
Transformation of carbon dots by ultraviolet irradiation, ozonation, and chlorination processes: kinetics and mechanisms†
Mengyao
Shen
,
Jingyi
Wu
,
Yuzheng
He
,
Guoguang
Liu
and
Yiping
Feng
 *
*
Guangzhou Key Laboratory Environmental Catalysis and Pollution Control, Guangdong Key Laboratory of Environmental Catalysis and Health Risk Control, School of Environmental Science and Engineering, Institute of Environmental Health and Pollution Control, Guangdong University of Technology, Guangzhou 510006, China. E-mail: ypfeng@gdut.edu.cn; Tel: +86 15989197900
First published on 1st December 2021
Abstract
As a new carbon nanomaterial, carbon dots (CDs) with unique physicochemical and luminescence properties have wide-ranging applications. Once released into water and wastewater treatment plants, CDs will undergo physicochemical transformations due to exposure to disinfection treatments. The present study examined the transformations of CDs under three typical disinfection treatments including ultraviolet (UV) irradiation, ozonation, and chlorination. Our results indicated that CDs can be degraded efficiently by UV irradiation, ozonation, and chlorination, implying the fundamental role of oxygen and chlorine related radicals in CDs degradation. The degradation of CDs by all three treatments followed pseudo-first-order reaction kinetics. Various characterization results revealed the structural and chemical property changes of CDs after treatment by the three disinfection processes. We proposed that under reactive species attack, CDs were firstly degraded into macromolecular species and gradually cleaved into micromolecular compounds with the release of CO2. Many chlorine substitution/addition by-products were identified after the chlorination treatment of CDs, suggesting the potential negative impacts and health risks of CDs after environment-relevant transformations. The CDs degradation efficiency differed slightly at the examined pH values of 5.0–9.0. Water constituents including humic acid and anions exhibited various effects on CDs degradation in the three disinfection processes. Nevertheless, high removal efficiencies of CDs by the three disinfection processes were observed in natural water samples. The results of this study highlighted the non-negligible influence of water disinfection treatment on the fate of CDs in aquatic environments and provided a quantitative framework for evaluating the fate of CDs during disinfection/oxidation.
Environmental significanceAs a new carbon nanomaterial, carbon dots (CDs) have attracted considerable application interest. Meanwhile, there has been increasing concern regarding the potential environmental impacts of CDs upon release. To date, the transformation and degradation of CDs under environmentally relevant conditions have remained poorly understood. This study indicated that CDs can be rapidly degraded by three typical disinfection treatments including ultraviolet irradiation, ozonation, and chlorination. The physicochemical properties of CDs are remarkably changed and potentially harmful degradation intermediates are produced by the three disinfection treatments. Overall, this research provided a quantitative framework for evaluating the fate of CDs during disinfection/oxidation and showed the significant influence of water disinfection treatment on CDs' fate in aquatic environments. |
1. Introduction
As a new type of carbon nanomaterial (CNM), carbon dots (CDs) have attracted tremendous interest due to their outstanding physicochemical features, e.g., excellent optical properties, superior fluorescence emissions, high dispersibility, and good biocompatibility.1 CDs could thus potentially replace semiconductor quantum dots that involve toxicity, high cost, and limited functionality.2 Recently, CDs have been widely developed and used in the fields of chemical sensing,3 biosensing,4 bioimaging,5 nanomedicine,3 electro- and photo-catalysis,6 and optoelectronic and energy device construction.7 The rapid increase in the production and use of CDs will inevitably lead to their release into aquatic environments, and the potential environmental impacts of CDs have also raised significant concerns.8 CDs have been found to induce toxic effects on various organisms, including mammalian cells,9–11 algae,12 and bacteria.13 In addition, several studies have also indicated that the presence of CDs facilitated the transport of heavy metals in porous media due to the high sorption capacity and high mobility of CDs,14,15 which would affect the risk of heavy metals in aquatic environments.Understanding the environment-relevant transformation of CDs is important to their environmental impact and health risk assessment. Recently, several studies have investigated the photochemical and biological transformations of CDs.16–19 It was found that both photochemical and biological reactions could degrade CDs more rapidly than other CNMs such as carbon nanotubes and graphene oxide,20–22 probably because the smaller size (<10 nm) and the abundant functional groups (e.g., –OH, –COOH, and –NH2) enabled higher reactivities for CDs.1 As reported, under the attack by reactive oxygen species, CDs can be degraded and finally mineralized into CO2 and H2O.19 Furthermore, during use and disposal, CDs will be discharged into sewage and water treatment plants and inevitably be exposed to disinfection in water and wastewater treatment.23 Considering the distinct toxicities of water disinfection by-products,24 it is essential to investigate the transformation of CDs and hence supply information for evaluating the toxicity change of CDs after water treatment processes.
Disinfection is a final treatment procedure for wastewater and drinking water treatment, where many nanoparticles could be transformed and changed by disinfectants before being discharged into natural water bodies.25 The most commonly used disinfectants (e.g., UV, chlorine, and O3) in water treatment possess strong oxidizing ability due to the release of reactive species (RS) including hydroxyl radicals (˙OH), superoxide radical anions (˙O2−), ClO−, and chlorine radicals (Cl˙).26 These RS are able to oxidize nanoparticles to change their physicochemical properties and generate diverse derivatized products.27–30 Under RS attack, primary nanoparticles are broken into small fragments and release CO2, as well as low molecular weight species rich in chloride, epoxy and hydroxyl functional groups.21,28,31,32 Such oxidation would enhance the water stability, mobility and transport of nanomaterials.29 In particular, Liu et al.,18 demonstrated that photodegradation of CDs produced 499 cytotoxicity-related molecules. Despite this, the transformation of CDs by various disinfection treatments has not been extensively studied, which provides essential information for evaluating the environmental behavior and fate of CDs in aquatic media.
In natural aquatic environments, the water constituents including ions and natural organic matter (NOM) will also affect the transformation of nanomaterials. Anions like Cl−, CO32−, and SO42− are able to react with primary radicals (e.g., HO˙) to form anionic radicals (e.g., Cl˙, CO3˙−, and SO4˙−).33,34 Therefore, such a radical-scavenging effect would affect the transformation of nanomaterials.35,36 In particular, except for consumption of radicals, NOM may coat the surface of nanomaterials and hence attenuate the decomposition of nanomaterials.31,37 Therefore, there is a need to evaluate the effect of water constituents on the degradation of CDs.
The objective of this study is to investigate the transformation and degradation of CDs by water disinfection treatment. The degradation kinetics, pathways, and mechanisms of CDs with three commonly used disinfectants including UV, O3, and chlorine were systematically investigated and compared. In natural waters, natural organic matter and inorganic anions are ubiquitously present and can potentially influence the reaction of nanomaterials.38 Therefore, the effects of pH, humic acid (HA), and inorganic anions (e.g., Cl−, NO3−, CO32−, SO42−, and HCO3−) on the transformation of CDs were also examined. CDs transformation intermediates in the three disinfection treatments were identified through liquid chromatography-tandem mass spectrometry (LC-MS) and the detailed transformation pathways and mechanisms were proposed. In addition, the degradation of CDs was also performed in different natural waters to simulate the degradation efficiency in real water/wastewater disinfection.
