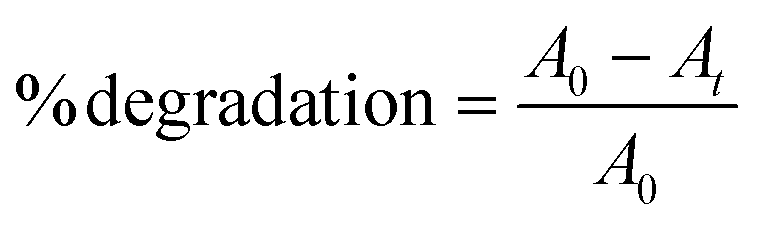DOI:
10.1039/D4NR01154F
(Paper)
Nanoscale, 2024,
16, 12445-12458
A novel GO hoisted SnO2–BiOBr bifunctional catalyst for the remediation of organic dyes under illumination by visible light and electrocatalytic water splitting†
Received
17th March 2024
, Accepted 7th May 2024
First published on 22nd May 2024
Abstract
It is imperative to develop affordable multi-functional catalysts based on transition metals for various applications, such as dye degradation or the production of green energy. For the first time, we propose a simple chemical bath method to create a SnO2–BiOBr–rGO heterojunction with remarkable photocatalytic and electrocatalytic activities. After introducing graphene oxide (GO) into the SnO2–BiOBr nanocomposite, the charge separation, electron mobility, surface area, and electrochemical properties were significantly improved. The X-ray diffraction results show the successful integration of GO into the SnO2–BiOBr nanocomposite. Systematic material characterization by scanning and transmission electron microscopy showed that the photocatalysts are composed of uniformly distributed SnO2 nanoparticles (∼11 nm) on the regular nanosheets of BiOBr (∼94 nm) and rGO. The SnO2–BiOBr–rGO photocatalyst has outstanding photocatalytic activity when it comes to reducing a variety of organic dyes like rhodamine B (RhB) and methylene blue (MB). Within 90 minutes of visible light illumination, degradation of a maximum of 99% for MB and 99.8% for RhB was noted. The oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) performance was also tested for the ternary nanocomposite, and significantly lower overpotential values of 0.34 and −0.11 V (vs. RHE) at 10 mA cm−2 were observed for the OER and HER, respectively. Furthermore, the Tafel slope values are 34 and 39 mV dec−1 for the OER and HER, respectively. The catalytic degradation of dyes with visible light and efficient OER and HER performance offer this work a broad spectrum of potential applications.
1. Introduction
The energy research program has been redirected towards advancing environmentally friendly and sustainable energy sources to address the challenges posed by climate change and global warming.1 Therefore, it is essential to switch to renewable energy sources like sunlight, wind, tides, and water. The expansion of industry has contaminated wastewater with organic dyes and other chemicals that cause cancer.2 Degrading the organic pigments and using the resultant water as a source for reactions that evolve oxygen (O2) and hydrogen (H2) are becoming more and more significant. Traditional methods of dye removal from wastewater, including adsorption, biological treatment, and physical separation, can often be inefficient, costly, or environmentally detrimental.3 To this end, photocatalytic water treatment utilizing solar light has attracted a lot of interest.4 Photocatalysis, which uses sunlight to break down organic pollutants into harmless chemicals, has become a viable approach to clean contaminated water.5 Due to its potential for environmental remediation, photocatalytic degradation of organic pollutants, such as RhB, MB, etc., in wastewater has attracted much interest.6 The wide use of organic dyes from various industries, including textile, printing, paper, and leather, is one of the most common problems. These dyes pose severe dangers to the environment and human health because of their resistance, toxicity, and ability to disrupt aquatic ecosystems.7 On the other hand, water splitting is gaining much attention for hydrogen and oxygen evolution by using electrocatalysts and/or photocatalysts. Electrocatalysts made from noble metals are crucial in electrochemical energy conversion processes. However, their widespread use is hindered by their high cost and limited natural availability. Therefore, there is a pressing need to develop durable and efficient electrocatalysts using affordable elements abundant in the Earth's crust.8 This necessity extends to catalytic reactions beyond the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER), such as dye degradation. Consequently, extensive research has been devoted to crafting versatile catalysts encompassing hydroxides, carbon-based substances, transition metal compounds, and various carbon materials.9 To this end, TiO2 has emerged as the foremost choice for photocatalytic applications due to its non-toxic nature, affordability, notable activity, and durability.10 Yet, its large band gap limits its activity only under ultraviolet irradiation, constituting approximately 4% of the solar spectrum. On the other hand, visible light comprises 40% of the solar spectrum. Therefore, designing a photocatalyst and/or electrocatalyst that shows its degradation activity in the visible region and significant oxygen and hydrogen evolution performance is the need of the hour.11
Recently, bismuth oxyhalides have attracted much interest due to their distinct layered structures and superior photocatalytic capabilities12 towards photocatalytic water splitting.13 BiOBr stands out because of its optimal band gap (∼2.57 eV).14 Different morphologies of BiOBr, ranging from nanoflakes and nanospheres to 3-D designs like nanoflowers, have been documented.15 To decrease the recombination rate of photogenerated electron and hole pairs in pure BiOBr, researchers have explored numerous heterojunctions, with BiPO4–BiOBr, Cu2S–BiOBr, BiOBr/C, and Bi2O3–BiOBr being notable examples.16–19 To this end, the potential application of BiOBr in the photocatalytic degradation of dyes, water splitting, antibiotics, phenols, etc., has been widely reported.20 On another front, tin dioxide (SnO2) is frequently utilized in photodetection and photoelectrochemistry due to its non-toxic nature, stable performance, and exceptional optoelectronic properties.21 Yet, its wider energy gap (Eg = 3.06 eV) combined with a greater likelihood of electron–hole pair recombination limits its use in photocatalysis.22 Tin dioxide (SnO2) and bismuth oxybromide (BiOBr) have each proved their potential as photocatalysts in the degradation of organic dyes among other photocatalysts.23 While SnO2 possesses excellent electronic properties and relatively high electron mobility, BiOBr stands out because of its suitable band gap and stability under visible light irradiation. However, like many semiconductor materials, they have some restrictions, such as insufficient light absorption and fast photogenerated electron and hole pair recombination.24 Therefore, new strategies have been adopted to overcome this problem, such as doping with a metal or non-metal, heterostructure formation, etc.
Doping semiconductors with metal or non-metal elements has become an effective strategy for the modulation of their electronic structure and, consequently, to expand their absorption of light to the visible spectrum. A graphene derivative, GO, which has a layered structure in two dimensions, has received much interest lately due to its capacity to boost photocatalytic and electrocatalytic performance.25 GO offers several advantages, such as increased light absorption, effective charge separation,66 and improved electron transfer.26 As per earlier reports, SnO2–BiOX (X = Br,27 I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24) has also been used for photocatalysis, but there are no results for MB dye degradation. One of the significant challenges faced by SnO2–BiOBr is that it has a high recombination rate for the photogenerated holes and electrons. This rapid recombination process reduces photocatalytic efficiency, as fewer charge carriers are available to participate in the photocatalytic reactions.28 The limited surface area of SnO2–BiOBr can be a drawback as it provides fewer active sites for photocatalytic and electrocatalytic reactions.67 As a result, doping GO into semiconductors like SnO2 and BiOBr may get over their inherent constraints and increase their photocatalytic performance and water splitting in concerted ways.19,29
24) has also been used for photocatalysis, but there are no results for MB dye degradation. One of the significant challenges faced by SnO2–BiOBr is that it has a high recombination rate for the photogenerated holes and electrons. This rapid recombination process reduces photocatalytic efficiency, as fewer charge carriers are available to participate in the photocatalytic reactions.28 The limited surface area of SnO2–BiOBr can be a drawback as it provides fewer active sites for photocatalytic and electrocatalytic reactions.67 As a result, doping GO into semiconductors like SnO2 and BiOBr may get over their inherent constraints and increase their photocatalytic performance and water splitting in concerted ways.19,29
In this study, we present a simple chemical bath method for a GO-doped SnO2–BiOBr nanocomposite, and this novel nanocomposite shows excellent photocatalytic and electrocatalytic performance. The distinct synthesis method allows SnO2 nanoparticles to be evenly distributed and integrated on the surface of the rGO and BiOBr matrix, creating a clearly defined heterojunction structure for visible spectrum activation. First, the nanocomposite is used as a photocatalyst to degrade RhB and MB dyes. Compared to the earlier reports, as shown in Table 1 our nanocomposite exhibits improved degradation performance for MB and RhB dyes under 90 minutes of visible light illumination. The combination of SnO2–BiOBr with GO has been proved, by many characterization studies, to increase the transient photocurrent response approximately ten times as compared to the SnO2–BiOBr heterojunction23 and also increase photogenerated charge carrier separation and their transportation, which lead to improved photo- and electrocatalytic efficiencies29 and extend their light absorption to the visible region. Finally, the nanocomposite was used as a photoelectrocatalyst, which enhanced its OER and HER performance with significantly lower overpotential and Tafel slope values in the presence of visible light. To our knowledge, there is no report on photocatalysis and photoelectrocatalysis using the SnO2–BiOBr–rGO nanocomposite and no other material shows all the properties together with this much enhancement in photo- and electrocatalytic properties.
Table 1 A table for comparing various earlier reported photocatalytic performances of SnO2, BiOBr, and GO-based composites for the photodegradation of MB dye
| Photocatalyst |
Photocatalyst amount (mg ml−1) |
MB concentration (ppm) |
Dye degradation (%) |
Irradiation time (min) |
Light source |
| This study |
0.4 |
20 |
99 |
90 |
Xenon lamp (>420 nm) |
| SnO2 |
0.3 |
10 |
90 |
30 |
UV48 |
| EDA–SnO2 |
0.1 |
20 |
96.3 |
90 |
250 W Hg lamp (400–700 nm)49 |
| GO/BiOBr |
0.5 |
10 |
98 |
30 |
Xenon lamp50 |
| SnO2–rGO |
0.4 |
10 |
93 |
50 |
Xenon lamp (>420 nm)51 |
| ZnO–BiOBr |
1 |
10 |
80 |
30 |
Xenon lamp52 |
| SnO2–MoS2 |
0.2 |
100 |
58.4 |
120 |
85 W CFL53 |
2. Materials used and synthesis route
2.1. Materials used
Potassium bromide (KBr) was obtained from Finar Chemicals. Stannic chloride pentahydrate extra pure (SnCl4·5H2O, ≥98%), sulphuric acid (98%), and rhodamine B (RhB) dye were purchased from Loba Chemie, and bismuth nitrate pentahydrate extra pure (Bi(NO3)3·5H2O, ≥98%), ammonium oxalate (AO) (99%), isopropyl alcohol (IPA) (99%) and methylene blue (MB) dye were purchased from SRL Chemicals. p-Benzoquinone (p-BQ) was purchased from TCI. De-ionized water (15 MΩ cm−1) was used for all the solutions, and ethylene glycol was purchased from EMPARTA with 99% purity.
2.2. Synthesis route
2.2.1 Synthesis route of SnO2.
2.5 millimolar (mM) SnCl4·5H2O and 2.5 mM NaOH were combined with 12.5 ml of DI and 12.5 ml of ethanol. After magnetic stirring for 1 hour, the mixture was then placed on a hydrothermal system for 12 hours at 180 °C. To remove unreacted precursors, the obtained product was centrifuged and then washed repeatedly with DI.24
2.2.2 Synthesis of BiOBr.
2.4 mM KBr and 2.4 mM Bi(NO3)3·5H2O were used. Both were added to a mixture containing 6 ml of DI and 14 ml of ethylene glycol.30 It was put on magnetic stirring for about an hour. The solution mixture was then dried in a hot air oven at 40 °C overnight to achieve a certain concentration. Then the resulting BiOBr powder sample was scrapped out from the Petri dish.
2.2.3 Modified Hummers’ method for GO.
Modified Hummers’ method was used for the synthesis of GO. 1.5 ml of H2SO4 and 1 g of graphite powder were mixed, and then 0.5 g of both K2S2O8 and P2O5 were added to the solution.31 This mixture was put on stirring and, after that, left overnight to dry to get a pre-oxidized graphite powder. 0.5 g of pre-oxidized graphite powder was taken, to which 13 ml of H2SO4 and 1.5 g of solid KMnO4 were added, giving a dark green color. Milli-Q water was added under slow stirring until the mixture changed from green to a chocolate brown color. Afterward, 2.5 ml of H2O2 solution was added so that the solution attained a bright yellow color. The bright yellow settled-down mixture was washed with 10% HCl and then with DI for pH control. The resultant dark green mixture was the obtained GO solution with a concentration of 15 mg ml−1.
