC exhibits a lower Young's modulus, most probably due to the absence of a cyclic structure in the polymer backbone.61
Polymer degradation
The degradation of CO2-based polycarbonates can result in the formation of cyclic carbonates, epoxides, or diols depending on the chosen conditions.41,46,62 Under different degradation conditions and depending on the monomer type, degradation can occur gradually over several weeks or rapidly within a few hours. Ideally, these degradation products can be utilized or polymerized thereafter or are metabolized by microorganisms in soil. Terpenes find wide application in medicine or for general biomedical purposes, either in their original form or as tailored derivatives for specific purposes.63 For instance, thymol, carvacrol, and menthol have a range of pharmaceutical applications with many different derivatives used.64–66 Hence, our objective for the presented polycarbonates was to obtain degradation products that are applicable for subsequent use. To focus on the degradation products rather than the degradation process itself, we selected harsh conditions to achieve full degradation. All three polymers, PCarGEC (Mn = 20.5 kg mol−1), PThyGEC (Mn = 21.5 kg mol−1), PMeGEC (Mn = 16.7 kg mol−1) were treated with an H2O/THF KOH (5 M) solution for 30 hours at 70 °C (see Scheme 2). All three experiments showed complete degradation of the respective polymer, and no evidence of the polymer was detectable by SEC analysis and 1H NMR characterization. Three different degradation product combinations are theoretically possible: epoxide monomer & CO2, cyclic carbonate, or diol & CO2. However, only the pure diol was observable via1H NMR and 13C NMR characterization (see ESI Fig. S12–S18 and S32†) with all signals assigned and no additional side products. Even if the cyclic carbonate or the epoxide are formed, they would undergo direct conversion into diols due the harsh basic conditions. The pure isolated diols are white solids in the case of CarPD and ThyPD, and a liquid for MePD. | ||
| Scheme 2 Polymer degradation of PCarGEC, PThyGEC and PMeGEC to terpene-based diols. | ||
While the terpene derivative diols have a similar terpene scent compared to their alcohol counterparts, the intensity is notably suppressed. Regarding circularity, although these diols are not reusable as monomers for copolymerization with CO2, they hold promising application possibilities. MePD, commercially known as agent 10, and its derivatives, for instance, exhibit potential use as effective cooling agents, surpassing other methanol combinations.67 Thymol and carvacrol derivatives with a similar structure to ThyPD and CarPD find applications as drug candidates or metabolic enzyme inhibitors,68 while thymol and carvacrol themselves are extensively employed in disease treatment like cancer or cardiometabolic diseases.64,66 Consequently, ThyPD and CarPD might exhibit similar properties or serve as precursors for more complex thymol/carvacrol-based structures. Overall, thermal recycling offers a promising opportunity to extend the lifespan of the presented polymers, avoiding chemical down cycling issues, instead creating low molecular products for alternative secondary applications without the need of any further purification. The bioactivity of the polymers and their degradation products are in focus of future investigations.
Conclusions
Biobased polycarbonates, derived from cyclic terpenoid-based glycidyl ethers and CO2, have been introduced. Utilizing a Co(Salen)Cl/[PPN]Cl catalytic system, all three epoxides based on menthol, carvacrol and thymol, were successfully copolymerized with CO2 within 24 hours at room temperature. We achieved molar masses up to 60 kg mol−1, with low dispersities confirmed by SEC (THF, PS). The molar masses can be controlled by initiator concentration or by the addition of chain transfer agents (CTAs). Notably, the absolute molar mass determined by universal calibration was found to be up to 18% higher due to the systematic underestimation of the side chains in the SEC (THF, PS). Transitioning to a TEB/[PPN]Cl catalytic system, recognized for its metal-free and eco-friendly nature, copolymerization was also possible. In this case, precise optimization of the catalyst ratios was necessary to attain high polymer selectivity and polycarbonate linkages. The properties of the novel polycarbonates are highly influenced by the terpene structure of the glycidyl ether monomer. A Tg range between 41 to 58 °C was achieved, while polymers with the potential for π–π stacking in their side chains exhibited higher Tg values (PCarGEC and PThyGEC). This distinction was also evident in the thermal degradation experiments, as polymers with π-system in their side chain reached T5% of 260 °C. Tensile testing was only successful for PThyGEC samples, demonstrating a high Young's modulus of 645 MPa and low flexibility. Investigating degradation possibilities, all polymers underwent degradation under strong basic conditions, yielding the respective diols and CO2. Considering the polymers’ end-of-life usage, CO2 is reusable in polycarbonate production and the diols may further serve as raw materials in the cosmetic and pharmaceutical industries.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the Max-Planck-Institute for Polymer Research in Mainz and especially Petra Räder for measuring the TGA of all polymers.References
- United Nations, Africa Renewal, 2015, 29, 8 CrossRef.
