
Ligand biodegradation-induced surface reconstruction of magnetite nanoparticles: potentially overlooked toxicity†
Huiru
Zhao
ab,
Hongyan
Meng
b,
Qiurong
Zhang
c,
Yining
Wu
a,
Haotian
Chen
d,
Xiaoman
Jiang
a and
Chengdong
Zhang
 *a
*a
aSchool of Environment, Beijing Normal University, Beijing 100857, China. E-mail: zhangchengdong@bnu.edu.cn
bCollege of Environmental Science and Engineering, Nankai University, Tianjin 300350, China
cInstitutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China
dSchool of Electronics Engineering and Computer Science, Peking University, Beijing 100871, China
First published on 24th November 2021
Abstract
The coating of nanoparticles (NPs) with biodegradable ligands has been considered an efficient way to improve the biocompatibility of NPs and to decrease their biopersistence. However, the role of ligand biodegradation in reshaping the core material and the resulting change in toxicity have not been fully explored. In this study, magnetite (Fe3O4) NPs coated with a high or low loading of hyaluronic acid (termed Fe3O4–HAH and Fe3O4–HAL) were synthesized. Fe3O4–HAR was obtained after the enzymatic degradation of Fe3O4–HAH by a hyaluronidase, which had a coating weight similar to that of Fe3O4–HAL, as verified by thermogravimetric analysis. Interestingly, among the three NPs, Fe3O4–HAR induced the highest rate of cellular damage. In particular, 2 times more hydroxyl radicals were detected in cells stressed by Fe3O4–HAR than in cells stressed by Fe3O4–HAH, despite the highest cellular uptake being observed for Fe3O4–HAH because of the ligand–receptor interactions between hyaluronic acid and membrane receptors. The structural characterization results revealed that ligand degradation induced notable surface reconstruction of the Fe3O4 core, showing a characteristic Fe3O4@Fe2O3 structure with surface-bound ferrous ions. This unique structure endowed Fe3O4–HAR with superior competency in initiating the Fenton reaction and accelerating the Fe(III)/Fe(II) cycle. Our results emphasize the importance of tracing the in vivo biotransformation of NPs from all of the different architectural parts, particularly deciphering the interplay among the core, shell, surface ligands, and surrounding ions.
Environmental significanceGenerally, coating with biodegradable ligands is regarded as an efficient way to enhance the biocompatibility of nanoparticles and reduce their potential persistence in vivo. However, how the biodegradation of surface coatings is implicated in core material reconstruction is not fully understood and may alter their surface reactivity and potential toxicity. Our results revealed that the enzymatic degradation of hyaluronic acid, a commonly used ligand, induced the surface reconstruction of magnetite nanoparticles. These reconstructed nanoparticles exhibited a high capability for producing hydroxyl radicals, thus boosting cytotoxicity. Therefore, it is imperative to monitor nanoparticle biotransformation from all of the different architectural parts, with particular concern on the interplay among the core, shell, surface coating, and surrounding solutes. |
1. Introduction
The design of nanoparticles (NPs) is important for their effective application, including tuning their intrinsic characteristics (i.e., chemical composition,1 size,2 and shape3) and employing a variety of surface ligands.4 Ligands play important roles in maintaining the colloidal stability of NPs and the encoding of various functionalities.5 For biomedical applications, biodegradability is highly desired, and various biodegradable ligands have been used to decorate NPs, such as poly(ethylene glycol) (which has a high degradation efficacy for low-molecular-weight structures),6 hyaluronic acid,7 and citric acid.8 However, the change in physicochemical properties of NPs due to ligand degradation may jeopardize the function of NPs and change their biodistribution, as well as cytotoxicity.7,9 From the perspective of nanosafety, it is important to consider the potential complications caused by ligand biotransformation.The current knowledge with respect to ligand biotransformation is described in the context of degradation mechanisms, resulting in changed NP properties and complicated biological outcomes. Ligand alteration may occur via hydrolysis in an acidic biological medium, cleavage by biogenerated reactive oxygen species (ROS), enzymatic degradation and displacement by other biomolecules.7,10,11 For example, the shell of an amphiphilic polymer on a gold NP surface, composed of poly(isobutylene-alt-maleic anhydride)-graft-dodecyl, underwent degradation by proteolytic enzymes in the liver.12 The citrate ligand, which was physically adsorbed on gold NPs, can be replaced by lipid molecules (i.e., 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) during interactions with the membrane.13 Consequently, the loss of the surface coating resulted in variations in the surface charges and topologies, decreased colloidal stability and agglomerated NPs.7,14 The important biological consequences of ligand degradation include impaired target function of NPs, change in persistence in the body,7 and increased toxicity.14 For instance, an intact dense polymer coating endows NPs with excellent colloidal stability, thus exhibiting a longer blood circulation time than NPs with degraded coatings or with less coating.15 A dense coating on a lead sulfide surface increased particle biocompatibility, whereas the loss of the coating may exaggerate the potential of lead ion release, thus causing long-term in vivo toxicity.16 Nevertheless, how the degradation of the surface coating influences the reshaping of the core material has not been fully explored. In particular, the dissolution of the core material, surface remodeling, passivation, and crystalline reorganization may occur simultaneously during surface ligand transformation. Systematic monitoring of core material changes during ligand transformation may provide useful information for assessing biodurability, biocompatibility and biosafety in the short and long term.
In this study, magnetite (Fe3O4) NPs were chosen because of their potential biomedical applications, such as magnetic bioseparation, detection, clinical diagnosis and therapy.7,17 Polyanionic hyaluronic acid (HA) was used as a coating ligand, which consists of two alternating units, namely, glucuronic acid and N-acetylglucosamine (Fig. S1†). HA is present in the skin, joints, and cornea at high concentrations, and it has been used to assemble nanoparticles to achieve highly efficient targeted therapy and imaging of tumors.18 Moreover, hyaluronidase (HAase), which can break down hyaluronic acid (Fig. S1†), is present in various organs and body fluids,19 and the enzyme activity varies considerably depending on the microenvironment pH, HA content, organ and pathological status.19 Hence, the effect of HA degradation by HAase on the changes in the Fe3O4 core was systematically investigated.
The aim of this study was 1) to investigate the alterations of the physicochemical properties and cytotoxicity of Fe3O4 NPs after enzymatic degradation of HA as the coating ligand and 2) to elucidate the reshaping progress of the Fe3O4 core during HA degradation. Our results emphasize the importance of assessing NP biotransformation, and considerable efforts should be made to comprehend the essential mechanisms relating to the fate of NPs in biological environments.