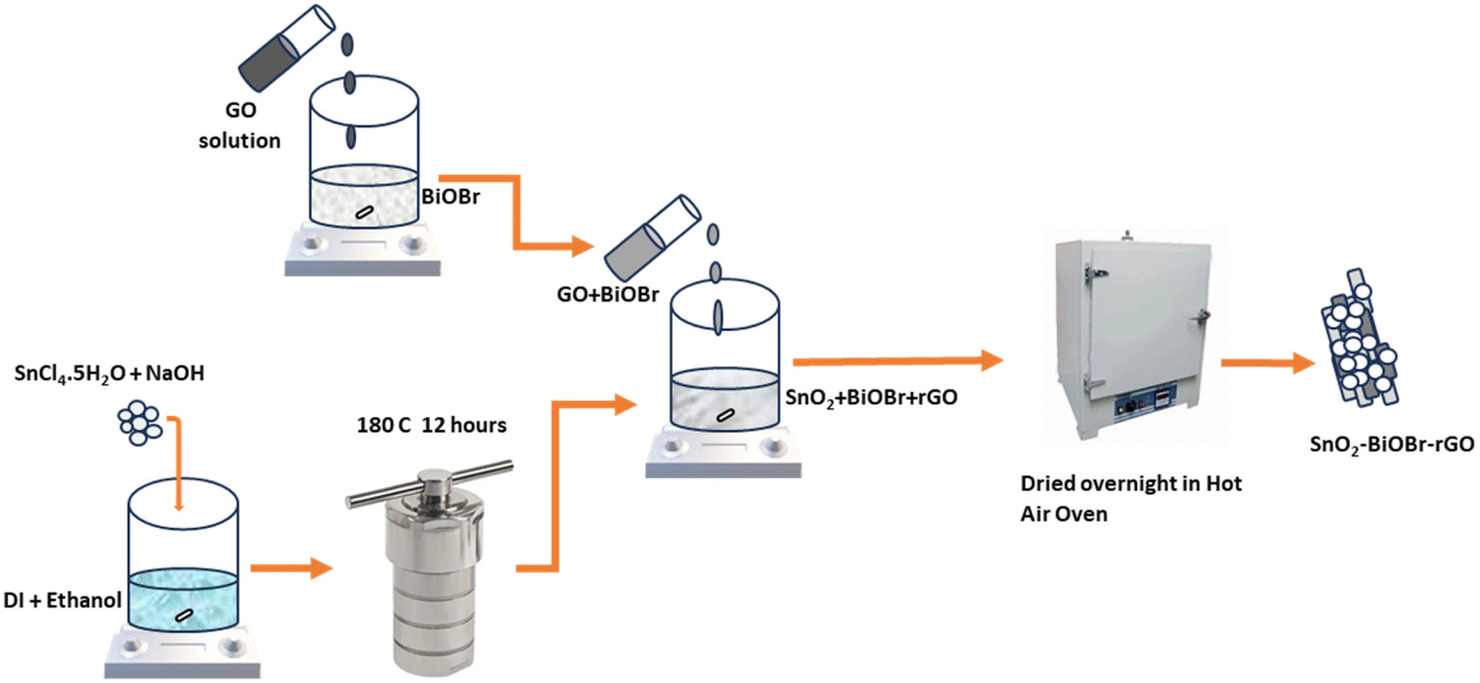
2.2.4 Nanocomposite preparation.
140.23 mg of SnO2 and 36 mg of GO (15 mg ml−1) were taken along with 4.8 ml of BiOBr (25 mg ml−1). The mixture of GO and BiOBr was put on magnetic stirring for 30 minutes. Then, this solution of GO and BiOBr was added drop by drop to the SnO2 solution under magnetic stirring and left in that state for 45 minutes. After that, the solution was mixed properly and put on overnight heating at 40 °C to get a powder form of the proposed nanocomposite (Scheme 1).
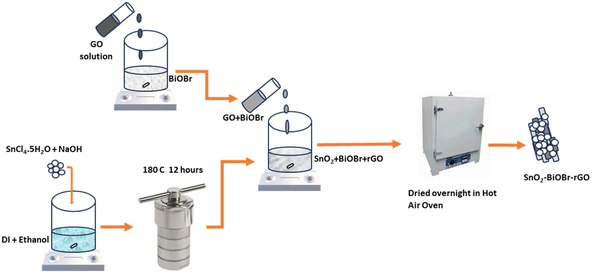 |
| | Scheme 1 Schematic showing the proposed synthesis route of the ternary nanocomposite. | |
2.3. Catalyst characterization
X-ray diffraction (XRD) analysis was performed by utilizing a Bruker D8 Advance diffractometer equipment with a scan rate of 3° min−1 to assess the crystallinity of the produced nanoparticles and composites. Field emission scanning electron microscopy (FESEM) (JEOL JSM 7610FPlus) was used to study the morphology. Surface analysis was performed by X-ray photoelectron spectroscopy (XPS) using a K-Alpha plus XPS (Thermo Fisher Scientific) and Al-Kα radiation (1486.6 eV). High-resolution transmission electron microscopy (HRTEM) imaging was conducted using a JEOL JEM2100 transmission electron microscope at 200 kV accelerating voltage. Diffuse reflectance spectroscopy (DRS) was recorded using an Agilent Cary 100 UV-Vis spectrophotometer with a 200–800 nm wavelength range. The electrochemical measurements were performed on an electrochemical workstation (Metrohm-Autolab PGSTAT302N) and a 350 W xenon lamp (ORIEL INSTRUMENTS, OPS-A1000). A similar light source is used for photocatalytic experiments with a 420 nm cut-off filter. The absorbance spectra were monitored with a Cary 4000 UV-Vis spectrophotometer.
2.4. Photocatalytic degradation of dyes
MB and RhB dyes were chosen as the model organic pollutants for degradation. The total amount of solution was 16 mL, and the concentrations for MB and RhB were 20 and 10 ppm, respectively. A xenon lamp with a 420 nm cut-off filter was used as the light source. The concentration of the photocatalysts was 0.4 mg ml−1. Initially, the solution was stirred for 30 minutes in the dark, followed by light illumination under ambient conditions. 0.5 ml aliquots were taken out after constant intervals and evaluated using a UV-vis spectrophotometer. The absorbance maxima of MB and RhB were noted at 664 and 554 nm, respectively. The dye degradation percentage (%) was calculated using eqn (1)| | 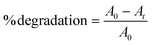 | (1) |
where A0 represents the initial absorbance, and At is that at time t.
2.5. Photoelectrochemical study
The photoelectrochemical characterization of the synthesized catalyst SnO2–BiOBr–rGO involved testing the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) using a three-electrode quartz cell setup in a Metrohm-Autolab PGSTAT302N electrochemical analyzer with a visible light source. A saturated calomel electrode (SCE), platinum wire, and prepared sample were used as reference, counter, and working electrodes, respectively. The electrolyte solution used for the measurements was 0.5 M H2SO4. 5 mg of the catalyst powder was dispersed in 1 mL of IPA and 5 μL of Nafion (1 wt%), followed by ultrasonication. After that, 10 μL from the prepared solution was dropcast on the conducting side of the ITO substrate within a 1 cm2 area. Electrodes were dried in a vacuum oven overnight at 40 °C. The linear sweep voltammetry (LSV) curves were taken at a 50 mV s−1 scan rate in the presence of visible light.
3. Results and discussion
3.1. Characterization of photocatalysts
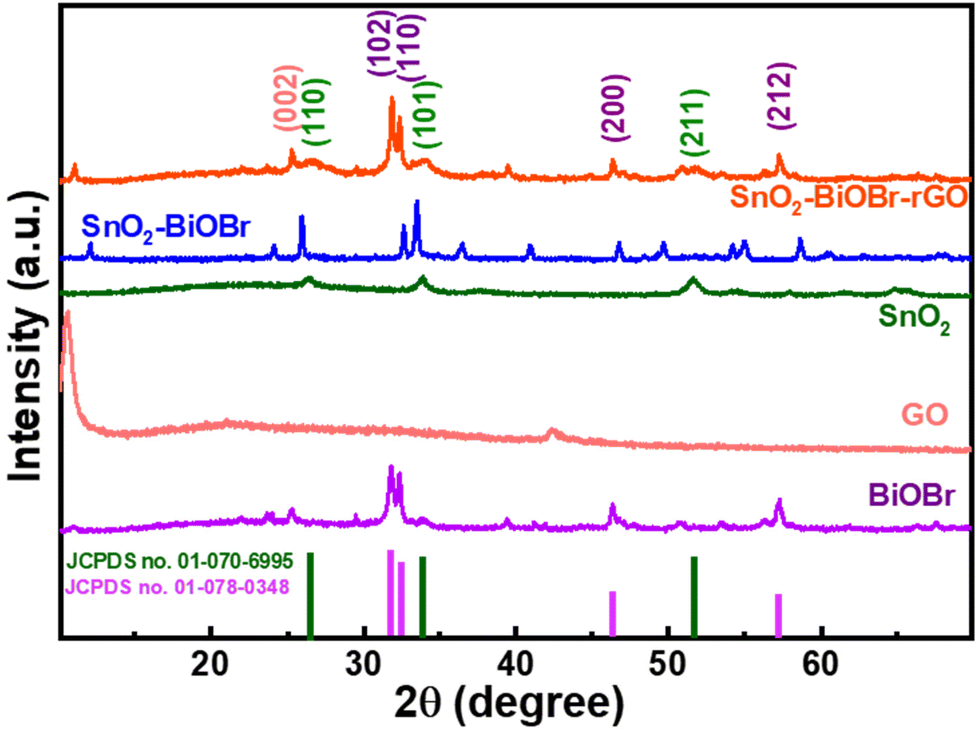
The crystalline nature of BiOBr, SnO2, GO, SnO2–BiOBr, and the SnO2–BiOBr–rGO nanocomposite was examined using a powder X-ray diffraction pattern. For the SnO2–BiOBr–rGO sample, peaks of individual constituents, including BiOBr, SnO2, and rGO can be observed with their corresponding planes.
The XRD analysis was performed in the 2θ range between 10° and 70°. BiOBr (JCPDS no. 01-078-0348)27 is confirmed as a pure crystalline material with sharp diffraction peaks in the X-ray diffractometry spectra. Diffraction peaks present at 2θ = 25.2°, 31.7°, 32.2°, 34.1°, 39.3°, 46.2°, 46.9°, 56.1°, and 57.1° corresponded to the (101), (102), (110), (003), (112), (200), (113), (114) and (212) Miller indices of BiOBr, which is attributed to the tetragonal structure of BiOBr. And GO shows diffraction peaks at 2θ = 11.02° corresponding to the (001) plane.32 All XRD peaks of SnO2 (JCPDS no. 01-070-6995) correspond to tetragonal SnO2. Pure SnO2 has three peaks, at 2θ = 26.6°, 33.9°, and 51.8°, for the diffraction planes (110), (101), and (211), respectively. For the final ternary composite, as shown in Fig. 1, the (002) plane corresponds to rGO,33 and (110), (101), (211) correspond to SnO2, and (102), (110), (200), (212) to BiOBr respectively. From the XRD analysis, we can also comment on the crystallinity of the samples. The more crystalline the sample, the less the value of the full width at half maximum (FWHM). As the final nanocomposite shows a lower value for FWHM as compared to the pristine samples as shown in Fig. S1,† it is more crystalline. As per earlier reports, the photocatalytic activity increases with the increase in the crystallinity of the sample.34 Also, the peak broadening may be due to the crystallite size; crystallite size refers to the size of the individual crystalline domains within a material. When crystallite size decreases, the peaks in the XRD pattern become broader. For SnO2 the size of the particles is very small due to which it has a broad peak as compared to BiOBr.
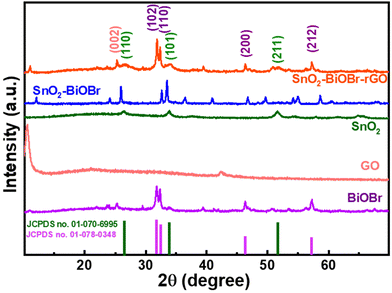 |
| | Fig. 1 Powder X-ray diffraction pattern of BiOBr, GO, SnO2, SnO2–BiOBr, and SnO2–BiOBr–rGO. | |
By looking at the functional groups present on the surface of the photocatalysts using FTIR analysis in the range of 550–4000 cm−1, it was possible to analyze further the chemical structures of pure SnO2, BiOBr, SnO2–BiOBr, and the SnO2–BiOBr–rGO nanocomposite. The spectra are shown in Fig. 2. The distinctive peak of the pure BiOBr sample is at 1372 cm−1, and the absorption bands between 550 and 1000 cm−1 are related to the stretching vibration of the Bi–O bond, while those between 1000 and 1500 cm−1 are related to the Bi–Br band.35 The O–Sn–O stretching vibrations are represented by a broad peak at 663 cm−1 in the SnO2 nanoparticles FTIR spectra.36 The successful synthesis of the SnO2–BiOBr composite was supported by the stretching vibration of O–Sn–O present at 663 cm−1. SnO2–BiOBr exhibits similar spectra to those of BiOBr. The vibrations of O–H of –COOH in GO are shown by the peaks centering at 3397 cm−1 and 1635 cm−1, respectively.37 SnO2–BiOBr materials with GO doping had considerably fewer GO functional groups. It is evident from this spectrum that the peak intensities of the GO-doped SnO2–BiOBr sample are comparable to those of the SnO2–BiOBr sample. The FTIR results demonstrate a potentially strong chemical interaction between rGO and SnO2–BiOBr, which may result from the novel synthesis process.