2. Materials and methods
2.1. Material and stock solution preparation
Carbon dots (CDs) were synthesized by a hydrothermal method according to our previous studies.39,40 Briefly, 3.0 g of citric acid and 1.0 g of urea were fully dissolved in 10 mL deionized (DI) water (18.2 mΩ cm, Milli-Q, Millipore, USA), and then the mixture was heated in a 50 mL Teflon-lined stainless steel autoclave at 180 °C for 5 h. The obtained CDs suspension was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 45 min to remove large particles. Then, to remove ultrafine particles (e.g., <1 nm), the CDs were dialyzed in DI water using a 3 KD cellulose membrane for 45 min through a centrifuge at 6000 rpm. The prepared CDs were characterized by various techniques as shown in Fig. S1.†
000 rpm for 45 min to remove large particles. Then, to remove ultrafine particles (e.g., <1 nm), the CDs were dialyzed in DI water using a 3 KD cellulose membrane for 45 min through a centrifuge at 6000 rpm. The prepared CDs were characterized by various techniques as shown in Fig. S1.†
2.2. Degradation experiments
All experiments were carried out in DI water and performed in triplicate. 0.1 mol L−1 NaOH or 0.1 mol L−1 H2SO4 was used to adjust the reaction solution pH, and unless indicated, the reaction pH was set as 7.0 ± 0.2. The reaction temperature was controlled at 25 ± 0.2 °C. Three disinfectants, UV, O3, and chlorine, were used to degrade CDs. Through the indigo carmine oxidation method,41 it was observed that the oxidation ability of a 1000 W mercury lamp was ≈200 mL min−1 O3 ≈ 0.8 mg L−1 available chlorine in the current study (Fig. S2†). The degradation of CDs was quantified by monitoring their absorbance at 332 nm (Fig. S1h†) using a UV-vis spectrophotometer (Mapada 3200 UV-vis, Shanghai, China). The initial concentration of CDs for the reaction was set as 10 mg L−1. UV irradiation experiments were conducted in an XPA-7 rotary photocatalytic reactor (Xujiang Machine Plant, Nanjing, China) with a 1000 W mercury lamp centered at 365 nm as the UV radiation source. Preliminary results showed that the visible-light irradiation (λ > 420 nm) from this mercury lamp only caused ca. 10% degradation of CDs, indicating that UV irradiation dominated the photodegradation of CDs.16 The reaction temperature was controlled at 25 ± 0.2 °C with a circulating water-cooling system, and the light intensity at 365 nm in the quartz tubes was measured as 5.7 × 10−5 Einstein per cm2 s−1. O3 treatment was performed on a 3S-A15 O3 generator (Quanju Technology, China) using pure oxygen (99.995%) as a precursor at a flow rate of 1.0 L min−1. Time-dependent O3 concentrations were determined as ca. 22.8 μmol L−1 using indigo as the reference compound.42,43 Chlorine treatment was performed by adding sodium hypochlorite (NaClO). Available chlorine in the solutions was determined using the spectrophotometric method and a residual chlorine meter. At the sampling time, 3 mL aliquots of the reaction solutions were rapidly transferred into a 4 mL cuvette, and the concentration of the residual CDs was determined at 332 nm using a spectrophotometer (Mapada 3200 UV-vis, Shanghai, China). Fig. S3† shows that the UV absorbance at λ332 varies slightly at high ionic strength and in natural waters used in this study, demonstrating that the use of absorbance at λ332 to estimate CDs concentration change during degradation is acceptable.152.3. Characterization
The structures and properties of CDs before and after treatment via the three disinfection processes were characterized by various instruments. The absorbance change of CDs was monitored using a UV-vis spectrophotometer (Mapada 3200 UV-vis, Shanghai, China). The fluorescence excitation–emission matrix (EEM) spectra of CDs were measured on a spectrofluorometer (F97 Pro, Shanghai Lengguang Technology, China). The EEM data were recorded at excitation wavelengths from 230 to 500 nm and emission wavelengths from 300 to 600 nm at regular steps of 10 nm. The residual total organic carbon (TOC) in reaction solutions at a predetermined sampling time was examined using a TOC analyzer (Shimadzu, TOC-L CPH, Japan). Fourier-transform infrared spectroscopy (FT-IR) spectra were acquired on a Bruker Vector-22 spectrometer (Bruker, Germany). Raman spectra were collected on a Renishaw inVia Raman microscope using an excitation wavelength of 514 nm. X-ray photoelectron spectroscopy (XPS) analyses were carried out on a Thermo Scientific K-Alpha X-ray photoelectron spectrometer, using non-monochromatized Al Kα X-rays as the excitation source and choosing C1s (284.8 eV) as the reference line. Transmission electron microscopy (TEM) images were collected on an FEI Talos F200S instrument (FEI Talos F200S, 200 kV).2.4. Product identification
In order to characterize the degradation products of CDs, 100 mL of reaction solutions were prepared. After 1 h exposure to UV, O3, and chlorine, the reaction sample was concentrated with a Waters Oasis HLB cartridge (6 mL, 500 mg) using a solid-phase extraction (SPE) process following our previous study.44 A previous study has demonstrated that SPE as an effective method can be used to separate and enrich degradation intermediates of CDs.18 In brief, the cartridge was initially activated with 5 mL methanol and 5 mL DI water. The reaction sample was then passed through the cartridge at a flow rate of 5.0 mL min−1. The extracts were eluted with 5 mL methanol, and the eluents were subsequently evaporated to 1.0 mL using a gentle nitrogen flow for ultra-performance liquid chromatography-tandem mass spectrometry (LC-MS) analysis. LC-MS analysis was performed on an HR/AM LC-MS (Thermo Scientific Q-Exactive) with an orbitrap resolution of 140000 FWHM (at an m/z of 200). Separation was carried out on a Hypersil GOLD C18 column (100 × 2.1 mm, 1.9 μm). DI water (with 0.1% formic acid) and methanol (v/v = 30/70) were used as the mobile phase for elution at a flow rate of 0.3 mL min−1. The injection volume was set as 5 μL and the column temperature was set at 35 °C. The mass spectrometer was operated in positive ionization mode. The temperatures of the capillary and aux. gas heaters were set to 320 °C and 350 °C, respectively.
3. Results and discussion
3.1. Characterization of CDs
The prepared CDs in the current study were characterized by various techniques, and their properties were consistent with those of typically used CDs.19,40 Briefly, the photograph in Fig. S1a† shows that the prepared CDs are green colored and show brighter blue emission under 365 nm UV lamp irradiation due to the introduction of amino groups (Fig. S1b†). The TEM image shows the spherical morphology (Fig. S1c†) of CDs, uniformly dispersed with an average size of ca. 3.25 nm (Fig. S1d†). High-resolution TEM (HR-TEM) displays the crystalline structure of CDs with a lattice spacing of 0.24 nm, corresponding well with the (100) lattice planes of graphite, indicating the crystalline graphitic carbon structure of the CDs.40 The XPS spectra in Fig. S1e† indicate that the CDs contain the C, N, and O elements. FTIR analysis shows that several functional groups including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1660 cm−1), C–NH–C (1108 cm−1), C–O/C–N (1400 cm−1), C–O–C (1055 cm−1), and –OH/N–H (3430 cm−1) are present on the surface of the CDs (Fig. S1f†).27,29,45,46 The UV-vis spectra (Fig. S1g†) display that the CDs have two intrinsic peaks centered at 240 nm and 332 nm, which can be respectively attributed to the π–π* transition of aromatic sp2 conjugated domains and the n–π* transitions of the –CONH– bond in aromatic structures.40,47 In addition, the absorbance at 332 nm for CDs increased linearly when the CDs concentrations were increased from 0 to 20 mg L−1 (Fig. S1h†). Therefore, the absorbance change at 332 nm was applied to quantify the degradation of CDs in this study.