2. Materials and methods
2.1 Materials
Ferric chloride hexahydrate (FeCl3·6H2O), ferrous dichloride tetrahydrate (FeCl2·4H2O) and HA (4.0 kDa) were purchased from Meilun Biology, China. HAase (from sheep testicular tissues) was purchased from Yuanye Biology, China. Ammonia water and hydrochloric acid (HCl) were obtained from Jiangtian Chemical Engineering, China. Standard iron solutions were from Luoen, China. Hydrogen peroxide solution (H2O2, 30% v/v) was obtained from Honeywell, USA. A hydroxyphenyl fluorescein (HPF) probe was purchased from Cayman, USA. Murine macrophage cell line J774.A1 was obtained from Tianjin Dingguo Biotechnology Co., Ltd. All reagents used in this study were of analytical grade or better.2.2 Synthesis of HA-coated Fe3O4 NPs
NPs were synthesized according to Huige's method with some modifications7 (Fig. 1). Briefly, 1.0 g or 0.4 g of HA was used to synthesize NPs with a high or low surface coating, which were termed Fe3O4–HAH and Fe3O4–HAL, respectively. HA was dissolved in 100 mL of water, and then nitrogen was bubbled through the solution for 30 min. The HA solution was boiled at 100 °C in an oil bath, followed by the addition of iron solution, which was prepared by dissolving FeCl3·6H2O (0.29 g, 0.54 mmol L−1) and FeCl2·4H2O (0.14 g, 0.36 mmol L−1) in dilute HCl solution (4 mL, 1.0 mol L−1). The process was performed in a nitrogen atmosphere with vigorous stirring. Ammonia solution (30 mL, 28% w/w) was added dropwise to adjust the pH to 9–10. Then, the mixture was refluxed for 40 min, cooled to room temperature and dialyzed for 72 h in a Spectra/Por dialysis bag (with a molecular weight cut off of 12–14 kDa) against deionized water. A total of eight exchanges were performed to remove unreacted reagents and soluble by-products. Finally, the substance in the dialysis bag (pH 7.0) was collected via filtration through a nylon membrane filter (with a diameter of 25 mm and a pore size of 0.22 μm) and then lyophilized for further use. | ||
| Fig. 1 Schematic of the synthesis of various NPs. | ||
To obtain the NPs after ligand biodegradation (termed Fe3O4–HAR), 4 g L−1 HAase (300–500 U mg−1) and 200 mg Fe L−1 Fe3O4–HAH were dissolved in 45 mL of pH 4.5 phosphate-buffered saline (PBS), and the mixture was shaken at 37 °C for 6 h. After that, the mixture was centrifuged, washed with 30 mL of ethanol three times, and then lyophilized for further use. If not indicated, the iron concentration (which was determined by inductively coupled plasma mass spectrometry (ICP-MS, Elan drc-e, PerkinElmer, USA)) instead of the overall NP concentration was used throughout the study. Particularly, all subsequent experiments involving the NPs were performed in the dark to exclude light interference.
2.3 Characterization of the various NPs
The morphologies of the NPs were visualized with transmission electron microscopy (TEM) at an accelerating voltage of 200 kV (JEM-2800F, Hitachi, Japan). The X-ray diffraction (XRD) patterns were obtained using a D/Max-2500 diffractometer (Rigaku, Tokyo, Japan) working at 40 kV and 40 mA with a CuKα radiation source. Thermogravimetric analysis (TGA) was performed at a heating rate of 10 °C min−1 in a nitrogen flux with simultaneous thermogravimetric/differential thermal analyzers (TGA/DSC1, Mettler Toledo, Switzerland). The Fourier transform infrared (FTIR) transmission spectra of the materials were obtained using a Bruker TENSOR 27 apparatus (Bruker Optics Inc., Karlsruhe, Germany). The particle size distributions of the NP suspensions and ζ potentials were measured by dynamic light scattering (DLS) using a ZetaPALS (Brookhaven Instruments, Holtsville, U.S.A.). Sonication (at 40 kHz for 10 min) was performed to ensure adequate dispersion before DLS and zeta potential measurements.2.4 Measurements of the cytotoxicity and intracellular contents of hydroxyl radicals (˙OH) and iron
J774.A1 cells were incubated with 20 mg Fe L−1 Fe3O4–HAH, Fe3O4–HAL and Fe3O4–HAR in Dulbecco's modified Eagle medium (DMEM, pH 7.4) for 12 h at 37 °C. Images of ROS generation were acquired on a confocal microscope (LSM 880, Jena, Germany) using the fluorescence probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA, KeyGEN BioTECH, Nanjing, China). The intracellular ROS content was quantified using an ROS detection kit (KeyGEN BioTECH, Nanjing, China). The total contents of glutathione (GSH) and malondialdehyde (MDA) were measured using a total glutathione assay kit (Beyotime, Shanghai, China) and a micro-MDA assay reagent kit (KeyGEN BioTECH, Nanjing, China), respectively. Cell viability was assessed using a Cell Counting Kit-8 (CCK-8, Beyotime, Shanghai, China).To determine the intracellular ˙OH content, cells were incubated with various NPs (20 mg-Fe/L) for 12 h, and after centrifugation (2500 rpm × 5 min), the cells were incubated with 2 μL of 5 mmol L−1 HPF (in 2 mL of DMEM) in the dark at 37 °C for 1 h. The cells were then washed with Hanks' balanced salt solution (HBSS) (pH = 7.4) three times. Finally, the fluorescence of the cells in 2 mL of HBSS was determined using a flow cytometer (Accuri C6 Plus, BD, Singapore) at an excitation wavelength of 488 nm and an emission wavelength of 520 nm.
To measure the iron content (termed Fetot), the cells were incubated with various NPs (at 20 mg Fe L−1) for 12 h, collected via centrifugation (2500 rpm × 5 min), washed with PBS three times and then resuspended in 1 mL of PBS solution. The cells were lysed by being frozen and thawed three times. After centrifugation, 1 mL of supernatant was mixed with 1 mL of nitric acid solution and 0.5 mL of H2O2 solution, and then the mixture was heated in a digestion device (COOLPEX, PreeKem, China) following a temperature program, which was set at 80 °C, 120 °C, 150 °C, and 180 °C for 3 min each, followed by 200 °C for 25 min. After that, the residue was cooled, transferred to a 10 mL volumetric flask, and filled up with ultrapure water. The Fetot, corresponding to both membrane-bound and internalized NPs, was determined by ICP-MS.
2.5 Abiotic ion dissolution, characterization of the NP surface composition and abiotic ˙OH production
Various NPs (200 mg Fe L−1) were suspended in PBS (pH 4.5) for 12 h, and the Fetot and ferrous ion (Fe2+) concentrations in the supernatant were determined with 1,10-phenanthroline, sodium acetate and hydroxylamine hydrochloride.20 To determine the Fe2+ concentration, 0.5 mL of supernatant was mixed with 0.3 mL of 2 g L−1 1,10-phenanthroline and 0.3 mL of 80 g L−1 sodium acetate, and then the mixture was left in the dark for 30 min. The absorbance at 510 nm was measured using an ultraviolet spectrophotometer (Cary 100, Agilent, Germany). To measure the Fetot, 0.3 mL of hydroxylamine hydrochloride (2 g L−1) was added to a 0.5 mL sample, which can reduce Fe3+ to Fe2+, and then the Fe2+ concentration was determined as described above. A calibration curve for an Fe2+-phenanthroline complex was also constructed (Fig. S2†), and the detection limit was approximately 0.04 mg L−1.The elemental composition on the surface of Fe3O4–HAR was determined with X-ray photoelectron spectroscopy (XPS) (PHI 5000 VersaProbe, Tokyo, Japan). Ion-beam etching was also employed to remove the surface layer, and the elemental composition in depth profiling was performed.