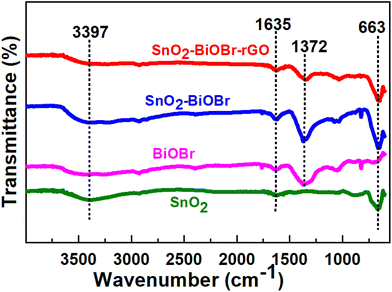 |
| | Fig. 2 FTIR spectra for BiOBr, SnO2, SnO2–BiOBr, and SnO2–BiOBr–rGO. | |
SEM imaging was carried out to identify typical surface morphologies. In Fig. 3(a), the SEM image of the SnO2 nanoparticles is shown, and we can calculate the particle size from this SEM image, which is around 11 nm, as depicted in the histogram in Fig. 3(a). Because of their small size, SnO2 particles are aggregated. Fig. 3(b) depicts BiOBr nanosheets, with these sheets having a size distribution of around 94 nm, as shown in the histogram in Fig. 3(b). As displayed in Fig. 3(c), SnO2 nanoparticles are uniformly present on the BiOBr sheets, and more sheets are present in the background when SnO2 is added to BiOBr. So, in the SnO2–BiOBr nanocomposite, the aggregation of SnO2 nanoparticles was less than that in pure SnO2. Still, we need more separation of charges, and for this, we add GO as Fig. 3(d) shows the final nanocomposite contains SnO2–BiOBr–rGO. The uniform distribution of SnO2 particles on the sheets of BiOBr and rGO prevents the aggregation of SnO2, causing the formation of efficient heterojunctions, which further facilitates charge separation under photocatalytic conditions.
 |
| | Fig. 3 FESEM images of (a) SnO2, (b) BiOBr (c) SnO2–BiOBr, (d) SnO2–BiOBr–rGO. | |
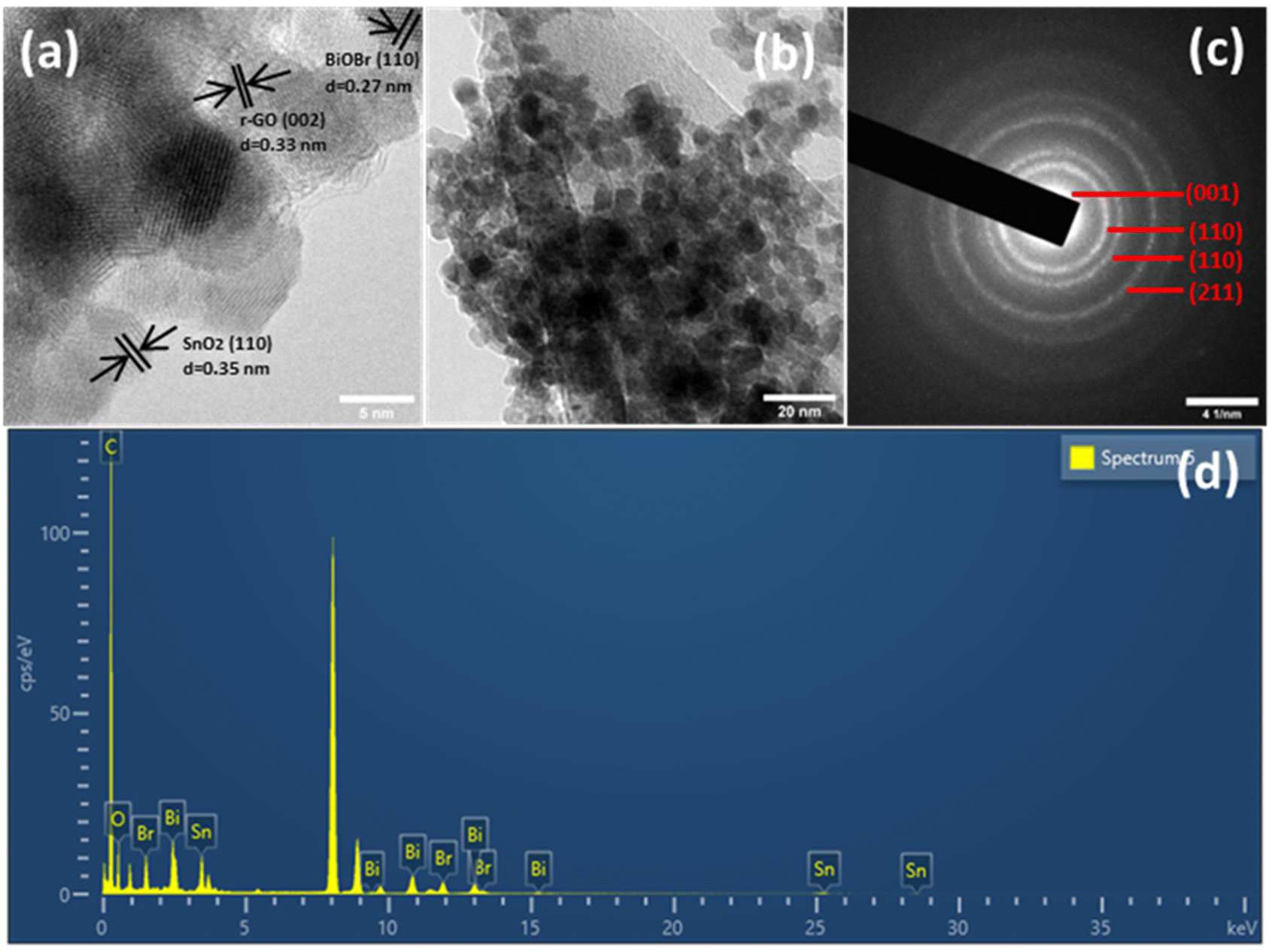
By HRTEM imaging of SnO2–BiOBr–rGO, as shown in Fig. 4(a), three sets are seen at the interfaces for various lattice images. The (110) crystal facets of SnO2, (110) of BiOBr, and (002) of rGO, respectively, were consistent with the lattice fringes of 0.35, 0.27, and 0.33 nm. SnO2 nanoparticles have an average diameter of around 11 nm and can be verified as distributed on the sheets of BiOBr and rGO sheets as shown in Fig. 4(b). Fig. 4(c) shows the SAED pattern of SnO2–BiOBr–rGO, which confirms the presence of (001), (110), (110), and (211) lattice planes of GO, SnO2, BiOBr, and SnO2, respectively, in the ternary nanocomposite SnO2–BiOBr–rGO. EDS analysis was also carried out to confirm the chemical composition of all the elements in SnO2–BiOBr–rGO quantitatively to be C (67.18%), O (5.21%), Br (4.33%), and Sn (8.89%), Bi (14.39%) weight percentage as shown in Fig. 4(d).
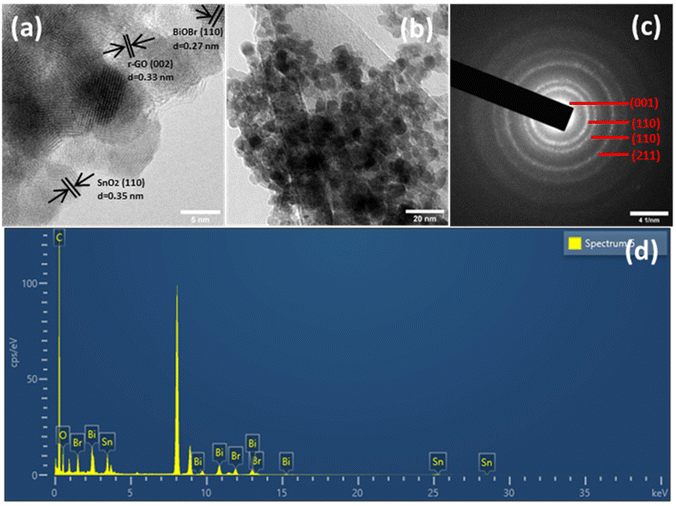 |
| | Fig. 4 (a) HRTEM image, and (b) TEM image (c) selected area electron diffraction (SAED) and (d) EDS spectrum of SnO2–BiOBr–rGO. | |
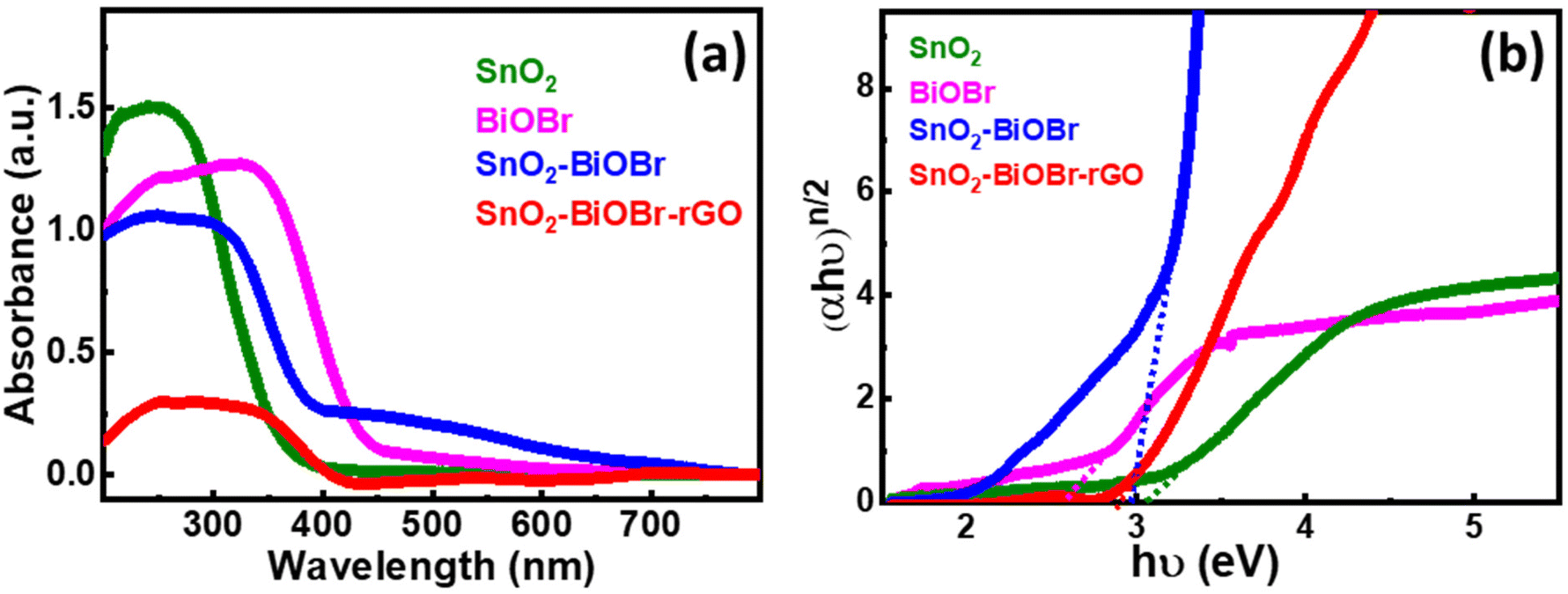
3.2. Optical characterization
In Fig. 5(a), SnO2 has an absorption edge at 365 nm wavelength. Also, BiOBr shows its absorption in the visible spectrum because its absorption edge is around 450 nm, and for SnO2–BiOBr and SnO2–BiOBr–rGO it is in the visible range. Fig. 5(b) shows the optical band edge calculated with the help of the Tauc equation: αhν = A(hν − Eg)n/2, where A is a constant; α, h, ν, and Eg are the absorption coefficient, Planck's constant, energy of a photon, and band gap energy, respectively; and n stands for optical transitions and can be equal to 1 and 4 for direct and indirect transition of a semiconductor respectively.