O (1660 cm−1), C–NH–C (1108 cm−1), C–O/C–N (1400 cm−1), C–O–C (1055 cm−1), and –OH/N–H (3430 cm−1) are present on the surface of the CDs (Fig. S1f†).27,29,45,46 The UV-vis spectra (Fig. S1g†) display that the CDs have two intrinsic peaks centered at 240 nm and 332 nm, which can be respectively attributed to the π–π* transition of aromatic sp2 conjugated domains and the n–π* transitions of the –CONH– bond in aromatic structures.40,47 In addition, the absorbance at 332 nm for CDs increased linearly when the CDs concentrations were increased from 0 to 20 mg L−1 (Fig. S1h†). Therefore, the absorbance change at 332 nm was applied to quantify the degradation of CDs in this study.
3.2. Transformation of CDs with UV, O3, and chlorine
As shown in Fig. 1, three disinfection processes including UV irradiation, ozonation, and chlorination were able to degrade CDs efficiently and caused the absorbance at 332 nm of CDs to gradually decrease. Both UV irradiation and ozonation caused ca. 90% of 10 mg L−1 CDs to be gradually degraded within 30 min treatment (Fig. 1a and b), while only 10% of CDs were transformed under the treatment with 1 mg L−1 Cl2 after 30 min (Fig. 1c). The degradation kinetics of CDs were further quantified by examining the absorbance change at 332 nm, and were fitted by the pseudo-first-order kinetics C = C0−kobs×t (R2 > 0.95, Fig. S4†). The obtained degradation rate constants kobs for CDs are presented in Fig. 1d. Apparently, kobs(UV) was similar to kobs(O3), but both were greatly higher than kobs(NaClO). The close kobs values for CDs implied that the transformation behaviors of CDs might be similar when treated with UV radiation and ozonation because both processes release the same radicals, e.g., hydroxyl radicals (HO˙) (Scheme S1†). However, kobs(NaClO) for CDs is much lower indicating the lower reactivity between CDs and chlorine-related active species. In addition, it can be observed that the degradation rate of CDs in chlorination became very slow after 2 minutes of treatment, which might be because of the higher reactivity of degradation intermediates towards chlorine-related active species than that of the CDs. Nevertheless, complete degradation of CDs can be achieved with the continual addition of NaClO, as indicated by the yellow line in Fig. 1c. | ||
| Fig. 1 UV-vis spectrum change of CDs after treatment by different disinfection processes: (a) UV irradiation, (b) ozonation, and (c) chlorination, and (d) the pseudo-first-order kinetic rate constants of CDs degradation for the three disinfection processes. All reactions were performed at a pH of 7.0 ± 0.2 at 25 ± 0.2 °C. Data points with the same letter have no significant difference from each other, p ≥ 0.05. | ||
3.3. Effects of the water matrix
The influence of solution pH ranging from 5.0 to 9.0, which represents the typical pH for most natural waters,48 on the transformation of CDs in the three disinfection processes was examined. As shown in Fig. 2a, the effect of pH (5.0–9.0) on CDs degradation in the UV irradiation and ozonation processes was indistinct, and it was slightly increased under alkaline conditions (pH 9.0). As shown in Scheme S1,† under UV illumination or O3 exposure, a basic solution containing abundant OH− would produce more HO˙,49 and hence facilitate the oxidation of CDs. However, higher kobs values were observed under both neutral and basic conditions (pH 7.0 and 9.0) in the chlorination process. It has been demonstrated that phenol groups are deprotonated to phenoxide at high pH, and the phenoxide ion has been shown to react faster with chlorine than unionized phenol.50 Similarly, the phenol groups on CDs were easier to be substituted by chlorine in neutral and alkaline media.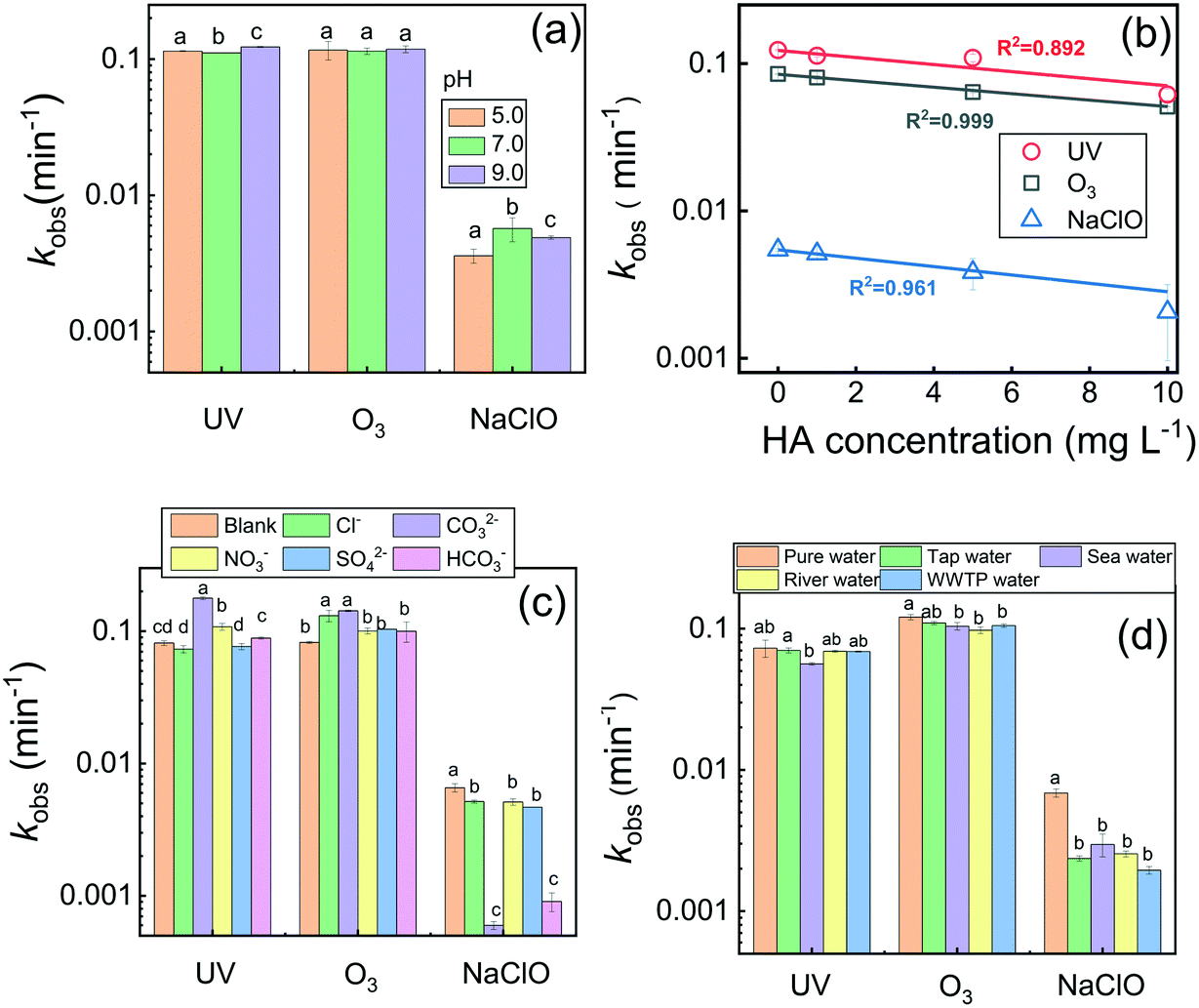 | ||
| Fig. 2 Influence of (a) solution pH, (b) humic acid (HA) concentration, (c) anions at 10 mM, and (d) different water matrices on the pseudo-first-order transformation rate constant (kobs) of CDs in the three disinfection processes. Data points with the same letter have no significant difference from each other, p ≥ 0.05. | ||
Natural water bodies are rich in natural organic matter (NOM), which is able to react with a variety of reactive oxygen species (ROS) and hence interfere with water treatment processes including disinfection. In this study, humic acid (HA, 0–10 mg L−1) was used as a representative of aquatic NOM to examine its influence on CDs degradation, and the results are presented in Fig. 2b. It should be noted that HA as background has been deducted before the measurement of CDs concentration by UV-vis spectroscopy. Obviously, kobs linearly decreased as the HA concentration increased from 0 to 10 mg L−1 in all three disinfection processes. The results indicated that the presence of HA inhibited the degradation of CDs, and the inhibition degree was positively correlated with the HA concentration. It is well-known that NOM is capable of quenching radicals and reversing intermediate radicals to parent compounds in radical-based oxidation reactions.51 Moreover, the carboxyl and hydroxyl functional groups on HA enable reactions with ROS including HO˙ (ref. 52) and ClO−.53 Therefore, such consumption of oxidants reduced the oxidation strength on CDs. In addition, the inner filter effect of HA also