- M. Stafford-Smith, D. Griggs, O. Gaffney, F. Ullah, B. Reyers, N. Kanie, B. Stigson, P. Shrivastava, M. Leach and D. O'Connell, Sustainability Sci., 2017, 12, 911–919 CrossRef PubMed.
- Polymers, Biopolymer-Based Materials towards the Sustainable Development Goals, available at: https://www.mdpi.com/journal/polymers/special_issues/8RII9XX1S7, accessed 02/2024.
- D. Kyriacos, Brydson's Plastics Materials, Butterworth-Heinemann, Oxford, 8th edn, 2017 Search PubMed.
- J. Maia, J. M. Cruz, R. Sendón, J. Bustos, J. J. Sanchez and P. Paseiro, Food Res. Int., 2009, 42, 1410–1414 CrossRef CAS.
- (a) Y. Liu and X.-B. Lu, Macromolecules, 2023, 56, 1759–1777 CrossRef CAS; (b) P. Wei, G. A. Bhat and D. J. Darensbourg, Angew. Chem., 2023, 135, e202307507 CrossRef; (c) G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC; (d) S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS.
- X. Li, L. Meng, Y. Zhang, Z. Qin, L. Meng, C. Li and M. Liu, Polymers, 2022, 14, 2159 CrossRef CAS PubMed.
- (a) M. M. Halmann and M. Steinberg, Greenhouse gas carbon dioxide mitigation. Science and Technology, Boca Raton, FL, 1999 Search PubMed; (b) T. Ema, Bull. Chem. Soc. Jpn., 2023, 96, 693–701 CrossRef CAS.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, Macromolecules, 2023, 56, 2362–2369 CrossRef CAS.
- D. K. Tran, A. Z. Rashad, D. J. Darensbourg and K. L. Wooley, Polym. Chem., 2021, 12, 5271–5278 RSC.
- J. Yang, J. Dong, Y. Wang, X. Zhang, B. Liu, H. Shi and L. He, Macromolecules, 2021, 54, 8503–8511 CrossRef CAS.
- S. Cui, Y. Qin and Y. Li, ACS Sustainable Chem. Eng., 2017, 5, 9014–9022 CrossRef CAS.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- (a) F. Parrino, A. Fidalgo, L. Palmisano, L. M. Ilharco, M. Pagliaro and R. Ciriminna, ACS Omega, 2018, 3, 4884–4890 CrossRef CAS PubMed; (b) C. Martín and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 CrossRef; (c) L. P. Carrodeguas, T. T. D. Chen, G. L. Gregory, G. S. Sulley and C. K. Williams, Green Chem., 2020, 22, 8298–8307 RSC; (d) C. Li, M. Johansson, P. Buijsen, G. Dijkstra, R. J. Sablong and C. E. Koning, Prog. Org. Coat., 2021, 151, 106073 CrossRef CAS; (e) S. Kernbichl and B. Rieger, Polymer, 2020, 205, 122667 CrossRef CAS; (f) C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed; (g) C. Li, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2015, 67, 449–458 CrossRef CAS; (h) C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 CrossRef CAS PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- A. Wambach, S. Agarwal and A. Greiner, ACS Sustainable Chem. Eng., 2020, 8, 14690–14693 CrossRef CAS.
- (a) Y. Zhi, X. Shan, S. Shan, Q. Jia, Y. Ni and G. Zeng, J. CO2 Util., 2017, 22, 299–306 CrossRef CAS; (b) W. Fan, Q. Jia, J. Mu and S. Shan, China Pat., CN103333329A, 2015 Search PubMed.
- S. Schüttner, C. Gardiner, F. S. Petrov, N. Fotaras, J. Preis, G. Floudas and H. Frey, Macromolecules, 2023, 56, 8247–8259 CrossRef.
- P. Holzmüller, C. Gardiner, J. Preis and H. Frey, Macromolecules, 2024, 57, 5358–5367 CrossRef.