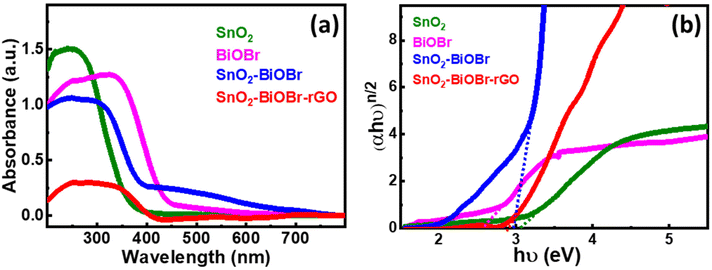 |
| | Fig. 5 (a) UV-visible-diffuse reflectance spectra for SnO2, BiOBr, SnO2–BiOBr, and SnO2–BiOBr–rGO. (b) Tauc plot of (αhν)n/2vs. hν (eV) for SnO2, BiOBr, SnO2–BiOBr, and SnO2–BiOBr–rGO. | |
From the literature, SnO2, BiOBr, and GO have n = 1. As shown in Fig. 5(b), the band gaps of SnO2, BiOBr, SnO2–BiOBr, and SnO2–BiOBr–rGO are 3.06,38 2.57,23 2.96, and 2.87 eV, respectively. By using Planck's equation for the energy (E) and wavelength (λ) relation, we can cross-verify that for the band gap of SnO2–BiOBr–rGO, the wavelength lies in the visible region as given below, where c is the speed of light and h is Planck's constant:
| 2.87 (eV) = 1242/λ (nm) and then the value is |
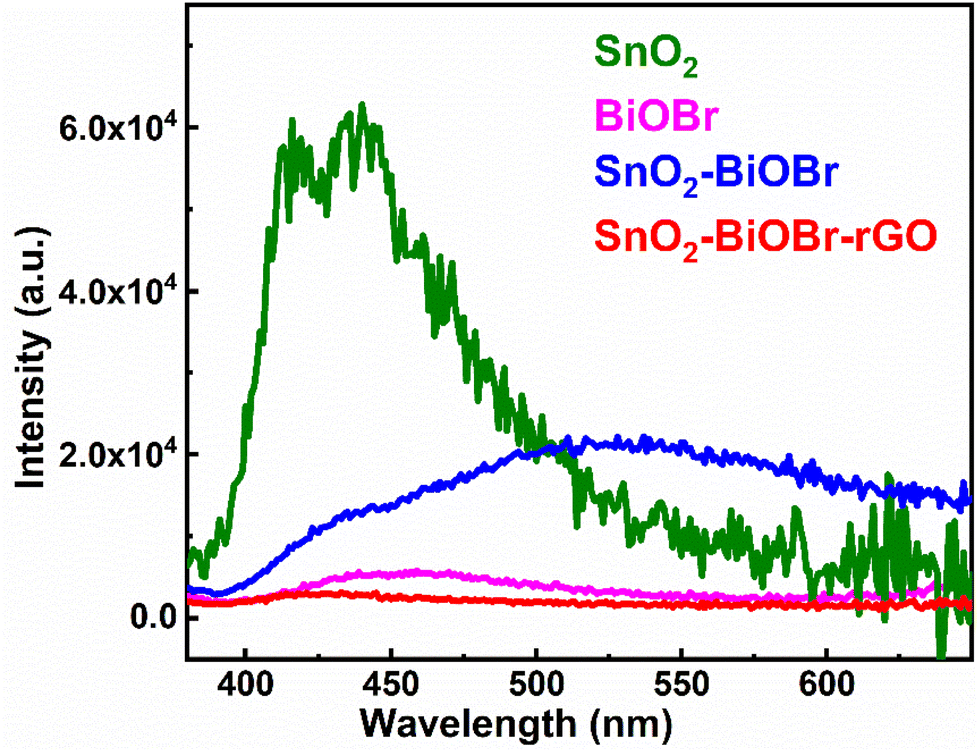
The recombination rate must be restricted, and photogenerated electron and hole pairs must be separated for better photocatalytic performance. Photoluminescence (PL) depends on the recombination rate; if it is low, then the intensity of the PL spectra is also decreased, which means it can be a promising photocatalyst.39 The results of the PL spectra were collected at the excitation wavelength 350 nm, as shown in Fig. 6. SnO2–BiOBr–rGO shows the highest charge separation efficiency as it has the lowest PL intensity.40
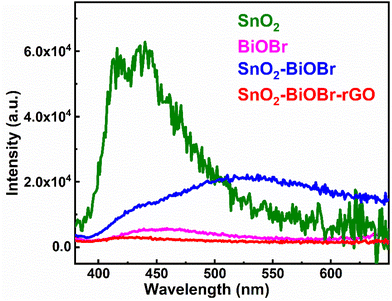 |
| | Fig. 6 Photoluminescence spectra of SnO2, BiOBr, SnO2–BiOBr, and SnO2–BiOBr–rGO. | |
3.3. Elemental analysis
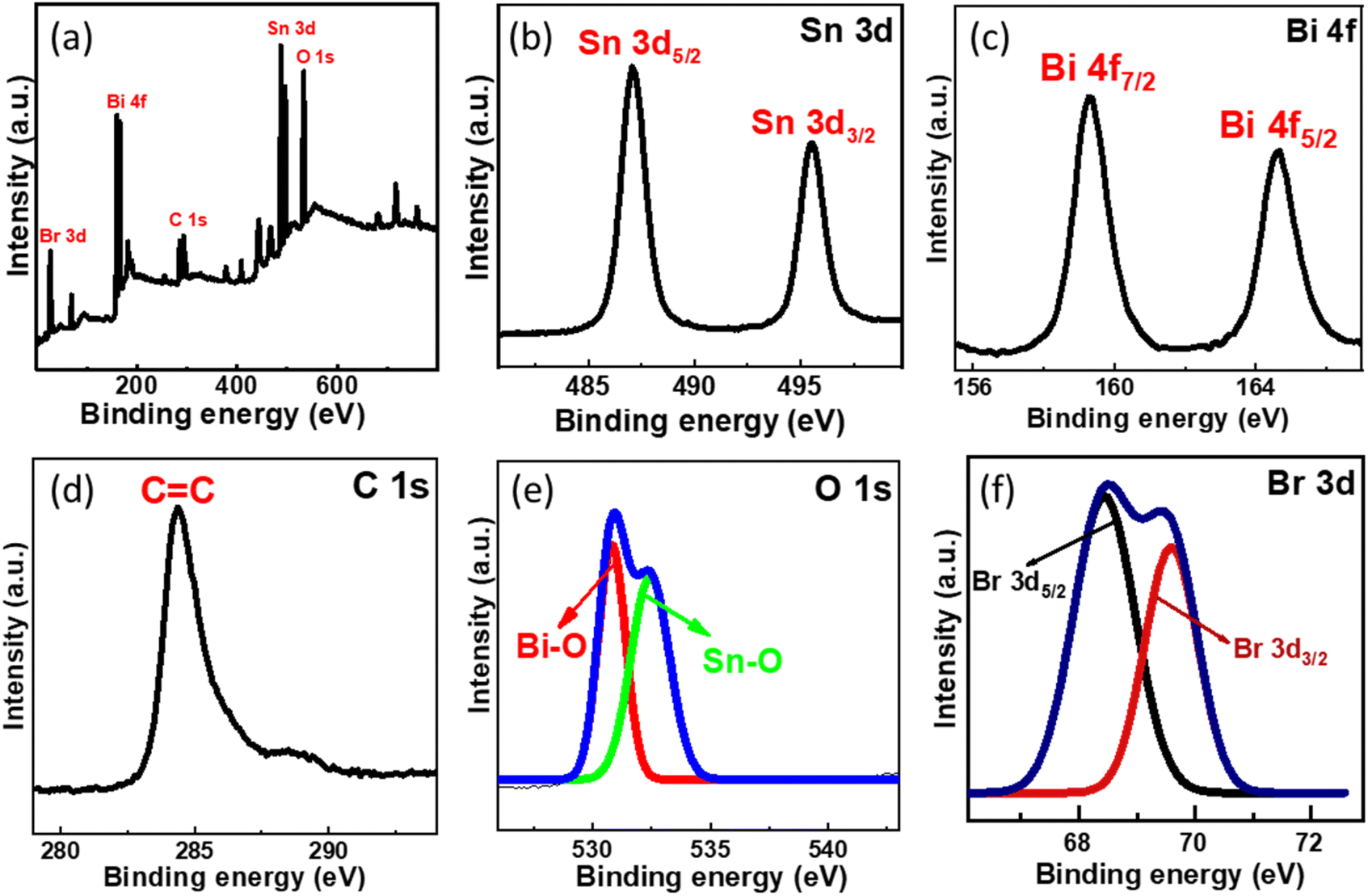
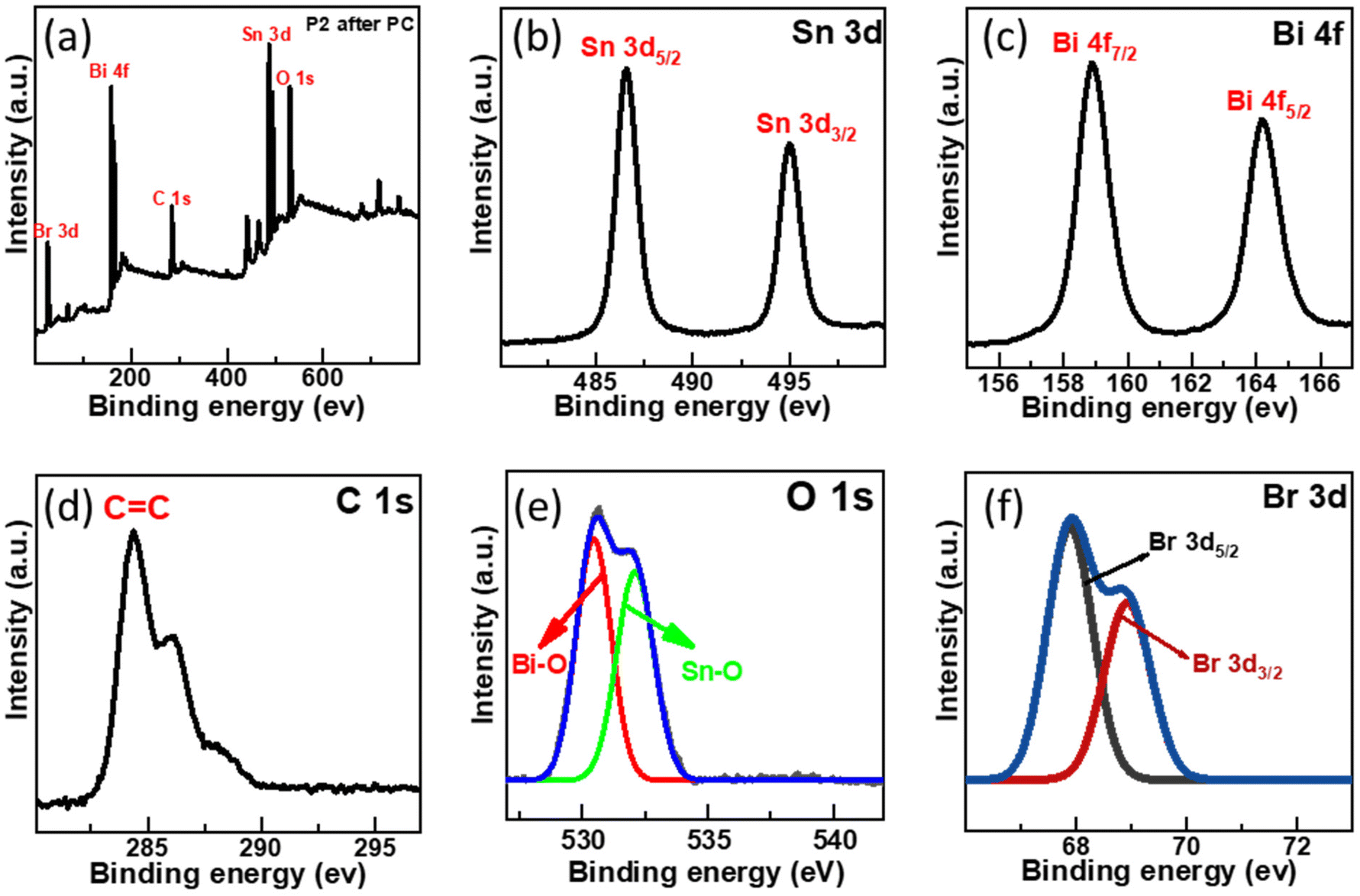
XPS analysis was conducted further to examine the chemical environment of the SnO2–BiOBr–rGO nanocomposite. Sn, Bi, C, O, and Br are all present in the SnO2–BiOBr–rGO ternary nanocomposite, as illustrated in Fig. 7(a), demonstrating that SnO2 was integrated with BiOBr and rGO sheets. The maxima peak of Sn 3d5/2 and Sn 3d3/2 at 487.08 and 495.38 eV confirms the Sn4+ oxidation state in the nanocomposite41 and it shows a slight peak shift towards lower binding energy as compared to pristine SnO2 as shown in Fig. S1.†
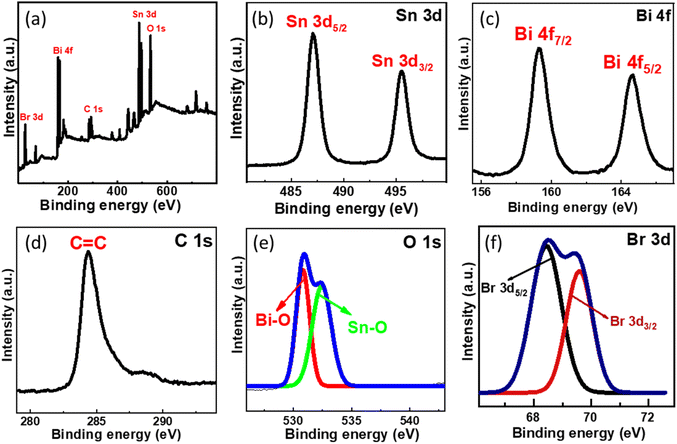 |
| | Fig. 7 (a) Survey XPS spectra of SnO2–BiOBr–rGO and high-resolution XPS spectrum of (b) Sn 3d, (c) Bi 4f, (d) C 1s, (e) O 1s, (f) Br 3d. | |
Two intense peaks present at 164.7 and 159.2 eV in the Bi 4f XPS spectrum of Fig. 7(c) are assigned for the Bi 4f5/2 and Bi 4f7/2 states, respectively, for BiOBr in the SnO2–BiOBr–rGO nanocomposite, showing that Bi was in the +3 state. In addition, when we compare it to BiOBr, the characteristic peak for Bi3+ moved somewhat (by about 0.3 eV) to higher energy locations,42 probably due to significant interactions among SnO2 and BiOBr. Br 3d and O 1s high-resolution spectra both showed similar binding-energy shifts.43 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is present in the composite as shown in Fig. 7(d), where the peak of C 1s is positioned at 284.38 eV.44 As shown in Fig. 7(e), the spectra of O 1s have two peaks that correspond to the Bi–O and Sn–O bonds, which is further confirmed by the image shown in Fig. S2.† The intensity peak at 530.88 eV is due to the Bi–O bond, while the one at 532.38 eV is caused by the Sn–O bond.45,46Fig. 7(f) shows intensity peaks at 68.69, and 69.38 eV for Br 3d5/2 and Br 3d3/2, respectively, for the Br 3d state in the nanocomposite with a shifting of approx. 0.4 eV towards a higher binding energy.23,47
C bond is present in the composite as shown in Fig. 7(d), where the peak of C 1s is positioned at 284.38 eV.44 As shown in Fig. 7(e), the spectra of O 1s have two peaks that correspond to the Bi–O and Sn–O bonds, which is further confirmed by the image shown in Fig. S2.† The intensity peak at 530.88 eV is due to the Bi–O bond, while the one at 532.38 eV is caused by the Sn–O bond.45,46Fig. 7(f) shows intensity peaks at 68.69, and 69.38 eV for Br 3d5/2 and Br 3d3/2, respectively, for the Br 3d state in the nanocomposite with a shifting of approx. 0.4 eV towards a higher binding energy.23,47
3.4. Photocatalysis for organic dye degradation
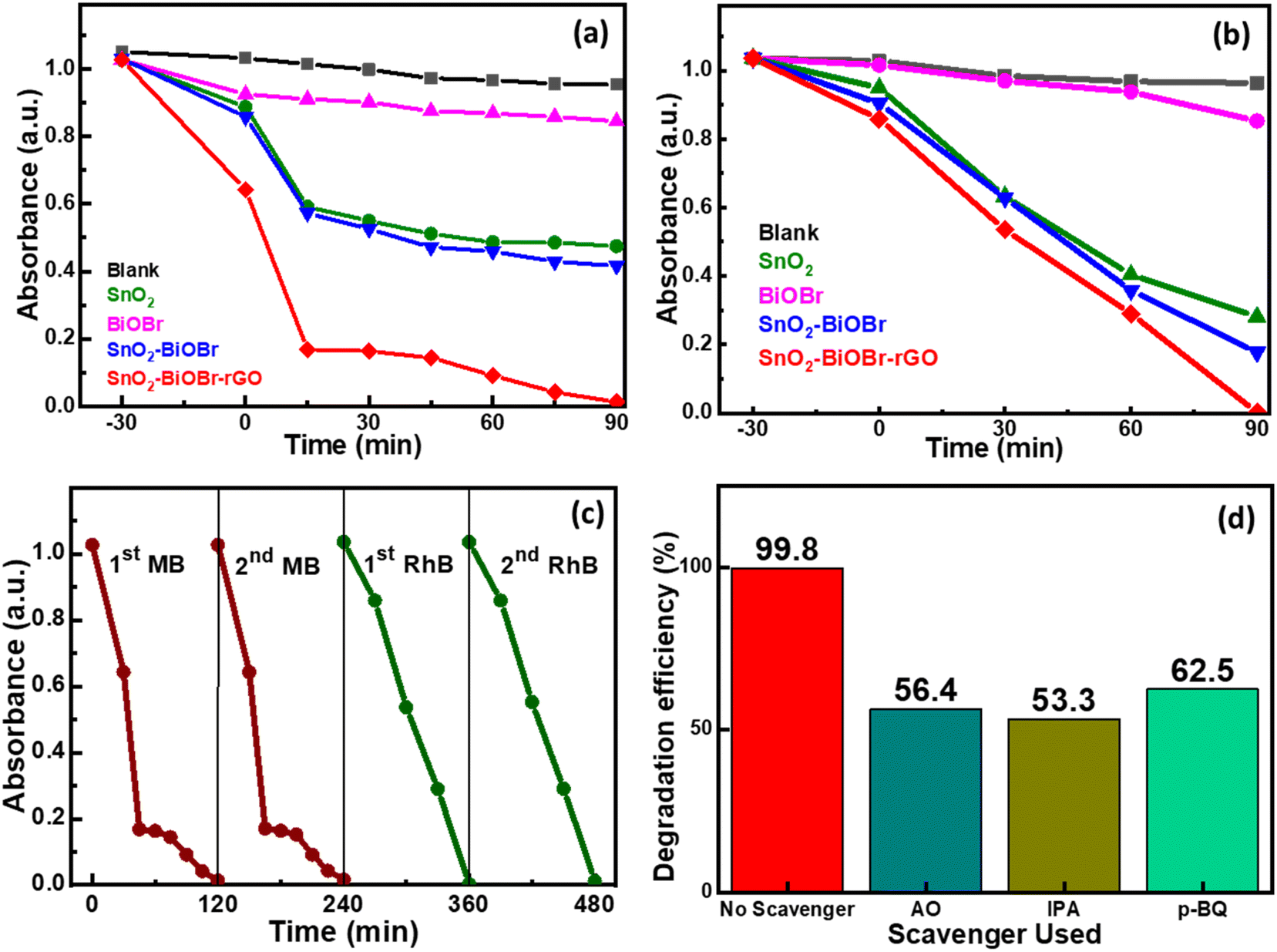