reduced the production of HO˙ (ref. 33) and hence decreased CDs degradation. Yuan et al.38 also found that the presence of HA as a photo-initiator promoted the UV transformation of silver nanoparticles but caused an inhibition effect in chlorination as a radical scavenger, indicating the complex role of NOM in the oxidation of different nanoparticles. The potential effects of inorganic anions including Cl−, CO32−, NO3−, SO42−, and HCO3− at 10 mM on the transformation of CDs in the three disinfection processes were also investigated. As shown in Fig. 2c, these anions showed distinctive influences on CDs degradation in the different treatments. In detail, CO32−, HCO3−, and NO3− significantly promoted the transformation of CDs under UV irradiation, while Cl− and SO42− showed a slight inhibition effect. However, all examined inorganic anions showed promotion effects on the degradation of CDs by ozonation treatment. As reported, the primary HO˙ would be partially transformed into secondary anionic radicals (e.g., CO3˙−, NO2˙, and SO4˙−) through the processes described in Scheme S2 (eqn (8)–(12)) in the ESI.†33,34 Therefore, the radical-scavenging effect was the main reason for the inhibition by these anions.44 Although some secondary anionic radicals have a lower reduction potential (e.g., CO3˙−, E0 = 1.78 V) than primary radicals (e.g., HO˙, E0 = 1.9–2.7 V), they are more stable and selective than primary radicals. For example, it has been reported that CO3˙− reacts rapidly with compounds containing electron-rich groups, such as nitrogen-containing substrates and aromatic compounds.33 The characterization shown in Fig. S1† indicated the crystalline graphitic carbon structure of the used CDs in this study, which have many oxygen- and nitrogen-functional groups. Therefore, the aromatic skeleton and electron-rich groups enabled the CDs to likely react with secondary anionic radicals and hence enhanced the degradation of CDs upon UV irradiation and ozonation.
In contrast, all anions inhibited CDs degradation in the chlorination process, especially CO32− and HCO3−. As shown in Scheme S2† (eqn (15)–(17)), CO32− and HCO3− can react with Cl˙ and ClO˙ radicals to form the secondary radical CO3˙−.54 Therefore, the radical-scavenging effect of CO32− and HCO3− caused the strong inhibition of CDs degradation.44 However, the generated CO3˙− as a selective oxidant might have a lower reactivity towards CDs than the primary Cl˙ and ClO˙ radicals.55 In addition, the addition of CO32− and HCO3− at 10 mM would elevate the pH value of the reaction solutions, thereby lowering the CDs transformation rates upon chlorination (Fig. 2a).
Furthermore, we also examined the degradation efficiency of CDs in different natural water matrices. The solution pH was adjusted to neutral to compare the effects of water constituents. Fig. 2d shows the degradation kinetic rates of CDs via the three disinfection processes in different water matrices. Generally, the CDs degradation efficiency was reduced in all natural water samples compared to that in DI water. Except in the chlorination treatment, the inhibition of natural water samples is negligible in the UV and O3 treatments. The constituents including anions and NOM (Table S1†) might account for the low degradation efficiency of CDs in natural water samples.
3.4. Characterization of CDs after treatments
Fig. S5† shows that the visual change of color and fluorescence intensity of CDs before and after being treated by the three disinfection processes. The color of the pristine CDs is green at 20 mg L−1, and then it turned brown after 30 min treatment by UV irradiation, ozonation, and chlorination. Moreover, fluorescence testing indicated that the bright blue emission of CDs became weak after treatment, which might be due to a reduction in the functional groups on the surface of CDs. The UV-vis spectrometry analysis (Fig. S6†) also showed that the two major peaks at 240 nm and 332 nm disappeared after UV irradiation and ozonation, indicating that the aromatic conjugated structure of CDs has been broken. The peak at 332 nm disappeared after the chlorination, whereas the peak at 240 nm was blue-shifted, which may be due to the decrease in the aromaticity and smaller size of the transformed CDs.32,56The morphology change of CDs after treatment by the three disinfection processes was observed by HR-TEM, as shown in Fig. S7.† The pristine CDs showed well-defined and homogeneous crystal lattices with a graphitic interplanar spacing of 0.24 nm (Fig. S7a†).17 However, we found that particles in the reactive groups present on the TEM grids are rare and hard to be observed compared to those in the control samples. There is a partial loss of the lattice structure for most of the treated CDs by all three disinfection processes, as indicated by the blue arrows in Fig. S7b and c,† while the remaining graphitic parts of the CDs still exhibited well-defined crystal lattices, as indicated by the yellow arrows in Fig. S7b and c.† This result confirmed the degradation of CDs, which resulted in graphitic structure damage as well as fragmentation into smaller pieces. From HR-TEM investigations, Martín et al.17 also observed that the lattice structure of graphene quantum dots was destroyed by human peroxidases in the presence of H2O2.