- (a) G. Shukla and R. C. Ferrier, J. Polym. Sci., 2021, 59, 2704–2718 CrossRef CAS; (b) A. Almena and M. Martín, Ind. Eng. Chem. Res., 2016, 55, 3226–3238 CrossRef CAS; (c) K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 3rd edn, 2008 Search PubMed.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean: Soil, Air, Water, 2008, 36, 657–661 CAS.
- P. Krafft, P. Gilbeau and B. Gosselin, US Pat., US9663427B2, 2017 Search PubMed.
- P. Krafft, P. Gilbeau, B. Gosselin and S. Claessens, US Pat., US2009/0270588 AI, 2009 Search PubMed.
- D. Cespi, R. Cucciniello, M. Ricciardi, C. Capacchione, I. Vassura, F. Passarini and A. Proto, Green Chem., 2016, 18, 4559–4570 RSC.
- (a) J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621–1637 CrossRef CAS PubMed; (b) C.-Q. Yang, X.-M. Wu, J.-X. Ruan, W.-L. Hu, Y.-B. Mao, X.-Y. Chen and L.-J. Wang, Phytochemistry, 2013, 96, 46–56 CrossRef CAS PubMed.
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC.
- J. Baranauskaitė, V. Jakštas, L. Ivanauskas, D. M. Kopustinskienė, G. Drakšienė, R. Masteikova and J. Bernatonienė, Nat. Prod. Res., 2016, 30, 672–674 CrossRef PubMed.
- D. Villanueva Bermejo, I. Angelov, G. Vicente, R. P. Stateva, M. Rodriguez García-Risco, G. Reglero, E. Ibañez and T. Fornari, J. Sci. Food Agric., 2015, 95, 2901–2907 CrossRef CAS PubMed.
- I. Batool, S. Nisar, L. Hamrouni and M. I. Jilani, Int. J. Chem. Biomol. Sci., 2018, 14, 71–76 Search PubMed.
- (a) S. E. Aly, A. S. Hathout and N. A. Abo-Sereih, J. Biol. Act. Prod. Nat., 2011, 1, 293–305 Search PubMed; (b) G.-W. Zheng, J. Pan, H.-L. Yu, M.-T. Ngo-Thi, C.-X. Li and J.-H. Xu, J. Biotechnol., 2010, 150, 108–114 CrossRef CAS PubMed.
- (a) A. Marchese, I. E. Orhan, M. Daglia, R. Barbieri, A. Di Lorenzo, S. F. Nabavi, O. Gortzi, M. Izadi and S. M. Nabavi, Food Chem., 2016, 210, 402–414 CrossRef CAS PubMed; (b) V. V. M. A. Souza, J. M. Almeida, L. N. Barbosa and N. C. C. Silva, J. Essent. Oil Res., 2022, 34, 181–194 CrossRef CAS; (c) J. A. Farco and O. Grundmann, Mini-Rev. Med. Chem., 2013, 13, 124–131 CrossRef CAS PubMed; (d) M. Sharifi-Rad, E. M. Varoni, M. Iriti, M. Martorell, W. N. Setzer, M. Del Mar Contreras, B. Salehi, A. Soltani-Nejad, S. Rajabi, M. Tajbakhsh and J. Sharifi-Rad, Phytother. Res., 2018, 32, 1675–1687 CrossRef CAS PubMed; (e) N. B. Rathod, P. Kulawik, F. Ozogul, J. M. Regenstein and Y. Ozogul, Trends Food Sci. Technol., 2021, 116, 733–748 CrossRef CAS.
- (a) V. H. Campos-Requena, B. L. Rivas, M. A. Pérez, C. R. Figueroa and E. A. Sanfuentes, LWT – Food Sci. Technol., 2015, 64, 390–396 CrossRef CAS; (b) E. Sanchez-Rexach, I. Martínez de Arenaza, J.-R. Sarasua and E. Meaurio, Eur. Polym. J., 2016, 83, 288–299 CrossRef CAS; (c) N. Petchwattana and P. Naknaen, Mater. Chem. Phys., 2015, 163, 369–375 CrossRef CAS; (d) R. Requena, M. Vargas and A. Chiralt, Food Hydrocolloids, 2018, 83, 118–133 CrossRef CAS; (e) R. Scaffaro, A. Maio, E. F. Gulino, M. Morreale and F. P. La Mantia, Materials, 2020, 13, 983 CrossRef CAS PubMed; (f) A. Sansukcharearnpon, S. Wanichwecharungruang, N. Leepipatpaiboon, T. Kerdcharoen and S. Arayachukeat, Int. J. Pharm., 2010, 391, 267–273 CrossRef CAS PubMed.