To test ˙OH generation, the NP suspensions (20 mg-Fe/L) were mixed with 5 μL of H2O2 solution (10 mmol L−1) and 2 μL of HPF solution (5 mmol L−1) for 30 min in the dark. After centrifugation, the fluorescence in the supernatant was recorded using a fluorescence spectrophotometer (F-7100, HITACHI, Japan) with an excitation wavelength of 488 nm and an emission wavelength of 515 nm. Moreover, dimethylpyridine nitrogen oxide (DMPO, 250 mmol L−1) was used as a spin trapping agent and was incubated with 20 mg Fe L−1 NPs for 30 min in the presence of 10 mmol L−1 H2O2. The electron paramagnetic resonance (EPR) spectra were measured with a Bruker EMX spectrometer (EMX Plus 6-1, Bruker, Germany).
2.6 Measurement of ion release during ligand degradation
The enzymatic degradation procedure was described previously, and the supernatant was collected at 0, 1, 2, 3, 4, 5 and 6 h. The concentrations of Fetot and Fe2+ were measured as described before. The supernatants of the Fe3O4–HAH suspension without added enzyme were also collected at the indicated times and subjected to ion measurements.2.7 Characterization of Fe2O3 surface layer formation and abiotic superoxide radical (˙O2−) production
Raman spectra were obtained using a Renishaw inVia Raman spectrometer (RM2000, London, U.K.) with a 633 nm He–Ne laser. The selected area electron diffraction (SAED) images and lattice images were obtained by high-resolution TEM (JEM-2100, JEOL, Japan).To test ˙O2− production, Fe3O4–HAL or Fe3O4–HAR (20 mg Fe L−1) was suspended in 2 mL of methanol with DMPO (250 mmol L−1) as a spin trapping molecule and then subjected to EPR spectroscopy. If needed, H2O2 solution at a final concentration of 10 mmol L−1 was added to the suspension during EPR analysis.
2.8 Statistical analysis
At least three replicates were run per treatment. All measurements were performed at least in triplicate, and the results are expressed as the mean ± standard deviation (SD). Significant differences were determined by an independent t test or one-way ANOVA. A p value less than 0.05 indicated a significant difference.3. Results and discussion
3.1 Characterization of the three NPs
The three NPs exhibited similar morphologies, physical sizes and crystalline structures. TEM images show that the three NPs were spherical with physical diameters of approximately 4–6 nm (Fig. 2a–c), which agreed well with previous studies showing that Fe3O4 NPs synthesized by the chemical co-precipitation method generally had small diameters of approximately 5–10 nm.21 Energy dispersive spectroscopy (EDS) (Fig. S3†) also verified the purity of the various Fe3O4 NPs. The XRD patterns revealed that the three NPs exhibited similar crystalline structures with characteristic peaks corresponding to the (220), (311), (400), (333), (440), and (533) facets (Fig. 2d), which corresponded well with the XRD pattern of magnetite (JCPD card no. 74-0748). The average grain size was quantitatively calculated by means of the Scherrer equation based on the XRD profiles, and the sizes were determined to be 5.10 nm, 5.60 nm and 5.20 nm for Fe3O4–HAH, Fe3O4–HAL, and Fe3O4–HAR, respectively, which coincided with the sizes determined by TEM counting. The TGA thermograms showed that more HA ligands were coated onto the F3O4–HAH surface relative to the lower weight loss for F3O4–HAL and F3O4–HAR (Fig. 2e). Similar retention rates were obtained for F3O4–HAL and F3O4–HAR (45.28% and 43.17%, respectively), indicating similar coating contents, whereas their different weight loss curves may point to varied HA structures after enzymatic degradation. The thermogram of HA alone was also provided in Fig. S4.†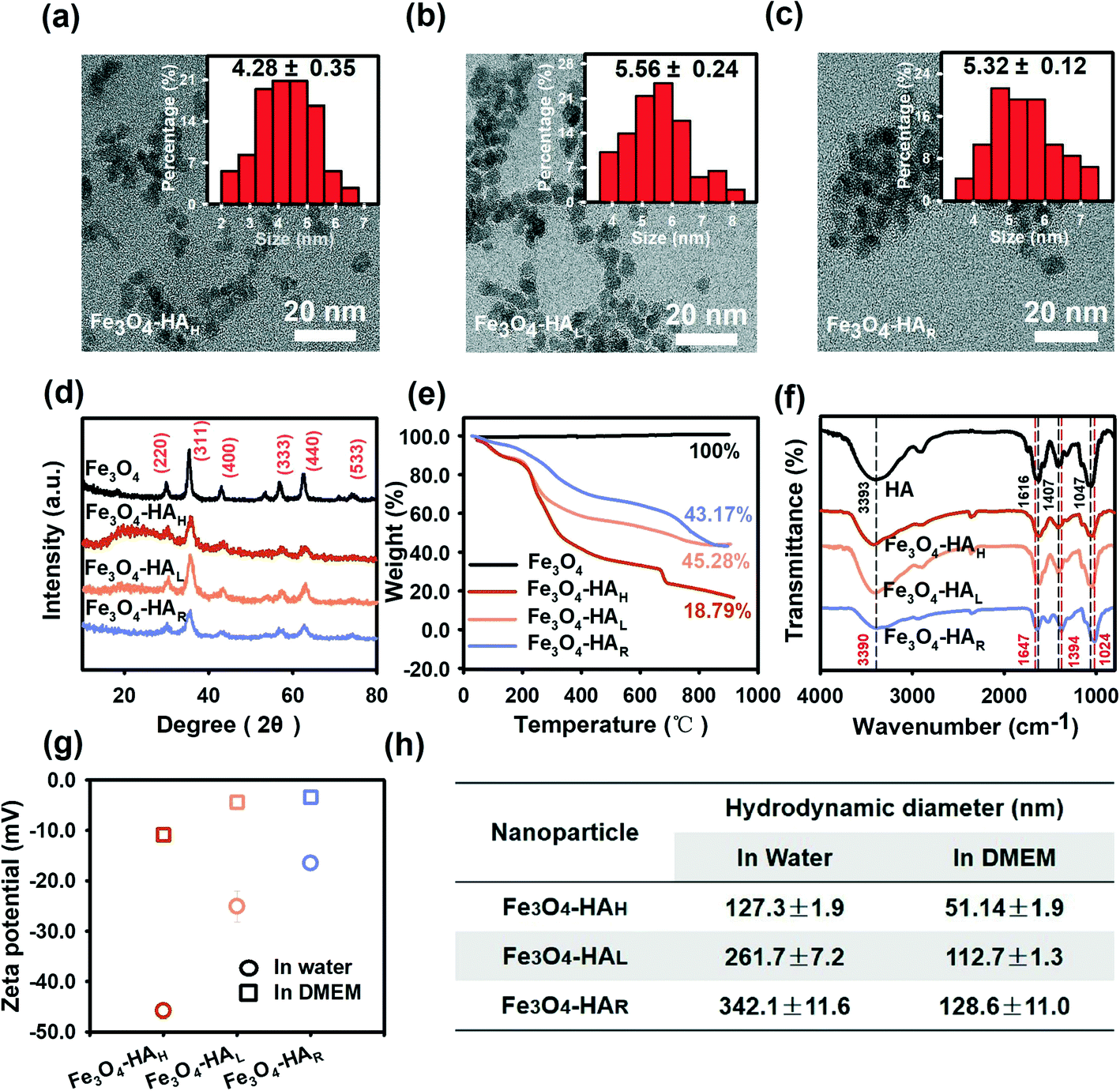 | ||