The chemical changes of CDs due to UV irradiation, ozonation, and chlorination were further examined by FT-IR, XPS, and fluorescence excitation–emission matrix (EEM) analysis, as shown in Fig. 3. The FT-IR spectra in Fig. 3a revealed the change in functional groups on the CDs via the three disinfection treatments. For example, the –CO (1660 cm−1), –NH (3129 cm−1), and –C–O/–C–N (1400 cm−1) peaks decreased significantly for UV irradiated-CDs, while a substantial increase of the –NH peak (3129 cm−1) was observed for ozonized-CDs. Moreover, the appearance of –C–Cl stretching (622 cm−1) in chlorinated-CDs was observed, confirming the reactions between CDs and ClO−. Previous studies also identified the formation of C–Cl bonds in the chlorination of C60 and graphene oxide.28,57
 | ||
| Fig. 3 Characterization of CDs before and after treatments by the three disinfection processes: (a) Fourier transform infrared (FT-IR) spectra, (b) XPS survey spectra, and (c–f) C1s, O1s, N1s, and Cl2p peak fitting spectra of CD samples, and (g) fluorescence excitation–emission matrix (EEM) spectra of CDs before and after treatment by the three processes. It should be noted that the pristine CDs were diluted 3 times. | ||
The chemical changes in CDs caused by UV irradiation, ozonation, and chlorination were also examined using XPS. The survey XPS spectra in Fig. 3b and peak fitting for C (Fig. 3c), O (Fig. 3d), N (Fig. 3e), and Cl (Fig. 3f) revealed the elemental change on CDs after all three treatments. The total N and O on CDs decreased from 10.49% to 9.22% and from 27.33% to 25.33%, respectively, after UV irradiation (Table 1), due to the loss of pyridine N-oxide (401.8 eV) and carbonyl content (CO, 288.1 eV) through photoreduction.19,21,28 This result was consistent with the oxygen- and nitrogen-functional group reduction in the FT-IR spectra (Fig. 3a). In contrast, the total O on CDs obviously increased to 12.21% after ozonation, which was ascribed to the introduction of carbonyl content (CO, 288.1 eV) by O3 oxidation.28 The chlorination treatment introduced 4.78% Cl to CDs, as shown in Fig. 3b and Table 1. The Cl2p peak can be fitted into two peaks, anionic Cl (198.2 eV) and C–Cl covalent bonds (200.2 eV), which are indicative of Cl covalently bonded to carbon on CDs.28,57,58 Moreover, the O1s peak fitting in Fig. 3d also indicated the generation of O–Cl bonds (535.7 eV), confirming again the introduction of Cl onto CDs through chlorination. Similarly, the introduction of Cl was also observed in the chlorination or photochlorination of C60,28,57 graphene oxide,29,31,59 and carbon nanotubes.30 The Raman spectra in Fig. S8† also revealed that the D (1353 cm−1) and G (1587 cm−1) bands of CDs became less intense after all three treatments, demonstrating again the loss of the CDs structure due to degradation.17
| Treatment | C1s peak fitting (%) | Total C (%) | Total O (%) | Total N (%) | Total Cl (%) | ||
|---|---|---|---|---|---|---|---|
| C–C/CC |
C–OH/C–O–C | CO |
|||||
| Pristine | 57.47 | 17.22 | 25.31 | 62.18 | 27.33 | 10.49 | — |
| UV | 61.42 | 17.64 | 20.84 | 65.45 | 25.33 | 9.22 | — |
| O3 | 53.46 | 12.40 | 34.14 | 56.88 | 30.90 | 12.21 | — |
| NaClO | 56.73 | 13.41 | 29.86 | 60.97 | 23.96 | 10.29 | 4.78 |
Furthermore, fluorescence excitation–emission matrix (EEM) analysis was applied to decompose the spectral feature differences between pristine and treated CDs. As shown in Fig. 3g, the strongest fluorescence emission band of pristine CDs located at 434 nm is observed by excitation at 350 nm, which is related to –CO and –C–NH surface groups.60 However, changes in the intensity and position of the emission maxima are observed after treatment. In detail, the maximum emission intensity of CDs was reduced by 70–85% after treatment due to the loss of aromatic structures. The three disinfection treatments caused the emission maxima to blue-shift. For example, as shown in Fig. S9,† when excited by 350 nm light which is attributed to the main n → π* absorption, the emission maxima (peak A) of pristine CDs blue-shifted from 434 nm to 430, 425, and 420 nm with UV irradiation, ozonation, and chlorination, respectively. As suggested, ozonation and chlorination of humic substances decreased the aromaticity and caused the breakdown of macromolecules into smaller fragments, and hence caused blue-shifted emissions.56,61 In addition, the O and Cl relevant groups on reacted CDs with strong electronegativity are electron-donating and hence contribute to the blue-shifted emissions. The emission peak of reacted CDs became wider than that of pristine CDs might because the particle size distribution became uniformly and chemical structure alteration after all three treatments.62 It should be noted that a new emission band (peak B), peaked at around 460 nm upon excitation at 400 nm, appeared after chlorination of CDs, which might be due to Cl functionalization.
3.5. Degradation intermediates and mechanisms
The possible degradation intermediates of CDs after treatment by the three disinfection processes were separated and identified by LC-MS. The representative HPLC chromatograms for the CDs degradation products via the three treatments showed a series of new peaks compared to that of unreacted CDs (Fig. S10†). The intensity of the total ion chromatograph of CDs before and after treatment is shown in Fig. S11.† The molecular weights of the pristine CDs ranged from m/z 100 to 500, corresponding to the typical CDs obtained through a hydrothermal method from citric acid and urea.19 However, the intensity of the total ion chromatograph decreased after the reaction along with some new peaks appearing, indicating the decomposition of CDs into lower molecular weight products. The products of CDs in UV irradiation and ozonation have similar molecular weights, which may be due to the same reactive radicals generated by both processes, such as HO˙ and O2˙− (Scheme S1†). According to the MS/MS spectra of the detected intermediates, the possible molecular formulas and structures of these intermediates were deduced, as shown in Fig. S12.† In addition, many chlorine substitution/addition by-products were identified after the chlorination treatment of CDs (Fig. S13†). Generally, these degradation products were characterized with an unsaturated benzene skeleton, as well as carbonyl and hydroxyl groups. The disinfection treatment also decreased the solution pH of CDs, indicating the generation of small molecular acid intermediates in reaction solutions.32 Total organic carbon (TOC) analysis revealed the TOC reduction in CDs suspensions after all three treatments, indicating that CDs were partially mineralized into CO2 during the reaction (Fig. S14†). Based on the above results and existing studies in the literature,19,29,45 the transformation mechanisms of CDs are proposed in Fig. 4. | ||
| Fig. 4 Proposed transformation mechanisms of CDs by UV irradiation, ozonation, and chlorination treatments. | ||
Radical scavenging experiments were conducted to verify the roles of reactive species during the CDs degradation.63,64 As shown in Fig. S15 and Table S2,† ˙OH, O2˙−, h+, and 1O2 all contributed to the degradation of CDs by UV irradiation, and O2˙− played a vital role for the photo-transformation of CDs, which is similar to a previous study.65 O2˙−, and 1O2 dominated the degradation of CDs in ozonation, while the contribution of ˙OH is negligible.66 Moreover, the addition of Na2S2O3 significantly inhibited the CDs degradation in chlorination, and the inhibition was increased as the Na2S2O3 concentration increased, indicating that chlorine-active species (Cl˙ and ClO˙) caused the chlorination of CDs.67,68
As presented in Scheme S1,† the reactive species (RS) including ˙O2−, ˙OH, HClO, ClO−, and Cl˙ accounted for CDs degradation. These RS will attack electron-rich groups like –COOH, –OH, and CC on CDs,21,22,29 resulting in the decomposition of CDs into macromolecular compounds firstly. Then, these macromolecular compounds will undergo ring-opening by the further attack of RS to generate micromolecular compounds and finally mineralize into CO2 and H2O. With the attack by chlorine-related radicals (e.g., HClO, ClO−, and Cl˙), C–Cl/C–ClO bonds were introduced on the reactive sites of the CDs fragments which result in the generation of chlorine substitution/addition by-products.