Two model organic dyes MB and RhB were selected to evaluate the photocatalytic performance of SnO2, BiOBr, SnO2–BiOBr, and the SnO2–BiOBr–rGO nanocomposite under visible light irradiation. At first, the solution was put on a magnetic stirrer for 30 minutes under dark conditions to attain the adsorption and desorption equilibrium. The changes in MB absorbance values and for RhB are shown in Fig. 8(a) and (b), respectively. Also, the reaction kinetics for the photodegradation of MB and RhB dyes are shown in ESI Fig. S3 and S4† respectively. As shown, SnO2–BiOBr–rGO has the highest degradation efficiency compared to others; for MB the degradation is ∼99%, and for RhB, it is ∼99.8% in 90 minutes. Hence, heterojunction formation has been proved to increase charge separation and hinder recombination.
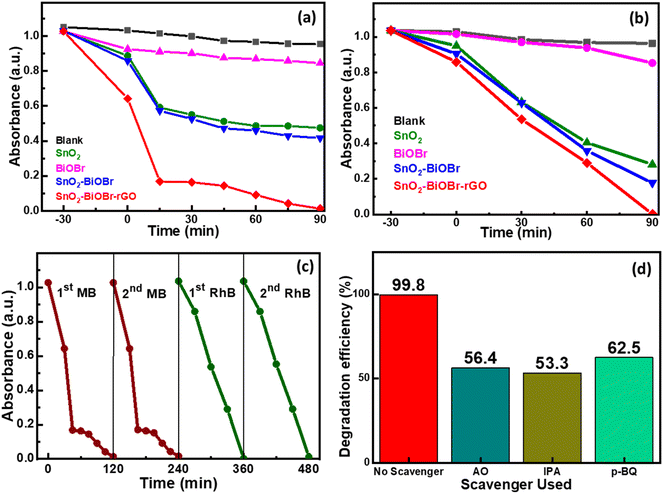 |
| | Fig. 8 Kinetics for (a) MB dye and (b) RhB dye degradation using SnO2, BiOBr, SnO2–BiOBr, and SnO2–BiOBr–rGO as photocatalysts. (c) Repeatability test for two cycles for MB and RhB dyes and (d) the effect of adding p-BQ, IPA, and AO as scavengers in RhB dye. | |
A photolysis experiment was also performed to check the self-degradation of dye under the same conditions as photocatalysis, which showed that no such self-degradation was observed. A repeatability experiment was also performed for MB and RhB dyes to test the reusability of the photocatalyst, as shown in Fig. 8(c). No noticeable change in the degradation rate was observed in the second run compared to the first run. To find the active species involved in the photocatalytic mechanism, scavenger experiments were performed. Three different radical scavengers, including 1,4-benzoquinone (p-BQ), ammonium oxalate (AO), and isopropyl alcohol (IPA) were used in the MB and RhB dye degradation process, and these are the scavengers of superoxide radicals (˙O2−), holes (h+), and hydroxyl radicals (˙OH), respectively.68Fig. 8(d) shows the percentage decline after adding different scavengers in the order IPA > AO > p-BQ. All three scavengers show a decrease in the degradation efficiency, which indicates that ˙O2−, h+, and ˙OH have roles in the degradation process. However, IPA shows the most significant decrease in the degradation rate compared to AO and p-BQ, suggesting that ˙OH is a more active species than h+ and after that ˙O2−.
Table 1 shows the MB degradation efficiencies from published research publications using various photocatalysts. The photocatalysts listed in the table have removed a smaller percentage of pollutants, as observed. Under visible light, only a modest concentration of contaminants, like 10 ppm, was degraded. Therefore, our newly synthesized SnO2–BiOBr–rGO nanocomposite outperformed previously reported photocatalysts considering its facile synthesis process: 99% degradation for MB and 99.8% for RhB under visible light irradiation.
3.5. Possible mechanism for the degradation process
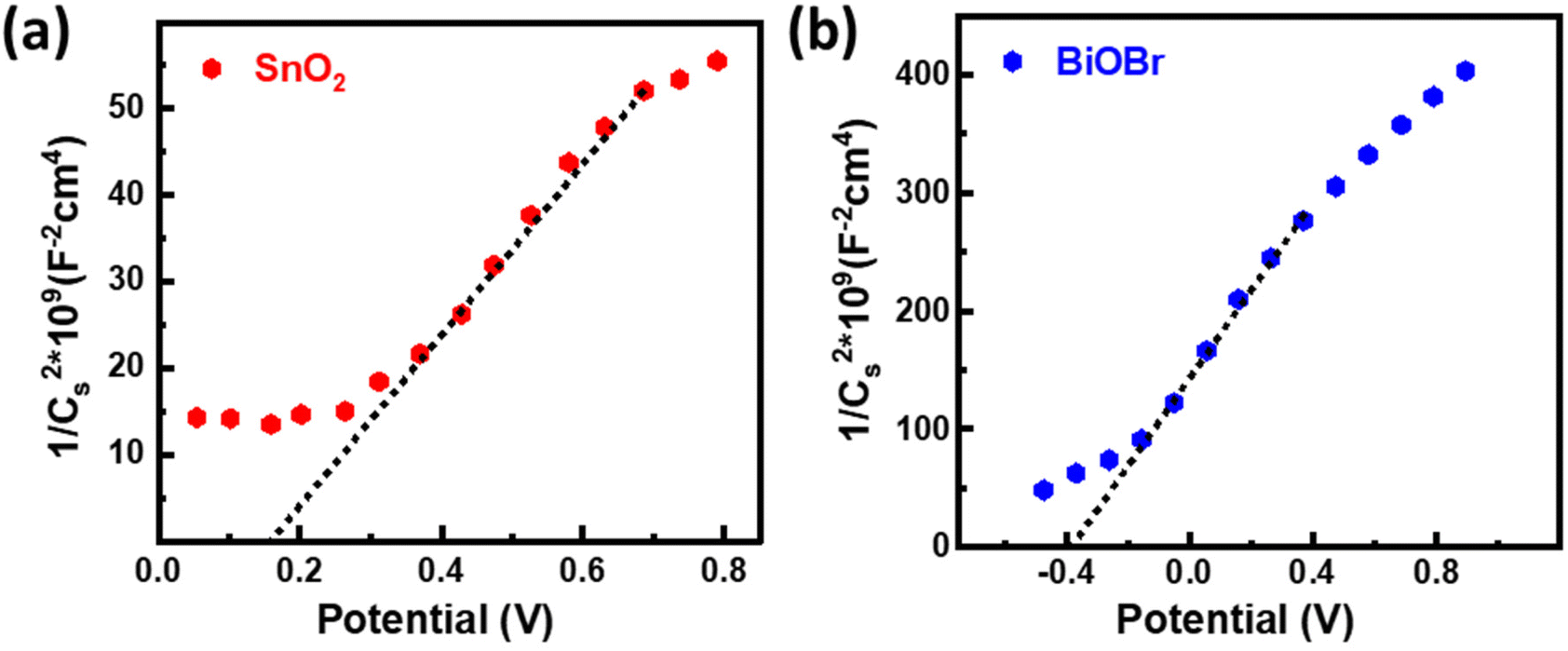
The energy band structure of photocatalysts offered a potential method for photoinduced electrons across heterogeneous interfaces. BiOBr–SnO2 shows type II heterostructure, which integrates many functional components in the nanoscale and significantly enhances photocatalytic performance.54 At the interface of SnO2–BiOBr, there is a banding effect due to the difference in chemical potential. This leads to band bending, which creates a built-in field that promotes the movement of photoinduced charge carriers in opposite directions, resulting in their spatial separation. Thus, type II heterostructure facilitates the charge separation and transfer efficiency of electron and hole pairs generated during light activation. Mott–Schottky's results can be used to identify the n- or p-type semiconductor nanomaterial and find the flat band potential.30 The positive slope confirms the n-type semiconductor of SnO2 and BiOBr for the 1/Cs2 potential curve as shown in Fig. 9. According to the conversion eqn (3),| | | EF (vs. NHE) = EF (vs. SCE) + 0.242 eV | (3) |
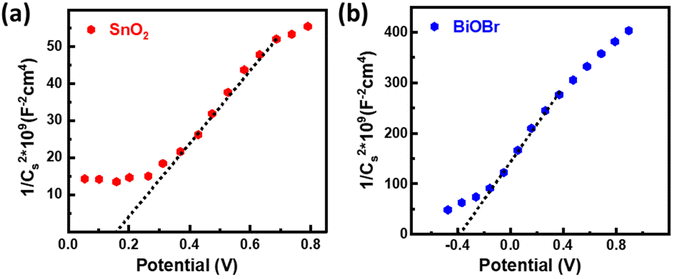 |
| | Fig. 9 Mott–Schottky plots for (a) SnO2 and (b) BiOBr. | |
the flat band potentials for SnO2 and BiOBr were 0.16 and −0.37 eV in SCE, respectively, and 0.402 and −0.128 eV in NHE.55 The value of flat band potential for the n-type is nearly 0.1 eV lower than that of the conduction band (CB) minima, and for the p-type, it is 0.1 eV higher than that of the valence band (VB) maxima.56 So, the conduction band potential (ECB) of SnO2 and BiOBr is equal to 0.502 eV and −0.028 eV. The valence band potential (EVB) of SnO2 and BiOBr can be calculated using eqn (4):
where
Eg is the band gap energy. From the equation,
EVB values are found to be 3.562 eV and 2.542 eV for SnO
2 and BiOBr, respectively as shown in ESI Fig. S5
†vs. NHE.