| Fig. 2 Characterization of various NPs. TEM images of (a) Fe3O4–HAH, (b) Fe3O4–HAL, and (c) Fe3O4–HAR, with insets showing the size distribution and mean size. (d) XRD patterns, (e) TGA thermograms, (f) FTIR spectra, (g) zeta potential and (h) DLS analysis (based on the intensity-weighted distribution) of Fe3O4–HAH, Fe3O4–HAL and Fe3O4–HAR. For the zeta potential and DLS analysis, 20 mg Fe L−1 NPs were suspended in water (pH 7.0) or DMEM (pH 7.4). All measurements were performed in triplicate, and the results are expressed as the mean values ± SDs. | ||
The varied structure of the coating ligand after degradation was supported by the FTIR spectra (Fig. 2f, also see Table S1† for the detailed wavelengths of each peak). Regarding free HA, the peaks at 3393 cm−1, 1616 cm−1, 1407 cm−1, and 1047 cm−1 were assigned to the stretching mode of the O–H bond, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of the amide,22 asymmetric vibration of COO−,22 and C–O stretching vibration,23 respectively, which were consistent with the reported HA structure.7 Almost identical ligand structures were observed for Fe3O4–HAH and Fe3O4–HAL, whereas peak shifts were detected for Fe3O4–HAR. For instance, the peak at 1616 cm−1 redshifted to 1647 cm−1, which was associated with the stretching vibration of the CO⋯H hydrogen bond between ligand fragments. The blueshifts from 1407 cm−1 to 1394 cm−1 and from 1047 cm−1 to 1024 cm−1 could be due to the interactions of iron ions with the O and N atoms of the surface ligand.24 Changes in the ligand functional groups also correlated well with the decreased net negative surface charge, thus inducing agglomeration in water as a result of reduced repulsive interactions (Fig. 2g and h). This trend was more evident when comparing Fe3O4–HAL to Fe3O4–HAR with similar loadings of the coating. Fe3O4–HAH exhibited the highest negative charge and the least aggregation in water, and the differences among the three NPs were alleviated in DMEM due to the presence of opposite ions. Thus, HA degradation concurrently changed the assembly pattern of the ligand and surface coordination chemistry.
O stretching band of the amide,22 asymmetric vibration of COO−,22 and C–O stretching vibration,23 respectively, which were consistent with the reported HA structure.7 Almost identical ligand structures were observed for Fe3O4–HAH and Fe3O4–HAL, whereas peak shifts were detected for Fe3O4–HAR. For instance, the peak at 1616 cm−1 redshifted to 1647 cm−1, which was associated with the stretching vibration of the CO⋯H hydrogen bond between ligand fragments. The blueshifts from 1407 cm−1 to 1394 cm−1 and from 1047 cm−1 to 1024 cm−1 could be due to the interactions of iron ions with the O and N atoms of the surface ligand.24 Changes in the ligand functional groups also correlated well with the decreased net negative surface charge, thus inducing agglomeration in water as a result of reduced repulsive interactions (Fig. 2g and h). This trend was more evident when comparing Fe3O4–HAL to Fe3O4–HAR with similar loadings of the coating. Fe3O4–HAH exhibited the highest negative charge and the least aggregation in water, and the differences among the three NPs were alleviated in DMEM due to the presence of opposite ions. Thus, HA degradation concurrently changed the assembly pattern of the ligand and surface coordination chemistry.
The enzymatic degradation of HA has been investigated in a few studies for the purpose of biomedical applications, production of low molecular weight HA, and elucidation of the physiological and pharmacological roles of HA.25 Generally, the degradation kinetics and fragmentation were enzyme-,26 pH-27 and time-dependent,28 and little structural change in the residues was perceived under mild conditions and for short degradation times.29 Herein, the structural change of HA coated on the Fe3O4 NPs after enzymatic degradation was associated with the modification of surface coordination with the iron core, which possibly stemmed from interactions with fragments.