4. Conclusions
This study investigated the transformations of CDs in typical disinfection processes including UV irradiation, ozonation, and chlorination. The results indicated that CDs can be rapidly degraded by UV irradiation, ozonation, and chlorination treatment, indicating that oxygen and chlorine based radicals are mainly responsible for CDs degradation. High degradation of CDs can be achieved at a pH of 5.0–9.0 in all three processes. As a representative natural organic matter, HA inhibited CDs oxidation with an inverse relationship with HA concentration. Both CO32− and NO3− significantly promoted the degradation of CDs in UV irradiation, while CO32− and Cl− played positive roles in CDs oxidation in ozonation. However, both CO32− and HCO3− greatly inhibited CDs oxidation in chlorination treatment. Nevertheless, high removal efficiencies of CDs by the three disinfection processes were observed in natural water samples. The characterization results indicated that the physicochemical properties of CDs were changed differently after treatment by the three disinfection processes including damage to crystal lattices, the elimination of O/N-functional groups, and the introduction of the Cl element. Product identification showed that the CDs degradation products were similar in all three oxidation processes. CDs were firstly degraded into small pieces and gradually cleaved into micromolecular compounds which contain unsaturated benzene, as well as carbonyl and hydroxyl groups. In particular, many chlorine substitution/addition by-products were identified after the chlorination treatment of CDs. Therefore, although this study indicated the reaction activity of CDs in typical disinfection processes, future studies are needed to determine whether the water disinfection treatment poses any ecological risks to the environment and human health.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21707019) and the Natural Science Foundation of Guangdong Province (2021A1515010019).References
- S. Y. Lim, W. Shen and Z. Q. Gao, Carbon quantum dots and their applications, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- J. B. Essner and G. A. Baker, The emerging roles of carbon dots in solar photovoltaics: A critical review, Environ. Sci.: Nano, 2017, 4, 1216–1263 RSC.
- X. L. Huang, F. Zhang, L. Zhu, K. Y. Choi, N. Guo, J. X. Guo, K. Tackett, P. Anilkumar, G. Liu, Q. M. Quan, H. S. Choi, G. Niu, Y. P. Sun, S. Lee and X. Y. Chen, Effect of injection routes on the biodistribution, clearance, and tumor uptake of carbon dots, ACS Nano, 2013, 7, 5684–5693 CrossRef CAS.
- G. A. Posthuma-Trumpie, J. H. Wichers, M. Koets, L. B. J. M. Berendsen and A. van Amerongen, Amorphous carbon nanoparticles: a versatile label for rapid diagnostic (immuno)assays, Anal. Bioanal. Chem., 2012, 402, 593–600 CrossRef CAS PubMed.
- S. J. Zhu, Q. N. Meng, L. Wang, J. H. Zhang, Y. B. Song, H. Jin, K. Zhang, H. C. Sun, H. Y. Wang and B. Yang, Highly photoluminescent carbon dots for multicolor patterning, sensors, and bioimaging, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, Recent progress in carbon quantum dots: synthesis, properties and applications in photocatalysis, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- X. M. Li, M. C. Rui, J. Z. Song, Z. H. Shen and H. B. Zeng, Carbon and graphene quantum dots for optoelectronic and energy devices: A review, Adv. Funct. Mater., 2015, 25, 4929–4947 CrossRef CAS.
- X. Liu, J. X. Li, Y. S. Huang, X. X. Wang, X. D. Zhang and X. K. Wang, Adsorption, aggregation, and deposition behaviors of carbon dots on minerals, Environ. Sci. Technol., 2017, 51, 6156–6164 CrossRef CAS.
- C. L. Li, C. M. Ou, C. Huang, W. C. Wu, Y. P. Chen, T. E. Lin, L. C. Ho, C. W. Wang, C. Shih and H. C. Zhou, Carbon dots prepared from ginger exhibiting efficient inhibition of human hepatocellular carcinoma cells, J. Mater. Chem. B, 2014, 2, 4564 RSC.
- T. Ku, F. Hao, X. Yang, Z. Rao and G. Jiang, Graphene quantum dots disrupt embryonic stem cell differentiation by interfering with the methylation level of Sox2, Environ. Sci. Technol., 2021, 55, 3144–3155 CrossRef CAS.
- Y. Wang, P. Anilkumar, L. Cao, J. H. Liu, P. G. Luo, K. N. Tackett, S. Sahu, P. Wang, X. Wang and Y. P. Sun, Carbon dots of different composition and surface functionalization: cytotoxicity issues relevant to fluorescence cell imaging, Exp. Biol. Med., 2011, 236, 1231 CrossRef CAS.
- K. Yao, X. Lv, G. Zheng, Z. Chen, Y. Jiang, X. Zhu, Z. Wang and Z. Cai, Effects of Carbon Quantum Dots on Aquatic Environments: Comparison of Toxicity to Organisms at Different Trophic Levels, Environ. Sci. Technol., 2018, 52, 14445–14451 CrossRef CAS.
- L. Hao, J. Huang, Y. Song, M. Zhang, H. Wang, F. Lu, H. Huang, L. Yang, X. Dai and Z. Gu, Degradable Carbon Dots with Broad-Spectrum Antibacterial Activity, ACS Appl. Mater. Interfaces, 2018, 10, 26936–26946 CrossRef PubMed.
- S. Kamrani, M. Rezaei, M. Kord and M. Baalousha, Co-transport and remobilization of Cu and Pb in quartz column by carbon dots, Sci. Total Environ., 2018, 626, 995–1004 CrossRef CAS PubMed.
- S. Kamrani, M. Rezaei, M. Kord and M. Baalousha, Transport and retention of carbon dots (CDs) in saturated and unsaturated porous media: Role of ionic strength, pH, and collector grain size, Water Res., 2018, 133, 338–347 CrossRef CAS.
- B. P. Frank, L. R. Sigmon, A. R. Deline, R. S. Lankone, M. J. Gallagher, B. Zhi, C. L. Haynes and D. H. Fairbrother, Photochemical transformations of carbon dots in aqueous environments, Environ. Sci. Technol., 2020, 54, 4160–4170 CrossRef CAS.