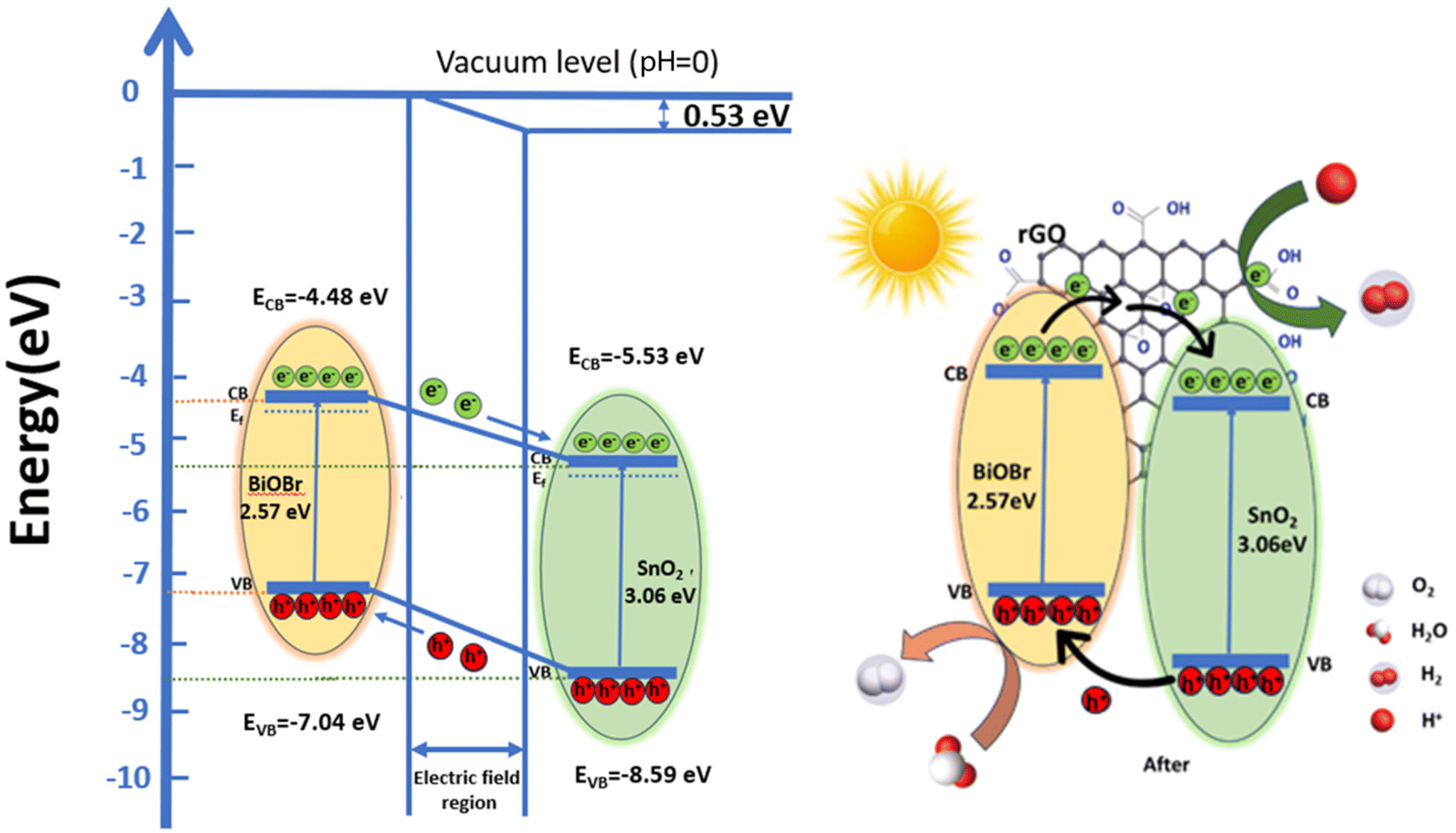
Based on these values, a band structure is proposed demonstrating the photocatalytic mechanism of the ternary nanocomposite in Fig. 10vs. vacuum. For BiOBr, the positions of CB are at −4.48 eV and VB at −7.04 eV, respectively. The CB and VB of SnO2 are −5.53 eV and −8.59 eV, respectively. Electrons move from BiOBr to SnO2 until they reach an equilibrium state.57 As a result, there is an excess of electrons on the SnO2 side which leads to the accumulation of a positive charge on the BiOBr side. This creates a gradient in the distribution of charges and ultimately results in the generation of an electric field at the interface between the two materials.58 This internal electric field extends the migration of charges at the interface of the junction as a result of band bending.59 This bending causes a shift in the band edges, illustrated in Fig. 10 relative to the vacuum level, and the built-in potential is determined to be 0.53 eV. In between the composite materials, rGO serves as a charge transporter.30 Its high electron absorption capacity and wide specific surface area allow it to transfer photogenerated electrons from BiOBr to rGO and subsequently to SnO2, improving charge separation and strengthening the inherent stability of both SnO2 and BiOBr. Furthermore, it aids in preventing photogenerated carrier recombination. Under irradiation, electrons move from the CB of BiOBr to the CB of SnO2, which are subsequently captured by rGO due to its good electron transfer ability and conductivity.29,70 Here, electrons are present on the rGO sheets, which can convert O2 to ˙O2− by reacting with them; these free radicals can be used to oxidize h+ ions to HO2− active radical and subsequently produce the ˙OH radicals.
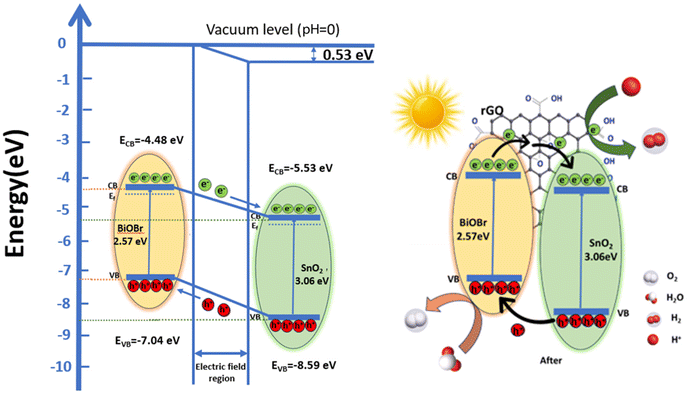 |
| | Fig. 10 Schematic illustration of energy level band bending diagram for BiOBr and SnO2 and a possible mechanism for organic dye degradation under visible light by the ternary nanocomposite (SnO2–BiOBr–rGO). | |
Furthermore, photogenerated holes in SnO2 are transferred to the BiOBr valence band, where they have the potential to convert OH− or H2O to ˙OH. This efficiently separates the electron–hole pairs, demonstrating that the heterostructure accepts rGO; otherwise, there is a strong likelihood that the holes and electrons will recombine. Therefore, by including GO, we may increase photocatalytic effectiveness and speed up dye molecule adsorption. To increase reactivity, the electrons on rGO sheets can be transported for dye degradation in the presence of light.69 Additionally, molecules of RhB can attach to aromatic areas on rGO sheets through π–π interactions.29 Further, h+ can break down pollutants into smaller molecules (such as turning RhB into CO2 and H2O) to have the impact of removing contaminants from the environment.30 Charge transfer in type II staggered heterojunction structures of SnO2–BiOBr with rGO produced a high electron–hole pair separation efficiency and increased photocurrent generation based on the energy band structure, as shown later. So, the photocatalytic activity of the ternary composite SnO2–BiOBr–rGO that was created has been significantly enhanced.
3.6. Elemental verification after photocatalysis
The XPS survey spectra revealed no noticeable shift in the photocatalyst ionic state even after the photocatalysis reaction, as illustrated in Fig. 11. Sn 3d high-resolution spectra exhibit a slight change from 487.08 and 495.38 eV to 486.53 and 494.98 eV, and Bi 4f high-resolution spectra show two peaks at 164.67 and 159.2 eV displaced to 164.2 and 158.9 eV. Br 3d peak energy also changes, going from 68.69 and 69.38 eV to 67.88 and 68.92 eV, respectively.60 These results indicated that Sn, Bi, and Br oxidation states did not change. These findings also showed that SnO2–BiOBr–rGO is highly stable and recyclable during the photocatalysis reaction.