3.2 Fe3O4–HAR induced high oxidative stress and cytotoxicity
Generally, iron NPs induce cellular damage via oxidative stress due to the potential initiation of Fenton or Fenton-like reactions. Macrophages serve as a first line of defense against exogenous particles in almost every organ system and have been extensively used as a common model in cytotoxicity studies. The CLSM images (Fig. 3a) show that a higher ROS content was induced in the J774 macrophage cell line by Fe3O4–HAR in comparison with Fe3O4–HAH and Fe3O4–HAL, and further quantification analysis evidenced that the ROS content in the Fe3O4–HAR-treated cells was almost 3 times higher than that in J774 cells alone (Fig. 3b). An approximately 50% increase in ROS induction was observed in the presence of Fe3O4–HAH or Fe3O4–HAL, while no difference was perceived between them. Consistent trends were found in terms of decreasing GSH content, increasing MDA levels, and decreasing cellular viability (Fig. 3c–e). Bearing in mind the possible involvement of the Fenton reaction, the intracellular ˙OH content was measured, and more than 2 times the ˙OH content was detected in the cells treated with Fe3O4–HAR relative to that in the J774 cells alone (Fig. 3f). Slight increases in the ˙OH content were noticed in the cells stressed by Fe3O4–HAL and Fe3O4–HAH, while no significant difference was detected between them. Apparently, different ˙OH radical production cannot be ascribed to diverse cellular uptakes since the highest rate of internalization was noted for Fe3O4–HAH due to the ligand–membrane receptor interactions between HA and CD4430 and good dispersion (Fig. 3g). Comparable rates of cellular uptake were detected for Fe3O4–HAL and Fe3O4–HAR because of similar ligand loadings. Although we cannot fully differentiate the intracellular NPs from surface-bound NPs based on the Fetot, it is generally thought that a higher level of adhesion ensures higher uptake efficiency.31 The iron content was visualized via Prussia blue staining (Fig. S5†). Collectively, the low cellular uptake, high content of generated ˙OH radicals, and high level of ROS induction suggested that Fe3O4–HAR may possess an excellent capability for producing ˙OH radicals. | ||
| Fig. 3 (a) CLSM images showing the ROS levels in the J774 cells treated with 20 mg Fe L−1 Fe3O4–HAH, Fe3O4–HAL or Fe3O4–HAR for 12 h in DMEM (pH 7). Quantification of the (b) ROS level, (c) GSH content, (d) MDA content, (e) cell viability, (f) intracellular ˙OH content and (g) Fetot. All experiments were carried out at least in triplicate, and the results are presented as the mean values ± SDs. Different letters indicate a significant difference (p < 0.05). | ||
It was suggested that surface coating influenced NP cytotoxicity via modifications in agglomerate size, cellular uptake, surface roughness, dissolution rate, and surface-related reactivity.32 Smaller NPs facilitate a higher rate of cellular uptake, thus potentially inducing higher toxicity. However, in this study, surface coating-related changes in the NP size and uptake showed negligible effects on cellular viability when comparing Fe3O4–HAH with Fe3O4–HAL. The possible reason was that the HA-coated Fe3O4 NPs were recognized to be biocompatible, and cellular uptake was not a determining factor on toxicity at low NP concentrations.7 Additionally, no damage to membrane integrity (caused by surface roughness) was perceived upon prompt contact with various NPs (as measured by lactate dehydrogenase release, data not shown). Fe3O4–HAL and Fe3O4–HAR with similar sizes and comparable ligand loadings revealed substantially distinctive radical production abilities. Thus, the capability of generating ˙OH radicals, which are closely related to ion dissolution behavior and surface-related reactivity, may be the foremost factor and will be evaluated under abiotic conditions, as described below.
3.3 Fe3O4–HAR released more Fe2+ and produced a large number of ˙OH radicals
The dissolution behaviors of the three NPs are compared in Fig. 4a, and the total amount of ions released by Fe3O4–HAH was higher than the amount released by Fe3O4–HAL and Fe3O4–HAR since Fe3O4–HAH was well dispersed. Generally, a higher dispersibility gives rise to a more exposed surface area, thus leading to a higher rate of dissolution.7,32 Interestingly, almost 100% of the released ions by Fe3O4–HAR were Fe2+ compared to 70% by Fe3O4–HAH and Fe3O4–HAL. The high content of Fe2+ that was detected may be ascribed to the higher solubility of Fe2+ over Fe3+ at this pH. Nevertheless, the markedly high level of Fe2+ by Fe3O4–HAR was possible due to the reversible adsorption of Fe2+ onto the NP surface, as evidenced by the following analysis. The change in surface stoichiometry was confirmed by XPS spectroscopy. As shown in Fig. 4b, the ratio of Fe(II)/Fe(III) on the Fe3O4–HAR surface changed to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.75, while the ratio remained at 1:2.01 after surface etching (Fig. 4c), which agreed with the stoichiometry of magnetite NPs.34
:1.75, while the ratio remained at 1:2.01 after surface etching (Fig. 4c), which agreed with the stoichiometry of magnetite NPs.34
 | ||
| Fig. 4 (a) Ion release (Fetot) from various NP suspensions and the percentage of Fe2+ in Fetot. Various NPs (200 mg Fe/L) were suspended in PBS (pH 4.5) at 37 °C for 12 h. XPS spectra of Fe3O4–HAR (b) before and (c) after surface etching. (d) UV-vis spectra showing the ˙OH radical production by various NPs (20 mg Fe L−1) incubated with 10 mmol L−1 H2O2 using HPF (5 mmol L−1) as a probe. Water instead of a NP suspension was used as a control. (e) EPR spectra showing ˙OH production from various NP suspensions (20 mg Fe L−1) in the presence of 10 mmol L−1 H2O2 using DMPO (250 mmol L−1) as a spin trapping reagent. (f) Contents of released Fetot and Fe2+ at different degradation times. Fe3O4–HAH (200 mg Fe L−1) was incubated with/without 4 g L−1 HAase in PBS (pH 4.5). All experiments were carried out at least in triplicate, and the results are presented as the mean values ± SDs. Different letters indicate a significant difference (p < 0.05). | ||
A high content of surface-adsorbed Fe2+ endowed Fe3O4–HAR with a high ability for producing ˙OH radicals since Fe2+ is superior to Fe3+ in mediating the Fenton reaction. Compared with Fe3O4–HAH and Fe3O4–HAL, Fe3O4–HAR produced nearly 1.5 times the number of ˙OH radicals when reacted with H2O2 (Fig. 4d), and this statement was further verified by the stronger peak of the DMPO–˙OH adducts generated by Fe3O4–HAR in the EPR spectra (Fig. 4e). It is worth mentioning that a significant amount of Fe2+ was released only during the enzymatic degradation of the ligands (Fig. 4f). One possible reason was that the ligand fragments (which contain COO-, OH- and C(O)N- groups) may promote ion dissolution (mostly Fe2+ ions) at the interface, and the decrease in the amount of Fe2+ after 4 h indicated the adsorption, oxidation, and precipitation of aqueous ions. Similar ligand-promoted dissolution has been reported in some studies.33,35 For instance, oxalate and ascorbic acid significantly promoted the dissolution of nanoscale hematite, which was suggested because metal–organic surface complexes destabilized the Fe–O bond and lowered the energy barrier for dissolution.33
The effect of ligand coating on core material dissolution has been suggested in some studies.36,37 Generally, the degradation of inorganic cores is governed by surface corrosion; thus, the protective efficiency of various coatings relies on their ability to prevent access to core surfaces. Therefore, relatively low dissolution rates were observed for the three NPs when considering high ligand loadings, even for Fe3O4–HAR. However, few studies have considered ligand degradation-induced core dissolution,38 in which ligand fragments with various complexation functional groups can strongly bind to the core surface, leading to Fe atoms being pulled out. Thus, a large number of ions were detected during ligand degradation. In particular, the co-presence of Fe2+ and Fe3O4 NPs can have a significant impact on the stoichiometry and reactivity of magnetite NPs, and the interplay mechanisms include reductive dissolution, the reversible uptake and release of Fe2+, bulk electron conduction, electron transfer, atom exchange, and recrystallization.35,39,40 To our knowledge, the contribution of these processes to the overall cytotoxicity has been decidedly overlooked. Our results reveal a piece of information by decoding the change in surface chemistry after ligand degradation.