- C. Martín, G. Jun, R. Schurhammer, G. Reina, P. Chen, A. Bianco and C. Menard-Moyon, Enzymatic degradation of graphene quantum dots by human peroxidases, Small, 2019, 15(52), 1905405 CrossRef.
- Y. Y. Liu, N. Y. Yu, W. D. Fang, Q. G. Tan, R. Ji, L. Y. Yang, S. Wei, X. W. Zhang and A. J. Miao, Photodegradation of carbon dots cause cytotoxicity, Nat. Commun., 2021, 12, 1–12 CrossRef.
- X. R. Chen, G. D. Fang, C. Liu, D. D. Dionysiou, X. L. Wang, C. Y. Zhu, Y. J. Wang, J. Gao and D. M. Zhou, Cotransformation of carbon dots and contaminant under light in aqueous solutions: A mechanistic study, Environ. Sci. Technol., 2019, 53, 6235–6244 CrossRef CAS PubMed.
- B. L. Allen, G. P. Kotchey, Y. Chen, N. Yanamala, J. Klein-Seetharaman, V. E. Kagan and A. Star, Mechanistic investigations of horseradish peroxidase-catalyzed degradation of single-walled carbon nanotubes, J. Am. Chem. Soc., 2009, 131, 17194 CrossRef CAS PubMed.
- W. C. Hou, I. Chowdhury, D. G. Goodwin, W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Photochemical transformation of graphene oxide in sunlight, Environ. Sci. Technol., 2015, 49, 3435–3443 CrossRef CAS.
- X. L. Qu, P. J. J. Alvarez and Q. L. Li, Photochemical transformation of carboxylated multiwalled carbon nanotubes: Role of reactive oxygen species, Environ. Sci. Technol., 2013, 47, 14080–14088 CrossRef CAS PubMed.
- U. Abd Rani, L. Y. Ng, C. Y. Ng and E. Mahmoudi, A review of carbon quantum dots and their applications in wastewater treatment, Adv. Colloid Interface Sci., 2020, 102124 CrossRef CAS PubMed.
- G. A. Boorman, V. Dellarco, J. K. Dunnick, R. E. Chapin, S. Hunter, F. Hauchman, H. Gardner, M. Cox and R. C. Sills, Drinking water disinfection byproducts: Review and approach to toxicity evaluation, Environ. Health Perspect., 1999, 107, 207–217 CAS.
- S. Pérez, M. Farré and D. Barceló, Analysis, behavior and ecotoxicity of carbon-based nanomaterials in the aquatic environment, TrAC, Trends Anal. Chem., 2009, 28, 820–832 CrossRef.
- H. He, P. Zhou, K. K. Shimabuku, X. Fang, S. Li, Y. Lee and M. C. Dodd, Degradation and deactivation of bacterial antibiotic resistance genes during exposure to free chlorine, monochloramine, chlorine dioxide, ozone, ultraviolet light, and hydroxyl radical, Environ. Sci. Technol., 2019, 53, 2013–2026 CrossRef CAS PubMed.
- T. Du, A. S. Adeleye, T. Zhang, N. Yang, R. Hao, Y. Li, W. Song and W. Chen, Effects of ozone and produced hydroxyl radicals on the transformation of graphene oxide in aqueous media, Environ. Sci.: Nano, 2019, 6, 2484–2494 RSC.
- Q. R. Zhang, M. L. Wang, C. H. Gu and C. D. Zhang, Water disinfection processes change the cytotoxicity of C60 fullerene: Reactions at the nano-bio interface, Water Res., 2019, 163, 114867 CrossRef CAS.
- Y. Li, N. Yang, T. T. Du, X. Z. Wang and W. Chen, Transformation of graphene oxide by chlorination and chloramination: Implications for environmental transport and fate, Water Res., 2016, 103, 416–423 CrossRef CAS.
- E. M. Verdugo, K. J. Nelson, C. M. Bako, R. L. Valentine and D. M. Cwiertny, Formation of trihalomethanes and haloacetic acids during chlorination of functionalized carbon nanotubes, Environ. Sci.: Nano, 2016, 3, 1327–1339 RSC.
- T. T. Du, A. S. Adeleye, A. A. Keller, Z. N. Wu, W. Han, Y. Y. Wang, C. D. Zhang and Y. Li, Photochlorination-induced transformation of graphene oxide: Mechanism and environmental fate, Water Res., 2017, 124, 372–380 CrossRef CAS PubMed.
- Y. Feng, M. Shen, Z. Xie, P. Chen, L.-Z. Zuo, K. Yao, W. Lv and G. Liu, Photochemical transformation of C3N4 under UV irradiation: Implications for environmental fate and photocatalytic activity, J. Hazard. Mater., 2020, 122557 CrossRef CAS.
- Y. Zhou, C. Chen, K. Guo, Z. Wu, L. Wang, Z. Hua and J. Fang, Kinetics and pathways of the degradation of PPCPs by carbonate radicals in advanced oxidation processes, Water Res., 2020, 185, 116231 CrossRef CAS.
- O. S. Keen, N. G. Loue and K. G. Linden, The role of effluent nitrate in trace organic chemical oxidation during UV disinfection, Water Res., 2012, 46, 5224–5234 CrossRef CAS PubMed.
- M. Zhang, X. Wang, H. Z. Hao, H. H. Wang, L. Duan and Y. Li, Formation of disinfection byproducts as affected by biochar during water treatment, Chemosphere, 2019, 233, 190–197 CrossRef CAS.
- Z. H. Yuan, Y. B. Chen, T. T. Li and C. P. Yu, Reaction of silver nanoparticles in the disinfection process, Chemosphere, 2013, 93, 619–625 CrossRef CAS.
- I. Chowdhury, W. C. Hou, D. Goodwin, M. Henderson, R. G. Zepp and D. Bouchard, Sunlight affects aggregation and deposition of graphene oxide in the aquatic environment, Water Res., 2015, 78, 37–46 CrossRef CAS PubMed.
- Z. Yuan, Y. Chen, T. Li and C. P. Yu, Reaction of silver nanoparticles in the disinfection process, Chemosphere, 2013, 93, 619–625 CrossRef CAS PubMed.
- Z. Xie, Y. Feng, F. Wang, D. Chen, Q. Zhang, Y. Zeng, W. Lv and G. Liu, Construction of carbon dots modified MoO3/g-C3N4 Z-scheme photocatalyst with enhanced visible-light photocatalytic activity for the degradation of tetracycline, Appl. Catal., B, 2018, 229, 96–104 CrossRef CAS.
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao and W. Lv, Facile synthesis of N-doped carbon dots/g-C3N4 photocatalyst with enhanced visible-light photocatalytic activity for the degradation of indomethacin, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS.
- R. E. Palma-Goyes, J. Silva-Agredo, I. González and R. A. Torres-Palma, Comparative degradation of indigo carmine by electrochemical oxidation and advanced oxidation processes, Electrochim. Acta, 2014, 140, 427–433 CrossRef CAS.
- H. Bader and J. Hoigné, Determination of ozone in water by the indigo method, Water Res., 1981, 15, 449–456 CrossRef CAS.
- M. M. Huber, S. Canonica, G. Y. Park and U. Von Gunten, Oxidation of pharmaceuticals during ozonation and advanced oxidation processes, Environ. Sci. Technol., 2003, 37, 1016–1024 CrossRef CAS PubMed.