- (a) M. Winnacker, S. Vagin, V. Auer and B. Rieger, Macromol. Chem. Phys., 2014, 215, 1654–1660 CrossRef CAS; (b) M. Winnacker, A. Tischner, M. Neumeier and B. Rieger, RSC Adv., 2015, 5, 77699–77705 RSC.
- (a) W. T. Diment, G. Rosetto, N. Ezaz-Nikpay, R. W. F. Kerr and C. K. Williams, Green Chem., 2023, 25, 2262–2267 RSC; (b) J. Shin, Y. Lee, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2012, 13, 3833–3840 CrossRef CAS PubMed.
- (a) S. Bedel, B. Lepoittevin, L. Costa, O. Leroy, D. Dragoe, J. Bruzaud, J.-M. Herry, M. Guilbaud, M.-N. Bellon-Fontaine and P. Roger, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1975–1985 CrossRef CAS; (b) E. H. Min, K. H. Wong, E. Setijadi, F. Ladouceur, M. Straton and A. Argyros, Polym. Chem., 2011, 2, 2045 RSC.
- K. M. Mydeen, J. P. Kanth, A. Hariharan, K. Balaji, S. Rameshkumar, G. Rathika and M. Alagar, J. Polym. Environ., 2022, 30, 5301–5312 CrossRef CAS.
- G. A. Parolin, L. F. D. Passero, J. H. G. Lago and L. O. Péres, Ind. Crops Prod., 2021, 171, 113935 CrossRef CAS.
- (a) I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed; (b) M. Grigore, Recycling, 2017, 2, 24 CrossRef; (c) V. M. Pathak and B. Navneet, Bioresour. Bioprocess., 2017, 4, 15 CrossRef; (d) N. Lucas, C. Bienaime, C. Belloy, M. Queneudec, F. Silvestre and J.-E. Nava-Saucedo, Chemosphere, 2008, 73, 429–442 CrossRef CAS PubMed.
- F. Siragusa, C. Detrembleur and B. Grignard, Polym. Chem., 2023, 14, 1164–1183 RSC.
- (a) A. Beharaj, E. Z. McCaslin, W. A. Blessing and M. W. Grinstaff, Nat. Commun., 2019, 10, 5478 CrossRef CAS PubMed; (b) F.-T. Tsai, Y. Wang and D. J. Darensbourg, J. Am. Chem. Soc., 2016, 138, 4626–4633 CrossRef CAS PubMed.
- K. D. Knight and M. E. Fieser, Inorg. Chem. Front., 2023, 11, 298–309 RSC.
- (a) P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS; (b) C.-C. Su, M. He, R. Amine, Z. Chen, R. Sahore, N. Dietz Rago and K. Amine, Energy Storage Mater., 2019, 17, 284–292 CrossRef.
- (a) G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS; (b) M. L. Smith, T. M. McGuire, A. Buchard and C. K. Williams, ACS Catal., 2023, 24, 15770–15778 CrossRef PubMed; (c) G. Rosetto, F. Vidal, T. M. McGuire, R. W. F. Kerr and C. K. Williams, J. Am. Chem. Soc., 2024, 146, 8381–8393 CrossRef CAS PubMed.
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Angew. Chem., 2022, 134, e202201785 CrossRef.
- (a) T. M. McGuire, A. C. Deac, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 18444–18449 CrossRef CAS PubMed; (b) Y. Yu, B. Gao, Y. Liu and X.-B. Lu, Angew. Chem., 2022, 134, e202204492 CrossRef.
- (a) C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS PubMed; (b) D. Ghosh and S. Agarwal, Polym. Chem., 2023, 14, 4626–4635 RSC; (c) D. H. Lamparelli, A. Villar-Yanez, L. Dittrich, J. Rintjema, F. Bravo, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed., 2023, 62, e202314659 CrossRef CAS PubMed.
- B. Liu, J. Chen, N. Liu, H. Ding, X. Wu, B. Dai and I. Kim, Green Chem., 2020, 22, 5742–5750 RSC.