3.4 Fe2O3 formation on the surface of Fe3O4–HAR facilitates the Fe(II)/Fe(III) cycle via ˙O2− generation
A close look at the XPS spectra (Fig. 4b) revealed the presence of the Fe2O3 layer at the NP interface. Generally, peaks at 710.7 eV and 724.3 eV were reported for Fe 2p3/2 and Fe 2p1/2, respectively.41 The peaks at 709.8 eV and 711.1 eV were attributed to Fe2+ and Fe3+ in the octahedral coordination of Fe3O4, respectively, while the peak at 712.8 eV was assigned to Fe3+ in the tetrahedral coordination of Fe3O4.42,43 Notably, a satellite peak at 717.9 eV was observed for Fe3O4–HAR but not for the etched NPs, and this peak was ascribed to Fe 2p3/2 for Fe2O3.43,44 The appearance of the Fe2O3 surface layer was also confirmed by Raman spectroscopy, HRTEM imaging and SAED (Fig. 5a–c). The peaks at 299 nm, 537 nm and 666 nm in the Raman spectra were the characteristic peaks of Fe3O4,45 and the appearance of new peaks at 231 nm, 410 nm, 616 nm and 1318 nm after ligand degradation was ascribed to Fe2O3 (ref. 46) (Fig. 5a). The lattice fringes along Fe3O4 (311) in the HRTEM image47 have a spacing of 0.25 nm, whereas a lattice spacing of 0.18 nm corresponds to Fe2O3 (024) (Fig. 5b). The corresponding SAED pattern of Fe3O4–HAR (Fig. 5c) exhibited visible diffraction rings corresponding to the (533), (220), and (311) planes of Fe3O4 and the (113) and (024) planes of Fe2O3.48 No such surface oxide formation was detected for Fe3O4–HAH and Fe3O4–HAL (Fig. S6†). Accordingly, Fe3O4–HAR was composed of an Fe3O4 core and a thin Fe2O3 surface layer with embedded Fe2+ ions (Fig. 5d). | ||
| Fig. 5 (a) Raman spectra of Fe3O4–HAR showing peaks corresponding to Fe3O4 and Fe2O3. (b) Lattice fringes and (c) SAED image of Fe3O4–HAR. (d) Proposed structure of Fe3O4–HAR. (e) EPR spectra of the DMPO–OOH adducts in the presence of Fe3O4–HAL or Fe3O4–HAR (20 mg Fe L−1 in methanol) with/without the addition of 10 mmol L−1 H2O2. DMPO (250 mmol L−1) was used as a spin trapping molecule. | ||
The ferric oxide layer in conjunction with surface-adsorbed Fe2+ can play an important role in accelerating the Fe(III)/Fe(II) cycle via ˙O2− generation, favoring the Fenton reaction.49 The ability to generate ˙O2− was tested for Fe3O4–HAR and Fe3O4–HAL in the presence and absence of H2O2. The EPR spectra showed representative peaks of DMPO–OOH adducts, and a stronger peak intensity was observed for Fe3O4–HAR than for Fe3O4–HAL with the addition of H2O2 (Fig. 5e). The peak was still discernible even in the absence of H2O2 for Fe3O4–HAR due to one electron transfer to molecular oxygen. It is worth mentioning that ˙O2− generation may also partially contribute to the observed high level of Fe2+ during ligand degradation. The mechanism is illustrated by the following equations (eqn (1)–(4)).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe3+ + H2O2 → ˙HO2 + Fe2+ + H+ Fe3+ + H2O2 → ˙HO2 + Fe2+ + H+ | (1) |
| Fe3+OH + Fe2+ + H2O ↔ Fe3+OFe2+OH + 2H+ | (2) |
| Fe3+OFe2+OH + O2 → Fe3+OFe3+OH + ˙O−2 | (3) |
| Fe3+ or Fe3+aq + ˙O−2 → Fe2+ or Fe2+aq + O2 | (4) |
In general, the passivation of Fe3O4 NPs during bio-nano interactions decreased surface-related activity and mitigated cellular toxicity, including surface oxidation, the absorption of protein or other biomolecules, and the decrease in ion leakage due to surface complexation with thiol groups from proteins.50,51 Nevertheless, our results revealed the more toxic effect of Fe3O4 after surface oxidation because the coexistence of the Fe3O4 core and oxidized Fe2O3 surface layer in conjunction with surface-adsorbed Fe2+ provided a unique reactive surface for the initiation of the Fenton reaction. In line with our studies, it has been shown that Fe3O4@Fe2O3 nanocomposites significantly improve the charge transport at the Fe3O4/Fe2O3 interface, boosting their catalytic activities.52 The surface-bound Fe2+ ions on the Fe2O3 shell benefit single-electron molecular oxygen activation, forming a large quantity of ˙O2−.53 Consequently, in view of the particular interest in Fe3O4 NPs in bioscience applications, more attention should be given to how passivation and depassivation occur in vivo and how ligand biodegradation concomitantly affects the rate and extent of these processes.
4. Conclusions
Overall, our results demonstrated that the enzymatic degradation of surface HA induced ligand-promoted ion dissolution, followed by the precipitation of Fe2O3. Then, a high level of aqueous Fe2+ was adsorbed onto the Fe3O2 surface layer, forming a complex Fe3O4@Fe2O3 structure with active surface-adsorbed Fe2+ (Fig. 6). This distinctive structure promoted their high competence for producing reactive ˙OH radicals, thus inducing more severe oxidative damage in macrophage cells. In this study, the enzymatic degradation of surface ligands was performed under relatively simple conditions; nonetheless, the reshaping, recrystallization and generation of secondary NPs would be more complicated if considering simultaneous degradation occurring on all the different architectural parts of the NPs, the coexistence of passivation and depassivation by-products and reactions with solution entities in biological environments (e.g., organic/inorganic ligands, redox molecules, and other nutrient metal ions). Overall, tracing the NP biotransformation both in vivo and in vitro would provide valuable information for understanding the fate and toxicity of engineered NPs, which will in turn, allow for the design of more rational and biocompatible NPs for biomedical applications. | ||
| Fig. 6 Diagram of the surface reconstruction for Fe3O4–HAR during enzymatic ligand degradation. | ||
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21777077 and 21976018). Partial support was also provided by the Fundamental Research Funds for the Central Universities.References
- S. Mourdikoudis, A. Kostopoulou and A. P. LaGrow, Magnetic nanoparticle composites: synergistic effects and applications, Adv. Sci., 2021, 8, 2004951–2005008 CrossRef CAS PubMed.
- J. Dolai, K. Mandal and N. R. Jana, Nanoparticle size effects in biomedical applications, ACS Appl. Nano Mater., 2021, 4, 6471–6496 CrossRef CAS.
- S. Fusco, H.-W. Huang, K. E. Peyer, C. Peters, M. Haeberli, A. Ulbers, A. Spyrogianni, E. Pellicer, J. Sort, S. E. Pratsinis, B. J. Nelson, M. S. Sakar and S. Pane, Shape-switching microrobots for medical applications: the influence of shape in drug delivery and locomotion, ACS Appl. Mater. Interfaces, 2015, 7, 6803–6811 CrossRef CAS PubMed.