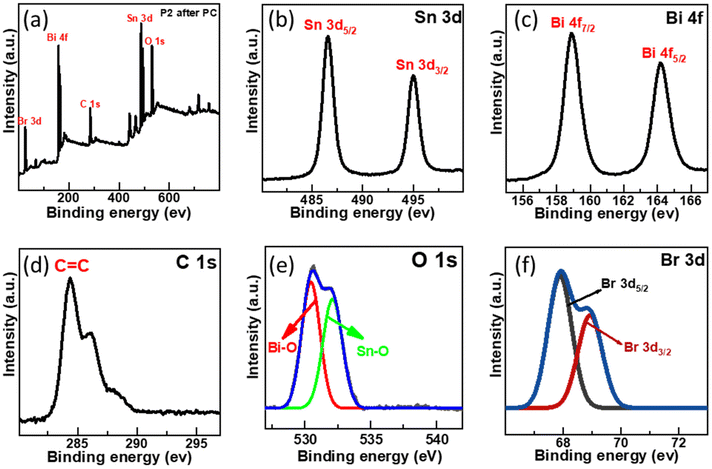 |
| | Fig. 11 (a) After photocatalytic treatment the survey scan of SnO2–BiOBr–rGO and high-resolution XPS spectrum of (b) Sn 3d, (c) Bi 4f, (d) C 1s, (e) O 1s, (f) Br 3d. | |
3.7. Photoelectrochemical oxygen and hydrogen evaluation study
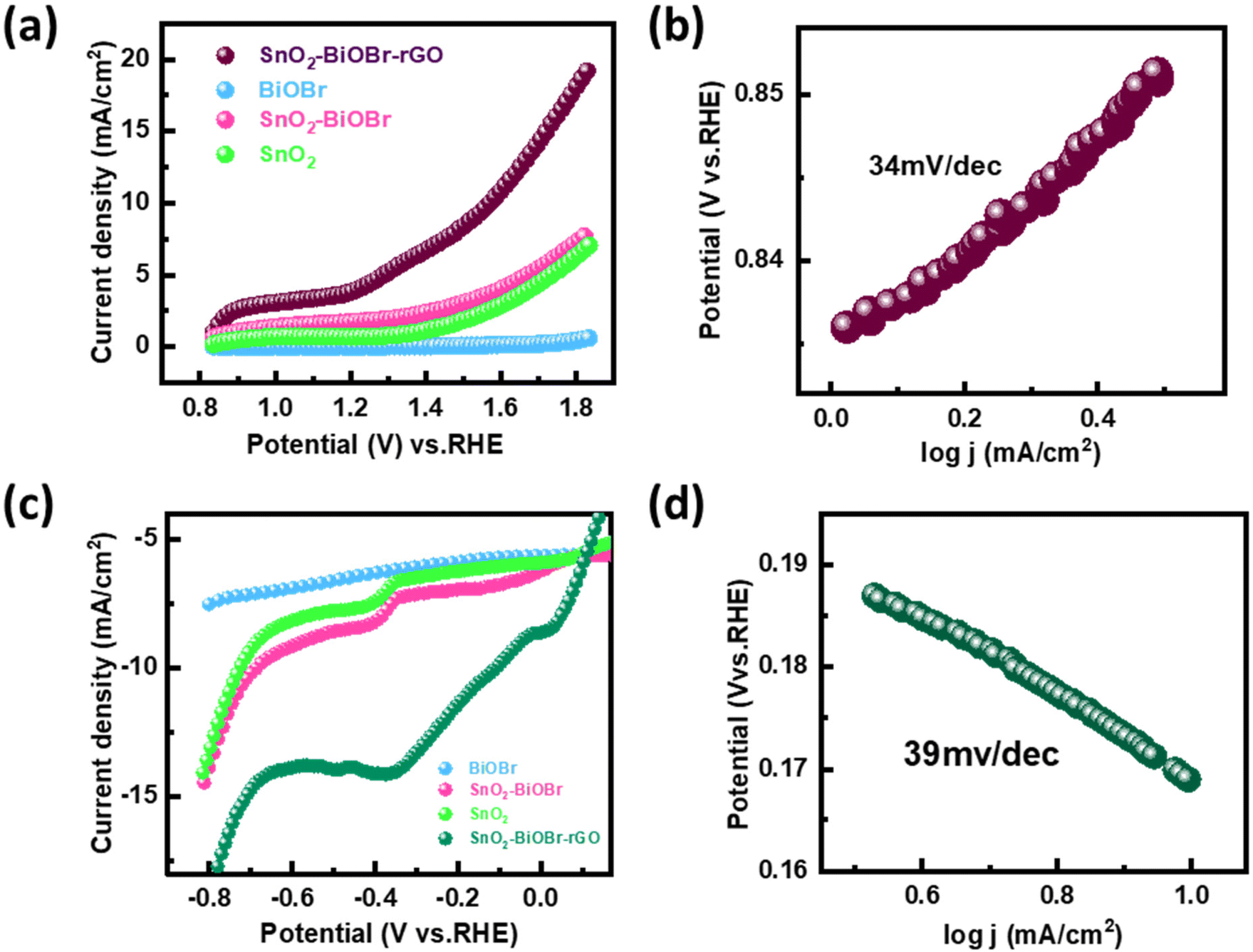
The performance of the synthesized SnO2–BiOBr–rGO sample was assessed for the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER) using a three-electrode quartz cell setup with 0.5 M H2SO4 as the electrolyte in the presence of light. The potential results, presented in Fig. 12, were measured against the reversible hydrogen electrode (RHE).61 The catalyst loading was 50 μg on the conducting side of an ITO substrate with a surface area of 1 cm2 from a 5 mg ml−1 solution. A platinum rod was used as the counter electrode, and a saturated calomel electrode (SCE) was used as the reference electrode. Linear sweep voltammetry (LSV) polarization curves were obtained for the OER in the potential range of 0.8 V to 1.9 V (vs. RHE) and for the HER in the potential range of −0.7 V to 0.1 V (vs. RHE) at a sweep rate of 50 mV s−1 under illumination by visible light. The polarization curves for both reactions showed that the SnO2–BiOBr–rGO sample exhibited significant enhancement in current density at the cathodic and anodic potentials, indicating high OER and HER activities62 as compared to the SnO2, BiOBr, SnO2–BiOBr. Therefore, the LSV plots demonstrated the remarkable OER and HER performance of the SnO2–BiOBr–rGO catalyst.63 The ternary nanocomposite SnO2–BiOBr–rGO exhibited an overpotential of about 0.34 V (vs. RHE) at 10 mA cm−2 for the oxygen evolution reaction (OER). The high catalytic activity of this nanocomposite was further supported by analyzing the Tafel slope, which shows an inherent property of active electrocatalyst materials correlated with the reaction kinetics at the electrode surface. The Tafel slope value of 34 mV dec−1, as depicted in Fig. 12(b), indicated favorable kinetics for the electrochemical OER, attributed to the high surface area and good electrical conductivity of the nanocomposite. For the hydrogen evolution reaction (HER), the nanocomposite showed an overpotential of about −0.11 V (vs. RHE) at 10 mA cm−2. The Tafel plot revealed a slope value of 39 mV dec−1, as shown in Fig. 12(d). These results indicate that the synthesized SnO2–BiOBr–rGO nanocomposite exhibits promising OER and HER catalytic activity with high stability in acidic media64 as compared to pristine SnO2, BiOBr, SnO2–BiOBr.
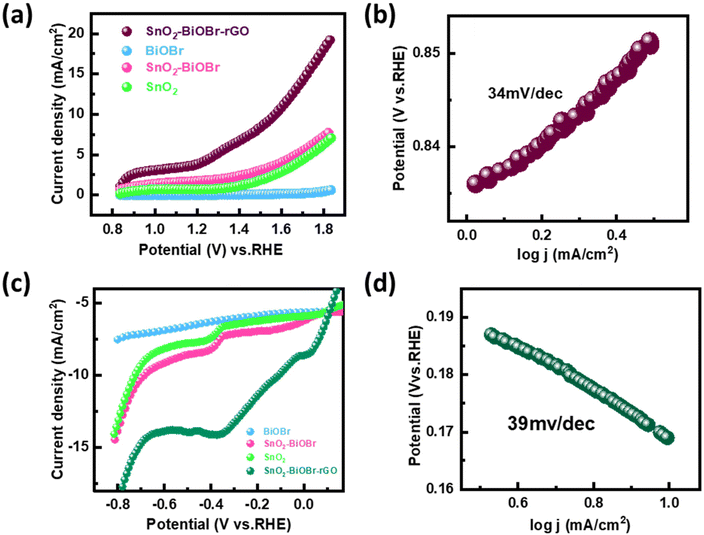 |
| | Fig. 12 (a) Linear sweep voltammetry (LSV) scan for OER performance of BiOBr, SnO2, SnO2–BiOBr, and SnO2–BiOBr–rGO under light irradiation (b) OER Tafel plot for SnO2–BiOBr–rGO (c) LSV scan for HER performance of BiOBr, SnO2, SnO2–BiOBr and SnO2–BiOBr–rGO under light irradiation (d) HER Tafel plot for the SnO2–BiOBr–rGO ternary nanocomposite. | |
3.8. Photoelectrochemical measurements
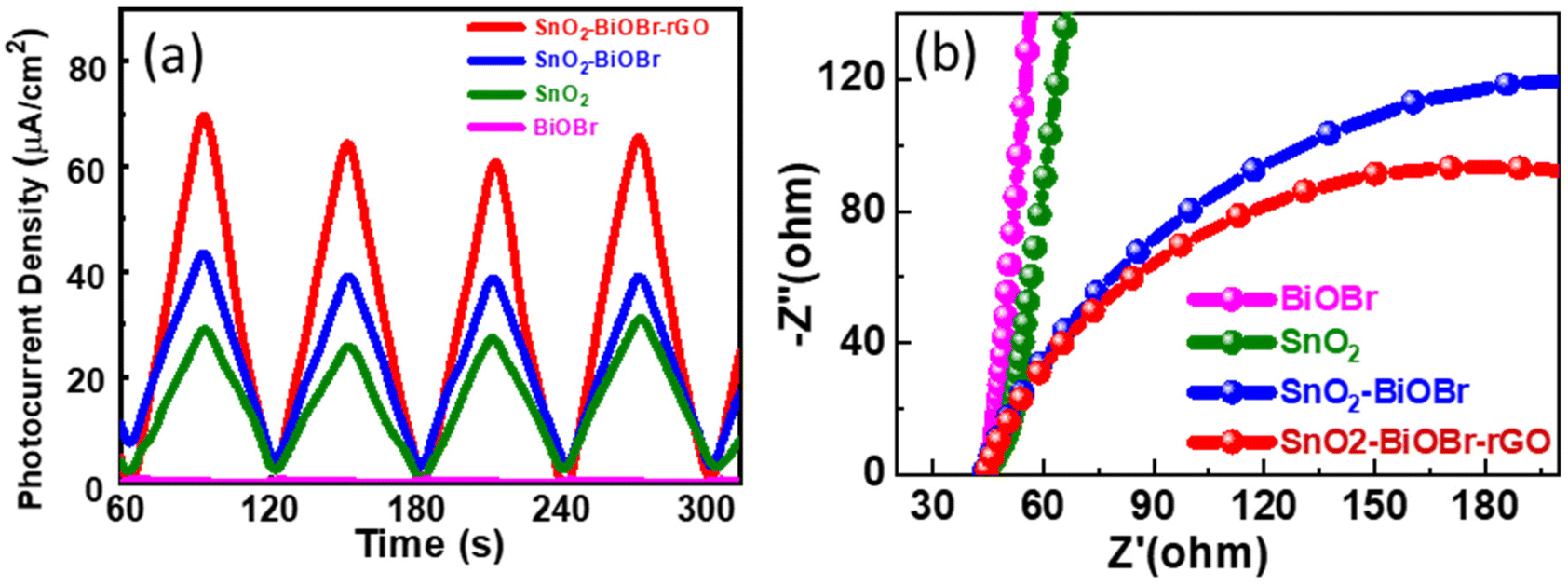
The charge separation efficiency can be determined from the photocurrent density vs. time curve. Fig. 13(a) shows the photocurrent response curves of pure SnO2, BiOBr, SnO2–BiOBr, and the SnO2–BiOBr–rGO nanocomposite at a constant onset potential 0.6 V for four continuous cycles under dark and light conditions. As shown in Fig. 13(a), the photocurrent density of the SnO2–BiOBr–rGO and SnO2–BiOBr composites is higher under the illumination of light than that of SnO2 (28.9 μA cm−2) and BiOBr (0.42 μA cm−2). When the light source is turned on, the maximum photocurrent density of SnO2–BiOBr–rGO is approx. 70.3 μA cm−2 as shown in the photocurrent response curves, much higher than that of pristine SnO2, BiOBr, and SnO2–BiOBr.65
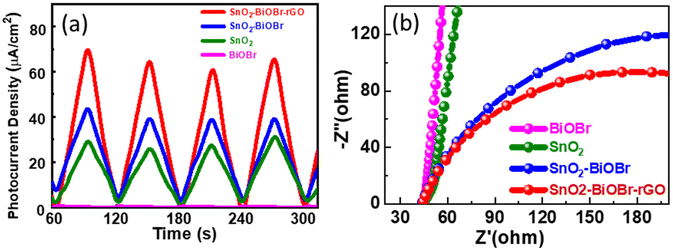 |
| | Fig. 13 (a) Photocurrent response of the SnO2, BiOBr, SnO2–BiOBr, SnO2–BiOBr–rGO and (b) EIS Nyquist plots under light irradiation for SnO2, BiOBr, SnO2–BiOBr, SnO2–BiOBr–rGO. | |
The electric field can justify at the interface between BiOBr and SnO2, the rGO, which helps in the charge separation of electrons and holes, and increases the photocurrent density. Photocurrent response studies reveal that a type II heterojunction exists between SnO2–BiOBr and GO, which helps to separate the photogenerated charge carriers and to enhance the degradation. It is well-known that GO can capture electrons due to its high conductivity. The photocurrent results also confirmed that the electron–hole pair recombination was prohibited due to junction formation. Furthermore, EIS measurement was also used to find the charge separation efficiency. It is well known that greater charge transfer efficiency is correlated with a smaller semicircle radius. Fig. 13(b) shows the EIS Nyquist plots of SnO2, BiOBr, SnO2–BiOBr, and SnO2–BiOBr–rGO. As SnO2–BiOBr–rGO shows the lowest electron transfer resistance, it has the lowest recombination of photogenerated carriers, which results in higher photocurrent response and photodegradation activity.
4. Conclusion
In conclusion, a visible light active ternary nanocomposite SnO2–BiOBr–rGO was synthesized using a facile non-hydrothermal procedure as a highly efficient catalyst with dual properties: one is the photocatalytic degradation of RhB and MB dyes and the second is the photoelectrocatalytic hydrogen and oxygen evolution. The uniform particle size of SnO2 makes its even distribution possible on the sheets of BiOBr which provides very high electron mobility but the recombination rate is high for the photogenerated carriers. The unique property of rGO is that it may readily collect photoelectrons and prevent the recombination of photogenerated charge carriers because it functions as an electron capture agent and converter and also provides more surface area to the active sites for photocatalysis and electrocatalysis. Therefore, when exposed to visible light, the SnO2–BiOBr–rGO composites showed enhanced photocatalytic performance and stability for the photodegradation of MB (99%) and RhB (99.8%). SnO2–BiOBr–rGO also shows a very low overpotential value of 0.34 and −0.11 V (vs. RHE) at 10 mA cm−2 for the OER and HER, respectively, and the Tafel slope values are 34 and 39 mV dec−1 for the OER and HER, respectively. Furthermore, EIS Nyquist and transient photocurrent response studies suggest that the SnO2–BiOBr–rGO composite has a substantially greater photoinduced charge separation efficiency. This work provides useful insights into the formation of heterostructures, as well as ways to tune the band gap for visible light activation and band alignment to efficiently perform the water splitting process. It opens up multiple avenues for multifunctional catalysts, such as in energy conversion, dye degradation, and storage, among other areas.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. Dhillon A. Naskar, and A. K. Basu would like to acknowledge the Institute of Nano Science and Technology Mohali for the instrumentation and financial support. A. Saha is thankful for the financial support from the SERB Start-up Grant (SRG/2019/001018) and CSIR-4M(FBR) grant. N. Kaushal acknowledges the DST-Inspire fellowship for its financial support (DST/Inspire/03/2019/001918).