- A. Akira, H. Toshimitsu, M. Takashi and A. Teruyoshi, Japanese Pat., JP20010124134, 2002 Search PubMed.
- (a) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed; (b) M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed.
- (a) D. J. Darensbourg, J. Chem. Educ., 2017, 94, 1691–1695 CrossRef CAS; (b) G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS.
- M. R. Ambler, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 191–201 CrossRef CAS.
- C. T. Cohen and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5182–5191 CrossRef CAS.
- C. Earl, I. H. Shah, S. Cook and C. R. Cheeseman, Sustainability, 2022, 14, 4124 CrossRef CAS.
- (a) D.-D. Zhang, X. Feng, Y. Gnanou and K.-W. Huang, Macromolecules, 2018, 51, 5600–5607 CrossRef CAS; (b) D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- Z. Chen, J.-L. Yang, X.-Y. Lu, L.-F. Hu, X.-H. Cao, G.-P. Wu and X.-H. Zhang, Polym. Chem., 2019, 10, 3621–3628 RSC.
- M. Trivedi, S. Patil, R. Mishra and S. Jana, J. Mol. Pharm. Org. Process Res., 2015, 03, 1000127 Search PubMed.
- Engineering solutions for CO2 conversion, ed. T. R. Reina, J. A. Odriozola and H. Arellano-Garcia, Wiley-VCH, John Wiley & Sons, Inc., Weinheim, Hoboken, NJ, 2021 Search PubMed.
- (a) J. K. Varghese, S. J. Na, J. H. Park, D. Woo, I. Yang and B. Y. Lee, Polym. Degrad. Stab., 2010, 95, 1039–1044 CrossRef CAS; (b) Y. Yu, B. Gao, Y. Liu and X.-B. Lu, Angew. Chem., 2022, 134, e202204492 CrossRef.
- (a) M. Jahangeer, R. Fatima, M. Ashiq, A. Basharat, S. A. Qamar, M. Bilal and H. M. Iqbal, J. Pure Appl. Microbiol., 2021, 15, 471–483 Search PubMed; (b) S. D. Tetali, Planta, 2019, 249, 1–8 CrossRef CAS PubMed.
- M. F. Nagoor Meeran, H. Javed, H. Al Taee, S. Azimullah and S. K. Ojha, Front. Pharmacol., 2017, 8, 380 CrossRef PubMed.
- G. P. P. Kamatou, I. Vermaak, A. M. Viljoen and B. M. Lawrence, Phytochemistry, 2013, 96, 15–25 CrossRef CAS PubMed.
- K. H. C. Baser, Curr. Pharm. Des., 2008, 14, 3106–3119 CrossRef CAS PubMed.
- (a) C. Fuganti, D. Joulain, F. Maggioni, L. Malpezzi, S. Serra and A. Vecchione, Tetrahedron: Asymmetry, 2008, 19, 2425–2437 CrossRef CAS; (b) A. Yokohama, M. Mitaka and T. Yokohama, US Pat., 4459425, 1984 Search PubMed; (c) A. O. Barel, M. Paye and H. I. Maibach, Cooling Ingredients and Their Mechanism of Action, Informa Healthcare, New York, N.Y., 2009 Search PubMed; (d) R. Eccles, J. Pharm. Pharmacol., 1994, 46, 618–630 CrossRef CAS PubMed; (e) M. Fujii, Y. Takeda, M. Yoshida, N. Utoguchi, M. Matsumoto and Y. Watanabe, Int. J. Pharm., 2003, 258, 217–223 CrossRef CAS PubMed.
- (a) A. Bytyqi-Damoni, A. Kestane, P. Taslimi, B. Tuzun, M. Zengin, H. G. Bilgicli and İ. Gulcin, J. Mol. Struct., 2020, 1202, 127297 CrossRef CAS; (b) M. Zengin, H. Genc, P. Taslimi, A. Kestane, E. Guclu, A. Ogutlu, O. Karabay and İ. Gulçin, Bioorg. Chem., 2018, 81, 119–126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on reagents, instrumentation, experimental and synthetic methods. Characterization of all monomers, copolymers, (NMR, SEC, DSC, MALDI-ToF). Retrosynthesis of presented bio-based polymers. See DOI: https://doi.org/10.1039/d4py00797b |
| This journal is © The Royal Society of Chemistry 2024 |