- Y. P. Feng, Q. Y. Song, W. Y. Lv and G. G. Liu, Degradation of ketoprofen by sulfate radical-based advanced oxidation processes: Kinetics, mechanisms, and effects of natural water matrices, Chemosphere, 2017, 189, 643–651 CrossRef CAS PubMed.
- Y. P. Feng, K. Lu, L. Mao, X. K. Guo, S. X. Gao and E. J. Petersen, Degradation of C-14-labeled few layer graphene via Fenton reaction: Reaction rates, characterization of reaction products, and potential ecological effects, Water Res., 2015, 84, 49–57 CrossRef CAS PubMed.
- V. Tucureanu, A. Matei and A. M. Avram, FTIR Spectroscopy for Carbon Family Study, Crit. Rev. Anal. Chem., 2016, 46, 502–520 CrossRef CAS.
- S. Cailotto, E. Amadio, M. Facchin, M. Selva, E. Pontoglio, F. Rizzolio, P. Riello, G. Toffoli, A. Benedetti and A. Perosa, Carbon dots from sugars and ascorbic acid: Role of the precursors on morphology, properties, toxicity, and drug uptake, ACS Med. Chem. Lett., 2018, 9, 832–837 CrossRef CAS PubMed.
- Y. P. Feng, X. T. Liu, K. A. Huynh, J. M. McCaffery, L. Mao, S. X. Gao and K. L. Chen, Heteroaggregation of graphene oxide with nanometer- and micrometer-sized hematite colloids: Influence on nanohybrid aggregation and microparticle sedimentation, Environ. Sci. Technol., 2017, 51, 6821–6828 CrossRef CAS PubMed.
- L. W. Chen, X. C. Li, J. Zhang, J. Y. Fang, Y. M. Huang and P. Wang, Production of hydroxyl radical via the activation of hydrogen peroxide by hydroxylamine, Environ. Sci. Technol., 2015, 49, 10373–10379 CrossRef CAS PubMed.
- A. L. Mackie, Y. R. Park and G. A. Gagnon, Chlorination kinetics of 11-Nor-9-carboxy-Δ9-tetrahydrocannabinol: effects of pH and humic acid, Environ. Sci. Technol., 2017, 51, 10711–10717 CrossRef CAS PubMed.
- S. Canonica and H. U. Laubscher, Inhibitory effect of dissolved organic matter on triplet-induced oxidation of aquatic contaminants, Photochem. Photobiol. Sci., 2008, 7, 547–551 CrossRef CAS PubMed.
- A. Latifoglu and M. D. Gurol, The effect of humic acids on nitrobenzene oxidation by ozonation and O3/UV processes, Water Res., 2003, 37, 1879–1889 CrossRef CAS.
- Y. Gao, J. Jiang, Y. Zhou, S.-Y. Pang, J. Ma, C. Jiang, Y. Yang, Z.-s. Huang, J. Gu and Q. Guo, Chlorination of bisphenol S: kinetics, products, and effect of humic acid, Water Res., 2018, 131, 208–217 CrossRef CAS PubMed.
- J. Y. Fang, Y. Fu and C. Shang, The Roles of Reactive Species in Micropollutant Degradation in the UV/Free Chlorine System, Environ. Sci. Technol., 2014, 48, 1859–1868 CrossRef CAS PubMed.
- Z. Wu, J. Fang, Y. Xiang, C. Shang, X. Li, F. Meng and X. Yang, Roles of reactive chlorine species in trimethoprim degradation in the UV/chlorine process: Kinetics and transformation pathways, Water Res., 2016, 104, 272–282 CrossRef CAS.
- J. Swietlik and E. Sikorska, Application of fluorescence spectroscopy in the studies of natural organic matter fractions reactivity with chlorine dioxide and ozone, Water Res., 2004, 38, 3791–3799 CrossRef CAS PubMed.
- J. W. Wu, D. Benoit, S. S. Lee, W. L. Li and J. D. Fortner, Ground state reactions of nC(60) with free chlorine in water, Environ. Sci. Technol., 2016, 50, 721–731 CrossRef CAS PubMed.
- J. K. Wassei, K. C. Cha, V. C. Tung, Y. Yang and R. B. Kaner, The effects of thionyl chloride on the properties of graphene and graphene-carbon nanotube composites, J. Mater. Chem., 2011, 21, 3391–3396 RSC.
- S. Y. An, J. W. Wu, Y. Nie, W. L. Li and J. D. Fortner, Free chlorine induced phototransformation of graphene oxide in water: Reaction kinetics and product characterization, Chem. Eng. J., 2020, 381, 122609 CrossRef CAS.
- K. Suzuki, L. Malfatti, M. Takahashi, D. Carboni, F. Messina, Y. Tokudome, M. Takemoto and P. Innocenzi, Design of carbon dots photoluminescence through organo-functional silane grafting for solid-state emitting devices, Sci. Rep., 2017, 7, 5469 CrossRef PubMed.
- G. V. Korshin, M. U. Kumke, C. W. Li and F. H. Frimmel, Influence of chlorination on chromophores and fluorophores in humic substances, Environ. Sci. Technol., 1999, 33, 1207–1212 CrossRef CAS.
- A. Barati, M. Shamsipur and H. Abdollahi, Carbon dots with strong excitation-dependent fluorescence changes towards pH. Application as nanosensors for a broad range of pH, Anal. Chim. Acta, 2016, 931, 25–33 CrossRef CAS PubMed.
- X. Zheng, Z. Wang, T. Chen, J. Ran and G. Liu, One-step synthesis of carbon nitride nanobelts for enhanced photocatalytic degradation of organic pollutants through peroxydisulfate activation, Environ. Sci.: Nano, 2020, 8, 245–257 RSC.
- C. Sichel, C. Garcia and K. Andre, Feasibility studies: UV/chlorine advanced oxidation treatment for the removal of emerging contaminants, Water Res., 2011, 45, 6371–6380 CrossRef CAS PubMed.
- X. Chen, G. Fang, C. Liu, D. D. Dionysiou, X. Wang, C. Zhu, Y. Wang, J. Gao and D. Zhou, Cotransformation of carbon dots and contaminant under light in aqueous solutions: A mechanistic study, Environ. Sci. Technol., 2019, 53, 6235–6244 CrossRef CAS PubMed.
- Y. Pi, J. Schumacher and M. Jekel, Decomposition of aqueous ozone in the presence of aromatic organic solutes, Water Res., 2005, 39, 83–88 CrossRef CAS PubMed.
- T. Yao, N. Yang, W. Chen and X. Wang, Transformation of graphene oxide by chlorination and chloramination: Implications for environmental transport and fate, Water Res., 2016, 103, 416–423 CrossRef PubMed.
- Q. Zhang, M. Wang, C. Gu and C. Zhang, Water disinfection processes change the cytotoxicity of C60 fullerene: Reactions at the nano-bio interface, Water Res., 2019, 163, 114867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Schemes S1 and S2, Fig. S1–S15, and Tables S1 and S2. See DOI: 10.1039/d1en00698c |
| This journal is © The Royal Society of Chemistry 2022 |