- Y. Li, T. M. Krentz, L. Wang, B. C. Benicewicz and L. S. Schadler, Ligand engineering of polymer nanocomposites: from the simple to the complex, ACS Appl. Mater. Interfaces, 2014, 6, 6005–6021 CrossRef CAS PubMed.
- D. Monego, T. Kister, N. Kirkwood, P. Mulvaney, A. Widmer-Cooper and T. Kraus, Colloidal stability of apolar nanoparticles: role of ligand length, Langmuir, 2018, 34, 12982–12989 CrossRef CAS PubMed.
- S. Alexander, Biopolymers, Wiley-VCH Verlag GmbH & Co. KGaA, Berlin, 2005 Search PubMed.
- H. Zhou, J. Tang, J. Li, W. Li, Y. Liu and C. Chen, In vivo aggregation-induced transition between T1 and T2 relaxations of magnetic ultra-small iron oxide nanoparticles in tumor microenvironment, Nanoscale, 2017, 9, 3040–3050 RSC.
- Q. Liang, J. Sherwood, T. Macher, J. M. Wilson, Y. Bao and C. J. Cassady, Citric acid capped iron oxide nanoparticles as an effective MALDI matrix for polymers, J. Am. Soc. Mass Spectrom., 2017, 28, 409–418 CrossRef CAS.
- H. Zhou, J. Ge, Q. Miao, R. Zhu, L. Wen, J. Zeng and M. Gao, Biodegradable inorganic nanoparticles for cancer theranostics: insights into the degradation behavior, Bioconjugate Chem., 2020, 31, 315–331 CrossRef.
- M. F. A. Lucas, M. Pavelka, M. E. Alberto and N. Russo, Neutral and acidic hydrolysis reactions of the third generation anticancer drug oxaliplatin, J. Phys. Chem. B, 2009, 113, 831–838 CrossRef PubMed.
- S. Athukorale, M. De Silva, A. LaCour, G. S. Perera, C. U. Pittman, Jr. and D. Zhang, NaHS induces complete nondestructive ligand displacement from aggregated gold nanoparticles, J. Phys. Chem. C, 2018, 122, 2137–2144 CrossRef.
- W. G. Kreyling, A. M. Abdelmonem, Z. Ali, F. Alves, M. Geiser, N. Haberl, R. Hartmann, S. Hirn, D. Jimenez de Aberasturi, K. Kantner, G. Khadem-Saba, J.-M. Montenegro, J. Rejman, T. Rojo, I. Ruiz de Larramendi, R. Ufartes, A. Wenk and W. J. Parak, In vivo integrity of polymer-coated gold nanoparticles, Nat. Nanotechnol., 2015, 10, 619–625 CrossRef PubMed.
- X. Wang, X. Wang, X. Bai, L. Yan, T. Liu, M. Wang, Y. Song, G. Hu, Z. Gu, Q. Miao and C. Chen, Nanoparticle ligand exchange and its effects at the nanoparticle-cell membrane interface, Nano Lett., 2019, 19, 8–18 CrossRef PubMed.
- R. Mo, T. Jiang, R. DiSanto, W. Tai and Z. Gu, ATP-triggered anticancer drug delivery, Nat. Commun., 2014, 5, 3364–3374 CrossRef PubMed.
- X. Cai, X. Liu, J. Jiang, M. Gao, W. Wang, H. Zheng, S. Xu and R. Li, Molecular mechanisms, characterization methods, and utilities of nanoparticle biotransformation in nanosafety assessments, Small, 2020, 16, 1907663–1907682 CrossRef PubMed.
- Y. Kong, J. Chen, H. Fang, G. Heath, Y. Wo, W. Wang, Y. Li, Y. Guo, S. D. Evans, S. Chen and D. Zhou, Highly fluorescent ribonuclease-a-encapsulated lead sulfide quantum dots for ultrasensitive fluorescence in vivo imaging in the second near-infrared window, Chem. Mater., 2016, 28, 3041–3050 CrossRef CAS PubMed.
- T. Gong, X. Song, L. Yang, T. Chen, T. Zhao, T. Zheng, X. Sun, T. Gong and Z. Zhang, Spontaneously formed porous structure and M1 polarization effect of Fe3O4 nanoparticles for enhanced antitumor therapy, Int. J. Pharm., 2019, 559, 329–340 CrossRef CAS PubMed.
- K. Y. Choi, H. Chung, K. H. Min, H. Y. Yoon, K. Kim, J. H. Park, I. C. Kwon and S. Y. Jeong, Self-assembled hyaluronic acid nanoparticles for active tumor targeting, Biomaterials, 2010, 31, 106–114 CrossRef CAS PubMed.
- E. J. Menzel and C. Farr, Hyaluronidase and its substrate hyaluronan: biochemistry, biological activities and therapeutic uses, Cancer Lett., 1998, 131, 3–11 CrossRef.
- N. Wang, L. Zhu, M. Lei, Y. She, M. Cao and H. Tang, Ligand-induced drastic enhancement of catalytic activity of nano-BiFeO3 for oxidative degradation of bisphenol A, ACS Catal., 2011, 1, 1193–1202 CrossRef.
- Z. Zarnegar and J. Safari, Modified chemical coprecipitation of magnetic magnetite nanoparticles using linear-dendritic copolymers, Green Chem. Lett. Rev., 2017, 10, 235–240 CrossRef.
- S. S. Suner, B. Ari, F. C. Onder, B. Ozpolat, M. Ay and N. Sahiner, Hyaluronic acid and hyaluronic acid: sucrose nanogels for hydrophobic cancer drug delivery, Int. J. Biol. Macromol., 2019, 126, 1150–1157 CrossRef PubMed.
- P. Mahlumba, P. Kumar, L. C. du Toit, M. S. Poka, P. Ubanako and Y. E. Choonara, Fabrication and characterisation of a photo-responsive, injectable nanosystem for sustained delivery of macromolecules, Int. J. Mol. Sci., 2021, 22, 3359–3380 CrossRef PubMed.
- M. R. Swart, B. C. B. Bezuidenhoudt, C. Marais and E. Erasmus, Spectroscopic characterisation of grubbs 2nd generation catalyst and its p-cresol derivatives, Inorg. Chim. Acta, 2021, 514, 120001–120009 CrossRef.
- M. Yazdani, A. Shahdadfar, C. J. Jackson and T. P. Utheim, Hyaluronan-based hydrogel scaffolds for limbal stem cell transplantation: a review, Cell, 2019, 8, 245–257 CrossRef PubMed.
- N. Hegyesi, E. Hodosi, P. Polyak, G. Faludi, D. Balogh-Weiser and B. Pukanszky, Controlled degradation of poly-epsilon-caprolactone for resorbable scaffolds, Colloids Surf., B, 2020, 186, 110678–110685 CrossRef CAS PubMed.