References
- J. Shah, A. Shukla and R. K. Kotnala, ACS Sustainable Chem. Eng., 2021, 9, 15229 CrossRef CAS.
- R. Jeyagopal, Y. Chen, M. Ramadoss, K. Marimuthu, B. Wang, W. Li and X. Zhang, Nanoscale, 2020, 12, 3879 RSC.
- A. Various, B. Likozar and A. K. D. Alsukaibi, Processes, 2022, 10, 1968 CrossRef.
- D. Chatterjee and S. Dasgupta, J. Photochem. Photobiol., C, 2005, 6, 186 CrossRef CAS.
- N. Kaushal, S. Sarraf, A. K. Basu, S. Mishra and A. Saha, Mater. Chem. Phys., 2024, 314, 128823 CrossRef CAS.
- M. M. Ali, D. J. Williams and M. S. Banu, J. Polym. Res., 2020, 27, 1 CrossRef.
- S. Sarraf, N. Kaushal, V. Chugh, A. Sundar, A. Saha and A. K. Basu, J. Mater. Sci., 2024, 59, 1513 CrossRef CAS.
- M. Fazil, S. M. Alshehri, Y. Mao and T. Ahmad, Langmuir, 2024, 40(8), 4063–4076 CrossRef CAS PubMed.
- J. R. Hemmerling, A. Mathur and S. Linic, Acc. Chem. Res., 2021, 54, 1992 CrossRef CAS PubMed.
- R. Li, T. Li and Q. Zhou, Catalysts, 2020, 10, 804 CrossRef CAS.
- J. C. G. Bünzli and A. S. Chauvin, Handb. Phys. Chem. Rare Earths, 2014, 44, 169 Search PubMed.
- G. X. Castillo-Cabrera, P. J. Espinoza-Montero, P. Alulema-Pullupaxi, J. R. Mora and M. H. Villacís-García, Front. Chem., 2022, 10, 900622 CrossRef PubMed.
- X. D. Dong, Y. M. Zhang and Z. Y. Zhao, Inorg. Chem., 2021, 60, 8461 CrossRef CAS PubMed.
- S. S. Imam, R. Adnan and N. H. Mohd Kaus, J. Environ. Chem. Eng., 2021, 9, 105404 CrossRef CAS.
- L. Meng, Y. Qu and L. Jing, Chin. Chem. Lett., 2021, 32, 3265 CrossRef CAS.
- W. An, W. Cui, Y. Liang, J. Hu and L. Liu, Appl. Surf. Sci., 2015, 351, 1131 CrossRef CAS.
- Y. Tang, D. Zhang, X. Pu, B. Ge, Y. Li and Y. Huang, J. Taiwan Inst. Chem. Eng., 2019, 96, 487 CrossRef CAS.
- X. Jiang, H. Tan, X. Shi, X. Cheng, W. Hu and X. Hu, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2022, 37, 801 CrossRef CAS.
- X. Zheng, L. Feng, Y. Dou, H. Guo, Y. Liang, G. Li, J. He, P. Liu and J. He, ACS Nano, 2021, 15, 13209 CrossRef CAS.
- J. Xu, L. Li, C. Guo, Y. Zhang and S. Wang, Chem. Eng. J., 2013, 221, 230 CrossRef CAS.
- A. Zuorro, J. R. Dominguez, J. García Rodríguez, J. A. Peres, Z. Frontistis, J. Luisa Alves do Nascimento, L. Chantelle, I. Maria Garcia dos Santos, A. Luiz Menezes de Oliveira and M. Cristina Ferreira Alves, Catalysts, 2022, 12, 428 CrossRef.
- L. Peng, Y. Xiao, X.-l. Wang, D.-w. Feng, H. Yu and X.-t. Dong, ChemistrySelect, 2019, 4, 8460 CrossRef CAS.
- H. Liu, C. Du, M. Li, S. Zhang, H. Bai, L. Yang and S. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 28686 CrossRef CAS.
- X. J. Wen, C. G. Niu, L. Zhang and G. M. Zeng, ACS Sustainable Chem. Eng., 2017, 5, 5134 CrossRef CAS.
- W. Zhang, J. Fu, Y. Wang, X. Zhang and J. Li, J. Phys. Chem. Solids, 2019, 127, 19 CrossRef CAS.
- L. K. Putri, L. L. Tan, W. J. Ong, W. S. Chang and S. P. Chai, Appl. Mater. Today, 2016, 4, 9 CrossRef.
- H. Liu, C. Du, M. Li, S. Zhang, H. Bai, L. Yang and S. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 28686 CrossRef CAS PubMed.
- X. J. Wen, C. G. Niu, L. Zhang and G. M. Zeng, ACS Sustainable Chem. Eng., 2017, 5, 5134 CrossRef CAS.
- C. Li, B. Wang, F. Zhang, N. Song, G. Liu, C. Wang and S. Zhong, J. Mater. Res. Technol., 2020, 9, 610 CrossRef CAS.
- Y. Liu, Y. Zhou, P. Zhu, X. Luo, J. Chen, Y. Li, Q. Qiu and T. Xie, J. Mol. Liq., 2023, 378, 121621 CrossRef CAS.
- R. S. Dey, H. A. Hjuler and Q. Chi, J. Mater. Chem. A, 2015, 3, 6324 RSC.
- K. Sa, P. C. Mahakul, B. V. R. S. Subramanyam, J. Raiguru, S. Das, I. Alam and P. Mahanandia, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 338, 012055 Search PubMed.
- G. Yasin, M. Arif, M. Shakeel, Y. Dun, Y. Zuo, W. Q. Khan, Y. Tang, A. Khan and M. Nadeem, Adv. Eng. Mater., 2018, 20, 1701166 CrossRef.
- M. Bellardita, A. Di Paola, B. Megna and L. Palmisano, J. Photochem. Photobiol., A, 2018, 367, 312 CrossRef CAS.
- W. Li, Y. Zou, X. Geng, F. Xiao, G. An and D. Wang, Mol. Catal., 2017, 438, 19 CrossRef CAS.
- H. J. Qin, Y. H. Zhang, Z. Wang and G. H. Yang, Catalysts, 2022, 12, 719 CrossRef CAS.
- K. Xiao, H. Huang, N. Tian and Y. Zhang, Mater. Res. Bull., 2016, 83, 172 CrossRef CAS.
- P. A. Luque, O. Nava, C. A. Soto-Robles, H. E. Garrafa-Galvez, M. E. Martínez-Rosas, M. J. Chinchillas-Chinchillas, A. R. Vilchis-Nestor and A. Castro-Beltrán, J. Mater. Sci.: Mater. Electron., 2020, 31, 16859 CrossRef CAS.
- M. K. Singh and M. S. Mehata, Opt. Mater., 2020, 109, 110309 CrossRef CAS.
- A. Senthilraja, B. Subash, B. Krishnakumar, D. Rajamanickam, M. Swaminathan and M. Shanthi, Mater. Sci. Semicond. Process., 2014, 22, 83 CrossRef CAS.
- A. Akhundi and A. Habibi-Yan Gjeh, Mater. Express, 2015, 5, 309 CrossRef CAS.
- Y. Miao, H. Yin, L. Peng, Y. Huo and H. Li, RSC Adv., 2016, 6, 13498 RSC.
- A. A. P. Khan, Sonu, A. Sudhaik, P. Raizada, A. Khan, M. A. Rub, N. Azum, M. M. Alotaibi, P. Singh and A. M. Asiri, Catal. Commun., 2023, 179, 106685 CrossRef CAS.
- K. Spilarewicz-Stanek, A. Jakimińska, A. Kisielewska, M. Dudek and I. Piwoński, Mater. Sci. Semicond. Process., 2021, 123, 105525 CrossRef CAS.
- X. J. Wen, C. G. Niu, L. Zhang and G. M. Zeng, ACS Sustainable Chem. Eng., 2017, 5, 5134 CrossRef CAS.
- S. Sharma, V. Dutta, P. Raizada, V. Kumar Thakur, A. K. Saini, D. Mittal, V. H. Nguyen, T. Ahamad, C. Chien Nguyen, S. Young Kim, Q. Van Le and P. Singh, Mater. Lett., 2022, 313, 131716 CrossRef CAS.
- A. A. Parwaz Khan, P. Singh, P. Raizada, A. Khan, A. M. Asiri and M. M. Alotaibi, Chemosphere, 2023, 316, 137839 CrossRef CAS.
- S. K. Tammina, B. K. Mandal and N. K. Kadiyala, Environ. Nanotechnol., Monit. Manage., 2018, 10, 339 Search PubMed.
- K. Bhuvaneswari, B. S. Nguyen, V. H. Nguyen, V. Q. Nguyen, Q. H. Nguyen, G. Palanisamy, K. Sivashanmugan and T. Pazhanivel, Mater. Lett., 2020, 276, 128173 CrossRef CAS.
- S. Vadivel, M. Vanitha, A. Muthukrishnaraj and N. Balasubramanian, J. Water Process Eng., 2014, 1, 17 CrossRef.
- Z. M. Alaizeri, H. A. Alhadlaq, S. Aldawood, M. J. Akhtar and M. Ahamed, Polymers, 2022, 14, 2036 CrossRef CAS.
- M. Cai, A. Shui and B. Du, Surf. Interfaces, 2023, 37, 102643 CrossRef CAS.
- A. Rani, K. Singh, A. S. Patel, A. Chakraborti, S. Kumar, K. Ghosh and P. Sharma, Chem. Phys. Lett., 2020, 738, 136874 CrossRef CAS.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326 RSC.
- X. Yao, H. Zhen, D. Zhang, J. Liu, X. Pu and P. Cai, Colloids Surf., A, 2022, 648, 129276 CrossRef CAS.
- B. A. Pinaud, Z. Chen, D. N. Abram and T. F. Jaramillo, J. Phys. Chem. C, 2011, 115, 11830 CrossRef CAS.
- R. Kumar, A. Sudhaik, Sonu, P. Raizada, V. H. Nguyen, Q. Van Le, T. Ahamad, S. Thakur, C. M. Hussain and P. Singh, Chemosphere, 2023, 337, 139267 CrossRef CAS PubMed.
- M. Malhotra, A. Sudhaik, Sonu, P. Raizada, T. Ahamad, V. H. Nguyen, Q. Van Le, R. Selvasembian, A. K. Mishra and P. Singh, Ind. Crops Prod., 2023, 202, 117000 CrossRef CAS.
- V. Dutta, S. Sonu, P. Raizada, V. K. Thakur, T. Ahamad, S. Thakur, P. Kumar Verma, H. H. P. Quang, V. H. Nguyen and P. Singh, Environ. Sci. Pollut. Res., 2023, 30, 124530 CrossRef CAS PubMed.
- Y. Guo, C. H. Lay, D. Zhou, S. Dong, J. Zhang and N. Ren, Environ. Sci. Pollut. Res., 2020, 27, 17516 CrossRef CAS PubMed.
- X. Wang, Z. Chen, J. Zheng, Y. Li, X. Peng, X. Zhang, H. Yin, X. Xiong, J. Duan, X. Li, Z. Wang, Z. Chen, J. Han, W. Xiao and Y. Yao, ACS Appl. Energy Mater., 2020, 3, 11848 CrossRef CAS.
- P. Senthilkumar, M. Mohapatra and S. Basu, RSC Adv., 2022, 12, 1287 RSC.
- R. Daiyan, X. Lu, W. H. Saputera, Y. H. Ng and R. Amal, ACS Sustainable Chem. Eng., 2018, 6, 1670 CrossRef CAS.
- Bhawna, S. Kumar, R. Sharma, A. Gupta, A. Tyagi, P. Singh, A. Kumar and V. Kumar, New J. Chem., 2022, 46, 4014 RSC.
- P. Li, H. Hu, J. Xu, H. Jing, H. Peng, J. Lu, C. Wu and S. Ai, Appl. Catal., B, 2014, 147, 912 CrossRef CAS.
- V. Chugh, A. Basu, A. Kaushik, Manshu, S. Bhansali and A. K. Basu, Nanoscale, 2024, 16, 5458–5486 RSC.
- N. Kaushal, A. Jain, A. Kumar, S. Sarraf, A. K. Basu, C.I. Raje and A. Saha, ChemPlusChem, 2023, 88, e202300125 CrossRef CAS PubMed.
- S. Sarraf, Manshu, V. Chugh, A. Saha and A. K. Basu, NanoWorld J, 2023, 9, S57–S61 Search PubMed.
- A. K. Basu, A. N. Sah, A. Pradhan and S. Bhattacharya, Sci. Rep., 2019, 9, 3686 CrossRef PubMed.
- A. K. Basu, P. S. Chauhan, M. Awasthi and S. Bhattacharya, Appl. Surf. Sci., 2019, 465, 56–66 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ce and
Aviru Kumar
Basu
ce and
Aviru Kumar
Basu