- H. Lenormand, F. Amar-Bacoup and J.-C. Vincent, pH effects on the hyaluronan hydrolysis catalysed by hyaluronidase in the presence of proteins. Part III. The electrostatic non-specific hyaluronan-hyaluronidase complex, Carbohydr. Polym., 2011, 86, 1491–1500 CrossRef CAS.
- P. Yuan, M. Lv, P. Jin, M. Wang, G. Du, J. Chen and Z. Kang, Enzymatic production of specifically distributed hyaluronan oligosaccharides, Carbohydr. Polym., 2015, 129, 194–200 CrossRef CAS.
- R. Nehme, R. Nasreddine, L. Orlic, C. Lopin-Bon, J. Hamacek and F. Piazza, Kinetic theory of hyaluronan cleavage by bovine testicular hyaluronidase in standard and crowded environments, Biochim. Biophys. Acta, Gen. Subj., 2021, 1865, 129837–129844 CrossRef CAS.
- A. Almalik, S. Karimi, S. Ouasti, R. Donno, C. Wandrey, P. J. Day and N. Tirelli, Hyaluronic acid (HA) presentation as a tool to modulate and control the receptor-mediated uptake of HA-coated nanoparticles, Biomaterials, 2013, 34, 5369–5380 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Aberg, Nanoparticle adhesion to the cell membrane and its effect on nanoparticle uptake efficiency, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- W. Nie, B. Zhang, X. Yan, L. Su, S. Wang, G. Han and D. Han, Degraded hyaluronic acid-modified magnetic nanoparticles, J. Nanomater., 2020, 2020, 1084890–1084898 Search PubMed.
- C. A. Lanzl, J. Baltrusaitis and D. M. Cwiertny, Dissolution of hematite nanoparticle aggregates: influence of primary particle size, dissolution mechanism, and solution pH, Langmuir, 2012, 28, 15797–15808 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- H. Peng, C. I. Pearce, A. T. N'Diaye, Z. Zhu, J. Ni, K. M. Rosso and J. Liu, Redistribution of electron equivalents between magnetite and aqueous Fe2+ induced by a model quinone compound AQDS, Environ. Sci. Technol., 2019, 53, 1863–1873 CrossRef CAS PubMed.
- M. N. Martin, A. J. Allen, R. I. MacCuspie and V. A. Hackley, Dissolution, agglomerate morphology, and stability limits of protein-coated silver nanoparticles, Langmuir, 2014, 30, 11442–11452 CrossRef CAS PubMed.
- O. Hakami, Y. Zhang and C. J. Banks, Influence of aqueous environment on agglomeration and dissolution of thiol-functionalised mesoporous silica-coated magnetite nanoparticles, Environ. Sci. Pollut. Res., 2015, 22, 3257–3264 CrossRef CAS.
- S. Bae, R. N. Collins, T. D. Waite and K. Hanna, Advances in surface passivation of nanoscale zerovalent iron: A critical review, Environ. Sci. Technol., 2018, 52, 12010–12025 CrossRef CAS PubMed.
- C. A. Gorski, R. M. Handler, B. L. Beard, T. Pasakarnis, C. M. Johnson and M. M. Scherer, Fe atom exchange between aqueous Fe2+ and magnetite, Environ. Sci. Technol., 2012, 46, 12399–12407 CrossRef CAS PubMed.
- V. Tishchenko, C. Meile, M. M. Scherer, T. S. Pasakarnis and A. Thompson, Fe2+ catalyzed iron atom exchange and re-crystallization in a tropical soil, Geochim. Cosmochim. Acta, 2015, 148, 191–202 CrossRef CAS.
- T. Yamashita and P. Hayes, Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- D. Wilson and M. A. Langell, XPS analysis of oleylamine/oleic acid capped Fe3O4 nanoparticles as a function of temperature, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS.
- W. Liu, B. Cheng, T. Miao, J. Xie, L. Liu, J. Si, G. Zhou, H. Qin and J. Hu, Room temperature electric field control of magnetic properties for the alpha-Fe2O3/Fe3O4 composite structure, J. Magn. Magn. Mater., 2019, 491, 165500–165507 CrossRef CAS.
- T. Yamashita and P. Hayes, Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials, Appl. Surf. Sci., 2009, 255, 8194–8194 CrossRef CAS.
- A. G. Nasibulin, S. Rackauskas, H. Jiang, Y. Tian, P. R. Mudimela, S. D. Shandakov, L. I. Nasibulina, J. Sainio and E. I. Kauppinen, Simple and rapid synthesis of alpha-Fe2O3 nanowires under ambient conditions, Nano Res., 2009, 2, 373–379 CrossRef.
- A. Asfaram, M. Ghaedi, A. Goudarzi and M. Rajabi, Response surface methodology approach for optimization of simultaneous dye and metal ion ultrasound-assisted adsorption onto Mn doped Fe3O4-NPs loaded on AC: kinetic and isothermal studies, Dalton Trans., 2015, 44, 14707–14723 RSC.
- Q. Xia, Z. Yao, D. Zhang, D. Li, Z. Zhang and Z. Jiang, Rational synthesis of micronano dendritic ZVI@Fe3O4 modified with carbon quantum dots and oxygen vacancies for accelerating Fenton-like oxidation, Sci. Total Environ., 2019, 671, 1056–1065 CrossRef.
- H. Wang, S. Liu, X. Yang, R. Yuan and Y. Chai, Mixed-phase iron oxide nanocomposites as anode materials for lithium-ion batteries, J. Power Sources, 2015, 276, 170–175 CrossRef.
- J. Shi, Z. Ai and L. Zhang, Fe@Fe2O3 core-shell nanowires enhanced fenton oxidation by accelerating the Fe(III)/Fe(II) cycles, Water Res., 2014, 59, 145–153 CrossRef.
- S. L. Gawali, S. B. Shelar, J. Gupta, K. C. Barick and P. A. Hassan, Immobilization of protein on Fe3O4 nanoparticles for magnetic hyperthermia application, Int. J. Biol. Macromol., 2021, 166, 851–860 CrossRef.
- A. Bajaj, B. Samanta, H. Yan, D. J. Jerry and V. M. Rotello, Stability, toxicity and differential cellular uptake of protein passivated-Fe3O4 nanoparticles, J. Mater. Chem., 2009, 19, 6328–6331 RSC.
- X. Tang, R. Jia, T. Zhai and H. Xia, Hierarchical Fe3O4@Fe2O3 core-shell nanorod arrays as high-performance anodes for asymmetric supercapacitors, ACS Appl. Mater. Interfaces, 2015, 7, 27518–27525 CrossRef CAS.
- Z. Ai, Z. Gao, L. Zhang, W. He and J. J. Yin, Core-shell structure dependent reactivity of Fe@Fe2O3 nanowires on aerobic degradation of 4-chlorophenol, Environ. Sci. Technol., 2013, 47, 5344–5352 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1en00724f |
| This journal is © The Royal Society of Chemistry 2022 |